

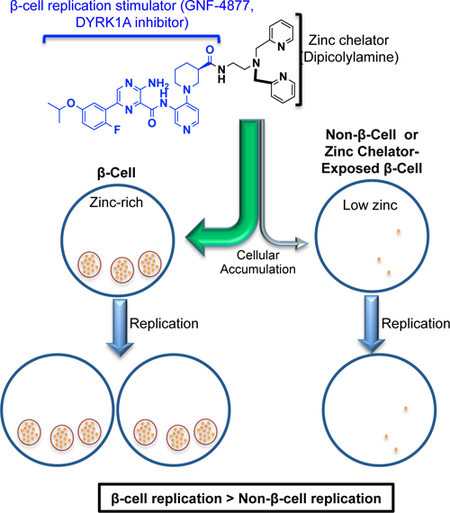
SUMMARY
Diabetes is a hyperglycemic condition characterized by pancreatic β-cell dysfunction and depletion. Whereas methods for monitoring β-cell function in vivo exist, methods to deliver therapeutics to β-cells are lacking. We leveraged the rare ability of β-cells to concentrate zinc to preferentially trap zinc-binding molecules within β-cells, resulting in β-cell-targeted compound delivery. We determined that zinc-rich β-cells and islets preferentially accumulated TSQ (6-Methoxy-8-p-Toluenesulfonamido-Quinoline) in a zinc-dependent manner compared to exocrine pancreas. Next, we asked whether appending a zinc-chelating moiety onto a β-cell replication-inducing compound was sufficient to confer preferential β-cell accumulation and activity. Indeed, the hybrid compound preferentially accumulated within rodent and human islets in a zinc-dependent manner and increased the selectivity of replication-promoting activity towards β-cells. These data resolve the fundamental question of whether intracellular accumulation of zinc-chelating compounds is influenced by zinc content. Furthermore, application of this principle yielded a proof-of-concept method for β-cell-targeted drug delivery and bioactivity.
Keywords: Diabetes, beta-cell, islet, beta-cell-replication, beta-cell regeneration, zinc, chelation, targeted compound delivery, targeting, tissue selectivity
eTOC Blurb
Lack of cell-type selectivity prevents translational use of β-cell replication-promoting compounds. Based upon the high zinc content of β-cells, Horton et al. explored functionalizing a β-cell-replicating molecule with a zinc-chelating moiety for targeted drug delivery. These studies validate a transferrable technology for β-cell-targeted drug delivery.
Graphical Abstract
INTRODUCTION
Normally, blood glucose levels are tightly regulated by the balanced activities of the pancreatic islet-derived hormones glucagon and insulin from α- and β-cells, respectively. With Diabetes mellitus, insulin production is insufficient and blood glucose levels are elevated. The adaptive increase in β-cell replication and mass that occurs with insulin-resistant states such as pregnancy and obesity has fostered an interest in therapeutically amplifying this homeostatic mechanism (Meier et al., 2008; Terauchi et al., 2007). Accordingly, substantial progress has been made toward identifying small molecule stimulators of β-cell replication including 5-iodotubericidin (Annes et al., 2012; Dirice et al., 2016), harmine (Wang et al., 2015), dipyridamole (Zhao et al., 2014), CC-401 (Abdolazimi et al., 2018), GNF-4877 (Shen et al., 2015), and others (Wang et al., 2009). However, the application of these compounds is limited by their indiscriminate replication-promoting activity. Given the central role of β-cells in maintaining glucose homeostasis, developing methods for β-cell-targeted drug delivery and imaging of β-cell mass in vivo has emerged as a research priority.
To realize the potential of β-cell-directed drug delivery and imaging, substantial effort has been made to identify β-cell-specific surface markers to leverage for this purpose (Ichise and Harris, 2010). Although a useful β-cell specific antigen has not yet emerged (Dorrell et al., 2011), a variety of surface receptors including the glucagon-like peptide 1 receptor (GLP-1R), GPR44, and the somatostatin receptor (SSTR2) (Braun, 2014) have been explored for targeting drugs to and/or imaging islets. For example, GLP-1R, a well-characterized G-protein-coupled receptor, was successfully used to selectively deliver estrogen to mouse β-cells (Finan et al., 2012). However, the application of GLP-1R-based strategies to humans may be limited by (a) the dose required for efficacy, which exceeded the maximum tolerated dose (~0.2 μg/kg (Nielsen and Baron, 2003) or 20 mcg/day; BYETTA® prescribing information (accessdata.fda.gov)) by several orders of magnitude, and (b) the insufficiently restricted expression of GLP-1R (Körner et al., 2007). Similarly, the low and insufficiently restricted expression of GPR44 and SSTR2 (up-regulated by islet tumors and useful in this context) are likely to prohibit their use for β-cell-targeted drug delivery (Bhandari et al., 2008; Eriksson et al., 2018; Hellström-Lindahl et al., 2016). Hence, currently recognized receptor-ligand based delivery strategies appear to have limited translational viability.
Strategies to achieve β-cell-targeted delivery and/or in vivo imaging have also attempted to leverage the unique biologic properties of β-cells. The potential of this strategy is heralded by the β-cell-selective toxins alloxan and streptozotocin, which enter β-cells through the partially selective Facilitated Glucose Transporter Member 2 (GLUT2) and capitalize on the β-cell’s susceptibility to reactive oxygen species and alkylating agents (Hammarström et al., 1967; Lenzen, 2008; Rakieten et al., 1963). Unfortunately, the Trojan horse use of GLUT2 has not yet proved adaptable for the delivery of other cargo. Similarly, (+)- dihydrotetrabenazine, a substrate for the Vesicular monoamine transporter 2 (VMAT2) transporter, has been explored for islet-specific imaging in vivo and drug delivery (Hao et al., 2016) However, VMAT2 expression may be insufficiently selective for β-cell-directed drug delivery (Eriksson et al., 2016). Likewise, the compound [11C]5-hydroxytryptophan, which is selectively accumulated in the endocrine pancreas via the large amino acid transporter (LAT), was used to assess β-cell mass (Per Lindström, 1981). However, rapid degradation of HTP-based probes by monoamine oxidase A may limit the feasibility of HTP-dependent islet targeting (Eriksson et al., 2014). These efforts highlight the multitude of opportunities and challenges associated with developing a strategy for β-cell-targeted compound delivery based upon their unique biologic properties.
To our knowledge, the exceptionally high intracellular zinc concentration of β-cells has not been explored for its drug-targeting potential. Indeed, β-cell insulin granules contain up to 20 mM zinc(II) compared to 2–10 μM in the zinc-rich compartments of most other cell types including the acinar pancreas; representing a >1,000-fold β-cell excess of zinc (Li, 2014; Maret, 2013). Maintenance of this level of zinc in islets depends upon a system of zinc transporters including ZnT8, which shows a highly restricted expression pattern (Chimienti et al., 2006). Visualization of β-cell zinc with cell-permeant metal chelators began with dithizone (DTZ), which displays shifted spectral properties following zinc-binding (Hibbard, 1937; McNary, 1954). Other examples include FluoZin, a dicarboxylic acid fluorescent indicator with nanomolar zinc affinity (Gee et al., 2002), and the ZinPyr/Newport Green family, derived from the dipicolylamine moiety of the strong chelator TPEN (N,N,N’,N’-tetrakis(2-pyridylmethyl)ethane-1,2-diamine) (Lukowiak et al., 2001; Walkup et al., 2000). Additionally, the zinc chelator TSQ (6-methoxy-8-p-toluenesulfonamidoquinoline) exhibits enhanced specificity for zinc over other divalent metals, enabling use of TSQ fluorescence intensity to estimate the zinc content of various tissues (Frederickson et al., 1987; Jindal et al., 1992). Despite the extensive work done to develop highly specific zinc-sensitive probes, the potential use of zinc chelators for β-cell-targeted compound delivery has not been explored. Indeed, it is unknown whether zinc-binding compounds preferentially accumulate within zinc-rich β-cells. Herein, we evaluate the potential use of zinc chelation-dependent β-cell-targeted drug delivery.
RESULTS
Islet staining with zinc-dependent probes is reversed by zinc chelation
The zinc-dependent probes DTZ and TSQ are commonly used to assess isolated islet purity (Fiedor et al., 1996; Jayaraman, 2008; Klochendler et al., 2016; Latif et al., 1988; McNary, 1954). Whereas isolated rat islets exposed to DTZ or TSQ (Figure 1A) display strong visible (pink-scarlet) (Figure S1A) and fluorescent (blue) staining (Figure 1B), respectively, relatively zinc-deficient exocrine tissue remains unstained. To determine whether this fluorescence was dependent on the high concentration of zinc, islets were also exposed to an excess of the strong, zinc-specific chelator TPEN (Meeusen et al., 2012). Indeed, exposure to TPEN removed detectable DTZ (Figures S1A, S1B) or TSQ staining (Figures 1B, S1C). Additionally, TPEN-sensitive TSQ staining was observed by Fluorescence-Activated Cell Scanning (FACS) in zinc-rich R7T1 β-cells (Efrat et al., 1995; Milo-Landesman et al., 2001) and in zinc-poor human embryonic kidney (HEK) 293T cells (Fukada and Kambe, 2014; Graham et al., 1977) (Figures 1C, S1D, S1E). As expected given the paucity of zinc in HEK293T cells, the TSQ/zinc fluorescence was minimal compared to that in R7T1 β-cells. Finally, to further confirm these effects were zinc-dependent, zinc content was measured directly by ICP-OES (Inductively coupled plasma-optical emission spectrometry) in HEK 293T cells, mouse exocrine pancreas, R7T1 β-cells, and mouse pancreatic islets (Figure 1D). As predicted, zinc in HEK 293T (34 nmol zinc/g protein) and exocrine tissue (31 nmol zinc/g protein) was low, and did not differ between the two (p=0.844). In contrast, R7T1 cells contained 12-fold higher zinc (380 nmol zinc/g protein, p=1.38 × 10−4) than exocrine tissue, while islets contained 23-fold higher zinc (717 nmol zinc/g protein, p=5.74 × 10−4) than exocrine tissue. Two non-exclusive mechanisms might contribute to the islet specificity of zinc-dependent visualization. First, as emphasized in the literature (Jindal et al., 1992; Nowakowski et al., 2015; Pearce et al., 2001), DTZ and TSQ only “turn on” when zinc bound; consequently, staining specificity reflects the high zinc content of β-cells. Second, DTZ and TSQ might preferentially accumulate in zinc-rich islet cells if the zinc:chelator interaction alters the balance between compound influx and efflux. While the former mechanism is well documented in the literature, the latter has not been experimentally addressed despite its potential relevance for β-cell-targeted drug delivery.
Figure 1. The spectral shift of TSQ used for islet visualization is reversible.

(A) Chemical structures of 6-methoxy-8-p-toluenesulfonamido-quinoline (TSQ, left) and N’,N’,N,N-Tetrakis(2-pyridylmethyl)-ethylenediamine (TPEN, right). (B) Representative images of TSQ-treated (10 μM) islets in the absence (left) and presence (right) of the Zn2+ chelator TPEN (1 mM). Staining was conducted in 6-well format (n=3) with 50 islets per well, and at least 5 images per condition were taken per well. Yellow arrows indicate unstained exocrine tissue. (C) Fluorescence quantification by FACS of R7T1 β-cells (left) and HEK 293T cells (right) following TPEN, TSQ, and TSQ/TPEN (green) exposure (n=2–4). (D) Zinc quantification by ICP-OES of HEK 293T, exocrine pancreas, R7T1 β-cells (R7T1), and pancreatic mouse islets (n=3–4). Data are represented as mean ± S.D. Comparisons with p > 0.05 are indicated by NS (not significant); 0.01<p<0.05 by *; 0.001<p<0.01 by **; 0.0001<p<0.001 by ***; and p<0.0001 by ****.
See also Figure S1.
R7T1 β-Cells demonstrate zinc content-dependent accumulation of TSQ
Next, we tested the hypothesis that β-cells accumulate TSQ in a zinc content-dependent manner by using zinc-rich R7T1 β-cells (Figure S2A). To directly measure cellular [TSQ], we developed a liquid chromatography/tandem mass spectrometry (LC-MS/MS) method using multiple reaction monitoring (MRM). This method offers reproducible quantification (Figure S2B) of TSQ and CC-401, which served as a non-zinc-chelating control for zinc-independent drug accumulation. Regrettably, DTZ did not ionize efficiently and was unsuitable for LC-MS/MS. R7T1 β-cells exposed to TSQ and vehicle (EtOH) accumulated significantly more TSQ (2.56 nmol TSQ/mg protein, p=3.35×10−5) than R7T1 β-cells exposed to TSQ and TPEN (1.52 nmol TSQ/mg protein) (Figure 2A). Importantly, accumulation of CC-401 was not affected by TPEN (Figure 2B), confirming the dependence of TSQ (a zinc chelator) but not CC-401 (a nonchelator) accumulation on intracellular zinc availability. To control for sample variability, TSQ accumulation was assessed after normalizing to CC-401 in each sample (Figure 2C). Robust zinc-dependent accumulation of TSQ was maintained in R7T1 β-cells (2.2-fold; EtOH vs. TPEN, p=1.92×10−5). Notably, zinc-dependent drug accumulation was not unique to R7T1 β-cells, as INS-1 β-cells demonstrated similar behavior (Figures S2C-2E) (Asfari et al., 1992). These results demonstrate that accumulation of the fluorescent zinc sensor TSQ is sensitive to the chelatable zinc content of β-cells.
Figure 2. TSQ accumulates in a Zn2+- and cell type-dependent manner.

(A) TSQ quantification (LC-MS/MS) from TSQ/CC-401-treated R7T1 β-cells (R7T1) without (blue) or with TPEN (red) co-treatment (n=20–32). (B) CC-401 quantification (LC-MS/MS) for the experiment in (A) monitored simultaneously with TSQ with vehicle (blue) or TPEN (red) co-treatment (n=20–32). (C) TSQ/CC-401 ratio for R7T1 with vehicle (blue) or TPEN (red) co-treatment (n=20–32). (D) TSQ quantification (LC-MS/MS) in primary mouse islet and exocrine tissues following TSQ/CC-401 treatment with vehicle (blue) or TPEN (red) co-treatment (n=24–29). (E) CC-401 quantification (LC-MS/MS) monitored simultaneously with TSQ in (D) (n=24–29). (F) TSQ/CC-401 ratio for mouse islet and exocrine tissues (D and E) (n=24–29). (G) TSQ quantification (LC-MS/MS) of TSQ/CC-401-treated human islets, co-treated with vehicle (blue), [TPEN]=100nM (red), or [TPEN]=10nM (green) (n=8–12). (H) CC-401 quantification (LC-MS/MS) determined simultaneously with TSQ in (G) following vehicle (blue), [TPEN]=100 nM (red), or [TPEN]=10 nM (green) co-treatment; (n=8–12). (I) TSQ/CC-401 ratio for human islets, co-treated with vehicle (blue), [TPEN]=100 nM (red), or [TPEN]=10 nM (green) (n=8–12). Data are represented as mean ± S.D. Comparisons with p > 0.05 are indicated by NS (not significant); 0.01<p<0.05 by *; 0.001<p<0.01 by **; 0.0001<p<0.001 by ***; and p<0.0001 by ****.
See also Figure S2.
Primary mouse and human islets exhibit zinc content-dependent TSQ accumulation
Next, we conducted a more thorough investigation of the hypothesis that islet-cells accumulate TSQ in a zinc content-dependent manner, extending the R7T1 β-cell work to freshly isolated mouse pancreatic islets and exocrine tissues. Islets accumulated significantly more TSQ than exocrine tissue (4.9 nmol/mg protein vs 2.6 nmol/mg protein; ~1.87-fold, p=0.007) (Fig 2D). Reduced TSQ accumulation by R7T1 β-cells compared to primary islets could relate to phenotypic alterations associated with immortalization. Importantly, the non-chelator CC-401 demonstrated no preference for islets versus exocrine tissue (p=0.12) (Fig 2E), indicating that primary islets preferentially accumulate TSQ. Indeed, islets showed a 2.4-fold (p=5.3×10−5) preference for TSQ compared to exocrine tissue (Fig 2F). Hence, primary mouse islets accumulated the zinc chelator TSQ to a greater extent than mouse exocrine tissue.
Next, we assessed whether the preferential TSQ accumulation in mouse islets depended on cellular zinc content. Accordingly, islet and exocrine [TSQ] and [CC-401] were compared following pre-incubation with vehicle (EtOH) or TPEN [1mM]. Consistent with the observations made on R7T1 β-cells, TPEN reduced islet [TSQ] 3.3-fold (p=5.5×10−5) (Fig 2D), and the TSQ/CC-401 ratio by 3.4-fold (p=1.6×10−8) (Fig 2F) indicating that islet TSQ accumulation is highly dependent upon the availability of zinc. Interestingly, exocrine [TSQ] was also reduced (to a lesser extent) by TPEN (1.9-fold; p=0.0022) (Fig 2D), suggesting a modest level of zinc-dependent TSQ accumulation in exocrine tissue. Although this effect was partially mitigated by normalization to [CC-401] (1.4-fold, p=3×10−4) (Fig 2F), exocrine zinc content was sufficient to increase TSQ accumulation. Importantly, TPEN did not affect [CC-401] in islets or in exocrine tissue (Figure 2E, p=0.12 for islets, p=0.093 for exocrine), confirming the specificity of TPEN’s effect on [TSQ]. Taken together, mouse islets accumulated more TSQ than exocrine pancreatic tissue, and this accumulation was dependent on zinc content.
Finally, we evaluated whether human islets exhibit zinc-dependent TSQ accumulation. Indeed, human islet [TSQ] was dose-dependently reduced by TPEN (4.4-fold [100 μM] TPEN, p=9.1×10−6; 1.6-fold [10 μM] TPEN, p=0.02) (Figure 2G). In contrast, TPEN had no effect on [CC-401] ([100 μM] TPEN, p=0.83; [10 μM] TPEN, p=0.13) (Figure 2H). Accordingly, human islets retained robust [CC-401]-normalized zinc-dependent TSQ accumulation (4.2-fold vs [100 μM] TPEN, p=1.4×10−6; 1.6-fold vs [10 μM] TPEN, p=0.0097) (Figure 2I). Hence, TSQ accumulation in islets in mice and humans was specifically dependent upon zinc binding.
Synthesis of a zinc-chelating DYRK1A inhibitor
The observation that TSQ preferentially accumulated in islets led us to hypothesize that the addition of a zinc-chelating moiety to a cargo compound would result in β-cell targeting of that compound. In particular, we postulated that the nonselective replication-promoting compound, GNF-4877, could be directed to β-cells by the addition of a zinc chelator moiety. To interrogate this hypothesis, we established a simple and robust method of assessing the zinc chelation activity of non-fluorescent small molecules. Fresh frozen pancreatic sections were stained with TSQ (Figure S3A) in presence of vehicle, chelator (TPEN), or experimental compound, and TSQ fluorescence was measured within the islet area (Figures S3A–C). Fortuitously, the nuclear dye DSC-1 (Nuclear Green) provided a TSQ-independent fluorescent method for visually identifying the islets area on pancreatic sections. Islet fluorescence directly correlated (R2=0.533) with TSQ concentration (Figure S3D). To confirm that TSQ-stained areas were indeed islets, pancreas sections from an established mouse model, INSCremTmG, were stained with TSQ (Xiao et al., 2013) (Figure 3A). These mice express mTomato in all insulin-negative cells, and EGFP in insulin-positive β- cells. Using this model, we confirmed that TSQ specifically stains β-cells. Using this assay, we compared the chelation activity of known chelators (TPEN and DPA), compounds of interest (CC-401 and GNF-4877), approved medications with purported zinc-chelation activity (gatifloxacin, SAHA, and oxytetracycline) (Brion et al., 1985; Li et al., 2007), and experimental GNF-4877 hybrid molecules synthesized as a part of this study (Figures 3B, 3C, S3A, and S3B). Despite not being known as strong zinc chelators, gatifloxacin, SAHA, and oxytetracycline were of interest due to their potential to accumulate in β-cells because of chelation activity (quinolones variably cause glucose dysregulation (Frothingham, 2005)). Hybrid molecules were generated by attaching a zinc-chelating moiety (DPA) or non-chelating isostere (DBA) to the GNF-4877 carboxylate without (4877-DPA, 4877-DBA) or with an extended spacer (4877-EXT-DPA, 4877-EXT-DBA). As anticipated CC-401, GNF-4877, and synthesized non-chelator hybrids 4877-DBA and 4877-EXTDBA displayed no zinc-chelating activity (Figure 3C). Similarly, gatifloxacin and SAHA were inactive, demonstrating weak, if any, zinc binding affinity based on their inability to displace TSQ. However, TPEN, DPA, oxytetracycline, and hybrid chelators all reduced TSQ fluorescence to background (Figure 3C). To further characterize the zinc chelating activity of these compounds, titration curves were performed with fixed [TSQ] of 10 μM and the IC50s and Hill slopes were calculated (Figures 3D and S3C). These results indicate a rank order of TPEN>DPA>4877-DPA>4877-EXT-DPA>oxytetracycline with regard to zinc binding. Interestingly, the Hill coefficient was greater for TPEN (3.7±1.8), DPA (7.7±5.1), and 4877-EXT-DPA (4.3±2.5) than for oxytetracycline (0.96±0.33) and 4877-DPA (1.0±0.48). These results demonstrated the distinct zinc chelating activities of the studied compounds, including confirmed inactivity of CC-401 and GNF-4877, and activity of synthesized 4877-chelator hybrids.
Figure 3. Frozen pancreas sections offer a platform to visualize islet Zn2+ content and assess compound zinc chelation activity.

(A) Fresh frozen INSCremTmG murine pancreatic section (10 μm thickness) expressing mTomato in INS− cells and GFP in INS+ cells stained with TSQ (10 μM). Islet area is outlined in yellow. (B) Structures of GNF-4877 (top left), the variable 4877 scaffold (top right), 4877-DPA, 4877-DBA, 4877-EXT-DPA, and 4877-EXT-DBA (bottom, from left) with either the chelating group dipicolylamine (DPA) appended or the non-chelating group dibenzylamine (DBA) appended. (C) Competition assay for Zn2+ binding with frozen sections treated sequentially with TSQ (10 μM) then vehicle, gatifloxacin (1 mM), CC-401 (1 mM), SAHA (1 mM), 4877-DBA (1 mM), GNF-4877 (1 mM), 4877-EXT-DBA (1 mM), TPEN (1 mM), oxytetracycline (1 mM), DPA (1 mM), 4877-DPA (1 mM) or 4877-EXT-DPA (0.5 mM). (D) Comparison of molecules’ zinc affinity on pancreas sections, characterized by IC50, Hill coefficient nH, and correlation coefficient R2. Data are represented as mean ± S.D.
See also Figure S3.
Next, we assessed whether preferential islet accumulation was a general characteristic of zinc-chelating compounds. Accordingly, we measured mouse islet accumulation of oxytetracycline, SAHA, GNF-4877, 4877-DPA, 4877-DBA, 4877-EXT-DPA, and 4877-EXT-DBA by LC-MS/MS following exposure in vitro (Figures 4A–C, S4A, S4B). Interestingly, only 4877-EXT-DPA recapitulated the ability of TSQ to accumulate in a Zn2+-dependent manner (~1.5-fold, p=0.029 for EtOH vs TPEN) (Figure 4A), while 4877-EXT-DBA (p=0.42) (Figure 4B), oxytetracycline (p=0.63), SAHA (p=0.15), and GNF-4877 (p=0.93) (Figures S4A, S4B) did not. When normalized to 4877-EXT-DBA to correct for inter-sample variability, 4877-EXT-DPA showed a 1.4-fold accumulation relative to TPEN treatment (p=0.0032) (Figure 4C). This effect was not present in exocrine cells (p=0.46). Additionally, 4877-EXT-DPA (normalized to 4877-EXT-DBA) showed greater accumulation in mouse islets than in exocrine tissue (1.3-fold, p=0.049). Next, we assessed whether these observations were recapitulated with human islets (n=3 donors). In agreement with mouse islet experiments, accumulation of the chelator 4877-EXT-DPA was reduced 41±9.6% (p=5.16 × 10−5) by TPEN treatment (Figure 4D) whereas the non-chelator 4877-EXT-DBA was modestly reduced 19±9.2% (p=0.047) by TPEN exposure (Figure 4E). Consequently, the relative accumulation of 4877-EXT-DPA to 4877-EXT-DBA was reduced 23±5.1% (p=3.04 × 10−5) by TPEN (Figure 4F). Taken together, these experiments established the general ability of zinc-chelating molecules with a steep Hill slope (nH>~3.5) (Figure 3D), to accumulate in a zinc-dependent manner. Both the islet-dependence and TPEN-dependence of 4877-EXT-DPA accumulation were consistent whether 4877-EXT-DPA was normalized to 4877-EXT-DBA or to CC-401 (Figures S4D-F), in both mouse and human islet experiments.
Figure 4. 4877-EXT-DPA accumulates in a zinc- and cell type-dependent manner.

(A) 4877-EXT-DPA quantification (LC-MS/MS) after co-treatment of 4877-EXT-DPA and 4877-EXT-DBA (10 μM each) in mouse islets and exocrine tissue, in the absence (blue) and presence (red) of TPEN (n=8–10). (B) 4877-EXT-DBA accumulation in mouse islets and exocrine tissue, monitored simultaneously with 4877-EXT-DPA in (A), in the absence (blue) and presence (red) of TPEN (n=8–10). (C) 4877-EXT-DPA/4877-EXT-DBA ratio in the absence (blue) and presence (red) of TPEN (n=8–10). (D) 4877-EXT-DPA accumulation after co-treatment of 4877-EXT-DPA and 4877-EXT-DBA (10 μM each) in human islets from 3 donors in 3 independent experiments, in the absence (blue) and presence (red) of TPEN (n=38–42). Data are normalized to the average TPEN-treated accumulation for each experiment. (E) 4877-EXT-DBA accumulation, monitored simultaneously with 4877-EXT-DPA in (D), in the absence (blue) and presence (red) of TPEN (n=38–42). Data are normalized to the average TPEN-treated accumulation for each experiment. (F) 4877-EXT-DPA/4877-EXT-DBA molar ratio in human islets, in the absence (blue) and presence (red) of TPEN (n=38–42). Data are represented as mean ± S.D.
See also Figure S4.
To assess the β-cell proliferation promoting potential of the synthesized compounds, we determined the impact of appended chelating and non-chelating moieties on affinity for the relevant β-cell replication target, DYRK1A. Importantly, 4877-EXT-DPA (Kd=1.1±0.2 nM) and 4877-EXT-DBA (Kd=6.4±1.5 nM) retained low nanomolar intrinsic DYRK1A-binding affinity (Figure 5A, DiscoverX), approximately 6-fold and 30-fold higher than GNF-4877 (0.18±0.048 nM). These data predicted that 4877-EXT-DPA and 4877-EXT-DBA were likely to retain β-cell replication-promoting activity. Next, we performed a cell-based DYRK1A-inhibition assay in zinc-low HEK 293T cells (Figures 5B, S5A) (Abdolazimi et al., 2018) to assess compound activity in a cellular context. A rank order of GNF-4877>4877-EXT-DPA>4877-EXT-DBA>4877-DBA>4877-DPA was observed. Consistent with its advanced lead status, GNF-4877 was the most potent (94±39 nM) DYRK1A inhibitor (Figure 5B). The relative inactivity of 4877-DBA (2,400±150 nM) and 4877-DPA (32,000±7800 nM) (Figure S5A), coupled with the more modestly reduced activity of 4877-EXT-DPA (457±32 nM) and 4877-EXT-DBA (1,800±80 nM) (Figure 5B) indicated that spacing the chelating/non-chelating group away from the GNF-4877 pharmacophore helped salvage cellular DYRK1A inhibition activity. Given its robust activity and chelation characteristics (Figures 3C, 3D), 4877-EXT-DPA and its non-chelating control 4877-EXT-DBA were further examined.
Figure 5. Attachment of a zinc-chelating moiety biases the replication-promoting activity of GNF-4877 toward zinc-rich β-cells.

(A) DYRK1A binding affinity of GNF-4877, 4877-EXT-DPA, and 4877-EXT-DBA (n=2 per concentration). (B) Dose response curves for GNF-4877, 4877-EXT-DPA, and 4877-EXT-DBA based on DYRK1A inhibition via a luciferase reporter in transfected HEK 293T cells (n=4 per concentration). (C) Representative images of dispersed rat islet cultures treated with 4877-EXT-DBA (3 μM) (top) or 4877-EXT-DPA (3 μM) (bottom) and stained (PDX-1, Insulin, and Ki-67). White arrows indicate replicating β-cells, yellow arrows indicate replicating non-β-cells. (D) Replication induced by DMSO, GNF-4877 (1–5 μM), 4877-EXT-DBA (3 μM), and 4877-EXT-DPA (3 μM) in co-cultured PDX-1 positive β-cells (blue) and non-β-cells (red). Data are pooled from two independent experiments of two rats each (n=8–11). (E) β-cell selectivity for the experiment in (D), defined as the ratio of β-cell replication to non-β-cell replication (n=8–11). (F) Human islet replication induction by DMSO, GNF-4877 (1–5 μM), 4877-EXT-DBA (3.3–10 μM), and 4877-EXT-DPA (3.3–10 μM). Data represent two donors in two independent experiments (n=17–25). (G) β-cell selectivity for human islets in (F). Data represent five pooled donors in five independent experiments (n=17–25). Data are represented as mean ± S.D.
See also Figure S5.
Attachment of a zinc chelator onto a replication-promoting molecule conveys preferential β-cell replication-promoting activity
The preferential islet accumulation of 4877-EXT-DPA raised the possibility that this compound might differentially promote replication of zinc-rich β-cells over zinc-poor islet-cell subtypes (Foster et al., 1993). To test this hypothesis, the in vitro β-cell and non-β-islet-cell replication rates of vehicle and compound treated rat islet-cells were measured (Figures 5C–D, S5B, and S5C). As anticipated, the control compound 4877-EXT-DBA modestly stimulated rat β-cell (4.5-fold, p=3.08×10−13 vs. DMSO) and non-β-cell (1.5-fold, p=6.0×10−4 vs. DMSO) replication (Figure 5D), with β-cell selectivity (β-cell induction/non-β-cell induction) of 3.1±0.5 (Figure 5E). In contrast, the chelator 4877-EXT-DPA induced substantially greater β-cell replication (26-fold, p=6.9×10−21 vs. DMSO) than in zinc-poor non-β-cells (3.4-fold, p=6.4×10−9 vs. DMSO) (Figure 5D), demonstrating a strong β-cell preference (β-cell selectivity=7.8±1.1) (Figure 5E). A similar result was obtained at lower concentration of 4877-EXT-DBA and 4877-EXT-DPA, supporting the robustness of this phenomenon (Figures S5D, S5E). As a control experiment, DPA’s effect on replication was determined. DPA did not stimulate β-cell replication, either alone or in combination with GNF-4877 (Figure S5F, S5G), indicating that a hybrid molecule is necessary for the β-cell-selective effect. These results indicated that the β-cell selectivity of replication-promoting activity was improved ~250% (p=9.1×10−9) by the presence of zinc-chelating activity (Figure 5E). Critically, the differential replication response was observed within the same well where compound exposure was equivalent, suggesting that β-cell zinc content promotes cell-autonomous intracellular compound accumulation in a replication-relevant cellular compartment.
Finally, we extended our findings to human islets by assessing cellular replication following treatment with DMSO, GNF-4877, 4877-EXT-DPA, or 4877-EXT-DBA (Figures 5F, S5H). As anticipated, GNF-4877 stimulated replication in both β-cells (2.6-fold, p=1.1×10−19 vs DMSO) and non-β-cells (3.4-fold, p=9.9×10−22 vs DMSO), showing 1.3-fold higher replication in non-β-cells (p=1.8×10−6; β-cell selectivity=0.78±0.17) (Figures 5G). Similarly, non-zinc-chelating 4877-EXT-DBA stimulated replication in both β-cells (2.2-fold, p=1.3×10−11) and non-β-cells (2.6-fold, p=3.6×10−14) (Figures 5F, S5H), with 1.2-fold higher replication in non-β-cells (p=0.024; β-cell selectivity=0.79±0.14; p=0.96 vs GNF-4877) (Figure 5G). By contrast, β-cell and non-β-cell replication induction by 4877-EXT-DPA was roughly equivalent (β-cells 2.6-fold vs DMSO, p=6.6×10−11; non-β-cells vs DMSO, p=2.9×10−11) (Figure 5F), leading to modestly, yet significantly, increased β-cell-selectivity (1.32-fold vs. GNF-4877, p=1.6×10−4; 1.32-fold vs. 4877-EXT-DBA, p=5.2×10−4)(Figure 5G). Therefore, appending the chelation group onto GNF-4877 biased replication-promoting activity towards rat (~250%) and human (~130%) β-cells compared to co-cultured non-β-islet-cells. Notably, the impact of the zinc-chelating moiety on the specificity of rodent and human replication-induction correlated with compound accumulation by rodent (~140%) and human islets (~120%) (Figure 4C,F), supporting the conclusion that β-cell selective replication resulted from zinc-dependent compound accumulation.
DISCUSSION
The unusual ability of islet β-cells to bio-concentrate zinc has been used for decades to identify islets with zinc-dependent dyes (Gee et al., 2002; Hibbard, 1937; Lukowiak et al., 2001; McNary, 1954). However, the fundamental question of whether zinc chelators preferentially accumulate within islets has remained unanswered. By coupling the fluorescent dye TSQ to quantitative LC-MS/MS, we demonstrate that islets do indeed preferentially accumulate zinc-chelating molecules. Critically, co-incubation with TPEN, a highly potent zinc chelator, reduced islet accumulation of zinc-chelating molecules but not non-chelators. Accordingly, the preferential accumulation of TSQ within rodent and human islets was zinc dependent. Furthermore, preferential islet drug accumulation was readily transferred to the replication-promoting molecule GNF-4877 through attachment of the potent zinc-chelating moiety DPA (4877-EXT-DPA). Remarkably, attachment of the zinc chelator conferred preferential replication-promoting activity towards β-cells over non-β-cells cultured in the same well. These studies provide a successful demonstration of a strategy for selectively targeting replication-promoting activity to the β-cell. This delivery strategy has general applicability to other β-cell-targeted therapeutics, e.g. calcium channel blockers, which have anti-apoptotic effect on β-cells, but are dose-limited by systemic effects (Xu et al., 2012).
Although this work represents an exciting advance in β-cell-directed compound delivery, several limitations are worth noting. First, attachment of a zinc-chelation moiety to the replication-promoting molecule GNF-4877 conferred zinc-dependent and preferential β-cell drug accumulation and activity; however, the magnitude of this effect was relatively small. The increase in β-cell replication selectivity (vs. non-β-cells) was ~2.5-fold (Figure 5E) for rodent islet cultures and lower for human islet cultures (Figure 5G). This observation is consistent with our limited medicinal chemistry effort, which focused on generating a bi-functional hybrid molecule (4877-EXT-DPA) that retained potent zinc-chelation and replication-promoting activities for proof of concept studies rather than optimization. Based upon (a) results obtained with TSQ, which displayed substantially greater preference for islets over other tissues (3.4-fold in rodent and 4.4-fold in human islets), (b) the correlation between zinc-dependent drug accumulation and β-cell replication selectivity and (c) the sigmoidal dose-response curve of compound-induced β-cell replication, modest chemical optimization of 4877-EXT-DPA would dramatically improve selectivity of β-cell replication promoting activity. Additionally, it may be possible to reduce drug accumulation in zinc-poor cells by modulating compound efflux, permeability, compartmentalization, and/or potency.
A second possible limitation of zinc chelation-dependent drug targeting is the potential to negatively impact normal cellular function, such as insulin storage in β-cells (Maret, 2013). In contrast to this expectation, mice treated for 5 weeks with a compound containing the same zinc-chelating DPA moiety used herein did not display observable toxicity, suggesting that zinc-chelating compounds may be well tolerated at therapeutically relevant doses (Rice et al., 2016). Still, it may be necessary to intermittently replete zinc stores in conjunction with in vivo administration of a zinc chelator. Third, several cell populations, in addition to β-cells, exhibit high intracellular zinc concentrations, including the prostate (Kelleher et al., 2011), pituitary gland (Danscher and Stoltenberg, 2005), paneth cells (Giblin et al., 2006), and a subset of glutamatergic neurons (Frederickson, 1989). Developing strategies to avoid these depots could be necessary for in vivo applications. Finally, we have not demonstrated in vivo utility of the chelator-based strategy for β-cell targeted drug delivery. Further experimentation is necessary to extend our proof-of-concept in vitro studies towards a safe, broadly applicable in vivo strategy for β-cell selective drug delivery. However, this work provides a critical first step towards validating a readily transferable β-cell-targeted drug-delivery strategy.
SIGNIFICANCE
Diabetes care would be transformed by the development of a regenerative therapeutic capable of expanding β-cell mass, increasing endogenous insulin production capacity and restoring glucose homeostasis. The recent discovery of highly potent β-cell replication-promoting compounds has brought this objective into view. However, available compounds are insufficiently β-cell selective. Consequently, developing a method for β-cell-targeted drug delivery has emerged as a critical hurdle to clinical translation. We find that the exceptionally high zinc content of β-cells (~1000× plasma) enables preferential accumulation of zinc-chelating compounds within β-cells. Furthermore, attachment of a zinc-chelating moiety to a non-selective replication-promoting compound biases bioactivity towards β-cells. Optimization of this technology could provide a general mechanism for engineering β-cell-directed therapeutics.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the Lead Contact, Justin Annes (jannes@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
ANIMAL MODELS
C57BL/6J wild-type adult male mice were obtained from Jackson Laboratories at 8–12 weeks of age. Male INSCre mice with a B6 background were crossed with ROSAmT/mG mice with a B6 background(Xiao et al., 2013). Mice were housed under normal conditions. Male Sprague-Dawley rats were obtained at 250–300 g adult weight and housed under normal conditions. Directly before the islet and exocrine pancreas isolation procedure, mice or rats were anesthetized in an isofluorane chamber and humanely euthanized by cervical dislocation. All protocols conform to the regulatory standards set forth by the Stanford University Administrative Panel on Laboratory Animal Care (APLAC).
CELL LINES
All cells (cell lines and primary cells) were cultured at 37°C, 5%CO2, and 21% O2 and manipulated in a sterile laminar flow hood.
HEK293T
HEK293T cells (female) were thawed from frozen stocks at 10% DMSO and maintained at low passage number (<40) in DMEM High Glucose with 10% Fetal Bovine Serum, Glutamax (2 mM), Penicillin (1 IU/mL)/Streptomycin(1 μg/mL), and 1 mM sodium pyruvate.
R7T1
R7T1 cells (karyotype not performed) were thawed from frozen stocks at 10% DMSO and maintained at low passage number (<20) in DMEM High Glucose with 10% Fetal Bovine Serum, Glutamax (2 mM), Penicillin (1 IU/mL)/Streptomycin(1 μg/mL), 1 mM sodium pyruvate, and 1 mg/mL Doxycycline. At 10 days prior to experiment, R7T1 culture media was changed to DMEM High Glucose with 10% Tetracycline-free Fetal Bovine Serum, Glutamax (2 mM), Penicillin (1 IU/mL)/Streptomycin(1 μg/mL), and 1 mM sodium pyruvate.
INS-1E
INS-1E cells (male) were thawed from frozen stocks and maintained at low passage number (<10) in RPMI1640 medium with 10% Fetal Bovine Serum with 50 μM 2-mercaptoethanol, Glutamax (2 mM), and Penicillin (1 IU/mL)/Streptomycin(1 μg/mL).
PRIMARY CELLS
Mouse islets and exocrine tissue were placed in DMEM Low Glucose with 10% Fetal Bovine Serum, Glutamax (2 mM), and Penicillin (1 IU/mL)/Streptomycin(1 μg/mL) and used directly. Rat islets were cultured 12–16 hrs in DMEM Low Glucose with 10% Fetal Bovine Serum, Glutamax (2 mM), and Penicillin (1 IU/mL)/Streptomycin(1 μg/mL). After dispersion to single cells, islet cells were cultured under the same conditions, with media changes every 48 hrs.
HUMAN CELLS
All human islet samples were obtained from accredited isolation programs (IIDP and IsletCore) as de-identified autopsy tissue and cultured under guidelines approved by the Stanford University Administrative Panel on Biosafety. Human islet donors were non-diabetic Caucasian males (average blood glucoses of 108, 149, 147.2 and 157.4 mg/dL, ages 27, 45, 30, and 28 with BMIs 29.8, 25,4, 34.7, and 30, respectively) and one Caucasian female (aged 60, BMI 26.0). Islets were obtained in CMRL media and exchanged into DMEM Low Glucose with 10% Fetal Bovine Serum, Glutamax (2 mM), Penicillin (1 IU/mL)/Streptomycin(1 μg/mL), and Exendin-4 (20 nM). After dispersion to single cells, islet cells were cultured under the same conditions, with media changes every 48 hrs.
METHOD DETAILS
PRIMARY ISLET REPLICATION ASSAY DETAILS
Rat islets were isolated using pancreatic perfusion via the common bile duct with collagenase A (Gotoh et al., 1987; Zhao et al., 2016). Following digestion, quenching, and centrifugation in Histopaque, islets were hand-picked and placed in Low Glucose DMEM. After 12–16 h, islets were digested to single cells using trypsin and repeated pipetting every 5 min for 20 mins. Trypsinized islet cells were dispersed at 25,000 cells/well into tissue-culture-treated 384-well plates pre-treated with conditioned media from 804G rat bladder carcinoma cells. After 48 h to adhere, islet media was changed and cells were treated with DMSO, 4877-EXT-DPA (1–3 μM), or 4877-EXT-DBA (1 μM) and cultured for 48 h. Cultures were fixed by addition of 8% PFA directly to media and incubation for 20 min at 25°C. Cultures were washed three times with PBS and antigen retrieved in freshly-made sodium citrate pH 6.0 buffer (0.75 mL) in cold formamide (14.25 mL) at 70°C for 45 min. Cultures were washed and blocked for 1 h with 6.25% donkey serum, 0.3% Triton-x 100 in PBS. Cultures were stained with PDX-1 (1:100) and Insulin (1:300) antibodies to identify β-cells and Ki-67 (1:200) to identify replication events, followed by 1 hr staining with secondary antibodies (Jackson Immunoresearch), and nuclear staining (Hoechst 33342, 5 μM). Automated analysis was performed with Cellomics ArrayScan VTI HCS Reader (ThermoFisher, >1,000 events per well), with nuclear staining (Hoechst 33342, Thermo #62249) used to exclude fibroblasts (larger nuclei), and average PDX and Insulin staining intensity distinguished β-cells from non-β-cells.
Human islets were cultured and identically to rat islets except that Exendin-4 (20 nM) was added to all cultures, and antibody staining did not utilize PDX to distinguish islets, relying instead on Insulin alone.
TSQ ACCUMULATION ASSAYS
Mouse islets were isolated by a similar procedure as above, hand-picked, and separated into microcentrifuge tubes on ice at 40–50 islets per tube in islet culture medium (low glucose DMEM containing 10% Fetal Bovine Serum). Exocrine tissue was collected from pelleted material after the Histopaque centrifugation step and immediately placed in islet culture medium. R7T1 and INS-1 β-cells were retrieved from adherent cell culture dishes, washed with PBS, trypsinized, and placed in islet culture medium. Aliquots of each were normalized for protein content using RIPA lysis and subsequent DC protein assay (Biorad). Samples were supplied with TPEN (1 mM) or EtOH in media, then incubated for 5 min at 37 °C. Samples were then tre ated with TSQ (10 μM), the control drug CC-401 (10 μM), and either TPEN or EtOH in media. After incubation at 37 °C for 30 min, samples were centrifuged and washed twice with PBS. The remaining volume (10 μL) of the sample was taken to the extraction step. Human islets from three different donors were treated identically to the mouse islets, except with TPEN at a concentration of 10 μM and 100 μM.
EXTRACTION AND QUANTIFICATION BY LC-MS/MS
Isolated islets, exocrine tissue, or cell culture samples in PBS (10 μL) were diluted in DMSO (5 μL) and vortexed briefly, then frozen at −20 °C. Fol lowing this step, sample (15 μL) was placed in MeCN (50 μL) containing the internal standard propranolol (500 nM). Samples were again vortexed, then centrifuged at 10,000 rpm for 1 min. Supernatant (50 μL) was diluted 1:1 in 0.1% formic acid. In parallel for each experiment, standard dilutions (1–8000 nM) were prepared in methanol and extracted along with samples. Samples were analyzed by LC-MS/MS on a QTrap 4000 (AB Sciex) triple quadrupole mass spectrometer, using a Phenyl-Hexyl column (Phenomenex). The flow rate was 0.500 mL/min in (A) 0.1% formic acid in water and (B) 0.1% formic acid in MeCN, with a rapid ramp from 0–95% B, followed by 10% B. TSQ and CC-401 mass spectra were monitored by multiple reaction monitoring (MRM) based on a method developed by infusion of the individual compounds (TSQ (Q1/Q3=329 Da/174 Da, DP=76.000 V, CE=25.000 V, CXP=10.000 V), CC-401 (Q1/Q3=389 Da/112.1 Da, DP=66.000 V, CE=41.000, CXP=6.000 V), 4877-EXT-DBA (Q1/Q3=719.384 Da/520.2 Da, DP=161.000 V, CE=45.000 V, CXP=14.000 V), and 4877-EXT-DPA (Q1/Q3=717.27 Da/91.2000 Da, DP=126.000 V, CE=129.000 V, CXP=6.000 V)). AbSciex Analyst software was utilized to automatically create a quantitation method based on the peak area of analytes in the calibration standards, using a linear fit. Nanomolar concentrations were converted to nmol/mg total protein by normalizing to the measures of total protein from the DC protein assay (BioRad).
DYRK1A INHIBITION ASSAY
Intrinsic binding to DYRK1A’s ATP-binding domain was tested by diluting compounds in DMSO and assaying in duplicate [0.1 nM] to [10 μM] and compared to DMSO-treated reference wells as previously described (KdELECT Kinase Assay, Eurofins) (Fabian et al., 2005). Briefly, a DNA-tagged DYRK1A construct was incubated with an immobilized ligand in the presence and absence of compounds, and the amount of ligand-bound DYRK1A was assessed by quantitative PCR. Data were analyzed by fitting to the Hill equation, with the Hill Slope set to −1.
DYRK-1A INHIBITION REPORTER CELL ASSAY
A previously-established DYRK1A inhibition assay (Abdolazimi et al., 2018) utilizing HEK 293T cells transfected with DYRK1A, NFAT-C1, NFAT-responsive firefly luciferase reporter, CMV-Renilla, and H2B Cherry at 70% confluence was utilized to assess cell-based activity of GNF-4877, 4877-EXT-DPA, 4877-EXT-DBA, 4877-DBA, and 4877-DPA. Cells were treated 24 h post transfection with compound concentrations from 1 nM to 90 uM in quadruplicate. After 48 h culture, cells were lysed and their luminescence (firefly and renilla) assessed using a dual-injection luminescence plate-reader.
ISLET VISUALIZATION IN CELL CULTURE
Islets were hand-picked and cultured overnight in islet culture medium, then transferred into PBS. Dithizone was used from a stock solution at 177 mM, suspended in 90% EtOH and alkanized to solubility with 1:500 NH4OH. For dithizone staining, islets were treated with either dithizone (17.7 μM) and TPEN (1 mM) or dithizone and EtOH and incubated at 37°C for 20 min. Islets were then visualized und er brightfield light. TSQ labeling was analogous, except that TSQ (SCBT) was used at 10 μM and visualized with a 385 nm laser.
ANALYSIS OF TSQ FLUORESCENCE BY FACS
After isolation from rats, islets were cultured overnight in DMEM Low Glucose with Penicillin (1 IU/mL) and Streptomycin (1 μg/mL). Islets were pelleted, at 200 rpm for 1 min, washed with PBS, trypsinized, washed with PBS, and resuspended in fluorescence-activated cell scanning (FACS) buffer (2% FBS in PBS), then separated into FACS tubes on ice at 40 islets per tube in 500 μL buffer. R7T1 β-cells were grown under growth-arresting conditions for 7 days prior to the experiment. Cells were plated in 12-well format at 1.3 million cells per well. Each well was trypsinized, washed with PBS, and resuspended in FACS buffer (2% FBS in PBS). After straining (70 μm) cells into FACS tubes, either TSQ alone (10 μM), TPEN alone (1 mM), or TSQ (10 μM) and TPEN (1 mM) was added and the tubes placed on ice for 15 min. Islets were stained for viability using propidium iodide. Live β-cells were gated and analyzed on the 355 nm UV laser (20 mW) with the 450/50 BP filter using the Hoechst fluorochrome settings. Live, singlet cells were gated and analyzed on using the same laser using the DAPI fluorochrome settings. Data were conducted in quadruplicate and represented as a histogram of count (y-axis) and DAPI (361 nm) fluorescence intensity (x-axis).
ANALYSIS OF ZINC BY ICP-OES
Isolated mouse islets, mouse exocrine tissue, and trypsinized R7T1 and HEK 293T cells were centrifuged at 800 rpm for 5 min. Samples were digested by adding concentrated HNO3 (228 uL) and heating samples 30 s at 100 °C, t hen freezing at −20 °C. On the day of the experiment, samples were thawed and diluted to 2% HNO3. An aliquot (10 uL) was taken and neutralized with 2 N NaOH (10 uL), and the concentration of protein was determined by DCA protein assay. The remainder of the solution was diluted to a final concentration of 2% HNO3 (8 mL) and the amount of zinc was determined by ICP-OES (Inductively coupled plasma - optical emission spectrometry) on a Thermo Scientific ICAP 6300 Duo View Spectrometer, running a diluted zinc standard in parallel to convert responses to concentrations. Data are represented as zinc normalized to total protein.
STAINING FROZEN SECTIONS WITH TSQ AND DTZ
In order to examine chelator staining in an ex vivo setting, mice were humanely sacrificed and pancreas and liver were harvested. Small portions of these organs were resected and frozen in O.C.T. compound (Fisher) and sectioned with a cryostat at a uniform thickness of 10 μm and mounted on glass microscope slides (Fisher), which were stored at −80°C. For dithizone (DTZ) staining, sections were demarcated with a hydrophobic barrier pen (Vector) and treated with 17.7 μM dithizone in PBS for 5 min without fixing. Dithizone containing solution was removed and replaced with either TPEN (1 mM) or EtOH in PBS for 5 min. Sections were washed once with PBS and analyzed under a fluorescent microscope (Leica). Staining with TSQ was identical, except that TSQ was used at 10 μM. In all cases, sections were co-stained with the green nuclear dye DCS1 (abcam) at 333 m to confirm that TSQ-labeled areas were indeed islets. The molecules gatifloxacin, CC-401, SAHA, 4877-DBA, GNF-4877, 4877-EXT-DBA, oxytetracycline, DPA, 4877-DPA, and 4877-EXT-DPA were compared in competition with TSQ at concentrations up to 1 mM to assess chelating characteristics. Pictures were taken at 20X magnification and fluorescence was quantified by outlining the islet highlighted by nuclear green staining using the Elliptical Marquee Tool (Adobe Photoshop) and determining the Blue channel staining intensity with the Histogram tool. This islet blue intensity was divided by an equivalently-sized ellipse in the non-islet portion of the image, and this value was normalized to the untreated (no TSQ) image (0%) and no inhibitor (10 uM TSQ) image (100%). To establish that TSQ staining reveals β-cells in the context of the pancreas, the pancreas from a INSCremTmG mouse (8 week old female) was sectioned (10 μm) and stained with TSQ (10 μM) without fixation or mounting, and images were taken at 20X magnification.
CHEMICAL SYNTHESIS
Reactions were performed under ambient atmosphere unless otherwise noted. Qualitative TLC analysis was performed on 250 mm thick, 60 Å, glass backed, F254 silica (Silicycle, Quebec City, Canada). Visualization was accomplished with UV light and exposure to p-anisaldehyde or KMnO4 stain solutions followed by heating. Flash chromatography was performed on a Teledyne Isco purification system using silica gel flash cartridges (SiliCycle®, SiliaSepTM 40–63 mm, 60Å). HPLC was performed on an Agilent 1260 Infinity preparative scale purification system using an Agilent PrepHT Zorbax Eclipse XDB-C18 reverse-phase column (21.2 × 250 mm). Structure determination was performed using 1H spectra that were recorded on a Bruker AV-500 spectrometer, and low-resolution mass spectra (ESI-MS) that were collected on a Shimadzu 20–20 ESI LCMS instrument.
4877-DPA SYNTHESIS
To a suspension of GNF-4877 (Shen et al., 2015) (40.5 mg, 0.0819 mmol) and Di-(2-picolyl)amine (S1, 19.2 mL, 0.107 mmol) in DMF (0.819 mL, 0.1 M) stirring at 23 °C was added HATU (40.5 mg, 0.107 mmol) follo wed by Hünig’s base (18.5 mL, 0.107 mmol). The resulting solution was stirred at 23 °C under air for 20 h. The reaction was poured into EtOAc (10 mL) and washed with water (10 mL, 1.0 M). The phases were separated and the aqueous phase was extracted with additional EtOAc (10 mL). The organics were combined, washed with LiCl (10% aq., 2 × 5 mL), brine (5 mL) dried over Na2SO4, filtered and the solvent was removed via rotary evaporation. The residue was purified by flash chromatography (0–10% MeOH/CH2Cl2 eluent) to afford 4877-DPA (42.5 mg, 77% yield, Rf = 0.17 in 1:1 EtOAc/hexanes).
4877-DPA CHARACTERIZATION
1H NMR (500 MHz, CD3OD) d 9.58 (s, 1H), 8.50 (d, J=2.2 Hz, 1H), 8.33 (d, J = 4.5 Hz, 1H), 8.27 (d, J = 5.4 Hz, 1H), 8.14 (d, J = 4.3 Hz, 1H), 7.66 (td, J = 7.7, 1.5 Hz, 1H), 7.41 (td, J = 7.6, 1.5 Hz, 1H), 7.38 (dd, J = 6.4, 3.1 Hz, 1H), 7.34 (d, J = 5.4 Hz, 1H), 7.23–7.17 (m, 1H), 7.14–7.09 (m, 1H), 7.07–7.02 (comp m, 2H), 6.99 (dt, J = 9.0, 3.3 Hz, 1H), 6.76 (d, J = 7.8 Hz, 1H), 4.63 (septet, J= 6.1 Hz, 1H), 4.37 (ABq, J = 15.7 Hz, Du = 302.5 Hz, 2H), 4.20 (ABq, J = 16.7 Hz, Du= 158.2 Hz, 2H), 3.67–3.60 (m, 1H), 3.27–3.15 (comp m, 2H), 3.14–3.04 (m, 1H), 2.71 (t, J = 11.5 Hz, 1H), 2.22–2.10 (m, 1H), 2.02–1.92 (comp m, 2H), 1.75 (qd, J = 13.8, 4.5 Hz, 1H), 1.34 (t, J = 6.1 Hz, 3H), 1.31 (t, J = 6.1 Hz, 3H); 13C NMR (126 MHz, CD3OD)δ 173.38, 163.37, 157.05, 155.33, 154.43, 154.16, 153.83, 152.51, 149.76, 148.88, 148.85, 138.56, 138.35, 128.45, 124.51, 124.40, 123.46, 122.29 (d, J=30.8 Hz), 121.43, 120.79, 117.03, 116.93 (d, J=30.8 Hz), 121.43, 120.79, 117.03. 116.93 (d, J = 24.6 Hz), 116.70, 115.41, 104.52, 69.67, 51.88, 50.12, 21.72, 21.53; LRMS (ESI+) m/z calc’d for (M – H)– [C37H38 FN9O3 – H]− : 674.3, found 674.2; HRMS (ESI+) found 676.3154.
4877-DBA SYNTHESIS
To a suspension of GNF-4877 (44.4 mg, 0.0898 mmol) and Di-benzylamine (S2, 22.4 mL, 0.117 mmol) in DMF (0.898 mL, 0.1 M) stirring at 23 °C was added HATU (44.4 mg, 0.117 mmol) followed by Hünig’s base (20.3 mL, 0.117 mmol). The resulting solution was stirred at 23 °C under a ir for 20 h. The reaction was poured into EtOAc (10 mL) and washed with water (10 mL, 1.0 M). The phases were separated and the aqueous phase was extracted with additional EtOAc (10 mL). The organics were combined, washed with LiCl (10% aq., 2 × 5 mL), brine (5 mL) dried over Na2SO4, filtered and the solvent was removed via rotary evaporation. The residue was purified by flash chromatography (20–75% EtOAc/hexanes then 5% MeOH/CH2Cl2 eluent) to afford 4877-DBA (21.4 mg, 35% yield, Rf = 0.60 in 1:1 EtOAc/hexanes).
4877-DBA CHARACTERIZATION
1H NMR (500 MHz, CD3OD) d 9.33 (2, 1H), 8.58 (d, J = 1.8 Hz, 1H), 8.29 (d, J = 6.2 Hz, 1H), 7.42 (dd, J = 6.4, 3.1 Hz, 1H), 7.38 (d, J = 6.4 Hz, 1H), 7.25–7.20 (comp m, 3H), 7.19–7.13 (comp m, 3H), 7.08 (dd, J = 10.6, 9.1 Hz, 1H), 7.00–6.93 (comp m, 3H), 6.83–6.78 (comp m, 2H), 4.67–4.61 (m, 1H), 4.60 (septet, J = 6.0 Hz, 1H), 4.25–4.19 (m, 1H), 4.36 (ABq, J = 14.8 Hz, Du=280.6 Hz, 2H), 4.11 (ABq, J = 16.6 Hz, Du=113.2 Hz, 2H), 3.64–3.51 (comp m, 2H), 3.22–3.10 (comp m, 2H), 3.10–3.02 (m, 1H), 2.09–1.91 (comp m, 3H), 1.88–1.75 (m, 1H), 1.28 (d, J = 6.0 Hz, 6H); 13C NMR (126 MHz, (CD3)2SO) δ 172.86, 164.30, 154.61, 154.21, 153.89 (d, J = 2.1 Hz), 152.70, 137.18, 136.63, 128.53, 128.38, 127.32, 127.00, 126.95, 126.27, 124.40 (d, J = 13.9 Hz), 123.43, 117.06, 116.90 (d, J = 24.5 Hz), 115.87 (d, J = 9.0 Hz), 114.03, 104.52, 69.74, 49.61, 47.56, 27.37, 24.24, 21.76, 21.63; LRMS (ESI+) m/z calc’d for (M + H)+ [C39H40FN7O3 + H]+ : 674.3, found 674.2; HRMS (ESI+) found 674.3249.
4877-EXT-DPA SYNTHESIS
To a solution of S3 (Vonlanthen et al., 2014) (105 mg, 0.150 mmol) in DMF (0.600 mL, 0.17 M) stirring at 23 °C was added Hüni g’s base (0.139 mL, 0.800 mmol) and the resulting solution was stirred for 5 min. GNF-4877 (49.5 mg, 0.100 mmol) was added followed by HATU (57.0 mg, 0.150 mmol), and the reaction was stirred at 23 °C under air for 18 h. The react ion was poured into EtOAc (20 mL) and washed with NaOH (10 mL, 1.0 M). The phases were separated and the aqueous phase was extracted with additional EtOAc (20 mL). The organics were combined, washed with brine (10 mL), dried over Na2SO4, filtered and the solvent was removed via rotary evaporation. The residue was dissolved in 3:1 MeOH/H2O (3.0 mL) and purified by preparative HPLC. Product containing fractions were combined and the MeCN was removed by rotary evaporation. The solution was basified to pH ~12 with NaOH (1.0 M, approx. 0.500 mL) and extracted with 10% MeOH/CH2Cl2 (3 × 15 mL). The combined organics were dried over Na2SO4, and concentrated under reduced pressure. The residue was dissolved in 50% MeCN/H2O (10 mL), diluted to 30% MeCN/H2O with HPLC grade H2O, frozen, then dried via lyophilization to afford 4877-EXT-DPA (53.1 mg, 74% yield) as a yellow powder.
4877-EXT-DPA CHARACTERIZATION
1H NMR (500 MHz, CD3OD) d 9.50 (s, 1H), 8.64 (s, 1H), 8.40 (d, J = 4.6 Hz, 2H), 8.25 (d, J = 5.4 Hz, 1H), 7.67 (t, J = 6.4 Hz, 2H), 7.47 (d, J = 7.8 Hz, 2H), 7.42 (dd, J = 6.3, 3.0 Hz, 1H), 7.22–7.19 (comp m, 3H), 7.19–7.13 (m, 1H), 6.97 (dt, J= 8.8, 3.4 Hz, 1H), 4.59 (septet, J = 6.0 Hz, 1H), 3.75 (s, 4H), 3.30–3.20 (comp m, 4H), 2.79 (t, J = 11.3 Hz, 1H), 2.72–2.63 (comp m, 2H), 2.61 (t, J = 6.2 Hz, 2H), 1.90–1.78 (comp m, 2H), 1.77–1.70 (m, 1H), 1.53 (qd, J = 14.0, 3.5 Hz, 1H), 1.28 (d, J = 6.0 Hz, 6H); 13C NMR (126 MHz, CD3OD) δ 175.29, 165.38, 160.22, 155.94, 155.84 (d, J = 9.3 Hz), 152.20, 149.55, 146.45, 138.55, 130.51, 125.80, 125.01, 123.79, 119.22, 117.90 (d, J = 24.5 Hz), 117.49 (d, J = 8.9 Hz), 116.66, 72.05, 60.95, 55.16, 54.78, 52.92, 38.25, 27.72, 26.11, 22.35, 22.33;LRMS (ESI+) m/z calc’d for (M + H)+ [C39H43FN10O3 + H]+ : 719.4, found 719.3; HRMS (ESI+) found 719.3576.
4877-EXT-DBA SYNTHESIS
To a solution of S4 (Andrews et al., 2010) (70.2 mg, 0.150 mmol) in DMF (0.600 mL, 0.17 M) stirring at 23 °C was added Hünig’s bas e (0.0958 mL, 0.550 mmol) and the resulting solution was stirred for 5 min. GNF-4877 (49.5 mg, 0.100 mmol) was added followed by HATU (57.0 mg, 0.150 mmol), and the reaction was stirred at 23 °C under air for 18 h. The reaction was poured into EtOAc (20 mL) and washed with NaOH (10 mL, 1.0 M). The phases were separated and the aqueous phase was extracted with additional EtOAc (20 mL). The organics were combined, washed with brine (10 mL), dried over Na2SO4, filtered and the solvent was removed via rotary evaporation. The residue was dissolved in 3:1 MeOH/H2O (3.0 mL) and purified by preparative HPLC. Product containing fractions were combined and the MeCN was removed via rotary evaporation. The solution was basified to pH ~12 with NaOH (1.0 M, approx. 0.500 mL) and extracted with 10% MeOH/CH2Cl2 (3 × 15 mL). The combined organics were dried over Na2SO4, and concentrated under reduced pressure. The residue was dissolved in 50% MeCN/H2O (10 mL), diluted to 30% MeCN/H2O with HPLC grade H2O, frozen, then dried via lyophilization to afford 4877-EXT-DBA (62.0 mg, 86% yield) as a yellow powder.
4877-EXT-DBA CHARACTERIZATION
1H NMR (500 MHz, CD3OD) d 9.52 (s, 1H), 8.64 (s, 1H), 8.25 (d, J = 5.4 Hz, 1H), 7.40 (dd, J = 6.4, 3.1 Hz, 1H), 7.32–7.26 (comp m, 4H), 7.26–7.21 (comp m, 4H), 7.20–7.10 (comp m, 4H), 6.96 (dt, J = 9.0, 3.4 Hz, 1H), 4.58 (septet, J = 6.1 Hz, 1H), 3.57–3.48 (comp m, 4H), 3.27–3.13 (comp m, 4H), 2.76–2.63 (comp m, 2H), 2.61–2.53 (m, 1H), 2.47 (t, J = 6.4Hz, 2H), 1.86–1.68 (comp m, 3H), 1.51–1.43 (m, 1H), 1.30–1.26 (comp m, 6H); 13C NMR (126 MHz, CD3OD) δ 175.17, 165.28, 156.70, 155.96, 155.83 (d, J = 2.6 Hz), 154.79, 152.06, 146.41, 140.57, 137.21, 137.19 (d, J = 3.6 Hz), 130.54, 130.06, 129.31, 128.10, 119.43, 117.91 (d, J = 24.5 Hz), 117.70 (d, J 8.4 Hz), 116.61, 72.26, 59.47, 55.01, 53.51, 52.85, 44.87, 38.18, 27.86, 26.08, 22.39, 22.34; LRMS (ESI+) m/z calc’d for (M + H)+ [C41H45FN8O3 + H]+ : 717.4, found 717.3; HRMS (ESI+) found 717.3671.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed using Prism 7.0 by Graphpad. Quantitative data are presented as mean ± standard deviation. Dose response curve fitting in Figure 3D and Figure 5B to calculate IC50s and Hill coefficients followed four-parameter [Inhibitor] vs. response. Dose response curve fitting for Figure 5A to calculate EC50’s followed four-parameter [Agonist] vs. response. Replicate number and definition of n is indicated in figure legends. Significance was based on pairwise Student’s t-test. Comparisons with p > 0.05 are indicated by NS (not significant); 0.01<p<0.05 by *; 0.001<p<0.01 by **; 0.0001<p<0.001 by ***; and p<0.0001 by ****.
DATA AND SOFTWARE AVAILABILITY
Raw data is deposited in Mendeley Data under: http://dx.doi.org/10.17632/3ycr7rs56f.1
NON-STANDARD SUPPLEMENTAL ITEMS
Contents of Chemical_Synthesis.zip
Synthesis of 4877-DPA, related to STAR methods Chemical Synthesis section describing 4877-DPA Synthesis
Synthesis of 4877-DBA, related to STAR methods Chemical Synthesis section describing 4877-DBA synthesis.
Synthesis of 4877-EXT-DPA, related to STAR methods Chemical Synthesis section describing 4877-EXT-DPA synthesis.
Synthesis of 4877-EXT-DBA, related to STAR methods Chemical Synthesis section describing 4877-EXT-DBA synthesis.
Supplementary Material
Highlights.
Zinc-chelating molecules preferentially accumulate within β-cells.
Compound accumulation is influenced by affinity for and availability of zinc.
Appending a zinc-chelating moiety to a cargo molecule enhances β-cell accumulation.
A hybrid zinc-chelating/replication-promoting compound has β-cell-biased activity.
ACKNOWLEDGEMENTS
We thank Julien Sage and Neali Armstrong for advice preparing the manuscript. Human islets were provided by the Integrated Islet Distribution Program (IIDP) established by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and by the IsletCore program established by the Alberta Diabetes Foundation and the University of Alberta. This work was supported in part by NIH P30 CA124435 utilizing the Stanford Cancer Institute Proteomics/Mass Spectrometry Shared Resource via the Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University Mass Spectrometry (SUMS). Other core facilities utilized were the Stanford Shared FACS Facility (S10RR027431), the Stanford Environmental Measurements Facility, and the Biomaterials and Advanced Drug Delivery Laboratory. Funding support came from the ChEM-H Chemistry/Biology Interface Predoctoral Training Program (TMH, GM120007) and Bio-X and Stanford Interdisciplinary Graduate Fellowship (TMH), Friedenrich Diabetes Fund (JPA), Endocrinology Training grant (PA, T32DK007217), Molecular Pharmacology training grant (HPM, T32GM113854), the SPARK Translational Research Program and Child Health Research Institute at Stanford University (JPA and SL, UL1TR001085); NIH NIDDK (JPA, R01DK101530 and P30DK116074).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Drugs@FDA: FDA Approved Drug Products.
REFERENCES
- Abdolazimi Y, Zhao Z, Lee S, Xu H, Allegretti P, Horton TM, Yeh B, Moeller HP, Nichols RJ, McCutcheon D, et al. (2018). CC-401 Promotes β-Cell Replication via Pleiotropic Consequences of DYRK1A/B Inhibition. Endocrinology 159, 3143–3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S, Burgess SJ, Skaalrud D, Kelly JX, and Peyton DH (2010). Reversal Agent and Linker Variants of Reversed Chloroquines: Activities against Plasmodium falciparum. J. Med. Chem 53, 916–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annes JP, Ryu JH, Lam K, Carolan PJ, Utz K, Hollister-Lock J, Arvanites AC, Rubin LL, Weir G, and Melton DA (2012). Adenosine kinase inhibition selectively promotes rodent and porcine islet β-cell replication. Proc. Natl. Acad. Sci. U. S. A 109, 3915–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asfari M, Janjic D, Meda P, Li G, Halban PA, and Wollheim CB (1992). Establishment of 2-mercaptoethanol-dependent differentiated insulin-secreting cell lines. Endocrinology 130,167–178. [DOI] [PubMed] [Google Scholar]
- Bhandari S, Watson N, Long E, Sharpe S, Zhong W, Xu S-Z, and Atkin SL (2008). Expression of Somatostatin and Somatostatin Receptor Subtypes 1–5 in Human Normal and Diseased Kidney. J. Histochem. Cytochem 56, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun M (2014). Chapter Seven - The Somatostatin Receptor in Human Pancreatic β-Cells In Vitamins & Hormones, Litwack G, ed. (Academic Press; ), pp. 165–193. [DOI] [PubMed] [Google Scholar]
- Brion M, Lambs L, and Berthon G(1985). Metal ion-tetracycline interactions in biological fluids. Part 5. Formation of zinc complexes with tetracycline and some of its derivatives and assessment of their biological significance. Agents Actions 17, 229–242. [DOI] [PubMed] [Google Scholar]
- Chimienti F, Devergnas S, Pattou F, Schuit F, Garcia-Cuenca R, Vandewalle B, Kerr-Conte J, Lommel LV, Grunwald D, Favier A, et al. (2006). In vivo expression and functional characterization of the zinc transporter ZnT8 in glucose-induced insulin secretion. J. Cell Sci 119, 4199–4206. [DOI] [PubMed] [Google Scholar]
- Danscher G, and Stoltenberg M (2005). Zinc-specific autometallographic in vivo selenium methods: tracing of zinc-enriched (ZEN) terminals, ZEN pathways, and pools of zinc ions in a multitude of other ZEN cells. J. Histochem. Cytochem. Off. J. Histochem. Soc 53, 141–153. [DOI] [PubMed] [Google Scholar]
- Dirice E, Walpita D, Vetere A, Meier BC, Kahraman S, Hu J, Dancík V, Burns SM, Gilbert TJ, Olson DE, et al. (2016). Inhibition of DYRK1A Stimulates Human β-Cell Proliferation. Diabetes 65,1660–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrell C, Grompe MT, Pan FC, Zhong Y, Canaday PS, Shultz LD, Greiner DL, Wright CV, Streeter PR, and Grompe M (2011). Isolation of mouse pancreatic alpha, beta, duct and acinar populations with cell surface markers. Mol. Cell. Endocrinol 339,144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efrat S, Fusco-DeMane D, Lemberg H, Emran O. al, and Wang X (1995). Conditional transformation of a pancreatic beta-cell line derived from transgenic mice expressing a tetracycline-regulated oncogene. Proc. Natl. Acad. Sci 92, 3576–3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson O, Selvaraju RK, Johansson L, Eriksson JW, Sundin A, Antoni G, Sörensen J, Eriksson B, and Korsgren O (2014). Quantitative Imaging of Serotonergic Biosynthesis and Degradation in the Endocrine Pancreas. J. Nucl. Med 55, 460–465. [DOI] [PubMed] [Google Scholar]
- Eriksson O, Laughlin M, Brom M, Nuutila P, Roden M, Hwa A, Bonadonna R, and Gotthardt M (2016). In vivo imaging of beta cells with radiotracers: state of the art, prospects and recommendations for development and use. Diabetologia 59, 1340–1349. [DOI] [PubMed] [Google Scholar]
- Eriksson O, Johnström P, Cselenyi Z, Jahan M, Selvaraju RK, Jensen-Waern M, Takano A, Winzell MS, Halldin C, Skrtic S, et al. (2018). In Vivo Visualization of β-Cells by Targeting of GPR44. Diabetes 67,182–192. [DOI] [PubMed] [Google Scholar]
- Fabian MA, W.H. B Iii, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, et al. (2005). A small molecule- kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol 23, 329–336. [DOI] [PubMed] [Google Scholar]
- Fiedor P, Wataszewski J, Oluwole SF, Licihska I, Mazurek AP, Hardy MA, and RowMski W(1996). A novel approach to in vivo visualization of human pancreatic islets. Transplant. Proc 28, 3514. [PubMed] [Google Scholar]
- Finan B, Yang B, Ottaway N, Stemmer K, Müller TD, Yi C-X, Habegger K, Schriever SC, García-Cáceres C, Kabra DG, et al. (2012). Targeted estrogen delivery reverses the metabolic syndrome. Nat. Med 18,1847–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster MC, Leapman RD, Li MX, and Atwater I (1993). Elemental composition of secretory granules in pancreatic islets of Langerhans. Biophys. J 64, 525–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson CJ (1989). Neurobiology of Zinc and Zinc-Containing Neurons. In International Review of Neurobiology, Smythies JR, and Bradley RJ, eds. (Academic Press; ), pp. 145–238. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Kasarskis EJ, Ringo D, and Frederickson RE (1987). A quinoline fluorescence method for visualizing and assaying the histochemically reactive zinc (bouton zinc) in the brain. J. Neurosci. Methods 20, 91–103. [DOI] [PubMed] [Google Scholar]
- Frothingham R (2005). Glucose homeostasis abnormalities associated with use of gatifloxacin. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am 41,1269–1276. [DOI] [PubMed] [Google Scholar]
- Fukada T, and Kambe T (2014). Zinc Signals in Cellular Functions and Disorders (Springer; ). [Google Scholar]
- Gee KR, Zhou Z-L, Qian W-J, and Kennedy R (2002). Detection and imaging of zinc secretion from pancreatic beta-cells using a new fluorescent zinc indicator. J. Am. Chem. Soc 124, 776–778. [DOI] [PubMed] [Google Scholar]
- Giblin LJ, Chang CJ, Bentley AF, Frederickson C, Lippard SJ, and Frederickson CJ (2006). Zinc-secreting Paneth cells studied by ZP fluorescence. J. Histochem. Cytochem. Off. J. Histochem. Soc 54, 311–316. [DOI] [PubMed] [Google Scholar]
- Gotoh M, Maki T, Satomi S, Porter J, Bonner-Weir S, Ochara CJ·, and Monaco AP (1987). REPRODUCIBLE HIGH YIELD OF RAT ISLETS BY STATIONARY IN VITRO DIGESTION FOLLOWING PANCREATIC DUCTAL OR PORTAL VENOUS COLLAGENASE INJECTION1. Transplantation 43, 725. [DOI] [PubMed] [Google Scholar]
- Graham FL, Smiley J, Russell WC, and Nairn R(1977). Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol 36, 59–74. [DOI] [PubMed] [Google Scholar]
- Hammarström L, Hellman B, and Ullberg S (1967). On the accumulation of alloxan in the pancreatic β-cells. Diabetologia 3, 340–345. [DOI] [PubMed] [Google Scholar]
- Hao X, Jin Q, Va P, Li C, Shen W, Laffitte B, and Wu TY-H (2016). Pancreas- Specific Delivery of β-Cell Proliferating Small Molecules. ChemMedChem 11,1129–1132. [DOI] [PubMed] [Google Scholar]
- Hellström-Lindahl E, Danielsson A, Ponten F, Czernichow P, Korsgren O, Johansson L, and Eriksson O (2016). GPR44 is a pancreatic protein restricted to the human beta cell. Acta Diabetol 53, 413–421. [DOI] [PubMed] [Google Scholar]
- Hibbard PL (1937). A dithizone method for measurement of small amounts of zinc. Ind. Eng. Chem. Anal. Ed 9,127–131. [Google Scholar]
- Ichise M, and Harris PE (2010). Imaging of β-Cell Mass and Function. J. Nucl. Med 51,1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaraman S (2008). A novel method for the detection of viable human pancreatic beta cells by flow cytometry using fluorophores that selectively detect labile zinc, mitochondrial membrane potential and protein thiols. Cytom. Part J. Int. Soc. Anal. Cytol 73, 615–625. [DOI] [PubMed] [Google Scholar]
- Jindal RM, Taylor RP, Gray DW, Esmeraldo R, and Morris PJ (1992). A new method for quantification of islets by measurement of zinc content. Diabetes 41, 1056–1062. [DOI] [PubMed] [Google Scholar]
- Kelleher SL, McCormick NH, Velasquez V, and Lopez V (2011). Zinc in Specialized Secretory Tissues: Roles in the Pancreas, Prostate, and Mammary Gland. Adv. Nutr. Int. Rev. J 2,101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klochendler A, Caspi I, Corem N, Moran M, Friedlich O, Elgavish S, Nevo Y, Helman A, Glaser B, Eden A, et al. (2016). The Genetic Program of Pancreatic β- Cell Replication In Vivo. Diabetes 65, 2081–2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Körner M, Stöckli M, Waser B, and Reubi JC (2007). GLP-1 Receptor Expression in Human Tumors and Human Normal Tissues: Potential for In Vivo Targeting. J. Nucl. Med 48, 736–743. [DOI] [PubMed] [Google Scholar]
- Latif ZA, Noel J, and Alejandro R (1988). A simple method of staining fresh and cultured islets. Transplantation 45, 827–830. [PubMed] [Google Scholar]
- Lenzen S (2008). The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia 51,216–226. [DOI] [PubMed] [Google Scholar]
- Li YV (2014). Zinc and insulin in pancreatic beta-cells. Endocrine 45,178–189. [DOI] [PubMed] [Google Scholar]
- Li Z-Q·, Wu F-J, Gong Y, Hu C-W·, Zhang Y-H, and Gan M-Y (2007). Synthesis, Characterization and Activity against Staphylococcus of Metal(II)-Gatifloxacin Complexes. Chin. J. Chem 25, 1809–1814. [Google Scholar]
- Lukowiak B, Vandewalle B, Riachy R, Kerr-Conte J, Gmyr V, Belaich S, Lefebvre J, and Pattou F (2001). Identification and purification of functional human beta- cells by a new specific zinc-fluorescent probe. J. Histochem. Cytochem. Off. J. Histochem. Soc 49, 519–528. [DOI] [PubMed] [Google Scholar]
- Maret W (2013). Zinc biochemistry: from a single zinc enzyme to a key element of life. Adv. Nutr. Bethesda Md 4, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNary WF (1954). Zinc-dithizone reaction of pancreatic islets. J. Histochem. Cytochem. Off. J. Histochem. Soc 2,185–194. [DOI] [PubMed] [Google Scholar]
- Meeusen JW, Nowakowski A, and Petering DH (2012). Reaction of Metal Binding Ligands with the Zinc Proteome: Zinc Sensors and TPEN. Inorg. Chem 51,3625–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, and Butler PC (2008). Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 57,1584–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milo-Landesman D, Surana M, Berkovich I, Compagni A, Christofori G, Fleischer N, and Efrat S (2001). Correction of hyperglycemia in diabetic mice transplanted with reversibly immortalized pancreatic beta cells controlled by the tet-on regulatory system. Cell Transplant 10, 645–650. [PubMed] [Google Scholar]
- Nielsen LL, and Baron AD (2003). Pharmacology of exenatide (synthetic exendin- 4) for the treatment of type 2 diabetes. Curr. Opin. Investig. Drugs Lond. Engl 2000 4, 401–405. [PubMed] [Google Scholar]
- Nowakowski AB, Meeusen JW, Menden H, Tomasiewicz H, and Petering DH (2015). Chemical-Biological Properties of Zinc Sensors TSQ and Zinquin: Formation of Sensor-Zn-Protein Adducts versus Zn(Sensor)2 Complexes. Inorg. Chem 54, 11637–11647. [DOI] [PubMed] [Google Scholar]
- Pearce DA, Jotterand N, Carrico IS, and Imperiali B (2001). Derivatives of 8- hydroxy-2-methylquinoline are powerful prototypes for zinc sensors in biological systems. J. Am. Chem. Soc 123, 5160–5161. [DOI] [PubMed] [Google Scholar]
- Lindström Per (1981). Further studies on 5-hydroxytryptamine transport in pancreatic islets and isolated β-cells. Br. J. Pharmacol 73, 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakieten N, Rakieten ML, and Nadkarni MV (1963). Studies on the diabetogenic action of streptozotocin (NSC-37917). Cancer Chemother. Rep 29, 91–98. [PubMed] [Google Scholar]
- Rice DR, Vacchina P, Norris-Mullins B, Morales MA, and Smith BD (2016). Zinc(II)-Dipicolylamine Coordination Complexes as Targeting and Chemotherapeutic Agents for Leishmania major. Antimicrob. Agents Chemother 60, 2932–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Taylor B, Jin Q, Nguyen-Tran V, Meeusen S, Zhang Y-Q, Kamireddy A, Swafford A, Powers AF, Walker J, et al. (2015). Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Nat. Commun 6, 8372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi Y, Takamoto I, Kubota N, Matsui J, Suzuki R, Komeda K, Hara A, Toyoda Y, Miwa I, Aizawa S, et al. (2007). Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance. J. Clin. Invest 117, 246–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonlanthen M, Connelly CM, Deiters A, Linden A, and Finney NS (2014). Thiourea-Based Fluorescent Chemosensors for Aqueous Metal Ion Detection and Cellular Imaging. J. Org. Chem 79, 6054–6060. [DOI] [PubMed] [Google Scholar]
- Walkup GK, Burdette SC, Lippard SJ, and Tsien RY (2000). A New Cell- Permeable Fluorescent Probe for Zn2+. J. Am. Chem. Soc 122, 5644–5645. [Google Scholar]
- Wang P, Alvarez-Perez J-C, Felsenfeld DP, Liu H, Sivendran S, Bender A, Kumar A, Sanchez R, Scott DK, Garcia-Ocaña A, et al. (2015). A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat. Med 21,383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Walker JR, Wang X, Tremblay MS, Lee JW, Wu X, and Schultz PG (2009). Identification of small-molecule inducers of pancreatic β-cell expansion. Proc. Natl. Acad. Sci. U. S. A 106,1427–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Chen Z, Shiota C, Prasadan K, Guo P, El-Gohary Y, Paredes J, Welsh C, Wiersch J, and Gittes GK (2013). No evidence for β cell neogenesis in murine adult pancreas. J. Clin. Invest 123, 2207–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Chen J, Jing G, and Shalev A (2012). Preventing β-Cell Loss and Diabetes With Calcium Channel Blockers. Diabetes 61,848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Low YS, Armstrong NA, Ryu JH, Sun SA, Arvanites AC, Hollister-Lock J, Shah NH, Weir GC, and Annes JP (2014). Repurposing cAMP-modulating medications to promote β-cell replication. Mol. Endocrinol. Baltim. Md 28,1682–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Abdolazimi Y, Armstrong NA, and Annes JP (2016). A High-content In Vitro Pancreatic Islet β-cell Replication Discovery Platform. JoVE J. Vis. Exp e54298–e54298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data is deposited in Mendeley Data under: http://dx.doi.org/10.17632/3ycr7rs56f.1