

Abstract
A new bench-stable trifluoromethylation reagent, phenyl bromodifluoroacetate, converts readily available alcohols to trifluoromethanes in a Cu-catalyzed deoxytrifluoromethylation reaction. This reaction streamlines access to target biologically active molecules, and should be useful for a variety of medicinal, agricultural, and materials chemists.
Graphical Abstract
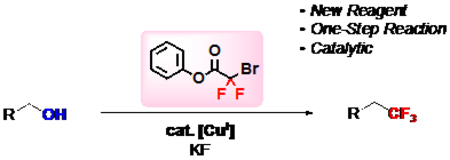
Introduction
Methods for directly converting common functional groups to fluorinated motifs enable the rapid and efficient discovery and development of new agrochemicals, materials, and pharmaceuticals.1 In particular, protocols that employ simple reagents and mild reaction conditions can facilitate both late-stage diversification of lead compounds and large-scale manufacture of chemicals. Given the ubiquity of alcohols in bioactive compounds, materials, and synthetic intermediates, methods that directly convert alcohols to useful fluorinated functional groups that alter physicochemical and biophysical properties of a parent compound are highly valuable.2 This powerful strategy has been realized for converting alcohols to monofluorinated analogs, which is an essential strategy to selectively and quickly incorporate fluorine into molecules (Figure 1B, left).1a For these one-step deoxyfluorination reactions of alcohols, many benchmark reagents are commercially available, and new methods are continuously being developed.3 However, analogous one-pot deoxytrifluoromethylation protocols for transforming alcohols to trifluoromethanes, another useful functional group for medicinal, agricultural and materials chemistries, remain extremely limited.
Figure 1:

a) Methods that convert common functional groups to fluorinated motifs are valuable to discovery and process chemists. b) While protocols that directly transform alcohols to alkyl fluorides are widely used, analogous one-pot methods to access trifluoromethanes from alcohols are rare. c) Development of a new reagent enables Cu-catalyzed deoxytrifluoromethylation.
Considering deoxyfluorination and deoxytrifluoromethylation reactions, intrinsic differences in stability and reactivity of –F and –CF3 render the latter reaction challenging. The deoxyfluorination strategy typically relies on in situ activation of alcohols to generate more electrophilic intermediates, and subsequent substitution by nucleophilic –F. Fluoride is a stable and small anion that can directly engage in nucleophilic substitutions, such as deoxyfluorination reactions. In contrast, –CF3 is unstable, bulky, and decomposes quickly in solution in the absence of a competent (usually sp2-hybridized) electrophile (Figure 1B, right).4 Because of this instability, –CF3 does not react through pure nucleophilic substitution mechanisms, but instead requires a transition metal to effectively generate a new C–CF3 bond.5 However, conditions for activating alcohols have historically not been compatible with transition metal-catalyzed reactions. As such, deoxytrifluoromethylation reactions of alcohols typically require multi-step transformations that inefficiently manipulate oxidation states, require excess time and labor, generate excess waste, decrease yields of desired products, and limit the use of functional groups that are sensitive to oxidation, reduction, and/or strong nucleophiles.6 Herein, we describe the development of a new reagent that, in concert with a Cu-based catalyst, enables the direct conversion of alcohols to trifluoromethanes under mild conditions.
Results and Discussion
To develop a one-step deoxytrifluoromethylation reaction, we aimed to develop a new reagent that would convert an alcohol to a suitable leaving group for reaction with Cu–CF3 under mild conditions (Figure 1C). Among several alcohol-derived electrophiles, we focused on pioneering work of Chen and co-workers,7 who established the Cu-mediated C–O bond activation of bromodifluoroacetates, and the compatibility of this system with Cu-mediated C–CF3 bond formation. Considering our previous work with bromodifluroacetates,8 we envisioned that a catalytic one-step deoxytrifluoromethylation reaction could be achieved by designing a stable reagent that would convert the starting alcohol to the corresponding bromodifluoroacetate derivative in situ, but that would not interfere with catalysis. We initially explored commercially available bromodifluoroacetates (e.g. MeO2CCF2Br and NaO2CCF2Br) as deoxytrifluoromethylation reagents; however, despite achieving catalytic turnover, thorough evaluation of reaction conditions did not provide a suitable system for deoxytrifluoromethylation (Figure 2A). We reasoned that water or an alcohol, generated from transesterification step, might adversely affect the final Cu-catalyzed decarboxylative trifluoromethylation step. To test this hypothesis, Cu-catalyzed trifluoromethylation of cinnamyl bromodifluoroacetate (3a) was conducted in the presence of an alcohol or H2O (1 equiv.). Compared to the control reaction, addition of MeOH or H2O decreased the yield of trifluoromethylated product (4a) from 75% to 19–24% (Figure 2B), thus confirming the incompatibility of simple H2O and MeOH leaving groups (LG–H).
Figure 2:

Design of Deoxytrifluoromethylation Reagent Required Identification of Benign Leaving Group.
Subsequent efforts identified different LG–H that would not interfere with the Cu-catalyzed process (Figure 2C). To identify such leaving groups (LG), we evaluated the effects of different additives (1.0 equiv.) on the conversion of cinnamyl bromodifluoroacetate 3a into trifluoromethyl products (Figure 2D).8a In these reactions, succinimide did not decrease the yield of trifluoromethyl product 4a, while phenol and trifluoroethanol slightly lowered the yields of 4a. Other N-based additives (e.g. 2-oxazolidinone, N-tosylaniline, and N–H heterocycles), thiols, and other alcohols, negatively affected the Cu-catalyzed process. Based on these additives, we prepared several bromodifluoroacetate (BDFA) -derived esters, and tested them as reagents in the deoxytrifluoromethylation reaction of cinnamyl alcohol 2a using optimized conditions (Table 1). Under these conditions, commercially available NaO2CCF2Br (1a), MeO2CCF2Br (1b), and EtO2CCF2Br (1c) still provided low yields of the desired product. Among the newly prepared reagents, phenyl bromodifluoroacetate (PhBDFA, 1d) afforded the best compromise between availability, stability, cost and yield of the desired cinnamyl trifluoromethane (4a). Substitutions on the phenyl ring of the PhBDFA (1d) to perturb electronic character (1d–1g) or steric hindrance (1h) did not improve the reaction. An exchange of the oxygen for the sulfur in compound 1i decreased the yield, which is consistent with our additive based screening. Similarly, bromodifluoroacetamide 1j provided similar yields as compared to the easier to synthesize bromodifluoroacetate esters. Moreover, reactions using 1j generated significant quantities of side products deriving from the allylation of N-tosylaniline. In addition, the bromodifluoroacylated succinimide was insufficiently stable for isolation, thus limiting the use of this presumably useful leaving group. Ultimately, PhBDFA (1d) was selected as the most suitable reagent for deoxytrifluoromethylation, given the inexpensive and readily available nature of phenol, and the reasonable stability of the phenyl ester toward storage.9
Table 1:
Phenyl Bromodifluoroacetate (PhBDFA) Provided the Trifluoromethyl Product 4a in Higher Yield than Other Bromodifluoroacylating Reagents
 |
Using phenyl bromodifluoroacetate (PhBDFA, 1d), representative allylic (2a–2i), propargylic (5a–5f) and benzylic (7a–7e) alcohols were trifluoromethylated in moderate to good yields (Tables 2–4). For allylic8a and propargylic8c substrates, direct deoxytrifluoromethylation typically provided higher yields than two-step protocols involving formation and purification of intermediate bromodifluoroacetic esters and subsequent Cu-catalyzed decarboxylative trifluoromethylation. Notably, many allylic and propargylic bromodifluoroacetates previously explored8a, c were unstable to basic conditions, silica gel, and cold storage (slowly decomposing in the refrigerator for weeks). Therefore, the one-step protocol offered distinct advantages by avoiding isolation of the instable intermediate bromodifluoroacetic esters. The direct deoxytrifluoromethylation displayed good functional group tolerance, with cinnamyl derivatives 2a–2e illustrating good compatibility with electron-donating, electron-withdrawing, and halogen functional groups. In addition, the reaction tolerated non-cinnamyl substrates, such as allylic alcohol 2f. Endocyclic alkenols, a structural unit present in several natural products, served as competent substrates, and delivered 4g–4h in good yields. As an example of such natural products, perillyl alcohol afforded trifluoromethyl product 4i in 57% of yield. In addition, selected propargylic alcohols 5a–5f were also deoxytrifluoromethylated to afford trifluoromethylallenes 6a–6f, bearing useful ester, amide, and chlorine groups.10 In contrast to allylic and propargylic alcohols, benzyl alcohols 7a–7e were more challenging substrates, and required higher temperature to afford synthetically useful yields with electron-rich rings (7a–7b). However, the reaction is compatible with a range of drug-like heterocycles, such as indole (7c), pyrazole (7d), and indazole (7e).6e Together, these examples showcase the potential of this deoxytrifluoromethylation reaction to provide valuable trifluoromethyl drug-like building-blocks from simple alcohols in a single-step.
Table 2:
Phenyl Bromodifluoroacetate Enables Cu-catalyzed One-pot Deoxytrifluoromethylation of Allylic Alcoholsa
 |
Standard reaction conditions: 2a–2i (0.50 mmol), 1d (1.0 mmol), CuI (0.050 mmol), DMEDA (0.055 mmol), HO2CCF2Br (0.25 mmol), KF (1.0 mmol), DMF (1.0 mL), 25 °C. Yield of material after chromatographic purification. For comparison, two-pot yields reported from reference 8a.
Reaction conducted on 5.0 mmol scale.
Yield determined by 19F NMR spectroscopy using α,α,α-trifluorotoluene as an internal standard.
Table 4:
Phenyl Bromodifluoroacetate Enables Cu-catalyzed One-pot Deoxytrifluoromethylation of Benzylic Alcoholsa
 |
Standard reaction conditions: 7a–7e (0.50 mmol), 1d (1.0 mmol), CuI (0.10 mmol), MeO2CCF2Br (0.20 mmol), KI (0.12 mmol), KF (2.0 mmol), DMF (0.25 mL), MeCN (0.25 mL), 70 °C. For comparison, two-pot yields reported from reference 8d.
To further illustrate the potential impact of the catalytic one-step deoxytrifluoromethylation reaction, we prepared trifluoromethylated intermediates 10 and 12 toward the syntheses of the COX-2-inhibitor L-784–512,11 and a fluorinated analog of Tebufenpyrad,12 respectively. Previously, allyl trifluoromethane 10 was prepared in a one-pot two-step procedure in 74% yield employing chlorodifluoroacetic anhydride [O(O2CCF2Cl)2] as the trifluoromethylating reagent; however, stoichiometric amounts of CuI were required to suppress the formation of the corresponding allyl chloride side-product (Scheme 1A). In contrast, use of PhBDFA (1d) as the trifluoromethylating reagent provided a similar yield with only a catalytic amount of copper salt. A more insightful comparison involved the synthesis of trifluoroethyl pyrazole 12 (Scheme 1B). The original route to this compound required four steps, including one final deoxygenation reaction with excess of undesirable Bu3SnH (Route i).12a In a second preparation of 12, our previous two-step reaction provided 12 in 60% yield (Route ii).8d Finally, when employing PhBDFA (1d), our catalytic conditions afforded the desired product directly from the alcohol in 57% yield (Route iii). This operationally straightforward one-step catalytic deoxytrifluoromethylation reaction provides direct access to a valuable fluorinated product from a simple alcohol precursor, while avoiding inefficient redox manipulations, chromatographic purifications, and generation of stoichiometric metallic byproducts. To further highlight the utility of the reaction, we carried out the deoxytrifluomethylation of cinnamyl alcohol 2a in multigram scale, as the scale up of related aromatic trifluoromethylation reactions using a related reagent (MeO2CCF2Cl) has proved problematic.13 In our case, no loss in the yield was observed relative to the small scales reactions, and 2.7 g of product was obtained in a single-reaction. In perspective, these examples illustrate the streamlined synthetic routes enabled by the catalytic one-step deoxytrifluoromethylation reaction, and its potential to be used in small multigram synthesis.
Scheme 1:

Catalytic One-Step Deoxytrifluoromethylation Reaction: (a–b) Shortens the Synthesis of Known Intermediates to the Synthesis of COX-2 Inhibitor L-784,512 and a Fluorinated Analog of Tebufenpyrad; (c) Is Equally Efficient on Multi-Gram Scale.
Several data support a mechanism involving a fast acylation/transesterification step prior to the Cu-catalyzed decarboxylative trifluoromethylation reaction (Scheme 2A). Using time-course analysis, we observed a quick acylation of cinnamyl alcohol 2a forming the corresponding bromodifluoroacetate 3a during the first 15 min of the Cu-catalyzed reaction, reaching a maximum concentration at ~2 h, and then converting to product 4a over the remaining course of the reaction (see SI for details). Further experiments identified KF as the key component in facilitating formation of the thermodynamically favored ester 3a. In contrast, excess BDFA, serving as a Brønsted acid, did not promote the transesterification step (Scheme 2B). Based on these results, relative to MeO2CCF2Br and other commercially available bromodifluoroacetates, the acylating ability of PhBDFA (1d) likely shifted the equilibrium of the transesterification step, thus increasing the concentration of the key bromodifluoroacetate 3a, and subsequently favoring the Cu-catalyzed trifluoromethylation (Scheme 2C). Such a facile acylation step supports an overall deoxytrifluoromethylation mechanism involving conversion of the alcohol to a bromodifluoracetate ester prior to a Cu-catalyzed decarboxylative trifluoromethylation, as previously suggested (Scheme 2A).7, 8e, 14
Scheme 2:

Control Experiments Suggest Rapid Formation of Bromodifluoroacetate 3a Prior to a Slow Cu-catalyzed Decarboxylative Trifluoromethylation Reaction
The ability of PhBDFA (1d) to serve as an effective trifluoromethylating reagent derived from its aforementioned acylating ability in addition to its stability under the reaction conditions (Scheme 2). Reagents previously used for trifluoromethylation of organic halides [RO2CCF2X (R = Na, Me, Et; X = Cl, Br)] decompose under mild conditions (>40 °C in the presence of copper)14–15 to release difluorocarbene, which polymerizes13 or reacts with alkenes or phenols.15b, 16 In contrast, minimal decomposition of the PhBDFA (1d) was observed (>82% of 1d recovered) under the reaction conditions (Scheme 2D). Moreover, the stability of PhBDFA (1d) discounted mechanisms involving initial outer-sphere thermal decomposition of 1d to generate difluorocarbene, and subsequent formation of the Cu–CF3, which has been implicated in trifluoromethylation reactions of organic halides.17 Overall, this analysis suggests that success of PhBDFA (1d), compared to MeO2CCF2Br, relies on the inertness of the leaving group (Figure 2D), and from equilibrium (Scheme 2C) and stability factors (Scheme 2D) of the newly designed reagent. Of note, the optimized conditions utilize MeO2CCF2Br or HO2CCF2Br additives to help generate the active Cu–CF3 catalyst. Though these species serve as poor reagents for esterifying the alcohol substrate (Figure 2A), they serve to convert the CuI precatalysts into the active Cu–CF3 species,8a and/or to regenerate Cu–CF3 that is lost throughout the reaction.8d Further, as previously reported,14 the CuI/KF-mediated conversion of MeO2CCF2Br or HO2CCF2Br to afford Cu–CF3 generates methyl iodide and CO2 as byproducts, which do not inhibit the catalytic reaction in a similar fashion to H2O and MeOH, which would be generated by transesterification.
Conclusion
In summary, a catalytic one-step deoxytrifluoromethylation reaction of readily available alcohols was developed using phenyl bromodifluoroacetate (1d, PhBDFA) as the key reagent. The new reaction provided diverse trifluoromethyl compounds in good yields and tolerated many useful functional groups. Moreover, its application to rapidly prepare intermediates in the synthesis of biologically active molecules illustrated the potential impact of this reaction. Initial mechanistic analysis suggested that the success of 1d, in comparison with commercially available bromodifluoroacetates, stems from both equilibrium and stability factors, in addition to its inertness under the catalytic conditions. The deoxytrifluoromethylation reaction complements the well-established deoxyfluorination reaction and opens up new possibilities to obtain fluoroalkylated products from readily available functional groups. Further studies to expand this reaction are undergoing in our lab.
Experimental Section
General Considerations
Unless otherwise noted, reactions were performed under an atmosphere of N2 using oven-dried glassware. Trifluoromethylation reactions in 1-dram vials sealed with a PTFE-lined screw cap. Reactions were setup inside a N2-filled glove box. All other reactions were performed in round-bottom flasks that were sealed with rubber septa using oven-dried glassware and standard Schlenk techniques. High-quality laboratory-grade polypropylene and polyethylene syringes bearing stainless steel needles were used to transfer air- and moisture-sensitive liquid reagents. Reactions were monitored by thin-layer chromatography (TLC) on UNIPLATE™ Silica Gel HLF 250 micron glass plates precoated with 230–400 mesh silica impregnated with a fluorescent indicator (250 nm), visualizing by quenching of fluorescence, or iodine. Purifications by column chromatography (SiO2) were conducted using an automated system or following Still’s general procedure. 19F NMR Yields and isolated yields reported in the manuscript represent an average of at least two independent runs. Yields reported in the supporting information refer to a single experiment.
Unless otherwise noted, reagents were purchased from commercial sources, and used as received. Commercial anhydrous potassium fluoride (KF) was further dried in high-vacuum (~1.5 Torr, 185 ºC) for 18 h, and stored in the glovebox. N,N’-dimethylethylenediamine (DMEDA) was distilled under nitrogen and stored in the glovebox. Anhydrous N,N-dimethylformamide (DMF), acetonitrile (CH3CN), methanol (MeOH), dichloromethane (DCM), tetrahydrofuran (THF), and triethylamine (NEt3) were dispensed from a solvent purification system, in which the solvent was dried by passage through two columns of activated alumina under argon.
Proton nuclear magnetic resonance (1H NMR) spectra and carbon nuclear magnetic resonance (13C NMR) spectra were recorded on Bruker AVIIIHD 400 spectrometer (400 and 100 MHz, respectively) or Bruker AVIII 500 with CPDUL cryoprobe spectrometer (500 and 125 MHz, respectively). Chemical shifts (δ) for protons are reported in parts per million (ppm) downfield from tetramethylsilane, and are referenced to proton resonance of residual CHCl3 in the NMR solvent (CHCl3: δ = 7.26 ppm). Chemical shifts (δ) for carbon are reported in ppm downfield from tetramethylsilane, and are referenced to the carbon resonances of the solvent residual peak (CDCl3: δ = 77.16 ppm). Fluorine nuclear magnetic resonance (19F NMR) spectra were recorded on a Bruker AVIIIHD 400 spectrometer (376 MHz). 19F NMR chemical shifts (δ) are reported in ppm upfield from trichlorofluoromethane (0 ppm). 19F NMR yields were determined from the crude mixtures using non-deuterated solvents and the relaxation delay time (D1) was set to 4 seconds to ensure reproducible results. NMR data are represented as follows: chemical shift (ppm), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, hept = heptet, m = multiplet), coupling constant in Hertz (Hz), integration.
Exact mass spectra were recorded using (1) electrospray ion source (ESI), positive mode, and time-of-flight (TOF) analyzer on a Waters LCT PremierTM mass spectrometer or (2) an atmospheric-pressure chemical ionization (APCI) ion source and TOF analyzer on a Waters Q-Tof PremierTM mass spectrometer, for which sample plus near mass internal exact mass standard were dissolved in hexane, and hexane or PhMe/hexane were used as the ionization solvent. Mass calibration was carried out directly before the measurement of the sample. Low-resolution mass spectra were recorded using an electron impact (EI) ion source and a tandem quadrupole analyzer on a Waters Quattro Micro GC mass spectrometer, injected via a gas chromatograph Agilent 6890N (GC/MS).
Quantitative gas chromatography (GC) analyses were recorded on an Agilent Technologies 7890A GC-system with Flame Induced Detector (FID) and a HP-5 column (0.32 mm × 30 m, film: 0.25 μm). The method used consisted of 70 ºC for 1 min, then 30 ºC/min to 250 ºC, and holding at 250 ºC for 1 min.
Infrared spectra were measured using attenuated total reflection (ATR) at a Fourier Transform Infrared Spectrometer. Uncorrected melting points were measured on Thomas Hoover Capillary Melting Point apparatus.
Preparation of Known Compounds
N-4-toluenesulfonylaniline,18 (E)-3-(4-methoxyphenyl)prop-2-en-1-ol (2b),8a (E)-3-(4-fluorophenyl)prop-2-en-1-ol (2d),8a (E)-3-(3-bromophenyl)prop-2-en-1-ol (2e),8a 3-(naphthalen-2-yl)prop-2-yn-1-ol (5a),8c 3-(2-methoxy-5-nitrophenyl)prop-2-yn-1-ol (5b),8c methyl 3-(3-hydroxyprop-1-yn-1-yl)benzoate (5c),8c tert-butyl 2-(3-hydroxyprop-1-yn-1-yl)-1H-indole-1-carboxylate (5d),8c 2,2,2-trifluoro-N-(4-(3-hydroxyprop-1-yn-1-yl)phenyl)acetamide (5d),8c 3-(3,4-dichlorophenyl)prop-2-yn-1-ol (5e),8c (1-phenyl-1H-pyrazol-4-yl)methanol (5f),8d (1-methyl-1H-indazol-3-yl)methanol (7e),19 (E)-2-methyl-3-(4-(methylsulfonyl)phenyl)prop-2-en-1-ol11 (9) and (5-(furan-2-yl)-1-methyl-1H-pyrazol-3-yl)methanol (11),12a were prepared according to previously literature reports. All compounds presented satisfactory analysis coherent with characterization data published previously.
Synthesis of Aryl Bromodifluoroacetates
General Procedure A:
An oven-dried two-neck flask equipped with a stirring bar, a gas outlet attached to the Schlenk line and a rubber septum under nitrogen was charged with bromodifluoroacetic acid (12.6 g, 72.0 mmol) and CH2Cl2 (150 mL). The resulting solution was cooled using an ice-water bath, and oxalyl chloride (5.7 mL, 66 mmol) was added. Next, DMF (0.23 mL, 3.0 mmol) was added dropwise (caution: rapid evolution of noxious gases). After stirring at low temperature for 10 min, the solution was stirred at room temperature until the gas evolution ceased (~3 h). Next, the reaction mixture was cooled using an ice-water bath, and phenol (60.0 mmol) was added in one portion. After 10 min stirring at low temperature, Et3N (10.0 mL, 72.0 mmol) was added, and the reaction was stirred for 30 min at low temperature, followed by stirring at room temperature for ~16 h. The reaction was quenched with HCl(aq) 1 mol L−1 (25 mL) and transferred to a separation funnel. The phases were separated, and the aqueous phase was extracted with CH2Cl2 (3 × 25 mL). The combined organic phases were washed with NaCl(sat) (1 × 25 mL), dried over anhydrous MgSO4, filtered, and concentrated. Purification by flash column chromatography provided the desired product.
Phenyl 2-bromo-2,2-difluoroacetate (1d)
General procedure A was followed using phenol (5.64 g, 60.0 mmol), bromodifluoroacetic acid (12.6 g, 72.0 mmol), oxalyl chloride (5.7 mL, 66 mmol), DMF (0.23 mL, 3.0 mmol), CH2Cl2 (0.15 L), and Et3N (10.0 mL, 72.0 mmol). Workup and chromatographic purification (100% hexanes) provided the title compound as a colorless oil (10.7 g, 71%).
1H NMR (CDCl3, 400 MHz) δ 7.49 – 7.43 (m, 2H), 7.37 – 7.30 (m, 1H), 7.25 – 7.18 (m, 2H). 13C{1H} NMR (CDCl3, 126 MHz) δ 158.1 (t, J = 32.3 Hz), 149.7, 130.0, 127.4, 120.6, 108.6 (t, J = 314.4 Hz). 19F NMR (CDCl3, 376 MHz) δ –61.6 (s, 2 F). IR (ATR) 3076, 1786, 1589, 1493, 1283, 1186, 1157, 1107, 945, 906, 839, 746, 704, 685 cm−1. HRMS (APCI, m/z): calcd for C8H5BrF2O2 [M]+ 249.9441, found 249.9433.
4-Methoxyphenyl 2-bromo-2,2-difluoroacetate (1e)
General procedure A was followed using 4-methoxyphenol (1.24 g, 10.0 mmol), bromodifluoroacetic acid (2.10 g, 12.0 mmol), oxalyl chloride (950 μL, 11.0 mmol), DMF (0.31 mL, 4.0 mmol), CH2Cl2 (0.030 L), and Et3N (1.4 mL, 10 mmol). Workup and chromatographic purification (100% hexanes) provided the title compound as a colorless oil (1.57 g, 56%).
1H NMR (CDCl3, 400 MHz) δ 7.17 – 7.10 (m, 2H), 6.98 – 6.91 (m, 2H), 3.82 (s, 3H). 13C{1H} NMR (CDCl3, 126 MHz) δ 158.4 (t, J = 32.1 Hz), 158.3, 143.2, 121.4, 114.9, 108.7 (t, J = 314.8 Hz), 55.7. 19F NMR (CDCl3, 376 MHz) δ –61.5 (s, 2 F). IR (ATR) 3005, 2963, 2839, 1786, 1599, 1504, 1466, 1443, 1286, 1250, 1180, 1109, 1032, 947, 854, 812, 750, 692 cm−1. HRMS (APCI, m/z): calcd for C9H7BrF2O3 [M]+ 279.9547, found 279.9536.
[1,1’-biphenyl]-4-yl 2-bromo-2,2-difluoroacetate (1f)
General procedure A was followed using 4-phenylphenol (5.10 g, 30.0 mmol), bromodifluoroacetic acid (3.90 g, 20.0 mmol), oxalyl chloride (2.1 mL, 24 mmol), DMF (0.62 mL, 8.0 mmol) CH2Cl2 (0.060 L), and Et3N (5.6 mL, 40 mmol). Instead of the work up described in General Procedure A, the reaction solvent was evaporated, and the crude mixture was washed with hexanes (100 mL). Next, a second filtration in SiO2 (10% of EtOAc in hexanes) was performed to provide the title compound as a white solid (5.5 g, 84%), contaminated with ~6% of 4-phenylphenol (estimated by 1H-NMR).
mp: 90 – 92 ºC (decomp.). 1H NMR (CDCl3, 400 MHz) δ 7.66 (d, J = 8.7 Hz, 2H), 7.61 – 7.56 (m, 2H), 7.46 (t, J = 7.4 Hz, 2H), 7.38 (t, J = 7.3 Hz, 1H), 7.30 (d, J = 8.7 Hz, 2H). 13C{1H} NMR (CDCl3, 126 MHz) δ 158.1 (t, J = 32.0 Hz), 149.1, 140.7, 139.9, 129.1, 128.7, 127.9, 127.3, 120.9, 108.6 (t, J = 314.3 Hz). 19F NMR (CDCl3, 376 MHz) δ –61.6 (s, 2 F). IR (ATR) 3040, 1794, 1518, 1485, 1217, 1199, 1107, 933, 862, 758, 685 cm−1. HRMS (APCI, m/z): calcd for C14H9BrF2O2 [M]+ 325.9754, found 325.9748.
4-(trifluoromethyl)phenyl 2-bromo-2,2-difluoroacetate (1g)
General procedure A was followed using 4-trifluoromethylphenol (1.62 g, 10.0 mmol), bromodifluoroacetic acid (2.10 g, 12.0 mmol), oxalyl chloride (950 μL, 11.0 mmol), DMF (0.31 mL, 4.0 mmol), CH2Cl2 (0.030 L), and Et3N (1.4 mL, 10 mmol). Workup and a fast filtration in SiO2 (10% of EtOAc in hexanes) provided the title compound as a colorless oil (1.36 g, 43%).
1H NMR (CDCl3, 400 MHz) δ 7.74 (d, J = 8.4 Hz, 1H), 7.37 (d, J = 8.3 Hz, 1H). 13C{1H} NMR (CDCl3, 126 MHz) δ 157.6 (t, J = 32.7 Hz), 152.0, 129.9 (q, J = 33.5 Hz), 127.5 (q, J = 3.6 Hz), 123.7 (q, J = 272.2 Hz), 121.3, 115.6, 108.3 (t, J = 314.3 Hz). 19F NMR (CDCl3, 376 MHz) δ –61.9 (s, 2 F), –63.0 (s, 3 F). IR (ATR) 1792, 1612, 1510, 1417, 1323, 1279, 1201, 1165, 1105, 1064, 1018, 941, 866, 812, 704 cm−1. HRMS (APCI, m/z): calcd for C9H4BrF5O2 [M]+ 317.9315, found 317.9312.
2,6-dimethylphenyl 2-bromo-2,2-difluoroacetate (1h)
General procedure A was followed using 2,6-dimethylphenol (1.83 g, 15.0 mmol), bromodifluoroacetic acid (1.93 g, 11.0 mmol), oxalyl chloride (1.0 mL, 12 mmol), DMF (0.31 mL, 4.0 mmol) CH2Cl2 (0.030 L), and Et3N (2.8 mL, 20 mmol). Workup and chromatographic purification (100% hexanes) provided the title compound as a colorless oil (1.6 g, 52%).
1H NMR (400 MHz, CDCl3) δ 7.16 – 7.08 (m, 3H), 2.22 (s, 6H). 13C{1H} NMR (126 MHz, CDCl3) δ 198.5 (t, J = 31.88 Hz), 188.0, 171.0, 170.3, 168.3, 149.6 (t, J = 314.04 Hz), 57.2. 19F NMR (CDCl3, 376 MHz) δ –60.9 (s, 2 F). IR (ATR) 3047, 2927, 1786, 1475, 1286, 1167, 1147, 1111, 945, 841, 770, 700 cm−1. HRMS (APCI, m/z): calcd for C10H9BrF2O2 [M]+ 277.9754, found 277.9743.
S-phenyl 2-bromo-2,2-difluoroethanethioate (1i)
General procedure A was followed using 4-methoxyphenol (1.10 g, 10.0 mmol), bromodifluoroacetic acid (2.10 g, 12.0 mmol), oxalyl chloride (950 μL, 11.0 mmol), DMF (0.31 mL, 4.0 mmol) CH2Cl2 (0.030 L), and Et3N (1.7 mL, 12 mmol). Workup and chromatographic purification (100% hexanes) provided the title compound as a colorless oil (0.4 g, 16%).
1H NMR (400 MHz, CDCl3) δ 7.67 – 7.39 (m, 1H). 13C{1H} NMR (126 MHz, CDCl3) δ 185.6 (t, J = 30.8 Hz), 134.8, 130.9, 130.0, 123.8, 113.2, (t, J = 319.7 Hz). 19F NMR (CDCl3, 376 MHz) δ –59.5 (s, 2 F). IR (ATR) 3065, 1712, 1479, 1442, 1192, 1144, 979, 815, 744, 687 cm−1. HRMS (APCI, m/z): calcd for C8H5BrF2OS [M]+ 265.9213, found 265.9207.
2-bromo-2,2-difluoro-N-phenyl-N-tosylacetamide (1j)
General procedure A was followed using N-p-toluenesulfonylaniline18 (0.54 g, 2.2 mmol), bromodifluoroacetic acid (0.35 g, 2.0 mmol), oxalyl chloride (0.18 mL, 2.1 mmol), DMF (5.0 μL, 0.1 mmol) CH2Cl2 (4.0 mL), and Et3N (0.39 mL, 2.8 mmol). Workup and chromatographic purification (20% EtOAc in hexanes) provided the title compound as a white solid (0.35 g, 43%).
mp: 136 – 138 ºC. 1H NMR (CDCl3, 400 MHz) δ 7.9 (d, J = 8.39 Hz, 2H), 7.6 – 7.5 (m, 1H), 7.5 – 7.4 (m, 2H), 7.4 (d, J = 8.66 Hz, 2H), 7.3 (d, J = 7.65 Hz, 2H), 2.5 (s, 3H). 13C{1H} NMR (CDCl3, 126 MHz) δ 157.9 (t, J = 28.19 Hz), 146.4, 134.3, 133.4, 131.1, 131.0, 129.9, 129.9, 129.4, 110.6 (t, J = 319.13 Hz), 22.0. 19F NMR (CDCl3, 376 MHz) δ –54.5 (s, 2 F). IR (ATR) 1720, 1361, 1166, 1117, 1084, 883, 398, 671, 572, 543 cm−1. HRMS (ESI, m/z): calcd for C15H12BrF2NO3S [M+Na]+ 425.9587, found 425.9557.
Initial Screening and Optimization of Reaction Conditions
Additive Screening Experiment
Experimental Procedure:
A 1 dram vial sealed with PTFE septum under N2 was charged with cinnamyl bromodifluoroacetate (3a) (58 mg, 0.20 mmol), KF (17 mg, 0.30 mmol), additive (0.20 mmol), DMF (0.10 mL), and a magnetic stir bar. The vial was placed on a pre-heated reaction block at 50 ºC, and an aliquot of the “Stock Solution of Activated Catalyst” (see infra) (0.10 mL) was transferred to the vial via syringe. After heating at 50 ºC for 14 h, the vial was removed from the heating block and cooled to room temperature. Next, the reaction mixture was diluted with EtOAc (2 mL), and dodecane (45 μL, 0.20 mmol) was injected as an internal standard, and the reaction mixture was stirred at room temperature for 10 minutes to ensure thorough mixing. An aliquot was taken from the vial, analyzed by GC-FID, and quantified using a standard curve. Results are reported based on GC-FID yields.
Experimental Procedure for the Preparation of “Stock Solution of Activated Catalyst”:
CuI (57 mg, 0.30 mmol), NaO2CCF2Br (0.15 g, 0.75 mmol) and KF (87 mg, 1.5 mmol) were added to a 10 mL round bottom flask. The flask was attached to a Schlenk line and evacuated and backfilled with N2 (3x). DMF (1.5 mL) was injected and the mixture was heated in an oil bath at 50 °C for 10 min.
General Optimization
General Procedure B:
A 1 dram vial was charged with CuI (1.9 mg, 0.010 mmol), KF (12 mg, 0.20 mmol), DMEDA (1.2 μL, 0.011 mmol), 0.10 mL of a freshly prepared solution of bromodifluoroacetic acid (8.8 mg, 0.050 mmol) in DMF (0.2 mL, final concentration = 0.25 mol L−1), and a magnetic stir bar. The reaction mixture was stirred for 2–5 min at room temperate until a blue/purple suspension formed, and at 50 ºC until the color changed to light yellow (~10 min). Next, the allylic alcohol (0.100 mmol) and phenyl bromodifluoroacetate (50.1 mg, 0.200 mmol) were dissolved in 0.1 mL of the bromodifluoroacetic solution in DMF, and the resulting solution was added to the reaction. Subsequently, the reaction vial was sealed with a PTFE-lined screw cap, and transferred out of the glove box. The sealed vial was placed on a pre-heated reaction block at 25 ºC and stirred for 24–48 h. Next, the reaction mixture was diluted with EtOAc (~3.0 mL), and 0.100 mmol naphthalene was added an internal standard, and the reaction mixture was stirred at room temperature for 10 minutes to ensure thorough mixing. An aliquot (130–150 μL) was taken from the vial, analyzed by GC-FID, and quantified using a standard curve. Results are reported based on GC-FID yields. Alternatively (or to double-check results), α,α,α-trifluorotoluene (12.3 μL, 0.100 mmol) was added as an internal standard, and the reaction mixture was stirred at room temperature for 5 minutes to ensure thorough mixing. An aliquot was taken from the vial for 19F NMR analysis. Results are reported based on 19F NMR yields.
Experimental Details and Characterization of Trifluoromethyl Compounds
Allyl Trifluoromethanes (Table 2)
General Procedure C:
A 1 dram vial was charged with CuI (9.5 mg, 0.050 mmol), KF (58 mg, 1.0 mmol), DMEDA (5.9 μL, 0.055 mmol), 0.30 mL of a freshly prepared solution of bromodifluoroacetic acid (44 mg, 0.25 mmol) in DMF (1.00 mL, final concentration = 0.25 mol L−1), and a magnetic stir bar. The reaction mixture was stirred for 2–5 min at room temperate until a blue/purple suspension formed, and at 50 ºC until the color changed to light yellow (~10 min). Next, the allylic alcohol (0.500 mmol) and phenyl bromodifluoroacetate (251 mg, 1.00 mmol) were dissolved in 0.70 mL of the bromodifluoroacetic acid solution in DMF, and the resulting solution was added to the reaction. Subsequently, the reaction vial was sealed with a PTFE-lined screw cap, and transferred out of the glove box. The sealed vial was placed on a pre-heated reaction block at 25 ºC and stirred for 36–48 h. Next, the reaction mixture was diluted with Et2O (~3.0 mL). α,α,α-Trifluorotoluene (30 μL, 0.25 mmol) was added as an internal standard, and the reaction mixture was stirred at room temperature for 10 minutes to ensure thorough mixing. An aliquot was taken from the vial for 19F NMR analysis. After determining the 19F NMR yield, the aliquot was recombined with the reaction mixture. Work up (Method A or B, see Note 1 below) and purification of the crude mixture through flash column chromatography provided the desired product (see Note 2).
Method A:
The crude reaction was further diluted with 50 mL of Et2O, and sequentially washed with H2O (3 × 10 mL), 1M NaOH (2 × 5 mL), H2O (1 × 10 mL) and NaCl(sat) (1 × 10 mL). The organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure (see Note 2).
Method B:
Some non-polar products co-eluted with phenyl bromodifluoroacetate during the chromatographic purification and required an alternative isolation procedure. The crude reaction was diluted with THF (10 mL), 1M NaOH (0.5 mL) was added, and the mixture was stirred for 2 h at room temperature. Next, the crude reaction was further diluted with 50 mL of Et2O, and sequentially washed with H2O (3 × 10 mL) and NaCl(sat) (1 × 10 mL). The organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure (see Note 2).
(E)-(4,4,4-trifluorobut-1-en-1-yl)benzene (4a)
General procedure C was followed using (E)-3-phenylprop-2-en-1-ol (2a) (0.67 g, 5.0 mmol), phenyl bromodifluoroacetate (1d) (2.5 g, 10 mmol), CuI (95 mg, 0.50 mmol), DMEDA (59 μL, 0.55 mmol), bromodifluoroacetic acid (0.44 g, 2.5 mmol), KF (0.58 g, 10 mmol) and DMF (10.0 mL). The reaction was stirred for 38 h. Work up according to Method A, except for the washing with 1M NaOH. Chromatographic purification (pentane) afforded the title compound as a colorless oil (0.70 g, 75%) containing residual CH2Cl2 (~0.14 g, estimated by 1H-NMR). Spectroscopic data agreed with the previous report.20
(E)-1-methoxy-4-(4,4,4-trifluorobut-1-en-1-yl)benzene (4b)
General procedure C was followed using (E)-3-(4-methoxyphenyl)prop-2-en-1-ol (2b) (82 mg, 0.50 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (9.5 mg, 0.050 mmol), DMEDA (5.9 μL, 0.055 mmol), bromodifluoroacetic acid (44 mg, 0.25 mmol), KF (58 mg, 1.0 mmol) and DMF (1.0 mL). The reaction was stirred for 44 h. Work up (Method A) and chromatographic purification (0–5% Et2O in pentane) afforded the title compound as a colorless oil (85 mg, 78%). Spectroscopic data agreed with the previous report.8a
(E)-1-nitro-4-(4,4,4-trifluorobut-1-en-1-yl)benzene (4c)
General procedure C was followed using (E)-3-(4-nitrophenyl)prop-2-en-1-ol (2c) (90 mg, 0.50 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (9.5 mg, 0.050 mmol), DMEDA (5.9 μL, 0.055 mmol), bromodifluoroacetic acid (44 mg, 0.25 mmol), KF (58 mg, 1.0 mmol) and DMF (1.0 mL). The reaction was stirred for 39 h. Work up (Method A) and chromatographic purification (0–10% EtOAc in hexanes) afforded the title compound as a yellow solid (83 mg, 72%). Spectroscopic data agreed with the previous report.8a
(E)-1-fluoro-4-(4,4,4-trifluorobut-1-en-1-yl)benzene (4d)
General procedure C was followed using (E)-3-(4-fluorophenyl)prop-2-en-1-ol8a (2d) (76 mg, 0.50 mmol phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (9.5 mg, 0.050 mmol), DMEDA (5.9 μL, 0.055 mmol), bromodifluoroacetic acid (44 mg, 0.25 mmol), KF (58 mg, 1.0 mmol) and DMF (1.0 mL). The reaction was stirred for 44 h. Work up (Method A) and chromatographic purification (pentane) afforded the title compound as a colorless oil (66 mg, 64%). Spectroscopic data agreed with the previous report.8a
(E)-1-bromo-3-(4,4,4-trifluorobut-1-en-1-yl)benzene (4e)
General procedure C was followed using (E)-3-(3-bromophenyl)prop-2-en-1-ol8a (2e) (106 mg, 0.50 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (9.5 mg, 0.050 mmol), DMEDA (5.9 μL, 0.055 mmol), bromodifluoroacetic acid (44 mg, 0.25 mmol), KF (58 mg, 1.0 mmol) and DMF (1.0 mL). The reaction was stirred for 44 h. Work up according to Method A, except for the washing with 1M NaOH. Chromatographic purification (pentane) afforded the title compound as a colorless oil (81 mg, 61%). Spectroscopic data agreed with the previous report.8a
(E)-(((5,5,5-trifluoropent-2-en-1-yl)oxy)methyl)benzene (4f)
General procedure C was followed using (Z)-4-(benzyloxy)but-2-en-1-ol (2f) (89 mg, 0.50 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (9.5 mg, 0.050 mmol), DMEDA (5.9 μL, 0.055 mmol), bromodifluoroacetic acid (44 mg, 0.25 mmol), KF (58 mg, 1.0 mmol) and DMF (1.0 mL). The reaction was stirred for 41 h. Work up (Method A) and chromatographic purification (0–5% Et2O in pentane or 0–5% EtOAc in hexanes) afforded the title compound as a colorless oil (94 mg, 81%). Spectroscopic data agreed with the previous report.8a
1-(2,2,2-Trifluoroethyl)cyclopent-1-ene (4g)
General procedure C was followed using cyclopent-1-en-1-ylmethanol (2g) (49 mg, 0.50 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (9.5 mg, 0.050 mmol), DMEDA (5.9 μL, 0.055 mmol), bromodifluoroacetic acid (44 mg, 0.25 mmol), KF (58 mg, 1.0 mmol) and DMF (1.0 mL). The reaction was stirred for 37 h. The product was too volatile for isolation. The yield was determined by 19F-NMR using α,α,α-trifluorotoluene. The identity of the product was confirmed by GC-MS analysis.
19F NMR (CDCl3, 376 MHz) δ –65.9 (t, J = 11.5 Hz, 3F). GC-MS 150 (15), 67 (100).
1-(2,2,2-Trifluoroethyl)cyclohex-1-ene
General procedure C was followed using cyclohex-1-en-1-ylmethanol (2h) (49 mg, 0.50 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (9.5 mg, 0.050 mmol), DMEDA (5.9 μL, 0.055 mmol), bromodifluoroacetic acid (44 mg, 0.25 mmol), KF (58 mg, 1.0 mmol) and DMF (1.0 mL). The reaction was stirred for 37 h. Product was too volatile for isolation. Yield determined by 19F-NMR using α,α,α-trifluorotoluene. The identity of the product was confirmed by GC-MS analysis.
19F NMR (CDCl3, 376 MHz) δ –65.9 (t, J = 11.2 Hz, 3F). GC-MS 164 (50), 149 (45), 136 (35), 81 (100), 67 (65).
4-(prop-1-en-2-yl)-1-(2,2,2-trifluoroethyl)cyclohex-1-ene (4i)
General procedure C was followed using perillyl alcohol (2i) (76 mg, 0.50 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (9.5 mg, 0.050 mmol), DMEDA (5.9 μL, 0.055 mmol), bromodifluoroacetic acid (44 mg, 0.25 mmol), KF (58 mg, 1.0 mmol) and DMF (1.0 mL). The reaction was stirred for 42 h. Work up (Method B) and chromatographic purification (pentane) afforded the title compound as a colorless oil (58 mg, 57%). Spectroscopic data agreed with the previous report.21
Allenyl Trifluoromethanes (Table 3)
Table 3:
Phenyl Bromodifluoroacetate Enables Cu-catalyzed One-pot Deoxytrifluoromethylation of Propargylic Alcoholsa
 |
Standard reaction conditions: 5a–5f (0.50 mmol), 1d (1.0 mmol), CuI (0.050 mmol), 1,10-phenanthroline (0.055 mmol), HO2CCF2Br (0.25 mmol), KF (1.0 mmol), DMF (1.0 mL), 50 °C. Yield of material after chromatographic purification. For comparison, two-pot yields reported from reference 8c.
General Procedure D:
A 1 dram vial was charged with CuI (9.5 mg, 0.050 mmol), KF (58 mg, 1.0 mmol), 1,10-phenantroline (9.9 mg, 0.055 mmol), 0.30 mL of a freshly prepared solution of bromodifluoroacetic acid (44 mg, 0.25 mmol) in DMF (1.00 mL, final concentration = 0.25 mol L−1), and a magnetic stir bar. The reaction mixture was stirred for 2–5 min at room temperate until a blue/purple suspension formed, and at 50 ºC until the color changed to light yellow (~10 min). Next, the propargylic alcohol (0.500 mmol) and phenyl bromodifluoroacetate (251 mg, 1.00 mmol) were dissolved in 0.70 mL of the bromodifluoroacetic acid solution in DMF, and the resulting solution was added to the reaction. Subsequently, the reaction vial was sealed with a PTFE-lined screw cap, and transferred out of the glove box. The sealed vial was placed on a pre-heated reaction block at 50 ºC and stirred for 24–38 h. Next, the reaction mixture was diluted with Et2O (~3.0 mL). α,α,α-Trifluorotoluene (30 μL, 0.25 mmol) was added as an internal standard, and the reaction mixture was stirred at room temperature for 10 minutes to ensure thorough mixing. An aliquot was taken from the vial for 19F NMR analysis. After determining the 19F NMR yield, the aliquot was recombined with the reaction mixture. The crude reaction was further diluted with 50 mL of Et2O, and sequentially washed with H2O (3 × 10 mL), 1M NaOH (2 × 5 mL), H2O (1 × 10 mL) and NaCl(sat) (1 × 10 mL). The organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure. Purification of the crude mixture through flash column chromatography provided the desired product
2-(1,1,1-trifluorobuta-2,3-dien-2-yl)naphthalene (6a)
General procedure D was followed using 3-(naphthalen-2-yl)prop-2-yn-1-ol8c (5a) (91 mg, 0.50 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (9.5 mg, 0.050 mmol), 1,10-phenantroline (9.9 mg, 0.055 mmol), bromodifluoroacetic acid (44 mg, 0.25 mmol), KF (58 mg, 1.0 mmol) and DMF (1.0 mL). The reaction was stirred for 37 h at 50 ºC. Work up and chromatographic purification (pentane) afforded the title compound as a colorless oil (82 mg, 69%). Spectroscopic data agreed with the previous report.8c
1-Methoxy-4-nitro-2-(1,1,1-trifluorobuta-2,3-dien-2-yl)benzene (6b)
General procedure D was followed using methyl 3-(3-hydroxyprop-1-yn-1-yl)benzoate8c (5b) (104 mg, 0.500 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (9.5 mg, 0.050 mmol), 1,10-phenantroline (9.9 mg, 0.055 mmol), bromodifluoroacetic acid (44 mg, 0.25 mmol), KF (58 mg, 1.0 mmol) and DMF (1.0 mL). The reaction was stirred for 37 h at 50 ºC. Work up and chromatographic purification (0–10% EtOAc in hexanes) afforded the title compound as a yellow oil (85 mg, 66%). Spectroscopic data agreed with the previous report.8c
Methyl 3-(3-hydroxyprop-1-yn-1-yl)benzoate (6c)
General procedure D was followed using methyl 3-(3-hydroxyprop-1-yn-1-yl)benzoate8c (5c) (190 mg, 1.00 mmol), phenyl bromodifluoroacetate (1d) (0.50 g, 2.0 mmol), CuI (19 mg, 0.10 mmol), 1,10-phenantroline (20 mg, 0.11 mmol), bromodifluoroacetic acid (88 mg, 0.50 mmol), KF (116 mg, 2.0 mmol) and DMF (2.0 mL). The reaction was stirred for 24 h at 50 ºC. Work up and chromatographic purification (0–10% EtOAc in hexanes) afforded the title compound as a yellow oil (180 mg, 70%). Spectroscopic data agreed with the previous report.8c
tert-Butyl 2-(1,1,1-trifluorobuta-2,3-dien-2-yl)-1H-indole-1-carboxylate (6d)
General procedure D was followed using tert-butyl 2-(3-hydroxyprop-1-yn-1-yl)-1H-indole-1-carboxylate8c (5d) (108 mg, 40 mmol), phenyl bromodifluoroacetate (1d) (0.20 g, 0.80 mmol), CuI (7.7 mg, 0.040 mmol), 1,10-phenantroline (7.9 mg, 0.088 mmol), bromodifluoroacetic acid (35 mg, 0.20 mmol), KF (46 mg, 0.8 mmol) and DMF (0.80 mL). The reaction was stirred for 24 h at 50 ºC. Work up and chromatographic purification (0–10% EtOAc in hexanes) afforded the title compound as a yellow oil (53 mg, 41%). Spectroscopic data agreed with the previous report.8c
2,2,2-Trifluoro-N-(4-(1,1,1-trifluorobuta-2,3-dien-2-yl)phenyl)acetamide (6e)
General procedure D was followed using 2,2,2-trifluoro-N-(4-(3-hydroxyprop-1-yn-1-yl)phenyl)acetamide8c (6e) (122 mg, 0.500 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (9.5 mg, 0.050 mmol), 1,10-phenantroline (9.9 mg, 0.055 mmol), bromodifluoroacetic acid (44 mg, 0.50 mmol), KF (58 mg, 1.0 mmol) and DMF (1.0 mL). The reaction was stirred for 24 h at 50 ºC. Work up and chromatographic purification (0–20% EtOAc in hexanes) afforded the title compound still contaminated with ~10% of phenol. After washing with 1M NaOH (3 × 5 mL), the pure product was obtained as an off-white solid (81 mg, 55%). Spectroscopic data agreed with the previous report.8c
1,2-dichloro-4-(1,1,1-trifluorobuta-2,3-dien-2-yl)benzene (6f)
General procedure D was followed using 3-(3,4-dichlorophenyl)prop-2-yn-1-ol8c (5f) (122 mg, 0.500 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (9.5 mg, 0.050 mmol), 1,10-phenantroline (9.9 mg, 0.055 mmol), bromodifluoroacetic acid (44 mg, 0.50 mmol), KF (58 mg, 1.0 mmol) and DMF (1.0 mL). The reaction was stirred for 24 h at 50 ºC. Work up and chromatographic purification (0–20% EtOAc in hexanes) afforded the title compound still contaminated with ~10% of phenol. After washing with 1M NaOH (3 × 5 mL), the pure product was obtained as a yellow solid (99 mg, 67%). Spectroscopic data agreed with the previous report.8c
Benzyl Trifluoromethanes (Table 4)
General Procedure E:
A 1 dram vial was charged with CuI (19 mg, 0.10 mmol), KF (0.12 mg, 2.0 mmol), KI (21 mg, 0.12 mmol), and a magnetic stir bar. Next, the benzylic alcohol (0.500 mmol) and phenyl bromodifluoroacetate (251 mg, 1.00 mmol) were dissolved in DMF (0.25 mL) and MeCN (0.25 mL), and the resulting solution was added to the reaction. Subsequently, the reaction vial was sealed with a PTFE-lined screw cap, and transferred out of the glove box. The sealed vial was placed on a pre-heated reaction block at 40 ºC and stirred for 30 min. Next, it was transferred to a second pre-heated reaction block at 70 ºC and stirred for 24 h. Next, the reaction was cooled to room temperature, and diluted with Et2O (~3.0 mL). α,α,α-Trifluorotoluene (30 μL, 0.25 mmol) was added as an internal standard, and the reaction mixture was stirred at room temperature for 10 minutes to ensure thorough mixing. An aliquot was taken from the vial for 19F NMR analysis. After determining the 19F NMR yield, the aliquot was recombined with the reaction mixture. The crude reaction was further diluted with 50 mL of Et2O, and sequentially washed with H2O (3 × 10 mL), 1M NaOH (2 × 5 mL) or Na2CO3 (3 × 5 mL), H2O (1 × 10 mL) and NaCl(sat) (1 × 10 mL). The organic phase was dried over Na2SO4, filtered and concentrated under reduced pressure. Purification of the crude mixture through flash column chromatography provided the desired product
2-(2,2,2-trifluoroethyl)naphthalene (8a)
General procedure E was followed using naphthalen-2-ylmethanol (7a) (79 mg, 0.500 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (19 mg, 0.10 mmol), methyl bromodifluoroacetate (38 mg, 0.20 mmol), KI (21 mg, 0.12 mmol), KF (0.12 g, 2.0 mmol) DMF (0.25 mL) and MeCN (0.25 mL). The reaction was stirred for 24 h at 70 ºC. Work up and chromatographic purification (pentane) afforded the product as a white solid (78 mg, 74%). Spectroscopic data agreed with the previous report.8d
1-(benzyloxy)-2-methoxy-4-(2,2,2-trifluoroethyl)benzene (8b)
General procedure E was followed using (4-(benzyloxy)-3-methoxyphenyl)methanol (7b) (123 mg, 0.500 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (19 mg, 0.10 mmol), methyl bromodifluoroacetate (38 mg, 0.20 mmol), KI (21 mg, 0.12 mmol), KF (0.12 g, 2.0 mmol) DMF (0.25 mL) and MeCN (0.25 mL). The reaction was stirred for 24 h at 70 ºC. Work up and chromatographic purification (0–10% EtOAc in hexanes) afforded the product as a white solid (93 mg, 62%). Spectroscopic data agreed with the previous report.22
1-Tosyl-3-(2,2,2-trifluoroethyl)-1H-indole (8c)
General procedure E was followed using (1-tosyl-1H-indol-3-yl)methanol23 (7c) (301 mg, 1.00 mmol), phenyl bromodifluoroacetate (1d) (0.50 g, 2.0 mmol), CuI (38 mg, 0.20 mmol), methyl bromodifluoroacetate (76 mg, 0.40 mmol), KI (42 mg, 0.25 mmol), KF (232 mg, 4.0 mmol), DMF (0.50 mL) and MeCN (0.50 mL). The reaction was stirred for 24 h at 70 ºC. Work up and chromatographic purification (0–5% EtOAc in hexanes) afforded the product as a white solid (217 mg, 61%). Spectroscopic data agreed with the previous report.24
(1-phenyl-1H-pyrazol-4-yl)methanol (8d)
General procedure E was followed using (1-phenyl-1H-pyrazol-4-yl)methanol8d (7d) (87 mg, 0.50 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (19 mg, 0.10 mmol), methyl bromodifluoroacetate (38 mg, 0.20 mmol), KI (21 mg, 0.12 mmol), KF (0.12 g, 2.0 mmol), DMF (0.25 mL) and MeCN (0.25 mL). Work up and chromatographic purification (10% EtOAc in hexanes) afforded the title compound as a white solid (43 mg, 39%). Spectroscopic data agreed with the previous report 8d
1-Methyl-3-(2,2,2-trifluoroethyl)-1H-indazole (8e)
General procedure E was followed using (1-methyl-1H-indazol-3-yl)methanol19 (7e) (81 mg, 0.50 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (19 mg, 0.10 mmol), methyl bromodifluoroacetate (38 mg, 0.20 mmol), KI (21 mg, 0.12 mmol), KF (0.12 g, 2.0 mmol), DMF (0.25 mL) and MeCN (0.25 mL). Work up and chromatographic purification (10% EtOAc in hexanes) afforded the title compound as a light-yellow solid (43 mg, 39%).
mp: 61 – 63 ºC. 1H NMR (CDCl3, 400 MHz) δ 7.71 (d, J = 8.2 Hz, 1H), 7.46 – 7.37 (m, 2H), 7.24 – 7.16 (m, 1H), 4.07 (s, 3H), 3.79 (q, J = 10.69 Hz, 2H). 13C{1H} NMR (CDCl3, 126 MHz) δ 141.1, 134.27 (q, J = 3.5 Hz), 126.7, 125.15 (q, J = 276.4 Hz), 123.2, 121.0, 120.3, 109.3, 35.6, 32.97 (q, J = 31.5 Hz). 19F NMR (CDCl3, 376 MHz) δ –65.4 (t, J = 10.6 Hz). IR (ATR) 3065, 2978, 2939, 2854, 1618, 1504, 1348, 1256, 1092, 918, 736 cm−1. HRMS (ESI, m/z): calcd for C10H10F3N2 [M+H]+ 215.0796, found 215.0793.
Synthetic Applications (Scheme 1)
(E)-1-(methylsulfonyl)-4-(4,4,4-trifluoro-2-methylbut-1-en-1-yl)benzene (10)
General procedure C was followed using (E)-2-methyl-3-(4-(methylsulfonyl)phenyl)prop-2-en-1-ol (9),11 (113 mg, 0.500 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (9.5 mg, 0.050 mmol), DMEDA (5.9 μL, 0.055 mmol), bromodifluoroacetic acid (44 mg, 0.25 mmol), KF (58 mg, 1.0 mmol) and DMF (1.0 mL). The reaction was stirred for 44 h. Work up and chromatographic purification (0–30% EtOAc in hexanes) afforded the title compound as a colorless oil (105 mg, 75%). Spectroscopic data agreed with the previous report.11b
5-(furan-2-yl)-1-methyl-3-(2,2,2-trifluoroethyl)-1H-pyrazole (12)
General procedure E was followed using (5-(furan-2-yl)-1-methyl-1H-pyrazol-3-yl)methanol12a (11) (89 mg, 0.500 mmol), phenyl bromodifluoroacetate (1d) (0.25 g, 1.0 mmol), CuI (19 mg, 0.10 mmol), methyl bromodifluoroacetate (38 mg, 0.20 mmol), KI (21 mg, 0.12 mmol), KF (0.12 g, 2.0 mmol), DMF (0.25 mL) and MeCN (0.25 mL). Work up according to General Procedure E was done using Na2CO3 (3 × 5 mL), because a decrease in the yield to 43% was observed when 1M NaOH was used. Chromatographic purification (0–30% EtOAc in hexanes) afforded the title compound as a light yellow oil (67 mg, 57%). Spectroscopic data agreed with the previous report.8d, 12a
Stability of the Phenyl Bromodifluoroacetate (1d)
The stability of phenyl bromodifluoroacetate (1d) was tested by storing it in 1-dram vials sealed with a PTFE-lined screw cap under three distinct conditions, namely 1) on the bench, 2) in a desiccator, and 3) in the freezer. The stability of the compound was evaluated by 1H- and 19F-NMR analysis (Figure S1 and S2), as well as the deoxytrifluoromethylation reaction of cinnamyl alcohol (2a) (Scheme S1). The compound was stable in all situations and showed no signs of decomposition by NMR. The reaction yields decreased as the reagent aged. However, the reaction yields were still satisfactory even after 6–8 months. During this work, the only decomposition pathway observed was the hydrolysis of the compound not properly protected against residual moisture. No light sensitivity was observed.
Mechanistic Experiments (Scheme 2)
Stability of Phenyl Bromodifluoroacetate (1d) Under Reaction Conditions
In the absence of alcohol, phenyl bromodifluoroacetate (1d) did not decompose when it was submitted to the standard reaction conditions, with either DMEDA or Phen as ligands. Test reactions were setup according to the General Procedure C (or D, if ligand was Phen), but the alcohol was not added. Both reactions were stirred at 50 ºC for 24 hours, and then analyzed by 19F-NMR using α,α,α-trifluorotoluene as standard.
Study of the Formation of Putative “Active Ester Intermediate” In Situ
Experimental Procedure for Addition of KF in the Presence of BDFA:
Inside the glovebox, a 1 dram vial was charged with the cinnamyl alcohol (2a) (67 mg, 0.50 mmol) and phenyl bromodifluoroacetate (1d) (251 mg, 1.00 mmol), naphthalene (12.8 mg, 0.100 mmol), 1.00 mL of a freshly prepared solution of bromodifluoroacetic acid (44 mg, 0.25 mmol) in DMF (1.00 mL, final concentration = 0.25 mol L−1), and a magnetic stir bar. The reaction mixture was stirred for 60 min at 25 ºC. Next, KF (58 mg, 1.0 mmol) was added, and the reaction mixture was kept stirring at 25 ºC. During the whole experiment, 50 μL aliquots were taken every 15–25 min and quenched with a few drops of Et2O. The samples were analyzed using GC-FID, and standard curves were used to calculated amount of each reaction component as a function of time.
Experimental Procedure for Addition of KF in the Absence of BDFA:
Inside the glovebox, a 1 dram vial was charged with the cinnamyl alcohol (2a) (67 mg, 0.50 mmol) and phenyl bromodifluoroacetate (1d) (251 mg, 1.00 mmol), naphthalene (12.8 mg, 0.100 mmol), 1.00 mL of DMF (1.00 mL, final concentration = 0.25 mol L−1), and a magnetic stir bar. The reaction mixture was stirred for 45 min at 25 ºC. Next, KF (58 mg, 1.0 mmol) was added, and the reaction mixture was kept stirring for another 60 min at 25 ºC. Next, bromodifluoroacetic acid (44 mg, 0.25 mmol) was added, and the reaction mixture was kept stirring at 25 ºC. During the whole experiment, 50 μL aliquots were taken every 15–25 min and quenched with a few drops of Et2O. The samples were analyzed using GC-FID, and standard curves were used to calculated amount of each reaction component as a function of time.
Time-Course Analysis of the Reaction
Experimental Procedure:
Inside the glovebox, deoxytrifluoromethylation reaction of cinnamyl alcohol 2a was prepared according General Procedure C. Naphthalene (12.8 mg, 0.100 mmol) was used as the internal standard. During the whole experiment, 50 μL aliquots were taken every 15–60 min and quenched with a few drops of Et2O. The samples were analyzed using GC-FID, and standard curves were used to calculate the amount of each reaction component as a function of time.
Supplementary Material
ACKNOWLEDGMENTS
We thank the donors of the Herman Frasch Foundation for Chemical Research (701-HF12), the National Science Foundation (CHE-1455163) for supporting this work. B.R.A. thanks the NIGMS Training Grant on Dynamic Aspects of Chemical Biology (T32 GM08545) for a graduate traineeship. NMR Instrumentation was provided by NIH Shared Instrumentation Grants S10OD016360 and S10RR024664, NSF Major Research Instrumentation Grants 9977422 and 0320648, and NIH Center Grant P20GM103418.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information. Supplementary experiments and copy of NMR spectra.
Two methods for the isolation of the products were used:
Extra care was taken to isolate products of lower molecular weight due to volatility. For NMR analysis of the crude reaction mixture, work up and purification using low boiling point solvents was preferred (Et2O, CH2Cl2 or pentane). Solvent evaporation was done under pressure of ~60–100 Torr and water-bath at 15–25 ºC.
REFERENCES:
- 1.a) Al-Maharik N; O’Hagan D, Organofluorine chemistry. Deoxyfluorination reagents for C-F bond synthesis. Aldrichimica Acta 2011, 44, 65–75; [Google Scholar]; b) Furuya T; Kamlet AS; Ritter T, Catalysis for fluorination and trifluoromethylation. Nature 2011, 473, 470; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liang T; Neumann CN; Ritter T, Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed 2013, 52, 8214–8264. [DOI] [PubMed] [Google Scholar]
- 2.Meanwell NA, Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem 2018, 61, 5822–5880. [DOI] [PubMed] [Google Scholar]
- 3.a) Nielsen MK; Ugaz CR; Li W; Doyle AG, PyFluor: A Low-Cost, Stable, and Selective Deoxyfluorination Reagent. J. Am. Chem. Soc. 2015, 137, 9571–9574; [DOI] [PubMed] [Google Scholar]; b) Li L; Ni C; Wang F; Hu J, Deoxyfluorination of alcohols with 3,3-difluoro-1,2-diarylcyclopropenes. Nat. Commun. 2016, 7, 13320; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Neumann CN; Ritter T, Facile C–F Bond Formation through a Concerted Nucleophilic Aromatic Substitution Mediated by the PhenoFluor Reagent. Acc. Chem. Res. 2017, 50, 2822–2833. [DOI] [PubMed] [Google Scholar]
- 4.a) Surya Prakash GK; Jog PV; Batamack PTD; Olah GA, Taming of fluoroform: Direct nucleophilic trifluoromethylation of Si, B, S, and C centers. Science 2012, 338, 1324–1327; [DOI] [PubMed] [Google Scholar]; b) Prakash GKS; Wang F; Zhang Z; Haiges R; Rahm M; Christe KO; Mathew T; Olah GA, Long-Lived Trifluoromethanide Anion: A Key Intermediate in Nucleophilic Trifluoromethylations. Angew. Chem. Int. Ed. 2014, 53, 11575–11578. [DOI] [PubMed] [Google Scholar]
- 5.Tomashenko OA; Grushin VV, Aromatic Trifluoromethylation with Metal Complexes. Chem. Rev. 2011, 111, 4475–4521. [DOI] [PubMed] [Google Scholar]
- 6.a) Yupu Q; Lingui Z; Brett RA; Ryan AA, Decarboxylative Fluorination Strategies for Accessing Medicinally- Relevant Products. Curr. Top. Med. Chem 2014, 14, 966–978; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Synese J; Narquizian R; Norcross RD; Pinard E Preparation of benzoylpiperazine derivatives a glycine transporter 1 (GlyT-1) inhibitors. WO2006072436A1, 2006; [Google Scholar]; c) Tam TF; Leung-Toung R; Wang Y; Zhao Y. Preparation of fluorinated derivatives of deferiprone as iron chelators for treating iron-overload diseases including neurodegenerative disorders. WO2008116301A1, 2008; [Google Scholar]; d) Chang J; Liu K; McEachem EJ; Mu C; Selenick HG; Shi F; Vocaldo DJ; Wang Y; Wei Z; Zhou Y; Zhu Y Preparation of pyranothiazole thioglycosides via cyclization reaction as selective glycosidase inhibitors. WO2012062157A1, 2012; [Google Scholar]; e) Oslob JD; McDowell RS; Johnson R; Yang H; Evanchik M Zaharia CA; Cai H; Hu LW (Acylaryl)imidazoles as modulators of lipid synthesis and their preparation. WO2014008197A1, 2014. [Google Scholar]
- 7.Duan J-X; Chen Q-Y, Novel synthesis of 2,2,2-trifluoroethyl compounds from homoallylic alcohols: a copper(I) iodide-initiated trifluoromethyl-dehydroxylation process. J. Chem. Soc., Perkin Trans 1 1994, 725–730. [Google Scholar]
- 8.a) Ambler BR; Altman RA, Copper-Catalyzed Decarboxylative Trifluoromethylation of Allylic Bromodifluoroacetates. Org. Lett 2013, 15, 5578–5581; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ambler BR; Peddi S; Altman RA, Copper-Catalyzed Decarboxylative Trifluoromethylation of Propargyl Bromodifluoroacetates. Synthesis 2014, 46, 1938–1946; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ambler BR; Peddi S; Altman RA, Ligand-Controlled Regioselective Copper-Catalyzed Trifluoromethylation To Generate (Trifluoromethyl)allenes. Org. Lett 2015, 17, 2506–2509; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Ambler BR; Zhu L; Altman RA, Copper-Catalyzed Synthesis of Trifluoroethylarenes from Benzylic Bromodifluoroacetates. J. Org. Chem 2015, 80, 8449–8457; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Ambler BR; Yang M-H; Altman RA, Metal-Catalyzed Decarboxylative Fluoroalkylation Reactions. Synlett 2016, 27, 2747–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.No decomposition of the PhBDFA 1d was noticeable through NMR analysis when it was stored for 6 months over the bench or in a desiccator or for 8 months in the freezer (see SI for details).
- 10.a) Hoffmann‐Röder A; Krause N, Synthesis and Properties of Allenic Natural Products and Pharmaceuticals. Angew. Chem. Int. Ed 2004, 43, 1196–1216; [DOI] [PubMed] [Google Scholar]; b) Holmes M; Nguyen KD; Schwartz LA; Luong T; Krische MJ, Enantioselective Formation of CF3-Bearing All-Carbon Quaternary Stereocenters via C–H Functionalization of Methanol: Iridium Catalyzed Allene Hydrohydroxymethylation. J. Am. Chem. Soc 2017, 139, 8114–8117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Tan L; Chen C.-y.; Larsen RD; Verhoeven TR; Reider PJ, An efficient asymmetric synthesis of a potent COX-2 inhibitor L-784,512. Tetrahedron Lett. 1998, 39, 3961–3964; [Google Scholar]; b) Chen CY; Larsen RD; Tan L -Process of making 3-aryloxy-4-arylfuran-2-ones useful as inhibitors of COX2. WO9915513A1, 1999. [Google Scholar]
- 12.a) Fustero S; Román R; Sanz-Cervera JF; Simón-Fuentes A; Bueno J; Villanova S, Synthesis of New Fluorinated Tebufenpyrad Analogs with Acaricidal Activity Through Regioselective Pyrazole Formation. J. Org. Chem. 2008, 73, 8545–8552; [DOI] [PubMed] [Google Scholar]; b) Román R; Navarro A; Wodka D; Alvim-Gaston M; Husain S; Franklin N; Simón-Fuentes A; Fustero S, Synthesis of Fluorinated and Nonfluorinated Tebufenpyrad Analogues for the Study of Anti-angiogenesis MOA. Org. Proc. Res. Dev. 2014, 18, 1027–1036. [Google Scholar]
- 13.Mulder JA; Frutos RP; Patel ND; Qu B; Sun X; Tampone TG; Gao J; Sarvestani M; Eriksson MC; Haddad N; Shen S; Song JJ; Senanayake CH, Development of a safe and economical synthesis of methyl 6-chloro-5-(trifluoromethyl)nicotinate: Trifluoromethylation on kilogram scale. Org. Proc. Res. Dev 2013, 17, 940–945. [Google Scholar]
- 14.Duan J-X; Su D-B; Chen Q-Y, Trifluoromethylation of organic halides with methyl halodifluoroacetates — a process via difluorocarbene and trifluoromethide intermediates. J. Fluorine Chem 1993, 61, 279–284. [Google Scholar]
- 15.a) Wheaton GA; Burton DD, Methyl chlorodifluoroacetate as a precursor for the generation of difluorocarbene. J. Fluorine Chem 1976, 8, 97–100; [Google Scholar]; b) Ni C; Hu J, Recent Advances in the Synthetic Application of Difluorocarbene. Synthesis 2014, 46, 842–863; [Google Scholar]; c) Yang Q; Cabrera PJ; Li X; Sheng M; Wang NX, Safety Evaluation of the Copper-Mediated Cross-Coupling of 2-Bromopyridines with Ethyl Bromodifluoroacetate. Org. Proc. Res. Dev. 2018, 22, 1441–1447. [Google Scholar]
- 16.a) Brahms DLS; Dailey WP, Fluorinated Carbenes. Chem. Rev 1996, 96, 1585–1632 [DOI] [PubMed] [Google Scholar]; b) Oshiro K; Morimoto Y; Amii H, Sodium Bromodifluoroacetate: A Difluorocarbene Source for the Synthesis of gem-Difluorocyclopropanes. Synthesis 2010, 2010, 2080–2084. [Google Scholar]
- 17.Qing-Yun C, Trifluoromethylation of organic halides with difluorocarbene precursors. J. Fluorine Chem. 1995, 72, 241–246. [Google Scholar]
- 18.Li Y; Hu B; Dong W; Xie X; Wan J; Zhang Z, Visible Light-Induced Radical Rearrangement to Construct C–C Bonds via an Intramolecular Aryl Migration/Desulfonylation Process. J. Org. Chem. 2016, 81, 7036–7041. [DOI] [PubMed] [Google Scholar]
- 19.Evertsson E; Inghardt T; Lindberg J; Linusson A; Giordanetto F Preparation of quinoline derivatives as MCH modulators. WO2005066132A1, 2005. [Google Scholar]
- 20.Parsons AT; Buchwald SL, Copper-catalyzed trifluoromethylation of unactivated olefins. Angew. Chem. Int. Ed. Engl 2011, 50, 9120–9123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Larsson JM; Pathipati SR; Szabó KJ, Regio- and Stereoselective Allylic Trifluoromethylation and Fluorination using CuCF3 and CuF Reagents. J. Org. Chem 2013, 78, 7330–7336. [DOI] [PubMed] [Google Scholar]
- 22.Qiao Y; Si T; Yang M-H; Altman RA, Metal-Free Trifluoromethylation of Aromatic and Heteroaromatic Aldehydes and Ketones. J. Org. Chem 2014, 79, 7122–7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rajan S; Puri S; Kumar D; Babu MH; Shankar K; Varshney S; Srivastava A; Gupta A; Reddy MS; Gaikwad AN, Novel indole and triazole based hybrid molecules exhibit potent anti-adipogenic and antidyslipidemic activity by activating Wnt3a/β-catenin pathway. Eur. J. Med. Chem 2018, 143, 1345–1360. [DOI] [PubMed] [Google Scholar]
- 24.Miyake Y; Ota S.-i.; Shibata M; Nakajima K; Nishibayashi Y, Copper-catalyzed nucleophilic trifluoromethylation of benzylic chlorides. Org. Biomol. Chem 2014, 12, 5594–5596. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.