

Abstract
A method for electrophilic sulfenylation by organophosphorus-catalyzed deoxygenative O-atom transfer from sulfonyl chlorides is reported. This C–S bond-forming reaction is catalyzed by a readily available small-ring phosphine (phosphetane) in conjunction with a hydrosilane terminal reductant to afford a general entry to sulfenyl electrophiles including valuable trifluoromethyl-, perfluoroalkyl-, and heteroaryl derivatives that are otherwise difficult to access. Mechanistic investigations indicate that the twofold deoxygenation of the sulfonyl substrate proceeds via the intervention of an off-cycle resting state thiophosphonium ion. The catalytic method represents an operationally simple protocol using a stable phosphine oxide as precatalyst and exhibits broad functional group tolerance.
Keywords: Redox chemistry, Oxygen Atom Transfer, Organocatalysis, Phosphetane, Sulfenylation
Organosulfur compounds display versatile redox reactivity, making them archetypal substrates for the development of catalytic O-atom transfer (OAT) methods.[1, 2] Historically, the oxygenative OAT to S (II) substrates has been the primary focus of synthetic efforts; indeed early transition metal-catalyzed sulfoxidation is now established as a preeminent route for the synthesis of S(IV) and S(VI) compounds, especially in stereoselective fashion (Figure 1A).[3] By contrast, the complementary deoxygenative OAT from high-valent organosulfur oxides has generally been viewed with less strategic synthetic importance.[4] One exception in this regard concerns the deoxygenation of sulfonyl derivatives; Sharpless recognized that transient organosulfur intermediates from the phosphine-mediated deoxygenation of sulfonyl chlorides can be trapped by external nucleophiles to effect desirable synthetic chemistry (Figure 1B, X =Cl).[5] In this vein, recent work by Shibata and Cahard,[6] Liu,[7] and Zhao[8] reflects the synthetic potential of this approach via the use of phosphorus derivatives as oxygen acceptors, allowing access to valuable and reactive sulfenyl electrophiles from the more readily handled sulfonyl congener. 9 The conceptual appeal of stoichiometric deoxygenative OAT by phosphine-mediated reduction of sulfonyl electrophiles is offset, though, by poor atom economy and low mass efficiency. These undesirable characteristics are exacerbated by the fact that the P(III) reagent, itself a potent nucleophile, consumes the electrophilic sulfenyl donor in competition with the target substrate to give undesired thiophosphonium ions (Figure 1B). In principle, a phosphine-catalyzed redox system for sulfonyl deoxygenation operating in the PIII/PV=O redox couple (Figure 1C) might improve the reaction mass efficiency and simultaneously limit the concentration of phosphine in solution available for unproductive capture of reactive sulfenylation intermediates. Further, the structural attributes enabling in situ reduction of a tetracoordinate phosphine oxide (i.e. catalyst turnover) might also permit conversion of structurally-related tetracoordinate thiophosphonium ions into catalytically active tricoordinate phosphines.
Figure 1.

(A) General oxygenative and deoxygenative O-atom transfer. (B) Stoichiometric deoxygenative O-atom transfer by using phosphines. (C) Novel phosphacatalytic deoxygenation of sulfonyl chlorides via PIII/PV=O redox cycling.
Catalytic chemistry driven by reversible interconversion of phosphines (R3PIII) and phosphine oxides (R3PV=O) is a developing modality in organophosphorus catalysis.[10,11] In this context, we have shown that a four-membered phosphacycloalkane (i.e. phosphetane 2, Table 1) in combination with a hydrosilane terminal reductant provides an efficient organocatalytic platform for OAT reactions. Such a phosphacatalytic system has been shown to promote efficient reductive OAT from carbonyl[12] and nitro groups[13] by cycling in the PIII/PV=O redox couple to reveal carbon- and nitrogen-based reactive intermediates, respectively. We envisioned advancing this biphilic organophosphorus-catalyzed OAT concept to encompass deoxygenative processing of sulfonyl moieties to furnish reactive sulfur(II)-based electrophilic intermediates.[14]
Table 1.
Phosphacycles as catalysts for deoxygenative trifluoromethylthiolation of Indole 1.a
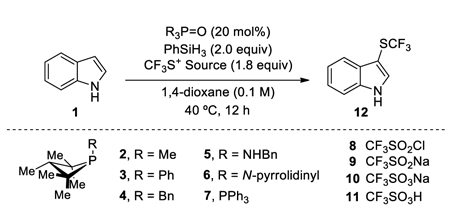
| Entry | R3P=O | CF3S+ Source | Silane | Yield (%) |
|---|---|---|---|---|
| 1 | 2•[O] | 8 | PhSiH3 | 99 |
| 2 | 2 | 8 | PhSiH3 | 99 |
| 3 | 2•[O] | 9 | PhSiH3 | 0 |
| 4 | 2•[O] | 10 | PhSiH3 | 0 |
| 5 | 2•[O] | 11 | PhSiH3 | 0 |
| 6 | 3•[O] | 8 | PhSiH3 | 80 |
| 7 | 4•[O] | 8 | PhSiH3 | 85 |
| 8 | 5•[O] | 8 | PhSiH3 | 90 |
| 9 | 6•[O] | 8 | PhSiH3 | 55 |
| 10 | 7•[O] | 8 | PhSiHs | 17 |
| 11 | 2•[O] | 8 | none | 0 |
| 12 | none | 8 | PhSiH3 | 0 |
Yield determined by 19F NMR spectroscopy of crude reaction mixture using α,α,α-trifluorotoluene as internal standard.
In this context, we elected to focus first on the development of a catalytic method for trifluoromethylsulfenylation by deoxygenation of trifluoromethylsulfonyl chloride (CF3SO2Cl) due to the well-established importance of fluoroalkylthioethers, especially trifluoromethylthioethers (R–SCF3), in agrichemical and pharmaceutical candidates.[15] With the aforementioned biphilic phosphetane-based catalytic system (20 mol% of phosphetane oxide 2·[O], 2 equiv of PhSiH3), the catalytic deoxygenation of CF3SO2Cl in 1,4-dioxane containing indole 1 resulted in regioselective C3-trifluoromethylsulfenylation product 12 in quantitative yield (Table 1, entry 1). Employing tricoordinate phosphine 2 as the catalyst (in lieu of phosphine oxide 2·[O]) provided product in comparable yield suggesting involvement of PIII species in the catalytic cycle (entry 2). Other commercially available CF3SO2-based reagents (sulfinate 9, sulfonate 10, sulfonic acid 11) proved ineffective (entries 3–5). Alteration of the identity of the exocyclic P-substituent of the four-membered ring catalyst from methyl to phenyl, benzyl, -NHBn or pyrrolidino moieties (entries 6–9) gives serviceable albeit inferior yields of 12. An attempt to use triphenylphosphine oxide 7·[O] as precatalyst resulted in only 17% product formation (entry 10). Conducting the reaction in absence of either phosphine oxide precatalyst 2·[O] or phenylsilane yielded no conversion to the product (entries 11,12), thus confirming the requirement of both phosphine oxide and silane reductant in these reactions. In the absence of indole, phosphetane oxide 2·[O] catalyzes reductive dimerization of CF3SO2Cl to the disulfide F3CS–SCF3.
The results of experiments to probe the scope of the catalytic sulfenylation reaction are shown in Table 2. Substitution throughout the indole core is well-tolerated, and electron-withdrawing as well as electron-donating groups could be used (12–28, Table 2A). Indoles with both free −NH 12 and N-Me substitution 13 are good substrates for the trifluoromethylsulfenylation reaction. Both 2-Me-indole (14) and 2-Ph-indole (15) were suitable substrates; sterically demanding 15 necessitated longer reaction time (12 h) compared to 14 (1 h). Electron-rich indoles with methoxy substitution at 4, 5, 6 or 7-positions are highly reactive substrates that formed the -SCF3 products (17–20) in 82–92% yield in 1 h of reaction time, while electron-deficient indoles (21–28) demanded longer reaction times (4–15 h), and in select cases slightly higher catalyst loading to form SCF3 products in 52–98% yield. Substrates with functional handles amenable to derivatization by cross-coupling reactions are well-represented (5-Bpin (16), 6-Cl (22), 4-Br (23), and 5-Br (24). Additionally, substrates containing a range of reducible functionalities including aldehyde (25), ester (26), nitro (27) and nitrile (28) groups, all yielded SCF3-products without incident. Apart from CF3SO2Cl, the catalytic deoxygenative transformation could be extended to perfluoroalkylsulfonyl chlorides including C4F9SO2Cl and C8F17SO2Cl to form the corresponding perfluoroalkylsulfenylated indoles in good yields (29 and 30, Table 2B).
Table 2.
Scope of catalytic double deoxygenation of sulfonyl chlorides for the synthesis of sulfenylindole derivatives.

|
25 mol% of catalyst loading was used.
This mild catalytic deoxygenative protocol could also be applied to a range of aryl (31–38) and alkyl (42) sulfonyl chlorides, thus establishing a simple and straightforward catalytic sulfenylation strategy (Table 2C, D). In general, the electron-deficient sulfonyl chlorides demonstrated higher reactivity towards catalytic deoxygenation (34–38) compared to electron-neutral (31, 33) and electron-rich sulfonyl chlorides (32). The catalytic protocol was similarly also compatible with a range of heteroarylsulfonyl chlorides containing thiophene 39, pyrazole 40 and oxazole rings 41 (Table 2E).
In order to gain insight into the reaction mechanism, in situ spectral monitoring of the catalytic reaction was performed. 31P NMR spectra (162 MHz, 25 °C) of a catalytic reaction (1.0 equiv of 1, 15 mol% of 2·[O], 2.0 equiv of PhSiH3, 1.8 equiv PhSO2Cl, 0.25 M in 1,4-dioxane) showed that phosphetane oxide anti- 2·[O] (δ 56.4 ppm) was consumed with concomitant generation of new resonances at δ 87.3 (major) and δ 94.4 ppm (minor) (Figure 2, A to B). Complete conversion of 2·[O] was observed around the 90 min mark, at which point the catalytic conversion of 1 continues and the resonances at δ 87.3 (major) and δ 94.4 ppm (minor) remain the only observable phosphorus-containing signals in solution. At an intermediate timepoint (t = 60 min), a small amount of epimer syn-2·[O] (δ 62.8 ppm) is noted; however, tricoordinated phosphorus species 2 was not observed at anytime during the reaction.
Figure 2.

Time-stacked in situ 31P NMR spectra during catalysis (T = 25 °C, 1,4-dioxane). (A) t = 0 min; (B) t = 60 min; (C) t = 90 min. Chemical shifts (δ): anti-2·[O], 56.4 ppm; ‘unknown’ peaks at 87.3 and 94.4 ppm.
In a separate experiment, in situ spectral monitoring (31P NMR, Figure 3) of a catalytic reaction with 15 mol% of tricoordinate anti-2 as precatalyst but conditions otherwise identical as above (1.0 equiv of 1, 2.0 equiv of PhSiH3, 1.8 equiv PhSO2Cl, 0.25 M in 1,4-dioxane at 25 °C) was performed. Complete conversion of anti-2 (δ 28.8 ppm) to a mixture of 2·[O] and the unknown species δ 87.3 ppm was observed immediately (t = 1 min) after PhSO2Cl addition. After additional 70 min, unknown resonances at δ 87.3 (major) and δ 94.4 ppm (minor) were the only observable P-containing species in solution.
Figure 3.

Time-stacked in situ 31P NMR spectra during catalysis (T = 25 °C, 1,4-dioxane). (A) 2; t = 0 min; (B) PhSiH3, PhSO2Cl; t = 1 min; (C) t = 70 min. Chemical shifts (δ): anti-2·[O], 56.4 ppm; anti-2, 28.8 ppm; ‘unknown’ peaks at 87.3 and 94.4 ppm.
The identity of the unknown species giving rise to the resonances at δ 87.3 (major) and δ 94.4 ppm (minor) was established to be phenylthiophosphetanium cation 2·[SPh]+ by independent synthesis from reaction of 2 with freshly prepared PhSCl. We thus inferred that 2·[SPh]+ might represent the active species responsible for direct sulfenyl transfer to the indole nucleophile. However, no reaction was observed between 2·[SPh]+ and indole 1 after 16 h of heating in 1,4-dioxane at 40 °C (Scheme 1, A). Evidently, 2·[SPh]+ is not a competent sulfenyl donor and must not be an “on-cycle” catalytic intermediate. Instead, data indicates that 2·[SPh]+ is an “off-cycle” resting state that can reenter the catalytic cycle by reaction with other catalytic components. Specifically, the treatment of 2·[SPh]+ with PhSiH3 converts quickly (t1/2<10 min) into tricoordinate phosphetane 2 (Scheme 1, B). Moreover, phenylthiophosphetanium cation 2·[SPh]+ was shown to be a catalytically competent precatalyst under standard conditions, quantitatively forming sulfenylindole 36 (Scheme 1, C).
Scheme 1.

Synthesis and reactivity of “off-cycle” phenylthiophosphetanium salt 2·[SPh]+. Reaction conditions: (a) indole (1, 1.0 equiv), dioxane, rt; (b) indole (1, 1.0 equiv), PhSiH3 (2.0 equiv), dioxane, rt; (c) indole (1, 1.0 equiv), PhSiH3 (2.0 equiv), PhSO2Cl (1.8 equiv), dioxane, rt.
Based on these experimental observations, we suggest a plausible reaction mechanism for phosphacatalytic deoxygenation/sulfenylation of indoles as illustrated in Figure 4. The reaction initiates with reduction of the precatalyst 2·[O] with phenylsilane to the active tricoordinate phosphetane 2 (step A), a step facilitated kinetically by the small ring size of the four-membered phosphacycle.[16] In accord with precedent, phosphetane 2 then operates on RSO2Cl to effect double deoxygenation, proceeding in a stepwise fashion via RSOCl by the accepted halophilic displacement pathway (step B-C).[17] We suggest that the identity of the active sulfenyl donor in the catalytic manifold may be I, formed from collapse of halophilic substitution intermediates RSO− and 2·[Cl]+. Indeed, in situ DART-MS analysis of a catalytic reaction with PhSO2Cl shows a peak at m/z = 283.13 amu consistent with a cation formulated as I, and the same cation is observed by DART-MS when 2·[O] is treated with PhSCl. In effect, cationic intermediate I may be viewed as a phosphine oxide Lewis base adduct of a sulfenium fragment. The enhancement of reactivity by Lewis base activation of electrophilic reagents (n→σ*) is known; 18 specifically, the work of Denmark provides precedent for Lewis base catalysis of sulfenyl transfer.[19] In this vein, reaction of I with an indole nucleophile would form product with cogeneration of HCl and regeneration of 2·[O] to close the catalytic cycle (step D). Alternatively, formation of the resting state thiophosphetanuim ion II may proceed directly from I (step E) or via sulfenyl chloride RSCl in an “off-cycle” pathway (step F), upon which II can rejoin the catalytic cycle by reduction with the terminal phenylsilane reductant (step G).
Figure 4.

Mechanistic proposal for phosphetane-catalyzed sulfenylation.
In summary, we have developed a catalytic deoxygenative protocol for general sulfenylation of indoles from readily available alkyl, aryl and heteroaryl sulfonyl chlorides including trifluoromethyl- and perfluoroalkylsulfonyl chlorides. This work represents a phosphacatalytic approach to double deoxygenation of sulfonyl chlorides that operates via PIII/PV=O redox cycling in the presence of a terminal hydrosilane reductant. While phosphetane 2 is most likely the active catalyst in the reaction, our mechanistic investigations have identified a novel thiophosphetanium cation II as the off-cycle catalyst resting state. The application of this phosphacatalytic sulfenylation system for other nucleophiles is currently in progress.
Supplementary Material
Acknowledgements
Financial support was provided by National Institute of General Medical Sciences of the US NIH (GM114547), and the MIT Charles E. Reed Faculty Initiative Fund. M.L. acknowledges the Belgian American Educational Foundation (BAEF) for a postdoctoral fellowship. S.-H. K.-L. thanks Prof. Pablo Mauleón and Universidad Autónoma de Madrid for a mobility grant, and Ministerio de Educación, Cultura y Deporte (MECD) for a FPU predoctoral fellowship. The authors acknowledge Liam Kelly (Jamison lab, MIT) for technical assistance.
References
- [1].Oae S, Organic Sulfur Chemistry, CRC Press, Boca Raton, 1991. [Google Scholar]
- [2].For general reviews on catalyzed oxygen-atom transfer, see:Holm RH, Chem. Rev 1987, 87, 1401–1449.Woo LK, Chem. Rev 1993, 93, 1125–1136.Shilov AE, Shteinman AA, Acc. Chem. Res 1999, 32, 763–771.Nam W, Acc. Chem. Res 2007, 40, 522–531.Srour H, Le Maux P, Chevance S, Simonneaux G, Coord. Chem. Rev 2013, 257, 3030–3050.Irie R, Uchida T, Matsumoto K, Chem. Lett 2015, 44, 1268–1283.Bryliakov KP, Chem. Rev 2017, 117, 11406–11459.
- [3].Pitchen P, Dunach E, Deshmukh MN, Kagan HB, J. Am. Chem. Soc 1984, 106, 8188–8193. [Google Scholar]; (b) Brunel JM, Kagan HB, Synlett 1996, 4, 404–406. [Google Scholar]; (c) Bolm C, Bienewald F, Angew. Chem. Int. Ed 1996, 34, 2640–2642. [Google Scholar]; (d) Saito B, Katsuki T, Tetrahedron Lett. 2001, 42, 3873–3876. [Google Scholar]; (d) Kagan HB, in Organosulfur Chemistry in Asymmetric Synthesis, Wiley-Blackwell, 2009, pp. 1–29. [Google Scholar]; (c) Dai W, Li G, Wang L, Chen B, Shang S, Lv Y, Gao S, RSC Adv. 2014, 4, 46545–46554. [Google Scholar]; (f) Li Z-Z, Yao S-Y, Wu J-J, Ye B-H, Chem. Commun 2014, 50, 5644–5647. [DOI] [PubMed] [Google Scholar]; (g) Devi T, Lee Y-M, Nam W, Fukuzumi S, J. Am. Chem. Soc 2018, 140, 8372–8375. [DOI] [PubMed] [Google Scholar]; (h) For a recent review, see: Han J, Soloshonok VA, Klika KD, Drabowicz J, Wzorek A, Chem. Soc. Rev 2018, 47, 1307–1350. [DOI] [PubMed] [Google Scholar]
- [4].For a general review on sulfoxide deoxygenations, see:Madesclaire M, Tetrahedron 1988, 44, 6537–6580.For some examples of reductive SO transformations, see:Pummerer R, Ber. Dtsch. Chem. Ges 1909, 42, 2282–2291.Burdon MG, Moffatt JG, J. Am. Chem. Soc 1965, 87, 4656–4658.Berger R, Ziller JW, Van Vranken DL, J. Am. Chem. Soc 1998, 120, 841–842.Hendrickson JB, Walker MA, Org. Lett 2000, 2, 2729–2731.Bur SK, Padwa A, Chem. Rev 2004, 104, 2401–2432.Colomer I, Velado M, Fernández de la Pradilla R, Viso A, Chem. Rev 2017, 117, 14201–14243.
- [5].Klunder JM, Sharpless KB, J. Org. Chem 1987, 52, 2598–2602. [Google Scholar]
- [6].Chachignon H, Maeno M, Kondo H, Shibata N, Cahard D, Org. Lett 2016, 18, 2467–2470. [DOI] [PubMed] [Google Scholar]
- [7].Jiang L, Yi W, Liu Q, Adv. Synth. Catal 2016, 358, 3700–3705. [Google Scholar]
- [8].(a) Zhao X, Lu X, Wei A, Jia X, Chen J, Lu K, Tetrahedron Lett. 2016, 57, 5330–5333. [Google Scholar]; (b) Zhao X, Li T, Yang B, Qiu D, Lu K, Tetrahedron 2017, 73, 3112–3117. [Google Scholar]; (c) Zhao X, Wei A, Lu X, Lu K, Molecules 2017, 22,1208. [Google Scholar]; (d) Lu K, Deng Z, Li M, Li T, Zhao X, Org. Biomol. Chem 2017, 15, 1254–1260. [DOI] [PubMed] [Google Scholar]
- [9].For other examples of sulfenylation via deoxygenation of high-valent organosulfur reagents, see:Bellale EV, Chaudhari MK, Akamanchi KG, Synthesis 2009, 2009, 3211–3213.Wu Q, Zhao D, Qin X, Lan J, You J, Chem. Commun 2011, 47, 9188–9190.Katrun P, Hongthong S, Hlekhlai S, Pohmakotr M, Reutrakul V, Soorukram D, Jaipetch T, Kuhakarn C, RSC Adv. 2014, 4, 18933–18938.Xiao F, Xie H, Liu S, Deng G-J, Adv. Synth. Catal 2014, 356, 364–368.Jiang L, Qian J, Yi W, Lu G, Cai C, Zhang W Angew. Chem. Int. Ed 2015, 54, 14965–14969.Wang D, Zhang R Lin S, Yan Z, Guo S, RSC Adv. 2015, 5, 108030–108033.Wu Z, Li Y-C, Ding W-Z, Zhu T, Liu S-Z, Ren X, Zou L-H, Asian J. Org. Chem 2016, 5, 625–628.Yu X, Wu Q, Wan H, Xu Z, Xu X, Wang D, RSC Adv. 2016, 6, 62298–62301.Ravi C, Mohan DC, Adimurthy S, Org. Biomol. Chem 2016, 14, 2282–2290.Qi H, Zhang T, Wan K, Luo M, J. Org. Chem 2016, 81, 4262–4268.Guo T, Wei X-N, Synlett 2017, 28, 2499–2504.Huang Z, Matsubara O, Jia S, Tokunaga E, Shibata N, Org. Lett 2017, 19, 934–937.Bu M, Lu G, Cai C, Org. Chem. Front 2017, 4, 266–270.Jiang L, Ding T, Yi W, Zeng X, Zhang W, Org. Lett 2018, 20, 2236–2240.
- [10].(a) Marsden SP Catalytic Variants of Phosphine Oxide-Mediated Organic Transformations In Sustainable Catalysis; Dunn PJ, Hii KK, Krische MJ, Williams MT, Eds.; John Wiley & Sons, Inc.: New York, 2013; pp. 339–361. [Google Scholar]; (b) Guo H, Fan YC, Sun Z, Wu Y, Kwon O Chem. Rev 2018, 118, 10049–10293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].For representative examples of PIII/PV=O redox cycling, see:O’Brien CJ, Tellez JL, Nixon ZS, Kang LJ, Carter AL, Kunkel SR, Przeworski KC, Chass GA, Angew. Chem. Int. Ed 2009, 48, 6836–6839.van Kalkeren HA, Leenders SHAM, A Hommersom CR, Rutjes FPJT, van Delft FL, Chem. Eur. J 2011, 17, 11290–11295.O’Brien CJ, Lavigne F, Coyle EE, Holohan AJ, Doonan BJ, Chem. Eur. J 2013, 19, 5854–5858.van Kalkeren HA, Bruins JJ, Rutjes FPJT, van Delft FL, Adv. Synth. Catal 2012, 354, 1417–1421.O’Brien CJ, Nixon ZS, Holohan AJ, Kunkel SR, Tellez JL, Doonan BJ, Coyle EE, Lavigne F, Kang LJ, Przeworski KC, Chem. Eur. J 2013, 19, 15281–15289.van Kalkeren HA, te Grotenhuis C, Haasjes FS, Hommersom C (Rianne) A., Rutjes FPJT, van Delft FL, Eur. J. Org. Chem 2013, 2013, 7059–7066.Lenstra DC, Rutjes FPJT, Mecinovic J, Chem. Commun 2014, 50, 5763–5766.Wang L, Wang Y Chen M, Ding M-W, Adv. Synth. Catal 2014, 356, 1098–1104Coyle EE, Doonan BJ, Holohan AJ, Walsh KA, Lavigne F, Krenske EH, O’Brien CJ, Angew. Chem. Int. Ed 2014, 53, 12907–12911.Saleh N, Voituriez A, J. Org. Chem 2016, 81, 4371–4377.Lee C-J, Chang T-H, Yu J-K, Madhusudhan Reddy G, Hsiao M-Y, Lin W, Org. Lett 2016, 18, 3758–3761.Saleh N, Blanchard F, Voituriez A, Adv. Synth. Catal 2017, 359, 2304–2315.Zhang K, Cai L, Yang Z, Houk KN, Kwon O, Chem. Sci 2018, 9, 1867–1872.
- [12].Zhao W, Yan PK, Radosevich AT, J. Am. Chem. Soc 2015, 137, 616–619. [DOI] [PubMed] [Google Scholar]
- [13].(a) Nykaza TV, Harrison TS, Ghosh A, Putnik RA, Radosevich AT, J. Am. Chem. Soc 2017, 139, 6839–6842. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nykaza TV, Ramirez A, Harrison TS, Luzung MR, Radosevich AT, J Am. Chem. Soc 2018, 140, 3103–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Nykaza TV, Cooper JC, Li G, Mahieu N, Ramirez A, Luzung MR, Radosevich AT, J. Am. Chem. Soc 2018, 140, 15200–15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chauhan P, Mahajan S, Enders D, Chem. Rev 2014, 114, 8807–8864. [DOI] [PubMed] [Google Scholar]
- [15].(a) Leroux F, Jeschke P, Schlosser M, Chem. Rev 2005, 105, 827–856. [DOI] [PubMed] [Google Scholar]; (b) Liang T, Neumann CN, Ritter T, Angew. Chem. Int. Ed 2013, 52, 8214–8264. [DOI] [PubMed] [Google Scholar]; (c) Swallow S Prog. Med. Chem 2015, 5, 65. [DOI] [PubMed] [Google Scholar]; (d) Kirsch P Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH: Weinheim, 2004. [Google Scholar]
- [16].(a) Marsi KL, J. Org. Chem 1974, 39, 265–267. [Google Scholar]; (b) Johnson MP, Trippett S, J. Chem. Soc., Perkin Trans 1 1982, 191–195. [Google Scholar]
- [17].Zefirov NS, Makhon’kov DI, Chem. Rev 1982, 82, 615–624. [Google Scholar]
- [18].(a) Denmark SE, Beutner GL, Angew. Chem. Int. Ed 2008, 47, 1560–1638. [DOI] [PubMed] [Google Scholar]; (b) Kalyani D, Kornfilt DJ-P, Burk MT, Denmark SE in Lewis Base Catalysis in Organic Synthesis, Wiley-VCH, Weinheim: 2016, p. 1153–1211. [Google Scholar]
- [19].For representative examples, see:Denmark SE, Hartmann E, Kornfilt DJP, Wang H, Nat. Chem 2014, 6, 1056–1064.Denmark SE, Kornfilt DJP, Vogler T, J. Am. Chem. Soc 2011, 133, 15308–15311.Denmark SE, Rossi S, Webster MP, Wang H, J. Am. Chem. Soc 2014, 136, 13016–13028.Denmark SE, Chi HM, J. Am. Chem. Soc 2014, 136, 3655–3663.Denmark SE, Chi HM, J. Am. Chem. Soc 2014, 136, 8915–8918.Denmark SE, Chi HM, J. Org. Chem 2017, 82, 3826–3843.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.