

Abstract
BACKGROUND:
Previous studies showed reduction of brain cannabinoid CB1 receptors in adults with cannabis and alcohol use disorders. Preclinical data suggest that these receptors also contribute to nicotine reward and dependence. Tobacco smoking may confound clinical studies of psychiatric disorders because many patients with such disorders smoke tobacco. Whether human subjects who smoke tobacco but are otherwise healthy have altered CB1 receptor binding in brain is unknown.
METHODS:
We measured CB1 receptors in brains of 18 healthy men who smoke tobacco (frequent chronic cigarette smokers), and 28 healthy men who do not smoke tobacco, using positron emission tomography and [18F]FMPEP-d2, a radioligand for CB1 receptors. We collected arterial blood samples during scanning to calculate the distribution volume (VT), which is nearly proportional to CB1 receptor density. Repeated-measures analysis of variance compared VT between groups in various brain regions.
RESULTS:
Brain CB1 receptor VT was about 20% lower in subjects who smoke tobacco than in subjects who do not. Decreased VT was found in all brain regions, but reduction did not correlate with years of smoking, number of cigarettes smoked per day, or measures of nicotine dependence.
CONCLUSIONS:
Tobacco-smoking healthy men have a widespread reduction of CB1 receptor density in brain. Reduction of CB1 receptors appears to be a common feature of substance use disorders. Future clinical studies on the CB1 receptor should control for tobacco smoking.
Keywords: Addiction, Brain imaging, Cannabinoid CB1 receptor, Positron emission tomography, Smoking, Tobacco
The brain cannabinoid system is involved in the addictive properties of a variety of substances of abuse (1). CB1 cannabinoid receptors are located presynaptically in multiple brain regions, including the ventral tegmental area in the midbrain, where release of gamma-aminobutyric acid (2) and glutamate are inhibited and dopamine release is modulated in response to many substances of abuse (3,4). We previously used positron emission tomography (PET) and an inverse agonist radioligand for CB1 receptors, [18F]FMPEP-d2 (5,6), to examine CB1 receptor binding in human brain in two substance use disorders: cannabis and alcohol. In chronic daily cannabis smokers, we found regionally selective and reversible downregulation of CB1 receptors; receptor binding was decreased in cortical regions but not in subcortical regions, and it returned to normal levels after about 4 weeks of monitored cannabis abstinence (7). In alcohol dependence, we found widespread and irreversible downregulation; receptor binding was decreased in both cortical and subcortical brain regions, and it remained similarly decreased after 2 to 4 weeks of monitored abstinence (8).
Tobacco is the most prevalent substance of abuse, with high addictive potential. Whether tobacco smoking affects CB1 receptor binding in human brain is unknown but is of importance for two reasons. First, preclinical and clinical observations suggest that CB1 receptors are involved in nicotine addiction. In animal studies, blockade of CB1 receptors reduced nicotine-seeking behaviors and nicotine-induced midbrain dopamine release (9). In humans, rimonabant, a CB1 receptor inverse agonist, was effective in smoking cessation (10,11). Second, many patients with psychiatric disorders smoke tobacco, which can be a significant confound in clinical studies examining CB1 receptor binding in such disorders. In the current study, we evaluated CB1 receptor binding in healthy men who smoke tobacco cigarettes, in comparison with men who do not smoke, using PET and [18F]FMPEP-d2.
METHODS AND MATERIALS
The National Institutes of Health Combined NeuroScience Institutional Review Board approved the protocol and the consent forms. Written informed consent was obtained from all subjects.
Subjects
Healthy men (n = 46) were free of somatic and psychiatric illness as confirmed by history, physical examination, structured diagnostic interviews (Structured Clinical Interview for DSM-IV, full version), electrocardiogram, and blood and urine tests. Subjects did not have current or lifetime history of substance use disorders (except for tobacco use disorder in the tobacco smokers) and had urine samples negative for cannabinoids, opiates, amphetamines, cocaine metabolites, and benzodiazepines during screening. We did not test for these urine markers on the day of the PET scan. Subjects had <10 lifetime exposures to cannabis and no use in the preceding 3 months. Recent heavy alcohol use was excluded by Alcohol Use Disorder Identification Test scores of ≤9 and no alcohol use during the 3 days prior to the PET scan. We did not systematically record caffeine consumption. In total, 18 subjects smoked cigarettes and 28 did not (Table 1). None of the nonsmokers was an ex-smoker. We recorded average number of cigarettes per day, age at onset of smoking, duration of smoking, and Fagerström Test for Nicotine Dependence scores. On average, smokers consumed 12 cigarettes per day (data available from 16 subjects) and had a Fagerström Test for Nicotine Dependence score of 4 (data available from 16 subjects), consistent with mild to moderate tobacco use disorder. Data from 32 (70%) of the subjects were published previously. We previously found that carriers of the C allele of a common single-nucleotide polymorphism, rs2023239, in the gene coding for the CB1 receptor (CNR1), have higher [18F]FMPEP-d2 binding than noncarriers. (8). We had these data available for 43 subjects (16 smokers and 27 nonsmokers). Genotyping was done as described previously (8).
Table 1.
Demographic, Clinical, and Radiochemical Information of the 46 Study Participants
| Smoker | Nonsmoker | p Value | |
|---|---|---|---|
| Number of Subjects | 18 | 28 | |
| Age, Years | 35 ± 10 | 32 ± 10 | .297 |
| Body Mass Index, kg/m2 | 27 ± 6 | 27 ± 4 | .692 |
| Racea | 6 C, 7 AA, 2 A | 15 C, 10 AA, 3 A | .757 |
| Amount of Tobacco Use, Cigarettes/Dayb | 12 ± 7 | NA | NA |
| Duration of Tobacco Use, Yearsc | 15 ± 9 | NA | NA |
| Age at Onset of Tobacco Use, Yearsc | 20 ± 8 | NA | NA |
| Fagerström Test for Nicotine Dependence Scoreb | 4 ± 2 | NA | NA |
| Injected Activity of [18F]FMPEP-d2, MBq | 182 ± 11 | 182 ± 8 | .940 |
| Injected Amount of [18F]FMPEP-d2, nmol | 2.7 ± 1.1 | 1.6 ± 0.5 | .001 |
| Fraction of Free [18F]FMPEP-d2 in Plasma, % | 0.48 ± 0.1 | 0.40 ± 0.2 | .154 |
Values are n or mean ± SD.
A, Asian; AA, African American; C, Caucasian; NA, not applicable.
Data missing from 3 subjects.
Data missing from 2 subjects.
Data missing from 4 subjects.
PET and Measurement of Parent Radioligand in Arterial Plasma
[18F]FMPEP-d2 was prepared as described previously (6) and in detail in our investigational new drug application (No. 105,198) submitted to the U.S. Food and Drug Administration (available at https://pdsp.unc.edu/databases/snidd/). The radioligand was obtained in high radiochemical purity (>99%) and had a molar activity of 107 ± 46 MBq/nmol at time of injection. The actual injected amount of [18F]FMPEP-d2 was somewhat higher in smokers than in nonsmokers (Table 1). However, the maximum mass dose (2 μg) was still within tracer dose limits with no significant expected CB1 receptor occupancy. With this maximum dose, we estimate the occupancy to be 0.1% using calculations and assumptions published previously (5).
After intravenous injection of [18F]FMPEP-d2 (Table 1), images were acquired for 120 minutes using an Advance camera (GE Healthcare, Milwaukee, WI) as described previously (5). We did not use motion correction during PET imaging. PET scans were performed at least 4 hours after last tobacco use. We did not record time of last tobacco exposure. Arterial blood samples were drawn as described previously (7). The plasma time-activity curve was corrected for the fraction of unchanged radioligand by radio-high-performance liquid chromatography separation (12), and the plasma-free fraction was measured by ultrafiltration (13).
For anatomical reference, 3D T1-weighted magnetic resonance images were acquired at 3T using either the GE Signa scanner (GE Healthcare) or the Philips Achieva scanner (Philips Healthcare, Andover, MA). These high-resolution anatomical images had a voxel size of 0.86 mm × 0.86 mm × 1.2 mm (transaxial acquisition; GE Healthcare) or 1 mm × 0.94 mm × 0.94 mm (sagittal acquisition; Philips Healthcare). Time of repetition, echo time length, and flip angle were 7.3 ms, 2.8 ms, and 6° for GE Healthcare and 8.1 ms, 3.7 ms, and 8° for Philips Healthcare, respectively. PET images were analyzed by applying a template of volumes of interest (14) as implemented in PMOD (version 3.0; PMOD Technologies, Zurich, Switzerland) (15) in the standard stereotactic space (16) as described previously (7). Distribution volume (VT) was estimated according to the two-tissue compartmental model (17) with concentration of parent radioligand in plasma as input function using PMOD as described previously (5). Statistical parametric mapping of VT values at voxel level was done using SPM8 as described previously (7), with body mass index (BMI) as a covariate. VT can also be conceptualized as ratio of area under the time-activity curve extrapolated into infinity of brain to plasma. To ensure that groups had similar input functions, plasma time-activity curve of the parent radioligand was normalized for injected dose and body weight, expressed as standardized uptake values, and extrapolated into infinity using rate constants from triexponential fitting. Area under this curve was then calculated and compared between groups.
Statistical Analysis
Data were analyzed using IBM SPSS Statistics 23.0 for Mac (version 24.0.0; IBM Corp., Armonk, NY). Variance was homogeneous across groups according to Levene’s test. Baseline characteristics of participants (Table 1) were compared using two-tailed t tests. To test whether CB1 receptors were decreased in subjects who smoke tobacco, we used a mixed-model two-way analysis of variance (ANOVA) with group status (smoker vs. nonsmoker) as between-subject factor and brain region as within-subject factor. BMI entered the model as a covariate because it affects VT (7). Correlations with clinical variables were assessed with Pearson’s correlation coefficients. Nominal data were compared using Pearson’s χ2 test. Any p values less than .05 were considered statistically significant in the ANOVA. In the presence of significant main effects and interactions, regional contrasts were examined using post hoc t tests of marginal means from the ANOVA, and effect sizes were calculated as absolute difference between group means divided by pooled estimate of standard deviation, which was calculated assuming that pooled estimate of variance is the sample size-weighted average of sample variances. Because these regional contrasts were assessed only after significant ANOVA findings, they were not corrected for multiple comparisons.
Because many patients with psychiatric and substance use disorders also smoke tobacco, we wondered whether tobacco smoking confounded our previous findings of CB1 receptor downregulation in individuals with cannabis or alcohol use disorder. To examine this, we combined all data from these three datasets (46 healthy subjects [18 tobacco smokers and 28 tobacco nonsmokers], 30 cannabis smokers [24 tobacco smokers and 6 tobacco nonsmokers], and 18 patients with alcohol dependence [11 tobacco smokers and 7 tobacco nonsmokers]) in an overall ANOVA model to assess the main effect of cigarette smoking across all subject groups. In this model, we looked at main effects of tobacco smoking, BMI, group status, and rs2023239 C allele carrier status.
RESULTS
The VT of [18F]FMPEP-d2 was lower in tobacco smokers than in nonsmokers (main effect of group: F = 8.30, p = .006). VT was lower in smokers in all brain regions, and the magnitude of this decrease differed significantly among brain regions (group × region interaction: F = 3.03, p = .029) (Figures 1 and 2 and Table 2). Decrease in BMI-adjusted VT ranged from 15% in the parietal cortex to 34% in white matter. The main effect of smoking was also significant when analyzed without correcting for BMI (F = 8.43, p = .006). Decreased VT in smokers was confirmed with an independent voxel-based whole-brain analysis (Figure 3). Among subjects who had available rs2023239 genotype data (n = 43 subjects), 8 of 27 non-smokers (29%) and 2 of 16 smokers (13%) carried the C allele (χ2 = 1.7, p = .199). When genotype entered the model, the main effect of smoking persisted (F = 5.26, p = .026), although the main effect of genotype did not reach statistical significance (F = 1.44, p = .238).
Figure 1.
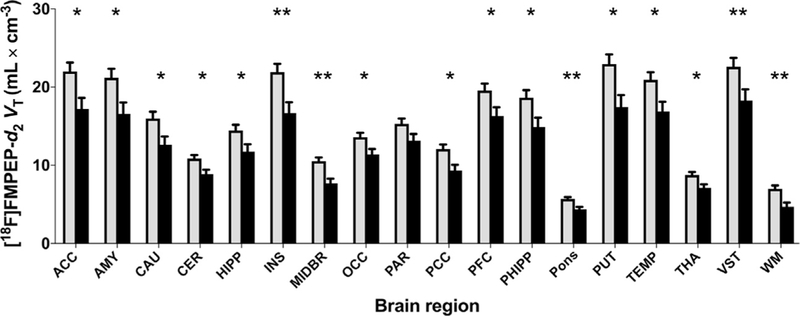
Distribution volume (VT) of [18F]FMPEP-d2 (a measure of cannabinoid CB1 receptor density) is lower in male tobacco smokers (black bars, n = 18) than in nonsmokers (gray bars, n = 28) in both cortical and subcortical regions. Values are estimated marginal means from the repeated-measures analysis of variance and are adjusted to an average body mass index of 26.8 kg/m2. Error bars are standard error of the mean. *p < .05; **p < .005; post hoc contrasts of marginal means from analysis of variance. ACC, anterior cingulate cortex; AMY, amygdala; CAU, caudate nucleus; CER, cerebellum; HIPP, hippocampus; INS, insula; MIDBR, midbrain; OCC, occipital cortex; PAR, parietal cortex; PCC, posterior cingulate cortex; PFC, prefrontal cortex; PHIPP, parahippocampal gyrus; PUT, putamen; TEMP, lateral temporal cortex; THA, thalamus; VST, ventral striatum; WM, white matter.
Figure 2.
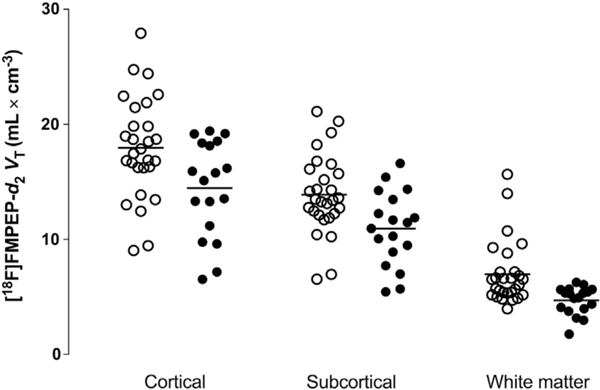
Individual body mass index–adjusted [18F]FMPEP-d2 distribution volume (VT) values in average cortical, subcortical, and white matter regions in nonsmokers (open circles) and smokers (closed circles).
Table 2.
Group Comparisons of Regional [18F]FMPEP-d2 Unadjusted VT Values
| Brain Region | % Difference | Effect Size | p Value |
|---|---|---|---|
| Anterior Cingulate Cortex | −23 | 0.81 | .012 |
| Amygdala | −23 | 0.77 | .016 |
| Caudate Nucleus | −22 | 0.77 | .017 |
| Cerebellum | −19 | 0.83 | .009 |
| Hippocampus | −19 | 0.70 | .029 |
| Insula | −25 | 0.92 | .004 |
| Midbrain | −28 | 1.12 | .001 |
| Occipital Cortex | −17 | 0.74 | .019 |
| Parietal Cortex | −15 | 0.62 | .050 |
| Pons | −24 | 1.04 | .001 |
| Posterior Cingulate Cortex | −24 | 0.92 | .005 |
| Prefrontal Cortex | −17 | 0.71 | .028 |
| Parahippocampal Gyrus | −21 | 0.75 | .019 |
| Putamen | −25 | 0.85 | .008 |
| Lateral Temporal Cortex | −20 | 0.79 | .014 |
| Thalamus | −20 | 0.81 | .011 |
| Ventral Striatum | −20 | 0.73 | .002 |
| White Matter | −34 | 1.02 | .002 |
Effect sizes were calculated as absolute difference of group mean values divided by a pooled estimate of standard deviation, which was calculated assuming that pooled estimate of variance is the sample size-weighted average of sample variances. The p values are from post hoc comparisons of estimated marginal means from the repeated-measures analysis of variance model (including body mass index as a covariate).
VT, distribution volume.
Figure 3.
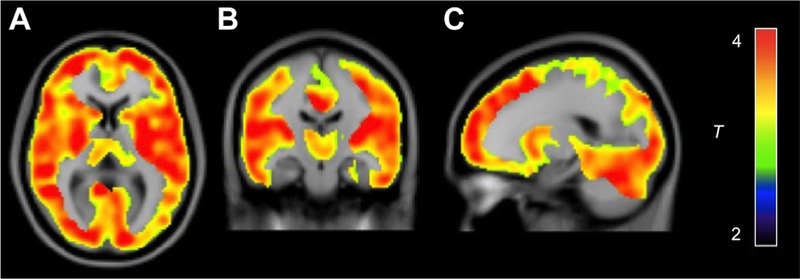
Whole-brain statistical parametric mapping analysis shows lower cannabinoid CB1 receptor density (distribution volume [VT]) in male tobacco smokers (n = 18) than in nonsmokers (n = 28) as a large single cluster. This cluster comprised 132,291 voxels with a maximum t value of 6.0 at [22, 10, 46] and a cluster-level familywise error corrected p value of <.001. Color bar represents t value in each voxel within the significant cluster. (A) Transaxial section. (B) Coronal section. (C) Sagittal section.
Decreased VT was not caused by higher plasma protein binding in smokers given that fraction of free radioligand in plasma was not significantly different between groups (Table 1). Area under the plasma time-activity curve of the parent radioligand extrapolated into infinity was similar between smokers (172 ± 53 standardized uptake values × minutes) and nonsmokers (167 ± 60 standardized uptake values × minutes) (mean ± SD; p = .789, t test), confirming similar input function. Although this measure was similar between groups, VT measurement is independent of the form of the input function and does not require it to be the same between groups to accurately quantify receptor availability.
Among smokers, whole brain VT did not correlate with number of cigarettes smoked per day (R = −.27, p = .319), score on the Fagerström Test for Nicotine Dependence (R = −.14, p = .611), lifetime duration of tobacco smoking (R =.21, p = .479), or age at onset of tobacco smoking (R = .18, p = .544).
In the combined analysis of 94 subjects (46 healthy subjects, 30 cannabis smokers, and 18 patients with alcohol dependence), we found a significant main effect of tobacco smoking on CB1 receptor VT (smaller VT in tobacco smokers; F = 4.10, p = .046) and the expected significant main effects of BMI (F = 8.90, p = .004) and substance use disorder status (F = 6.38, p = .003). When genotype entered the model (n = 91 subjects), the main effect of tobacco smoking diminished somewhat (F = 2.66, p = .106) and the main effect of genotype was significant (F = 4.45, p = .038). Tobacco smoking had no effect among cannabis smokers (F = 0.02, p = .894) or individuals with alcohol dependence (F = 0.40, p = .537) as also reported previously. Thus, tobacco smoking produced no additive CB1 receptor downregulation in individuals with cannabis or alcohol use disorder (Supplemental Figure S1). However, this post hoc cross-sectional comparison should be interpreted with caution because the original studies were not specifically designed to examine effects of tobacco smoking. Therefore, we cannot definitively rule out the possibility that tobacco smoking affected comparisons between healthy subjects and cannabis smokers or patients with alcohol dependence.
DISCUSSION
We found about 20% lower VT of [18F]FMPEP-d2 in tobacco-smoking healthy subjects compared with nonsmoking subjects, consistent with CB1 receptor downregulation. This reduction was significant in all brain regions, although some regions had larger reduction than others. Together with our previous data from individuals with cannabis or alcohol use disorder, these data support a role for brain CB1 receptors in various substance use disorders.
Previous animal studies on the effects of chronic nicotine exposure on CB1 receptor density produced inconsistent findings. Some studies on juvenile or adolescent rats have shown decreased hippocampal CB1 receptor messenger RNA (18), decreased cingulate cortex CB1 receptor density (19), and decreased striatal CB1 receptor density as well as increased hippocampal CB1 receptor density (20). One study found increased CB1 receptor binding after adolescent exposure but unchanged binding after adult exposure (21). In contrast to juvenile or adolescent rodents, adult rodents generally were not found to have significant changes in CB1 receptors after chronic nicotine exposure (22,23). A previous micro-PET study with [18F]MK-9470, another radioligand for CB1 receptors, did not find significant changes in binding in rat brain after chronic nicotine exposure (24). These discrepancies in previous preclinical studies may be explained by differences in measuring techniques and age at nicotine exposure. With regard to adolescent versus adult exposure, we found no correlations between CB1 receptor binding and age at onset of tobacco smoking. That is, we found no evidence that CB1 receptor downregulation was more pronounced in subjects who started smoking at a younger age (<18 years; n = 6 subjects), although all subjects had adult exposure whether or not they were already exposed during adolescence.
Lower CB1 receptor binding may be a common imaging biomarker for several substances of abuse (25). These receptors are found throughout the brain, and the changes are often seen in widespread areas such as those found in tobacco smokers in the current study and in alcohol-dependent individuals previously (8). Such widespread downregulation may reflect increased overall tone of endogenous cannabinoids, which could be viewed as a neuroprotective compensatory mechanism. Chronic nicotine administration may downregulate CB1 receptors by chronic overstimulation by endogenous cannabinoids (9), similar to what is hypothesized to occur with chronic alcohol consumption (8,26). In contrast, cannabis smokers have CB1 receptor downregulation only in neocortical brain regions but not in the basal ganglia, midbrain, or cerebellum (7). This regional dissimilarity between substance use disorders may be partly explained by different mechanisms of action of these drugs; the psychoactive ingredient of cannabis, Δ9-tetrahydrocannabinol, binds to CB1 receptors directly, whereas nicotine and alcohol affect CB1 receptors indirectly via endogenous cannabinoids (1).
In contrast to our finding in healthy subjects, a recent PET study found higher brain CB1 receptor binding in patients with schizophrenia who smoked tobacco compared with patients who did not smoke, although both patient cohorts had lower CB1 receptor binding than healthy nonsmoking control subjects (27). Together with our previous findings of no effects of tobacco smoking in subjects with cannabis use disorder (7) or alcohol use disorder (8), this pattern of results suggests that tobacco smoking has different effects on brain CB1 receptors in healthy subjects than in subjects with neuropsychiatric illness.
A potential site for a functional connection between nicotine and endocannabinoid neurotransmission is the midbrain dopamine neurons projecting to ventral parts of the striatum, including the nucleus accumbens, considered to be a critical part of the brain reward circuit (4). Nicotinic acetylcholine receptor subtype α4β2 on gamma-aminobutyric acidergic and dopaminergic neurons and receptor subtype α7 on glutamatergic terminals are targets for mediating rewarding actions of nicotine (28). Acute nicotine exposure increases dopamine concentration in the nucleus accumbens, an effect that can be reduced by blocking CB1 receptors (29). In the midbrain ventral tegmental area, acute nicotine exposure increases endo-cannabinoid levels (30), providing further evidence of endo-cannabinoid modulation of nicotine reward. In the midbrain, CB1 receptors are located in presynaptic terminals of both excitatory and inhibitory neurons, so functional responses to increased endocannabinoids may be complex (28). In addition, non-cannabinoid receptor targets are likely affected by endogenous cannabinoids (28).
Clinical trials showed the efficacy of rimonabant, a CB1 receptor inverse agonist, in promoting smoking cessation (10,11), supporting the role of CB1 receptors in nicotine dependence. However, clinical use of this drug is limited by psychiatric side effects. Although CB1 blockade may acutely reduce nicotine-induced mesolimbic dopamine release (29), it might not reverse all cannabinoid effects of nicotine because CB1 receptors are downregulated. Enhancing endogenous cannabinoid function in withdrawal and chronic abstinence might intuitively seem to be a way to compensate for such downregulation, although we do not yet know whether CB1 receptors return to normal levels after abstinence from nicotine. Preclinical studies investigating the impact of blocking the enzyme that breaks down some endocannabinoids (fatty acid amide hydrolase) do not fully agree on the utility of this approach (9), which may reflect the complexity of endo-cannabinoid modulation of drug action.
We did not find correlations between amount of tobacco exposure and CB1 receptor downregulation. A potential limitation of this study is that we did not measure concentrations of nicotine or any of its metabolites, such as cotinine, which would give more accurate estimates of prior tobacco exposure than retrospective self-reports of years of smoking and number of cigarettes per day.
Decreased VT of [18F]FMPEP-d2 likely represents decreased number of CB1 receptors rather than occupancy of receptors by endocannabinoids. As we argued previously (7,8), about 90% of VT represents specific binding in monkey brain (5). In rodent brain, binding of [11C]MePPEP, a close radioligand analog of [18F]FMPEP-d2, could not be displaced by endogenous cannabinoids (31), suggesting that decreased VT of [18F]FMPEP-d2 observed in the current study is not due to increased levels of endocannabinoids. Finally, the outcome measure VT corrects for possible group differences in peripheral distribution and metabolism of the radioligand.
The current study has several limitations. First, we studied only male subjects, as we had done previously in studies on cannabis use (7) and alcohol dependence (8); therefore, whether these results are generalizable to women remains to be determined. Studying only male subjects limits our understanding of endocannabinoid function in substance use disorders. Second, altered nondisplaceable binding may have confounded our measurements because VT is a composite measure of specific and nondisplaceable binding. However, the contribution of nondisplaceable binding is rather small (~10%) (5) and does not vary substantially among brain regions, such that it is unlikely to fully explain the 20% to 30% lower VT in tobacco smokers than in non-smokers. Third, although we excluded significant cannabis or alcohol exposure, we did not systematically assess caffeine consumption. Caffeine may influence CB1 receptors via adenosine receptors (32).
In summary, we showed that tobacco smoking is associated with widespread downregulation of CB1 receptors in human brain. This finding adds to the growing evidence for CB1 receptor abnormalities in substance use disorders and suggests that future clinical studies on this receptor target should carefully control for tobacco smoking.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Intramural Research Programs of the National Institute of Mental Health (Project Nos. ZIAMH002852 [to RBI] and ZIAMH002793 [to VWP]) and National Institute on Drug Abuse (NIDA) (Project No. DA000413 [to MAH]) under Clinical Protocol NCT00816439. JH was supported by personal grants from the Academy of Finland, Finnish Cultural Foundation, Finnish Foundation for Alcohol Studies, Finnish Medical Foundation, Instrumentarium Foundation, Jalmari and Rauha Ahokas Foundation, Paulo Foundation, Research Foundation of Orion Corp., and Yrjö Jahnsson Foundation.
We thank Kimberly Jenko, Kacey Anderson, and David Clark for assisting in the measurements of radioligand in plasma; Maria D. Ferraris Araneta, Gerald Hodges, William C. Kreisl, and Barbara Scepura, as well as Kathleen Demuth and the NIDA staff, for subject recruitment and care; the National Institutes of Health PET Department for imaging; and PMOD Technologies for providing its image analysis and modeling software.
Footnotes
DISCLOSURES
The authors report no biomedical financial interests or potential conflicts of interest.
Contributor Information
Jussi Hirvonen, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda; Department of Radiology, University of Turku, Turku, Finland..
Paolo Zanotti-Fregonara, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda; Houston Methodist Research Institute, Houston, Texas.
David A. Gorelick, Chemistry and Drug Metabolism Section, National Institute on Drug Abuse, National Institutes of Health Department of Psychiatry, School of Medicine, University of Maryland, Baltimore, Maryland.
Chul Hyoung Lyoo, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda.
Denise Rallis-Frutos, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda.
Cheryl Morse, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda.
Sami S. Zoghbi, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda
Victor W. Pike, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda
Nora D. Volkow, Office of the Director, National Institute on Drug Abuse, National Institutes of Health
Marilyn A. Huestis, Chemistry and Drug Metabolism Section, National Institute on Drug Abuse, National Institutes of Health, Lambert Center for the Study of Medicinal Cannabis and Hemp, Thomas Jefferson University, Philadelphia, Pennsylvania
Robert B. Innis, Molecular Imaging Branch, National Institute of Mental Health, National Institutes of Health, Bethesda
REFERENCES
- 1.Parsons LH, Hurd YL (2015): Endocannabinoid signalling in reward and addiction. Nat Rev Neurosci 16:579–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin CS, Clifford PR, Maisto SA, Earleywine M, Kirisci L, Longabaugh R (1996): Polydrug use in an inpatient treatment sample of problem drinkers. Alcohol Clin Exp Res 20:413–417. [DOI] [PubMed] [Google Scholar]
- 3.Herkenham M, Lynn AB, Little MD, Johnson MR, Melvin LS, de Costa BR, Rice KC (1990): Cannabinoid receptor localization in brain. Proc Natl Acad SciUSA 87:1932–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vries T, Schoffelmeer A (2005): Cannabinoid CB receptors control conditioned drug seeking. Trends Pharmacol Sci 26:420–426. [DOI] [PubMed] [Google Scholar]
- 5.Terry GE, Hirvonen J, Liow JS, Zoghbi SS, Gladding R, Tauscher JT, et al. (2010): Imaging and quantitation of cannabinoid CB1 receptors in human and monkey brains using 18F-labeled inverse agonist radioligands. J Nucl Med 51:112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donohue SR, Krushinski JH, Pike VW, Chernet E, Phebus L, Chesterfield AK, et al. (2008): Synthesis, ex vivo evaluation, and radiolabeling of potent 1,5-diphenylpyrrolidin-2-one cannabinoid subtype-1 receptor ligands as candidates for in vivo imaging. J Med Chem 51:5833–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hirvonen J, Goodwin RS, Li CT, Terry GE, Zoghbi SS, Morse C, et al. (2012): Reversible and regionally selective downregulation of brain cannabinoid CB1 receptors in chronic daily cannabis smokers. Mol Psychiatry 17:642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirvonen J, Zanotti-Fregonara P, Umhau JC, George DT, Rallis-Frutos D, Lyoo CH, et al. (2013): Reduced cannabinoid CB1 receptor binding in alcohol dependence measured with positron emission tomography. Mol Psychiatry 18:916–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gamaleddin IH, Trigo JM, Gueye AB, Zvonok A, Makriyannis A, Goldberg SR, Le Foll B (2015): Role of the endogenous cannabinoid system in nicotine addiction: Novel insights. Front Psychiatry 6:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cahill K, Ussher MH (2011): Cannabinoid type 1 receptor antagonists for smoking cessation. Cochrane Database Syst Rev 3:CD005353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson JD, Cinciripini PM, Karam-Hage M, Aubin HJ, Dale LC, Niaura R, et al. (2018): Pooled analysis of three randomized, double-blind, placebo controlled trials with rimonabant for smoking cessation. Addict Biol 23:291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gandelman MS, Baldwin RM, Zoghbi SS, Zea-Ponce Y, Innis RB (1994): Evaluation of ultrafiltration for the free-fraction determination of single photon emission computed tomography (SPECT) radiotracers: Beta-CIT, IBF, and iomazenil. J Pharm Sci 83:1014–1019. [DOI] [PubMed] [Google Scholar]
- 13.Zoghbi SS, Shetty HU, Ichise M, Fujita M, Imaizumi M, Liow JS, et al. (2006): PET imaging of the dopamine transporter with 18F-FECNT: A polar radiometabolite confounds brain radioligand measurements. J Nucl Med 47:520–527. [PubMed] [Google Scholar]
- 14.Tzourio-Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, et al. (2002): Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. NeuroImage 15:273–289. [DOI] [PubMed] [Google Scholar]
- 15.Burger C, Mikolajczyk K, Grodzki M, Rudnicki P, Szabatin M, Buck A (1998): Java tools for quantitative post-processing of brain PET data. J Nucl Med 39:277p–278p. [Google Scholar]
- 16.Friston KJ, Holmes AP, Worsley KJ, Poline JP, Frith C, Frackowiak RSJ (1995): Statistical parametric maps in functional imaging: A general linear approach. Hum Brain Mapp 2:189–210. [Google Scholar]
- 17.Innis RB, Cunningham VJ, Delforge J, Fujita M, Gjedde A, Gunn RN, et al. (2007): Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J Cereb Blood Flow Metab 27:1533–1539. [DOI] [PubMed] [Google Scholar]
- 18.Aydin C, Oztan O, Isgor C (2012): Long-term effects of juvenile nicotine exposure on abstinence-related social anxiety-like behavior and amygdalar cannabinoid receptor 1 (CB1R) mRNA expression in the novelty-seeking phenotype. Behav Brain Res 228:236–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mateos B, Borcel E, Loriga R, Luesu W, Bini V, Llorente R, et al. (2011): Adolescent exposure to nicotine and/or the cannabinoid agonist CP 55,940 induces gender-dependent long-lasting memory impairments and changes in brain nicotinic and CB1 cannabinoid receptors. J Psychopharmacol 25:1676–1690. [DOI] [PubMed] [Google Scholar]
- 20.Marco EM, Granstrem O, Moreno E, Llorente R, Adriani W, Laviola G, Viveros MP (2007): Subchronic nicotine exposure in adolescence induces long-term effects on hippocampal and striatal cannabinoid-CB1 and mu-opioid receptors in rats. Eur J Pharmacol 557:37–43. [DOI] [PubMed] [Google Scholar]
- 21.Werling LL, Reed SC, Wade D, Izenwasser S (2009): Chronic nicotine alters cannabinoid-mediated locomotor activity and receptor density in periadolescent but not adult male rats. Int J Dev Neurosci 27:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonzalez S, Fernandez-Ruiz J, Sparpaglione V, Parolaro D, Ramos JA (2002): Chronic exposure to morphine, cocaine or ethanol in rats produced different effects in brain cannabinoid CB1 receptor binding and mRNA levels. Drug Alcohol Depend 66:77–84. [DOI] [PubMed] [Google Scholar]
- 23.Balerio GN, Aso E, Berrendero F, Murtra P, Maldonado R (2004): Delta9-tetrahydrocannabinol decreases somatic and motivational manifestations of nicotine withdrawal in mice. Eur J Neurosci 20:2737–2748. [DOI] [PubMed] [Google Scholar]
- 24.Gérard N, Ceccarini J, Bormans G, Vanbilloen B, Casteels C, Goffin K, et al. (2010): Influence of chronic nicotine administration on cerebral type 1 cannabinoid receptor binding: An in vivo micro-PET study in the rat using [18F]MK-9470. J Mol Neurosci 42:162–167. [DOI] [PubMed] [Google Scholar]
- 25.Hirvonen J (2015): In vivo imaging of the cannabinoid CB1 receptor with positron emission tomography. Clin Pharmacol Ther 97:565–567. [DOI] [PubMed] [Google Scholar]
- 26.Caille S, Alvarez-Jaimes L, Polis I, Stouffer DG, Parsons LH (2007): Specific alterations of extracellular endocannabinoid levels in the nucleus accumbens by ethanol, heroin, and cocaine self-administration. J Neurosci 27:3695–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ranganathan M, Cortes-Briones J, Radhakrishnan R, Thurnauer H, Planeta B, Skosnik P, et al. (2016): Reduced brain cannabinoid receptor availability in schizophrenia. Biol Psychiatry 79:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pavon FJ, Serrano A, Sidhpura N, Polis I, Stouffer D, de Fonseca FR, et al. (2018): Fatty acid amide hydrolase (FAAH) inactivation confers enhanced sensitivity to nicotine-induced dopamine release in the mouse nucleus accumbens. Addict Biol 23:723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheer JF, Wassum KM, Sombers LA, Heien MLAV, Ariansen JL, Aragona BJ, et al. (2007): Phasic dopamine release evoked by abused substances requires cannabinoid receptoractivation. J Neurosci 27:791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buczynski MW, Polis IY, Parsons LH (2013): The volitional nature of nicotine exposure alters anandamide and oleoylethanolamide levels in the ventral tegmental area. Neuropsychopharmacology 38:574–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Terry G, Liow J, Chernet E, Zoghbi S, Phebus L, Felder C, et al. (2008): Positron emission tomography imaging using an inverse agonist radioligand to assess cannabinoid CB1 receptors in rodents. Neuro-Image 41:690–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rossi S, De Chiara V, Musella A, Mataluni G, Sacchetti L, Siracusano A, et al. (2010): Effects of caffeine on striatal neurotransmission: Focus on cannabinoid CB1 receptors. Mol Nutr Food Res 54:525–531. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.