

SUMMARY
EndoC-βH1 is emerging as a critical human β cell model to study the genetic and environmental etiologies of β cell (dys)function and diabetes. Comprehensive knowledge of its molecular landscape is lacking, yet required, for effective use of this model. Here, we report chromosomal (spectral karyotyping), genetic (genotyping), epigenomic (ChIP-seq and ATAC-seq), chromatin interaction (Hi-C and Pol2 ChIA-PET), and transcriptomic (RNA-seq and miRNA-seq) maps of EndoC-βH1. Analyses of these maps define known (e.g., PDX1 and ISL1) and putative (e.g., PCSK1 and mir-375) β cell-specific transcriptional cis-regulatory networks and identify allelic effects on cis-regulatory element use. Importantly, comparison with maps generated in primary human islets and/or β cells indicates preservation of chromatin looping but also highlights chromosomal aberrations and fetal genomic signatures in EndoC-βH1. Together, these maps, and a web application we created for their exploration, provide important tools for the design of experiments to probe and manipulate the genetic programs governing β cell identity and (dys)function in diabetes.
In Brief
EndoC-βH1 is becoming an important cellular model to study genes and pathways governing human β cell identity and function, but its (epi)genomic similarity to primary human islets is unknown. Lawlor et al. complete and compare extensive EndoC and primary human islet multiomic maps to identify shared and distinct genomic circuitry.
Graphical Abstract
INTRODUCTION
Type 2 diabetes (T2D) is a complex disease characterized by elevated blood glucose levels. Ultimately, T2D results when pancreatic islets are unable to produce and secrete enough insulin to compensate for insulin resistance in peripheral tissues of the body. Individual genetic variation combined with dietary and environmental stressors contribute to disease risk and pathogenesis (Lawlor et al., 2017b; Mohlke and Boehnke, 2015). Genome-wide association studies have identified hundreds of genetic loci associated with T2D and related traits, but extensive work remains to identify the causal or functional variants, define their target genes, and determine the roles of these genes in β cell identity and function. Several studies have employed (epi) genomic and transcriptomic profiling of human islets (van de Bunt et al., 2015; Fadista et al., 2014; Varshney et al., 2017), purified b cells (Ackermann et al., 2016; Blodgett et al., 2015), and single-cell populations (Lawlor et al., 2017a; Segerstolpe et al., 2016; Xin et al., 2016) to identify changes in transcriptional regulation and gene expression associated with β cell (dys)function and T2D. However, the molecular and physiologic consequences of these alterations and their causal link to β cell failure and T2D pathogenesis remain largely undefined.
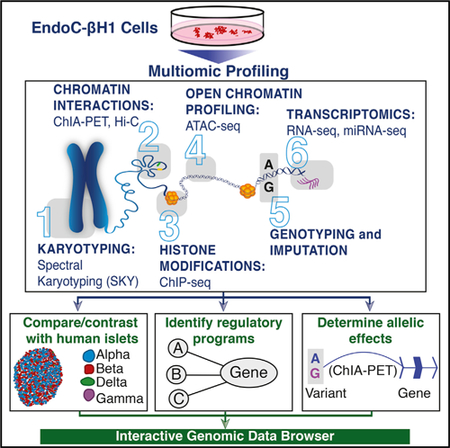
With the recent creation of an immortalized human β cell line, EndoC-βH1 (Ravassard et al., 2011), islet researchers now possess a tool to experimentally interrogate the molecular mechanisms governing human β cell identity and (dys)function. Since the initial report of their creation, studies utilizing EndoC-βH1 to build insights into human β cell regulation and function have grown steadily. These studies have demonstrated that the physiology (e.g., response to glucose and insulin secretion) of EndoC-βH1 cells resembles that of their primary islet counterparts (Andersson et al., 2015; Krizhanovskii et al., 2017; Oleson et al., 2015; Teraoku and Lenzen, 2017) and that EndoC-βH1 can be used to identify novel genes involved in human insulin secretion (Ndiaye et al., 2017). To motivate further functional studies of human β cell molecular biology and guide the development of cellular models (e.g., for small molecule screening; Tsonkova et al., 2018), extensive characterization of the EndoC-βH1 molecular landscape is needed. Here, we completed multiomic profiling of EndoC-βH1 cells to extensively map (1) chromosomal (spectral karyotyping), (2) 3D epigenomic and/or chromatin looping (Hi-C [Belton et al., 2012] and ChIA-PET [Li et al., 2014]), (3) histone modification (ChIP-seq), (4) chromatin accessibility (ATAC-seq) (Buenrostro et al., 2013), (5) genetic (dense genotyping and imputation), and (6) transcriptomic (RNA-seq and miRNA-seq) signatures of EndoC-βH1. With these high-resolution maps, we sought to (1) identify gene regulatory programs central to human β cell identity and function; (2) nominate putative functional variants, putative molecular mechanisms, and target genes underlying T2D, glucose, and insulin genetic associations; and (3) build a publicly available web application for interactive, intuitive exploration of these data. By comparing these multiomic profiles to those generated from human islets in this study (Hi-C) and parallel studies (Khetan et al., 2018), we identified shared and unique cis-regulatory elements (cis-REs) and gene expression features. Taken together, these data, the insights gleaned from their analysis, and the research support provided by the web application serve as a high-content resource to enable and guide future functional assessment and molecular studies of β cell (dys)function.
RESULTS
Chromosomal and Genetic Heterogeneity in EndoC-βH1
To pursue a precise, comprehensive understanding of the regulatory networks that govern EndoC-βH1 and/or islet β cell identity and function, we first investigated the chromosomal complement and stability of this cell line using spectral karyotyping (SKY) (Figure 1A). SKY analysis of 14 EndoC-βH1 metaphase spreads revealed that the number of chromosomes was pseudo-diploid (n = 46–48) (Figure S1). Nearly all metaphases (n = 13/14) had a normal XY sex complement, with only one having a missing Y chromosome, (metaphase 2; Table S1).
Figure 1. Extensive Karyotyping and Genotyping of EndoC-βH1.

(A) SKY of EndoC-βH1 for a representative metaphase.
(B) Summary of the frequency of chromosomal abnormalities across 14 metaphases. Black boxes indicate the presence of an event, while white boxes indicate an absence.
(C) Bar plots highlighting the risk allele burden of NHGRI-EBI GWAS Catalog diabetes-associated GWAS loci in EndoC-βH1. T1D, type 1 diabetes; T2D, type 2 diabetes. Glucose traits include fasting plasma-glucose- and fasting glucose-related traits interacting with BMI from the NHGRI-EBI GWAS catalog (MacArthur et al., 2017). Insulin traits include proinsulin and fasting insulin traits interacting with BMI.
(D) Chromosome cartoons illustrating EndoC-βH1 genotypes and the reported locus at glucose trait GWAS SNPs. Cases in which independent association signals mapped to the same locus are indicated by the locus name followed by parentheses containing numbers of SNPs with each risk genotype. Chromosomes 10 and 20 are marked with asterisks to indicate that the previously observed copy-number alterations (illustrated in Figure 1B) may obfuscate interpretation of variant genotypes on these chromosomes.
The most common autosomal aberrations in EndoC-βH1 included chromosome 20 gains (n = 11/14 metaphases) and chromosome 10 losses (n = 10/14). Both of these were independently detected as copy-number changes by comparative genomic hybridization (CGH) analysis of the cell line (Univercell Biosolutions, 2011). As summarized in Figure 1B and Table S1, we also noted recurrent 10;17 (11/14 metaphases), 7;18 (10/14 metaphases), 3;17 (7/14 metaphases), and 3;21 (7/14 meta-phases) chromosomal translocations as well as rarer events including 12;22 (metaphase 1) and 3;5 (metaphase S2.5) translocations and chromosome 12 losses (2/14 metaphases; Table S1). Together, these results emphasize that although EndoC-βH1 is largely diploid, vigilance and caution are warranted when completing and interpreting studies of genes or cis-REs on chromosomes 3, 7, 10, 17, 18, 20, and 21. We advise investigators to specifically assess copy-number variation at loci of interest, particularly in the regions identified herein as unstable or variable among the population.
Delineation of T2D- and Related-Metabolic-Trait-Associated GWAS SNP Genotypes in EndoC-βH1
Genome-wide association studies (GWASs) have identified hundreds of index and linked SNPs representing putative causal variants at hundreds of loci (Mahajan et al., 2018) associated with T2D genetic risk and changes in associated quantitative traits (e.g., fasting glucose, insulin, and proinsulin levels). We completed dense genotyping and imputation of EndoC-βH1 (STAR Methods) to determine the genotypes at ~2.5 million sites genome-wide (minor allele frequency, MAF >1%), including disease-associated SNPs. First, we overlapped EndoC-βH1 genotypes with National Human Genome Research Institute/European Bioinformatics Institute (NHGRI/EBI) GWAS catalog (STAR Methods) single lead SNPs associated with glucose levels (fasting glucose), insulin levels (fasting insulin and proinsulin levels), type 1 diabetes (T1D), or T2D (MacArthur et al., 2017). EndoC-βH1 exhibited homozygous non-risk genotypes at >50% of these SNPs (Figure 1C; Table S2). For ~20% of analyzed GWAS loci, EndoC-βH1 possessed a heterozygous genotype, including rs10830963 at the MTNR1B locus (chr11) and rs11920090 at the SLC2A2 locus (chr3) (Figure 1D; Table S2). Overlap with T2D-associated SNPs (n = 6,725 index and linked [R2 >0.8] SNPs, representing 403 unique signals) reported in the most recent meta-analysis (Mahajan et al., 2018) revealed a similar genotype distribution, in which EndoC-βH1 was heterozygous for ~30% (n = 119/403; Table S2) of T2D signals. These unique signals represent attractive candidates for (epi)genome editing to experimentally determine T2D-associated allelic effects on cis-RE use in human β cells.
The EndoC-βH1 Epigenome and Transcriptome Largely Resemble Those of Primary Islets but Retain Fetal or Progenitor Islet Cell Signatures
To identify the genome-wide location of EndoC-βH1 cis-REs (Figure 2A), we generated chromatin accessibility maps using ATAC-seq and defined chromatin states (ChromHMM) by completing and integrating ChIP-seq profiles for multiple histone modifications. ATAC-seq identified 127,894 open chromatin sites in EndoC-βH1. Qualitative comparison of EndoC-βH1 open chromatin and chromatin state maps to those in primary human islets (Khetan et al., 2018; Varshney et al., 2017) revealed that the genomic architecture for well-known islet-specific loci such as PCSK1 (Figure 2A), PDX1, and NKX6–1 was remarkably similar in both, suggesting EndoC-βH1 cells effectively recapitulate b cell cis-regulatory landscapes.
Figure 2. Multiomic Comparative Analysis of EndoC-βH1 and Human Pancreatic Islets.

(A) Integrated view of the EndoC-βH1 and human islet (epi)genomic and transcriptomic features surrounding the PCSK1 locus on chromosome 5. Histone modification ChIP-seq data from EndoC-βH1, human islets, and five Epigenome Roadmap cell types and/or tissues (Roadmap Epigenomics Consortium et al., 2015) were jointly analyzed to determine ChromHMM-based chromatin states in a uniform manner.
(B) Spearman correlation between EndoC-βH1 ATAC-seq profiles and their corresponding profiles from islets, sorted α or β cells, and other cell types and tissues (STAR Methods). α, primary islet α cells; β, primary islet β cells; CD4T, CD4+ T immune cell; GM12878, B-lymphoblast cell line; skeletal, skeletal muscle; PBMC, peripheral blood mononuclear cells. EndoC-βH1 exhibits greatest similarity to islets and their cellular constituents.
(C) Heatmap illustrating Z scores of HOMER enrichment p values for TF motifs in cell-type-specific OCRs.
(D) Comparison of chromatin states between EndoC-βH1 and human islets. Blue box highlights putative enhancer cis-REs in both EndoC-βH1 and human islets; orange box indicates putative EndoC-βH1 enhancers that are repressed in islets.
(E) TF motifs enriched in genomic regions containing putative enhancer cis-REs in both EndoC-βH1 and islets (blue) or EndoC-βH1 only (orange). Points in gray denote TFs that are not enriched in either category.
We further compared each ATAC-seq dataset to those from primary islets, sorted β or α cells, and other primary cell types, including adipocyte, skeletal muscle, peripheral blood mononuclear cells (PBMCs), and CD4+ T cells (STAR Methods). We identified a total of 269,701 open chromatin regions (OCRs) across all cell types analyzed (STAR Methods). Among all studied cell types, EndoC-βH1 ATAC-seq profiles most resembled those of β cells (Figure 2B; Spearman R = 0.67), islets (Figure 2B; Spearman R = 0.64), and α cells (Figure 2B; Spearman R = 0.62) (Spearman’s test p values < 2.225 e−308).
Next, we compared OCRs and chromatin states to determine where and to what extent EndoC-βH1 chromatin states recapitulated those of human islets, their constituent cell types, or other metabolic tissues. EndoC-βH1, islet, and β cell OCRs were commonly enriched for binding sites of transcription factors (TFs) implicated in islet cellular identity and function (Figure 2C; group III: FOXA2, FOXO1, RFX, NKX6–1, and PDX1). EndoC-βH1 OCRs also showed exclusive enrichment of sequence motifs that correspond to TFs reported to regulate pluripotency and pancreatic progenitor states (Figure 2C; group IV: HNF6, SOX2, and OCT4), perhaps reflecting the fetal origin and/or derivation of EndoC-βH1. At EndoC-βH1 ATAC-seq OCRs, promoter annotations (from ChromHMM; STAR Methods) were widely conserved between EndoC-βH1 and other cell types, including islets as expected (Figure S2A; centered Pearson correlation >0.95; STAR Methods). In contrast, enhancers, which often encode cell-specific transcriptional regulatory elements (Heinz et al., 2015), at EndoC-βH1 ATAC-seq OCRs were most comparable between islet and EndoC-βH1 (Figure S2A, black point; centered Pearson correlation ~0.71).
To further assess similarities and differences between islet and EndoC-βH1 epigenomes, we investigated the proportions and features of chromatin states that were preserved or disparate between them. Unsurprisingly, a large proportion of promoters (11,907/19,482; ~61%) were preserved (Figure 2D) and contained motifs for a variety of TFs from the ETS family (e.g., ELK4, ETS, and ELF1; Table S3) with established roles in cellular differentiation, proliferation, and apoptosis (Findlay et al., 2013). Regions annotated as repressed in both islets and EndoC-βH1 were enriched for CTCF and BORIS binding motifs (Table S3), DNA-binding proteins known to bind and establish transcriptional insulators at chromatin territory boundaries. 16,351 out of 51,325 putative enhancers (defined via ChromHMM) were shared between islets and EndoC-βH1 (Figure 2D, blue box) and showed strong enrichment for general (ATF3, AP-1, and JUN) TFs (Figure 2E, blue dots; Table S3) relative to all enhancer regions. Interestingly, we observed a substantial number of EndoC-βH1 enhancers that were annotated as quiescent or repressed in islets (n = 19,380) (Figure 2D, orange box). Relative to all enhancers, these sites were enriched for sequence motifs of TFs controlling pluripotency (OCT2 and NANOG) (Sokolik et al., 2015; Tantin, 2013), pancreatic development or lineage specification (HNF6 and ISL1) (Zhang et al., 2009), and β cell fate determination (PDX1 and NKX6–1) (Thompson and Bhushan, 2017) (Figure 2E, orange dots; Table S3). Based on these findings, it is possible that these regions may represent fetal or developmental cis-REs that are active in the fetal-derived EndoC-βH1 and inactive in adult islets composed of mature β cells. Nonetheless, a significant number of cis-REs (n = 16,351) are conserved between EndoC-βH1 and human islets.
Next, we measured EndoC-βH1 gene expression using RNA-seq and compared it to RNA-seq profiles of islets and other cell types and/or tissues (Figure S2B). As anticipated, the EndoC-βH1 transcriptome most strongly correlated with transcriptomes of islets (R = 0.87) and primary β cells (R = 0.86) among all tissues or cells tested. Of the 27,564 protein coding and/or large intergenic noncoding RNA (lincRNA) genes considered, 11,554 were expressed in EndoC-βH1 and 12,231 genes were expressed in islet (with 10,473 genes expressed in both). Similarly, EndoC-βH1 small non-coding RNA (miRNA) profiles resembled human islets more than other profiled tissues (adipose, skeletal muscle; Figure S2C) in principal-component analysis (PCA). In particular, PC1 loadings were highly correlated with key islet miRNAs, including mir-375 (Table 1), a critical regulator of b cell mass and identity (Eliasson, 2017), while PC2 stratified primary tissue (adipose, skeletal muscle, and islet) from immortalized cells (EndoC-βH1). Consistent with the PCA, miRNA expression levels in EndoC-βH1 and islets were highly correlated (R = 0.779; Figure S2D), and the vast majority of the most highly expressed miRNAs in EndoC-βH1 have been reported previously to be enriched in primary human β cells relative to whole islets (Table 1) (van de Bunt et al., 2013). Together, the chromatin accessibility, chromatin state, gene expression, and small RNA expression analyses reveal substantial conservation between the transcriptional regulatory and gene expression landscapes of EndoC-βH1 and primary islets.
Table 1.
Top 25 Expressed miRNAs in EndoC-βH1 Cells
| miRNA | EndoC-βH1 Rank | EndoC-βH1 RPMMM | β Cell Enrichment / Depletion |
|---|---|---|---|
| hsa-miR-375 | 1 | 162076.925 | 2.84 |
| hsa-miR-127–3p | 2 | 90512.35 | - |
| hsa-miR-27b-3p | 3 | 85043.195 | 3.09 |
| hsa-miR-192–5p | 4 | 70126.5525 | 2.01 |
| hsa-miR-192–5p_+_1 | 5 | 37621.0275 | - |
| hsa-miR-182–5p | 6 | 36662.2475 | 2.15 |
| hsa-miR-22–3p | 7 | 29623.8 | 2.87 |
| hsa-miR-191–5p | 8 | 25424.1575 | 3.02 |
| hsa-miR-26a-2–5p | 9 | 15578.9175 | 2.94 |
| hsa-miR-26a-1–5p | 10 | 15496.7075 | 2.94 |
| hsa-miR-141–3p | 11 | 13656.6825 | 2.84 |
| hsa-miR-92a-1–3p | 12 | 13615.6425 | 2.30 |
| hsa-miR-30d-5p | 13 | 11108.9275 | 0.75 |
| hsa-miR-654–3p | 14 | 11107.385 | - |
| hsa-miR-148a-3p | 15 | 10651.885 | 2.22 |
| hsa-miR-200b-3p | 16 | 10266.2575 | 1.48 |
| hsa-miR-92a-2–3p | 17 | 10161.165 | 2.30 |
| hsa-miR-381 | 18 | 9342.8875 | 1.08 |
| hsa-miR-25–3p | 19 | 9230.2225 | 1.98 |
| hsa-miR-181a-1–5p | 20 | 8702.47 | 1.81 |
| hsa-miR-181a-2–5p | 21 | 8702.3125 | 1.81 |
| hsa-miR-21–5p | 22 | 8439.755 | 0.21 |
| hsa-miR-183–5p | 23 | 8338.08 | 1.58 |
| hsa-miR-92b-3p | 24 | 7531.295 | 4.63 |
| hsa-miR-125a-5p | 25 | 6743.8375 | 1.24 |
Table consisting of the top 25 expressed miRNAs in EndoC-βH1 cells. Each miRNA rank was determined by its corresponding expression level (reads per million mapped miRNA [RPMMM]). β cell enrichment / depletion was determined by dividing the counts of each miRNA in β cells (from van de Bunt et al., 2013) by the counts in islets.
Hi-C Profiling of EndoC-βH1 and Human Islets Reveals β Cell-Specific Chromatin Looping Domains
Next, we sought to determine spatial chromatin organization and identify chromatin domains both in EndoC-βH1 and islets using Hi-C. We generated Hi-C maps with ~6 billion reads each for EndoC-βH1 and human islet cells. The maps have 1.9 billion contacts and 1.5 billion contacts, respectively. Using Juicer (Durand et al., 2016a) (STAR Methods), we identified 9,100, 2,580, and 9,448 Hi-C loops in EndoC-βH1, human islet, and GM12878 (a human lymphoblastoid cell line) (Rao et al., 2014), respectively. The reduced number of Hi-C loops identified in primary islet may be attributed to lower unique read depth (~3.7 billion unique reads in islet cells versus ~4.7 billion unique reads in EndoC-βH1) (Table S1). Together, this represents 19,428 independent DNA loops. Aggregate peak analyses (APAs) (Rao et al., 2014) (Figure 3A, top plots) revealed that chromatin looping sites (anchors) were comparable in EndoC-βH1, islets, and GM12878 for the majority of (>90%) the total chromatin loops (n = 19,428/21,128). Consistent with previous studies (Rao et al., 2014; Vietri Rudan et al., 2015), CTCF and CTCFL DNA-binding motifs were overwhelmingly enriched (p < 1e-229 and p < 1e-114, respectively) among all Hi-C anchor sequences (Table S4), verifying that general 3D chromatin structures and loops are preserved between different mammalian tissues and cell types. Importantly, however, we detected 1,078 and 117 chromatin loops that were exclusively present in EndoC-βH1 and islet, respectively, compared to GM12878 (Figure 3A, bottom plots).
Figure 3. Generating a Genome-wide Map of Looping in EndoC-βH1 and Human Pancreatic Islets (Hi-C).

(A) Aggregate peak analysis (APA) plots showing the total signal across all loops (top three panels) and EndoC-βH1-specific loops (bottom three panels) in EndoC-βH1 (left), human islet (center), and GM12878 (right) cells. Of note, islets exhibit similar contact point enrichments at EndoC-βH1-specific peaks compared to GM12878.
(B) Cartoon illustrating the different classes of Hi-C loops between example common (gray peaks) or cell-specific (black peaks) ATAC-seq OCRs for two different theoretical cell types.
(C) TF motifs enriched in GM12878 (blue) or EndoC-βH1 (red) Hi-C looping anchors that overlap cell-specific ATAC-seq peaks loop classes B and C in panel B above).
(D) Hi-C contact maps highlighting a specific loop at the SLC30A8 locus (denoted by dotted black circle) observed in both EndoC-βH1 (left) and primary human islets (center) but absent in GM12878 (right).
(E) Multiomics view of Hi-C, ChIA-PET (Pol2), chromatin states, ATAC-seq, RNA-seq, and gene tracks at the SLC30A8 neighborhood containing the Hi-C contact point highlighted in (D). Tracks corresponding to EndoC-βH1, human islet, and GM12878 are colored red, black, and blue, respectively. Dark blue boxes below each gene name represent the reference transcript annotations derived from Gencode v19. The red arrow at the bottom of the image indicates the putative EndoC-βH1- and islet-specific promoter for SLC30A8. The black arrow indicates the putative embryonic stem cell and K562 cell-specific promoter for SLC30A8 (Roadmap Epigenomics Consortium et al., 2015).
To further study cell-specific loops, we subdivided EndoC-βH1 and GM12878 differential Hi-C loops into three classes based on the cell type specificity of the ATAC-seq OCRs they bring into physical proximity (Figure 3B): (A) loops between two non-specific OCRs, (B) loops between two cell-specific OCRs, or (C) loops between one cell-specific OCR and one non-specific OCR. Class B/C loops were classified as cell-specific and further studied. Comparison of EndoC-βH1-specific (n = 315) and GM12878-specific (n = 308) loops revealed a strong bias for cell-specific TF binding at anchor sites (Figure 3C). In EndoC-βH1-specific anchors, we observed enrichment for TFs involved in β cell differentiation and function (NKX6–1, FOXA2, and FOXA1) (Thompson and Bhushan, 2017) as well as OCT4, a key regulator for early embryo development (Le Bin et al., 2014; Wu and Schöler, 2014), while GM12878-specific anchors were enriched for TFs necessary for B cell proliferation and activation (MEF2C and NFAT) (Herglotz et al., 2016; Peng et al., 2001). Furthermore, genes adjacent to EndoC-βH1-specific anchors (STAR Methods) were most enriched (hypergeometric false discovery rate [FDR]-adjusted p value < 0.05) for islet-associated gene ontology (GO) terms, including insulin secretion, glucose homeostasis, and neuronal or endocrine development (Figure S3A; complete results are shown in Table S4). For several genes affiliated with these GO terms, such as SLC30A8, which encodes a zinc efflux transporter involved in zinc ion sequestering and insulin secretion (Mitchell et al., 2016), we observed striking similarities in Hi-C contact frequencies between islet and EndoC-βH1 cells (Figure 3D). In contrast, we observed far fewer chromatin loops and large spans of polycomb-repressed and/or quiescent chromatin for this locus in GM12878 cells (Figure 3E).
Approximately 50% (4,543/9,100) of EndoC-βH1 loop anchors overlapped EndoC-βH1 ATAC-seq OCRs, 44% (n = 1,987/4,543) of which occurred between promoter and enhancer elements (Figure S3B). Of these 4,543 loops, 587 were specifically present in EndoC-βH1 yet absent in GM12878 cells (EndoC-βH1 specific); the remaining 3,956 were captured in both EndoC-βH1 and GM12878 (non-specific). We observed a substantially higher proportion (64%; 376/587) of EndoC-βH1-specific loops that overlapped EndoC-βH1 stretch enhancers (Parker et al., 2013) (Fisher’s exact test p value < 4.23 e-42) compared to that of nonspecific loops (34%; 1,358/3,956). To examine the functional specificity of these loops, we overlapped chromatin state (ChromHMM) information from EndoC-βH1 and 27 other tissue or cell types for all EndoC-βH1 Hi-C loops. For each cell type, we determined the percentage of Hi-C anchors that contained the same chromatin state as EndoC-βH1 (STAR Methods). Islets had the highest percentage of chromatin states identical to) EndoC-βH1 at Hi-C anchor sites among all tested tissues or cell types, especially promoter-enhancer loops (Figure S3C; orange line plot). These findings enumerate regions of cell-specific chromatin looping associated with islet development and function and indicate that EndoC-βH1 forms cell-type-specific chromatin domains or territories highly similar to primary human islets.
EndoC-βH1 Pol2 ChIA-PET Identifies β Cell cis-Regulatory Hubs
To map functional cis-regulatory networks, we completed RNA polymerase (Pol2) ChIA-PET (Li et al., 2017b) in EndoC-βH1, identifying 25,336 putative Pol2-mediated chromatin interactions. We further filtered these interactions, retaining only those for which both interacting sites (ChIA-PET anchors) overlapped EndoC-βH1 ATAC-seq OCRs, resulting in 16,756 putative cis-regulatory interactions (STAR Methods). As shown in Figure 4A, the overwhelming majority of Pol2-mediated chromatin interactions linked active enhancer and active promoter chromatin states to themselves and each other. Importantly, ChIA-PET detected EndoC-βH1-specific interactions (Figure 4B; compare EndoC-βH1, GM12878, and K562 ChIA-PET tracks) coinciding with those previously reported in targeted 4C-seq analyses of human islets (Pasquali et al., 2014), including the ISL1 (Figure 4B; n = 8 sites denoted by asterisks) and PDX1 (Figure S4A; n = 9 sites) loci.
Figure 4. RNA Polymerase 2 ChIA-PET Identifies Chromatin Interactions in EndoC-βH1.

(A) Heatmap showing the chromatin states of EndoC-βH1 ChIA-PET interaction nodes.
(B) Example of a Pol2 ChIA-PET interaction between active enhancer (blue box) and active promoter (green box) cis-REs in the ISL1 locus on chromosome 5. Asterisks under EndoC-βH1 ChIA-PET interactions (red) indicate interacting sites in the ISL1 locus detected in human islet 4C-seq analyses (Pasquali et al., 2014).
(C) ChIA-PET network connectivity of gene promoters in EndoC-βH1 containing at least three interactions with other regulatory elements. For each gene, the number of connections between other regulatory elements (e.g., active enhancer and weak enhancer) and the proportion of total links in which the chromatin states are EndoC-βH1-specific (blue) or identical in both human islet and EndoC-βH1 (green) are shown in bar plots on the right. The remaining proportions that are neither EndoC-βH1 specific nor common to islet and EndoC-βH1 are not shown. Red font denotes loci containing genes crucial for β cell identity and development.
(D) Top: Bar plot illustrating the proportions of chromatin states at the Pol2 ChIA-PET interacting sites (nodes) shared between EndoC-βH1, islets, and additional Epigenomics Roadmap tissues and cell lines. Bottom: Heatmap demonstrating the chromatin states of EndoC-βH1 Pol2 ChIA-PET interacting sites (nodes) in islets (left) or stomach smooth muscle (right).
See also Figure S4.
In addition to replicating interactions previously studied by 4C-seq in human islets, Pol2 ChIA-PET identified hundreds of additional promoter-promoter and promoter-enhancer interactions genome-wide (Figure 4C; STAR Methods). These include extensive Pol2 interactions in loci containing genes crucial for b cell identity and development such as PDX1, ISL1, NKX6–1, MAFB, and miR375 (Figure 4C, red text). As shown in Figures 4C and S4A, multiple interactions were detected between the PDX1 promoter and classically described essential PDX1 transcriptional enhancer sequences (n = 7/14 enhancer interactions), which contain binding sites for islet TFs such as FOXA2 (Gao et al., 2008; Gerrish et al., 2004). During embryonic development, C57BL/6 mouse pancreata displayed a transition in expression of MafB to MafA (Nishimura et al., 2006), suggesting that these two factors are tightly involved in β cell differentiation and function. The high degree of connectivity in MAFB (versus that of MAFA) may therefore reflect the fetal or naive state of EndoC-βH1 cells. miR375, a small non-coding RNA, possessed multiple connections to active promoter and enhancer elements (Figure 4C; red text), consistent with its role as a post-transcriptional regulator of genes involved in β cell development or differentiation and insulin secretion or exocytosis (Eliasson, 2017).
Interestingly, INSM1, a gene necessary for pancreatic endocrine cell differentiation (Osipovich et al., 2014), harbored the most connections in EndoC-βH1 (n = 97 total interactions, n = 7 between active promoters and enhancers; Figure S4B). Other genes linked by ChIA-PET interactions are involved in insulin processing and secretion, including PCSK1, one of the prohormone convertases that catalyzes (pro)insulin processing; RIMBP2, whose protein mediates formation of a complex for polarized accumulation and exocytosis of insulin granules (Fan et al., 2017); RGS7, a critical regulator of muscarinic-stimulated insulin secretion (Wang et al., 2017); and CDC42, which is essential for second-phase insulin secretion (Wang et al., 2007). Additionally, genes implicated in the protection and management of stress were highly connected in EndoC-βH1 ChIA-PET interactions. Notable candidates were ZFAND2B, whose induction helps protect against human β amyloid peptide toxicity or accumulation in a C. elegans transgenic Alzheimer’s disease model (Hassan et al., 2009); SUSD4, a complement inhibitor and tumor suppressor that modulates endoplasmic reticulum stress; and CD59, which is required for mediating exocytosis events facilitating insulin secretion (Blom, 2017; Krus et al., 2014). Finally, TSHZ1, a PDX1 target gene whose expression levels were notably lower in human islet donors with T2D (Raum et al., 2015), harbored five links to active enhancer elements in EndoC-βH1, suggesting that perturbation of the cis-regulatory networks identified herein may contribute to T2D pathogenesis.
Finally, we sought to study to what extent the putative cis-regulatory networks detected in EndoC-βH1 may be preserved in islets and other cell types. Due to limited availability of ChIA-PET data in human islets and other relevant tissues, we decided to use the chromatin interaction sites determined by EndoC-βH1 ChIA-PET and compare the functional annotations (ChromHMM state annotations) at these loci across 27 different tissue or cell types. Overall, aggregate counts of these chromatin state interactions for each cell type were most similar between EndoC-βH1 and islet (Figure 4D, green bar plots; STAR Methods). ChIA-PET interactions between regions annotated as active promoters in EndoC-βH1 were similarly annotated as active promoters across the 27 other cell or tissue types (Figure 4D, Act. promoter 3 Act. promoter, red line plot; STAR Methods). In contrast, the majority of active enhancers interacting in EndoC-βH1 were marked as active enhancers only in human islets (Figure 4D, Act. Enhancer × Act. Enhancer, yellow line plot). Multidimensional scaling of all cell or tissue chromatin state annotations at EndoC-βH1 ChIA-PET interacting sites reaffirmed high similarity between EndoC-βH1 and islets (Figure S4C), consistent with strong conservation of active enhancer state annotations, as previously observed (Figure 4D, line plots). These results suggest that these interactions may represent β cell cis-regulatory hubs. Indeed, anchors for 41% of ChIA-PET interactions (6,904/16,756) overlapped islet stretch enhancers, suggesting that these interactions may encompass key islet functional chromatin domains.
Integration of EndoC-βH1 Genotype and 3D Genomic Interaction Maps Identifies Allelic Imbalance at β Cell-Specific cis-REs
We and others have demonstrated that genetic variants, including those associated with T2D and other quantitative measures of islet (dys)function, can alter cis-RE use (chromatin accessibility quantitative trait loci [caQTL]) (Khetan et al., 2018) and target gene expression (expression quantitative trait loci [eQTL]; van de Bunt et al., 2015; Fadista et al., 2014). Recently, approaches have been used to assess allelic effects on these molecular features at heterozygous sites within a single sample. We applied these allelic imbalance (AI) analyses in EndoC-βH1 to identify genetic variants that alter b cell cis-REs and target gene expression. To identify instances of AI in EndoC-βH1, we examined the allelic bias of ~2 million heterozygous SNPs (STAR Methods) within OCRs (ATAC-seq peaks), active enhancer elements (H3K27ac peaks), or expressed genes (RNA-seq; Figure 5A). Less than 10% of all SNPs occurring in OCRs and enhancer elements showed significant AI (Figure 5A; part I; FDR <10%). Approximately 25% of SNPs exhibited gene expression AI (Figure 5A). When considering variants with adequate coverage in both EndoC-βH1 ATAC-seq OCRs and H3K27ac-marked enhancer regions (n = 1,734 SNPs), we noted a positive correlation (R = 0.2) in the corresponding AI ratios (Figure S5A), suggesting the potential for coordinate regulation of chromatin accessibility and histone modification at these cis-regulatory sites. In total, 119 out of 403 T2D-associated signals (~30%) overlapped EndoC-βH1 cis-REs (Table S2); 34 out of 119 of these unique signals (~29%) were heterozygous in EndoC-βH1 and potentially amenable to allelic analyses. GREGOR (Schmidt et al., 2015) enrichment analysis of these same 403 signals identified significant overlap (p value < 1 e-7) in EndoC-βH1 ATAC-seq (n = 67 SNPs) OCRs. Moreover, 24,102 out of 126,013 of T2D-associated 99% genetic credible set SNPs (19%) (Mahajan et al., 2018), representing 327 out of 380 distinct association signals (86%), overlapped an EndoC-βH1 ATAC-seq OCR and/or H3K27ac peak. These overlaps suggest that EndoC-βH1 may be a useful tool to dissect the function of cis-REs implicated in T2D genetic risk.
Figure 5. Allelic Effects on EndoC-βH1 Transcriptional Regulatory Features.

(A) EndoC-βH1 genotype information was integrated with ATAC-seq, H3K27ac, and RNA-seq data to identify sequence variants altering cis-RE accessibility and/or activity (ATAC-seq and H3K27ac) or mRNA levels (RNA-seq) in EndoC-βH1. Pie charts summarize the proportions of variants exhibit significant AIs (blue; FDR < 10%) in each of the corresponding sequencing profiles.
(B) Cartoon representation of approach to identify systematic allelic effects on EndoC-βH1 cis-regulatory networks.
(C) Multiomic view highlighting allelic effects on the SAMD5 locus cis-regulatory network in EndoC-βH1. A variant site exhibiting significant AI in H3K27 acetylation (denoted by blue arrow) is linked (red ChIA-PET interaction) to the transcription start site (TSS) of SAMD5. Within the SAMD5 locus, five transcribed SNPs exhibited significant allelic bias in gene expression (RNA-seq) in a direction consistent with the H3K27ac allelic bias.
(D) Left: Bar plots summarizing the proportions of variants with ATAC-seq/H3K27ac imbalance (blue bars; FDR < 10%) that overlap ChIA-PET interacting loci. Right: Pie charts specifying the chromatin state (ChromHMM) annotations of the overlapping variants.
Next, we leveraged information from EndoC-βH1 ChIA-PET interactions to determine potential allelic effects on cis-regulatory networks and target gene expression. To achieve this, we (1) identified SNPs overlapping enhancers with AI (H3K27ac and/or ATAC-seq), (2) determined if the SNP-containing enhancer linked (via ChIA-PET) to the transcription start site (TSS) of a gene, and (3) assessed if SNPs in the promoter or transcribed region of the predicted target gene exhibited AI in H3K27ac and/or ATAC-seq or RNA-seq, respectively (Figure 5B). For example, rs2294805 exhibited AI in an EndoC-βH1 enhancer downstream of SAMD5 and was linked to this gene’s TSS by a ChIA-PET interaction (Figure 5C). Notably, 5 out of 11 transcribed SAMD5 SNPs exhibited significant AI in RNA-seq gene expression data. In all cases, one parental allele (denoted in blue) was consistently over-represented in both H3K27ac ChIP-seq and RNA-seq data. Although the exact role of SAMD5 in human islets has not been described, expression of this gene is high in adult α cells but absent in adult β cells (Lawlor et al., 2017a; Segerstolpe et al., 2016; Wang et al., 2016). SAMD5 has been recently identified as a marker of peribiliary gland (PBG) cells (Yagai et al., 2017), and PBG stem cells have been documented to differentiate into glucose-responsive pancreatic islets (Cardinale et al., 2011). These data identify SAMD5 as one of the most highly connected loci in EndoC-βH1 (Figures 4C and S4B; n = 35 ChIA-PET connections between the gene and active enhancers; n = 5 interactions to other active promoters) and highlight a potential cis-regulatory hub for fetal β and islet cell development. Further manipulation of the cis-regulatory network in this locus may provide greater insight into its putative roles in islet cell differentiation and function.
Overall, 2,500 out of 5,515 (~45%) and 8,794 out of 43,492 (~20%) of heterozygous SNPs passing coverage thresholds (STAR Methods) in ATAC-seq and H3K27ac cis-REs, respectively, overlapped a ChIA-PET anchor (Figure 5D, bar plots). For both datasets, <5% of SNPs demonstrated significant AI. AI SNPs were present in active promoter and enhancer regions of the genome (Figure 5D, pie charts marked with blue arrows). Enhancers exhibited enriched AI compared to promoters. For example, in the ATAC-seq data, 49.5% of AI SNPs occurred in enhancers compared to 30.9% of non-AI SNPs (Fisher’s exact test p value = 0.0002824 for comparison of enhancer versus promoter counts). Similarly, 54.6% of H3K27ac AI SNPs occurred in enhancers compared to 43% of non-AI SNPs (Fisher’s exact test p value = 3.343e-05 for comparison of enhancer versus promoter counts) (Figure 5D).
Enhancers govern cell-type-specific gene expression patterns (Heinz et al., 2015). We identified 50 and 185 enhancer SNPs with significant ATAC-seq and H3K27ac AI, respectively (Figure 5D, yellow portion of pie charts marked with blue arrows). We used ChIA-PET interactions to link enhancers showing AI to promoters of the genes they might regulate (Table S5). This identified enhancer-promoter links for 21 out of 50 (42%) ATAC-seq and 91 out of 185 (~49%) H3K27ac AI enhancer SNPs, respectively. To assess if observed allelic effects on EndoC-βH1 cis-regulatory networks extended to primary islets, we examined the eQTL direction of effect of these SNPs on steady-state expression of their predicted target genes in human islets (Varshney et al., 2017). Islet eQTL Z scores (STAR Methods) were most correlated to the H3K27ac allelic ratio at SNPs where the human islet eQTL target gene (i.e., the gene whose expression in human islets is influenced by a genetic variant) matched that of the EndoC-βH1 ChIA-PET target gene (i.e., the gene linked to the enhancer by EndoC-βH1 ChIA-PET as depicted in Figure 5B) (n = 42/91) (Figure S5B, red points; R = 0.32), as opposed to those genes in the locus that were not linked to the enhancer by ChIA-PET (Figure S5B, gray points; R = 0.17).
Importantly, these analyses suggest that integrated EndoC-βH1 omics analyses provide molecular insights into diabetes genetics (e.g., GWAS). For example, T2D-associated index and 99% credible set SNP rs57235767, for which the “C” risk allele exhibited reduced EndoC-βH1 H3K27ac counts, exhibited consistent downregulation of the ChIA-PET-predicted target gene C11orf54 (Figure S5B, asterisk) expression in islet cohorts (Fadista et al., 2014; Varshney et al., 2017). Similarly, the T2D risk allele of rs3807136, a 99% credible set SNP in linkage disequilibrium (r2 > 0.8) with the index and 99% credible set SNP rs2268382, displayed a higher proportion of H3K27ac counts in EndoC-βH1 and increased expression of the predicted target gene, CEP41. These human islet SNP-gene interactions (that are also recapitulated in EndoC-βH1 cells) represent high-priority targets for (epi)genomic modification and should assist efforts to decrypt the genes contributing to T2D pathogenesis.
DISCUSSION
Here, we report extensive multiomic mapping and integrated analysis of cytogenetic (karyotyping), large-scale chromatin structural conformation (Hi-C), cis-regulatory networks (ChIAPET), histone mark (ChIP-seq), chromatin accessibility (ATAC-seq), genetic (genotyping), and gene expression (RNA-seq) information in EndoC-βH1 human β cells. For convenient and interactive browsing of this data, we have created an R shiny web application (Chang et al., 2018) available at https://shinyapps.jax.org/endoc-islet-multi-omics/. These data and the browser application should serve as a resource for future studies to explore the complex β cell regulatory programs uncovered in this study and guide targeted studies of regulatory networks, genes, and pathways of interest.
SKY revealed chromosomal heterogeneity among individual cells in the EndoC-βH1 population. These included copy number variation, such as chromosome 20 gain and chromosome 10 loss, that has been identified independently by array CGH analyses, as well as previously unappreciated structural alterations, including chromosome 10:17 and 3:21 translocations, that are frequent within the population. Consistent with SKY, we observed enhanced contact frequency between chromosomes 3 and 21 in EndoC-βH1 Hi-C maps (Juicebox [Durand et al., 2016b]; http://aidenlab.org/juicebox/?juiceboxURL=http://bit.ly/2NxWcDp), suggesting that these two technologies may complement one another to identify cell line abnormalities. As evidenced by other less prevalent chromosomal aberrations among the population, it is possible that this cell line may continue to evolve with continued passaging. Thus, caution should be taken, and these aberrations should be considered, in future (epi)genome editing or EndoC-βH1 molecular and functional experiments involving genes or regulatory elements on these chromosomes.
Overall, comparative analyses of omics profiles indicate substantial similarity among EndoC-βH1, islet, and primary β cell transcriptomes (Figure 2B; Pearson R > 0.86). EndoC-βH1 open chromatin profiles were modestly correlated with islet (R = 0.64) and primary β (R = 0.67) cells, highlighting potential drift between the cell line and primary islet cells at the level of chromatin accessibility. This includes ~19,000 putative EndoC-βH1 enhancers annotated as quiescent or polycomb repressed in human islets. Interestingly, these sites contain potential binding sites for TFs with important roles in β cell development and pancreatic precursor fates and functions (e.g., NKX6–1, PDX1, ISL1, and HNF6) (Thompson and Bhushan, 2017) and pluripotency (e.g., NANOG and OCT2) (Sokolik et al., 2015; Tantin, 2013). Thus, these discordant features may reflect the fetal nature of EndoC-βH1 cells, their transformed state, or both.
Using Hi-C to map higher-order chromatin structure in EndoC-βH1 and a corresponding map from a human islet donor, we defined islet and β cell chromatin domains and territories. Consistent with previous findings (Rao et al., 2014), the overall spatial chromatin organization was similar across EndoC-βH1, islet, and GM12878 cells (Figure 3A; top panel), and all Hi-C anchors were enriched for TFs with general roles in chromatin organization (e.g., CTCF and BORIS; Table S4). Importantly, however, Hi-C analyses also identified ~1,078 islet and β cell-specific chromatin domains, several of which were evident in both EndoC-βH1 and primary human islets (Figure 3). These cell-specific chromatin territories were enriched for β cell-specific TFs (Figure 3C) and brought into close physical proximity genes linked to islet-associated biological process GO terms (Figure S3A) compared to those of GM12878.
Here, we also report a Pol2 ChIA-PET map of chromatin interactions in EndoC-βH1, which further refined chromatin territories to reveal functional EndoC-βH1 cis-regulatory networks. In addition to validating chromatin interactions previously reported in 4C-seq analyses at select loci in human islets, Pol2 ChIA-PET identified hundreds of interactions genome-wide between active promoter and enhancer regions potentially involved in the regulation and transcription of dozens of β cell-specific loci (Figures 4A–4C). Comparison of Pol2 ChIA-PET interaction locations in EndoC-βH1, GM12878, and K562 revealed that the overwhelming majority of interactions at these loci were unique to EndoC-βH1. Due to high cell input requirements (~100 million cells) for current ChIA-PET library construction protocols (Li et al., 2017b), we were unable to validate these findings in human islets. However, consistent with our previous observations between islet and EndoC-βH1 Hi-C maps (Figure S3C), we noted that chromatin states of ChIA-PET interaction nodes in EndoC-βH1 were most conserved in islet (Figure 4D, Figure S4C) compared to those of 27 other cell or tissue types. Thus, the cis-regulatory programs we define for EndoC-βH1 should provide valuable insights into important transcriptional hubs that drive islet and b cell identity and function.
GWASs have identified hundreds of loci that contribute to genetic risk of T2D and other quantitative measures of islet dysfunction, such as glucose, insulin, and proinsulin levels (Fuchsberger et al., 2016; Mahajan et al., 2018). We and others have linked a subset of SNPs and loci to altered cis-RE activity and steady-state islet gene expression (van de Bunt et al., 2015; Fadista et al., 2014; Khetan et al., 2018; Varshney et al., 2017). For the majority of loci, challenges remain to define the (1) causal or functional SNP, (2) determine its molecular effect on cis-RE activity, and (3) identify the putative target gene(s). By combining our dense genotype and 3D chromatin interaction (ChIA-PET) networks in EndoC-βH1, we sought to identify SNPs with imbalanced expression or cis-RE use and link them physically with their target genes. Using this approach, we linked 91 out of 185 (H3K27ac imbalanced) and 21 out of 50 (ATAC-seq imbalanced) SNPs to potential target genes (e.g., rs2294805 to SAMD5 in Figure 5B). Our ability to assess chromatin interactions between diabetes-associated SNPs and their target genes was modest. Nonetheless, we identified two candidate SNPs (rs57235767, rs3807136) that demonstrated consistent directions-of-effect on measures of cis-RE activity (e.g., H3K27ac) and target gene expression in EndoC-βH1 and human islets (Figure S5B) (Varshney et al., 2017). Several factors could underlie the modest frequency of diabetes-associated GWAS SNPs linked by Pol2 ChIA-PET interactions: (1) limited sensitivity of ChIA-PET technology, (2) condition or disease specificity of GWAS SNP effects on cis-RE use or activity, or (3) condition-specific Pol2 interactions between cis-REs and their target genes.
In summary, this study provides an integrated multiomic analysis of EndoC-βH1, a human pancreatic β cell line with increasing utility and importance to the β cell and diabetes communities. Integrated analysis of chromatin interaction and gene expression information identified chromosomal territories and cis-regulatory networks governing β cell identity and function. Overall, comparison of EndoC-βH1 (epi)genomic and 3D chromatin profiles with those of human islets verified common signatures of gene expression, TF binding, and cis-RE use. These analyses also highlighted genomic discrepancies between EndoC-βH1 and their primary cell counterparts, possibly reflecting the fetal or embryonic origin of the cell line and/or its transformed state. Integration of EndoC-βH1 cis-regulatory maps with genome-wide genotype information nominated target genes and identified SNP allelic effects on transcriptional regulatory networks, including a subset of T2D-associated SNPs. Together, the data and tools provided here should serve as helpful guides for rational design of targeted and hypothesis-driven studies of candidate genes, pathways, or cis-REs to determine their roles in β cell (dys)function and diabetes.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Stitzel (michael.stitzel@jax.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
EndoC-βH1 cell culture and processing
EndoC-βH1 cells provided by EndoCells/INSERM were cultured and passaged as previously described (Ravassard et al., 2011). Briefly, cells were seeded at a density of approximately 600,000 cells/cm2 on tissue culture-treated plates pre-coated overnight with extracellular matrix (Sigma) and fibronectin (Sigma) in EndoC-βH1 complete medium. Cells were passaged approximately every 7 days. Cells were harvested at various passages and distinct sites (e.g., NHGRI, JAX-GM) for karyotyping, genotyping, ATAC-seq, ChIP-seq, RNA-seq, Hi-C, and Pol2 ChIA-PET analyses.
Human islet acquisition and procurement
The single human pancreatic islet sample (obtained from a cadaveric donor; UNOS ID: ADAC418; sex: Female; age: 43 years old) used in this study was obtained from the National Disease Research Interchange (NDRI) and processed according to NHGRI institutional review board-approved protocols. The islet was shipped overnight from the distribution center. On receipt, we pre-warmed the islet to 37°C in shipping media for 1–2 h before harvest; ~50,000 islet equivalents (IEQs) were harvested for Hi-C.
METHOD DETAILS
Spectral karyotyping (SKY)
Spectral karyotyping of EndoC-βH1 was completed to identify structural and numerical chromosome aberrations using standard procedures as previously described. In brief, EndoC-βH1 cells were cultured to 80% confluence. Metaphase spreads were prepared from these cells after mitotic arrest with Colcemid (0.015 mg/mL, 16 to 18 hours) (GIBCO, Gaithersburg, MD), hypotonic treatment(0.075 mol/L KCl, 20 minutes, 37°C), and fixation with methanol–acetic acid (3:1). Commercial SKY probe and software (Applied Spectral Imaging INC, Carlsbad, CA) was used to identify and visualize the individually colored chromosomes obtained from two slides’ worth of metaphase spreads from the same passage.
Genotyping, imputation, and phasing of EndoC-βH1
EndoC-βH1 was genotyped with the HumanOmni2.5–4v1_H BeadChip Array (Illumina, San Diego, CA, USA). We mapped the Illumina array probe sequences to the hg19 genome assembly and excluded likely problematic ones as described in (Varshney et al., 2017).
We applied the following filtering criteria to remove additional SNP probes prior to pre-phasing of the array genotypes: 1) we assessed allele frequency of the SNPs using combined genotypes of EndoC-βH1 and 163 other samples that were genotyped on similar chips; and 2) we removed SNPs with an alternate allele frequency difference with 1000G EUR samples > 20%, or palindromic SNPs with a minor allele frequency > 20%, genotype missingness > 2.5%, Hardy-Weinberg p value < 10−4. At the end, a total of 1,851,388 SNPs were used in pre-phasing and imputation.
We performed pre-phasing and imputation separately on autosomal and chrX markers using the Michigan Imputation Server (Das et al., 2016). We used Eagle v2.3 (Loh et al., 2016) for autosomal chip marker pre-phasing and SHAPEIT v2.r790 (Delaneau et al., 2011) for chrX markers. We subsequently used minimac3 (Das et al., 2016) for imputation of missing genotypes using the Haplotype Reference Consortium (HRC version, hrc.r1.1.2016) panel (McCarthy et al., 2016).
GWAS SNP pruning
Lists of reference SNP identifiers were obtained from the NHGRI-EBI Catalog of SNPs (https://www.ebi.ac.uk/gwas/; accessed January 19th, 2017) for Type 2 diabetes, Type 1 diabetes, fasting glucose traits, fasting insulin traits, and proinsulin level categories. For each disease category, GWAS SNPs were pruned using PLINK version 1.9 (Purcell et al., 2007) to identify SNPs in high linkage disequilibrium (LD) (R2 > 0.8) using the parameters “–maf 0.05–clump–clump-p1 0.0001–clump-p2 0.01–clump-r2 0.8–clump-kb 1000” to ensure that each variant haplotype was tested only once during the enrichment analysis. For each SNP pair in LD (R2 > 0.8) the SNP with the least significant p value was discarded. T2D-associated SNPs from (Mahajan et al., 2018) were obtained and pruned using the same methodology described above.
ATAC-seq
EndoC-βH1 ATAC-seq libraries were prepared as previously described (Varshney et al., 2017) and sequenced on an Illumina NextSeq 500 with 2 3 125 bp cycles. Raw sequence fastq files for adipocyte tissue, bulk islet (Khetan et al., 2018), islet beta and alpha (GSE76268) (Ackermann et al., 2016), peripheral mononuclear blood cells (PBMC) (Ucar et al., 2017), skeletal muscle (Scott et al., 2016), GM12878 and CD4+ T cells (GSE47753) (Buenrostro et al., 2013) were obtained from their corresponding studies. Paired-end ATAC-seq reads were quality trimmed using Trimmomatic version 0.33 (Bolger et al., 2014) and parameters “TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36.” Trimmed reads were aligned to human genome (hg19) using BWA version 0.7.12 (Li, 2013), specifically using the bwa mem –M option. Duplicate reads were removed using “MarkDuplicates” from Picard-tools version 1.95 (The Broad Institute, 2013). Quality of aligned reads were examined using Qualimap version 2.2.1 and default parameters “bamqc –bam –gd hg19.” (Okonechnikov et al., 2016). After preprocessing and quality filtering, peaks were called on alignments with MACS version 2.1.0 (Zhang et al., 2008) using the parameters “-g ‘hs’–nomodel–keep-dup all–broad–broad-cutoff 0.05 -f BAMPE.” Peaks located in blacklisted regions of the genome were removed. Remaining overlapping peaks from all cell types were merged with BEDTools version 2.26.0 (Quinlan and Hall, 2010) to generate a single peak set (n = 269,701). Raw read counts in these peaks for each cell type were determined using the R package DiffBind_2.4.8 (Ross-Innes et al., 2012). Spearman rank-order correlation was calculated for cell types using the merged peaks with deepTools version 2.4.2 (Ramírez et al., 2014) and parameters “multiBamSummary BED-file –BED.”
ChIP-seq
CTCF, H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K79me2, H3K4me3, H3K9me3 ChIP-seq was performed as previously described (Stitzel et al., 2010) and sequenced on an Illumina HiSeq 2000 using 2 × 100 bp cycles. Harmonized ChromHMM (Ernst and Kellis, 2017) states for EndoC-βH1 and NIH Roadmap cells/tissues were determined as previously described: “Chromatin states were determined by applying the ChromHMM (version 1.10) hidden Markov model algorithm at 200-bp resolution to five chromatin marks and input. We ran ChromHMM with a range of possible states and selected a 13-state model, because it most accurately captured information from higher-state models and provided sufficient resolution to identify biologically meaningful patterns in a reproducible way” (Varshney et al., 2017).
Transcription factor motif enrichment analysis
“findMotifsGenome.pl” (HOMER version 4.6 (Heinz et al., 2010)) script with parameters “hg19 -size 200” was used to determine TF motifs enriched in ATAC-seq OCRs for each cell type (Figure 2C). In each analysis, all merged OCRs (n = 269,701) were provided as background (e.g., EndoC-βH1 called OCRs (foreground) versus all merged OCRs (background)). The same parameters were used to identify enriched motifs in either “Enhancers in Both EndoC-βH1 and Islet” (n = 16,351) or “Enhancers in EndoC-βH1 Only” (n = 19,380) compared to all enhancers (n = 51,325) (Figure 2D). The same HOMER script and parameters were also used to identify enriched motifs in EndoC-βH1 (n = 315) versus GM12878 (n = 308) cell-specific Hi-C loops.
Similarity of cell/tissue type chromatin state (ChromHMM) annotations at EndoC-βH1 ATAC-seq OCRs
EndoC-βH1 ATAC-seq OCRs (n = 127,894) were overlapped with chromatin state (ChromHMM) annotations from EndoC-βH1, human islet, adipocyte, skeletal muscle, GM12878, and PBMC cells provided in (Varshney et al., 2017). Next, only OCRs that intersected a ChromHMM annotation from all tissue/cell types (n = 127,887/127,894) were retained (union set). Within a tissue/cell type, or instances where multiple chromatin state elements intersected an EndoC-βH1 OCR, annotations were prioritized as follows: promoter, enhancer, transcription, repressed, or low signal. At each OCR, cell/tissue ChromHMM annotations were compared to those of EndoC-βH1 and assigned a binary classification (1 = the annotations were the same, 0 = the annotations were different). Aggregated counts of pairwise chromatin state annotations based on EndoC-βH1 OCRs were then computed for each tissue/cell type, and a resulting similarity matrix was calculated using the “simil” function within the proxy version 0.4 R package (Meyer and Buchta, 2018).
RNA-seq
Total RNA was extracted and purified from EndoC-βH1 using Trizol as previously described (Varshney et al., 2017). All sequencing was performed on an Illumina NextSeq 500 with 2 × 101 bp cycles. Raw fastq files for human islets (Khetan et al., 2018), islet beta and alpha (Ackermann et al., 2016), PBMC (GSE90275), skeletal muscle (GSE78611), adipocyte (GSE93486) (ENCODE Project Consortium, 2012), GM12878 (GSE30400) (Rozowsky et al., 2011), and CD4+ T cell (GSE18927) (Schultz et al., 2015) were obtained from the associated databases. Paired-end RNA-seq reads were trimmed using Trimmomatic with the same parameters as used for ATAC-seq reads. Trimmed reads were aligned to human genome (hg19) using STAR version 2.53 (Dobin et al., 2013) with default parameters and expression levels of all genes were determined using QoRTs version 1.2.42 (Hartley and Mullikin, 2015) with default parameters and Gencode v19 transcript annotations. A total of 27,564 protein-coding genes and long intergenic non-coding RNAs (lincRNAs) were considered in the study.
miRNA-seq
Total RNA was extracted and purified from 2000–3000 islet equivalents (IEQ) or 2 × 106 EndoC-βH1 cells using Trizol (Life Technologies). RNA quality was confirmed with Bioanalyzer 2100 (Agilent); islet samples with RNA integrity number (RIN) greater than 6.5 were prepared for miRNA sequencing; EndoC- βH1 cells RNA RIN scores were > 9.0. miRNA libraries were prepared at the NIH Intramural Sequencing Core (NISC) from 1 μg total RNA using Illumina’s TruSeq Small RNA Library Kit according to the manufacturer’s guidelines, except a 10% acrylamide gel was used for better separation of library from adapters. Libraries were pooled in groups of about 8 for gel purification. Single-end 51 base sequencing was performed on Illumina HiSeq 2500 sequencers in Rapid Mode using version 2 chemistry. Data was processed using RTA version 1.18.64 and CASAVA 1.8.2. All resulting data was processed with miRquant 2.0 (Kanke et al., 2016).
Hi-C
Hi-C libraries were generated as described in (Rao et al., 2014) and analyzed using the Juicer Tools version 1.75 pipeline (Durand et al., 2016a). We sequenced 6,065,763,792 Hi-C read pairs in EndoC-βH1 cells, yielding 1,909,699,446 Hi-C contacts; we also sequenced 6,009,242,588 Hi-C read pairs in islet cells, yielding 1,516,995,339 Hi-C contacts. Loci were assigned to A and B compartments at 500 kB resolution. Loops were annotated using HiCCUPS at 5kB and 10kB resolutions with default Juicer parameters. This yielded a list of 9,100 loops in EndoC-βH1 cells and 2,580 loops in Islet cells. GM12878 loop calls (n = 9,448 loops) were downloaded from Gene Expression Omnibus (GSE63525). Differential loop calling with HiCCUPS at 5kb and 10kb identified 1,120 loops as significantly enriched for EndoC-βH1 cells and 829 loops as significantly enriched for GM12878 cells. Similar comparison of islet and EndoC-βH1 loops determined 935 loops as significantly enriched for EndoC-βH1 and 49 loops as being significantly enriched for islet. Aggregate peak analysis (APA) plots were calculated using Juicer and the “apa” command using default parameters. Visualization of Hi-C maps was performed using Juicebox version 1.6.11 (Durand et al., 2016b) with the “Observed/Expected” view and “Balanced” (Knight-Ruiz) normalization. All the code used in the above steps is publicly available at (https://github.com/theaidenlab). Genomic Regions Enrichment of Annotations Tool (GREAT; (McLean et al., 2010) was used to identify pathways enriched in the single nearest genes (whose TSS was within 2 kb) of EndoC-βH1-specific anchors.
ChIA-PET
EndoC-βH1 RNA Polymerase 2 (Pol2) ChIA-PET libraries were generated and sequenced reads were processed and analyzed according to the protocol in (Li et al., 2017b). ChIA-PET interactions were identified using ChIA-PET2 (Li et al., 2017a) using the “bridge linker mode” option. Corresponding ChIA-PET interactions for K562 (GSE39495) and GM12878 (GSE72816) cells were obtained from Gene Expression Omnibus. ChIA-PET and Hi-C loops were further filtered using the Bioconductor package InteractionSet_1.8.0 (Lun et al., 2016) to retain only those in which both interacting sites (anchors) overlapped OCRs. ChIA-PET anchors were annotated to the nearest gene. To assess the physical connectivity between genes and their putative regulatory regions as captured by ChIA-PET interactions, the number of distinct links between anchors annotated to each gene were counted and categorized by their chromatin state and regulatory function. Counting was carried out both between each gene promoter and all linked regulatory regions (Figure 4C), and also between all annotated anchors regardless of their chromatin state and all linked regulatory regions (Figure S4B). For example, consider anchors A1 and A2, both overlapping enhancer regions annotated to gene i. These anchors respectively link to anchors B1, located in a TSS, and B2, in an enhancer. In this scenario, a connectivity degree of two would be computed for gene i, corresponding to an enhancer-TSS and a TSS-TSS interaction, respectively.
The functional specificity of EndoC-βH1 ChIA-PET interactions was investigated by overlapping interaction anchors on ChromHMM chromatin states computed from EndoC-βH1 data as well as 27 other tissue/cell types (Varshney et al., 2017), and calculating the rate of conservation of chromatin states of both anchors. For example, a rate of 80% enhancer-enhancer conservation would mean that 8 out of 10 interactions of this type in EndoC-βH1 are also found in another cell type. The resulting proportions were computed for all interactions combined, and for specific relevant regulatory interactions (Figure 4D, line plots). In addition, aggregated counts of pairwise chromatin state interactions based on EndoC-βH1 ChIA-PET interactions were computed for the same cell types as above, and pairwise distances (D) between the resulting 29 count matrices were computed and plotted as scaled similarity values relative to EndoC-βH1 (i.e., 1-D/Dmax), so that D = 0 for EndoC-βH1 interactions and D = Dmax for the most divergent cell type (Figure 4D, bar plot). These same methods were used to determine the functional specificity of EndoC-βH1 Hi-C interactions.
ATAC-seq allelic bias analysis
All allelic bias analyses were performed using WASP (van de Geijn et al., 2015) (version 0.2.2 after GitHub commit 5a52185 and bug fix in pull request #67). For the EndoC-βH1 ATAC-seq allelic bias analyses, after original BWA mapping, reads were filtered to properly-paired, high-quality autosomal reads using SAMtools (v. 1.3.1; flags -f 3 -F 4 -F 8 -F 256 -F 2048 -q 30) (Li et al., 2009). Remapping and filtering as part of the WASP pipeline utilized the same parameters. As the last step of the WASP pipeline, duplicate removal was performed using WASP’s rmdup_pe.py script. In order to avoid double-counting SNPs covered by both reads in a pair, overlapping read pairs were clipped using bamUtil’s clipOverlap (https://genome.sph.umich.edu/wiki/BamUtil:_clipOverlap; v. 1.0.14). The four replicate libraries were then merged using SAMtools merge.
For each SNP, we determined the number of reads containing each allele (requiring base quality of at least 20). We excluded SNPs with total coverage less than 10, as well as SNPs in regions blacklisted by the ENCODE Consortium because of poor mappability (wgEncodeDacMapabilityConsensusExcludable.bed and wgEncodeDukeMapabilityRegionsExcludable.bed). Allelic bias testing was performed using a two-tailed binomial test, using an adjusted expectation for the null to account for residual reference bias as described in (Scott et al., 2016). Briefly, for each of the 16 reference-alternate allele pairs (e.g., AG and GA are separate allele pairs), we calculated the expected fraction of reference alleles (fracRef) under the null as the sum of the reference allele counts divided by the sum of the total allele counts for SNPs of that allele pair. To prevent SNPs of high coverage from biasing the expected fracRef, we down-sampled SNPs with coverage in the top 25th percentile to the median coverage, and used these downsampled reference and total allele counts when calculating the expected fracRef. We used the observed allele-pair specific fracRef as the true fracRef under the null hypothesis of no allelic bias in the binomial test. Multiple testing correction was performed using the Benjamini-Hochberg correction (FDR < 10%).
ChIP-seq allelic bias analysis
For the EndoC-βH1 ChIP-seq allelic bias analyses, paired-end libraries were processed as follows. Adapters were trimmed using cta(v. 0.1.2) and reads mapped using BWA mem (-M flag; v. 0.7.12). Reads were filtered to properly-paired, high-quality autosomal reads using SAMtools (flags -f 3 -F 4 -F 8 -F 256 -F 2048 -q 30). Single-end libraries were mapped using BWA aln (v. 0.7.12; default parameters) and filtered using SAMtools (flags -F 4 -F 256 -F 2048 -q 30). Remapping and filtering as part of the WASP pipeline utilized the same parameters as for the original mapping. For paired-end libraries, overlapping read pairs were clipped using bamUtil’s clipOverlap. Replicates were merged using SAMtools merge. Allele counting and allelic bias testing was performed as described above for ATAC-seq.
RNA-seq allelic bias analysis
For the EndoC-βH1 RNA-seq allelic bias analyses, after original STAR mapping, reads were filtered to properly-paired, high-quality autosomal reads using SAMtools (v. 1.3.1; flags -f 3 -F 4 -F 8 -F 256 -F 2048 -q 255). Remapping and filtering as part of the WASP pipeline utilized the same parameters. In order to avoid double-counting SNPs covered by both reads in a pair, overlapping read pairs were clipped using bamUtil’s clipOverlap. Allele counting and allelic bias testing was performed as described above for ATAC-seq.
Comparison of islet eQTL and EndoC-βH1 biased SNPs allelic effect
Human islet eQTL data were obtained from (Varshney et al., 2017). For EndoC-βH1 biased (H3K27ac) enhancer SNPs that were also linked (via ChIA-PET chromatin interaction) to a target gene (n = 91/185 SNPs in Figure 5C), all corresponding islet eQTL SNP-gene pairs were retrieved. For 42/91 SNPs linked to target genes, the H3K27ac allelic effect bias was calculated assuming that the EndoC-βH1 effect allele was the same as the islet eQTL effect allele. Allelic effect bias was calculated by dividing the effect allele coverage (either reference or alternate allele) by the total coverage of the SNP. A scalar value of 0.5 was subtracted from this value to determine whether the effect allele had an increased (positive value), decreased (negative value), or no (zero) bias in H3K27ac coverage. Randomly selected eQTL SNP-gene pairs that did not have corresponding connections (via ChIA-PET chromatin interaction) to a target gene were considered as a null/background set.
QUANTIFICATION AND STATISTICAL ANALYSIS
Visualization of all Hi-C, ChIA-PET, chromatin state (ChromHMM), ATAC-seq, and RNA-seq data examined in this study (Figures 1, 2, 3, 4, and 5) were produced with R using ggplot2_3.0.0 (Wickham, 2016), pheatmap_1.0.10 (Kolde, 2018), Sushi_1.18.0 (Phanstiel et al., 2014), as well as Juicer Tools (Figure 3) (Durand et al., 2016a) and Juicebox (Figure 3) (Durand et al., 2016b).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| H3K27ac-human antibody | Abeam | Abeam Cat# ab4729; RRID:AB_2118291 |
| H3K4me3-mouse antibody | Abeam | Abeam Cat# ab12209; RRID:AB_442957 |
| ENCODE Project Antibody validation H3K4me1 | Abeam | Abeam Cat# ab8895; RRID:AB_306847 |
| ENCODE Project Antibody validation H3K36me3 | Abeam | Abeam Cat# ab9050; RRID:AB_306966 |
| H3K27me3-celegans antibody | Abeam | Abeam Cat# ab6002; RRID:AB_305237 |
| H3K79me2-human antibody | Abeam | Abeam Cat# ab3594; RRID:AB_303937 |
| H3K9me3-human,H3K9me3-mouse antibody | Abeam | Abeam Cat# ab8898; RRID:AB_306848 |
| Rabbit Anti-CTCF Polyclonal Antibody, Unconjugated | Abeam | Abeam Cat# ab70303; RRID:AB_1209546 |
| RNA Polymerase II 8WG16 Monoclonal Antibody, anti-RNA Polymerase II | Covance Research Products | Covance Research Products Cat# MMS-126R; RRID:AB_10013665 |
| Biological Samples | ||
| Human pancreatic islet | NDRI | UNOS ID: ADAC418 |
| Critical Commercial Assays | ||
| SKY Probe | Applied Spectral Imaging INC | Cat# FPRPR0028 |
| HumanOmni2.5–4v1_H BeadChip Array | Illumina | Cat# WG-311–2514 |
| TruSeq ChIP Sample Prep Kit | Illumina | Cat# IP-202–1024 |
| TruSeq Stranded mRNA Kit | Illumina | Cat# 20020595 |
| TruSeq Small RNA Library Kit | Illumina | Cat# RS-200–0036 |
| Deposited Data | ||
| Raw and analyzed data | This paper | NCBI BioProject: PRJNA480287; European Variation Archive: ERZ674947; NCBI GEO:GSE118588 |
| Islet ATAC-seq and RNA-seq | Khetan et al., 2018 | NCBI SRA:SRP117935 |
| Purified beta and alpha cell ATAC-seq and RNA-seq | Ackermann et al., 2016 | NCBI GEO: GSE76268 |
| Peripheral mononuclear blood cells (PBMC) ATAC-seq | Ucar et al., 2017 | European Genome-phenome Archive: EGAS00001002605 |
| PBMC RNA-seq | ENCODE Project Consortium, 2012 | NCBI GEO: GSE90275 |
| Skeletal muscle ATAC-seq | Scott et al., 2016 | dbGap: phs001068.v1.p1 |
| Skeletal muscle RNA-seq | ENCODE Project Consortium, 2012 | NCBI GEO: GSE78611 |
| GM12878 and CD4+ T ATAC-seq | Buenrostro et al., 2013 | NCBI GEO: GSE47753 |
| GM12878 RNA-seq | Rozowsky et al., 2011 | NCBI GEO: GSE30400 |
| CD4+ T RNA-seq | Schultz et al., 2015 | NCBI GEO: GSE18927 |
| Adipocyte RNA-seq | ENCODE Project Consortium, 2012 | NCBI GEO: GSE93486 |
| GM12878Hi-C | Rao et al., 2014 | NCBI GEO: GSE63525 |
| K562 RNA Pol2 ChlA-PET | Li et al., 2012 | NCBI GEO: GSE39495 |
| GM12878 RNA Pol2 ChlA-PET | Tang et al., 2015 | NCBI GEO: GSE72816 |
| Experimental Models: Cell Lines | ||
| EndoC-betaH1: Cell line | EndoCells/INSERM | RRID: CVCL_L909 |
| Software and Algorithms | ||
| Michigan Imputation Server | Das et al., 2016 | https://imputationserver.sph.umich.edu/index.html |
| Eagle v2.3 | Loh et al., 2016 | https://data.broadinstitute.org/alkesgroup/Eagle/ |
| SHAPEIT v2.r790 | Delaneau et al., 2011 | http://mathgen.stats.ox.ac.uk/genetics_software/shapeit/shapeit.html |
| minimac3 | Das et al., 2016 | https://genome.sph.umich.edu/wiki/Minimac3 |
| PLINK version 1.9 | Purcell et al., 2007 | http://zzz.bwh.harvard.edu/plink/ |
| Trimmomatic version 0.33 | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| BWA version 0.7.12 | Li, 2013 | http://bio-bwa.sourceforge.net |
| Samtools version 1.3.1 | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Picard-tools version 1.95 | The Broad Institute, 2013 | https://broadinstitute.github.io/picard/ |
| Qualimap version 2.2.1 | Okonechnikov et al., 2016 | http://qualimap.bioinfo.cipf.es |
| MACS version 2.1.0 | Zhang et al., 2008 | http://liulab.dfci.harvard.edu/MACS/ |
| BEDTools version 2.26.0 | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| deepTools version 2.4.2 | Ramírez et al., 2014 | https://deeptools.readthedocs.io/en/develop/ |
| ChromHMM version 1.17 | Ernst and Kellis, 2017 | http://compbio.mit.edu/ChromHMM/ |
| HOMER version 4.6 | Heinz et al., 2010 | http://homer.ucsd.edu/homer/index.html |
| STAR version 2.53 | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| QoRTs version 1.2.42 | Hartley and Mullikin, 2015 | https://hartleys.github.io/QoRTs/ |
| miRquant 2.0 | Kanke et al., 2016 | https://github.com/Sethupathy-Lab/miRquant |
| Juicer Tools version 1.75 | Durand et al., 2016a | https://github.com/aidenlab/juicer/wiki/Juicer-Tools-Quick-Start |
| Juicebox version 1.6.11 | Durand et al., 2016b | https://www.aidenlab.org/juicebox/ |
| GREAT version 3.0 | McLean et al., 2010 | http://great.stanford.edu/public/html/ |
| ChlA-PET2 version 0.9.2 | Li et al., 2017a | https://github.com/GuipengLi/ChlA-PET2 |
| WASP version 0.2.2 | van deGeijn et al., 2015 | https://github.com/bmvdgeijn/WASP |
| Other | ||
| Haplotype Reference Consortium (HRC version, hrc.r1.1.2016) panel | McCarthy et al., 2016 | http://www.haplotype-reference-consortium.org/ |
| NHGRI-EBI Catalog of SNPs | N/A | https://www.ebi.ac.uk/gwas/ |
| R Shiny application for browsing human islet and EndoC-βH1 genomic data | This paper | http://shinyapps.jax.org/endoc-islet-multi-omics |
Highlights.
Comprehensive multiomic maps of EndoC-βH1 human b cell line and primary islets
Sequence motifs enriched in EndoC-specific enhancers reflect its precursor state
Identification of regulatory hubs preserved between EndoC-βH1 and human islets
Identified SNP alleles (including T2D GWAS) altering cis-regulatory signatures
ACKNOWLEDGMENTS
We thank Jane Cha for aid in graphic design. We thank all members of the Stitzel, Ucar, Parker, Sethupathy, Collins, Ruan, and Aiden labs for helpful discussion and feedback on this study and manuscript. This study was funded by NIH grants R00DK092251 (M.L.S.) and R00DK099240 (S.C.J.P.), NIH intramural support from project ZIA-HG000024 (F.S.C.), and the American Diabetes Association Pathway to Stop Diabetes Initiator (grant 1–14-INI-07 to S.C.J.P.) and Accelerator (grant 1–16-ACE-47 to P.S. and grant 1–18-ACE-15 to M.L.S.) awards. This study was also supported by the NIH (training grant T32 HG00040 to P.O. and grants DP2OD008540, U01HL130010, and UM1HG009375), the NSF (grant PHY-1427654), the USDA (grant 2017–05741), the Welch Foundation (grant Q-1866), NVIDIA, IBM, Google, the Cancer Prevention Research Institute of Texas (grant R1304), and the McNair Medical Institute (E.L.A.). Opinions, interpretations, conclusions, and recommendations are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or American Diabetes Association.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession numbers for EndoC-βH1 Hi-C, ChIA-PET, ChIP-seq, ATAC-seq, RNA-seq, miRNA-seq as well as human islet Hi-C data reported in this paper are: NCBI BioProject accession PRJNA480287, NCBI Gene Expression Omnibus accession GSE118588. The accession number for EndoC-βH1 genotype data is: European Variation Archive accession ERZ674947 and project PRJEB27824. The R shiny web application to visualize and interact with the multi-omic data generated in this study is freely available at https://shinyapps.jax.org/endoc-islet-multi-omics/. Interactive Hi-C maps are available at aidenlab.org/juicebox.
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and five tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.083.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ackermann AM, Wang Z, Schug J, Naji A, and Kaestner KH (2016). Integration of ATAC-seq and RNA-seq identifies human a cell and b cell signature genes. Mol. Metab 5, 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson LE, Valtat B, Bagge A, Sharoyko VV, Nicholls DG, Ravassard P, Scharfmann R, Spégel P, and Mulder H (2015). Characterization of stimulus-secretion coupling in the human pancreatic EndoC-βH1 β cell line. PLoS ONE 10, e0120879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belton J-M, McCord RP, Gibcus JH, Naumova N, Zhan Y, and Dekker J (2012). Hi-C: a comprehensive technique to capture the conformation of genomes. Methods 58, 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blodgett DM, Nowosielska A, Afik S, Pechhold S, Cura AJ, Kennedy NJ, Kim S, Kucukural A, Davis RJ, Kent SC, et al. (2015). Novel observations from next-generation RNA sequencing of highly purified human adult and fetal islet cell subsets. Diabetes 64, 3172–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom AM (2017). The role of complement inhibitors beyond controlling inflammation. J. Intern. Med 282, 116–128. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardinale V, Wang Y, Carpino G, Cui C-B, Gatto M, Rossi M, Berloco PB, Cantafora A, Wauthier E, Furth ME, et al. (2011). Multipotent stem/progenitor cells in human biliary tree give rise to hepatocytes, cholangiocytes, and pancreatic islets. Hepatology 54, 2159–2172. [DOI] [PubMed] [Google Scholar]
- Chang W, Cheng J, Allaire JJ, Xie Y, McPherson J (2018). shiny: Web Application Framework for R. R package version 1.2.0 https://CRAN.R-project.org/package=shiny.
- Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, et al. (2016). Next-generation genotype imputation service and methods. Nat. Genet 48, 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O, Marchini J, and Zagury J-F (2011). A linear complexity phasing method for thousands of genomes. Nat. Methods 9, 179–181. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Shamim MS, Machol I, Rao SSP, Huntley MH, Lander ES, and Aiden EL (2016a). Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst 3, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, and Aiden EL (2016b). Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst 3, 99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliasson L (2017). The small RNA mir-375 - a pancreatic islet abundant miRNA with multiple roles in endocrine b cell function. Mol. Cell. Endocrinol 456, 95–101. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, and Kellis M (2017). Chromatin-state discovery and genome annotation with ChromHMM. Nat. Protoc 12, 2478–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadista J, Vikman P, Laakso EO, Mollet IG, Esguerra JL, Taneera J, Storm P, Osmark P, Ladenvall C, Prasad RB, et al. (2014). Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc. Natl. Acad. Sci. USA 111, 13924–13929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, Matsunaga K, Wang H, Ishizaki R, Kobayashi E, Kiyonari H, Mukumoto Y, Okunishi K, and Izumi T (2017). Exophilin-8 assembles secretory granules for exocytosis in the actin cortex via interaction with RIM-BP2 and myosin-VIIa. eLife 6, e26174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay VJ, LaRue AC, Turner DP, Watson PM, and Watson DK (2013). Understanding the role of ETS-mediated gene regulation in complex biological processes. Adv. Cancer Res 119, 1–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ, Ma C, Fontanillas P, Moutsianas L, McCarthy DJ, et al. (2016). The genetic architecture of type 2 diabetes. Nature 536, 41–47, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao N, LeLay J, Vatamaniuk MZ, Rieck S, Friedman JR, and Kaestner KH (2008). Dynamic regulation of PDX1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev 22, 3435–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrish K, Van Velkinburgh JC, and Stein R (2004). Conserved transcriptional regulatory domains of the pdx-1 gene. Mol. Endocrinol 18, 533–548. [DOI] [PubMed] [Google Scholar]
- Hartley SW, and Mullikin JC (2015). QoRTs: a comprehensive toolset for quality control and data processing of RNA-Seq experiments. BMC Bioinformatics 16, 224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan WM, Merin DA, Fonte V, and Link CD (2009). AIP-1 ameliorates b-amyloid peptide toxicity in a Caenorhabditis elegans Alzheimer’s disease model. Hum. Mol. Genet 18, 2739–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Romanoski CE, Benner C, and Glass CK (2015). The selection and function of cell type-specific enhancers. Nat. Rev. Mol. Cell Biol 16, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herglotz J, Unrau L, Hauschildt F, Fischer M, Kriebitzsch N, Alawi M, Indenbirken D, Spohn M, Müller U, Ziegler M, et al. (2016). Essential control of early B-cell development by Mef2 transcription factors. Blood 127, 572–581. [DOI] [PubMed] [Google Scholar]
- Kanke M, Baran-Gale J, Villanueva J, and Sethupathy P (2016). miRquant 2.0: an Expanded Tool for Accurate Annotation and Quantification of MicroRNAs and their isomiRs from Small RNA-Sequencing Data. J. Integr. Bioinform 13, 307. [DOI] [PubMed] [Google Scholar]
- Khetan S, Kursawe R, Youn A, Lawlor N, Jillette A, Marquez EJ, Ucar D, and Stitzel ML (2018). Type 2 diabetes-associated genetic variants regulate chromatin accessibility in human islets. Diabetes 67, 2466–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde R (2018). pheatmap: Pretty Heatmaps. R package version 1.0.10 https://CRAN.R-project.org/package=pheatmap.
- Krizhanovskii C, Kristinsson H, Elksnis A, Wang X, Gavali H, Bergsten P, Scharfmann R, and Welsh N (2017). EndoC-βH1 cells display increased sensitivity to sodium palmitate when cultured in DMEM/F12 medium. Islets 9, e1296995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krus U, King BC, Nagaraj V, Gandasi NR, Sjölander J, Buda P, Garcia-Vaz E, Gomez MF, Ottosson-Laakso E, Storm P, et al. (2014). The complement inhibitor CD59 regulates insulin secretion by modulating exocytotic events. Cell Metab 19, 883–890. [DOI] [PubMed] [Google Scholar]
- Lawlor N, George J, Bolisetty M, Kursawe R, Sun L,S. V, Kycia I, Robson P, and Stitzel ML (2017a). Single cell transcriptomes identify human islet cell signatures and reveal cell-type-specific expression changes in type 2 diabetes. Genome Res 27, 208–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor N, Khetan S, Ucar D, and Stitzel ML (2017b). Genomics of islet (dys)function and type 2 diabetes. Trends Genet 33, 244–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bin GC, Muñoz-Descalzo S, Kurowski A, Leitch H, Lou X, Mansfield W, Etienne-Dumeau C, Grabole N, Mulas C, Niwa H, et al. (2014). Oct4 is required for lineage priming in the developing inner cell mass of the mouse blastocyst. Development 141, 1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv, doi: 1303.3997v2. [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, et al. (2012). Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Cai L, Chang H, Hong P, Zhou Q, Kulakova EV, Kolchanov NA, and Ruan Y (2014). Chromatin interaction analysis with paired-end tag (ChIA-PET) sequencing technology and application. BMC Genomics 15, S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Chen Y, Snyder MP, and Zhang MQ (2017a). ChIA-PET2: a versatile and flexible pipeline for ChIA-PET data analysis. Nucleic Acids Res 45, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Luo OJ, Wang P, Zheng M, Wang D, Piecuch E, Zhu JJ, Tian SZ, Tang Z, Li G, and Ruan Y (2017b). Long-read ChIA-PET for base-pair-resolution mapping of haplotype-specific chromatin interactions. Nat. Protoc 12, 899–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh P-R, Palamara PF, and Price AL (2016). Fast and accurate long-range phasing in a UK Biobank cohort. Nat. Genet 48, 811–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun ATL, Perry M, and Ing-Simmons E (2016). Infrastructure for genomic interactions: Bioconductor classes for Hi-C, ChIA-PET and related experiments. F1000Res 5, 950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, Junkins H, McMahon A, Milano A, Morales J, et al. (2017). The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res 45 (D1), D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, Payne AJ, Steinthorsdottir V, Scott RA, Grarup N, et al. (2018). Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat. Genet 50, 1505–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, et al. ; Haplotype Reference Consortium (2016). A reference panel of 64,976 haplotypes for genotype imputation. Nat. Genet 48, 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, and Buchta C (2018). proxy: Distance and similarity measures. R package version 0.4–22 https://CRAN.R-project.org/package=proxy.
- Mitchell RK, Hu M, Chabosseau PL, Cane MC, Meur G, Bellomo EA, Carzaniga R, Collinson LM, Li W-H, Hodson DJ, and Rutter GA (2016). Molecular genetic regulation of Slc30a8/ZnT8 reveals a positive association with glucose tolerance. Mol. Endocrinol 30, 77–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohlke KL, and Boehnke M (2015). Recent advances in understanding the genetic architecture of type 2 diabetes. Hum. Mol. Genet 24 (R1), R85–R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ndiaye FK, Ortalli A, Canouil M, Huyvaert M, Salazar-Cardozo C, Lecoeur C, Verbanck M, Pawlowski V, Boutry R, Durand E, et al. (2017). Expression and functional assessment of candidate type 2 diabetes susceptibility genes identify four new genes contributing to human insulin secretion. Mol. Metab 6, 459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura W, Kondo T, Salameh T, El Khattabi I, Dodge R, Bonner-Weir S, and Sharma A (2006). A switch from MafB to MafA expression accompanies differentiation to pancreatic b-cells. Dev. Biol 293, 526–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonechnikov K, Conesa A, and García-Alcalde F (2016). Qualimap 2: advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleson BJ, McGraw JA, Broniowska KA, Annamalai M, Chen J, Bush-kofsky JR, Davis DB, Corbett JA, and Mathews CE (2015). Distinct differences in the responses of the human pancreatic β-cell line EndoC-βH1 and human islets to proinflammatory cytokines. Am. J. Physiol. Regul. Integr. Comp. Physiol 309, R525–R534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipovich AB, Long Q, Manduchi E, Gangula R, Hipkens SB, Schneider J, Okubo T, Stoeckert CJ Jr., Takada S, and Magnuson MA (2014). Insm1 promotes endocrine cell differentiation by modulating the expression of a network of genes that includes Neurog3 and Ripply3. Development 141, 2939–2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SCJ, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, Black BL, et al. ; NISC Comparative Sequencing Program; National Institutes of Health Intramural Sequencing Center Comparative Sequencing Program Authors; NISC Comparative Sequencing Program Authors (2013). Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc. Natl. Acad. Sci. USA 110, 17921–17926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquali L, Gaulton KJ, Rodríguez-Seguí SA, Mularoni L, Miguel-Escalada I, Akerman İ, Tena JJ, Morán I, Gómez-Marín C, van de Bunt M, et al. (2014). Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat. Genet 46, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng SL, Gerth AJ, Ranger AM, and Glimcher LH (2001). NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity 14, 13–20. [DOI] [PubMed] [Google Scholar]
- Phanstiel DH, Boyle AP, Araya CL, and Snyder MP (2014). Sushi.R: flexible, quantitative and integrative genomic visualizations for publication-quality multi-panel figures. Bioinformatics 30, 2808–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, and Sham PC (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). deep-Tools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res 42, W187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raum JC, Soleimanpour SA, Groff DN, Coré N, Fasano L, Garratt AN, Dai C, Powers AC, and Stoffers DA (2015). Tshz1 regulates pancreatic b-cell maturation. Diabetes 64, 2905–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravassard P, Hazhouz Y, Pechberty S, Bricout-Neveu E, Armanet M, Czernichow P, and Scharfmann R (2011). A genetically engineered human pancreatic β cell line exhibiting glucose-inducible insulin secretion. J. Clin. Invest 121, 3589–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium; Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, et al. (2012). Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozowsky J, Abyzov A, Wang J, Alves P, Raha D, Harmanci A, Leng J, Bjornson R, Kong Y, Kitabayashi N, et al. (2011). AlleleSeq: analysis of allele-specific expression and binding in a network framework. Mol. Syst. Biol 7, 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt EM, Zhang J, Zhou W, Chen J, Mohlke KL, Chen YE, and Willer CJ (2015). GREGOR: evaluating global enrichment of trait-associated variants in epigenomic features using a systematic, data-driven approach. Bioinformatics 31, 2601–2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz MD, He Y, Whitaker JW, Hariharan M, Mukamel EA, Leung D, Rajagopal N, Nery JR, Urich MA, Chen H, et al. (2015). Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 523, 212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott LJ, Erdos MR, Huyghe JR, Welch RP, Beck AT, Wolford BN, Chines PS, Didion JP, Narisu N, Stringham HM, et al. (2016). The genetic regulatory signature of type 2 diabetes in human skeletal muscle. Nat. Commun 7, 11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segerstolpe Å, Palasantza A, Eliasson P, Andersson E-M, Andréasson A-C, Sun X, Picelli S, Sabirsh A, Clausen M, Bjursell MK, et al. (2016). Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab 24, 593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolik C, Liu Y, Bauer D, McPherson J, Broeker M, Heimberg G, Qi LS, Sivak DA, and Thomson M (2015). Transcription factor competition allows embryonic stem cells to distinguish authentic signals from noise. Cell Syst 1, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stitzel ML, Sethupathy P, Pearson DS, Chines PS, Song L, Erdos MR, Welch R, Parker SCJ, Boyle AP, Scott LJ, et al. ; NISC Comparative Sequencing Program (2010). Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab 12, 443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Luo OJ, Li X, Zheng M, Zhu JJ, Szalaj P, Trzaskoma P, Magalska A, Wlodarczyk J, Ruszczycki B, et al. (2015). CTCF-mediated human 3D genome architecture reveals chromatin topology for transcription. Cell 163, 1611–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tantin D (2013). Oct transcription factors in development and stem cells: insights and mechanisms. Development 140, 2857–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teraoku H, and Lenzen S (2017). Dynamics of insulin secretion from EndoC-βH1 β-cell pseudoislets in response to glucose and other nutrient and nonnutrient secretagogues. J. Diabetes Res 2017, 2309630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Broad Institute (2013). Picard-Tools: A set of command line tools (in Java) for manipulating high-throughput sequencing (HTS) data and formats such as SAM/BAM/CRAM and VCF http://broadinstitute.github.io/picard/.
- Thompson P, and Bhushan A (2017). β Cells led astray by transcription factors and the company they keep. J. Clin. Invest 127, 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsonkova VG, Sand FW, Wolf XA, Grunnet LG, Kirstine Ringgaard A, Ingvorsen C, Winkel L, Kalisz M, Dalgaard K, Bruun C, et al. (2018). The EndoC-βH1 cell line is a valid model of human β cells and applicable for screenings to identify novel drug target candidates. Mol. Metab 8, 144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ucar D, Márquez EJ, Chung C-H, Marches R, Rossi RJ, Uyar A, Wu T-C, George J, Stitzel ML, Palucka AK, et al. (2017). The chromatin accessibility signature of human immune aging stems from CD8+ T cells. J. Exp. Med 214, 3123–3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Univercell Biosolutions (2011). EndoC-BH1: a functional immortalized human β cell http://www.univercell-biosolutions.com/human-heart-cells-and-stem-cells-production.
- van de Bunt M, Gaulton KJ, Parts L, Moran I, Johnson PR, Lindgren CM, Ferrer J, Gloyn AL, and McCarthy MI (2013). The miRNA profile of human pancreatic islets and β-cells and relationship to type 2 diabetes pathogenesis. PLoS ONE 8, e55272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Bunt M, Manning Fox JE, Dai X, Barrett A, Grey C, Li L, Bennett AJ, Johnson PR, Rajotte RV, Gaulton KJ, et al. (2015). Transcript expression data from human islets links regulatory signals from genome-wide association studies for type 2 diabetes and glycemic traits to their downstream effectors. PLoS Genet 11, e1005694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Geijn B, McVicker G, Gilad Y, and Pritchard JK (2015). WASP: allele-specific software for robust molecular quantitative trait locus discovery. Nat. Methods 12, 1061–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney A, Scott LJ, Welch RP, Erdos MR, Chines PS, Narisu N, Albanus RD, Orchard P, Wolford BN, Kursawe R, et al. ; NISC Comparative Sequencing Program (2017). Genetic regulatory signatures underlying islet gene expression and type 2 diabetes. Proc. Natl. Acad. Sci. USA 114, 2301–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vietri Rudan M, Barrington C, Henderson S, Ernst C, Odom DT, Tanay A, and Hadjur S (2015). Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep 10, 1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Oh E, and Thurmond DC (2007). Glucose-stimulated Cdc42 signaling is essential for the second phase of insulin secretion. J. Biol. Chem 282, 9536–9546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YJ, Schug J, Won K-J, Liu C, Naji A, Avrahami D, Golson ML, and Kaestner KH (2016). Single-cell transcriptomics of the human endocrine pancreas. Diabetes 65, 3028–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Pronin AN, Levay K, Almaca J, Fornoni A, Caicedo A, and Slepak VZ (2017). Regulator of G-protein signaling Gβ5-R7 is a crucial activator of muscarinic M3 receptor-stimulated insulin secretion. FASEB J 31, 4734–4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2: Elegant graphics for data analysis Springer-Verlag, New York: R package version 3.1.0. https://ggplot2.tidyverse.org. [Google Scholar]
- Wu G, and Schöler HR (2014). Role of Oct4 in the early embryo development. Cell Regen. (Lond.) 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Y, Kim J, Okamoto H, Ni M, Wei Y, Adler C, Murphy AJ, Yancopoulos GD, Lin C, and Gromada J (2016). RNA sequencing of single human islet cells reveals type 2 diabetes genes. Cell Metab 24, 608–615. [DOI] [PubMed] [Google Scholar]
- Yagai T, Matsui S, Harada K, Inagaki FF, Saijou E, Miura Y, Nakanuma Y, Miyajima A, and Tanaka M (2017). Expression and localization of sterile a motif domain containing 5 is associated with cell type and malignancy of biliary tree. PLoS ONE 12, e0175355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ables ET, Pope CF, Washington MK, Hipkens S, Means AL, Path G, Seufert J, Costa RH, Leiter AB, et al. (2009). Multiple, temporal-specific roles for HNF6 in pancreatic endocrine and ductal differentiation. Mech. Dev 126, 958–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.