

 BTK is an effective target for the treatment of B-cell malignant tumors and autoimmune diseases.
BTK is an effective target for the treatment of B-cell malignant tumors and autoimmune diseases.
Abstract
BTK is an effective target for the treatment of B-cell malignant tumors and autoimmune diseases. In this work, a series of 2-phenyl pyrimidine derivatives were prepared and their preliminary in vitro activities on B-cell leukemia cells as well as the BTK enzyme were determined. The results showed that compound 11g displayed the best inhibitory activity on BTK with an inhibition rate of 82.76% at 100 nM and excellent anti-proliferation activity on three B-cell leukemia lines (IC50 = 3.66 μM, 6.98 μM, and 5.39 μM against HL60, Raji and Ramos, respectively). Besides, the flow cytometry analysis results indicated that 11g inhibited the proliferation of the Raji cells in a dose- and time-dependent manner, and blocked the Ramos cells at the G0/G1 phase, which is in accordance with the positive control ibrutinib. The mechanism investigation demonstrated that 11g could inhibit the phosphorylation of BTK and its downstream substrate phospholipase γ2 (PLCγ2). All these results showed that 11g was a promising lead compound that merited further optimization as a novel class of BTK inhibitor for the treatment of B-cell lymphoblastic leukemia.
Introduction
Bruton's tyrosine kinase (BTK) is a member of the TEC family of non-receptor tyrosine kinase which is predominantly expressed in all hematopoietic cells except plasma and T cells.1 It is a crucial kinase in the B-cell receptor (BCR) signaling pathway.2,3 The overexpression of BTK usually causes severe leukemia and B-cell-related lymphoma. Therefore, the development of drug molecules targeting BTK has become an attractive therapeutic approach.
Since BTK was discovered in the 1990s,4 many small-molecule BTK inhibitors have been developed. Based on their binding mode with BTK, researchers found that the non-catalytic cysteine (Cys481) residue in the extended hinge region is the key binding site for the irreversible BTK inhibitors such as ibrutinib (approved), acalabrutinib (approved), ONO-4059 (phase II), spebrutinib (phase II) and HM71224 (phase II) (Fig. 1). They generally contain a pyrimidine core with an acrylamide group which can bind to the Cys481 residue covalently as a Michael acceptor and irreversibly inhibit BTK.5,6 Ibrutinib, a highly potent oral small-molecule inhibitor, is a first-generation BTK inhibitor. It was approved by the FDA for the treatment of mantle cell lymphoma (MCL) in 2013, and was further used as a monotherapy to treat chronic lymphocytic leukaemia (CLL) in 2014.7 In 2015 and 2017, ibrutinib was approved for the treatment of Waldenstrom's macroglobulinaemia (WM) and chronic graft-versus-host disease (cGVHD), respectively.8 Despite the great success achieved by ibrutinib, its off-target effect also leads to the clinically observed adverse effects including arthralgias, rash, atrial fibrillation, diarrhea, ecchymosis and massive hemorrhage due to ibrutinib's irreversible binding with other kinases such as epidermal growth factor receptor (EGFR), ITK, and TEC.9 Moreover, most of the BTK inhibitors mentioned above have been reported to have limited efficacy and strong side effects. Hence, there is still an increasing need to discover novel BTK inhibitors which could be developed into therapeutic candidates for the treatment of B-cell malignancies.10
Fig. 1. Chemical structures of the reported BTK inhibitors.

Previous structure and activity relationship (SAR) explorations showed that a pyrimidine core and a typical N-phenylacrylamide pharmacophore are important for novel inhibitors to bind with BTK. CGI-560 (Fig. 1) was obtained by screening from compound libraries and exhibited modest BTK potency (half-maximal inhibitory concentration (IC50) = 400 nM) but promising selectivity (>ten-fold) over an initial panel of 16 kinases.11 Based on this, we hope to splice some pharmacophores on this skeleton to improve the selectivity as well as inhibition for BTK, and then obtain the novel BTK inhibitor. First, we introduced the key acrylamide group and acyl side chain of the reported BTK ligand PLS-123 to substitute the side chains at the two ends of CGI-560. Moreover, Dan Zhao et al. reported the structural optimizations of diphenylpyrimidine derivatives based on spebrutinib, which showed that the chloro substituent at the C-5 position of the pyrimidine core is more favorable than the fluoro atom due to stronger hydrophobicity in the binding site.12 So we introduced the chloro atom at the C-6 position after the skeleton transition. Finally, we design a series of 2-phenyl pyrimidine derivatives as novel potent BTK inhibitors which retain the pyrimidine core and typical N-phenylacrylamide functionality (Fig. 2).
Fig. 2. Rational design strategy of our compounds.

The syntheses of novel 2-phenyl pyrimidine 11a–11i are summarized in Scheme 1. 4-Nitrobenzaldehyde as the starting material was condensed with hydroxylamine hydrochloride under reflux to afford benzonitrile 2 which was transformed to compound 3 by reacting with ammonium chloride. The pyrimidine nucleus 4 was prepared through the condensation reaction of compound 3 and diethyl malonate, and then it was reacted with POCl3 to produce the key intermediate 5 which was reduced by Fe to yield compound 6. Substitution of compound 6 with acryloyl chloride generated compound 7. On the other hand, 4-nitrobenzoyl chloride was reacted with different amines or phenylamine to provide compounds 9a–9i, following a reduction reaction by Fe to get the different side chains (10a–10i). Finally, the Pd-catalyzed Buchwald–Hartwig amination of 7 with different side chains (10a–10i) afford target compounds 11a–11i.
Scheme 1. Reagents and conditions: (a) hydroxylamine hydrochloride, DMSO, 100 °C, 0.5 h, 82.0%; (b) methanol, sodium methoxide, rt, 5 h; ammonium chloride, rt, 4 h, 97.0%; (c) ethanol, diethyl malonate, sodium methoxide, N2, reflux, 4 h, 87.4%; (d) POCl3, DMF, 100 °C, 8 h, 46.3%; (e) methanol : water (1 : 1), Fe, NH4Cl, 70 °C, 5 h, 60.7%; (f) DMF, acryloyl chloride, triethylamine, 0 °C, 1 h, 56.5%; (g) THF, triethylamine, different amines or phenylamine, 0 °C, 0.5 h, 91.5%; (h) methanol : water (1 : 1), Fe, NH4Cl, 70 °C, 0.5 h, 69.2%; (i) anhydrous dioxane, Pd(PPh3)2Cl2, K2CO3, 100 °C, 8 h, 20.5–34.0%.

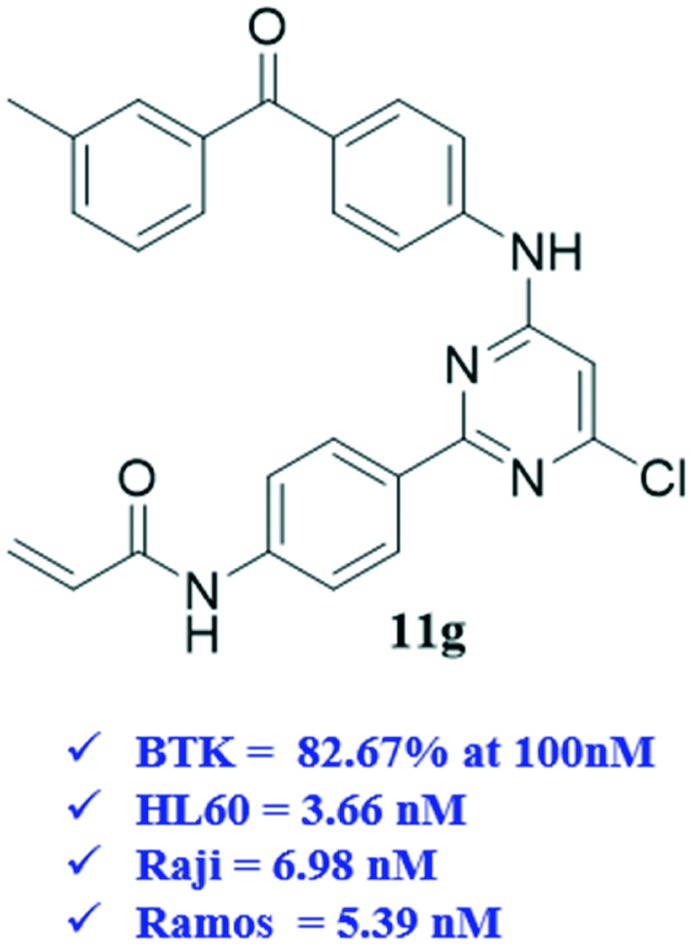
The activity against BTK of the synthesized molecules was evaluated using an ADP-Glo™ kinase assay system.13–15 Three B-cell leukemia lines (Raji, HL60 and Ramos) which over-expressed the BTK enzyme were used for the cell proliferation evaluations through the MTT method. For comparison, ibrutinib was tested as the positive control. As shown in Table 1, most of our designed compounds showed good inhibition (>50%) against BTK. In particular, 11e, 11g and 11h could strongly inhibit the BTK activity, which displayed 82.7–83.9% inhibition in contrast with that of ibrutinib (99.4%). The kinase-based test results confirmed that the large-size substituent groups (analogues 11d, 11e, 11g and 11h) are more favorable than the small-size groups (analogues 11a–11c). It was not beneficial for the inhibition with the disubstituted arylamine as R (analogues 11f and 11i). As a global observation, most of the target compounds inhibit the proliferations of these leukemia lines at concentrations ranging from 1 to 20 μmol L–1. Compound 11g, which has a 3-methyl phenylcarbamoyl substituent at the C-4 aniline moiety of the pyrimidine core, possessed the most promising anti-proliferative activity on the three leukemia lines. Particularly, it is higher than ibrutinib for its activity against the Raji cells (IC50 = 6.98 μM and 14.5 μM, respectively), suggesting that a new action mechanism for them to be produced interferes with the B-cell lymphoblastic leukemia cells. Nevertheless, the fluorine-substituted and methoxy-substituted C-4 aniline moieties almost lost their activity against the B-cell leukemia lines (IC50 not determined) in spite of their high capability to inhibit the BTK activity. We speculated that the low water solubility might be the main reason for their low anti-B lymphoma cell activity. In contrast, 11b, 11c, 11d and 11f, which only possessed moderate activity against BTK (Inh% = 40–60%), were able to strongly inhibit the proliferation of the three leukemia lines with an IC50 value ranging from 1 to 20 μmol L–1.
Table 1. SAR of the synthesized compounds.
| Compound | R | HL60 IC50 (μM) | Raji IC50 (μM) | Ramos IC50 (μM) | BTK a Inh% at 100 nm |
| 11a |
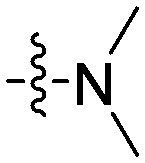
|
>50 | 42.9 | >50 | ND b |
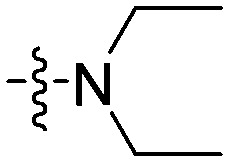
| 11b |
|
9.34 | 12.5 | 11.2 | 40.3 |
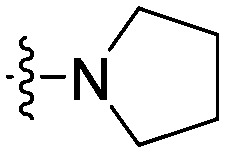
| 11c |
|
15.8 | 19.2 | 10.4 | 59.8 |
| 11d |
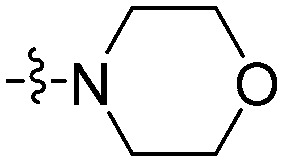
|
5.94 | 14.7 | 7.24 | 60.7 |
| 11e |
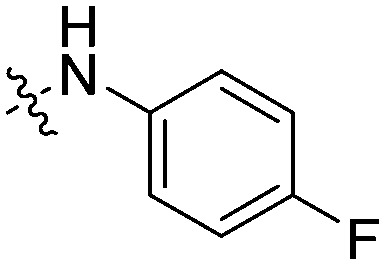
|
ND | ND | ND | 83.9 |
| 11f |
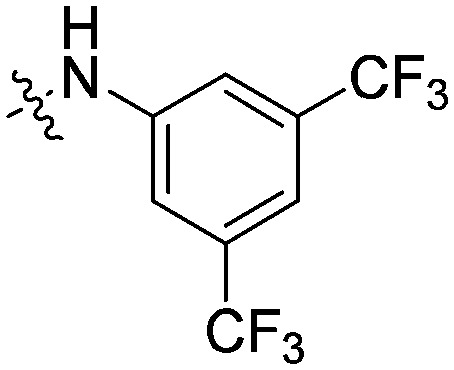
|
4.79 | 9.36 | 6.18 | 56.5 |
| 11g |
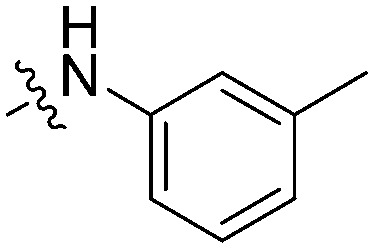
|
3.66 | 6.98 | 5.39 | 82.7 |
| 11h |
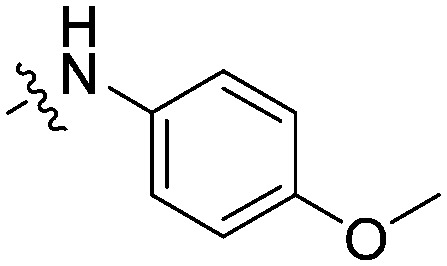
|
ND | ND | ND | 83.9 |
| 11i |
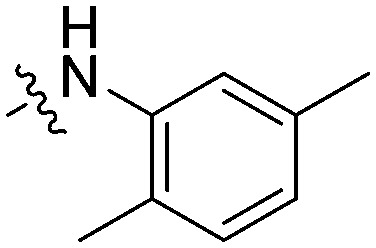
|
14.9 | 20.9 | 16.8 | ND |
| Ibrutinib | 3.57 | 14.5 | 2.31 | 99.4 |
aPercentage inhibition rate of each compound on BTK at 100 nM was measured using the ADP-Glo™ kinase assay system, shown as means of three experiments.
bNot determined.
Additionally, the effect of treatment time and drug concentration was further explored for compound 11g on Raji cell viability. As a result, all of these compounds inhibited the proliferation of the Raji cells in a dose- and time-dependent manner, and almost completely blocked the Raji cells (90%) at a concentration of 40 μM after 72 h (Fig. 3). Moreover, 11g displayed better anti-proliferation activity than positive control ibrutinib in a time-dependent manner at a concentration of 5 μM.
Fig. 3. The effects of treatment time and drug concentration for compound 11g on Raji cell viability.

Flow cytometry assays by incubating Ramos cells with 5 μM compound 11g for 48 h were also performed. Excitingly, the result shown in Fig. 4 indicate that inhibitor 11g induced Ramos cell apoptosis with 38.5% which is higher than the reference agent ibrutinib (7.83%) at the concentration of 5 μM. In addition, to investigate the anti-proliferative mechanism of our designed compounds on B-cell leukemia cells, we further evaluated the effect of the most active inhibitor 11g on the distribution of the Ramos cells in the cell cycle with ibrutinib as the positive control. As shown in Fig. 5, after incubation with 5 μmol L–1 inhibitor 11g and ibrutinib for 48 h, the DNA contents of the Ramos cells were 36.27% and 30.5% in the G0/G1 phase, respectively. Apparently, the newly synthesized molecule 11g blocked the Ramos cells at the G0/G1 phase, which is in accordance with the reference compound ibrutinib.
Fig. 4. Compound 11g induced Ramos cell apoptosis in vitro. The cells were incubated at the concentration of 5 μM for 48 h, and DMSO and ibrutinib were treated as the blank and positive controls, respectively.

Fig. 5. Cell cycle analysis of the treatment of the Ramos cells with compound 11g at the concentration of 5 μM for 48 h, and DMSO and ibrutinib were treated as the blank and positive controls, respectively.

The BCR signaling pathway is relevant to several severe leukemias and lymphomas. The BCR is the first aggregate with CD79 which contains immunoreceptor tyrosine-based activation motifs (ITAM). After that, ITAM are phosphorylated primarily by Src-family tyrosine kinases, such as Lyn. The Src-family kinases and phosphorylated ITAM serve as a scaffolding platform for engaging and activating SH2 domains containing kinases, including Syk, which leads to BTK being recruited to the cell membrane and phosphorylated in its SH2 domain. The activated BTK then phosphorylates its substrate phospholipaseγ2 (PLCγ2), leading to activation of the downstream signaling pathways, such as NF-κB and STAT3.16 In fact, over-expression of BTK may cause the disorder of the B-cell signaling pathway, leading to B-cell malignancies and autoimmune diseases.17 Thus we performed the western blotting assay to detect the phosphorylation of BTK and its substrate PLCγ2. The result showed that 11g could inhibit their phosphorylation, though it is weaker than the positive control ibrutinib (Fig. 6).
Fig. 6. Effect of 11g on BTK and PLCγ2 phosphorylation in the Ramos cells with ibrutinib as the positive control.

Conclusions
In summary, we designed and synthesized a series of 2-phenyl pyrimidine derivatives based on the skeleton of highly selective but modestly potent BTK inhibitor CGI-560, then a variety of pharmacophores were used to improve their BTK inhibition. Most of them exhibited good inhibitory activities in both cellular and enzymatic assays. In particular, compound 11g with a 3-methyl phenylcarbamoyl substituent at the C-4 aniline moiety of the pyrimidine core displayed stronger activity than the positive control (ibrutinib) against Raji cells. The further mechanism study showed that compound 11g could not only induce cell cycle arrest and apoptosis in B-cell leukemia lines but also inhibit the phosphorylation of BTK and its substrate PLCγ2. Overall, 11g could serve as a promising highly selective lead candidate for further study.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We are grateful to the Program of Jiangsu Key Laboratory of Drug Design and Optimization, China Pharmaceutical University, the postgraduate innovation fund of Huahai medical corporation and the college students' innovation and entrepreneurship training program (201810316104) for the financial support of this research.
Footnotes
†Electronic supplementary information (ESI) available. See DOI: 10.1039/c8md00413g
References
- De Lucca G. V., Shi Q., Liu Q., Batt D. G., Beaudoin Bertrand M., Rampulla R., Mathur A., Discenza L., D'Arienzo C., Dai J., Obermeier M., Vickery R., Zhang Y., Yang Z., Marathe P., Tebben A. J., Muckelbauer J. K., Chang C. J., Zhang H., Gillooly K., Taylor T., Pattoli M. A., Skala S., Kukral D. W., McIntyre K. W., Salter-Cid L., Fura A., Burke J. R., Barrish J. C., Carter P. H., Tino J. A. J. Med. Chem. 2016;59:7915–7935. doi: 10.1021/acs.jmedchem.6b00722. [DOI] [PubMed] [Google Scholar]
- Khan W. N. Immunol. Res. 2001;23:147–156. doi: 10.1385/IR:23:2-3:147. [DOI] [PubMed] [Google Scholar]
- Satterthwaite A. B., Witte O. N. Immunol. Rev. 2010;175:120–127. [PubMed] [Google Scholar]
- Buggy J. J., Elias L. Int. Rev. Immunol. 2012;31:119–132. doi: 10.3109/08830185.2012.664797. [DOI] [PubMed] [Google Scholar]
- Cohen M. S., Zhang C., Shokat K. M., Taunton J. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y., Yang H., Wang C., Meng Q., Li L., Sun H., Zhen Y., Liu K., Li Y., Ma X. Bioorg. Med. Chem. 2017;25:765–772. doi: 10.1016/j.bmc.2016.11.054. [DOI] [PubMed] [Google Scholar]
- Sachdeva M., Dhingra S. Int. J. Appl. Basic Med. Res. 2015;5:54–57. doi: 10.4103/2229-516X.149245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C., Tian D., Ren X., Ding S., Jia M., Xin M., Thareja S. Eur. J. Med. Chem. 2018:315–326. doi: 10.1016/j.ejmech.2018.03.062. [DOI] [PubMed] [Google Scholar]
- Xue Y., Song P. R., Song Z. L., Wang A. L., Tong L. J., Geng M. Y., Ding J., Liu Q. S., Sun L. P., Xie H., Zhang A. J. Med. Chem. 2018;61:4608–4627. doi: 10.1021/acs.jmedchem.8b00441. [DOI] [PubMed] [Google Scholar]
- Chen H., Song P., Diao Y., Hao Y., Dou D., Wang W., Fang X., Wang Y., Zhao Z., Ding J., Li H., Xie H., Xu Y. MedChemComm. 2018;9:697–704. doi: 10.1039/c8md00019k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di P. J., Huang T., Balazs M., Barbosa J., Barck K. H., Bravo B. J., Carano R. A., Darrow J., Davies D. R., Deforge L. E. Nat. Chem. Biol. 2011;7:41–50. doi: 10.1038/nchembio.481. [DOI] [PubMed] [Google Scholar]
- Zhao D., Huang S., Qu M., Wang C., Liu Z., Li Z., Peng J., Liu K., Li Y., Ma X. Eur. J. Med. Chem. 2017;126:444–455. doi: 10.1016/j.ejmech.2016.11.047. [DOI] [PubMed] [Google Scholar]
- Somberg R., Pferdehirt B. and Kupcho K., Cell, 2003.
- Zegzouti H., Zdanovskaia M., Hsiao K., Goueli S. A. Assay Drug Dev. Technol. 2009;7:560. doi: 10.1089/adt.2009.0222. [DOI] [PubMed] [Google Scholar]
- Cizdziel J. V., Tolbert C., Brown G. Spectrochim. Acta, Part B. 2010;65:176–180. [Google Scholar]
- Wang Y., Zhang L. L., Champlin R. E., Wang M. L. Clin. Pharmacol. Ther. 2015;97:455–468. doi: 10.1002/cpt.85. [DOI] [PubMed] [Google Scholar]
- Satterthwaite A. B., Witte O. N. Immunol. Rev. 2000;175:120–127. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.