

Abstract
Colorectal cancer (CRC) is the third most diagnosed cancer in the western world, affecting 1 out of approximately 22 people in their lifetime. Several epidemiological studies suggest a positive association between high plasma cholesterol levels and colorectal cancer. However, the molecular mechanisms by which cholesterol may alter the risk of colorectal cancer (CRC) are ill-defined as the cholesterol lowering drugs statins do not appear to decrease a patient’s risk of developing colorectal cancer. Cholesterol is metabolized to active derivatives including cholesterol oxidization products (COP), known as oxysterols, which have been shown to alter cellular proliferation. These metabolites and not cholesterol per se, may therefore affect the risk of developing colorectal cancer. The cholesterol metabolite or the oxysterol 27-hydroxycholesterol (27-OHC) is the most abundant oxysterol in the plasma and has been shown to be involved in the pathogenesis of several cancers including breast and prostate cancer. However, the role of 27-OHC in colorectal cancer has not been investigated. We treated Caco2 and SW620, two well characterized colon cancer cells with low, physiological and high concentrations of 27-OHC, and found that 27-OHC reduces cellular proliferation in these cells. We also found that the effects of 27-OHC on cell proliferation are not due to cellular cytotoxicity or apoptotic cellular death. Additionally, 27-OHC-induced reduction in cell proliferation is independent of actions on its target nuclear receptors, liver-X-receptors (LXR) and estrogen receptors (ER) activation. Instead, our study demonstrates that 27-OHC significantly decreases AKT activation, a major protein kinase involved in the pathogenesis of cancer as it regulates cell cycle progression, protein synthesis, and cellular survival. Our data shows that treatment with 27-OHC substantially decreases the activation of AKT by reducing levels of its active form, p-AKT, in Caco2 cells but not SW620 cells. All-together, our results show for the first time that the cholesterol metabolite 27-OHC reduces cell proliferation in colorectal cancer cells.
Keywords: 27-hydroxycholesterol, AKT, cholesterol, Colorectal Cancer, oxysterols, proliferation
1. Introduction
In the United States, Colorectal Cancer (CRC) is the third most diagnosed cancer, affecting 1 out of approximately 22 people in their lifetime. Globally, it is the third most common type of cancer, leading to an estimated 1.4 million patients being diagnosed in 2016 [1] with more than 600,000 deaths [1, 2]. There are several risk factors for CRC including age [3], family history of polyps [4], alcohol consumption [5], consumption of westernized diet [6, 7], and inflammatory bowel disease [8]. Cholesterol has also been considered a risk factor for cancer, but data about the involvement of cholesterol levels in CRC are still controversial. Several epidemiological studies suggested that higher cholesterol levels were associated with increased risk of developing cancer [9]. Several studies have shown that lowering cholesterol levels with statins, reduced the risk of some cancers [10]. Specifically in CRC, an observational study found a decreased risk of CRC with statin therapy [11, 12]. However, other recent studies reported that lower cholesterol levels are associated with an increased risk of CRC [13].
Cholesterol is a precursor to several important molecules such as bile acids, steroid hormones, vitamin D, and oxysterols. Oxysterols are cholesterol oxidation products formed by the addition of a polar group such as hydroxyl, keto, or epoxy groups [14, 15]. Oxysterols are not only metabolites that regulate cholesterol concentrations, but active molecules that regulate several functions including plasma membrane permeability and signal transduction [16], apoptosis [17, 18], and immune system [19–21]. The most abundant oxysterol in the plasma is 27-hydroxycholesterol (27-OHC) [22]. 27-OHC is a macrophage synthesized cholesterol metabolite that can enhance inflammation in macrophages which are detected in abundance in atheromatous lesions [23]. Multiple lines of study have demonstrated that cholesterol-enriched diets increase plasma 27-OHC levels [24–29]. This increase in plasma 27-OHC upon high cholesterol diet feeding was observed despite significant heterogeneity in these studies with regards to the relative fraction and percentage of cholesterol content present in the respective diets used in these studies that varied from 0.25% to 2.0% w/w. Furthermore, this phenomenon of high cholesterol diet-induced increase in 27-OHC was recapitulated in different animal species that included rabbits [24], rats [29], mice [26–28], as well non-human primates [25]. 27-OHC is a ligand for liver-X-receptor (LXR), a nuclear receptor that has a variety of functions [30]. For the last decade, 27-OHC has also been shown to act as a selective estrogen receptor modulator (SERM) [31–33]. This discovery implicated 27-OHC in ER-positive carcinogenesis in breast cancer [27]. We and others have also demonstrated that 27-OHC can be deleterious in prostate cancer cells [34, 35]. However, because cholesterol metabolism is suspectedto influence a variety of cancer progression, little is known about 27-OHC involvement in CRC. We determined in the present study the effects of 27-OHC on cell proliferation in two major cell line models for CRC, Caco2 and SW620 cells. We also determined the extent to which 27-OHC effects involve LXR, ER and the AKT pathway.
2. Material and Methods
2.1. Materials:
Caco2 cells (cat # HTB-37™), MTT Cell Proliferation Assay (cat # 30–1010K), and SW620 cells (cat # CCL-227™) (ATCC, Manassas, VA). DMEM media, Leibovitz’s L-15 media, 100U/ml penicillin, 100ug/ml streptomycin, and 0.25ug/ml amphotericin (Life Technologies, Carlsbad, CA). FBS (Atlanta Biologicals, Flowery Branch, GA). GW3965 (cat # 2474), 27-hydroxycholesterol (cat # 3907), and estradiol (cat # 2824) (R&D systems from Minneapolis, MN). 6α-epoxycholesterol-3-sulfate (cat # C4136–000) (Steraloids, Newport, RI). Afuresertib (cat # 17988) (Cayman Chemicals from Ann Arbor, MI). CytoTox 96® non-radioactive cytotoxicity assay kit (cat # G1782) and DeadEnd Fluorometric TUNEL assay kit (cat # G3250) (Promega, Valencia, CA). Hard Set mounting medium with DAPI (cat # H-1500) (Vector Laboratories, Burlingame, CA). DNase (cat #AM2222), Alexa Fluor 594nm (cat #A11037), Halt™ proteases and phosphatase inhibitor cocktail (cat #78446) (Fisher Scientific, Hampton, NH). Immun-Blot® PVDF membrane for protein blotting (cat # 1620177), Clarity™ Western ECL substrate (BioRAD, Hercules, CA). The antibodies used in this study are included in Table 1 below.
Table 1.
Antibodies and their sources and applications used in the present study.
| Name | Catalog # | Species | Application |
|---|---|---|---|
| ABCA1 | Novus biologicals: 105SS | Rb | Western Blotting |
| ABCG1 | Thermo Fisher: PA5–13462 | Rb | Western Blotting |
| Actin | Santa Cruz: sc-47778 | Rb | Western Blotting |
| AKT | Cell Signaling 9272S | Rb | Western Blotting |
| p-AKT | Cell signaling 4060S | Rb | Western Blotting |
| ERα | Santa Cruz: sc-787 | Ms | Western Blotting |
| ERα | Thermo Fisher: PA5–16440 | Rb | Immunofluorescence |
| ERβ | Thermo Fisher: PA1310B | Rb | Western Blotting |
| LXRα | Thermo Fisher: PA1330 | Rb | Western Blotting |
| LXRα | Thermo Fisher: PA1333 | Rb | Western Blotting |
2.2. Cell Culture:
Caco2 non-metastatic cells were grown in DMEM media containing 10% FBS. SW620 metastatic cells were grown in Leibovitz’s L-15 containing 10% FBS. 100U/ml penicillin, 100ug/ml streptomycin, and 0.25ug/ml amphotericin were added. Cells were maintained at 5% CO2 at 37°C. Cells were treated with (0, 0.5, 1, 10, 50, or 100 μM) 27-hydroxycholesterol (27-OHC), 2nM estradiol (E2), 100nM ICI 182 780, 10μM GW3965, and/or 10μM of 5α−6α-epoxycholesterol-3-sulfate (ECHS). Circulating 27-OHC levels are 0.15–0.73 μM, and these concentrations can be in the millimolar range in some pathological situations such as atherosclerosis [14]. Our study was approved by the Institutional Biosafety Committee of the School of Medicine at the University of North Dakota.
2.3. MTT assay:
14,000 cells were seeded in a 96-well plate and grown overnight. Next morning, different concentrations of 27-OHC (0–300 μM) was added and incubated for 24hrs. After 24hrs, MTT assay was performed following manufacture’s protocol. Briefly, 10ul of MTT reagent was added and cells were incubated at 37°C until purple precipitate formed (2–4hrs). Media was removed and 100ul of detergent reagent was added and was incubated at room temperature in dark for 2 hours. Plate was read using a microplate reader at 570nm. Ethanol (ETOH) was used as a vehicle control with treatment was made to 1 and the other samples were normalized to the ETOH treatment and the fold change was determined.
2.4. LDH assay:
14,000 cells were seeded in a 96-well plate and grown overnight. Next morning, different concentrations of 27-OHC (0–300 μM) was added and incubated for 24hrs. After 24hrs, LDH assay was performed using CytoTox 96® non-radioactive cytotoxicity assay following manufacture’s protocol. 50ul aliquots of media were transferred to a new 96-well plate and 50ul of CytoTox 96® Reagent was added for 30 minutes in the dark at room temperature. 50ul of Stop solution was added and the absorbance was read at 490nm. Ethanol treatment was made to 1 and the other samples were normalized to the Ethanol treatment and the fold change was determined.
2.5. TUNEL assay:
To measure apoptosis, TUNEL assay was performed using DeadEnd Fluorometric TUNEL assay following manufacturer’s protocol. Cells were fixed with 4% formaldehyde on a cover slip and permeabilized with 0.2% triton X-100 solution. The cells were then incubated with terminal deoxynucleotidyl transferase, fluorescein-12-dUTP for 1 hour at 37°C in the dark. The slides were mounted using DAPI containing solution to stain the nuclei. The slides were then visualized with a fluorescence microscopy. DNase was used as a positive TUNEL control.
2.6. Immunofluorescence:
100,000 cells were seeded on a coverslip and were rinsed with PBS and fixed and permeabilize with 4% formaldehyde, 0.1% triton X-100 in PBS. The coverslips were blocked with 10% goat serum. Primary antibody was added overnight at 4°C with anti-estrogen receptor alpha (see table 1) at 1:100 dilution. The estrogen receptor alpha was conjugated with Alexa Fluor 594nm and was mounted with Hard Set mounting medium with DAPI. Slides were visualized using Zeiss LSM-510 Meta Confocal Microscope.
2.7. Western Blotting:
Cells were harvested by a PBS wash and the addition of RIPA buffer with Halt™ proteases and phosphatase inhibitor cocktail followed by spinning at 14,000g for 10 minutes at 4°C. The supernatant was then used for further processing. Bovine Serum Albumin (BSA) assay used to quantify the protein concentrations of the lysates using standard protocol. 10ug to 35ug of sample were separated using a 10 percent SDS-PAGE gels and transferred to a PVDF membrane. The membrane was incubated with antibodies (See table 1). The membrane was then exposed to Clarity™ Western ECL substrate. The blots were then visualized using Omega Lum™ G imaging system (Aplegen). Quantification of the bands developed through densitometry using Image J software
2.8. Statistical analysis:
All the assays were performed in triplicates. The level of significance was determined using either unpaired t-test or one way analysis of variance (one-way ANOVA). Post-hoc Tukey test was used to determine statistical significance. Statistical testing was performed using GraphPad Prism software 7. All data is presented as a mean ± SEM. The values of treated cells are relative to control.
3. Results
3.1. 27-OHC decreases cell proliferation in colon cancer cell lines
Two colon cancer cell lines were utilized, non-metastatic Caco2 and metastatic SW620. To determine the role of 27-OHC in CRC, cell proliferation was measured after different concentrations of 27-OHC for 24 hours using a MTT assay. We found that concentrations at and above 10μM that there was a significant decrease in cell proliferation relative to vehicle control (Figure 1A and 1B). To determine whether this decrease in cell proliferation is associated with cellular cytotoxicity, LDH assay was performed in both cell lines. We found that the 27-OHC induced decrease in cell proliferation and cell viability was not associated with cell death or cytotoxicity at physiological concentrations, in both the Caco2 cells and SW620 cells. We subsequently determined whether it was possible to elicit cytotoxicity in these cells with 27-OHC. We found that 27-OHC, at higher supra-physiological concentration of 300μM, did cause cell death in Caco2 cells, but not SW620 cells (Figure 1C and 1D). Our data suggests that the 27-OHC induced decrease is cell proliferation and cell viability is not a result of cellular cytotoxicity.
Figure 1.

27-hydroxycholesterol decreases cell proliferation in Caco2 and SW620 cells. MTT assay (A and B) and LDH assay (C and D) show that 24 hours treatment with 27-OHC reduces cell proliferation and does trigger cellular cytotoxicity in Caco2 at 300μM but not in SW620 cells respectively. Results are fold change relative to vehicle control. **p <0.01, ***p<0.001.
To determine whether the reduction in cell proliferation emanates from significant cell death following treatment with 27-OHC, a Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed. This assay detects DNA fragmentation and represents a characteristic hallmark of apoptosis. Our data shows that at concentrations that reduce cell proliferation, 27-OHC doesn’t induce apoptotic cell death in either Caco2 (Figure 2A) or SW620 (Figure 2B) cells, suggesting that the reduced cell proliferation by 27-OHC is not due to apoptotic cell death.
Figure 2.

27-OHC treatment doesn’t cause apoptotic cell death. TUNEL assay shows that treatment for 24h with 27-OHC doesn’t trigger cell death in Caco2 cells (A) or SW620 cells (B). DAPI (blue) is a counter stain for nuclei and green represents TUNEL incorporation. DNase was used as positive TUNEL control. Bar=100μm.
3.2. 27-OHC decreases cell proliferation independently of ER or LXR activation
We determined the protein levels of 27-OHC receptors, ERαβ and LXRαβ, in 27-OHC treated Caco2 and SW620 cell lines. Our data shows that both cell lines express ER and LXR both isoform α and β. Protein levels of LXRα, LXRβ, ERα, and ERβ didn’t differ between vehicle and 27-OHC treatments (Figure 3A–D).
Figure 3.

Both Caco2 and SW620 cells express ER and LXR. Western blot analysis show that both Caco2 (A) and SW620 (C) cells express ERα, ERβ, LXRα and LXRβ isoforms. Optical density analysis shows that the protein levels of ERα, ERβ, LXRα and LXRβ is similar in both vehicle and 27-OHC (10μM) -treated cells in Caco2 (B) and SW620 (D) cells. There was no significant difference between vehicle treated and 27-OHC treated for all groups.
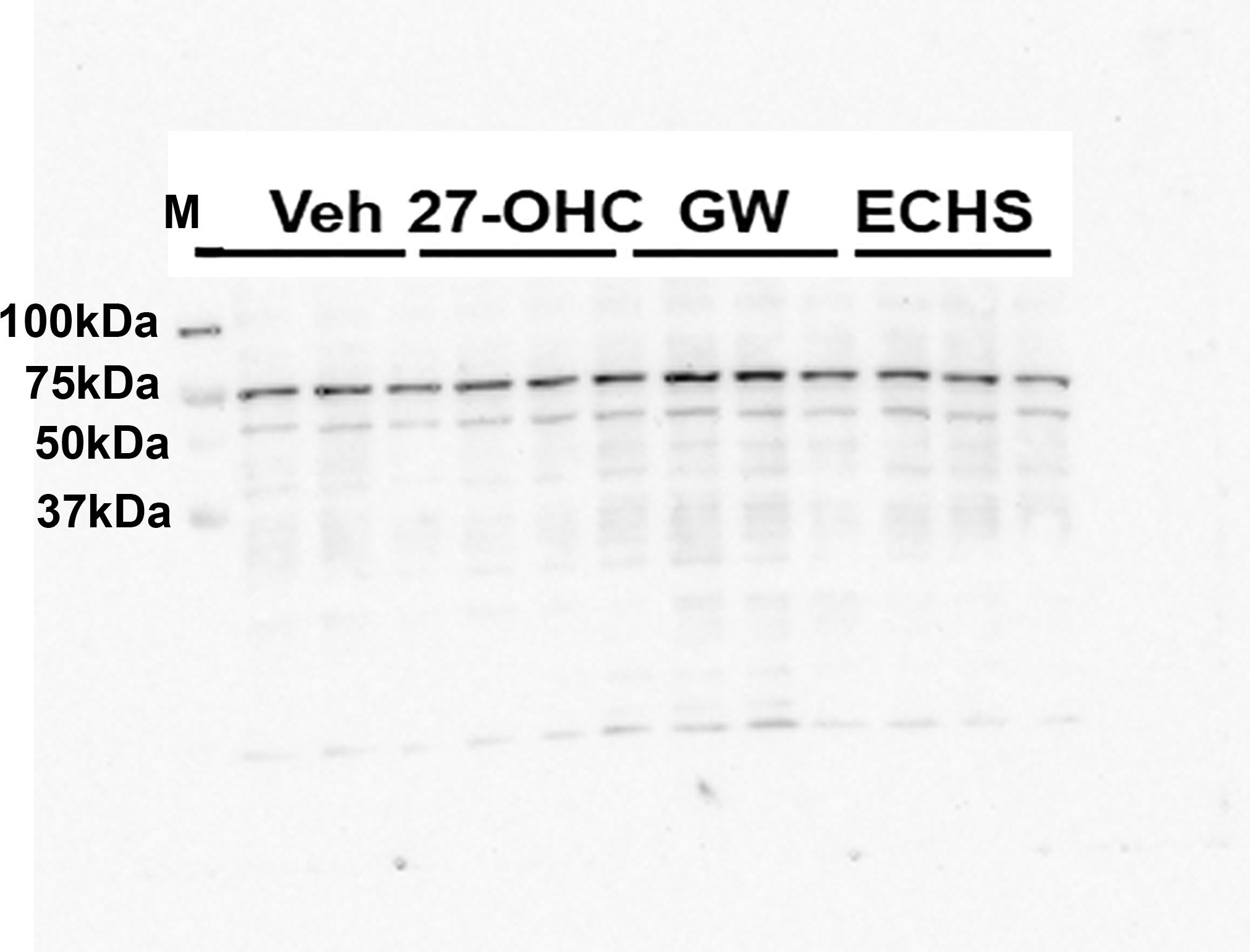
To determine the extent to which 27-OHC effects on cell proliferation and cell viability emanate from effects on LXR or ER, we treated cells with agonists and antagonists of ER and LXR. We found that while 27-OHC reduces cell proliferation and cell viability, neither the ER agonist estradiol (E2) nor the ER antagonist ICI 182 780 affects cell proliferation and cell viability; as well neither LXR agonist GW3965 nor the LXR antagonist ECHS affected cell proliferation and cell viability in Caco2 cells (Figure 4A) or in SW620 cells (Figure 4B). Since changes in cellular viability and cellular proliferation are associated with changes in cytotoxicity and given that we did not observe changes in cellular viability and cellular proliferation with the with agonists and antagonists of ER and LXR, as determined by MTT reduction assay (Figure 4A, 4B), we did not determine cell death or cytotoxicity exhibited by these agonists and antagonists of ER and LXR. Furthermore, as the LXR agonist and the LXR antagonist, GW3965 and ECHS respectively, did not cause any changes in cell proliferation and cell viability (Figure 4A, 4B), it was necessary to establish that the LXR transcriptional machinery was responsive in Caco-2 as well as SW620 cells, and that the lack of effects of 27-OHC on LXR transcriptional activity were indeed valid and not a consequence of unresponsive LXR transcriptional machinery. To this end, we determined the effects of the LXR agonist, GW3965, on the expression of LXR target genes, ABCA1 and ABCG1 (Figure 4C–4F). Our data shows that the concentration of GW3965 used (10μM) is above the minimal effective concentration to induce LXR target genes, ABCA1 and ABCG1, as determined by immunoblotting and densitometric analysis (Figure 4C–4F). While the LXR agonist, GW3965, upregulated the protein levels of the ABCA1 and ABCG1 in Caco2 cells, but exclusively ABCG1 in SW620 cells (Figure 4C–4F), the LXR antagonist, ECHS, did not downregulate the protein levels of ABCA1 or ABCG1 in Caco2 and SW620 cells (Figure 4C-F). Interestingly, GW3965 at the same aforementioned concentration (10μM) did not affect cell viability and cell proliferation as determined by the MTT reduction assay in Caco2 and SW620 cells (Figure 4A, 4B). This data suggests that the LXR is transcriptionally responsive and that the concentration of GW3965 (10μM) is above the minimal effective concentration to activate LXR, while not inducing any changes in cellular proliferation and cell viability. To further verify whether the 27-OHC-induced effects on cell proliferation are associated with LXR activation, we determined the expression levels of LXR target genes, ABCA1 and ABCG1 in 27-OHC treated cells. 27-OHC did not upregulate the expression levels of LXR target genes, ABCA1 and ABCG1 in Caco2 and SW620 (Figure 4C–4F), while eliciting cell proliferation and cell viability changes (Figure 4A, 4B). Our data suggests that the 27-OHC-induced decrease in cell proliferation and cell viability is independent of LXR activation.
Figure 4.

While 27-OHC reduces cell proliferation in Caco2 (A) and SW620 (B) cells, treatments with either the ER agonist E2, the ER antagonist ICI 182 780, the LXR agonist GW3965 (GW), or the LXR antagonist ECHS didn’t affect cell proliferation either when administered alone or in presence of 27-OHC (10 μM). Western blots and optical density analysis show that treatment with the LXR agonist GW3965 increased the protein levels of ABCG1 and ABCA1, downstream target proteins to LXR activation, in Caco2 (C,E) and increased ABCG1, not ABCA1, in SW620 cells (D,F). *p <0.05, ** p<0.01, and *** p<0.001. All data is fold change relative to vehicle control.
Analogously, as the ER agonist and the ER antagonist, estradiol (E2) and ICI 182780 respectively, did not cause any changes in cell proliferation and cell viability (Figure 4A,4B), it was necessary to establish that the ER transcriptional machinery was responsive in Caco-2 as well as SW620 cells. We determined the integrity of the ER transcriptional activity in the Caco2 and SW620 cell-lines, using estradiol (E2) as the positive control for ERα activation. We determined the estradiol-induced nuclear localization of ERα as a degree of ER activation as well as a measure for demonstrating that ER signaling is responsive in these cell-lines. We found that estradiol (2nM), elicited a pronounced translocation of ERα from the cytosol into the nucleus in Caco2 cells (Figure 5A, arrows), demonstrating that the ER signaling is responsive in these cell-lines as well as the concentration of estradiol is above the minimal effective concentration. Conversely to Caco2 cells, in SW620 cells treated with E2, the ERα did not translocate into the nucleus and remained distributed in the cytoplasm (Figure 5B). Treatment with 27-OHC or with the vehicle did not elicit any effect on ERα translocation from the cytosol into the nucleus in either Caco2 or SW620 cells (Figure 5A, 5B), thereby demonstrating that 27-OHC elicited no effects on the activation status of ER. Our data suggests that the 27-OHC-induced decrease in cell proliferation and cell viability is independent of ER activation.
Figure 5.

27-OHC didn’t lead to ER alpha translocation to the nucleus. Treatment with estradiol (E2) leads to ERα translocation into the nucleus of Caco2 (A, white arrows) but not SW620 (B) cells. Treatment with either vehicle or 27-OHC didn’t lead to nuclear localization of ERα as evidenced by the presence of immunoreactivity to ERα antibody (red) in the cytoplasm but not in the nucleus (counter stained with DAPI (blue) in Caco2 (A) and SW620 (B) cells. Bar=10μm.
3.3. AKT signaling may be involved in Caco2 but not SW620 cells
Because AKT is a signaling molecule involved in regulating cellular proliferation, we determined the extent to which AKT signaling is involved in the 27-OHC-induced decrease in cell proliferation. We determined the protein levels of the activated, phosphorylated form of AKT (p-AKT at serine 473), along with AKT levels. Our data shows a significant decrease in p-AKT and increase in total AKT levels in Caco2 cells treated with 27-OHC compared to vehicle treatment (Figure 6A-B). In SW620 there was no significant difference in p-AKT or AKT levels (Figure 6A-B). To determine the functional consequence of the 27-OHC-induced reduction in p-AKT levels, we determined whether AKT inhibition effects cell proliferation and cell viability. To this end, we used a pan AKT inhibitor, Afuresertib [36, 37] that competitively inhibits AKT kinase activity. We found that the AKT inhibitor, Afuresertib, significantly decreases proliferation and cell viability of Caco2 cells as determined by MTT reduction assay (Figure 6C). Furthermore, we found that the AKT inhibitor, Afuresertib, caused a decrease in total AKT levels as determined by immunoblotting and densitometric analysis (Figure 6D, 6E). Our data shows that AKT activation plays a critical role in cell proliferation and cell viability of Caco2 cells and could be an underlying target of the 27-OHC-induced decrease in cell proliferation and cell viability
Figure 6.

27-OHC decreases p-AKT. Western blot analysis (A) and optical density analysis (B) showing reduced levels of active p-AKT in Caco2 but not in SW620 cells following treatment with 27-OHC (10μM). Treatment with the AKT inhibitor afuresertib (AF) led to a decrease in cell proliferation in Caco2 cells as shown with an MTT assay compared to untreated (UT) or vehicle treated (Veh) cells (C). A representative Western blot (E) and optical density analyses (F,G,H) showing an increase in p-AKT and a decrease in AKT levels with afuresertib in Caco2 cells. * p <0.05; **p < 0.01.
4. Discussion
In this study, we determined the effects of the oxysterol 27-OHC on cell proliferation in Caco2 and SW620 colorectal cancer cells. The MTT assay shows that while physiological concentrations of 27-OHC didn’t affect proliferation, supra Physiological concentrations (10, 50 and 100μM) substantially reduced basal cell proliferation. The reduction in cell proliferation with 27-OHC is not associated with cytotoxicity or apoptotic cell death. The effects of 27-OHC on proliferation appear to be independent of its cognate receptors LXR or ER. We also show that in Caco2, but not SW620 cells that the reduction in cell proliferation is associated with a decrease in p-AKT levels. We also show that using an AKT inhibitor significantly decreased cellular proliferation.
27-hydroxycholesterol is the most abundant oxysterol in the plasma [22]. Several studies implicated this oxysterol in several different types of cancers including breast and prostate cancers [34, 38, 39]. Oxysterols have been also involved in several diseases, such as atherosclerosis [40], Alzheimer’s disease [41, 42], and Huntington disease [43]. Oxysterols play a role in cancer cell proliferation both through proproliferative and antiproliferative effects depending on the oxysterol and the type of cancer. Similar to our findings with 27-OHC, the oxysterol 7-beta hydroxycholesterol has been showed to decrease cell proliferation in Caco2 cells [44]. Additionally, treatment with Caco2 cells with another oxysterol, 25-hydroxycholesterol, led to increased IL-8 and IL-1β levels [45]. However, there has been no study investigating the role of 27-OHC in cell proliferation in colorectal cell lines as this oxysterol in the plasma.
Colorectal Cancer can develop after periods of chronic inflammation, such as inflammatory bowel diseases including Crohn’s disease [8]. Oxysterols have been shown to be involved in both innate and adaptive immunity. Oxysterols increase inflammatory cytokines and chemokines such as TNF-α, Il-1β, Il-8. Oxysterols synthesis is a component of the regulatory network that modulates the extent of innate immune reactions as well as the nature and intensity of adaptive responses [46], and inhibition of 25-hydroxylase in macrophages increased IL-1 via SREBP downregulation [21]. 25-hydroxycholesterol has also been shown to regulate B cell proliferation and IgA levels [19]. 27-hydroxycholesterol has also been shown to inhibit viral infections [20].
27-OHC has been shown to act as a selective estrogen receptor modulator (SERM) [31–33]. There appears to be protective association between hormone replacement therapy and colorectal cancer, specifically through activation of ERβ [47]. For example, transfection of colon cancer line SW489 with ERβ resulted in a decrease in cell proliferation and cell cycle arrest with xenografts weighing 70% less than control [48]. 27-hydroxycholesterol is also a ligand for liver-x-receptor [30]. Liver-x-receptor are known to play a role in colon cancer progression. Indeed, it has been shown that LXR activation through addition of the agonist GW3965 reduced proliferation in human colorectal cancer HT29 cell line [49], however, this effect was not seen in Caco2 or SW620 cells. Administration of the LXR specific agonist, GW3965 also significantly reduced expression of proliferation in mouse colon [50].
Our results also demonstrate that treatment with the ER agonist estradiol did not influence cell proliferation in both cell lines, suggesting that 27-OHC-induced cell proliferation in CRC cells is independent of effects on ER. Previous studies showed that, E1, but not E2, significantly decreases proliferation when added exogenously to the colonic epithelial cell line, SW620 cells [51]. In different cell models however, 27-OHC has been shown to increase proliferation in endometrium through ER and LXR pathways [52]. Although we show a decrease in cell proliferation, the cellular mechanisms by which 27-OHC affects proliferation is yet to be determined. As the effects of 27-OHC on proliferation appear to be independent of 27-OHC receptors, LXR and ER, other signaling pathways such as the AKT pathways mat be involved in 27-OHC effects. We show a decrease in p-AKT levels following 27-OHC treatment in Caco2 not SW620 cells. Our current results are in accordance with previous data showing reduced p-AKT levels in organotypic hippocampal slices from mice incubated with 27-OHC [53] and other labs that showed that 27-OHC leads to a decrease in p-AKT levels [54]. In other cell models however, such as macrophages and lymphocytes, 27-OHC has been shown to increase p-AKT levels [55, 56]. We further show that there was a decrease in total AKT levels with afuresertib and a decrease in cellular proliferation, suggesting that AKT may be mediating the decrease in cell proliferation in Caco2 cells. Whether effects on p-AKT mediate 27-OHC effects on cell proliferation in Caco2 cells is still to be confirmed and cellular mechanisms mediating 27-OHC-induced proliferation in SW620 cells identified. Emerging evidence has shown that cholesterol oxidation products and dietary oxysterols induce inflammation in the colonic epithelial cells, thereby disrupting tight junctions and compromising the “physical barrier” functional aspect of the colonic epithelial cell physiology. In light of these findings, future studies are warranted to determine the effects of 27-OHC on molecular and transcriptional mechanisms that mediate migration and invasiveness of colon cancer. Our current on-going work is also delving into the effects of 27-OHC on the mechanisms that underlie phenomenon of epithelial-mesenchymal transition which could be as a result of loss of tight junctions and barrier integrity
5. Conclusion
In conclusion, our results suggest that at high concentrations of 27-OHC, similar to those found in conditions such as atherosclerosis, may confer protection by reducing cell proliferation in vitro. To the best of our knowledge, our results are the first to demonstrate such results in Caco2 and SW620 colorectal cell models. We also show that the 27-OHC effects are not associated with cytotoxicity or apoptotic cell death. The decrease in cell proliferation we demonstrate appears to be independent of effects on activation of LXR and ER, the two type receptors to which 27-OHC binds to. Such data suggest that cellular mechanisms other than the genomic pathways downstream to ER and LXR activation is involved in the 27-OHC effects. Further studies are needed to identify these cellular mechanisms to increase our understanding of the potential role of the endogenous cholesterol oxidation product 27-OHC in the etiopathogenesis of CRC.
Supplementary Material
{kind=link}
Highlights:
27-OHC decreases cell proliferation in Colon Cancer cells
27-OHC decreases cell proliferation independently of ER and LXR activation.
27-OHC leads to a decrease in p-AKT in Caco2 cells but not SW620 cells
27-OHC differentially affects colon cancer cells
Article 02: Untitled
Acknowledgements:
The authors acknowledge use of the Imaging and Image Analysis Core Facility which is supported in part by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant numbers P30GM103329 and 5P20GM113123.
Funding sources: This work was supported by a Grant from the National institute of health (R01AG045264) to Dr. Othman Ghribi.
Abbreviations
- 27-OHC
27-hydroxycholesterol
- ABCA1
ATP-binding cassette, sub-family A, member 1
- ABCG1
ATP-binding cassette, sub-family G, member 1
- AF
Afuresertib
- BSA
Bovine Serum Albumin
- COP
cholesterol oxidation products
- CRC
Colorectal Cancer
- DMSO
dimethyl sulfoxide
- ECHS
5α−6α-epoxycholesterol-3-sulfate
- E2
Estradiol
- ER
Estrogen Receptors
- ETOH
Ethanol
- GW
GW3965
- LDH
Lactate dehydrogenase
- LXR
Liver X Receptor
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- SERM
Selective Estrogen Receptor Modulator
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TUNEL
Terminal deoxynucleotidyl transferase dUTP nick end labeling
- Veh
Vehicle
Footnotes
Conflict of interest: The authors do not have any conflicts of interests to report
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Verma M, Kumar V, Epigenetic Biomarkers in Colorectal Cancer, Mol Diagn Ther, 21 (2017) 153–165. [DOI] [PubMed] [Google Scholar]
- [2].Dolatkhah R, Somi MH, Kermani IA, Ghojazadeh M, Jafarabadi MA, Farassati F, Dastgiri S, Increased colorectal cancer incidence in Iran: a systematic review and meta-analysis, BMC Public Health, 15 (2015) 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gandhi J, Davidson C, Hall C, Pearson J, Eglinton T, Wakeman C, Frizelle F, Population-based study demonstrating an increase in colorectal cancer in young patients, British Journal of Surgery, 104 (2017) 1063–1068. [DOI] [PubMed] [Google Scholar]
- [4].Giuseppe C, Antonella V, Role of family history and tumor location on prognosis of patients with colorectal cancer and synchronous metastases, Int J Colorectal Dis, 32 (2017) 1069–1072. [DOI] [PubMed] [Google Scholar]
- [5].Wang Y-M, Zhou Q-Y, Zhu J-Z, Zhu K-F, Yu C-H, Li Y-M, Systematic Review with Meta-Analysis: Alcohol Consumption and Risk of Colorectal Serrated Polyp, Dig. Dis. Sci, 60 (2015) 1889–1902. [DOI] [PubMed] [Google Scholar]
- [6].Chan DSM, Lau R, Aune D, Vieira R, Greenwood DC, Kampman E, Norat T, Red and processed meat and colorectal cancer incidence: meta-analysis of prospective studies, PLoS ONE, 6 (2011) e20456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Park Y, Lee J, Oh JH, Shin A, Kim J, Dietary patterns and colorectal cancer risk in a Korean population: A case-control study, Medicine (Baltimore), 95 (2016) e3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jewel Samadder N, Valentine JF, Guthery S, Singh H, Bernstein CN, Wan Y, Wong J, Boucher K, Pappas L, Rowe K, Bronner M, Ulrich CM, Burt RW, Curtin K, Smith KR, Colorectal Cancer in Inflammatory Bowel Diseases: A Population-Based Study in Utah, Dig. Dis. Sci, 62 (2017) 2126–2132. [DOI] [PubMed] [Google Scholar]
- [9].Shafique K, McLoone P, Qureshi K, Leung H, Hart C, Morrison DS, Cholesterol and the risk of grade-specific prostate cancer incidence: evidence from two large prospective cohort studies with up to 37 years’ follow up, BMC Cancer, 12 (2012) 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jacobs EJ, Newton CC, Thun MJ, Gapstur SM, Long-term Use of Cholesterol-Lowering Drugs and Cancer Incidence in a Large United States Cohort, Cancer Research, 71 (2011) 1763–1771. [DOI] [PubMed] [Google Scholar]
- [11].Cardwell CR, Hicks BM, Hughes C, Murray LJ, Statin use after colorectal cancer diagnosis and survival: a population-based cohort study, J. Clin. Oncol, 32 (2014) 3177–3183. [DOI] [PubMed] [Google Scholar]
- [12].Liu Y, Tang W, Wang J, Xie L, Li T, He Y, Deng Y, Peng Q, Li S, Qin X, Association between statin use and colorectal cancer risk: a meta-analysis of 42 studies, Cancer Causes Control, 25 (2014) 237–249. [DOI] [PubMed] [Google Scholar]
- [13].Mamtani R, Lewis JD, Scott FI, Ahmad T, Goldberg DS, Datta J, Yang Y-X, Boursi B, Disentangling the Association between Statins, Cholesterol, and Colorectal Cancer: A Nested Case-Control Study, PLOS Medicine, 13 (2016) e1002007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Brown AJ, Jessup W, Oxysterols and atherosclerosis, Atherosclerosis, 142 (1999) 1–28. [DOI] [PubMed] [Google Scholar]
- [15].Kulig W, Cwiklik L, Jurkiewicz P, Rog T, Vattulainen I, Cholesterol oxidation products and their biological importance, Chemistry and Physics of Lipids, 199 (2016) 144–160. [DOI] [PubMed] [Google Scholar]
- [16].Olkkonen VM, Hynynen R, Interactions of oxysterols with membranes and proteins, Molecular Aspects of Medicine, 30 (2009) 123–133. [DOI] [PubMed] [Google Scholar]
- [17].Aupeix K, Weltin D, Mejia JE, Christ M, Marchal J, Freyssinet JM, Bischoff P, Oxysterol-induced apoptosis in human monocytic cell lines, Immunobiology, 194 (1995) 415–428. [DOI] [PubMed] [Google Scholar]
- [18].Salvayre R, Auge N, Benoist H, Negre-Salvayre A, Oxidized low-density lipoprotein-induced apoptosis, Biochim. Biophys. Acta, 1585 (2002) 213–221. [DOI] [PubMed] [Google Scholar]
- [19].Bauman DR, Bitmansour AD, McDonald JG, Thompson BM, Liang G, Russell DW, 25-Hydroxycholesterol secreted by macrophages in response to Toll-like receptor activation suppresses immunoglobulin A production, Proc. Natl. Acad. Sci. U.S.A, 106 (2009) 16764–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Civra A, Cagno V, Donalisio M, Biasi F, Leonarduzzi G, Poli G, Lembo D, Inhibition of pathogenic non-enveloped viruses by 25-hydroxycholesterol and 27-hydroxycholesterol, Scientific Reports, 4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Reboldi A, Dang EV, McDonald JG, Liang G, Russell DW, Cyster JG, Inflammation. 25-Hydroxycholesterol suppresses interleukin-1-driven inflammation downstream of type I interferon, Science, 345 (2014) 679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Duane WC, Javitt NB, 27-hydroxycholesterol: production rates in normal human subjects, J. Lipid Res, 40 (1999) 1194–1199. [PubMed] [Google Scholar]
- [23].Kim S-M, Jang H, Son Y, Lee S-A, Bae S-S, Park YC, Eo S-K, Kim K, 27-hydroxycholesterol induces production of tumor necrosis factor-alpha from macrophages, Biochem. Biophys. Res. Commun, 430 (2013) 454–459. [DOI] [PubMed] [Google Scholar]
- [24].Brooks SW, Dykes AC, Schreurs BG, A High-Cholesterol Diet Increases 27-Hydroxycholesterol and Modifies Estrogen Receptor Expression and Neurodegeneration in Rabbit Hippocampus, J. Alzheimers Dis, 56 (2017) 185–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kushwaha RS, Guntupalli B, Rice KS, Carey KD, McGill HC, Effect of Dietary Cholesterol and Fat on the Expression of Hepatic Sterol 27-Hydroxylase and Other Hepatic Cholesterol-Responsive Genes in Baboons (Papio Species), Arteriosclerosis, Thrombosis, and Vascular Biology, 15 (1995) 1404–1411. [DOI] [PubMed] [Google Scholar]
- [26].Nelson ER, DuSell CD, Wang X, Howe MK, Evans G, Michalek RD, Umetani M, Rathmell JC, Khosla S, Gesty-Palmer D, McDonnell DP, The Oxysterol, 27-Hydroxycholesterol, Links Cholesterol Metabolism to Bone Homeostasis Through Its Actions on the Estrogen and Liver X Receptors, Endocrinology, 152 (2011) 4691–4705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V, Umetani M, Geradts J, McDonnell DP, 27-Hydroxycholesterol Links Hypercholesterolemia and Breast Cancer Pathophysiology, Science, 342 (2013) 1094–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Takeshi H, Yuji M, Akira H, Yasuhiko H, Tadashi I, Yoshifumi S, Teruo M, Yasushi M, Serum concentration of 27‐hydroxycholesterol predicts the effects of high‐cholesterol diet on plasma LDL cholesterol level, Hepatology Research, 39 (2009) 149–156. [DOI] [PubMed] [Google Scholar]
- [29].Xiaona Z, Chenyan L, Yu A, Quanri L, Hongguo R, Lingwei T, Ying W, Yushan W, Rong X, Increased Levels of 27-Hydroxycholesterol Induced by Dietary Cholesterol in Brain Contribute to Learning and Memory Impairment in Rats, Molecular Nutrition & Food Research, 62 (2018) 1700531. [DOI] [PubMed] [Google Scholar]
- [30].Fu X, Menke JG, Chen Y, Zhou G, MacNaul KL, Wright SD, Sparrow CP, Lund EG, 27-Hydroxycholesterol Is an Endogenous Ligand for Liver X Receptor in Cholesterol-loaded Cells, Journal of Biological Chemistry, 276 (2001) 38378–38387. [DOI] [PubMed] [Google Scholar]
- [31].DuSell CD, Umetani M, Shaul PW, Mangelsdorf DJ, McDonnell DP, 27-Hydroxycholesterol Is an Endogenous Selective Estrogen Receptor Modulator, Molecular Endocrinology, 22 (2008) 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Umetani M, Domoto H, Gormley AK, Yuhanna IS, Cummins CL, Javitt NB, Korach KS, Shaul PW, Mangelsdorf DJ, 27-Hydroxycholesterol is an endogenous SERM that inhibits the cardiovascular effects of estrogen, Nature Medicine, 13 (2007) 1185–1192. [DOI] [PubMed] [Google Scholar]
- [33].Umetani M, Shaul PW, 27-Hydroxycholesterol: the first identified endogenous SERM, Trends in Endocrinology & Metabolism, 22 (2011) 130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Raza S, Meyer M, Goodyear C, Hammer KDP, Guo B, Ghribi O, The cholesterol metabolite 27-hydroxycholesterol stimulates cell proliferation via ERβ in prostate cancer cells, Cancer Cell Int., 17 (2017) 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Raza S, Meyer M, Schommer J, Hammer KDP, Guo B, Ghribi O, 27-Hydroxycholesterol stimulates cell proliferation and resistance to docetaxel-induced apoptosis in prostate epithelial cells, Med. Oncol, 33 (2016) 12. [DOI] [PubMed] [Google Scholar]
- [36].Yamaji M, Ota A, Wahiduzzaman M, Karnan S, Hyodo T, Konishi H, Tsuzuki S, Hosokawa Y, Haniuda M, Novel ATP-competitive Akt inhibitor afuresertib suppresses the proliferation of malignant pleural mesothelioma cells, Cancer Medicine, 6 (2017) 2646–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Nitulescu GM, Margina D, Juzenas P, Peng Q, Olaru OT, Saloustros E, Fenga C, Spandidos DA, Libra M, Tsatsakis AM, Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review), International Journal of Oncology, 48 (2016) 869–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Hashimoto Y, Sugiura H, Togo S, Koarai A, Abe K, Yamada M, Ichikawa T, Kikuchi T, Numakura T, Onodera K, Tanaka R, Sato K, Yanagisawa S, Okazaki T, Tamada T, Kikuchi T, Hoshikawa Y, Okada Y, Ichinose M, 27-Hydroxycholesterol accelerates cellular senescence in human lung resident cells, American Journal of Physiology-Lung Cellular and Molecular Physiology, 310 (2016) L1028–L1041. [DOI] [PubMed] [Google Scholar]
- [39].Raza S, Ohm JE, Dhasarathy A, Schommer J, Roche C, Hammer KDP, Ghribi O, The cholesterol metabolite 27-hydroxycholesterol regulates p53 activity and increases cell proliferation via MDM2 in breast cancer cells, Mol. Cell. Biochem, 410 (2015) 187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Umetani M, Ghosh P, Ishikawa T, Umetani J, Ahmed M, Mineo C, Philip W Shaul, The Cholesterol Metabolite 27-Hydroxycholesterol Promotes Atherosclerosis via Proinflammatory Processes Mediated by Estrogen Receptor Alpha, Cell Metabolism, 20 (2014) 172–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lütjohann D, Papassotiropoulos A, Björkhem I, Locatelli S, Bagli M, Oehring RD, Schlegel U, Jessen F, Rao ML, von Bergmann K, Heun R, Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients, J. Lipid Res, 41 (2000) 195–198. [PubMed] [Google Scholar]
- [42].Mateos L, Ismail M-A-M, Gil-Bea F-J, Leoni V, Winblad B, Björkhem I, Cedazo-Mínguez A, Upregulation of brain renin angiotensin system by 27-hydroxycholesterol in Alzheimer’s disease, J. Alzheimers Dis, 24 (2011) 669–679. [DOI] [PubMed] [Google Scholar]
- [43].Leoni V, Long JD, Mills JA, Di Donato S, Paulsen JS, P.-H.s. group, Plasma 24S-hydroxycholesterol correlation with markers of Huntington disease progression, Neurobiology of Disease, 55 (2013) 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Roussi S, Winter A, Gosse F, Werner D, Zhang X, Marchioni E, Geoffroy P, Miesch M, Raul F, Different apoptotic mechanisms are involved in the antiproliferative effects of 7beta-hydroxysitosterol and 7beta-hydroxycholesterol in human colon cancer cells, Cell Death Differ., 12 (2005) 128–135. [DOI] [PubMed] [Google Scholar]
- [45].Bai B, Yamamoto K, Sato H, Sugiura H, Tanaka T, Combined effect of 25-hydroxycholesterol and IL-1beta on IL-8 production in human colon carcinoma cell line (Caco-2), Inflammation, 29 (2005) 141–146. [DOI] [PubMed] [Google Scholar]
- [46].Park K, Scott AL, Cholesterol 25-hydroxylase production by dendritic cells and macrophages is regulated by type I interferons, J. Leukoc. Biol, 88 (2010) 1081–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Barzi A, Lenz AM, Labonte MJ, Lenz HJ, Molecular Pathways: Estrogen Pathway in Colorectal Cancer, Clinical Cancer Research, 19 (2013) 5842–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Hartman J, Edvardsson K, Lindberg K, Zhao C, Williams C, Ström A, Gustafsson J-A, Tumor repressive functions of estrogen receptor beta in SW480 colon cancer cells, Cancer Research, 69 (2009) 6100–6106. [DOI] [PubMed] [Google Scholar]
- [49].Lo Sasso G, Bovenga F, Murzilli S, Salvatore L, Di Tullio G, Martelli N, D’Orazio A, Rainaldi S, Vacca M, Mangia A, Palasciano G, Moschetta A, Liver X receptors inhibit proliferation of human colorectal cancer cells and growth of intestinal tumors in mice, Gastroenterology, 144 (2013) 1497–1507, 1507.e1491-1413. [DOI] [PubMed] [Google Scholar]
- [50].Vedin L-L, Gustafsson J-Å, Steffensen KR, The oxysterol receptors lxrα and lxrβ suppress proliferation in the colon: LXRs INHIBIT PROLIFERATION IN COLON CELLS, Molecular Carcinogenesis, 52 (2013) 835–844. [DOI] [PubMed] [Google Scholar]
- [51].English MA, Kane KF, Cruickshank N, Langman MJS, Stewart PM, Hewison M, Loss of Estrogen Inactivation in Colonic Cancer, The Journal of Clinical Endocrinology & Metabolism, 84 (1999) 2080–2085. [DOI] [PubMed] [Google Scholar]
- [52].Gibson DA, Collins F, Cousins FL, Esnal Zufiaurre A, Saunders PT, The impact of 27-hydroxycholesterol on endometrial cancer proliferation, Endocr. Relat. Cancer, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Sharma S, J. Prasanthi RP, Schommer E, Feist G, Ghribi O, Hypercholesterolemia-induced Aβ accumulation in rabbit brain is associated with alteration in IGF-1 signaling, Neurobiology of Disease, 32 (2008) 426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Ismail M-A-M, Mateos L, Maioli S, Merino-Serrais P, Ali Z, Lodeiro M, Westman E, Leitersdorf E, Gulyás B, Olof-Wahlund L, Winblad B, Savitcheva I, Björkhem I, Cedazo-Mínguez A, 27-Hydroxycholesterol impairs neuronal glucose uptake through an IRAP/GLUT4 system dysregulation, The Journal of Experimental Medicine, (2017) jem.20160534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Kim S-M, Kim B-Y, Lee S-A, Eo S-K, Yun Y, Kim C-D, Kim K, 27-Hydroxycholesterol and 7alpha-hydroxycholesterol trigger a sequence of events leading to migration of CCR5-expressing Th1 lymphocytes, Toxicology and Applied Pharmacology, 274 (2014) 462–470. [DOI] [PubMed] [Google Scholar]
- [56].Vurusaner B, Gamba P, Testa G, Gargiulo S, Biasi F, Zerbinati C, Iuliano L, Leonarduzzi G, Basaga H, Poli G, Survival signaling elicited by 27-hydroxycholesterol through the combined modulation of cellular redox state and ERK/Akt phosphorylation, Free Radical Biology and Medicine, 77 (2014) 376–385. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.