

Abstract
The cellular functions of RNA are not limited to their role as blueprints for protein synthesis. In particular, noncoding RNA, such as, snRNAs, lncRNAs, miRNAs, play important roles. With increasing numbers of RNAs being identified, it is well known that the transcriptome outnumbers the proteome by far. This emphasizes the great importance of functional RNA characterization and the need to further develop tools for these investigations, many of which are still in their infancy. Fluorescent light‐up aptamers (FLAPs) are RNA sequences that can bind nontoxic, cell‐permeable small‐molecule fluorogens and enhance their fluorescence over many orders of magnitude upon binding. FLAPs can be encoded on the DNA level using standard molecular biology tools and are subsequently transcribed into RNA by the cellular machinery, so that they can be used as fluorescent RNA tags (FLAP‐tags). In this Minireview, we give a brief overview of the fluorogens that have been developed and their binding RNA aptamers, with a special focus on published crystal structures. A summary of current and future cellular FLAP applications with an emphasis on the study of RNA–RNA and RNA–protein interactions using split‐FLAP and Förster resonance energy transfer (FRET) systems is given.
Keywords: fluorescent light-up aptamers (FLAPs), fluorogenic probes, RNA structures, RNA imaging, RNA–protein interactions
1. Introduction
RNA has been recognized as a major player in cellular function for decades now; however, robust methods to directly measure and understand the different functions of RNA in cells are still under development.1, 2, 3, 4 RNAs are in constant dynamic interaction with other biomolecules such as proteins5, 6, 7, 8, 9, 10, 11 and other RNAs (e.g., snRNAs, lncRNAs, miRNAs),12, 13, 14 thereby significantly altering cellular function and cell fate.15, 16, 17, 18 State‐of‐the‐art methods to detect an RNA of interest (ROI) and its cellular interactions include fluorescence in situ hybridization (FISH)19, 20 and the MS2‐green fluorescent protein (MS2‐GFP) system where the interaction between an RNA derived from enterobacteria phage MS2 and the MS2 coat protein fused to GFP is used to detect an ROI.21, 22, 23, 24 Although FISH enables the detection of single RNA molecules in the cell, it involves fixation and therefore only static measurements of the ROI and its interactions.25 By contrast, the MS2‐GFP system allows spatiotemporal measurements,26 but it generates high background fluorescence and depends on large modifications that could interfere with the biological function of the ROI.23, 27
The use of GFP as a genetically encodable reporting system for proteins in cells has revolutionized the field of protein biochemistry and cellular biology.28, 29, 30, 31, 32, 33 As an intrinsically fluorescent protein that is encodable on the DNA level and therefore can be produced by the biochemical machinery of the cell itself, it has inspired and driven the development of genetically encodable fluorescent reporters on the RNA level. Since intrinsically fluorescent RNAs are not known, the fluorescent reporter instead consists of a cell‐permeable small‐molecule fluorogenic dye (fluorogen) specifically bound to an RNA structure (Figure 1 A).34 Once the RNA is transcribed and folded into the correct conformation, it binds the fluorogen and enhances its intrinsically very low fluorescence over many orders of magnitude even in living cells (Figure 1 B).35, 36 Hereafter, we call these RNA aptamers fluorescent light‐up aptamers (FLAPs). FLAPs are significantly smaller than localization tools such as the MS2‐GFP system and should have a lower propensity to interfere with cellular functions. Herein, we will review FLAPs that have been successfully utilized in the cell. A brief summary of the fluorogens that have been developed and the corresponding selected RNA aptamers, with a special focus on available crystal structures, will be given. We will then highlight the current and potential future cellular applications of FLAPs with emphasis on the study of RNA–RNA and RNA–protein interactions using split‐FLAP and Förster resonance energy transfer (FRET) systems.
Figure 1.
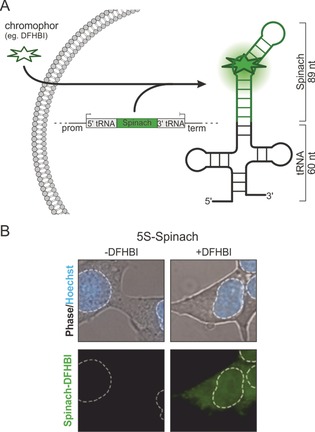
Schematic overview of FLAP. A) A DNA construct carrying the genetic information for an RNA scaffolding structure (tRNA) and the aptamer (Spinach in green) is transferred into a cell and transcribed into RNA by the cellular transcriptional machinery. A small molecule fluorogen, which is highly cell permeable and non‐toxic, is applied and binds to the aptamer RNA. This leads to a drastic increase in fluorescence. We refer to these systems as fluorescence light‐up aptamers (FLAPs). B) Spinach, the first non‐toxic FLAP, was fused to the 5S rRNA and expressed in HEK293T cells. Images were taken without and after the addition of DFHBI fluorogen and monitored. An overlay of phase contrast and nuclear staining using Hoechst shows the location of the cell and its nucleus (dashed white line). Image modified from Paige et al.36
2. Development of FLAPs
The first fluorogen found to have enhanced fluorescence upon binding to an RNA aptamer was the triphenylmethane dye malachite green (MG), which was reported in 2003 by Tsien and co‐workers.35 However, since MG has been shown to produce reactive oxygen species under irradiation,37, 38 other fluorogens have been developed. Optimal fluorogens should be cell permeable and nontoxic and should have low to no intrinsic and unspecific fluorescence in the cell to minimize background and off‐target fluorescence. The binding affinity of the fluorogen to its aptamer is crucial since this step results in bright fluorescence due to confinement in the conformational state in which fluorescence is the major pathway for excited‐state relaxation (conditional fluorophore).36 Currently used fluorogens are mainly derived from the fluorophore moiety, 4‐hydroxybenzlidene imidazolinone (HBI), found in (e)GFP (Figure 2 A), but also include asymmetrical cyanine dyes (Figure 2 B) and fluorophore–quencher conjugates.
Figure 2.

Fluorogens for FLAPs. A) GFP‐derived fluorogens: 3,5‐dimethoxy‐4‐hydroxybenzylidene imidazolinone (DMHBI), 3,5‐difluoro‐4‐hydroxybenzylidene imidazolinone (DFHBI), (Z)‐4‐(3,5‐difluoro‐4‐hydroxybenzylidene)‐2‐methyl‐1‐(2,2,2‐trifluoroethyl)‐1H‐imidazol‐5(4H)‐one) (DFHBI‐1T), and 3,5‐difluoro‐4‐hydroxybenzyli‐dene‐imidazolinone‐2‐oxime (DFHO). B) Asymmetrical cyanine dyes: biotin‐modified thiazol orange derivatives (TO1‐biotin and TO3‐biotin), an oxazole yellow derivative (YO3‐biotin), a dimethylindole red analogue (DIR‐Pro), and an oxazole thiazole blue analogue (OTB‐SO3). C) Fluorophore‐quencher conjugates: sulforhodamine–dinitroaniline (SR‐DN).
The associated fluorogenic aptamers are selected in vitro from a large pool of random RNA sequences through a process termed systematic evolution of ligands by exponential enrichment (SELEX).39, 40 They are single‐stranded RNAs that bind the fluorogen with high selectivity and affinity based on adaptive recognition.35, 41, 42 A fundamental issue with in vitro SELEX is the artificial environment during the selection process resulting in aptamers that often show divergent properties in the cellular environment (e.g., decreased fluorescence due to incorrect folding).43, 44, 45 Therefore, further optimization by directed evolution or rational design based on the crystal structure, if available, is usually necessary.46 In addition, FLAPs are expressed on an aptamer scaffold (e.g., tRNALys 3, F30, Figure 1) for efficient transcription and folding.47, 48 More recently, strategies have been developed to select aptamers under more cell‐like conditions45 to minimize alternative non‐fluorescent RNA conformations and improve folding of the fluorescent aptamer in the cell and for fluorescence brightness49, 50, 51 rather than solely fluorogen affinity.52
2.1. FLAPs for GFP‐derived Fluorogens
In 2011, Jaffrey and co‐workers reported the first GFP‐derived fluorogens, 3,5‐dimethoxy‐4‐hydroxybenzylidene imidazolinone (DMHBI) and (Z)‐4‐(3,5‐difluoro‐4‐hydroxybenzylidene)‐1,2‐dimethyl‐1H‐imidazol‐5(4H)‐one (DFHBI), which is present in its phenolate form (Figure 2 A).36 A 98‐nt FLAP, termed Spinach (Figure 2 A, 3) was selected for DFHBI and showed roughly 50 % of the fluorescent brightness of eGFP when bound to the fluorogen (Figure 3 and Table 1).36, 61 It was successfully tagged to 5S ribosomal RNA (rRNA), which could be visualized in the presence of DFHBI in HEK293T cells in real time (Figure 1 B) as granules or accumulated in the nucleus.36, 46 Further development of the FLAP to Spinach2 (96 nt) in 2013 using systematic mutagenesis led to increased thermal stability in living cells.44 Spinach2 was used to localize CGG RNA repeats in COS‐7 cells in the presence of DFHBI.44 The same researchers continued pioneering GFP‐derived fluorogens. The derivate (Z)‐4‐(3,5‐difluoro‐4‐hydroxybenzylidene)‐2‐methyl‐1‐(2,2,2‐trifluoroethyl)‐1H‐imi‐dazol‐5(4H)‐one) (DFHBI‐1T) bears a 1,1,1‐trifluoroethyl substituent at the N1 position of the imidazolone ring (Figure 2 A).56 Bound to Spinach2, the resulting FLAP showed enhanced brightness due to the lower background fluorescence of the unbound fluorogen and increased fluorescence of the complex, leading to approximately 88 % brightness of eGFP (Figure 3 and Table 1).56
Figure 3.
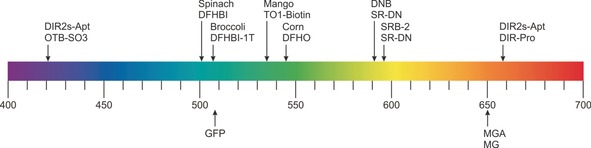
Overview of available FLAPs. The spectral range of emission wavelengths (arrows) of various FLAPs and their fluorogens.
Table 1.
Overview of spectral characteristics and fluorogen–aptamer affinities of FLAPs compared to the fluorescent proteins GFP and eGFP.
| Aptamer | Fluorogen | K D [nm] | λ ex [nm] | λ em [nm] | ϵ [a] [M−1 cm−1] | Φ [b] | Brightness[c] | Length [nt][d] | QYI [e] | Publication year | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| GFP | N/A | 395 | 508 | 27 600 | 0.79 | 21 800 | N/A | N/A | 1994 | 36, 53 | |
| eGFP | N/A | 489 | 508 | 55 000 | 0.60 | 33 000 | N/A | N/A | 1996 | 36, 54, 55 | |
| MGA | MG | 117 | 630 | 650 | 15 0000 | 0.19 | 28 500 | 38 | 2360 | 2003 | 35 |
| Spinach | DFHBI | 537 | 469 | 501 | 24 300 | 0.72 | 17 500 | 98 | 1030 | 2011 | 36 |
| Spinach2 | DFHBI | 430 | 454 | 498 | 26 100 | 0.70 | 18 300 | 95 | 1000 | 2013 | 44 |
| Spinach2 | DFHBI‐1T | 560 | 482 | 505 | 31 000 | 0.94 | 29 100 | 95 | 940 | 2014 | 56 |
| Broccoli | DFHBI‐1T | 360 | 472 | 507 | 29 600 | 0.94 | 27 800 | 49 | 940 | 2014 | 45 |
| Corn | DFHO | 70 | 505 | 545 | 29 000 | 0.25 | 7300 | 2×36 | 420 | 2017 | 52 |
| Mango | TO1‐Biotin | 3 | 510 | 535 | 77 500 | 0.14 | 10 900 | 29 | 700 | 2014 | 57 |
| DIR2s‐Apt | DIR‐Pro | 252 | 600 | 658 | 16 4000 | 0.33 | 54 100 | 57 | N/A | 2017 | 58 |
| DIR2s‐Apt | OTB‐SO3 | 662 | 380 | 421 | 73 000 | 0.51 | 37 200 | 57 | N/A | 2017 | 58 |
| SRB‐2 | SR‐DN | 1400 | 579 | 596 | N/A | 0.65 | N/A | 54 | 24 | 2013 | 59 |
| DNB | SR‐DN | 800 | 572 | 591 | 50 300 | 0.98 | 49 300 | 75 | 36 | 2015 | 60 |
[a] Extinction coefficient (ϵ). [b] Quantum yield (Φ). [c] Brightness=ϵ⋅Φ. [d] Length (nt) of the core aptamer sequence. [e] Quantum yield increase (QYI)=Φ bound/ Φ free.
In 2014, the co‐crystal structure of Spinach–DFHBI was solved, which facilitated a rational optimization of the aptamer.62 While a core structure was identified, in which mutations lead to dramatic fluorescence reduction, the flanking sequences could be readily substituted by shorter stabilizing A‐form duplexes. This resulted in the optimized 51‐nt FLAP, termed Baby Spinach, which was used to tag and image 16S rRNA in E. coli.62, 63 Another variant, termed iSpinach (69 nt), was optimized by Ryckelynck and co‐workers in 2016 using random mutagenesis with microfluidic‐assisted compartmentalization for in vitro applications.49, 64
In an effort to make the selection of FLAPs more efficient, Jaffrey and co‐workers integrated fluorescence enhancement into their SELEX process and used fluorescence‐activated cell sorting (FACS) with E. coli cells.45 This selection method resulted in Broccoli (49 nt) in 2014, which showed superior properties for applications in cells (no requirement for high‐magnesium medium or a tRNA scaffold) and around 84 % of eGFP brightness with DFHBI‐1T (Figure 3 and Table 1). To gain more brightness (almost 2‐fold), two Broccoli sequences were fused together (dimeric Broccoli, dBroccoli, 92 nt). Both Broccoli and dBroccoli were used to tag 5S rRNA in HEK293T cells.
The newest member of the FLAP family is Corn (36 nt) and was selected after several rounds of SELEX, directed evolution, and additional selection in E. coli through FACS by Jaffrey and co‐workers in 2017.52 The fluorogen 3,5‐difluoro‐4‐hydroxybenzylidene‐imidazolinone‐2‐oxime (DFHO, Figure 2 A) is an analogue of the intrinsic fluorescent moiety in red fluorescent protein (RFP).56 It was anticipated that the N‐hydroxyl imine substituent at the 2‐position of the imidazolinone ring would hinder cis–trans isomerization and thus photobleaching, which is often observed with DFHBI or DFHBI‐T1 in their complexes with Spinach and Broccoli, respectively. Although Corn–DFHO has only 22 % eGFP brightness (Figure 3 and Table 1), it indeed showed improved photostability properties compared with dBroccoli–DFHBI in fusion with U6 RNA in HEK293T cells. It was used to perform quantitative measurements of polymerase III transcription in HEK293T cells.52
2.2. FLAPs for Cyanine Fluorogens
In an effort to produce a superior FLAP that combines high affinity and brightness to analyze low‐abundant ROIs (Table 1), Unrau and co‐workers performed a completely new SELEX against a biotin‐modified thiazol orange derivative (TO1‐biotin, Figure 2 B) in 2014.57 RNA Mango was selected as a FLAP with a K D value of 3.2 nm and was found to also bind TO3‐biotin (where the methylchinoline (MQ) and the benzothiazole (BzT) aromatic moieties are connected by a three‐ instead of a one‐carbon linker in TO1‐biotin) with a slightly lower affinity (K D≈7 nm). The 29‐nt core sequence of RNA Mango was tagged to E. coli S6 RNA, and its function tested through in vitro binding studies to RNA polymerase. In 2018, the selection round in which Mango was found was rescreened applying a competitive ligand‐binding microfluidic technique. New Mango derivatives had improved fluorescent properties and binding affinities and were used to localize U6 snRNA, 5S rRNA, and the box C/D scaRNA in live mammalian cells.51
In 2017, dimethylindole red and oxazole thiazole blue derivatives were revisited by Armitage and co‐workers as fluorogens for FLAPs to emit fluorescence at opposite wavelengths.58, 65 A promiscuous FLAP was selected that binds both dimethylindole red (DIR‐Pro) and oxazole thiazole blue (OTB‐SO3, Figure 2 B), each producing very different emission wavelengths.58, 65 Both complexes exceed eGFP in brightness (164 % and 113 %, respectively, Figure 3 and Table 1) and have good photostability properties. DIRs‐Apt was fused to an aptamer binding to the epidermal growth factor receptor (EGFR), applied extracellularly, and finally used to visualize mammalian cells.58
2.3. FLAPS for Fluorophore–Quencher Conjugates
Another type of small molecule that has found application as a fluorogen for FLAPs in cells are fluorophore–quencher conjugates. The fluorescence is internally quenched through contact of the two moieties to produce a fluorogenic dye. Although they are rather big, these turn‐on probes present a potential strategy to vary excitation and emission wavelengths of the aptamer–fluorogen complex. In a particular example, dinitroanilin is attached via a triethylene glycol linker to sulforhodamine (Figure 2 C).59, 60 Initially in 2013, a RNA aptamer was selected for the sulforhodamine moiety of the sulforhodamine–dinitroaniline (SR‐DN) conjugate (Figure 2 C). Despite its weak affinity (K D=1.2 μm), a 100‐fold fluorescence increase was measured in vitro upon binding to the aptamer SRB‐2. This effect might come from the chosen experimental settings in vitro, however, this FLAP was also successfully utilized in vivo for imaging in E. coli.59 In 2015, a new selection was performed against the dinitroaniline moiety of the SR‐DN conjugate. The resulting FLAP was again tested and visualized in E. coli and exceeded eGFP brightness by around 50 % (Figure 3 and Table 1).60
3. 3D Crystal Structures of Spinach, Mango, and Corn
Crystallization of RNA molecules is known within the structural community to be challenging for several reasons.66 Nevertheless three‐dimensional (3D) structures of RNA molecules with atomic resolution are one of the main ways to gain in‐depth insight into these highly flexible molecules. For FLAPs, obtaining crystal structures has had a huge impact because it has resolved the exact coordination of their fluorogens as well as their overall conformation. The first crystal structure of a FLAP was achieved with the malachite‐green aptamer (MGA) in complex with tetramethylrhosamine.67 Until now, only three additional FLAP structures (plus their variations) have been solved, namely those of Spinach, Mango, and Corn (Table 2).
Table 2.
Overview of available FLAP crystal structures.
| Aptamer | Fluorogen | Res. [Å][a] | Year | PDB ID | Ref. |
|---|---|---|---|---|---|
| MGA | TMR | 2.80 | 2000 | 1f1t | 67 |
| Spinach | DFHBI | 2.19 | 2014 | 4kzd | 68 |
| Spinach | apo | 2.40 | 2014 | 4kze | 68 |
| Spinach | BrBI | 2.45 | 2014 | 4q9q | 68 |
| Spinach | DFHBI‐1T | 3.12 | 2014 | 4q9r | 68 |
| Spinach | DFHBI & Ba‐ions | 2.80 | 2014 | 4ts0 | 62 |
| Spinach | DFHBI | 2.80 | 2014 | 4ts2 | 62 |
| iSpinach | DFHBI | 2.00 | 2017 | 5ob3 | 64 |
| Mango | TO1‐Biotin & Ir‐ions | 1.70 | 2017 | 5c3f | 70 |
| Corn | DFHO & Ir‐ions | 2.51 | 2017 | 5bjp | 71 |
| Corn | DFHO | 2.35 | 2017 | 5bjo | 71 |
| Spinach | apo | 2.09 | 2018 | 6b3k | 66 |
| Spinach | apo | 1.64 | 2018 | 6b14 | 66 |
3.1. Strategies for Spinach Crystallization
In 2014, two independent groups solved the X‐ray structure of Spinach using two different strategies. Huang et al. inserted a 7‐nt recognition motif at the tip of a flexible loop to bind the aptamer to an auxiliary Fab BL3‐6 antibody fragment (Figure 4 B).68 Fab BL3‐6 has been used before as a crystallization chaperone to assist RNA crystallization and served as a molecular replacement model for solving the final structures.69 Interestingly, Fab BL3‐6 builds up to 99 % of the buried surface area within the crystal lattice, resulting in a RNA aptamer structure that is less influenced by the crystal packing itself. The same research group was able to show that a improved Fab3‐6 protein binding resulted in an even better crystal structure with the so far highest resolution of 1.64 Å (Table 2).66 Warner et al. independently solved a Spinach structure using no assisting protein chaperone.62 Here, the initial published Spinach sequence was split into two halves at a flexible loop (L3‐loop), resulting in two hybridizing Spinach RNA strands (Figure 4 C). These were synthesized individually and subsequently hybridized. Throughout the RNA strand design, hybridizing stem regions P1 and P3 were stabilized by rational point mutations resulting in stronger G:C base pairing (Figure 5 A,B). For the final structure determination, a three‐step process was needed: Initial phase information for solving the structures was obtained by BaCl2 co‐crystallization and subsequent SAD‐phasing. Subsequently, the register of the RNA sequence was checked by the site‐specific incorporation of 5‐iodouracil and its resulting anomalous signal. Finally, the anomalous signals of the co‐crystallized bromo‐substituted DFHBI derivative (DBrHBI) confirmed the orientation of the bound fluorogen.
Figure 4.

Comparison of spinach structures. A) Secondary‐structure prediction by mfold using default settings for RNA.100 The previously reported36 secondary structure for spinach could be reproduced as top third solution (ΔG=−35.50 kcal mol−1). B–D) Spinach X‐ray crystal structure variants. B) Fab BL3‐6 assisted approach, PDB ID: 4kzd.62 C) Split hybridization approach, PDB ID: 4ts0.68 D) iSpinach, PDB ID: 5ob3.64 Dotted line indicates loop used for separation of the two strands during construct design. Red region shows 7‐nt Fab BL3‐6 recognition motif. Green=bound DFHBI fluorogen.
Figure 5.
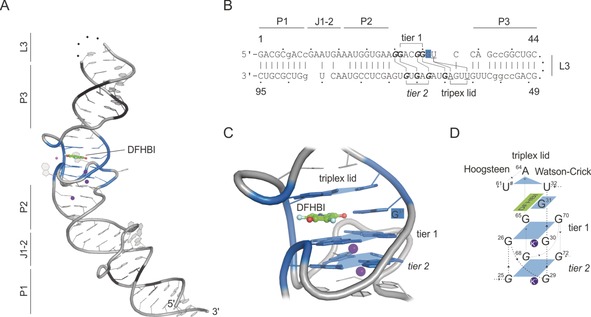
Spinach–DFHBI crystal structure (PDB ID: 4ts2).62 A) Crystal structure of Spinach; names of structural features are indicated. Black=substituted nucleotides for stabilization through Watson–Crick base pairs; Green=DFHBI, pink and purple spheres=Mg2+ and K+ ions; blue colored region=DFHBI‐binding region including G‐quadruplex; dotted line=cleavage site for crystal construct design (L3 loop region). B) Sequence of the crystallized Spinach sequence by Warner et al.62 Lower case=substituted nucleotides for stabilization through Watson–Crick base pairs; bold=G‐quadruplex forming nucleotides; underlined=triplex‐lid‐forming nucleotides. C) Close‐up view of the fluorogen‐binding region. G‐quadruplex tiers are shown, and shaped platforms are indicated by blue planes. Each plane coordinates a K+ ion (purple). DFHBI is shown in addition to its coordinating G31. The closing triplex lid is highlighted on top of DFHBI. D) Schematic overview of Spinach–DFHBI fluorogen‐binding region. Numbers indicate the nucleotide numbers from Spinach according to (B).
Fernandez‐Millan and colleagues were able to minimize the length and improve the folding and stability, thereby its spectral characteristics. The new aptamer was called iSpinach, and its structure could be solved without cleavage, sequence insertions or auxiliary protein binders (Figure 4 D).49, 64
3.2. Comparison of Spinach Crystal Structures
All solved spinach structures resulted in an overall very similar elongated fold not foreseen in secondary‐structure predictions (Figure 4).36 It is comprised of a double‐stranded basal stem, harboring the 5′‐ and 3′‐ends, the fluorogen‐binding region, and the apical stem region. The basal and apical stems are crucial for proper overall folding of the aptamer and positioning and rigidification of the fluorogen‐binding site (Figure 4 A,B). The more hydrogen bridges that are formed in the stems, the more rigidly the core in between the basal and the apical stem can host the fluorogen. To enhance folding and stability, the general strategy within these regions is to strengthen the stem by introducing G:C base pairs as well as deleting unpaired or additional nucleotides (such as the J1 & J2 region from the parental Spinach sequence). As a result, the basal stem could be shortened in iSpinach while maintaining its stability (Figure 4 D).62, 64 The L3‐loop at the tip of the Spinach structure is known to be flexible and plays an important role in all strategies for structure determination. Warner et al. used the L3‐loop during construct design to split Spinach into the two strands that were then synthesized and subsequently hybridized to give a Spinach structure with an absent L3 loop (Figure 5 A).62 Huang et al. used the L3 entry point to insert their 7‐nt Fab3‐6 chaperon recognition motif, thereby making it suitable for their RNA chaperoning crystallization setup (Figure 4 B).68 Fernandez et al. used this loop to optimize intramolecular contacts of the loop nucleotides, thereby strengthening the apical region. Their structure resolves the loop in its rigidified conformation (Figure 4 D).64 Affinity maturation of the Fab3‐6 crystallization chaperon resulted in the highest resolution structure up to now (1.64 Å, PDB ID: 6b14).66
A closer look at the actual DFHBI‐binding region reveals a stack of two G‐quadruplex tiers (1 & 2) towards the basal stem. Each is built up in a classical manner by four guanosines complexing a potassium ion (Figure 5 A,C). Remarkably, the G‐quadruplexes are formed in a non‐consecutive manner, resulting in large interconnecting loops between two G bases.62 This makes the formation rather complicated and difficult to predict (Figure 5 B,C). The two G‐quadruplexes build the platform for planar fluorogen‐binding. On the side of the apical stem, a triplex lid composed of a Watson–Crick (A64–U32) and a Hoogsteen base pair (A64–U61) sandwiches the fluorogen into place. Side contacts are formed between backbone 2′‐OH groups and more importantly to an unpaired base of G31 that lies in plane with DFHBI (Figure 5 C,D).
Interestingly, as different as all Spinach sequences are, these three basic features, 1) G‐quadruplex base, 2) flanking, unpaired and in‐plane G31, and 3) a triplex lid (Figure 5 D), are shared by all Spinach‐derived structures.
3.3. Mango: A Red FLAP
Mango is the first FLAP that utilizes fluorophores derived from thiazole orange and therefore provides sufficiently bright, red‐shifted emission (Figure 6 A).57 Its crystal structure was solved with an additional 4‐bp duplex stem to stabilize the core sequence of 29 nt. Mango crystalizes with two molecules per asymmetric unit as a homodimer, but was shown be mostly monomeric in solution (Figure 6 B).70 Its central fluorogen‐binding platform is again a G‐quadruplex, this time consisting of three individual tiers, connected by six short connecting loops (1–2 nt, Figure 6 C) that minimize the overall functional size of the Mango core sequence down to 23 nt. The thiazole orange binds together with a biotin moiety on top of the G‐quadruplex, thereby covering the whole surface of this region without any further nucleobases in plane. Three nucleotides form a lid‐like structure and assist fluorogen binding by sandwiching it from the top. In Spinach, the three lid bases form one entity through hydrogen‐bond formation, which acts like a closed surface flanking the fluorogen (Figure 6 B,C). In contrast, each individual lid base in Mango stabilizes a different moiety of TO1‐biotin: U15 stabilizes the biotin moiety, A20 the methylchinoline (MQ), and A26 the benzothiazole (BzT) of TO1‐biotin (Figure 6 C).
Figure 6.
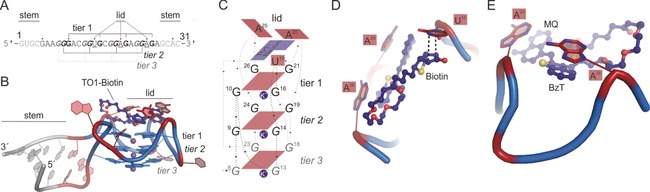
Structural overview of Mango–TO1‐biotin (PDB ID: 5bjo).70 A) Sequence of Mango core sequence (23 nt) plus a 4‐nt fusion on 5′‐ and 3‐ end forming a 4‐bp RNA duplex stem. Guanosines of the three G‐quadruplex tiers are indicated. Underlined nt form the lid structure. B) Crystal structure of one Mango monomer of the ASU. Light grey=4‐bp‐stabilizing RNA duplex; Blue=G‐quadruplex tiers (1–3), violet spheres=potassium ions; dark blue=TO1‐biotin fluorogen; bases of the lid structure are labeled. C) Schematic overview of the Mango TO1‐biotin‐binding region. Numbers indicate the nucleotide numbers from Mango according to (A). D,E) Two perspectives of TO1‐biotin binding to the lid structure composed of U15 stabilizing the biotin moiety through hydrogen bonds (black dashed lines) and A20 and A26 forming π–π interactions with the methylchinoline (MQ with A20) and the benzothiazole (BzT with A26) moiety of TO1‐biotin.
3.4. Corn Structural Comparison
Corn is a light‐up aptamer composed of just 36 nt.71 However, its functional unit is a homodimer, thus making it similar to the size of other Spinach‐based aptamers. Its 3D structure resembles a stem‐loop with a short but stable stem (Figure 7 A, P1), a linker region J1, and a loop region that folds back onto itself (tip). By folding back onto itself, Corn exposes a stack of G‐quadruplexes within the kink region. The sequence of nucleotides composing the G‐quadruplexes is non‐consecutive, but in contrast to Spinach G‐quadruplexes, the interlinking loops are small (1 nt), with the biggest being the folding back tip (5 nt, Figure 7 B).71 The DFHO fluorogen is coordinated on top of this G‐quadruplex region, thereby locking it into its planar conformation. In Spinach, the fluorogen DFHBI is sandwiched between the G‐quadruplex and a triplex lid (Figure 7 C,D). The crystal structure of Corn revealed that there is no lid region present. Still the coordination from the top is complemented by a second aptamer to form a homodimer with the fluorogen bound within the homodimer interface. This results in a second G‐quadruplex that forms the lid and locks the fluorogen into place (Figure 7 C,D). Nucleotides A24 and *A14 flank the sides of the fluorogen‐binding region. The oxime group of DFHO forms a hydrogen bond to *A14, which is not completely in plane with the G‐quadruplex. The role of G31 in Spinach, which is completely in plane with DFHBI and lies on top of the G‐quadruplex plane, is partially covered by A24 in Corn. Interestingly, A24 is turned perpendicular to the G‐quadruplex plane. This could be due to the fact that the additional interaction with *A14 and the oxime group push the fluorogen a bit further out, so that A24 adopts a different conformation than G31 does in Spinach–DFHBI (Figure 8). This also causes DFHO to be coordinated more central with respect to the G‐quadruplex, whereas DFHBI is shifted towards one half of the plane on top of G26 and G65.
Figure 7.
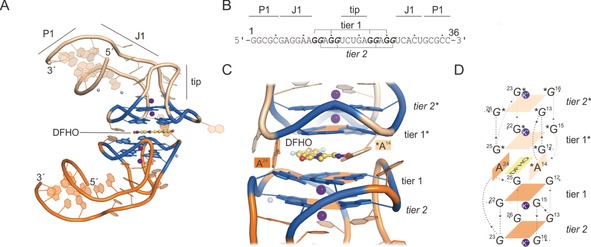
Corn–DFHO crystal structure (PDB ID: 5bjo).71 A) Crystal structure of Corn; names of structural features are indicated. Orange=aptamer A; light orange=aptamer B of the homodimer complex; purple spheres=Mg2+ and K+ ions; blue colored region=DFHO‐binding region including G‐quadruplex. B) Sequence of the crystallized Corn sequence by Warner et al.71 Bold=G‐quadruplex‐forming nucleotides. C) Close‐up of the fluorogen‐binding region. G‐quadruplex tiers are shown, and shaped platforms are indicated by orange planes. Each plane coordinates a potassium ion (purple). DFHO is shown in addition to its flanking nucleotides A24 of aptamer one and A14* of the second aptamer copy. D) Schematic overview of Corn–DFHO binding region. Numbers indicate the nucleotide numbers according to (B).
Figure 8.

Fluorogen coordination in Spinach–DFHBI (A) and Corn–DFHO (B). Dashed lines indicate hydrogen‐bond formation to flanking nucleotides.
Taken together, in all solved crystal structures, a quadruplex serves as the platform on which the fluorogen is bound. Structural analyses of the latest FLAPs revealed that G‐quadruplexes in particular seem to provide the basis for fluorogen binding. Spinach comprises a rather complex sequence with large interlinking loops. In Mango, these loop structures for G‐quadruplex formation are minimized, whereas the G‐quadruplex itself consists of three tiers. In Corn, this structural feature already seems rather simple, nevertheless, it might have potential for further minimization. So far, the following structural features seem favored for FLAPs:
G‐quadruplex build‐up from at least two tiers located on a strong base (strong duplex, additional planar multiplexed strand structures)
A fluorogen‐sandwiching top lid (can be another G‐quadruplex)
Fluorogen flanking nucleotides that lock it into place on the G‐quadruplex.
4. Cellular Applications of FLAPs
Numerous fluorogenic aptamers have been explored, but only a few have actually made it into cells since FLAPs were initially designed for in vitro applications.72, 73, 74 Robust expression, correct folding, sufficient brightness, and low photobleaching are only a few FLAP properties that determine their applicability in cells.63 Usually, newly developed FLAPs intended for cellular applications are tested in live cells (bacteria or cultured mammalian cells) either in isolation or as a tag to an RNA of interest (FLAP‐tag, Figure 9 A). These applications are mentioned in Section 2 on developments of FLAPs. In addition, FLAPs have been tested for quantitative detection of low‐abundant RNAs in live cells, where sufficient brightness becomes a challenge.75 Guet et al. used Spinach–DFHBI to localize the low‐abundance polymerase II derived mRNAs GAL1, ASH1, and STL1 in live yeast (S. cerevisiae) cells.76 Here, adaptation of the imaging workflow compensated for the lower brightness compared to MS2‐ or PP7‐based systems with GFP and allowed precise, quantitative, and dynamic studies of mRNA biogenesis and trafficking. In another approach, Pothoulakis et al. fused Spinach to a translated mRNA coding for monomeric red fluorescent protein (mRFP1) to measure mRNA and protein levels in E. coli simultaneously. The results highlight the fact that RNAs have much faster turnover in cells than proteins, and stress the importance of dynamic analysis techniques for RNA imaging.77, 78
Figure 9.
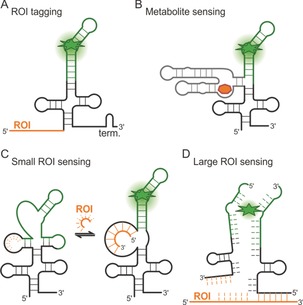
FLAP fusions to other RNAs for application in live cells. A) FLAP‐tagged RNA of interest (ROI). B) Bifunctional FLAPs through fusion to a metabolite‐sensing aptamer or riboswitch. C) Small RNA‐sensing FLAP. D) mRNA‐sensing split‐FLAP.
FLAPs can also be expressed as fusions with a sensor RNA unit that is able to detect an endogenous biomolecule, such as a metabolite, protein, or RNA. The analyte binds to the sensor unit (e.g., aptamer, riboswitch, reverse complimentary sequence) and induces a structural rearrangement, causing the FLAP unit to fold into its fluorogen‐binding tertiary structure, thereby resulting in fluorescence increase in presence of the fluorogen. In this way, the analyte can act either in analogy to an allosteric effector or cause strand displacement. (Figure 9 B). Similarly, an RNA strand is displaced through the binding of an RNA of interest (e.g., miRNA), which consequently can bind the fluorogen (Figure 9 C).
The first bifunctional in vitro fluorescent aptamers were developed in 2004 by Kolpashchikov and co‐workers.79 Via a communication module, an MG aptamer was attached to an analyte‐binding aptamer selective for adenosine triphosphate (ATP), flavin mononucleotide (FMN), or theophylline (TH). Jaffrey and co‐workers developed Spinach into the first bifunctional sensor FLAP for S‐adenosylmethionine (SAM) and adenosine 5′‐diphosphate (ADP), which was genetically encoded in E. coli for real‐time detection and imaging of the metabolites.80, 81 Streptavidin and MS2 coat protein (MCP) were also imaged in E. coli with appropriate Spinach sensor RNAs.82 While these approaches used target‐binding aptamers that were selected by SELEX,83 which can be challenging, Kellenberger et al. fused Spinach to variants of a natural GEMM‐I riboswitch to produce a biosensor for cyclic di‐guanosine monophosphate (GMP) and cyclic AMP‐GMP in E. coli.84, 85 In 2015, Spinach2 was fused to cyclic di‐AMP‐binding riboswitches to visualize levels in L. monocytogenes and to screen for diadenylate cyclase activity of enzymes.86 A modified Spinach2‐S‐adenosyl‐l‐homocysteine (SAH) riboswitch fusion was used to detect SAH in live E. coli and to screen inhibitors of methyltransferases in vitro.87 Spinach riboswitches involving strand displacement upon analyte binding were developed for thiamine 5′‐pyrophosphate (TPP) and expressed and imaged in E. coli.88 The above mentioned promiscuous aptamer DIR2s‐Apt fused to the epidermal growth factor receptor (EGFR) represents a bifunctional aptamer that was used to differentiate between cell‐surface and internalized EGFR on mammalian cells by applying the two fluorogens (DIR‐Pro and OTB‐SO3) at different times.58
FLAPs have also been applied as sensors for endogenous RNAs (mRNAs and micro RNAs). A modified Spinach FLAP was engineered to offer a reverse complementary binding site for miRNA miR‐122 by Meyers and co‐workers in 2017.89 Upon binding of miR‐122, the binding site for DFHBI is reconstituted and fluorescence generated. Sufficient brightness for measurements in live HEK293T cells, however, were only possible with a six‐tandem repeat.89 Ying et al. engineered another miRNA‐sensing FLAP by fusing the sulforhodamine (SR)‐binding FLAP to a miRNA‐binding sequence.90 Here too, miR‐21 binding leads to re‐folding of the SR‐binding site, and upon binding of SR to unquenching of the SR–dinitroaniline conjugate (SR‐DN). This miR‐21 sensor was genetically encoded and used to quantitatively detect miR‐21 in different live mammalian (tumor) cells. To detect endogenous mRNA in mammalian cells, Wang et al. very recently developed a split‐Broccoli system.91, 92 Here, the endogenous mRNA serves as a template to reconstitute Broccoli from two halves that adjacently bind to the mRNA of interest (Figure 9 D). This approach leads to lower background fluorescence and sufficient brightness for real‐time imaging of β‐actin mRNA distribution in live HeLa cells.
5. FLAPs to Study Bimolecular RNA Interactions
Interactions between biomacromolecules are of great importance both for the understanding of cellular functions and also for the design of interaction modulators.93−96 FLAPs have great potential as tools to analyze RNA interactions. While until very recently, their usage to study bimolecular interaction was little to non‐existent, four studies have now been published exemplifying how FLAPs could be used in the future.
5.1. RNA–RNA Interaction Studies
The Corn aptamer forms a homodimer with DFHO as the fluorogen binding within its interaction interface (Figure 10). Therefore, homodimerization is an essential feature in forming the fluorogen‐binding site.97 Corn dimerizes with high affinity (K D=1 nm) in a quasisymmetrical manner on both sides of the flat surface of DFHO (Figure 10 A). Since the system already consists of two molecules of RNA, studying RNA–RNA interactions by fusing Corn to two interacting RNAs of interest seems like an evident application, but this has not been shown so far. The strong homodimerization could prove to be counterproductive in this context. A second FLAP, split‐dBroccoli, is a variant of dBroccoli and once cut in two halves its re‐assembly can be monitored through fluorescence in vitro as well as in vivo.98 Split‐dBroccoli harbors two binding sites for its fluorogen DFHBI‐T1, which makes it brighter compared to other systems, but also rather large (Figure 10 B). The system was successfully used to detect cellular RNA–RNA interactions: one part of the system was fused to a “toehold RNA”, which hides a ribosome‐binding site and the other half was fused to a “trigger RNA”. Binding of the trigger TNA to the toehold RNA not only resulted in reconstitution of split‐dBroccoli and fluorescence activation, but also accessibility of the ribosome‐binding site, which enabled translation of a downstream reporter gene.98 This experiment shows nicely how FLAPs can help to understand cellular processes in real time. However, once the strong self‐assembly of these complementation systems is overcome, their utilization will gain even more impact.
Figure 10.

Complementation systems for the detection of RNA–RNA interactions. A) Schematic representation of the Corn homodimer with its according fluorogen (yellow=DFHO). The two ROIs need to be fused to a unique Corn derivative (black and grey) to avoid spontaneous self‐assembly. Interaction of the two ROIs will then trigger Corn fluorescence. B) Schematic representation of split‐dBroccoli with its according fluorogen (green=DFHBI‐1T). The two ROIs need to be fused to one of the two dBroccoli strands. Integration of base pair mismatches between both dBroccoli strands will avoid spontaneous self‐assembly. Interaction of the two ROIs will then trigger dBroccoli fluorescence. C) Scheme of a Spinach–Mango fusion construct with the corresponding fluorogens [Spinach (green): DFHBI‐1T, Mango (red): YO3‐biotin] used in a FRET setup (apta‐FRET). Rational fusion construct design brings both FLAPs into close proximity and induces FRET fluorescence. Microscopy images (black box) of bacteria expressing this apta‐FRET construct show each individual FLAP fluorescence (green and red image) and a FRET signal of the acceptor fluorescence (greyscale image). For a bimolecular interaction technique, the two ROIs need to be fused to one of these FLAPs. Proximity induced by ROI interaction triggers FRET fluorescence. Scale bar: 10 μm, orange and blue indicate the two interacting ROIs, image modified from Jepsen et al.97
Another method for measuring bimolecular interactions is to use proximity‐based Förster resonance energy transfer (FRET) from a fluorescent donor to acceptor molecules. Herein, the bimolecular interaction of two molecules of interest brings the two fluorophores into close proximity, so that the direct transfer of the excited donor electron can excite an electron of the acceptor molecule. Two observable readouts are commonly used: 1) the fluorescence emission of the donor molecule decreases as the acceptor molecule takes over the energy of the excited electron, and 2) the acceptor molecule can also release this energy through fluorescence of a usually red‐shifted wavelength. FLAPs are fluorophores that in principle can undergo such mechanism. Two aptamers with suitable spectral characteristics (Spinach and Mango) were fused together in a system called “apta‐FRET” (Figure 10 C).71 The fusion of the two, as well as several steps of construct optimization (kissing loop structure, variations of scaffolding RNA‐duplex) for close proximity and correct dipole orientation, fluorophore optimization for better spectral overlap (oxazole yellow derivative YO3‐biotin as Mango fluorogen instead of TO1‐biotin, Figure 2 B), resulted in an RNA construct with two FLAPs that undergo FRET using Mango fluorescence as a read‐out of the acceptor.71 Apta‐FRET could be shown in vitro and also in vivo when expressed in bacteria (Figure 10 D). Several system variants were successfully tested, including small‐molecule sensing by integration of a specific riboswitch into the construct or target‐RNA sensing by inserting a RNA‐recognition sequence. This demonstrates the potential of FLAPs, especially for studying bimolecular interactions in vivo.71
5.2. RNA–Protein Interaction Studies
Until now, all described systems that either have or could have the potential to be utilized in bimolecular interaction studies have been designed for RNA–RNA interactions. In cellular processes, the interaction of RNA molecules with the proteome is an essential interplay. Using FLAP‐tags in combination with biological protein fluorophores opens up a new dimension of possible investigations. Spinach as a green fluorophore was used together with mCherry as a red protein fluorophore in a FRET‐based system.99 Therein, a RNA–protein interaction (PP7‐coat‐protein/pp7‐RNA of the Pseudomonas aeroguinosa bacteriophage) was used to demonstrate energy transfer from Spinach fused to pp7‐RNA as the donor towards mCherry fused to the PP7‐coat as the acceptor fluorophore protein (Figure 11 A). Since no acceptor fluorescence was observable, specific donor quenching of Spinach was used as readout. Spinach is a medium‐sized FLAP (ca. 80 nt compared to Mango, which is ca. 30 nt) and its structure is quite elongated. It tolerates fusions into the loop at the tip of the structure (L3‐loop), where the pp7‐RNA was integrated for this study (Figure 11 B). When comparing sizes of the protein fusions to the size of the RNA fusions in a model, it becomes clear that tag size and positioning for both the RNA and the protein side are limiting. However, no self‐assembly of the FRET system itself was observed. The RNA–protein interaction could also be quantified, even in cell lysates. This is the first example of an RNA–protein interaction that is completely genetically encodable and could be transferred into cells on DNA level.99 Optimizing spectral characteristics as well as minimizing tag sizes (such as the use of Mango) will help to broaden the applicability of the system and introduce acceptor fluorescence as a readout.
Figure 11.
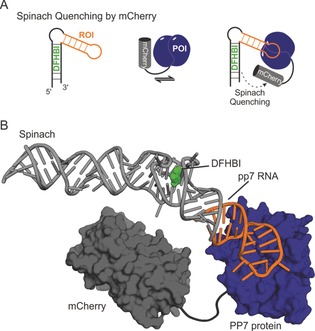
Spinach donor‐quenching for a FRET based RNA–protein interaction assay. A) An ROI (orange) is fused to Spinach as a fluorescence donor (black). A POI (dark blue) is fused to the mCherry protein (dark grey) as a quencher. Upon interaction of RNA and protein of interest, Spinach donor‐quenching occurs due to close proximity of the two fluorophores. B) Structural model of biomolecules used in (A). The model was constructed from X‐ray crystal structures of the PP7‐protein (dark blue surface) bound to its pp7‐RNA (orange stick model, PDB ID: 2qux),101 Spinach–DFHBI (light grey stick model, DFHBI shown as a green space‐filling, PDB ID: 4ts2),62 and mCherry (dark grey surface, PDB ID: 2h5q).102
Taken together, these studies demonstrate that FLAPs already show great potential as tools to analyze bimolecular interactions, including RNA–RNA and RNA–protein interactions. Therefore, the various available systems are currently in the process of being improved even further. A larger set of available wavelengths with suitable brightness, especially together with biological protein fluorophores tailored for the usage in FRET‐based systems, will help to provide improved signal‐to‐noise ratios as well as robustness in terms of applicability.
6. Conclusions
Since the methods for identifying new aptamer sequences, such as SELEX and evolved versions of it including its combination with FACS, for example, are established, the main focus in terms of development is on spectral improvement of the resulting FLAPs. The spectral variation in terms of suitable wavelength is quite broad, but their applicability is usually extremely limited due to very low quantum yields and extinction coefficients. Therefore, the development of new families of small‐molecule fluorogens with improved fluorescent characteristics coupled with state‐of‐the‐art aptamer identification techniques for high‐affinity aptamers will broaden the applicability of FLAPs in vitro and in vivo in the future. Currently, FLAPs are mainly used in the areas of RNA localization and sensing of cellular metabolites or small compounds. Improving the fluorescence intensity as well as rigidification and miniaturization of the structure are promising ways to enhance their utilization. Many biological areas such as RNA transcription and gene regulation, and RNA processing or RNA trafficking, for example, of mRNAs, viral RNAs, and long‐noncoding RNAs, would greatly benefit from these developments. With the first FLAP‐FRET system for bimolecular interaction analysis having been successful, the biochemical toolbox is growing. Future applications for the analysis of RNA–protein interactions need protein fluorophores and FLAPs that are tailored for one another to achieve an optimized FRET response. This will open the potential usage of FLAPs for protein–RNA interactions in biological fields such as cellular biology, signal transduction, and gene regulation. Indeed, one of the major current restrictions of FLAPs is the requirement for the addition of external fluorogens. By contrast, GFP forms its fluorogen autonomously. It can therefore be used intensively in fields such as developmental biology for the spatiotemporal analysis of protein expression even in whole organisms. Compared to GFP, the applicability of FLAPs is currently limited to cultured cells. Improvements to FLAPs for future applications will surely be appreciated by the entire RNA community.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Saskia Neubacher studied chemistry at the Free University Berlin (Germany). In 2012, she completed her PhD with Prof. Christoph Arenz at the Humboldt University Berlin (Germany), where her studies focused on the maturation of miRNAs. After working as Associate Editor for Wiley‐VCH, she returned to the laboratory as a project leader in the group of Prof. Tom Grossmann at the VU University Amsterdam (The Netherlands). Her research interests include the study and modulation of RNA–protein interactions.

Biographical Information
Sven Hennig studied biochemistry at the Ruhr‐University Bochum and received his PhD from the MPI for molecular physiology in 2008 under the supervision of E. Wolf . After postdoctoral research with C. Ottmann in 2008 (CGC, Dortmund, Germany) and A. H. Fox (UWA, Perth, Australia) in 2010, he started his own research group on the modulation of transcription factor complexes at the CGC (Dortmund, Germany) in 2012. He has been assistant professor for structural chemical biology and head of X‐ray crystallization at the VU University Amsterdam since 2017. His research focuses on structure determination of protein–RNA complexes and modulation of the transcriptome.

Acknowledgements
We thank Dr. Philipp Thiel (Eberhard Karls University Tübingen, Germany) for the assistance (BALLView) in the design of the frontispiece.
S. Neubacher, S. Hennig, Angew. Chem. Int. Ed. 2019, 58, 1266.
References
- 1. König J., Zarnack K., Luscombe N. M., Ule J., Nat. Rev. Genet. 2012, 13, 77–83. [DOI] [PubMed] [Google Scholar]
- 2. Mercer T. R., Dinger M. E., Mattick J. S., Nat. Rev. Genet. 2009, 10, 155–159. [DOI] [PubMed] [Google Scholar]
- 3. Holt C. E., Schuman E. M., Neuron 2013, 80, 648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McHugh C. A., Russell P., Guttman M., Chen M., Manley J., Licatalosi D., Darnell R., Wang Z., Gerstein M., Snyder M., et al., Genome Biol. 2014, 15, 1–10. [Google Scholar]
- 5. Ghildiyal M., Zamore P. D., Nat. Rev. Genet. 2009, 10, 94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Glisovic T., Bachorik J. L., Yong J., Dreyfuss G., FEBS Lett. 2008, 582, 1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McLoughlin N. M., Mueller C., Grossmann T. N., Cell Chem. Biol. 2018, 25, 19–29. [DOI] [PubMed] [Google Scholar]
- 8. Lunde B. M., Moore C., Varani G., Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pospiech N., Cibis H., Dietrich L., Müller F., Bange T., Hennig S., Sci. Rep. 2018, 8, 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gerstberger S., Hafner M., Tuschl T., Nat. Rev. Genet. 2014, 15, 829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Burd C. G., Dreyfuss G., Science 1994, 265, 615–621. [DOI] [PubMed] [Google Scholar]
- 12. Licatalosi D. D., Darnell R. B., Nat. Rev. Genet. 2010, 11, 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baek D., Villén J., Shin C., Camargo F. D., Gygi S. P., Bartel D. P., Nature 2008, 455, 64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo H., Ingolia N. T., Weissman J. S., Bartel D. P., Nature 2010, 466, 835–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smith M. A., Mattick J. S., in Bioinformatics (Ed.: J. Keith), Humana Press, New York, 2017, pp. 65–85. [Google Scholar]
- 16. Blythe A. J., Fox A. H., Bond C. S., Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 46–58. [DOI] [PubMed] [Google Scholar]
- 17. Houseley J., Tollervey D., Cell 2009, 136, 763–776. [DOI] [PubMed] [Google Scholar]
- 18. Liu Y., Beyer A., Aebersold R., Cell 2016, 165, 535–550. [DOI] [PubMed] [Google Scholar]
- 19. Femino A. M., Fay F. S., Fogarty K., Singer R. H., Science 1998, 280, 585–590. [DOI] [PubMed] [Google Scholar]
- 20. Dunagin M., Cabili M. N., Rinn J., Arjun R., in Nucl. Bodies Noncoding RNAs (Eds.: S. Nakagawa, T. Hirose), Humana Press, New York, 2015, pp. 3–19. [Google Scholar]
- 21. Bertrand E., Chartrand P., Schaefer M., Shenoy S. M., Singer R. H., Long R. M., Mol. Cell 1998, 2, 437–445. [DOI] [PubMed] [Google Scholar]
- 22. SenGupta D. J., Zhang B., Kraemer B., Pochart P., Fields S., Wickens M., Proc. Natl. Acad. Sci. USA 1996, 93, 8496–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lim B., Curr. Opin. Biotechnol. 2018, 52, 49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ray S., Widom J. R., Walter N. G., Chem. Rev. 2018, 118, 4120–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Urbinati C. R., Long R. M., Wiley Interdiscip. Rev. RNA 2011, 2, 601–609. [DOI] [PubMed] [Google Scholar]
- 26. Pijuan-Sala B., Guibentif C., Göttgens B., Nat. Rev. Mol. Cell Biol. 2018, 19, 399–412. [DOI] [PubMed] [Google Scholar]
- 27. Dictenberg J., Trends Biotechnol. 2012, 30, 621–626. [DOI] [PubMed] [Google Scholar]
- 28. Prendergast F. G., Mann K. G., Biochemistry 1978, 17, 3448–3453. [DOI] [PubMed] [Google Scholar]
- 29. Miyawaki A., J. Electron Microsc. 2013, 62, 63–68. [Google Scholar]
- 30. Nienhaus K., Nienhaus G. U., Chem. Soc. Rev. 2014, 43, 1088–1106. [DOI] [PubMed] [Google Scholar]
- 31. Ormö M., Cubitt A. B., Kallio K., Gross L. A., Tsien R. Y., Remington S. J., Science 1996, 273, 1392–1395. [DOI] [PubMed] [Google Scholar]
- 32. Pollok B., Trends Cell Biol. 1999, 9, 57–60. [DOI] [PubMed] [Google Scholar]
- 33. Tsien R. Y., Annu. Rev. Biochem. 1998, 67, 509–544. [DOI] [PubMed] [Google Scholar]
- 34. Sando S., Narita A., Hayami M., Aoyama Y., Chem. Commun. 2008, 33, 3809–3940. [DOI] [PubMed] [Google Scholar]
- 35. Babendure J. R., Adams S. R., Tsien R. Y., J. Am. Chem. Soc. 2003, 125, 14716–14717. [DOI] [PubMed] [Google Scholar]
- 36. Paige J. S., Wu K. Y., Jaffrey S. R., Science 2011, 333, 642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Grate D., Wilson C., Proc. Natl. Acad. Sci. USA 1999, 96, 6131–6136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Engelhart A. E., Nat. Chem. Biol. 2017, 13, 1140–1141. [DOI] [PubMed] [Google Scholar]
- 39. Tuerk C., Gold L., Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- 40. Ellington A. D., Szostak J. W., Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- 41. Hermann T., Patel D. J., Science 2000, 287, 820–826. [DOI] [PubMed] [Google Scholar]
- 42. Da Costa J. B., Andreiev A. I., Dieckmann T., Biochemistry 2013, 52, 6575–6583. [DOI] [PubMed] [Google Scholar]
- 43. Martell R. E., Nevins J. R., Sullenger B. A., Mol. Ther. 2002, 6, 30–34. [DOI] [PubMed] [Google Scholar]
- 44. Strack R. L., Disney M. D., Jaffrey S. R., Nat. Methods 2013, 10, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Filonov G. S., Moon J. D., Svensen N., Jaffrey S. R., J. Am. Chem. Soc. 2014, 136, 16299–16308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ketterer S., Fuchs D., Weber W., Meier M., Nucleic Acids Res. 2015, 43, 9564–9572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Filonov G. S., Kam C. W., Song W., Jaffrey S. R., Filonov G. S., Kam C. W., Song W., Jaffrey S. R., Chem. Biol. 2015, 22, 649–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ponchon L., Dardel F., Nat. Methods 2007, 4, 571–576. [DOI] [PubMed] [Google Scholar]
- 49. Autour A., Westhof E., Ryckelynck M., Nucleic Acids Res. 2016, 44, 2491–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gotrik M., Sekhon G., Saurabh S., Nakamoto M., Eisenstein M., Soh H. T., J. Am. Chem. Soc. 2018, 140, 3583–3591. [DOI] [PubMed] [Google Scholar]
- 51. Autour A., Jeng S. C. Y., Cawte A. D., Abdolahzadeh A., Galli A., Panchapakesan S. S. S., Rueda D., Ryckelynck M., Unrau P. J., Nat. Commun. 2018, 9, 656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Song W., Filonov G. S., Kim H., Hirsch M., Li X., Moon J. D., Jaffrey S. R., Nat. Chem. Biol. 2017, 13, 1187–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chalfie M., Tu Y., Euskirchen G., Ward W. W., Prashert D. C., Science 1994, 263, 802–805. [DOI] [PubMed] [Google Scholar]
- 54. Heim R., Cubitt A. B., Tsien R. Y., Nature 1995, 373, 663–664. [DOI] [PubMed] [Google Scholar]
- 55. Cormack B. P., Valdivia R. H., Falkow S., Gene 1996, 173, 33–38. [DOI] [PubMed] [Google Scholar]
- 56. Song W., Strack R. L., Svensen N., Jaffrey S. R., J. Am. Chem. Soc. 2014, 136, 1198–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dolgosheina E. V., Jeng S. C. Y., Panchapakesan S. S. S., Cojocaru R., Chen P. S. K., Wilson P. D., Hawkins N., Wiggins P. A., Unrau P. J., ACS Chem. Biol. 2014, 9, 2412–2420. [DOI] [PubMed] [Google Scholar]
- 58. Tan X., Constantin T. P., Sloane K. L., Waggoner A. S., Bruchez M. P., Armitage B. A., J. Am. Chem. Soc. 2017, 139, 9001–9009. [DOI] [PubMed] [Google Scholar]
- 59. Sunbul M., Jäschke A., Angew. Chem. Int. Ed. 2013, 52, 13401–13404; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13643–13646. [Google Scholar]
- 60. Arora A., Sunbul M., Jäschke A., Nucleic Acids Res. 2015, 43, e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Han K. Y., Leslie B. J., Fei J., Zhang J., Ha T., J. Am. Chem. Soc. 2013, 135, 19033–19038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Warner K. D., Chen M. C., Song W., Strack R. L., Thorn A., Jaffrey S. R., Ferré-D′Amaré A. R., Nat. Struct. Mol. Biol. 2014, 21, 658–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Okuda M., Fourmy D., Yoshizawa S., Nucleic Acids Res. 2017, 45, 1404–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Fernandez-Millan P., Autour A., Ennifar E., Westhof E., Ryckelynck M., RNA 2017, 23, 1788–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Constantin T. P., Silva G. L., Robertson K. L., Hamilton T. P., Fague K., Waggoner A. S., Armitage B. A., Org. Lett. 2008, 10, 1561–1564. [DOI] [PubMed] [Google Scholar]
- 66. Koirala D., Shelke S. A., Dupont M., Ruiz S., DasGupta S., Bailey L. J., Benner S. A., Piccirilli J. A., Nucleic Acids Res. 2018, 46, 2624–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Baugh C., Grate D., Wilson C., J. Mol. Biol. 2000, 301, 117–128. [DOI] [PubMed] [Google Scholar]
- 68. Huang H., Suslov N. B., Li N. S., Shelke S. A., Evans M. E., Koldobskaya Y., Rice P. A., Piccirilli J. A., Nat. Chem. Biol. 2014, 10, 686–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Koldobskaya Y., Duguid E. M., Shechner D. M., Suslov N. B., Ye J., Sidhu S. S., Bartel D. P., Koide S., Kossiakoff A. a., Piccirilli J. a., Nat. Struct. Mol. Biol. 2011, 18, 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. R. J. Trachman III , Demeshkina N. A., Lau M. W. L., Panchapakesan S. S. S., Jeng S. C. Y., Unrau P. J., Amaré A. R. F., Nat. Chem. Biol. 2017, 13, 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Warner K. D., Sjekloa L., Song W., Filonov G. S., Jaffrey S. R., Ferré-D′Amaré A. R., Nat. Chem. Biol. 2017, 13, 1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kolpashchikov D. M., J. Am. Chem. Soc. 2005, 127, 12442–12443. [DOI] [PubMed] [Google Scholar]
- 73. Lee J., Lee K. H., Jeon J., Dragulescu-Andrasi A., Xiao F., Rao J., ACS Chem. Biol. 2010, 5, 384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ying Z.-M., Tu B., Liu L., Tang H., Tang L.-J., Jiang J.-H., Chem. Commun. 2018, 54, 3010–3013. [DOI] [PubMed] [Google Scholar]
- 75. Shin I., Ray J., Gupta V., Ilgu M., Beasley J., Bendickson L., Mehanovic S., Kraus G. A., Nilsen-hamilton M., Nucleic Acids Res. 2014, 42, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Guet D., Burns L. T., Maji S., Boulanger J., Hersen P., Wente S. R., Salamero J., Dargemont C., Nat. Commun. 2015, 6, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pothoulakis G., Ceroni F., Reeve B., Ellis T., ACS Synth. Biol. 2014, 3, 182–187. [DOI] [PubMed] [Google Scholar]
- 78. Schoenberg D. R., Maquat L. E., Nat. Rev. Genet. 2012, 13, 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Stojanovic M. N., Kolpashchikov D. M., J. Am. Chem. Soc. 2004, 126, 9266–9270. [DOI] [PubMed] [Google Scholar]
- 80. Paige J. S., Nguyen-Duc T., Song W., Jaffrey S. R., Science 2012, 335, 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Strack R. L., Song W., Jaffrey S. R., Nat. Protoc. 2014, 9, 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Song W., Strack R. L., Jaffrey S. R., Nat. Methods 2013, 10, 873–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Schwalbe H., Buck J., Fürtig B., Noeske J., Wöhnert J., Angew. Chem. Int. Ed. 2007, 46, 1212–1219; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 1232–1240. [Google Scholar]
- 84. Kellenberger C. A., Chen C., Whiteley A. T., Portnoy D. A., Hammond M. C., J. Am. Chem. Soc. 2015, 137, 6432–6435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kellenberger C. A., Wilson S. C., Hickey S. F., Gonzalez T. L., Su Y., Hallberg Z. F., Proc. Natl. Acad. Sci. USA 2015, 112, 5383–5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kellenberger C. A., Chen C., Whiteley A. T., Portnoy D. A., Hammond M. C., J. Am. Chem. Soc. 2015, 137, 6432–6435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Su Y., Hickey S. F., Keyser S. G. L., Hammond M. C., J. Am. Chem. Soc. 2016, 138, 7040–7047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. You M., Litke J. L., Jaffrey S. R., Proc. Natl. Acad. Sci. USA 2015, 112, E2756. [Google Scholar]
- 89. Huang K., Doyle F., Wurz Z. E., Tenenbaum S. A., Hammond K., Caplan J. L., Meyers B. C., Nucleic Acids Res. 2017, 45, e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ying Z., Wu Z., Tu B., Tan W., Jiang J., J. Am. Chem. Soc. 2017, 139, 9779–9782. [DOI] [PubMed] [Google Scholar]
- 91. Wang Z., Luo Y., Xie X., Hu X., Song H., Zhao Y., Shi J., Wang L., Glinsky G., Chen N., et al., Angew. Chem. int. Ed. 2018, 57, 972–976; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 984–988. [Google Scholar]
- 92. Zhang J., Fei J., Leslie B. J., Han K. Y., Kuhlman T. E., Ha T., Sci. Rep. 2015, 5, 17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Khalil A. M., Rinn J. L., Semin. Cell Dev. Biol. 2011, 22, 359–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pelay-Gimeno M., Glas A., Koch O., Grossmann T. N., Angew. Chem. Int. Ed. 2015, 54, 8896–8927; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 9022–9054. [Google Scholar]
- 95. Romano M., Buratti E., J. Biomol. Screening 2013, 18, 967–983. [DOI] [PubMed] [Google Scholar]
- 96. Guan L., Disney M. D., ACS Chem. Biol. 2012, 7, 73–86. [DOI] [PubMed] [Google Scholar]
- 97. Jepsen M. D. E., Sparvath S. M., Nielsen T. B., Langvad A. H., Grossi G., Gothelf K. V., Andersen E. S., Nat. Commun. 2018, 9, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Alam K. K., Tawiah K. D., Lichte M. F., Porciani D., Burke D. H., ACS Synth. Biol. 2017, 6, 1710–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Roszyk L., Kollenda S., Hennig S., ACS Chem. Biol. 2017, 12, 2958–2964. [DOI] [PubMed] [Google Scholar]
- 100. Zuker M., Nucleic Acids Res. 2003, 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chao J. A., Patskovsky Y., Almo S. C., Singer R. H., Nat. Struct. Mol. Biol. 2008, 15, 103–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Shu X., Shaner N. C., Yarbrough C. A., Tsien R. Y., Remington S. J., Biochemistry 2006, 45, 9639–9647. [DOI] [PubMed] [Google Scholar]