

Abstract
Metabolomics deals with the whole ensemble of metabolites (the metabolome). As one of the ‐omic sciences, it relates to biology, physiology, pathology and medicine; but metabolites are chemical entities, small organic molecules or inorganic ions. Therefore, their proper identification and quantitation in complex biological matrices requires a solid chemical ground. With respect to for example, DNA, metabolites are much more prone to oxidation or enzymatic degradation: we can reconstruct large parts of a mammoth's genome from a small specimen, but we are unable to do the same with its metabolome, which was probably largely degraded a few hours after the animal's death. Thus, we need standard operating procedures, good chemical skills in sample preparation for storage and subsequent analysis, accurate analytical procedures, a broad knowledge of chemometrics and advanced statistical tools, and a good knowledge of at least one of the two metabolomic techniques, MS or NMR. All these skills are traditionally cultivated by chemists. Here we focus on metabolomics from the chemical standpoint and restrict ourselves to NMR. From the analytical point of view, NMR has pros and cons but does provide a peculiar holistic perspective that may speak for its future adoption as a population‐wide health screening technique.
Keywords: fingerprinting, metabolomics, NMR, omic sciences, profiling
1. What is Metabolomics
Metabolomics is the accepted name for the ‐omic science that deals with the characterization of the metabolome, in turn defined as the whole set of metabolites in a certain biological system such as a cell, a tissue, an organ, or an entire organism. The term metabonomics, as distinct from metabolomics, is also used when we refer to the science of understanding the network of interactions that metabolites undergo in a living system and is therefore closer to the concept of systems biology. Metabolomics deals with the objects, metabonomics with their interconnections and functions.1 In practice the two terms are mostly used interchangeably.
Metabolomics is downstream with respect to the other ‐omic sciences, and this has important implications, as illustrated in Figure 1. Metabolomics is much more influenced by environmental factors, and this makes metabolomic data more difficult to interpret but also richer of information about the health status of an individual. While the genome is (almost) invariant throughout the lifespan of an individual, the metabolome may change as a result of lifestyle, stress and, most importantly, onset of pathologies. On the other hand, while the number of biological objects increases from 104–105 genes to ca. 107 proteins, it decreases down to 103–104 metabolites1, 2 (Figure 1), thus condensing the information. The distribution of metabolites in a biological fluid (urine) is shown as an example in Figure 2.
Figure 1.
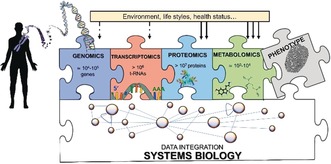
The flow of information in systems biology proceeds from the genome to the transcriptome, the proteome and finally to the metabolome. From left to right they are increasingly variable during an individual lifespan, and all concur to the phenotype.
Figure 2.
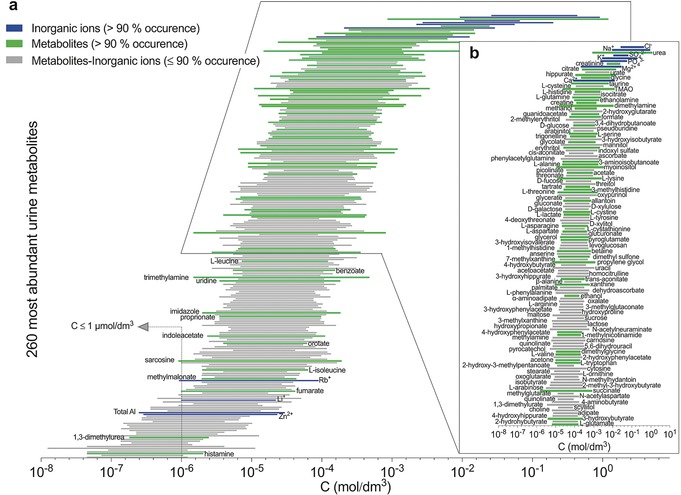
a) Concentration ranges of the 260 most abundant metabolites in urine as determined by LC‐MS, NMR, GC‐MS, ICP‐MS and HPLC.18 Metabolites are sorted according to their mean absolute concentration values in urine. Green and blue bars highlight the organic metabolites and the inorganic ions that appear at high occurrence in urine, respectively; gray bars those at lower occurrence.18 b) Enlargement of the first 136 urine metabolites, with mean concentration value >30 μΜ. Adapted from Ref. 14.
Although the majority of studies of metabolomics deal with humans, and therefore with human health and diseases, metabolomics also applies to animals,3 plants4 and microorganisms,5, 6 and therefore also impacts on other important fields such as agriculture, food production and environment.7, 8, 9 There are very many review articles that, as it can be imagined, describe metabolomics from many different viewpoints.10 The present one attempts at looking at metabolomics mainly from a chemical viewpoint. We are well aware that the whole is greater than the sum of all its parts, that is, understanding metabolomics implies a systemic view of the biological entity and cannot be limited to correctly identifying and quantifying as many chemical objects as possible in a system. On the other hand, expertise in analytical chemistry, medicinal chemistry, chemometrics, molecular reactivity, good laboratory practice, and in at least one of the two major instrumental metabolomic techniques (MS and NMR) is essential to properly collect, handle and store the metabolomic samples, perform the experiments, analyze the results, and even detect flaws that might escape an eye that is not chemically trained. Metabolomics can bring to great discoveries, but we need to be sure that the discovery is based on solid ground.
Of the two main analytical techniques, MS and NMR,11 we will only deal with NMR which is our main field of research, but we think that the two techniques are very complementary, and the weaknesses of one are compensated by the strengths of the other,10, 12, 13 as illustrated in Table 1. The main reason to stick to one of them is that, because the merits of the two techniques are intrinsically different, doing metabolomics by NMR results in developing an “NMR‐vision” of metabolomics that is different from the “MS‐vision”. By doing this we hope to contribute to increase the “biodiversity” of metabolomic approaches, to the advantage of the scientific communities (the medical community above all) that are progressively attracted by it but still need to appreciate and exploit the fact that metabolomics comes in very different flavors.
Table 1.
| Technology | NMR | MS |
|---|---|---|
| Reproducibility | Very high | Fair |
| Detection limit | Micromolar range | Picomolar range |
| Sample preparation | Minimal | Several steps: often requires chromatographic separation and sample derivatization |
| Volume of the original sample used | 0.1–0.5 mL | 0.01–0.2 mL |
| Types of molecules detected | Any molecules containing NMR active nuclei | Most organic and some inorganic |
| Types of experiments | All metabolites above detection limit are observed simultaneously | Several, tailored for specific chemical species |
| Ambiguous/false identification | Origin: compounds with degenerate chemical shifts, chemical shift variability due to experimental conditions (pH, temperature, ionic strength), presence of only one singlet signal. Experimental approaches: 2D experiments14 | Origin: compounds (e.g. isomers) that can match a given atomic composition or a parent ion mass.15 Experimental approaches: LC‐MS/MS |
2. Metabolomics by NMR
A major application of NMR in metabolomics is the detection of all small molecules in a sample that are above a given concentration threshold (Table 1) using one‐dimensional 1H pulse sequences. Two main approaches are employed: i) solution NMR, for the detection of soluble metabolites in biofluids, cell lysates or polar/apolar tissue extracts and ii) HR‐MAS (High Resolution Magic Angle Spinning), for the measurement of metabolites in semi‐solid samples, like intact tissues.
2.1. Types of Samples
The types of samples are many; the most common and significant are listed in Table 2, Figures 3 and 4. The samples can be classified in terms of molecular/biochemical complexity, from cells to tissues and biofluids. The NMR detectable part of the metabolome corresponds to tens‐hundreds of molecules, mainly belonging to the class of amino acids, carbohydrates, alcohols and organic acids (Figure 3). NMR can also contribute to the definition of the overall lipid composition of a biosystem, the so‐called lipidomics.16, 17 Sample‐specific considerations can be applied. Different types of cells are characterized by different metabolomes, but the measurable molecules are end‐products or intermediates of the main metabolic pathways. Intensity variations in intracellular metabolites (endo‐metabolome) induced by treatment with drugs, overexpression of a protein, genetic manipulation, etc. can be directly related to the up‐ or down‐regulation of specific pathways. For a comprehensive picture of the biochemistry of cells, the complementary information provided by analyzing culture media (exo‐metabolome) is extremely important.27 The NMR spectra of tissues reflect organ‐specific biochemistry (aerobic respiration, lipid metabolism, etc.) but are also affected by the heterogeneity of the tissue composition, the contribution from the extracellular matrix and the tissue microenvironment, so that their biochemical interpretation is more difficult than in cultured cells. Nevertheless, tissue samples are extremely valuable as direct reporters of the diseased organ, where variations in the metabolome with respect to a healthy status are expected to be most evident. The latter feature is shared with some compartmentalized biofluids; for example, cerebrospinal fluid (CSF) reflects the biochemistry of the central nervous systems, and exhaled breath condensate (EBC) that of respiratory pathways. The saliva metabolome not only changes in correlation with oral disorders but also in the presence of distant pathologies.28 An important contribution to the metabolome of fecal extracts originates from metabolites resulting from the gut microbiota.
Table 2.
Type of samples, pre‐analytical procedures, NMR sample preparation, NMR spectral acquisition.
| Type of | Pre‐analytical procedures[a] | Analytical procedures | |
|---|---|---|---|
| sample | NMR sample preparation[b] | NMR spectral acquisition[c] | |
| URINE | Collect the first urine of the morning after a minimum of 8 h fasting. Specify if collected in different daytimes or not fasting. Centrifuge the urine within 120 min after the collection at 1000–3000 RCF for 5 min at +4 °C and/or filtrate the samples by 0.20 μm cut‐off filter. Recover the urine in sterile condition making 1 mL aliquots. Store at −80 °C.29 |
Thaw at room temperature and shake. Centrifuge at 14000 RCF for 5 min at 4 °C.30 Add 630 μl of sample to 70 μL of potassium phosphate buffer (1.5 m K2HPO4, 100 % (v/v) 2H2O, 2 mm NaN3, 5.8 mm TMSP; pH 7.4).[d] Transfer 600 μL of each mixture into a 5 mm NMR tube.30 |
31Recommended magnetic field: 600 MHz Recommended acquisition temperature: 300 K 1D NOESY‐presat (Bruker: noesygppr1d) Scans: 32 Data points: 65 536 Spectral width: 12 019.230 Hz Acquisition time: 2.73 s Relaxation delay: 4 s Mixing time: 0.01 s 2D J‐RES (Bruker: jresgpprqf) Scans: 2 Data points direct dimension (F2): 8192 Data points indirect dimension (F1): 40 Spectral width direct dimension (F2): 10 026.738 Hz Spectral width indirect dimension (F2): 78.000 Hz Increment for delay: 12820.51 μs Acquisition time direct dimension (F2): 0.41 s Acquisition time indirect dimension (F1): 0.26 s Relaxation delay: 2 s |
| BLOOD |
SERUM
Collect blood into anticoagulant free tubes. Allow the blood to clot in an upright position for 30–60 min at RT. Spin centrifuge within 30 min from collection at 1500 RCF for 10 min at RT.29 PLASMA Collect blood into tubes contain an anticoagulant (preferred EDTA; avoid heparin). Centrifuge within 30 min from collection at 820 RCF for 10 min at 4 °C.29 |
Thaw at room temperature and shake. Add 350 μl of sample to 350 μL of sodium phosphate buffer (70 mm Na2HPO4; 20 % (v/v) 2H2O, 6.1 mm NaN3; 4.6 mm TMSP; pH 7.4).[d] Transfer 600 μL of each mixture into a 5 mm NMR tube.30 |
31Recommended magnetic field: 600 MHz Recommended acquisition temperature: 310 K 1D NOESY‐presat (Bruker: noesygppr1d) Scans: 32 Data points: 98 304 Spectral width: 18 028.846 Hz Acquisition time: 2.73 s Relaxation delay: 4 s Mixing time: 0.01 s 1D CPMG (Bruker: cpmgpr1d) Scans: 32 Data points: 73 728 Spectral width: 12 019.230 Hz Acquisition time: 3.07 s Relaxation delay: 4 s Total spin‐echo delay: 80 ms 1D Diffusion‐edited (Bruker: ledbpgppr2s1d) Scans: 32 Data points: 98 304 Spectral width: 18 028.846 Hz Acquisition time: 2.73 s Relaxation delay: 4 s Square gradient strength: 80 % of the maximum gradient strength (53.5 G cm−1) Square gradient length: 1.5 ms Diffusion time: 0.119 s 2D J‐RES (Bruker: jresgpprqf) For all the parameters see urine. |
| For both serum and plasma: recover supernatant in sterile condition making 0.5 mL aliquots. Store at −80 °C.29 | |||
| SALIVA | Collect saliva after refraining from eating, drinking, smoking, or using oral hygiene products for at least 1 h. Rinse the mouth twice with water before spitting the saliva in sterile tubes making 1 mL aliquots. Freeze asap at −20 °C and then store in liquid nitrogen.32 |
Thaw at room temperature and shake. Centrifuge at 14000 RCF for 5 min at +4 °C. Add 630 μl of sample to 70 μL of potassium phosphate buffer (1.5 m K2HPO4, 100 % (v/v) 2H2O, 2 mm NaN3, 5.8 mm TMSP; pH 7.4). Transfer 600 μL of each mixture into a 5 mm NMR tube.33 |
Recommended magnetic field: 600 MHz Recommended acquisition temperature: 300 K 1D NOESY‐presat (Bruker: noesygppr1d) Scans: 128 For all the other parameters see urine. 2D J‐RES (Bruker: jresgpprqf) For all the parameters see urine. |
| CSF | Collect CSF via lumbar puncture in sterile polypropylene tubes. Centrifuge asap at 2000 RFC for 10 min at RT. Collect the supernatant in sterile condition making 0.5 mL aliquots. Store at −80 °C.34 |
Thaw at room temperature and shake. Add 750 μL of sample to 150 μL of potassium phosphate buffer (0.9 m K2HPO4, 60 % (v/v) 2H2O, 1.2 mm NaN3, 3.5 mm TMSP; pH 7.4).[d] Transfer 600 μL of each mixture into a 5 mm NMR tube. |
Recommended magnetic field: 600 MHz Recommended acquisition temperature: 300 K 1D NOESY‐presat (Bruker: noesygppr1d) Scans: 256 For all the other parameters see blood. 2D J‐RES (Bruker: jresgpprqf) For all the parameters see urine. |
| EBC | Collect EBC after refraining from eating for at least 3 h using a condenser equipped with a saliva trap. Rinse the mouth twice with water before breathing through a mouthpiece for 15 min making a 1.5 mL aliquot. Transfer the EBC into glass vials closed with 20 mm butyl rubber lined with polytetrafluoroethylene septa and crimped with perforated aluminum seals. Before sealing, remove volatile substances by a gentle nitrogen gas flow for 3 min. Freeze asap in liquid nitrogen and then store at −80 °C.35 |
Thaw at room temperature and shake. Centrifuge at 14000 RCF for 5 min at +4 °C. Add 540 μl of sample to 60 μL of 2H2O containing 5.8 mm TMSP. Transfer 600 μL of each mixture into a 5 mm NMR tube. |
Recommended magnetic field: 600 MHz Recommended acquisition temperature: 300 K 1D NOESY‐presat (Bruker: noesygppr1d) Scans: 512 For all the other parameters see urine. 2D J‐RES (Bruker: jresgpprqf) For all the parameters see urine. |
| FECES | Collect feces in sterile containers. Add 2 mL of PBS/2H2O buffer (0.1 m, pH 7.4) to 1 g of each feces sample and homogenize the mixture by vortexing for 60 s.36 Sonicate for 30 min and centrifuge at 10000 RCF for 5 min at RT. Collect the supernatant in sterile condition making 1 mL aliquots. Store at −80 °C storage.37 |
Thaw at room temperature and shake. Centrifuge at 14000 RCF for 5 min at +4 °C. Add 630 μl of sample to 70 μL of potassium phosphate buffer (1.5 m K2HPO4, 100 % (v/v) 2H2O, 2 mm NaN3, 5.8 mm TMSP; pH 7.4). Transfer 600 μL of each mixture into a 5 mm NMR tube. |
Recommended magnetic field: 600 MHz Recommended acquisition temperature: 300 K 1D NOESY‐presat (Bruker: noesygppr1 d) Scans: 128–256 For all the other parameters see urine. 2D J‐RES (Bruker: jresgpprqf) For all the parameters see urine. |
| CELLS |
ENDO‐METABOLOME
Place cell plates into ice and rinse with PBS. Scrape and collect cells with PBS supplemented with 1 % Protease Inhibitor Cocktail and 1 % Phosphatase Inhibitor Cocktail. Sonicate and ultra‐centrifugate for 1 h at 4 °C. Collect the supernatant in sterile condition making 0.6 mL aliquots. Store at −80 °C freezer.23 |
Thaw in ice and shake. Add 540 μl of sample to 60 μL of 2H2O containing 5.8 mm TMSP. Transfer 600 μL of each mixture into a 5 mm NMR tube.23 |
Recommended magnetic field: 900 MHz Recommended acquisition temperature: 300 K 1D NOESY‐presat (Bruker: noesygppr1d) Scans: 128–256 Data points: 110 060 Spectral width: 17 942.584 Hz Acquisition time: 3.07 s Relaxation delay: 4 s Mixing time: 0.01 s |
|
1D CPMG
(Bruker: cpmgpr1d) Scans: 128–256 Data points: 110 060 Spectral width: 17 942.584 Hz Acquisition time: 3.07 s Relaxation delay: 4 s Total spin‐echo delay: 80 ms |
|||
|
EXO‐METABOLOME
Removal of the growing medium. Collect the medium in sterile condition making 1 mL aliquots. Store at −80 °C. |
Thaw at room temperature and shake. Add 350 μl of sample to 350 μL of sodium phosphate buffer (70 mm Na2HPO4; 20 % (v/v) 2H2O, 6.1 mm NaN3; 4.6 mm TMSP; pH 7.4). Transfer 600 μL of each mixture into a 5 mm NMR tube. |
Recommended magnetic field: 900 MHz Recommended acquisition temperature: 310 K 1D NOESY‐presat (Bruker: noesygppr1d) Scans: 32–64 Data points: 11 0060 Spectral width: 17 942.584 Hz Acquisition time: 3.07 s Relaxation delay: 4 s Mixing time: 0.01 s |
|
| TISSUES | Collect tissues in the most aseptic conditions possible. Cut tissue samples ≤0.5 cm in any single dimension. Snap freeze in liquid nitrogen within 30 min from collection. Stored in liquid nitrogen vapor. |
Trim frozen tissue samples (10–15 mg) to fit HR‐MAS ZrO2 rotor insert capacity. Fill the insert with 2H2O containing 5.8 mm TMSP. Cover the rotor inserts with plug and plug‐restraining screw and insert it into the 4 mm rotor for HR‐MAS.38 |
31, 38Recommended magnetic field: HR‐MAS 600 MHz Recommended acquisition temperature: 277 K 1D ZGPR Scans: 128–256 Data points: 32 768 Spectral width: 12 019.230 Hz Acquisition time: 1.36 s Relaxation delay: 2 s Rotor speed: 4 MHz 1D CPMG (Bruker: cpmgpr1d) Scans: 128–256 Data points: 32 768 Spectral width: 12 019.230 Hz Acquisition time: 3.07 s Relaxation delay: 1.36 s Total spin‐echo delay: 94 ms Rotor speed: 4 MHz |
[a] It is extremely important to: 1) minimize as much as possible the time between sample collection, processing and storage keeping the samples at 4 °C, if not differently specified; 2) use additive‐free tubes and laboratory materials to avoid sample contamination. [b] Minimize as much as possible the time between sample preparation and NMR spectral acquisition. [c] 1) Acquire NMR spectra in automatic mode using a refrigerated (4–6 °C) sample changer. 2) Acquire NMR spectra of samples in a totally random way, interleaving the samples of different groups to avoid batch effects. [d] According to Bruker guidelines.
Figure 3.
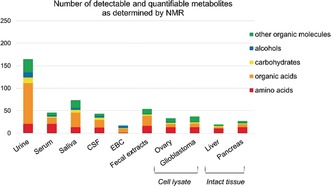
Number of detectable and quantifiable metabolites with ≥50 % occurrence in the 1H NMR spectra of different biological fluids (urine,18 serum,19 saliva,20 CSF,21 EBC,22 fecal extracts, cell lysates (e.g. ovary and glioblastoma cells)23, 24 and intact tissues (e.g. liver and pancreas).25, 26
Figure 4.
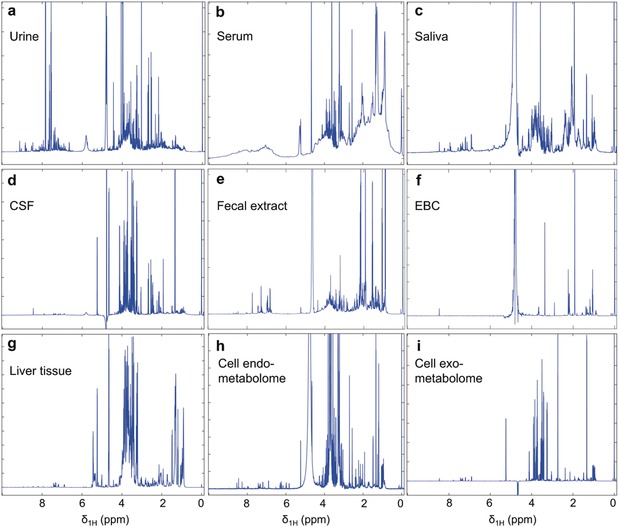
1H NMR spectra of: a) urine, b) serum, c) saliva, d) CSF, e) fecal extract, f) EBC, g) liver tissue, h) cell lysate (endo‐metabolome) and i) cell media (exo‐metabolome). The NMR spectra are recorded at 600 MHz, except for cell lysates and media (900 MHz).
Systemic biofluids, such as urine or blood serum and plasma, in general have a fainter biochemical correlation with a diseased organ or apparatus, but present two main advantages: the simple, noninvasive or minimally invasive collection, and the ability to reflect the overall response of the individual to a disease status. On the other hand, urine and blood are largely different in terms of chemical composition, with urine metabolites being heavily influenced by environmental factors such as food and liquid intake, while blood has a better defined and stable metabolome.
From the above considerations it is clear that preserving the chemical composition of the in vivo metabolome, and ensuring spectral reproducibility, are key factors for the significance of metabolomic analysis. In general, the entire workflow can be subdivided into two main phases, the pre‐analytical and the analytical phases. Both require specific procedures for accurate downstream analyses but are characterized by different degrees of development.
2.1.1. SOPs for Pre‐Analytical Treatment
Molecular analyses of biological samples are necessarily preceded by the so‐called pre‐analytical phase, which includes several steps such as primary sample collection, processing, transport, and storage. The impact of non‐appropriate pre‐analytical procedures on downstream metabolomic analyses can be heavy and is one of the main reasons that makes it difficult to compare metabolomic data collected in multicenter studies. Some of the molecules forming the metabolome are indeed very sensitive to sample conditions, and their levels can change drastically from collection to analyses. It is therefore important to develop simple and validated standard operating procedures (SOPs) for each type of sample to be strictly followed to ensure that the subsequent assay will determine the metabolome in the original sample and not an artificial profile generated during the pre‐examination process.
The development of validated procedures requires the systematic simulation of different pre‐analytical situations. In this way one can identify the critical steps and parameters that may influence the levels of the metabolites that are prone to degradation (with consequent accumulation of their degradation products). Systematic studies exist for some of the most common samples;39, 40 they have revealed enzymatic activities in the samples as main sources of variations. For blood derivatives, the presence of cells during sample processing heavily affects the concentration of glucose and lactate; therefore, serum and plasma harvesting should be initiated within 30 min from blood collection. Residual cellular activity is a problem also in urine; mild centrifugation and/or filtration upon sample collection are therefore required. While maintaining samples at low temperature throughout the pre‐analytical process is obviously an efficient strategy to slow down degradation reactions, one should avoid sample freezing before removal of cellular components, to prevent cell breaking and consequent release of enzymes into the biofluid.41 In tissues, it is obviously impossible to preclude cellular/enzymatic activities, and the best strategy consists in flash freezing samples in liquid nitrogen upon collection. This treatment reduces to a minimum the detrimental effects of the so‐called post‐resection or cold ischemia. More difficult to avoid is the impact of the intraoperative warm ischemia, that also alters the levels of metabolites associated to oxidative stress and apoptosis.42 If warm ischemia times can hardly be standardized during surgical procedures, their accurate annotation is recommended. Finally, particular attention should be devoted in the selection of sample collection vials that should not contain stabilizers or other contaminants that can interfere with the NMR analysis, giving rise to detectable NMR signals.43
A noteworthy example of activities focused on pre‐analytics was conducted within the FP7 project SPIDIA (standardization and improvement of generic pre‐analytical tools and procedures for in vitro diagnostics), which has led to the first example of technical specifications for pre‐examination processes in metabolomics.29
2.1.2. SOPs for Analytical Treatment and NMR Acquisition
The analytical step initiates with the NMR sample preparation, usually starting from cryopreserved samples. To ensure intra‐ and inter‐laboratory comparability, several efforts have been made to develop standardized procedures for i) NMR sample preparation and ii) spectral acquisition. The former is designed to obtain NMR‐quality samples; buffering is required to easily reference chemical shift values to existing databases; dilution has to be defined for subsequent absolute quantification. The latter relies on instrumental optimization, NMR pulse sequence selection, and choice of acquisition parameters. Instrument producers have been very active in the field. The selection of a “recommended” magnetic field facilitates comparison of spectra acquired in different laboratories and with available spectral databases (see Section 3.3.2). The use of 600 MHz spectrometers represents the best compromise between a good spectral sensitivity and resolution and affordable instrumental cost, and it is therefore considered the standard field for biofluid and tissue analyses. The economical aspect is essential for a technique that aims at being translated into clinical practices. Higher fields are often used to achieve better performances, especially with non‐clinical samples, such as cell cultures. Instrumental optimization includes an ample range of aspects: probe design, sample changer and software tools for automatic acquisition (temperature control, optimization of shim, water suppression, etc.) of about hundred samples. In most biofluids, low mass metabolites coexist with high mass biomolecules, such as lipids, proteins, and lipoproteins. Three NMR pulse sequences are used in these cases to selectively observe the different components: i) the nuclear Overhauser effect spectroscopy (NOESY) pulse sequence yields a spectrum in which both signals of metabolites and high molecular weight molecules are visible; the Carr‐Purcell–Meiboom–Gill (CPMG) pulse sequence enables the selective observation of small molecule components in solutions containing macromolecules (via T2 filtering); the DIFFUSION‐EDITED sequence permits the selective observation of macromolecular components in solutions containing small molecules. As summarized in Table 2, for several sample types, it is a common practice to acquire all three spectra. In defining the total acquisition time, one shall consider the stability of the sample under the selected experimental conditions. It should be pointed out that some authors prefer to physically remove macromolecular components via centrifugation with 3000 MWCO Amicon Ultra‐0.5 filters followed by NOESY acquisition rather than relying of CMPG filtering.44, 45 The recording temperature is clearly an issue that, in the case of HR‐MAS, sums up to destructive friction/centrifugation effects induced by spinning the sample in the rotor. Thus, the selection of recycling times between different scans is again a compromise between magnetization recovery (ideally 5×T1 of the slowest relaxing signals) and acquisition of a number of scans sufficient for good S/N before sample degradation. Typical spectra in Table 2 require 4–32 min acquisition. Refrigerated automatic sample changers, where NMR tubes are kept at 6 °C until acquisition, have been developed to facilitate the automatic acquisition of large sets of samples. Along the lines of a general standardization of the entire process involving metabolomic studies, there are ongoing efforts in the community to achieve open data standards and accessible repositories that allow researchers to store, exchange, and compare metabolomic data with pertinent metadata information.39, 46 For example, MetaboLights (https://www.ebi.ac.uk/metabolights/) or Metabolomics Workbench (http://www.metabolomicsworkbench.org) are repositories of metabolomic experiments and derived information.
3. Analysis of the Spectra
3.1. Processing
In the metabolomic work‐flow, the data processing step follows the acquisition of the raw spectra and is used to transform the data in a form suitable for subsequent statistical analyses (Figure 5). Each NMR spectrum must be properly adjusted for phase and baseline. Both operations are usually performed automatically; manual adjustment is discouraged for metabolomic data, because it can introduce operator‐biased artefacts. The spectra need also to be properly aligned to a reference signal, using a chemical shift standard, ideally not interacting with any sample component (for instance deuterated trimethylsilylpropanoic acid (TMSP) may bind macromolecules), thus in samples like serum/plasma, tissues and cell extracts an alternative internal reference is preferred (e.g. the anomeric signal of glucose).47 For the same reason, use of TMSP as a standard for quantification is discouraged. For the absolute quantification of metabolites, alternative approaches have been proposed, such as the production of an artificial NMR signal based upon PULCON method48 or the ERETIC method.49
Figure 5.
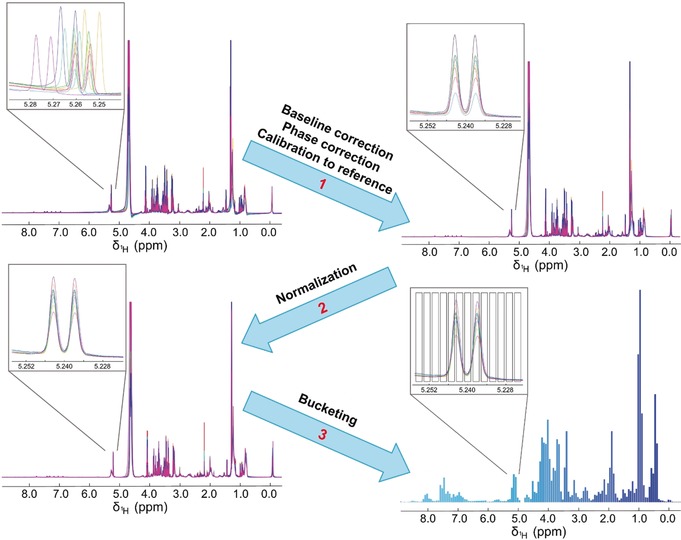
Key stages of NMR spectral processing: 1) baseline correction, phase correction, calibration to an internal reference peak (e.g. glucose, TMSP, etc.); 2) normalization; 3) bucketing.
3.2. Normalization for Different Types of Spectra/Samples
In order to compare signal—or bucket—intensities in different samples, one should refer to the same amount of total sample. In biofluids this seems an obvious task, as one prepares the NMR sample starting from the same volume of the original fluid (Table 2). In practice, a preliminary step of correction for dilution effects is needed due to the presence of large physiological variations in concentration. For examples, urines exhibit significant metabolite concentration fluctuations depending on the hydration state of the individual (in turn related to water/food intake, physical exercise and sweating, etc.),50 therefore, a global intensity correction by means of normalization is a critical initial step. For biofluids under stricter physiological control (like serum/plasma) normalization is less crucial, but could be beneficial to compensate for non‐physiological sources of variations emerging from (small) experimental inaccuracies and/or technical artifacts.51 Total area normalization is a common practice but there are more sophisticated and reliable methods. In the quest for the “optimal” normalization method, a plethora of strategies/algorithms have been developed. Table S1 (Supporting information) shows and briefly describes 23 state‐of‐the‐art methods belonging to five main categories, including those not explicitly developed for metabolomics but borrowed from other fields. Although several comparisons are reported in the literature, a definitive consensus on which normalization method to use in which case is still lacking. For biofluids, Probabilistic Quotient Normalization (PQN),52 Quantile Normalization,53 Cubic Splines Normalization (CSN),54 and Pairwise Log‐Ratios (PLR),55 are generally described as good choices50, 51, 55, 56 that outperform total area normalization. For instance, PQN has been successfully used with EBC,57, 58 saliva,33 and urine59 samples. For tissues, cell lysates or cell/tissue extracts, exact quantification of the starting material is not straightforward. For cells, it can be assumed that an increase of cell number produces a linear increase of metabolite signal intensities, thus data normalization is usually performed to the number of cells.60, 61 Alternatively, one could refer to the total DNA or total protein content.61 On the same line, for tissues, normalization according to the sample weight is recommended.62 If this information is not available, total area normalization represents a good method, as the total spectral area can be considered proportional to the number of cells or the weight of tissues.
3.3. Different Experimental Approaches in Metabolomics
Depending on the biological question at issue, metabolomic analysis can be planned with two different methodological approaches: targeted and untargeted analyses (Figure 6). The targeted approach involves the monitoring of a panel of metabolites selected a priori on the basis of known metabolic pathways or pre‐identified biomarkers that are undoubtedly associated with the disease or condition of interest.11 These selected metabolites must be unambiguously assigned and quantified in the samples. Conversely, untargeted metabolomics provides a global view of a sample by analyzing all (or as many as possible) measurable analytes present, including unknown chemicals.63 The latter purpose can be achieved via two strategies: fingerprinting or profiling. Fingerprinting is a global, rapid evaluation of an NMR spectrum as a whole that can be considered a “fingerprint” of all (assigned or unassigned) detectable metabolites present in that biological sample.64 This can be achieved only by transforming NMR spectra in matrices of data for example, through bucketing, that is, a procedure used to reduce the total number of variables and to compensate for small misalignments in the spectra. One bucket (or bin) is a little portion of the spectrum, usually with a width of 0.02 or 0.04 ppm. The integrated spectral intensity within each bin is then calculated to obtain the variables used for feeding the statistical algorithms. Although many more sophisticated algorithms for bucketing exist, this equidistant binning is the most commonly used method65 and often works quite well despite its simplicity.66 Alternatively, some practitioners prefer to work with full resolution spectra, totally avoiding binning. In such cases, specific algorithms67 for peak alignment are needed (e.g. the very efficient icoshift68), to assure that peaks can be compared across multiple spectra. The fingerprinting approach is essentially utilized to provide sample classification. Indeed, metabolic fingerprinting aims at discriminating specimens in relation to different biological conditions (e.g. presence/absence of disease, before/after treatment), in turn characterizing a specific health state with a unique metabolic pattern.11 Metabolic profiling, in contrast, deals with the determination of the concentrations of all quantifiable metabolites in a biological sample. Profiling provides considerably more meaningful data from a biochemical perspective since it also enables the identification of metabolites and metabolic pathways associated with a specific physiological or pathological condition. However, it is important to mention that the spectral processing required to deconvolute 1D NMR spectra, in order to obtain concentrations, may not be straightforward and is not yet completely automated. The molecules quantifiable via profiling are significantly less numerous than those contributing to the fingerprint (in the case of urine <50 %).64 The latter, therefore, is the best tool for sample classification and to build statistical models.
Figure 6.
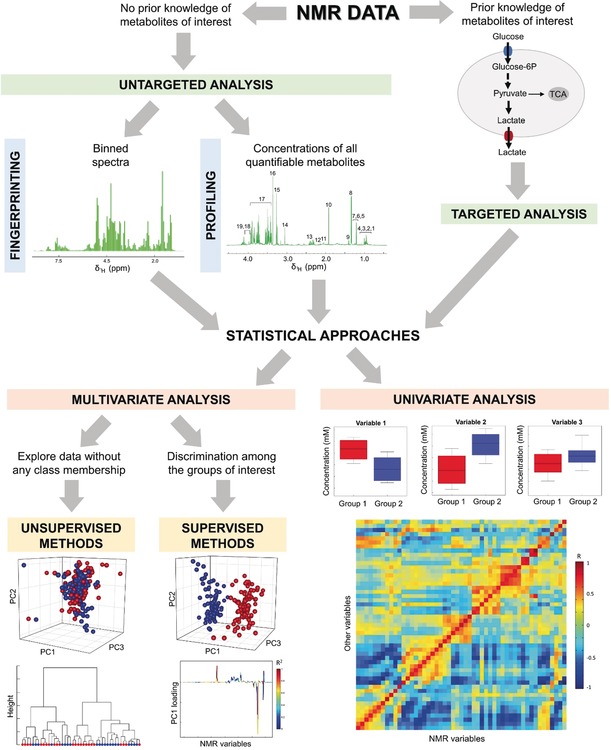
Different metabolomic strategies. If knowledge of the metabolites or metabolic pathways of interest is available, a targeted approach, which involves the analysis of only these specific metabolites, is the preferred choice. In the absence of prior knowledge, the problem can be addressed by analyzing the spectrum, with a so‐called untargeted approach. Untargeted analysis can be achieved via metabolic fingerprinting or profiling. The former is a global evaluation of all of the features of a binned spectrum without identification of single metabolites; the latter deals with the analysis of all quantifiable metabolites. Each of these three different sets of data can be addressed by either multivariate or univariate analyses. Multivariate methods are routinely used to visualize biological data, to identify possible clusters, and to build predictive models; these can be divided into two main categories: unsupervised analyses to explore data without any class membership and supervised analyses to discriminate among known groups of interest. Conversely, univariate methods are used to identify metabolites and thus metabolic pathways that are altered or correlated with specific biological conditions.
3.3.1. Multivariate Analysis for Classification and Modeling
Starting from the available data (full or binned NMR spectra, or a list of concentrations) arranged in rows (samples) and columns (variables) in a data matrix, the principal aims of metabolomics can be summarized in four goals: i) visualize the overall differences, trends, relationships, and correlations among different samples; ii) detect whether there is a significant difference between investigated groups (e.g., healthy vs. diseased subjects); iii) highlight the spectral regions mostly contributing to these differences and, iv) construct a predictive model for the correct classification of new samples.69 Multivariate statistics is the key to achieve these goals, by either i) unsupervised or ii) supervised methods. Unsupervised methods are utilized to summarize, explore, and discover clusters or trends in the data unlabeled with any class membership; therefore, no prior assumptions or knowledge of the data are needed.70 Unsupervised methods usually represent the first step in data analysis, helping to visualize the data and to discover possible outliers. One effective class of exploratory approaches involve the projection of the data in a new space using just few dimensions (data reduction). This includes classical methods, such as Principal Component Analysis (PCA),71, 72, 73, 74 and Independent Component Analysis (ICA),75, 76 as well as promising new ones developed for metabolomics, such as Group‐Wise Principal Component Analysis (GPCA).77 Another interesting class of approaches is data clustering, which consists in the assignment of a set of samples into subsets (so‐called clusters) so that samples belonging to the same cluster are similar in some sense (i.e. they share similar metabolic features). Classical clustering methods, such as K‐Means (KM)78, 79, 80 and Partition Around Medoids (PAM),81 have played a historical role in the data mining field. More recent methods include Spectral Clustering (SC)82, 83 and KODAMA.84, 85 Another option to obtain visually interpretable maps that capture inherent relationships among observables are the Self‐Organizing Maps (SOM),86, 87 developed in the neural networks field.
In contrast, supervised approaches use a priori knowledge to generate models that are tightly focused on the effects of interest. The goal of supervised data analysis is to find a rule to extend the knowledge already available from pre‐existing samples to new samples, so that predictions can be made (i.e. to classify normal vs. abnormal samples for diagnosis and/or prognosis). In doing so, the characteristics of the training set (data already available) are defined, “learned” by the statistical method, and applied to predict the test set (the unlabeled samples to be classified). Supervised analysis includes methods based on projection and data reduction, such as Partial Least Squares (PLS)88, 89, 90, 91 and its variant Orthogonal Partial Least Squares (OPLS);92 methods based on machine learning, such as K‐Nearest Neighbors (K‐NN);93, 94 and methods based on neural networks architectures, such as the potentially ground‐breaking paradigm of deep learning.95, 96 An overview of the main multivariate statistical techniques employed in metabolomics is reported in Table S2. It is worth noting that all the multivariate algorithms can be used on full resolution spectra, on the bin data matrix, or a panel of quantified metabolites, depending on the chosen approach.
To demonstrate the performance of a predictive model, proper validation of the results is performed using cross‐validation strategies,97 or an external and completely independent dataset. Moreover, a permutation test is often advocated to calculate the statistical significance of the results. Several measures of the model quality exist, such as those derived from the confusion matrix (i.e. sensitivity, specificity, and accuracy). The use of independent training and validation sets is the preferred approach in the medical community, but requires collecting samples from different hospitals, even from different countries. In this way, weaknesses due to for example, not perfectly identical SOPs (see Section 2) can become apparent. Conversely, artificially dividing in two groups samples collected under identical conditions is not affected by this type of problems. Thus, the selection of the best approach strongly depends on the aim of the research: cross‐validation on a unique collection of samples should be recommended for exploratory studies, where the aim is to see whether a fingerprint for a specific condition exists. Independent validation is then recommended to further demonstrate that the fingerprint is still strong also in the presence of “environmental noise”. Immediately jumping to this second strategy may lead to conclude that a fingerprint does not exists while it may be only obscured by sub‐optimal procedures that could be easily fixed.
3.3.2. Identification, Quantification, Univariate Analysis
The process of metabolite identification in NMR spectra, especially for less common biospecimens, is not straightforward due to the high complexity of their 1H‐NMR spectra. Usually many resonances can be directly assigned in one‐dimensional spectra based on chemical shifts and multiplicity. This task is facilitated by databases, often freely available: in particular, for human metabolites, the Human Metabolome Database (HMDB)98, 99, 100 is becoming the de‐facto standard reference database. For doubtful cases, the addition of standard molecules (spiking) to the NMR sample could be of help. However, two‐dimensional spectra are often required to assign new metabolites, as briefly summarized below (and reviewed in more details in Ref. 101): i) 1H‐1H J‐resolved (J‐RES), to provide information about multiplicity and coupling patterns, ii) 1H‐1H Correlation Spectroscopy (COSY) and 1H‐1H Total Correlation Spectroscopy (TOCSY), to provide, respectively, short and long range scalar connectivities, iii) natural‐abundance heteronuclear experiments that use 1H in combination with other nuclei, for example, 13C‐1H Heteronuclear Single Quantum Coherence Spectroscopy (HSQC), Heteronuclear Multiple‐Quantum Correlation (HMQC) and Heteronuclear Multiple‐Bond Correlation Spectroscopy (HMBC), to obtain information on the direct scalar coupling between 13C and 1H nuclei.101 The quest for a completely automatic assignment tool is an active research field, with promising achievements (Figure 7, see also Section 7).14 Of course, as shown in Table 1, NMR and MS are two complementary approaches and combined MS/NMR can be used to correctly identify a wide range of metabolites.102, 103
Figure 7.
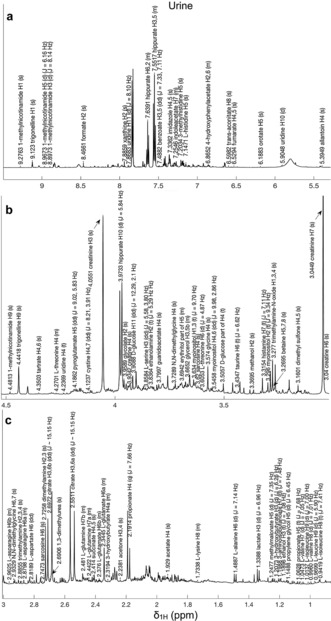
The automated assignment of several 1H NMR signals of >60 urine metabolites, performed by the urine shift predictor.14 The urine spectrum is divided into 3 regions: a) 9.5–5.5 ppm, b) 4.5–3.0 ppm and c) 3.0–0.9 ppm.
Further, while NMR spectroscopy is an intrinsically quantitative technique (signals are proportional to the concentration of nuclei), the ability to accurately and reproducibly quantify signals from metabolites in complex mixtures is complicated by spectral crowding and signal overlapping, as well as from raising of the baseline caused by the presence of large molecular mass components. In order to solve the latter problem, a sample treatment has been proposed for serum/plasma, that includes a centrifugation step at the level of sample preparation to remove large molecules.44 In our view, any sample manipulation represents a potential source of experimental errors; therefore, we prefer broad signal suppression by CMPG. Nevertheless, a careful estimation of the effect of spectral acquisition parameters on signal intensity for the different metabolites shall be conducted. Spectral crowding and signal overlap are unavoidable in the absence of chemical separation.
Tools for (semi) automatic quantitation have been developed. Among them there are BATMAN,104 BAYESIL,44 ASICS105 and the NMR Suite Software Package (Chenomx Inc., Edmonton, Canada). The first three are mostly automated computational tools based on Bayesian inference (BATMAN and BAYESIL) or linear models (ASICS). ASICS is freely available. BATMAN is also freely available, it performs quite well, but it is computationally very demanding. BAYESIL is faster, but it is commercial and requires a dedicated kit for the preparation of the samples. The B.I. (Bruker IVDr) platform for analysis and quantification of metabolites in biofluids (urine, CSF, serum/plasma) has been recently released by Bruker BioSpin.
NMR Suite is another commercial software. It is a complete computer‐assisted tool for analysis and deconvolution of the spectra, permitting the user‐guided fitting, integration, and quantitation of the selected peaks. However, being mostly manual, it is time consuming and it needs a skilled operator. As witnessed by the total number of citations, BATMAN, BAYESIL and NMR Suite (61, 50, 90 Web of Science citations, respectively) are the most commonly used packages by practitioners.
In order to find a metabolite (or a panel of metabolites) that can be considered as a possible biomarker for the specific pathology or condition under investigation, each metabolite needs to be analyzed independently of all the others without considering any possible interactions. Univariate statistical analysis is the key to achieve this goal: statistical tests, correlation analysis and Receiver Operating Characteristic (ROC) curves are the most used approaches.
On the biological assumption that metabolite concentrations are not normally distributed, non‐parametric tests are often utilized. The Wilcoxon–Mann–Whitney106 test is the non‐parametric analogue of the classical t‐test to compare the distribution of a metabolite in two groups. The null hypothesis (H0) claims that two randomly selected samples from two populations actually belong to the same population; the alternative hypothesis (Ha) states that H0 is false and the two population are distinct. A variant of this test also exists, called the Wilcoxon signed ranks test,107 that is suited to compare two matched samples from a repeated measurement in the presence of a paired study design. When the groups are more than two, Kruskal–Wallis test108 (the analogue of the parametric Analysis of Variance) or Friedman test109 (for paired samples) are employed. A summary of the main statistical tests for univariate analysis of metabolites is reported in Table S3.
The output of all these tests is a P‐value, that expresses the probability of obtaining a result equal to or more extreme than what was actually observed, assuming the null hypothesis true. Conventionally, when the P‐value is less than the significance level of 0.05 or 0.01 the null hypothesis is rejected, and the metabolite is deemed statistically different in the groups of interest. When several metabolites are tested together, to avoid random false positives, multiple testing corrections need to be adopted: Bonferroni110 and Benjamini–Hochberg111 are the most widespread methods. However, P‐values need to be taken with care: they are often misused, misunderstood and misinterpreted;112 for this reason statisticians have proposed replacing (or accompanying) P‐values with effect size113 (a standardized measure of the magnitude of the observed phenomenon) as an alternative (complementary) measure of evidence.
ROC curves are graphical representations that illustrate the diagnostic ability of a binary classifier (e.g. the concentration of a biomarker to pinpoint a disease or the efficacy of a composite prognostic score to catch the risk of developing a disease). The curve is created by plotting sensitivity versus one minus specificity for all possible thresholds of the test. Accuracy is measured by the area under the ROC curve (AUC). An area of 1 represents a perfect test; an area of 0.5 represents a worthless test. ROC analysis could also be employed to check the performance of a multivariate supervised classifier. In this case, the cross‐validated output of the classifier (a continuous response vector) is used as the input for the calculation of the ROC curve. Again, an AUC near 1 indicates a good classifier. ROC analysis is of particular importance because it provides a simple tool to select optimal models and to discard suboptimal ones, thus permitting a direct cost/benefit analysis of a diagnostic test.
Pearson correlations can be calculated to test whether there is an association (linear dependence), between metabolites and clinical data or biological features. Correlations are expressed by a coefficient (R) which ranges between +1 (totally correlated), 0 (no correlation) and −1 (totally anticorrelated). From metabolite to metabolite correlation maps, metabolic networks with a biological meaning can be inferred.114, 115, 116, 117
4. Biomedical and Biochemical Significance
Metabolomics is focused on the analysis of intermediates and end products of metabolism in the form of endogenous (gene‐derived metabolites), exogenous (environmentally derived metabolites) and gut microbiota‐derived metabolites. For this reason, metabolites, unlike genes and proteins, are easier to correlate with the phenotype and act as direct signatures of biochemical activity, since they play a central role in disease development, cellular signaling and physiological control.118 Metabolomics monitors the global outcome of all exogenous and endogenous factors, without making assumptions about the effect of any single contribution to that outcome. This makes metabolomics a perfect instrument to investigate and understand the molecular mechanisms of human health and disease.1 Metabolomics can contribute to precision medicine for the comprehension of individual susceptibility to drug administration, nutrition and life style interventions, influence of environmental factors, as well as to the characterization of the metabolic signature of diseases for diagnostic and prognostic purposes; and to the understanding of the biochemical causes at the basis of different pathophysiological conditions.
4.1. Individual Fingerprint
One of the main findings of metabolomics is that each individual is “chemically different” from any other in terms of small molecules composition of its systemic biofluids like urine, blood and saliva.33, 119, 120 Our research group has significantly contributed to define and characterize this strong chemical signature, called individual metabolic phenotype or metabotype.119, 121, 122, 123, 124, 125 In urine, a biofluid whose composition is strongly affected by daily variations, the presence of an invariant part of the human metabolic phenotype, characteristic of each individual has been established through multivariate statistical analysis of several samples collected on different days. This has led to the definition of the “metabolic space”, where samples from each subject occupy a well‐defined and circumscribed sub‐space and each individual can be discriminated from all others with near 100 % accuracy (Figure 8 a). The individual metabolic phenotype is the result of a combination of many factors such as genotype, host metabolism, gut microflora composition, dietary habits, physical activity etc. The collection of multiple urine samples is fundamental to identify the invariant part from the day‐to‐day variability of the subjects. We have shown that around 20 urine samples per individual, collected in different days, are enough to achieve an individual discrimination of 97–98 %. By using 40 urine samples per person values above 99 % can be obtained.119, 121, 122 Using multiple urine collections in the same day for a few days (e.g., 4–5 samples per day, for 10 days) is substantially equivalent to 1 sample per day for 40 days.125 In addition to being characteristic of each individual at any time point, metabotypes in urine are stable over a time scale of 8–10 years. Over this time‐frame, significant metabotype drifts were observed only after the onset of important pathophysiological conditions; the “normal” individual metabotype was regained if the condition was reversed (Figure 8 b).122 These results clearly underline the enormous value of this approach to monitor subjects during their life‐span, in order to detect early disease onset, its progression, response to therapy or dietary intervention, etc., with obvious applications in the context of precision medicine.
Figure 8.
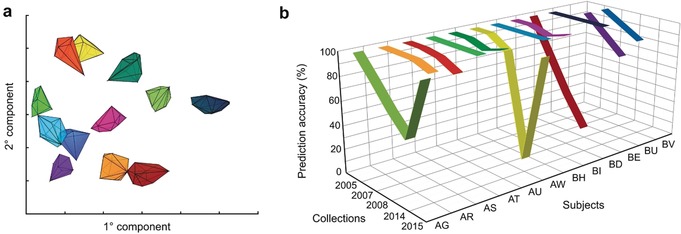
The NMR‐derived urine individual metabolic phenotype and its stability over time. a) Multiple urine samples collected from 12 healthy donors (each identified by a given color) over a period of 20 days occupy a well‐defined portion of the metabolic space (PCA‐CA score plot), thus indicating that intraindividual variations are much smaller than interindividual differences. This is due to an invariant part of the metabolome characteristic of each individual, which identifies the individual phenotype. b) The individual phenotype over the time scale of 10 years is very stable in the absence of physiopathological conditions that can cause abrupt deviations (subjects AG, AW, BD). If this condition is over, the individual phenotype reverts back (AG, AW); adapted from Ref. 122.
A strong signature of the individual phenotypes has been also characterized in blood.120 Due to its nature, blood is free from most of the daily variations observable in urine, and the intra‐individual variability is very low. Thus, good discrimination among individuals can be obtained using fewer samples per person. A high degree of stability of the individual phenotype in serum over 7 years has been also demonstrated.126
A clear individual metabolic phenotype exists also in saliva33 and human breath,127 although it is slightly less strong than the urinary or blood phenotype.
The “individual metabolic phenotype” of urine and blood, and possibly that of other biofluids, possesses suitable characteristics for personalized healthcare solutions, being typical for each subject, stable over time and essentially independent of lifestyle and dietary intake,119, 121, 122, 125, 128, 129, 130 thus allowing us to monitor individual status and response to different stimuli.131, 132, 133
4.2. Metabolomics and Diseases
For metabolomics applications in medicine, the ability to monitor the individual metabolic fingerprint parallels the ability to identify clear disease signatures. Classically, metabolomics has been used to characterize metabolic profiles of diseases, with the intent of discovering new biomarkers and identifying biochemical pathways involved in disease pathogenesis. NMR metabolomics has already increased our understanding of cellular and physiological metabolism, helping to identify many unexpected biochemical causes for several important chronic and complex diseases,134, 135 such as cancer,23, 136, 137, 138, 139, 140, 141 cardiovascular diseases,115, 142, 143, 144, 145, 146 diabetes,147, 148, 149, 150 and obesity.151, 152, 153 One of the most striking aspects of metabolomics is the ability to detect at the systemic level alterations in the metabolome, which correlate with pathological states even for those diseases that are not immediately associated to metabolism. Such sensitivity, most probably involving immune mechanisms, is particularly promising to monitor the individual response to illness. NMR metabolomics has the most ambitious objective of detecting early metabolic perturbations even before the manifestation of disease symptoms. Indeed, the goal of precision medicine is to customize subjects’ therapeutical treatments according to their specific omic profiles/fingerprints.13 NMR metabolic profiling in large‐scale epidemiologic studies, though is still recent, has uncovered novel biomarkers for various diseases, has contribute to their etiologic understanding, and has shown the ability to predict disease risks and the effects of drug administration, with the potential to translate into multiple clinical settings.140, 154, 155, 156, 157
The literature available concerning metabolomics applied to clinical examples is extremely wide; we are presenting here a limited number of examples of application to a few diseases using different types of samples.
Coeliac disease (CD) is a multifactorial and complex disorder involving genetic and environmental factors that induce autoimmune response and nutrients malabsorption, thus having great potential impact on metabolism. Our group has extensively described the NMR metabolic signature of CD in serum and urine by identifying important alterations of the metabolic profiles of patients with respect to healthy controls, especially for what concerns energy and ketone body metabolism, and gut microbiota alterations.158, 162 Furthermore, our statistical model has demonstrated to be effective in monitoring the adherence and efficacy of the gluten free diet in CD patients (Figure 9 a).
Figure 9.
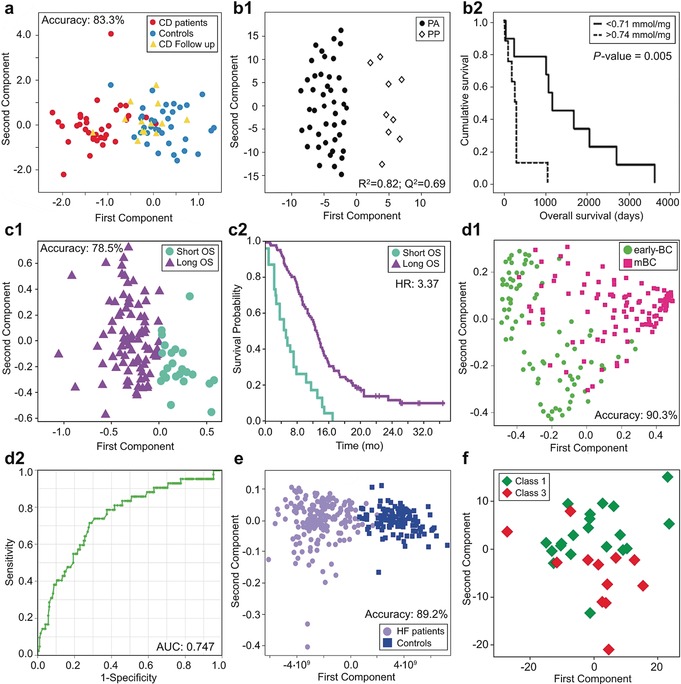
Applications in the biomedical field. a) Monitoring the efficacy of the gluten free diet in CD patients: predictive clustering of CD patients after 12 months on a gluten‐free diet (yellow triangles) using a PLS‐RCC model built on CD patients (red circles) and healthy controls (light blue circles).158 b) Metabolomic profiling of tumor tissues predicting clinical outcome of pancreatic adenocarcinoma patients.26 b1) OPLS‐DA model discriminates pancreatic adenocarcinoma tissues (black circles) with respect to pancreatic healthy tissues (white squares), R2 and Q2 were used to measure model quality: R2> 0.7 and Q2>0.5 can be considered as a good predictor. b2) Ethanolamine concentration (the threshold value was 0.740 nmol mg−1) as a single metabolic biomarker for the prediction of overall survival in patients with PA. Kaplan–Meier curves show differences between long‐term (black line) and short‐term (segmented line) survival patients. c) Prediction of overall survival in patients with mCRC. c1) PLS‐CA clustering for long OS (purple triangles) and short OS (aquamarine circles) mCRC patients. c2) Kaplan–Meier curves showing survival probability based on the 1H‐NMR metabolomic model.159 d) Identification of early‐BC patients at increased risk of disease recurrence via serum metabolomics. d1) Clustering of serum metabolomic profiles between early‐BC (green circles) and mBC (pink squares) patients using a Random Forest (RF) classifier in the training set. d2) Prediction of relapse in the test set containing 192 relapse early‐BC patients and 42 early‐BC patients free from disease up to 6 years (ROC curve).160 e) Discrimination between HF patients (lilac circles) and healthy subjects (dark blue squares) using an OPLS‐DA.143 f) Pharmaco‐metabonomic phenotyping: a scores plot from PCA of the pre‐dose of paracetamol urine spectra. NMR data discriminates low histology damage (Class 1, green squares) and severe histology damage (Class 3, red squares).161
A striking finding was that potential coeliac people, that is, those with typical antibody signature of CD but without symptoms and without or slight intestinal damage, are already classified as coeliac from a metabolomic point of view, suggesting that the CD metabotype precedes the manifestation of the disease.2
Cancer is probably the most studied pathology so far via NMR metabolomics;163, 164, 165, 166, 167, 168 Cancer patients present metabolic profiles that are different from those of healthy controls and patients with benign diseases; moreover, the site, the stage, and the location of the tumors may differently affect the metabolome.169 The most fascinating aspect of metabolomic research is the challenge of translating these evidences into diagnostic or prognostic models capable of ensuring early diagnosis or prediction of recurrence and of the final outcome of the patients with results comparable or even better with respect to classical methodologies. Metabolomic analysis of intact tissues via HR‐MAS offers the possibility to obtain information directly at the level of the organ affected by the tumor, and these ones could have prognostic values. HR‐MAS NMR analysis of pancreatic intact tissue can provide important information for the characterization of pancreatic adenocarcinomas (PA) and could discriminate PA from healthy pancreatic parenchyma (PP) (Figure 9 b1). Ethanolamine resulted as a possible single metabolic biomarker (Figure 9 b2) for the prediction of long‐term survival.26 The analysis of systemic biofluids such as blood can give fundamental information regarding the patient prognosis. Metastatic colorectal cancer patients (mCRC) and healthy controls were clearly discriminated by multivariate statistical analysis of the serum NMR data; mCRC patients presented various metabolic alterations regarding energy metabolism and inflammatory response.159 Furthermore, mCRC patients clustered according to their overall survival (Figure 9 c1) with a better performance with respect to traditional biomarkers (hazard ratio=3.37).
Breast cancer (BC) is a complex and heterogeneous disease which has been extensively characterized by many platforms such as clinicopathological risk factors and various ‐omic techniques including metabolomics, which has shown significant promises in diagnosis, prognosis, and patient management.170, 171, 172, 173, 174, 175 In this precision medicine era, however, development of tailored oncological treatments for BC and accurate instruments to identify patients at high risk of disease recurrence are lagging.176 Our group has demonstrated that NMR serum metabolomics could contribute significantly to this aim: in the first single center pilot study, relapse was predicted with quite good accuracy in both training set (90 % sensitivity, 67 % specificity, and 73 % predictive accuracy, AUC: 0.863), and validation set (82 % sensitivity, 72 % specificity, and 75 % predictive accuracy, AUC: 0.824).177 The results have been reproduced in a multicenter study by analyzing 699 serum samples collected in the framework of an international phase III clinical trial (Figure 9, d1, d2).160
Cardiovascular diseases provide another excellent target for metabolomics, especially for what concerns early diagnosis. For example, heart failure (HF) is a complex, chronic, progressive syndrome in which the heart muscle is unable to pump enough blood to cope with the body needs for blood and oxygen. Unfortunately, HF is asymptomatic in its first stages, when medical intervention would still be effective; therefore, early assessment of this disease is a crucial and challenging task.178 We have shown that NMR serum metabolomics is an excellent instrument for the discrimination between HF patients and healthy controls (Figure 9 e). Even more important, the metabolomic fingerprint does not change with disease progression; therefore, it could really represent a useful tool for the purpose of early diagnosis.143
Metabolomics also offers a cost‐effective and productive route to uncover new targets for drug discovery, and to predict and monitor individual response to drug treatment (pharmacometabolomics).13, 58, 179, 180, 181, 182 For these purposes, animal models represent the preferred choice, especially for first explorative studies. Since animal models ensure a high rate of standardization, they enable a clearer identification of the intervention effects using a reduced number of subjects with higher discrimination power (often even unsupervised analysis is effective in identifying clusters of interest). As an example, Clayton and co‐workers in their study on paracetamol (acetaminophen) administration showed that NMR pharmacometabolomic phenotyping could be successfully used.161 They analyzed pre‐ and post‐dose urine samples from 65 rats given a single toxic‐threshold dose of paracetamol, observing that pre‐dose NMR discrimination is related to the post‐dose variation in histopathology (Figure 9 f).
For the sake of completeness, it is important to underline that the use of the metabolomic approach is not confined to human medicine. Several potential applications in the veterinary industry are possible, in particular in the framework of disease diagnosis and investigation, optimization of health and production, drug discovery and animal welfare.3, 183, 184, 185, 186, 187
4.3. Pathway Analysis
Once the biomarkers, or metabolic profiles, whose concentrations are altered due to the biological processes under investigation have been identified, pathway analysis can be performed with the final aim of obtaining a mechanistic explanation of the changes observed. Pathway analysis strengthens the information generated by metabolomic analysis. In the framework of diseases, for instance, the identification of the altered metabolites has been successfully correlated with the biological pathways and processes involved in pathogenesis and disease progression.149, 158, 188, 189 In particular, pathway analysis has been applied in several cancer studies to unravel the processes at the basis of the oncogenic phenotype.137, 190
Online biological databases, such as KEGG191 (Kyoto Encyclopedia of Genes and Genomes) Pathway Database, provide information of a large number of metabolic pathways and can be easily used to identify and visualize the metabolites involved in several biological processes. Furthermore, the development of metabolite set enrichment analysis (MSEA) methods192 strongly helps the identification and the functional and/or biological interpretation of patterns of metabolite concentration changes in a biologically meaningful context. Publicly available tools implementing these methods are available.193 Among these tools, MetaboAnalyst—a comprehensive tool for NMR‐ and MS‐ based metabolomic analysis and interpretation194 includes sections for metabolic pathway analysis and metabolite set enrichment analysis. These approaches examine the metabolites present in the biological matrix at a particular time point, thus providing steady state levels of metabolites.
Stable isotope‐resolved metabolomics (SIRM) provides a more dynamic assessment of specific metabolic pathways using stable isotopes (13C, 15N or 2H).195 In this approach, isotopically enriched precursors such as 1,2‐13C2‐glucose or 13C5, 15N2‐ glutamine, are administered to a biological system, enabling the analysis of the fate of individual atoms of a particular metabolite, and thereby a robust tracing of metabolic pathways and of their fluxes in cells, animals and humans (fluxomics). During the last years, the use of isotopic labelling tracers has been successfully applied to unveil the different metabolic pathways activated under different conditions in cancer, providing important information for the development of new therapeutic strategies.196, 197, 198
According to the type of samples, the biological interpretation of changes in concentration levels of metabolites through pathway analysis may be tangled. The simpler the system studied, the easier the reconstruction of the biological pathways involved in the investigated process. For instance, in cultured cell models the biological explanation is the most immediate. Our research group investigated the metabolic changes induced by the oncogenic enzyme Sphingosine Kinase 1 (SK1) in ovarian cancer.23 Using an NMR‐untargeted approach, we demonstrated that SK1 expression is sufficient to alter radically the metabolomic profile of ovarian cancer cells influencing both the glycolytic pathway and the tricarboxylic acid cycle (TCA) (Figure 10). With this approach we have highlighted the occurrence of SK1‐induced Warburg effect, and pointed out that SK1 expression also modulates the pentose phosphate pathway and nucleotide biosynthesis, both fundamental to satisfy the anabolic demand of high proliferating cancer cells.
Figure 10.
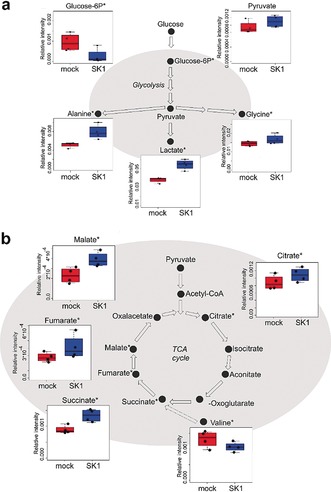
SK1 expression induces a metabolic switch known as the Warburg effect in A2780 ovarian cancer cells.23 From the comparison between A2780 mock (blue) and SK1‐expressing cells (red) emerges that expression of SK1 induced a high glycolytic rate (a), characterized by increased levels of lactate, and decreased oxidative metabolism, associated with the accumulation of intermediates of the TCA (b). Changes in metabolite levels caused by SK1 overexpression were statistically significant by paired Wilcoxon test, *P‐value<0.05.
5. General Considerations on Experimental Design and Significance of the Results
As in any other scientific field, before initiating a metabolomic study it is of primary importance to set up a correct experimental design, especially regarding the total number of samples to collect, the type of samples to be analyzed, the number of different groups to be examined, the inclusion and exclusion criteria, the choice of the control group, the kind of clinical design (prospective, retrospective, paired/unpaired). Well planned studies ensure that the experiments are efficient and help to avoid biases in the statistical analysis.199
Having in mind a clear object of the study and a well posed biological question is of course of great help in deciding an appropriate experimental plan. However, metabolomics, in its untargeted incarnation, does not require any initial hypothesis; therefore, it may happen that it is not the original needs that drive the experimental steps, but, conversely, metabolomics is applied to samples already collected, and often for different purposes. For this reason, researchers in metabolomics are faced with challenges posed, for instance, by samples not properly collected and/or stored (e.g. according to SOPs designed for different purposes), causing unwanted sources of variation, lack of correct statistical power analysis or small number of samples for statistical analysis. Power analysis200 is sometimes forgotten by metabolomic practitioners, but it is a pillar of any good experimental design. Roughly speaking, statistical power is a measure of the chance to detect in the experiment statistically significant effects provided that the said effects really exist. Power is (also) a function of the sample size, so the most common aim of a power analysis is to determine the minimum number of samples needed for the study.201 Human clinical studies often require large cohorts, due to the high interindividual variability202 that could obliterate the investigated effects. The most common clinical design for human metabolomic studies is the simple comparison of samples collected from two groups of donors (e.g. a diseased group vs. healthy controls). This makes sense and can works reasonably well; however, interindividual variations are an unavoidable source of noise. A better approach would be to compare the same healthy individual with its diseased counterpart (Figure 11), by planning a long‐lasting cohort study where a number of healthy individuals are recruited and followed for many years, ideally collecting repeated samples until at least some of them develop the disease of interest (or any disease, see below). This approach has the great advantage to eliminate interindividual variation. Further, using repeated samples, it is not only possible to define the disease fingerprint, but also its dynamic evolution in time, allowing us to trace the path of each individual from healthiness to pathology, thereby opening the door to a predictive medicine strategy able to detect a disease before the appearance of the symptoms.
Figure 11.
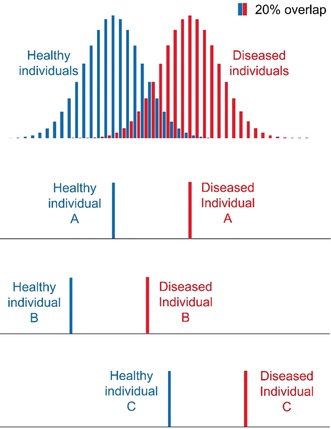
A single metabolite that is significant for the discrimination of healthy and diseased populations still may not provide a clear‐cut answer (20 % false positives and false negatives in this example, upper panel). The ambiguity originates from inter‐individual variability, represented by the width of the two Gaussian distributions. Conversely, it is more likely that an individual can be correctly classified if the level of the metabolite is quantified before and after the onset of the disease. While individual A is correctly classified as healthy and subsequently as diseased also according to population statistics, individuals B and C would not be classified correctly if evaluated only by population statistics.
A further point speaks in favor of metabolomics in this respect. When used for epidemiological purposes, it will permit the buildup of fingerprints for many different diseases (especially the most common) as long as they appear in the cohort. Therefore, subsequent samples, either from the same or from different individuals, could be compared with an increasing number of fingerprints, and therefore allow for early diagnosis of an increasing number of diseases.
Another comment is due to the concept itself of univariate analysis applied to untargeted metabolomics: in essence, the aim of univariate analysis is that of finding a specific biomarker. Indeed, as metabolomics can quantitate many metabolites at once, it has been sometimes termed a “biomarker discovery” tool. However, as such, the results of metabolomics have been often disappointing. In fact, a statistically valid criterion such as a low P‐value or a large effect size can only tell that the concentration of a certain metabolite is statistically different between two groups. Assuming two Gaussian concentration distributions for the two groups, the region of overlap between the two curves is very often too large (Figure 11), that is, the accuracy in distinguishing if a sample belongs to one group or the other is often modest (in the example, 80 %, that is, 20 % of false positives or false negatives). A good biomarker should provide a much higher accuracy. Unfortunately, it is becoming evident that there are not so many metabolites that are as good as, for instance, glucose for diabetes. However, a much more robust separation could be made if a panel of all metabolites that are statistically significant between the two groups is considered to be a single biomarker. In fact, two biomarkers that fail in 20 % of the cases, when taken together, would only fail in 4 % of the cases, and three of them would bring the failures down to less than 1 %. Basically, this reasoning brings us back to profiling and multivariate analysis (Section 3.3). Taken to the extreme, fingerprinting and multivariate analysis would always be the most efficient method, that is, a fingerprint would be, by definition, the best biomarker. While the above considerations indicate fingerprinting as the way to go, it should be considered that any method to be used in clinics will need extensive validation and ultimately approval by regulatory agencies. Up to now, researchers have experienced difficulties in having an analytical method considered by regulatory agencies if not based on the quantitation of one or more well identified metabolites. This is an attitude that may change in the future, under the growing evidence that fingerprints, once obtained under rigorous SOPs, are by themselves reliable analytical quantities.
6. Other Fields of Application: Foodomics as an Example
The food and beverage industry is constantly growing, and the global expenditure on this sector is twelve times bigger than the global expenditure on the health care sector.203, 204 It is therefore not surprising that the traditional use of NMR as an analytical tool for quality control of food stuff has recently evolved into a key tool for the new field of “foodomics”, a discipline that studies food and nutrition in relation to consumer's well‐being.205 Foodomics can be applied to study and characterize each step of the production/consumption chain in a comprehensive, integrated and high‐throughput approach, to improve also consumer's well‐being, health, and confidence.
The applications of metabolomics in foodomics can be summarized in three main areas (Figure 12): i) in the field of human health, which can be divided in food consumption monitoring studies, where, given a particular diet, the consequent metabolome changes are investigated,8, 206, 207, 208, 209 and in treating/preventing diseases by improving and monitoring patient diet;158 ii) in the food resources area, which can be investigated through the analysis of the composition of food from animal and plant origin and defined also on the basis of climate, land and cultural practices (“terroir”) that contribute to the traceability and thus to the characterization of the genuine product; iii) in food processing, which includes the characterization of the most influencing pre‐ and post‐production manipulations on the original product, such as altered growing conditions (e.g. feed, GMOs, chemicals, pesticides etc.), the effect of packaging and storage,210, 211 and, last but not least, safety and authenticity control. Post‐harvest manipulations, such as storage, can widely affect food profiles. For example, the molecular effect of controlled atmosphere, a common practice applied to extend the storage life of fruit, was studied on apples (Malus domestica).210
Figure 12.
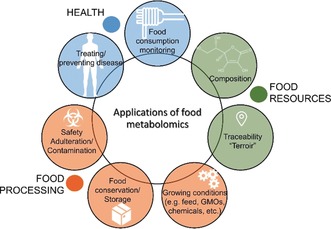
The main applications of food metabolomics. Food consumption monitoring and treating/preventing diseases, for the human health field (blue); chemical and molecular analyses of food composition and its characteristic ecosystem that contributes to the definition of traceability/authenticity of food resource (green); food processing area for the characterization of the effect of pre‐ and post‐harvest manipulations on food (orange).
Despite the fact that conventional quality, safety and authenticity control in food is based on targeted strategies, high resolution 1H‐NMR has found its way into routine food analysis,214 offering several advantages if performed under well‐defined instrumental specifications and SOPs, generating an extremely reproducible food fingerprint and fully quantitative data by means of a single experiment with minimal or no sample preparation. Thus, the fingerprint of a food matrix, analyzed with statistical methods, can reveal some latent information such as the provenance of milk samples from different farms even when all located in the same small geographical area,215 the origin of fruit juice212 and the grape variety for wine213 (Figure 13).
Figure 13.
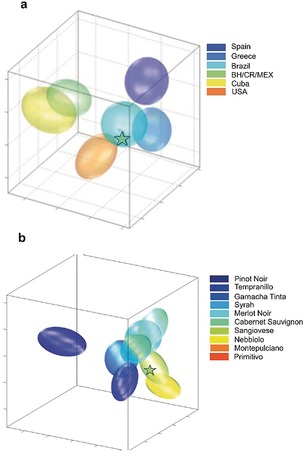
3D projections of the model space with the ellipsoids of possible groups available in the databases. a) Extract from JuiceScreenerTM, estimation of the origin of an orange juice.212 b) Extract from WinescreenerTM report of a Sangiovese sample (star) with respect to other Italian wines. Classification model for the wine type assignment, representing the probability of classification for every group.213
A platform for 1H‐NMR based food quality and authenticity control, the FoodScreenerTM platform, has already been introduced by Bruker BioSpin based on an Avance 400 MHz spectrometer, for the analysis and characterization of fruit juices and wines, and with application to honey samples being currently in development.214
7. Technical Improvements
As for other omics, the future of metabolomics is driven by technology developments. Herein, we will summarize a few recent technical advances that could expand the common metabolomic routine analyses by NMR, by providing alternative protocols for sample preparation and methods for facilitating metabolite assignment in the NMR spectra of biofluids.
As far as sample collection is concerned, one of the main problems in following SOPs is associated to situations where sample collection at home is needed: samples like urine and saliva can be collected directly by the donor, but problems may arise if filtering or keeping the samples at the right temperature is needed. Recently, we proposed a complementary pre‐analytical method relying on the gelification of biofluids and subsequent analysis by HR‐MAS NMR spectroscopy.216 Urine gelification is rapid and reproducible and permits preservation of the sample for 24 h without the need of refrigeration (Figure 14 a).
Figure 14.
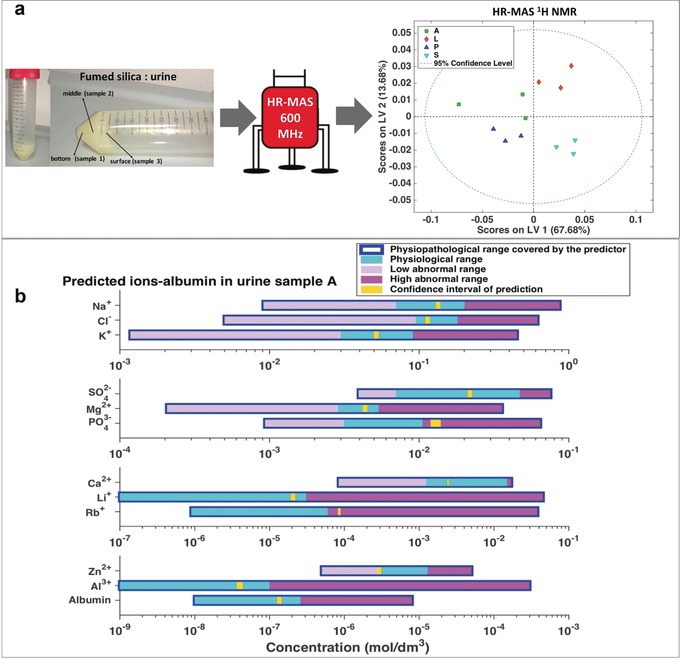
Examples of technical improvement. a) Gelification of urine by silica particles provides a ready‐to‐use HR‐MAS sample that could be exploited for metabolomic routine studies. b) Prediction of 11 inorganic ions and albumin by urine shift predictor.14
Moreover, this approach has a substantial advantage in cases of low biofluids abundance (e.g. newborns, small animals), since the HR‐MAS sample only requires ≈40 μL of gelified urine. In order to improve the identification of new metabolites in biological matrixes, a separation protocol has been proposed,217 where the solution of an NMR metabolomic mixture is passed through a set of filters, loaded with either anionic or cationic ion exchange resin beads (IERB). This method provides a separation of the metabolites depending on their specific physical‐chemical properties.217 As a result, the 1H‐13C HSQC 2D spectra of the flow through and the eluate could be used for a better assignment of the metabolites.
A novel approach for the automated and accurate identification of urine metabolites was recently introduced by our group. This has been achieved by constructing a large number (ca. 4000) of artificial urine samples where the concentrations of the most abundant metabolites and of several inorganic ions were varied to sparsely but efficiently populate an N‐dimensional concentration matrix. By the use of statistical machine learning tools, we constructed an algorithm that, based upon the chemical shifts of only five signals, can accurately predict the chemical shifts of many other metabolites in the urine sample (so far 90 spin systems chemical shifts of 63 metabolites) (Figure 7). This predictor, besides allowing for easy quantitation of many metabolites whose signals are visible in the spectrum, provides estimates of the concentrations of 11 NMR “invisible” inorganic ions (Figure 14 b). Our software estimates the inorganic ions (including trace metal ions) in one single, inexpensive experiment, thus making NMR a significant tool in the recently introduced field of ionomics.218 This strategy could be expanded to other metabolomic fluids, such as juices, soft drinks etc.
8. Summary and Outlook
This Review aimed at highlighting the many chemical aspects of metabolomics. This was not meant to imply that the ultimate goal of metabolomics research is chemistry, but rather that chemistry is needed for a better understanding of the biomedical goals and can even be inspirational for defining the strategy of how to best achieve these goals. By restricting ourselves to metabolomics by NMR we hope we have shown that the horizon of metabolomics—in particular of its untargeted fingerprinting version, which is best performed by NMR—can be actually enlarged towards population‐wide metabolomic screening, especially as we expect further technological advancements, of which Section 7 provides some examples. As highlighted in Section 4, metabolomics is being gradually appreciated as the science that is closest to provide early diagnosis of diseases. The medical community is progressively appreciating that searching for disease‐specific biomarkers is probably no longer the way to go: single biomarkers seldom have enough discriminating power, while a panel of biomarkers taken together has a better chance to provide higher discriminating power. By extrapolating this concept (Section 5), a whole fingerprint, that virtually contains “all” detectable metabolites, will have the highest possible discriminating power. This extrapolation requires a leap of faith, that is, that an NMR spectrum is equivalent to a complete panel of perfectly quantitated metabolites. Only a chemist with enough NMR knowledge could convincingly make the case in front of non‐chemists, that presently would rather trust a table of chemical substances with the associated concentrations.
But how far are we from actually adopting metabolomics by NMR as a population‐wide screening method? From the examples reported in Section 4, and from many other examples in the recent literature, it appears that i) for several diseases there are already NMR fingerprints that are characteristic of each of these diseases; ii) by comparing the NMR spectrum of a “healthy” subject with these fingerprints, an early diagnosis can be made with an accuracy of 70–85 %; iii) recurrences in some types of cancers can be predicted earlier than by any other method, again with ca. 80 % accuracy. The conceptual distance from the present situation to the adoption of metabolomics at the level of national health systems is given by the need to move further in two directions: towards defining fingerprints for a larger number of diseases, and towards increasing the number of subjects from which the fingerprint is built, to increase robustness. It should be noted that, as the population screening will progress, the system will become more and more informative (more diseases will be fingerprinted, and therefore more could be detected from the same NMR spectrum) and more reliable (accuracy will further increase). It should also be noted that even an accuracy of 80 % would be more than enough to prescribe, to individuals with a tentative early diagnosis of a given pathology, a specific check‐up for that disease (e.g. echocardiography for heart failure, or antibody tests for celiac disease, etc.).
The next question to answer is the cost of adopting such a population‐wide NMR screening: although NMR instruments are expensive (a 600 MHz dedicated instrument costs more than 1 million € + VAT), they are long‐lived and can work almost continuously for months, provided the automatic sample changer is refilled constantly. They do not need to be recalibrated nearly as often as MS instruments, and sample preparation is extremely simple. Overall, the cost per sample when all operations are optimized, including initial investment, maintenance and upgrading, personnel and consumables, would probably be lower than 15 €. Therefore, a metabolomic NMR analysis on, say, a serum sample would add only a modest amount to the cost of a blood test for a panel of analytes as it is currently done for periodic check‐ups.
The last question is the feasibility: for instance, to screen 400 million Europeans once every two years, 200 million spectra per year would need to be recorded. A single instrument can run about 120 spectra per day, that is, around 40 000 spectra per year, so ca. 5000 NMR instruments will be needed. They will have to be spread across the continent, in multiple copies in major and middle‐sized hospital, as well as in public and private clinical analysis laboratories, because clinical analyses are typically performed locally to avoid shipping of massive numbers of samples. Five thousand spectrometers may seem as a very large number but is about the same as the number of MRI scanners in use in Europe, each of which costing significantly more than one million €. When MRI started to appear as a potentially useful technique, it took only a few years for MRI scanners to spread and grow to the levels of today.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Claudio Luchinat is Professor of Chemistry at the University of Florence, co‐founder and Director of the Center of Magnetic Resonance (CERM), and President of the Interuniversity Consortium on Magnetic Resonance of Metallo Proteins (CIRMMP). His research interests include bioinorganic chemistry, NMR‐based structural biology, and NMR of paramagnetic species. In the last decade his research has also focused on NMR‐based metabolomics, demonstrating the existence of individual metabolic fingerprints and of specific metabolic fingerprints of several diseases. The NMR metabolomics team from left to right: Leonardo Tenori, Gaia Meoni, Veronica Ghini, Claudio Luchinat, Paola Turano, Alessia Vignoli, Panteleimon G. Takis, Cristina Licari.

Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The current NMR metabolomic activities at CERM are co‐funded by the European Commission H2020 projects SPIDIA4P (contract no. 733112), Phenomenal (contract no. 654241), Propag‐ageing (contract no. 634821), the ERA‐NET project ITFoC, the FP7 EC project Pathway‐27 (contract no. 311876) and Fondazione CRF. VG is the recipient of a post‐doctoral fellowship 2018 provided by Fondazione Veronesi. We thank Manfred Spraul and Hartmut Schaefer (Bruker Biospin) for a long‐standing collaboration. Samples for the development of pre‐analytical SOPs were provided by the da Vinci European Biobank (daVEB, DOI: https://doi.org/10.5334/ojb.af).
A. Vignoli, V. Ghini, G. Meoni, C. Licari, P. G. Takis, L. Tenori, P. Turano, C. Luchinat, Angew. Chem. Int. Ed. 2019, 58, 968.
References
- 1. Nicholson J. K., Lindon J. C., Nature 2008, 455, 1054–1056. [DOI] [PubMed] [Google Scholar]
- 2. Calabrò A., Gralka E., Luchinat C., Saccenti E., Tenori L., J. Autoimmune Dis. 2014, 2014, e756138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kirwan J., In Practice 2013, 35, 438–445. [Google Scholar]
- 4. Pontes J. G. M., Brasil A. J. M., Cruz G. C. F., de Souza R. N., Tasic L., Anal. Methods 2017, 9, 1078–1096. [Google Scholar]
- 5. Tang J., Curr. Genomics 2011, 12, 391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ramirez-Gaona M., Marcu A., Pon A., Guo A. C., Sajed T., Wishart N. A., Karu N., Djoumbou Feunang Y., Arndt D., Wishart D. S., Nucleic Acids Res. 2017, 45, D440–D445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hall R. D., Brouwer I. D., Fitzgerald M. A., Physiol. Plant. 2008, 132, 162–175. [DOI] [PubMed] [Google Scholar]
- 8. Münger L. H., Trimigno A., Picone G., Freiburghaus C., Pimentel G., Burton K. J., Pralong F. P., Vionnet N., Capozzi F., Badertscher R., et al., J. Proteome Res. 2017, 16, 3321–3335. [DOI] [PubMed] [Google Scholar]
- 9. Kim S., Kim J., Yun E. J., Kim K. H., Curr. Opin. Biotechnol. 2016, 37, 16–23. [DOI] [PubMed] [Google Scholar]
- 10. Markley J. L., Brüschweiler R., Edison A. S., Eghbalnia H. R., Powers R., Raftery D., Wishart D. S., Curr. Opin. Biotechnol. 2017, 43, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Klupczyńska A., Dereziński P., Kokot Z. J., Acta Pol. Pharm. 2015, 72, 629–641. [PubMed] [Google Scholar]
- 12. Lindon J. C., Nicholson J. K., Annu. Rev. Anal. Chem. 2008, 1, 45–69. [DOI] [PubMed] [Google Scholar]
- 13. Wishart D. S., Nat. Rev. Drug Discovery 2016, 15, 473–484. [DOI] [PubMed] [Google Scholar]
- 14. Takis P. G., Schäfer H., Spraul M., Luchinat C., Nat. Commun. 2017, 8, 1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scalbert A., Brennan L., Fiehn O., Hankemeier T., Kristal B. S., van Ommen B., Pujos-Guillot E., Verheij E., Wishart D., Wopereis S., Metabolomics 2009, 5, 435–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barbosa B. S., Martins L. G., Costa T. B. B. C., Cruz G., Tasic L., Methods Mol. Biol. 2018, 1735, 365–379. [DOI] [PubMed] [Google Scholar]
- 17. Li J., Vosegaard T., Guo Z., Prog. Lipid Res. 2017, 68, 37–56. [DOI] [PubMed] [Google Scholar]
- 18. Bouatra S., Aziat F., Mandal R., Guo A. C., Wilson M. R., Knox C., Bjorndahl T. C., Krishnamurthy R., Saleem F., Liu P., et al., PloS One 2013, 8, e73076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Psychogios N., Hau D. D., Peng J., Guo A. C., Mandal R., Bouatra S., Sinelnikov I., Krishnamurthy R., Eisner R., Gautam B., et al., PLoS ONE 2011, 6, e16957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dame Z. T., Aziat F., Mandal R., Krishnamurthy R., Bouatra S., Borzouie S., Guo A. C., Sajed T., Deng L., Lin H., et al., Metabolomics 2015, 11, 1864–1883. [Google Scholar]
- 21. Wishart D. S., Lewis M. J., Morrissey J. A., Flegel M. D., Jeroncic K., Xiong Y., Cheng D., Eisner R., Gautam B., Tzur D., et al., J. Chromatogr. B 2008, 871, 164–173. [DOI] [PubMed] [Google Scholar]
- 22. Airoldi C., Ciaramelli C., Fumagalli M., Bussei R., Mazzoni V., Viglio S., Iadarola P., Stolk J., J. Proteome Res. 2016, 15, 4569–4578. [DOI] [PubMed] [Google Scholar]
- 23. Bernacchioni C., Ghini V., Cencetti F., Japtok L., Donati C., Bruni P., Turano P., Mol. Oncol. 2017, 11, 517–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cuperlovic-Culf M., Ferguson D., Culf A., Morin P., Touaibia M., J. Biol. Chem. 2012, 287, 20164–20175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martínez-Granados B., Monleón D., Martínez-Bisbal M. C., Rodrigo J. M., del Olmo J., Lluch P., Ferrández A., Martí-Bonmatí L., Celda B., NMR Biomed. 2006, 19, 90–100. [DOI] [PubMed] [Google Scholar]
- 26. Battini S., Faitot F., Imperiale A., Cicek A. E., Heimburger C., Averous G., Bachellier P., Namer I. J., BMC Med. 2017, 15, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Palama T. L., Canard I., Rautureau G. J. P., Mirande C., Chatellier S., Elena-Herrmann B., Analyst 2016, 141, 4558–4561. [DOI] [PubMed] [Google Scholar]
- 28. Romano F., Meoni G., Manavella V., Baima G., Tenori L., Cacciatore S., Aimetti M., J. Periodontol. 2018, 10.1002/JPER.18-0097. [DOI] [PubMed] [Google Scholar]
- 29.CEN/TS 16945, Molecular in vitro diagnostic examinations. Specifications for pre-examination processes for metabolomics in urine, venous blood serum and plasma, 2016.
- 30. Bernini P., Bertini I., Luchinat C., Nincheri P., Staderini S., Turano P., J. Biomol. NMR 2011, 49, 231–243. [DOI] [PubMed] [Google Scholar]
- 31. Beckonert O., Keun H. C., Ebbels T. M. D., Bundy J., Holmes E., Lindon J. C., Nicholson J. K., Nat. Protoc. 2007, 2, 2692–2703. [DOI] [PubMed] [Google Scholar]
- 32. Navazesh M., Ann. N. Y. Acad. Sci. 1993, 694, 72–77. [DOI] [PubMed] [Google Scholar]
- 33. Wallner-Liebmann S., Tenori L., Mazzoleni A., Dieber-Rotheneder M., Konrad M., Hofmann P., Luchinat C., Turano P., Zatloukal K., J. Proteome Res. 2016, 15, 1787–1793. [DOI] [PubMed] [Google Scholar]
- 34. Teunissen C. E., Petzold A., Bennett J. L., Berven F. S., Brundin L., Comabella M., Franciotta D., Frederiksen J. L., Fleming J. O., Furlan R., et al., Neurology 2009, 73, 1914–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. de Laurentiis G., Paris D., Melck D., Maniscalco M., Marsico S., Corso G., Motta A., Sofia M., Eur. Respir. J. 2008, 32, 1175–1183. [DOI] [PubMed] [Google Scholar]
- 36. Lamichhane S., Yde C. C., Schmedes M. S., Jensen H. M., Meier S., Bertram H. C., Anal. Chem. 2015, 87, 5930–5937. [DOI] [PubMed] [Google Scholar]
- 37. Deda O., Gika H. G., Wilson I. D., Theodoridis G. A., J. Pharm. Biomed. Anal. 2015, 113, 137–150. [DOI] [PubMed] [Google Scholar]
- 38. Beckonert O., Coen M., Keun H. C., Wang Y., Ebbels T. M. D., Holmes E., Lindon J. C., Nicholson J. K., Nat. Protoc. 2010, 5, 1019–1032. [DOI] [PubMed] [Google Scholar]
- 39. Gardner A., Parkes H. G., Carpenter G. H., So P.-W., J. Proteome Res. 2018, 17, 1521–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brunius C., Pedersen A., Malmodin D., Karlsson B. G., Andersson L. I., Tybring G., Landberg R., Bioinformatics 2017, 33, 3567–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emwas A.-H., Luchinat C., Turano P., Tenori L., Roy R., Salek R. M., Ryan D., Merzaban J. S., Kaddurah-Daouk R., Zeri A. C., et al., Metabolomics 2015, 11, 872—894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cacciatore S., Hu X., Viertler C., Kap M., Bernhardt G. A., Mischinger H.-J., Riegman P., Zatloukal K., Luchinat C., Turano P., J. Proteome Res. 2013, 12, 5723–5729. [DOI] [PubMed] [Google Scholar]
- 43. Ghini V., Unger F. T., Tenori L., Turano P., Juhl H., David K. A., Metabolomics 2015, 11, 1769–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.S. Ravanbakhsh, P. Liu, T. Bjorndahl, R. Mandal, J. R. Grant, M. Wilson, R. Eisner, I. Sinelnikov, X. Hu, C. Luchinat, et al., ArXiv14091456 Cs Q-Bio, 2014.
- 45. Gowda G. A. N., Raftery D., Anal. Chem. 2014, 86, 5433–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wishart D. S., Feunang Y. D., Marcu A., Guo A. C., Liang K., Vázquez-Fresno R., Sajed T., Johnson D., Li C., Karu N., et al., Nucleic Acids Res. 2018, 46, D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pearce J. T. M., Athersuch T. J., Ebbels T. M. D., Lindon J. C., Nicholson J. K., Keun H. C., Anal. Chem. 2008, 80, 7158–7162. [DOI] [PubMed] [Google Scholar]
- 48. Wider G., Dreier L., J. Am. Chem. Soc. 2006, 128, 2571–2576. [DOI] [PubMed] [Google Scholar]
- 49. Akoka S., Barantin L., Trierweiler M., Anal. Chem. 1999, 71, 2554–2557. [DOI] [PubMed] [Google Scholar]
- 50. Wu Y., Li L., J. Chromatogr. A 2016, 1430, 80–95. [DOI] [PubMed] [Google Scholar]
- 51. Hochrein J., Zacharias H. U., Taruttis F., Samol C., Engelmann J. C., Spang R., Oefner P. J., Gronwald W., J. Proteome Res. 2015, 14, 3217–3228. [DOI] [PubMed] [Google Scholar]
- 52. Dieterle F., Ross A., Schlotterbeck G., Senn H., Anal. Chem. 2006, 78, 4281–4290. [DOI] [PubMed] [Google Scholar]
- 53. Lee J., Park J., Lim M., Seong S. J., Seo J. J., Park S. M., Lee H. W., Yoon Y.-R., Anal. Sci. 2012, 28, 801–805. [DOI] [PubMed] [Google Scholar]
- 54. Åstrand M., J. Comput. Biol. 2003, 10, 95–102. [DOI] [PubMed] [Google Scholar]
- 55. Filzmoser P., Walczak B., J. Chromatogr. A 2014, 1362, 194–205. [DOI] [PubMed] [Google Scholar]
- 56. Kohl S. M., Klein M. S., Hochrein J., Oefner P. J., Spang R., Gronwald W., Metabolomics 2012, 8, 146–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bertini I., Luchinat C., Miniati M., Monti S., Tenori L., Metabolomics 2014, 10, 302–311. [Google Scholar]
- 58. Montuschi P., Santini G., Mores N., Vignoli A., Macagno F., Shohreh R., Tenori L., Zini G., Fuso L., Mondino C., et al., Front. Pharmacol. 2018, 10.3389/fphar.2018.00258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vignoli A., Rodio D. M., Bellizzi A., Sobolev A. P., Anzivino E., Mischitelli M., Tenori L., Marini F., Priori R., Scrivo R., et al., Anal. Bioanal. Chem. 2017, 409, 1405–1413. [DOI] [PubMed] [Google Scholar]
- 60. León Z., García-Cañaveras J. C., Donato M. T., Lahoz A., Electrophoresis 2013, 34, 2762–2775. [DOI] [PubMed] [Google Scholar]
- 61. Muschet C., Möller G., Prehn C., de Angelis M. H., Adamski J., Tokarz J., Metabolomics 2016, 12, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Xiao X., Hu M., Liu M., Hu J., Metabolomics 2016, 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.L. D. Roberts, A. L. Souza, R. E. Gerszten, C. B. Clish, Curr. Protoc. Mol. Biol 2012, CHAPTER, Unit30.2. [DOI] [PMC free article] [PubMed]
- 64. Griffiths W. J., Metabolomics, Metabonomics and Metabolite Profiling, Royal Society of Chemistry, London, 2008. [Google Scholar]
- 65. Smolinska A., Blanchet L., Buydens L. M. C., Wijmenga S. S., Anal. Chim. Acta 2012, 750, 82–97. [DOI] [PubMed] [Google Scholar]
- 66. Emwas A.-H., Saccenti E., Gao X., McKay R. T., dos Santos V. A. P. M., Roy R., Wishart D. S., Metabolomics 2018, 14, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vu T. N., Laukens K., Metabolites 2013, 3, 259–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Savorani F., Tomasi G., Engelsen S. B., J. Magn. Reson. 2010, 202, 190–202. [DOI] [PubMed] [Google Scholar]
- 69. Lindon C. J., Nicholson J. K., Holmes E., The Handbook of Metabonomics and Metabolomics, Elsevier, Amsterdam, 2007. [Google Scholar]
- 70. Ren S., Hinzman A. A., Kang E. L., Szczesniak R. D., Lu L. J., Metabolomics 2015, 11, 1492–1513. [Google Scholar]
- 71. Wold S., Esbensen K., Geladi P., Chemom. Intell. Lab. Syst. 1987, 2, 37–52. [Google Scholar]
- 72. Abdi H., Williams L. J., Wiley Interdiscip. Rev. Comput. Stat. 2010, 2, 433–459. [Google Scholar]
- 73. Bujak R., Markuszewski M. J., in Identif. Data Process. Methods Metabolomics, Future Science Ltd, London, 2015, pp. 82–95. [Google Scholar]
- 74. Ringnér M., Nat. Biotechnol. 2008, 26, 303–304. [DOI] [PubMed] [Google Scholar]
- 75. Hyvärinen A., Oja E., Neural Netw. 2000, 13, 411–430. [DOI] [PubMed] [Google Scholar]
- 76. Baptiste M. V., Rousseau F. R., de Tullio P., Govaerts B., J. Biom. Biostat. 2017, 1–8.30555734 [Google Scholar]
- 77. Camacho J., Rodríguez-Gómez R. A., Saccenti E., J. Comput. Graph. Stat. 2017, 26, 501–512. [Google Scholar]
- 78. Hartigan J. A., Wong M. A., J. R. Stat. Soc. Ser. C 1979, 28, 100–108. [Google Scholar]
- 79. Srinivasan A., Galbán C. J., Johnson T. D., Chenevert T. L., Ross B. D., Mukherji S. K., Am. J. Neuroradiol. 2010, 31, 736–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cuperlović-Culf M., Belacel N., Culf A. S., Chute I. C., Ouellette R. J., Burton I. W., Karakach T. K., Walter J. A., Magn. Reson. Chem. 2009, 47, S96–104. [DOI] [PubMed] [Google Scholar]
- 81. Kaufman L., Rousseeuw P. J., Finding Groups in Data: An Introduction to Cluster Analysis, Wiley, Hoboken, 2009. [Google Scholar]
- 82. Shi J., Malik J., IEEE Trans. Pattern Anal. Mach. Intell. 2000, 22, 888–905. [Google Scholar]
- 83. Tiwari P., Rosen M., Madabhushi A., Med. Phys. 2009, 36, 3927–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cacciatore S., Luchinat C., Tenori L., Proc. Natl. Acad. Sci. USA 2014, 111, 5117–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cacciatore S., Tenori L., Luchinat C., Bennett P. R., MacIntyre D. A., Bioinformatics 2017, 33, 621–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kohonen T., Neural Netw. 2013, 37, 52–65. [DOI] [PubMed] [Google Scholar]
- 87. Zheng H., Ji J., Zhao L., Chen M., Shi A., Pan L., Huang Y., Zhang H., Dong B., Gao H., Oncotarget 2016, 7, 59189–59198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Geladi P., Chemom. Intell. Lab. Syst. 1992, 15, vii–viii. [Google Scholar]
- 89. Wold S., Eriksson L., Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar]
- 90. Abdi H., Wiley Interdiscip. Rev. Comput. Stat. 2010, 2, 97–106. [Google Scholar]
- 91. Gromski P. S., Muhamadali H., Ellis D. I., Xu Y., Correa E., Turner M. L., Goodacre R., Anal. Chim. Acta 2015, 879, 10–23. [DOI] [PubMed] [Google Scholar]
- 92. Trygg J., Wold S., J. Chemom. 2002, 16, 119–128. [Google Scholar]
- 93. Cover T., Hart P., IEEE Trans. Inf. Theory 1967, 13, 21–27. [Google Scholar]
- 94. Beckonert O., Bollard E., Ebbels T. M. D., Keun H. C., Antti H., Holmes E., Lindon J. C., Nicholson J. K., Anal. Chim. Acta 2003, 490, 3–15. [Google Scholar]
- 95. Schmidhuber J., Neural Netw. 2015, 61, 85–117. [DOI] [PubMed] [Google Scholar]
- 96. Alakwaa F. M., Chaudhary K., Garmire L. X., J. Proteome Res. 2018, 17, 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Westerhuis J., Hoefsloot H., Smit S., Vis D., Smilde A., van Velzen E., van Duijnhoven J., van Dorsten F., Metabolomics 2008, 4, 81–89. [Google Scholar]
- 98. Wishart D. S., Tzur D., Knox C., Eisner R., Guo A. C., Young N., Cheng D., Jewell K., Arndt D., Sawhney S., et al., Nucleic Acids Res. 2007, 35, D521–D526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Wishart D. S., Knox C., Guo A. C., Eisner R., Young N., Gautam B., Hau D. D., Psychogios N., Dong E., Bouatra S., et al., Nucleic Acids Res. 2009, 37, D603–D610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wishart D. S., Jewison T., Guo A. C., Wilson M., Knox C., Liu Y., Djoumbou Y., Mandal R., Aziat F., Dong E., et al., Nucleic Acids Res. 2013, 41, D801–D807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dona A. C., Kyriakides M., Scott F., Shephard E. A., Varshavi D., Veselkov K., Everett J. R., Comput. Struct. Biotechnol. J. 2016, 14, 135–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bingol K., Bruschweiler-Li L., Yu C., Somogyi A., Zhang F., Brüschweiler R., Anal. Chem. 2015, 87, 3864–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Boiteau R. M., Hoyt D. W., Nicora C. D., Kinmonth-Schultz H. A., Ward J. K., Bingol K., Metabolites 2018, 10.3390/metabo8010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Hao J., Liebeke M., Astle W., De Iorio M., Bundy J. G., Ebbels T. M. D., Nat. Protoc. 2014, 9, 1416–1427. [DOI] [PubMed] [Google Scholar]
- 105. Tardivel P. J. C., Canlet C., Lefort G., Tremblay-Franco M., Debrauwer L., Concordet D., Servien R., Metabolomics 2017, 13, 109. [Google Scholar]
- 106. Neuhäuser M., in Int. Encycl. Stat. Sci., Springer, Berlin, Heidelberg, 2011, pp. 1656–1658. [Google Scholar]
- 107. Snedecor G. W., Cochran W. G., Statistical Methods, Iowa State University Press, Ames, Iowa, 1989. [Google Scholar]
- 108. Kruskal W. H., Wallis W. A., J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar]
- 109. Conover W. J., Practical Nonparametric Statistics , 3rd ed., Wiley, New York, 1999. [Google Scholar]
- 110. Bonferroni C. E., in Studi Onore Profr. Salvatore Ortu Carboni, Bardi, Rome, 1935, pp. 13–60. [Google Scholar]
- 111. Benjamini Y., Hochberg Y., J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar]
- 112. Vidgen B., Yasseri T., Front. Phys. 2016, 10.3389/fphy.2016.00006. [DOI] [Google Scholar]
- 113. Sullivan G. M., Feinn R., J. Grad. Med. Educ. 2012, 4, 279–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Saccenti E., Menichetti G., Ghini V., Remondini D., Tenori L., Luchinat C., J. Proteome Res. 2016, 15, 3298–3307. [DOI] [PubMed] [Google Scholar]
- 115. Saccenti E., Suarez-Diez M., Luchinat C., Santucci C., Tenori L., J. Proteome Res. 2015, 14, 1101–1111. [DOI] [PubMed] [Google Scholar]
- 116. Suarez-Diez M., Adam J., Adamski J., Chasapi S. A., Luchinat C., Peters A., Prehn C., Santucci C., Spyridonidis A., Spyroulias G. A., et al., J. Proteome Res. 2017, 16, 2547–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Rosato A., Tenori L., Cascante M., De Atauri Carulla P. R., Martins Dos Santos V. A. P., Saccenti E., Metabolomics 2018, 14, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Patti G. J., Yanes O., Siuzdak G., Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Assfalg M., Bertini I., Colangiuli D., Luchinat C., Schäfer H., Schütz B., Spraul M., Proc. Natl. Acad. Sci. USA 2008, 105, 1420–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Holmes E., Loo R. L., Stamler J., Bictash M., Yap I. K. S., Chan Q., Ebbels T., De Iorio M., Brown I. J., Veselkov K. A., et al., Nature 2008, 453, 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Bernini P., Bertini I., Luchinat C., Nepi S., Saccenti E., Schäfer H., Schütz B., Spraul M., Tenori L., J. Proteome Res. 2009, 8, 4264–4271. [DOI] [PubMed] [Google Scholar]
- 122. Ghini V., Saccenti E., Tenori L., Assfalg M., Luchinat C., J. Proteome Res. 2015, 14, 2951–2962. [DOI] [PubMed] [Google Scholar]
- 123. Saccenti E., Menichetti G., Ghini V., Remondini D., Tenori L., Luchinat C., J. Proteome Res. 2016, 15, 3298–3307. [DOI] [PubMed] [Google Scholar]
- 124. Saccenti E., Tenori L., Verbruggen P., Timmerman M. E., Bouwman J., van der Greef J., Luchinat C., Smilde A. K., PLOS ONE 2014, 9, e106077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wallner-Liebmann S., Gralka E., Tenori L., Konrad M., Hofmann P., Dieber-Rotheneder M., Turano P., Luchinat C., Zatloukal K., Genes Nutr. 2015, 10, 441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yousri N. A., Kastenmüller G., Gieger C., Shin S.-Y., Erte I., Menni C., Peters A., Meisinger C., Mohney R. P., Illig T., et al., Metabolomics 2014, 10, 1005–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Martinez-Lozano Sinues P., Kohler M., Zenobi R., PLoS ONE 2013, 8, e59909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Krug S., Kastenmüller G., Stückler F., Rist M. J., Skurk T., Sailer M., Raffler J., Römisch-Margl W., Adamski J., Prehn C., et al., FASEB J. 2012, 26, 2607–2619. [DOI] [PubMed] [Google Scholar]
- 129. Nicholson G., Rantalainen M., Maher A. D., Li J. V., Malmodin D., Ahmadi K. R., Faber J. H., Hallgrímsdóttir I. B., Barrett A., Toft H., et al., Mol. Syst. Biol. 2011, 7, 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Stella C., Beckwith-Hall B., Cloarec O., Holmes E., Lindon J. C., Powell J., van der Ouderaa F., Bingham S., Cross A. J., Nicholson J. K., J. Proteome Res. 2006, 5, 2780–2788. [DOI] [PubMed] [Google Scholar]
- 131. Pinto J., Barros A. S., Domingues M. R. M., Goodfellow B. J., Galhano E., Pita C., Almeida M. D. C., Carreira I. M., Gil A. M., J. Proteome Res. 2015, 14, 1263–1274. [DOI] [PubMed] [Google Scholar]
- 132. Diaz S. O., Pinto J., Barros A. S., Morais E., Duarte D., Negrao F., Pita C., Almeida M. D. C., Carreira I. M., Spraul M., et al., J. Proteome Res. 2016, 15, 311–325. [DOI] [PubMed] [Google Scholar]
- 133. Scalabre A., Jobard E., Demède D., Gaillard S., Pontoizeau C., Mouriquand P., Elena-Herrmann B., Mure P.-Y., J. Proteome Res. 2017, 16, 3732–3740. [DOI] [PubMed] [Google Scholar]
- 134. Rodríguez D. A., Alcarraz-Vizán G., Díaz-Moralli S., Reed M., Gómez F. P., Falciani F., Günther U., Roca J., Cascante M., Metabolomics 2012, 8, 508–516. [Google Scholar]
- 135. Bjerrum J. T., Wang Y., Hao F., Coskun M., Ludwig C., Günther U., Nielsen O. H., Metabolomics 2015, 11, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Claudino W. M., Quattrone A., Biganzoli L., Pestrin M., Bertini I., Di L. A., J. Clin. Oncol. 2007, 25, 2840–2846. [DOI] [PubMed] [Google Scholar]
- 137. Spratlin J. L., Serkova N. J., Eckhardt S. G., Clin. Cancer Res. 2009, 15, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Carrola J., Rocha C. M., Barros A. S., Gil A. M., Goodfellow B. J., Carreira I. M., Bernardo J., Gomes A., Sousa V., Carvalho L., et al., J. Proteome Res. 2011, 10, 221–230. [DOI] [PubMed] [Google Scholar]
- 139. Hasim A., Ma H., Mamtimin B., Abudula A., Niyaz M., Zhang L.-W., Anwer J., Sheyhidin I., Mol. Biol. Rep. 2012, 39, 8955–8964. [DOI] [PubMed] [Google Scholar]
- 140. Tiziani S., Lodi A., Khanim F. L., Viant M. R., Bunce C. M., Günther U. L., PLoS ONE 2009, 10.1371/journal.pone.0004251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Yang Y., Liu Y., Zheng L., Zhang Q., Gu Q., Wang L., Wang L., Int. J. Biol. Sci. 2015, 11, 595–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Deidda M., Piras C., Bassareo P. P., Cadeddu Dessalvi C., Mercuro G., IJC Metab. Endocr. 2015, 9, 31–38. [Google Scholar]
- 143. Tenori L., Hu X., Pantaleo P., Alterini B., Castelli G., Olivotto I., Bertini I., Luchinat C., Gensini G. F., Int. J. Cardiol. 2013, 168, e113–e115. [DOI] [PubMed] [Google Scholar]
- 144. Brindle J. T., Nicholson J. K., Schofield P. M., Grainger D. J., Holmes E., Analyst 2003, 128, 32–36. [DOI] [PubMed] [Google Scholar]
- 145. Brindle J. T., Antti H., Holmes E., Tranter G., Nicholson J. K., Bethell H. W. L., Clarke S., Schofield P. M., McKilligin E., Mosedale D. E., et al., Nat. Med. 2002, 8, 1439–1444. [DOI] [PubMed] [Google Scholar]
- 146. Bernini P., Bertini I., Luchinat C., Tenori L., Tognaccini A., J. Proteome Res. 2011, 10, 4983–4992. [DOI] [PubMed] [Google Scholar]
- 147. Mäkinen V.-P., Tynkkynen T., Soininen P., Peltola T., Kangas A. J., Forsblom C., Thorn L. M., Kaski K., Laatikainen R., Ala-Korpela M., et al., J. Proteome Res. 2012, 11, 1782–1790. [DOI] [PubMed] [Google Scholar]
- 148. Deja S., Barg E., Młynarz P., Basiak A., Willak-Janc E., J. Pharm. Biomed. Anal. 2013, 83, 43–48. [DOI] [PubMed] [Google Scholar]
- 149. Liu X., Gao J., Chen J., Wang Z., Shi Q., Man H., Guo S., Wang Y., Li Z., Wang W., Sci. Rep. 2016, 6, 30785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Dani C., Bresci C., Berti E., Ottanelli S., Mello G., Mecacci F., Breschi R., Hu X., Tenori L., Luchinat C., J. Matern.-Fetal Neonatal Med. 2014, 27, 537–542. [DOI] [PubMed] [Google Scholar]
- 151. Gralka E., Luchinat C., Tenori L., Ernst B., Thurnheer M., Schultes B., Am. J. Clin. Nutr. 2015, 102, 1313–1322. [DOI] [PubMed] [Google Scholar]
- 152. Calvani R., Brasili E., Praticò G., Sciubba F., Roselli M., Finamore A., Marini F., Marzetti E., Miccheli A., J. Clin. Gastroenterol. 2014, 48, S5–7. [DOI] [PubMed] [Google Scholar]
- 153. Elliott P., Posma J. M., Chan Q., Garcia-Perez I., Wijeyesekera A., Bictash M., Ebbels T. M. D., Ueshima H., Zhao L., van Horn L., et al., Sci. Transl. Med. 2015, 7, 285ra62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Würtz P., Kangas A. J., Soininen P., Lawlor D. A., Davey Smith G., Ala-Korpela M., Am. J. Epidemiol. 2017, 186, 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Rankin N. J., Preiss D., Welsh P., Burgess K. E. V., Nelson S. M., Lawlor D. A., Sattar N., Atherosclerosis 2014, 237, 287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Giskeødegård G. F., Madssen T. S., Euceda L. R., Tessem M.-B., Moestue S. A., Bathen T. F., NMR Biomed. 2018, e3927. [DOI] [PubMed] [Google Scholar]
- 157. Everett J. R., Prog. Nucl. Magn. Reson. Spectrosc. 2017, 102–103, 1–14. [DOI] [PubMed] [Google Scholar]
- 158. Bertini I., Calabrò A., De Carli V., Luchinat C., Nepi S., Porfirio B., Renzi D., Saccenti E., Tenori L., J. Proteome Res. 2009, 8, 170–177. [DOI] [PubMed] [Google Scholar]
- 159. Bertini I., Cacciatore S., Jensen B. V., Schou J. V., Johansen J. S., Kruhoffer M., Luchinat C., Nielsen D. L., Turano P., Cancer Res. 2012, 72, 356–364. [DOI] [PubMed] [Google Scholar]
- 160. Hart C. D., Vignoli A., Tenori L., Uy G. L., Van To T., Adebamowo C., Hossain S. M., Biganzoli L., Risi E., Love R. R., et al., Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Andrew Clayton T., Lindon J. C., Cloarec O., Antti H., Charuel C., Hanton G., Provost J.-P., Le Net J.-L., Baker D., Walley R. J., et al., Nature 2006, 440, 1073–1077. [DOI] [PubMed] [Google Scholar]
- 162. Bernini P., Bertini I., Calabrò A., la Marca G., Lami G., Luchinat C., Renzi D., Tenori L., J. Proteome Res. 2011, 10, 714–721. [DOI] [PubMed] [Google Scholar]
- 163. Čuperlović-Culf M., NMR Metabolomics in Cancer Research, Elsevier, Amsterdam, 2012. [Google Scholar]
- 164. Reed M. A. C., Singhal R., Ludwig C., Carrigan J. B., Ward D. G., Taniere P., Alderson D., Günther U. L., Neoplasia 2017, 19, 165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Monteiro M. S., Barros A. S., Pinto J., Carvalho M., Pires-Luís A. S., Henrique R., Jerónimo C., Bastos M. D. L., Gil A. M., Guedes D. P., Sci. Rep. 2016, 10.1038/srep37275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Rocha C. M., Barros A. S., Goodfellow B. J., Carreira I. M., Gomes A., Sousa V., Bernardo J., Carvalho L., Gil A. M., Duarte I. F., Carcinogenesis 2015, 36, 68–75. [DOI] [PubMed] [Google Scholar]
- 167. Fages A., Duarte-Salles T., Stepien M., Ferrari P., Fedirko V., Pontoizeau C., Trichopoulou A., Aleksandrova K., Tjønneland A., Olsen A., et al., BMC Med. 2015, 13, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Stenson M., Pedersen A., Hasselblom S., Nilsson-Ehle H., Karlsson B. G., Pinto R., Andersson P.-O., Leuk. Lymphoma 2016, 57, 1814–1822. [DOI] [PubMed] [Google Scholar]
- 169. Palmnas M. S. A., Vogel H. J., Metabolites 2013, 3, 373–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Jobard E., Pontoizeau C., Blaise B. J., Bachelot T., Elena-Herrmann B., Trédan O., Cancer Lett. 2014, 343, 33–41. [DOI] [PubMed] [Google Scholar]
- 171. Asiago V. M., Alvarado L. Z., Shanaiah N., Gowda G. A. N., Owusu-Sarfo K., Ballas R. A., Raftery D., Cancer Res. 2010, 70, 8309–8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Choi J. S., Baek H.-M., Kim S., Kim M. J., Youk J. H., Moon H. J., Kim E.-K., Nam Y. K., PLoS ONE 2013, 10.1371/journal.pone.0083866. [DOI] [Google Scholar]
- 173. Oakman C., Tenori L., Biganzoli L., Santarpia L., Cappadona S., Luchinat C., Di L. A., Int. J. Biochem. Cell Biol. 2011, 43, 1010–1020. [DOI] [PubMed] [Google Scholar]
- 174. Oakman C., Tenori L., Claudino W. M., Cappadona S., Nepi S., Battaglia A., Bernini P., Zafarana E., Saccenti E., Fornier M., et al., Ann. Oncol. 2011, 22, 1295–1301. [DOI] [PubMed] [Google Scholar]
- 175. Jobard E., Trédan O., Bachelot T., Vigneron A. M., Aït-Oukhatar C. M., Arnedos M., Rios M., Bonneterre J., Diéras V., Jimenez M., et al., Oncotarget 2017, 8, 83570–83584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. McCartney A., Vignoli A., Hart C., Tenori L., Luchinat C., Biganzoli L., Di Leo A., Breast 2017, 34, S13–S18. [DOI] [PubMed] [Google Scholar]
- 177. Tenori L., Oakman C., Morris P. G., Gralka E., Turner N., Cappadona S., Fornier M., Hudis C., Norton L., Luchinat C., et al., Mol. Oncol. 2015, 9, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Tripoliti E. E., Papadopoulos T. G., Karanasiou G. S., Naka K. K., Fotiadis D. I., Comput. Struct. Biotechnol. J. 2017, 15, 26–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Mercier K. A., Al-Jazrawe M., Poon R., Acuff Z., Alman B., Sci. Rep. 2018, 8, 584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Merz A. L., Serkova N. J., Biomark. Med. 2009, 3, 289–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Zhang L., Hatzakis E., Patterson A. D., Curr. Pharmacol. Rep. 2016, 2, 231–240. [Google Scholar]
- 182. Carrigan J. B., Reed M. A. C., Ludwig C., Khanim F. L., Bunce C. M., Günther U. L., ChemPlusChem 2016, 81, 453–459. [DOI] [PubMed] [Google Scholar]
- 183. Basoglu A., Baspinar N., Tenori L., Hu X., Yildiz R., Metabolomics: open access 2014, 4, 134. [Google Scholar]
- 184. Basoglu A., Baspinar N., Tenori L., Vignoli A., Yildiz R., Metabolomics 2016, 12, 128. [Google Scholar]
- 185. Basoglu A., Baspinar N., Tenori L., Vignoli A., Gulersoy E., Biol. Trace Elem. Res. 2017, 1–8. [DOI] [PubMed] [Google Scholar]
- 186.A. Basoglu, I. Sen, G. Meoni, L. Tenori, A. Naseri, “NMR-Based Plasma Metabolomics at Set Intervals in Newborn Dairy Calves with Severe Sepsis,” 10.1155/2018/8016510 can be found under https://www.hindawi.com/journals/mi/2018/8016510/, 2018. [DOI] [PMC free article] [PubMed]
- 187. Dervishi E., Zhang G., Mandal R., Wishart D. S., Ametaj B. N., Anim. Int. J. Anim. Biosci. 2018, 12, 1050–1059. [DOI] [PubMed] [Google Scholar]
- 188. Caracausi M., Ghini V., Locatelli C., Mericio M., Piovesan A., Antonaros F., Pelleri M. C., Vitale L., Vacca R. A., Bedetti F., et al., Sci. Rep. 2018, 8, 2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Schicho R., Shaykhutdinov R., Ngo J., Nazyrova A., Schneider C., Panaccione R., Kaplan G. G., Vogel H. J., Storr M., J. Proteome Res. 2012, 11, 3344–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Cano K. E., Li L., Bhatia S., Bhatia R., Forman S. J., Chen Y., J. Proteome Res. 2011, 10, 2873–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Kanehisa M., Goto S., Nucleic Acids Res. 2000, 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Xia J., Wishart D. S., Nucleic Acids Res. 2010, 38, W71–W77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Ramirez-Gaona M., Marcu A., Pon A., Grant J., Wu A., Wishart D. S., J. Vis. Exp. 2017, 10.3791/54869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Xia J., Wishart D. S., Curr. Protoc. Bioinforma. 2016, 55, 1410.1–14.10.91. [DOI] [PubMed] [Google Scholar]
- 195. Fan T. W.-M., Lorkiewicz P. K., Sellers K., Moseley H. N. B., Higashi R. M., Lane A. N., Pharmacol. Ther. 2012, 133, 366–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Lane A. N., Fan T. W.-M., Bousamra M., Higashi R. M., Yan J., Miller D. M., OMICS J. Integr. Biol. 2011, 15, 173–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Bruntz R. C., Lane A. N., Higashi R. M., Fan T. W.-M., J. Biol. Chem. 2017, 292, 11601–11609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Le A., Lane A. N., Hamaker M., Bose S., Gouw A., Barbi J., Tsukamoto T., Rojas C. J., Slusher B. S., Zhang H., et al., Cell Metab. 2012, 15, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Oberg A. L., Vitek O., J. Proteome Res. 2009, 8, 2144–2156. [DOI] [PubMed] [Google Scholar]
- 200. Cohen J., Statistical Power Analysis for the Behavioral Sciences, L. Erlbaum Associates, Mahwah, NJ, 1988. [Google Scholar]
- 201. McHugh M., Biochem. Medica 2008, 263–274. [Google Scholar]
- 202. Vignoli A., Tenori L., Luchinat C., Saccenti E., J. Proteome Res. 2018, 17, 97–107. [DOI] [PubMed] [Google Scholar]
- 203.“Food & Beverages—worldwide | Statista Market Forecast,” can be found under https://www.statista.com/outlook/253/100/food-beverages/worldwide.
- 204.“Wellness Now a $3.72 Trillion Global Industry,” can be found under https://www.globalwellnessinstitute.org/wellness-now-a-372-trillion-global-industry/.
- 205. Cifuentes A., J. Chromatogr. A 2009, 1216, 7109. [DOI] [PubMed] [Google Scholar]
- 206. Savorani F., Rasmussen M. A., Mikkelsen M. S., Engelsen S. B., Food Res. Int. 2013, 54, 1131–1145. [Google Scholar]
- 207. Zotti M., Coco L. D., Pascali S. A. D., Migoni D., Vizzini S., Mancinelli G., Fanizzi F. P., Heliyon 2016, 2, e00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Gao Q., Praticò G., Scalbert A., Vergères G., Kolehmainen M., Manach C., Brennan L., Afman L. A., Wishart D. S., Andres-Lacueva C., et al., Genes Nutr. 2017, 12, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Rådjursöga M., Karlsson G. B., Lindqvist H. M., Pedersen A., Persson C., Pinto R. C., Ellegård L., Winkvist A., Food Chem. 2017, 231, 267–274. [DOI] [PubMed] [Google Scholar]
- 210. Cukrov D., Zemiani M., Brizzolara S., Cestaro A., Licausi F., Luchinat C., Santucci C., Tenori C., Van Veen H., Zuccolo A., et al., Front. Plant Sci. 2016, 10.3389/fpls.2016.00146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Santucci C., Brizzolara S., Tenori L., Food Anal. Methods 2015, 8, 2135–2140. [Google Scholar]
- 212. Spraul M., Schütz B., Humpfer E., Mörtter M., Schäfer H., Koswig S., Rinke P., Magn. Reson. Chem. 2009, 47, S130–S137. [DOI] [PubMed] [Google Scholar]
- 213. Minoja A. P., Napoli C., Food Res. Int. 2014, 63, 126–131. [Google Scholar]
- 214. Humpfer E., Schütz B., Fang F., Cannet C., Mörtter M., Schäfer H., Spraul M., Magn. Reson. Food Sci. 2015, 77–83. [Google Scholar]
- 215. Tenori L., Santucci C., Meoni G., Morrocchi V., Matteucci G., Luchinat C., Food Res. Int. 2018, 10.1016/j.foodres.2018.06.066. [DOI] [PubMed] [Google Scholar]
- 216. Takis P. G., Tenori L., Ravera E., Luchinat C., Anal. Chem. 2017, 89, 1054–1058. [DOI] [PubMed] [Google Scholar]
- 217. Zhang B., Yuan J., Brüschweiler R., Chem. Eur. J. 2017, 23, 9239–9243. [DOI] [PubMed] [Google Scholar]
- 218. Konz T., Migliavacca E., Dayon L., Bowman G., Oikonomidi A., Popp J., Rezzi S., J. Proteome Res. 2017, 16, 2080–2090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary