

SUMMARY
Alternative translation initiation and stop codon readthrough in a few well-studied cases have been shown to allow the same transcript to generate multiple protein variants. Because the brain shows a particularly abundant use of alternative splicing, we sought to study alternative translation in CNS cells. We show that alternative translation is widespread and regulated across brain transcripts. In neural cultures, we identify alternative initiation on hundreds of transcripts, confirm several N-terminal protein variants, and show the modulation of the phenomenon by KCl stimulation. We also detect readthrough in cultures and show differential levels of normal and readthrough versions of AQP4 in gliotic diseases. Finally, we couple translating ribosome affinity purification to ribosome footprinting (TRAP-RF) for cell-type-specific analysis of neuronal and astrocytic translational readthrough in the mouse brain. We demonstrate that this unappreciated mechanism generates numerous and diverse protein isoforms in a cell-type-specific manner in the brain.
Graphical Abstract
In Brief
By deep sequencing ribosome-bound mRNA fragments, Sapkota et al. demonstrate non-canonical translation initiation and stop codon readthrough that diversify proteins in the mouse brain. They also show that neuronal stimulation regulates the choice of initiation sites, whereas gliotic diseases differentially regulate the levels of normal and readthrough AQP4 isoforms.
INTRODUCTION
“One gene, one protein” has long been an obsolete axiom. Cells use alternative splicing, where a single gene generates multiple transcripts, each of which may be translated into a protein isoform (Graveley, 2001). To further expand proteomic landscapes, cells can also utilize alternative translation initiation sites (TISs) and termination sites on a given transcript (Dabrowski et al., 2015; Kozak, 1999). Alternative splicing is well studied and known to be modulated by neural activity and disease states and in a cell-type-specific manner in the CNS (Hermey et al., 2017; Licatalosi and Darnell, 2006; Parikshak et al., 2016; Quesnel-Vallières et al., 2016; Zhang et al., 2014b). However, alternative initiation and termination have not yet been systematically surveyed across the CNS or assessed for variation across neurons and astrocytes. In other tissues, genomic and proteomic studies have revealed a widespread use of alternative TISs (Slav-off et al., 2013; Ingolia et al., 2011; Vanderperre et al., 2013; Wan and Qian, 2014; Zupanic et al., 2014; Menschaert et al., 2013; Lee et al., 2012) and stop codon readthrough (Dunn et al., 2013; Jungreis et al., 2011). In the CNS, only single-gene studies have been done, which have identified alternative open reading frames for individual transcripts and demonstrated their functional roles in homeostasis and disease (Bergeron et al., 2013; Chua et al., 2012; De Bellis et al., 2017; Delmas et al., 1992; Green et al., 2016; Simkin et al., 2008; Studtmann et al., 2014; Thomas et al., 2008; Vanderperre et al., 2011).
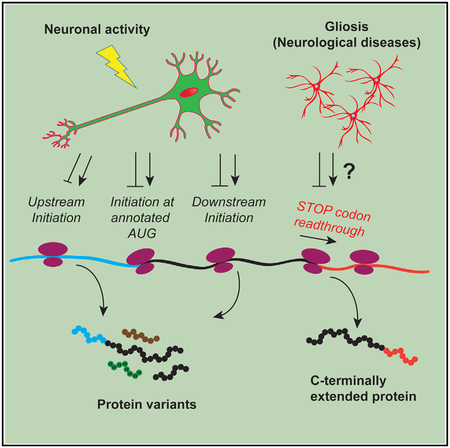
With alternative initiation and termination, “one transcript, one protein” also becomes an untrue axiom. In conventional eukaryotic initiation, a 43S preinitiation complex scans the 5′ UTR until it encounters an AUG at the annotated TIS (aTIS), where the 60S ribosomal subunit joins it to form an 80S initiation complex (Jackson et al., 2010). However, an 80S complex may assemble at an upstream TIS (uTIS) if the 5′ UTR has an optimal AUG (or near-cognate NUG) or at a downstream TIS (dTIS) if the aTIS is suboptimal (Kozak, 1999). Similarly, in conventional termination, the 80S complex concludes translation at the first in-frame stop codon. For some transcripts, however, it may read past that stop codon at some frequency and conclude translation at a second stop codon in the 3′ UTR (Dabrowski et al., 2015). As expected, alternative initiation may generate N-terminal protein isoforms when in frame or entirely different protein species when out of frame, whereas stop codon read-through can give rise to C-terminally extended protein isoforms (Figure 1A).
Figure 1. RF of Homoharringtonine-Treated Neuron-Glia Culture Reveals Translation Initiation Sites.

(A) uTISs, aTISs, and dTISs and the corresponding protein products are depicted. uTISs and dTISs give rise to N-terminal variants of the protein when in frame, but code for completely new polypeptide sequences when out of frame. Stop codon readthrough that generates a C-terminal extension is also depicted. In this study, Figures 1, 2, and 3 concern TISs, and Figures 4, 5, and 6 concern readthrough.
(B) Experimental workflow for the in vitro study. Mixed neurons and glia from post-natal day (P)0 mice were cultured for 7 days in vitro, exposed to KCl or no KCl, and subjected to RF and TIS-mapping RF as shown.
(C) Average ribosomal density across transcripts shows a run-off of elongating ribosomes from the proximal 300-nt region of the coding sequence in the HHT-treated sample as compared to the no-HHT sample. Plot from the KCl-untreated cultures is shown; KCl-treated cultures gave the same density distributions.
(D) The number of high-fidelity TISs in the 426 most robustly expressed transcripts. Most of the transcripts showed >1 TIS. Only the top-five TISs were called.
(E) Codon composition and frame status of TISs across transcripts. TIS codons are shown as ATG or a cognate thereof (NTG). Numbers inside the bars indicate the percentages of in-frame TISs.
RF, ribosome footprinting; HHT, homoharringtonine; TIS, translation initiation site; u/a/dTIS, upstream/annotated/downstream TIS. Also see Figures S1 and S2 and Table S1.
Here we profiled and examined the regulation of alternative TISs and stop codon readthrough in CNS cells. We screened for uTISs and dTISs genome-wide in neuron-glia cultures using ribosome footprinting (RF), determined a subset of these protein products in neurons using liquid chromatography-tandem mass spectrometry (LC-MS/MS), and then examined how their use is regulated by KCl depolarization. We also identified transcripts evidencing stop codon readthrough in cultures and then focused on one of the candidates, Aqp4, to determine whether the resulting extended protein is regulated by diseases. Finally, we coupled translating ribosome affinity purification to RF (TRAP-RF) to determine the extent to which neurons and astrocytes use readthrough in vivo.
RESULTS
RF of Homoharringtonine-Treated Neuron-Glia Cultures Reveals Use of Novel TISs In Vitro
To investigate alternative TISs, we used mouse brain-derived primary neuron-glia mixed cultures as our experimental platform because they offer two advantages. First, such cultures can be rapidly and synchronously treated with homoharringtonine (HHT) to arrest initiating ribosomes while allowing elongating ribosomes to run off the transcript (Ingolia et al., 2011). Non-neural cell cultures treated successively with HHT and cycloheximide (CHX), a drug that immobilizes elongating ribosomes (Schneider-Poetsch et al., 2010), have been used to identify TISs previously (Ingolia et al., 2011). Second, KCl treatment of neuronal cultures induces membrane depolarization, inflow of Ca2+ through L-type Ca2+ channels, and expression of c-Fos and other activity-dependent genes, thus offering a simple yet effective paradigm for studying activity-dependent gene regulation (Bading et al., 1993; Kim et al., 2010; Greer and Greenberg, 2008).
Accordingly, we utilized neuron-glia cultures with or without HHT and KCl treatments and subjected them to RF following CHX treatment (Figure 1B). We first ensured our data had the characteristics of bona fide ribosome footprints—specifically, we confirmed that ribosomal P sites primarily correspond to the correct reading frame and display the characteristic 3-nt periodicity in the coding sequence (CDS) when compared to the UTRs (Figure S1). Next, we examined footprint distribution with HHT treatment, which revealed a near-complete run-off of ribosomes from the proximal 300-nt region of the CDS 2 min after introducing the drug (Figure 1C). We focused on this 300-nt region of the most robustly expressed transcripts and developed a set of stringent criteria (see STAR Methods) to detect high-fidelity TISs in both KCl-treated and -untreated cultures. We detected TISs in 426 transcripts, most of which possessed multiple TISs, with 89.7% having two or more and only 11.3% possessing a single one (Table S1; Figure 1D). Although previous high-throughput studies on TISs have used non-neural cells, we compared our list of alternative TISs to published lists generated from mouse embryonic stem cells (Ingolia et al., 2011) or various human cultured cells (Vanderperre et al., 2013) and found that more than 50% of the transcripts in our list overlapped with the lists from the previous studies. These results convinced us that RF on HHT-treated neuron-glia culture combined with a stringent analysis is able to detect alternative TISs in neural transcripts.
Because the number, composition, and frame of TISs are important determinants of the proteomic complexity of a cell, we examined these features across the transcripts for which we detected high-fidelity TISs (Figure 1E). On average, a transcript contained 3.18 TISs—with 1.33 uTISs, 0.94 dTIS, and 0.91 aTIS. The aTIS-per-transcript value of <1 resulted from the absence of an initiation peak at the annotated ATG of some transcripts, suggesting that these transcripts might be misannotated or the dominant protein isoform is not the canonical one in neural cells. ATGs detected at aTISs were always in frame, consistent with their presence as a key contributor to the original annotation. Similarly, near-cognate codons such as CTG, TTG, and GTG detected at aTISs were in frame >99% of the time. On the other hand, ATGs and cognate codons detected at uTISs and dTISs were out of frame ≥47% of the time. These data suggest that a brain transcript may use multiple TISs, each at a rate that is regulated in a transcript-specific manner, which can generate multiple protein variants or completely different peptide sequences.
LC-MS Detects Protein Products of Alternative Initiation
We next asked whether a subset of the alternative TISs results in a detectable amount of polypeptides, and were interested to identify which of these were specifically coming from neurons. We noted that most uTISs and dTISs are close to their corresponding aTISs, suggesting that a biochemical assay such as a western blot would not clearly resolve the alternative and normal protein isoforms. Therefore, we examined a quantitative proteome of cultured primary mouse cortical neurons using LCMS. In spite of the different sensitivities of LC-MS and RF, we clearly detected variant N-terminal peptides resulting from 10 of the predicted alternative TISs (Table 1). These included a peptide from an in-frame uTIS lying at the −63rd nt of Rtn1, which encodes reticulon 1, an endoplasmic reticulum protein expressed in neuroendocrine cells and potentially involved in vesicle secretion (Chiurchiù et al., 2014), and a peptide from an in-frame dTIS at the 15th nt of Uchl1, which encodes ubiquitin C-terminal hydrolase L1, a neuron-specific deubiquitinating protease with protective roles in neurodegeneration and Alzheimer’s disease (Bilguvar et al., 2013; Zhang et al., 2014a) (Figures 2A and 2B). We also examined the proteome of the mouse brain reported by others (Sharma et al., 2015) and found variant N-terminal peptides corresponding to 18 alternative TISs—5 as confirmed by neuronal LC-MS, and 13 additional (Table S2). Thus, at least a subset of the TISs detected by our algorithm is utilized in the CNS to produce detectable protein, and alternative translation does contribute to the diversity in the brain proteome.
Table 1.
Peptide Products of Alternative Translation Initiation Sites Detected by Mass Spectrometry Analysis of Neuronal Cultures
| Transcript ID | Gene Symbol | Alternative Initiation Site | Frame |
|---|---|---|---|
| ENSMUST00000031131 | Uchl1 | +15 | in frame |
| ENSMUST00000031565 | Fascn1 | +12 | in frame |
| ENSMUST00000106255 | Cap1 | +9 | in frame |
| ENSMUST00000028981 | Mapre1 | +12 | in frame |
| ENSMUST00000112229 | Gpm6b | +12 | in frame |
| ENSMUST00000174548 | Hnrnpl | +102 | in frame |
| ENSMUST00000007980 | Hnrnpa0 | −54 | in frame |
| ENSMUST00000067664 | Ywhae | −66 | in frame |
| ENSMUST00000143971 | Minos1 | −21 | in frame |
| ENSMUST00000021497 | Rtn1 | −63 | in frame |
+ and − indicate downstream and upstream relative to the canonical initiation site, respectively. Also see Table S2.
Figure 2. Mass Spectrometry Detects the Peptides Resulting from Alternative TISs.

(A and B) Peptides resulting from an in-frame uTIS (Rtn1) (A) and an in-frame dTIS (Uchl1) (B). Upper: the percentage of ribosomes at different TISs (red arrowheads correspond to alternative TISs whose novel products are detected; zeros correspond to aTISs). Middle: the peptide sequences with the amino acids corresponding to alternative TISs and aTISs in red and green, respectively. Bottom: the tandem mass spectra and mass-to-charge ratios of b and y product ions confirming peptide sequences. Alternative TISs for Rtn1 and Uchl1 lie at the −63rd and +15th nt relative to the aTIS, respectively.
(C and D) Conservation of uTISs with respect to 5′ UTR (C) and of dTISs with respect to CDS (D). Violin plots show the average of the PhastCons scores for the 3 nt of the ATG, CTG, GTG, or TTG present at the uTISs or dTISs or elsewhere in the 5′ UTR or CDS. Wilcoxon test was used to assess the statistical significance.(E) Conservation of the uTISs and dTISs of mass spectrometry-confirmed peptide products. Plot shows the average of the PhastCons scores of the 3 nucleotides. Also see Tables S1 and S2.
To determine whether the identified alternative TISs might represent evolutionarily important adaptations, we examined their conservation across phyla. We used PhastCons (Siepel et al., 2005) in a multi-species alignment of 60 vertebrates to compute conservation scores for the NTGs (ATG, CTG, GTG, or TTG) detected as alternative TISs, and tested the hypothesis that these were more conserved than their background sequence (the entire 5′ UTRs for uTISs and the entire CDSs for dTISs). We also computed the score for the NTGs not detected as alternative TISs and the respective 5′ UTRs or CDSs. Surprisingly, regardless of alternative TISs or not and in frame or not, 5′ UTR NTGs were less conserved whereas CDS NTGs were generally more conserved than their respective backgrounds (Figures 2C and 2D). However, NTGs detected both as uTISs or dTISs were not more conserved than NTGs detected in their respective backgrounds. When we specifically examined the conservation of the ten alternative TISs for which we detected the corresponding peptides with LC-MS (Table 1), we found eight of them to be highly conserved, one only moderately conserved, and one not conserved (Figure 2E). In sum, our analyses suggest that specific alternative TISs, particularly those that are robust enough to generate detectable protein, may be highly conserved. Yet, the significantly decreased conservation of NTGs in 5′ UTRs, even for the many that show initiation, also suggests that some modifications of TISs may be a substrate for evolution of gene regulation across species.
TISs Reveal the Complexity of Initiation on Neural Transcripts
We wanted to examine whether the identified TISs provide support for the well-accepted scanning model of initiation, which postulates that the small ribosomal subunit scans the 5′ UTR of a transcript and triggers initiation at an AUG, or near cognate, surrounded by specific nucleotides (Kozak sequence) (Kozak, 1999). To this end, we quantified the Kozak strength of each TIS by using the Kozak consensus sequence empirically derived from a synthetic translation assay as a benchmark (Sample et al., 2018) and the relative usage of each TIS by dividing the ribosomal occupancy at that TIS over the total ribosomal occupancy across all TISs on a given transcript. Overall, aTISs possessed significantly stronger Kozak sequences than uTISs and dTISs (Figure S2A). Comparing the two quantities across all TISs in all transcripts, we found that TISs with stronger Kozak scores—especially aTISs, as expected—are generally used more frequently than those with weaker Kozak scores (Figure S2B). Next, comparing uTIS Kozak scores with aTIS usage, we observed that uTISs with stronger Kozak scores are strongly suppressive toward aTISs (Figure S2C), consistent with the idea that they can act as a sink for the scanning ribosomal subunits that would otherwise proceed downstream (Kozak, 1999). Finally, we found that 5′ UTRs possess more uTISs as a function of their length (Figure S2D), and consequently longer 5′ UTRs are more suppressive toward aTISs than shorter 5′ UTRs (Figure S2E). Based on a multivariate analysis, the aTIS Kozak score and uTIS Kozak score are the best predictors of aTIS usage (R-squared 0.518985) (Figure S2F). These results reveal an interplay between uTISs and aTISs that is likely to regulate the amount of canonical proteins synthesized from neural transcripts.
Similarly, we found that aTISs with stronger Kozak scores exert suppression toward dTISs; however, this effect was more modest (Figure S2G), suggesting additional factors might regulate dTIS usage. Codon bias is known to influence local ribosomal traffic (Tuller et al., 2010), and therefore post-aTIS codon bias can arguably affect the access of ribosomes to dTISs (e.g., an inefficiently translating ribosome downstream of the first aTIS might block access to subsequent dTISs by scanning ribosomes). When we compared the post-aTIS and post-dTIS regions for codon composition, we indeed found specific codons and amino acids to be significantly different between the two (not shown). However, aside from the expected increase in ATGs at aTISs relative to dTISs, the remaining observations (depleted AGG and CAC) did not clearly match any expectations from codon usage biases. Although these observations do not rule out the possibility of additional mechanisms that might regulate dTIS usage, they show that the codons immediately beyond an aTIS may be skewed more than expected by chance and potentially regulate ribosomal encounter with a dTIS.
Transcripts Change TIS Usage in Response to Neuronal Depolarization
Neuronal activity induces de novo protein synthesis—both from existing mRNAs and via induction of a specific transcriptional program (Costa-Mattioli et al., 2009; Dalal et al., 2017; Flavell and Greenberg, 2008). However, it is unclear whether it can also alter TIS usage on a given transcript. Therefore, we asked whether KCl depolarization of our neuron-glia culture model regulates the use of TISs. For this purpose, we removed transcripts that had only one TIS and/or no aTIS from our list of transcripts. We found that, of the remaining transcripts with at least one alternative TIS, 32.5% exhibited a significant change in the ribosomal occupancies of their aTISs, uTISs, or dTISs, with a majority showing increased use of aTISs in response to KCl depolarization (Figure 3A). More importantly, we observed that an increased use of aTISs was accompanied by a corresponding decreased use of alternative TISs and vice versa, with a few exceptions where changes at both TISs went in the same direction. These results indicate that specific transcripts use their TISs differentially in response to neuronal depolarization.
Figure 3. Neuronal Activity Modulates the Use of TISs.

(A) Heatmap shows transcripts with significant changes (false discovery rate ≤0.05) in the usage of aTISs, uTISs, or dTISs in response to KCl depolarization of neuron-glia culture. Usage of a TIS was computed as the P site counts at that TIS divided by the total P site counts across all TISs for a given transcript. For transcripts with multiple uTISs and dTISs, combined changes over all uTISs and dTISs is shown. Chi-square was used to test for the difference of P site counts across TISs.
(B) Representative tracts are shown to highlight the abundance of ribosomes at TISs and the use of different TISs in response to KCl depolarization. Numbers on the x axis indicate nucleotide positions, with zero being the aTIS, and numbers on the y axis indicate the percentages of ribosomes. Arrowheads point to the uTISs and dTISs.
(C) Boxplot quantifies the KCl-mediated shifts in TIS use of the transcripts shown in (B). n = 3, paired t test. Error bar, SEM. Also see Table S1.
We then examined the transcripts exhibiting depolarization-regulated use of TISs. The increased use of aTISs by the majority of transcripts was consistent with an increase in de novo protein synthesis from the canonical open reading frame following stimulation. Transcripts that showed this included Grina and Hspa4, which code for an NMDA receptor subunit and a heat shock protein involved in the folding of other proteins, respectively (Garcia-Quintanilla and Miranzo-Navarro, 2016; Stetler et al., 2010) (Figures 3B and 3C). On the other hand, among the few transcripts that showed an increased use of uTISs was Kif1b, which codes for a kinesin superfamily protein involved in transporting synaptic vesicles (Hirokawa and Take-mura, 2005) (Figures 3B and 3C). Interestingly, Gfap, which codes for an intermediate filament in astrocytes (Middeldorp and Hol, 2011), also exhibited a modest regulation of its uTIS. Notably, as shown in Figure 3A, we found that although some of the uTISs and dTISs with altered usage were in frame, most were out of frame, suggesting the regulated production of not only N-terminal protein variants but also novel polypeptides following activity. Overall, these findings show that stimulation alters the use of TISs of several transcripts that are involved in diverse functions, indicating a regulated production of alternative protein isoforms in neural cells.
RF Identifies Novel C-Terminal Extensions Mediated by Stop Codon Readthrough In Vitro
As illustrated in Figure 1A, stop codon readthrough is another mode of alternative translation, and it generates a C-terminally extended protein variant. Recently, RF was utilized to identify readthrough in Drosophila embryos (Dunn et al., 2013). Shortly thereafter, bioinformatics approaches predicted at least 7 read-through events in human transcripts, of which 4 were validated as capable of >1% readthrough using luciferase assays in HeLa cells (Loughran et al., 2014). These included the Aqp4 transcript, which encodes a water channel that is highly expressed in astroglia. A genome-wide screen for transcripts exhibiting readthrough, however, has not yet been conducted in CNS cells.
To profile readthrough in our neuron-glia cultures, we used the RF dataset from HHT-untreated cultures and counted the footprints mapping to the 3′ UTR of robustly expressed transcripts. We carefully excluded any transcripts where alternative splicing or stop codon SNPs might give rise to such footprints (see STAR Methods for details). Overall, we identified 18 transcripts with at least 1% readthrough (Table S3). These include Aqp4, as well as malate dehydrogenase 1 (Mdh1) (Figure 4A). For an independent validation of these findings, we cloned a cassette encompassing the distal CDS and the readthrough region of candidate transcripts into a dual-luciferase vector so that Renilla luciferase was constitutively expressed whereas firefly luciferase was expressed only if the stop codon was read through (Fixsen and Howard, 2010) (Figure 4B). Comparison of the relative luciferase activities confirmed that the mouse Aqp4 sequence permits readthrough of 12.5% when compared to a positive control sequence where the stop codon was mutated to a sense codon (Figure 4C). This is a level similar to that previously reported for human AQP4 in HeLa cells (Loughran et al., 2014). Likewise, the Mdh1 sequence permits readthrough at 5.5%, whereas the Map2 sequence is not read through. The addition of an extra stop codon beyond the original stop codons abrogated the read-through of both Aqp4 and Mdh1, indicating luciferase expression is not due to a cryptic promoter or internal ribosome entry site sequence.
Figure 4. RF of Cycloheximide-Treated Neuron-Glia Culture Reveals Stop Codon Readthrough In Vitro.

(A) A schematic of footprint mapping and genome browser tracts with examples of readthrough are shown. Aqp4 and Mdh1 display footprints mapping to the readthrough region, whereas Map2 does not.
(B) Dual-luciferase assay assessing the permissiveness of sequences for readthrough. A cassette spanning the distal CDS and the readthrough region is cloned between the Renilla luciferase (RL) and firefly luciferase (FL) such that the latter is expressed in transfected cells only if ribosomes read past the cassette. The stop codon is mutated to a sense codon in the positive control, whereas an extra stop codon is added in the negative control.
(C) Dual-luciferase assay shows that Aqp4 and Mdh1 undergo readthrough at the rates of 12.5% and 5.5%, respectively, and that Map2 does not undergo readthrough. Readthrough rate is calculated as shown in (B) (n = 3; error bar, SEM).
CDS, coding sequence; RA, Renilla activity; FA, firefly activity; Ct, control. Also see Table S3 and Figure S3.
For another independent validation of readthrough, we analyzed our neuronal LC-MS data but were unable to detect any extended peptides (see Discussion). We then resorted to mouse brain MS data from others (Sharma et al., 2015), and could detect readthrough peptides for Aqp4, Ttr (transthyretin), and Map1lc3a (microtubule-associated protein 1 light chain 3 alpha) (Figure S3). We noticed that whereas read-through of the latter two transcripts was not detected by our neuron-glia RF, that of several others that are strongly expressed in the brain and well known to undergo readthrough, including MDH1 (Hofhuis et al., 2016), was not detected by MS, suggesting MS negative results should be interpreted with caution. Interestingly, an arginine was incorporated in lieu of the TGA stop codon of Aqp4, suggesting that other amino acids besides those predicted by tRNA wobbling such as tryptophan (TGG) or a cysteine (TGT, TGC) can be incorporated, and similar to what has previously been seen by Hofhuis et al. for Mdh1. Together, our results show the possibility of several readthrough events in the brain, highlight the conflict between RF and MS in detecting readthrough, and provide a case of how the stop codon might be recoded in readthrough.
Readthrough Localizes AQP4 to the Perivascular Region in the CNS
We next wanted to test whether readthrough is of functional significance. We focused on AQP4, an astrocyte membrane protein shown to have a readthrough version in other species by previous studies (De Bellis et al., 2017; Loughran et al., 2014). To detect readthrough-extended AQP4 (designated AQP4X hereafter), we developed a rabbit polyclonal antibody using a peptide from the readthrough region as an epitope. The antibody recognized AQP4X but not AQP4 in immunofluorescence staining of cell cultures expressing the respective version of AQP4, thus demonstrating sensitivity and specificity (Figure S4). Western blot on mouse brain lysates revealed that anti-AQP4X recognizes the expected 35-kDa band in the adult brain but not in the developing brains of postnatal day 9 or younger mice (Figure S5A). This is interesting because astrocyte maturation, particularly the endfeet at the blood-brain barrier, are not complete until 3 weeks of age (Caley and Maxwell, 1970). In line with western blot, immunofluorescence staining showed that AQP4X is abundant in the adult mouse brain (Figure S5B). We noted that AQP4X is significantly concentrated perivascularly and adjacent to the endothelial cell marker PECAM-1, whereas AQP4 is also abundant farther from the blood vessels (Figures S5B and S5C). Similar results were also recently reported by others with an independently raised antibody in rats (De Bellis et al., 2017). Thus, readthrough confers a conserved perivascular localization signal to AQP4.
Because commercial anti-AQP4 antibodies also recognize AQP4X, our immunofluorescence data above could not resolve whether AQP4 also is perivascular to some extent. To address this, we transduced mouse brains with adeno-associated virus 9 (AAV9) expressing cMyc-Aqp4X− (Aqp4 mRNA with an additional stop codon to prevent readthrough) or cMyc-Aqp4X+ (Aqp4 stop codon mutated to allow 100% readthrough) under the astrocytic promotor Gfap and examined cMyc epitope expression from the two viruses using immunofluorescence staining (Figure S5D). Although virally expressed proteins showed slightly unusual subcellular expression patterns (compare Figures S5B and S5D), possibly due to overexpression relative to binding partners, AAV9-Gfap::cMycAqp4X− still clearly resulted in significantly less perivascular cMyc than AAV9-Gfap::cMycAqp4X+ (Figure S5E). These results are consistent with the above data on endogenous AQP4 and AQP4X, and suggest that AQP4X is relatively enriched in the perivascular region.
AQP4 is expressed in numerous tissues and required for their normal functioning (Mobasheri et al., 2007; Nagelhus and Ottersen, 2013). Therefore, we also asked whether its readthrough is a brain-specific or a ubiquitous phenomenon. By immunofluorescence staining, we detected AQP4X in the retina as well as the kidneys (Figures S5F and S5G). However, whereas AQP4X was mostly perivascular, and hence distinct from AQP4 in the retina, it was completely overlapping with AQP4 in the kidneys. Thus, AQP4 readthrough is not a brain-specific phenomenon; however, whether it alters subcellular localization depends on the tissue.
AQP4 and AQP4X Are Differentially Regulated by Gliosis across Multiple Disease Models
Currently, it is unclear whether readthrough always occurs at a consistent rate or is regulated in response to conditions in the brain. The AQP4 gene has been implicated in diverse (patho) physiological conditions in the brain, including brain water homeostasis, edema, and ischemia (reviewed in Nagelhus and Ottersen, 2013). Given the largely mutually exclusive localizations of AQP4 and AQP4X in the brain, we first examined whether they are independently regulated in ischemia. We induced ischemia in adult mice by transient middle cerebral artery occlusion (tMCAO). In mice, MCAO results in relatively reproducible infarcts in the lateral caudatoputamen and frontoparietal cortex (Sicard and Fisher, 2009). We performed immunostaining for AQP4 and AQP4X in ischemic mouse brains and quantified their signals in gliotic peri-infarct regions. We found that compared to the regions in the contralesional hemisphere, GFAP-labeled peri-infarct regions showed a 2.5-fold upregulation of AQP4 but only a disproportionate 1.3-fold upregulation of AQP4X (Figures 5A and 5B). This difference suggested that AQP4X generation, degradation, post-translational modification, or folding can be differentially regulated during pathological processes.
Figure 5. AQP4 and AQP4X Are Differentially Upregulated in Gliosis.

(A, C, and E) Immunostaining for AQP4, AQP4X, and GFAP in brain sections from mice with middle cerebral artery occlusion (A), infantile Batten disease (C), and injury-induced gliosis (E). Normal and gliotic hemispheres of the same sections are imaged in (A) and (E), and age-matched wild-type littermate controls are used in (C).
(B, D, and F) Fluorescence intensities in (A), (C), and (E) quantified in (B), (D), and (F), respectively, show that AQP4 is significantly more upregulated than AQP4X by gliosis. Regions of interest were drawn to cover the gliotic area and the contralateral normal area in (A), whereas whole images were considered in (B) and (C). 3 mice, 6 sections/mouse, paired t test; error bar, SEM.
(G) Barplot compiles the relative upregulations of AQP4 and AQP4X from (B), (D), and (F).
Scale bars, 20 μm. Also see Figures S4 and S5.
To determine whether this regulation was driven by the ischemia itself or was more parsimoniously explained as part of gliosis, we used mice lacking palmitoyl protein thioesterase-1 (PPT1), a lysosomal enzyme that is absent in human patients with infantile Batten disease (Gupta et al., 2001). PPT1−/− mice exhibit several key features of this disease, including neurode-generation and gliosis (Bible et al., 2004; Gupta et al., 2001). We compared the expression levels of AQP4 and AQP4X in PPT1−/− and wild-type brains using immunofluorescence staining and observed that they were indeed differentially regulated in response to gliosis in the knockout brains. Whereas AQP4 expression was 2.5-fold upregulated, AQP4X was 1.5-fold upregulated (Figures 5C and 5D).
We also examined whether injury-induced gliosis also exerts a similar regulatory effect on AQP4 and AQP4X, using tissue from mice having undergone stereotactic injection, which can also induce gliosis. We marked the site of injection using AAV9 expressing cMyc-tagged Cyan Fluorescent Protein (CFP) under the Gfap promoter (Sakers et al., 2017). A robust expression of cMyc and CFP (data not shown) and mild gliosis were evident along the injected line after 1 month (Figure 5E). We then repeated the immunostaining as above and found gliotic regions to express AQP4 at ~3-fold and AQP4X at 1.5-fold more intensely as compared to the uninjected contralateral side (Figures 5E and 5F). Overall, our results indicate that diseases and conditions accompanied by gliosis differentially regulate the two isoforms of AQP4 (Figure 5G).
TRAP-RF Identifies Novel, Cell-Type-Specific, C-Terminal Extensions Mediated by Stop Codon Readthrough In Vivo
We finally sought to rule out the possibility that many alternate translation events are due to the unnatural conditions of cell culture, and to determine whether the phenomenon is regulated in a cell-type-specific manner. Therefore, we assessed alternative translation in vivo, specifically focusing on readthrough, which, unlike TISs, can be detected without pharmacological manipulation of the brain. Further, to analyze read-through in a cell-type-specific manner, we used mouse lines that express GFP-tagged ribosomes in either neurons (Snap25::Rpl10a-Egfp) or astrocytes (Aldh1l1::Rpl10a-Egfp), thus allowing the affinity purification of neuronal or astrocytic ribosome-bound mRNA (Dougherty et al., 2012; Doyle et al., 2008). We subjected the transgenic brains to TRAP followed by RF, an approach we call TRAP-RF (Figure 6A), similar to one recently described by Gonzalez et al. (2014). The resulting footprints mapping to the transcriptome were highly reproducible between replicates (Figures 6B and 6C) and showed 3-nt periodicity in the CDS (Figures S6A and S6B), indicating a good reliability of the approach we have developed.
Figure 6. TRAP-RF Detects Cell-Type-Specific Stop Codon Readthrough In Vivo.

(A) TRAP-RF workflow. mRNAs from transgenic brains expressing GFP-tagged ribosomes in neurons (Snap25::Rpl10a-Egfp) or astrocytes (Aldh1l1::Rpl10a-Egfp) are affinity purified with anti-GFP-conjugated magnetic beads, and subjected to on-bead digestion with RNase and the subsequent RF protocol as shown.
(B and C) Log2 (reads per kilobase million [RPKM]) of ribosome footprints mapping to the coding sequence (B) and proximal 3′ UTR (C) was reproducible between the replicate samples for both Snap25::Rpl10a-Egfp and Aldh1l1::Rpl10a-Egfp.
(D) Comparison of TRAP-RF samples between neurons and astrocytes shows the expected enrichment of neuronal (green) and astrocytic (red) transcripts identified in previous experiments (Dougherty et al., 2012).
(E) Of the 50 transcripts detected undergoing ≥1% readthrough in the brain, 21 are neuronal, 19 are astrocytic, and the remaining 10 are non-cell-type specific.
(F) Of the 18 transcripts with ≥1% readthrough in neuron-glia culture, 13 do so in vivo as well.
IP, immunoprecipitation. Also see Table S4 and Figures S3 and S6.
We confirmed enrichment of neuronal and astrocyte marker genes in Snap25::Rpl10a-Egfp and Aldh1l1::Rpl10a-Egfp TRAP-RF data, respectively, matching prior non-footprinted experiments using these lines (Dougherty et al., 2012; Doyle et al., 2008), indicating we were indeed profiling ribosomes enriched from the cell types of interest (Figure 6D). To be sure this was not due to any TRAP-specific artifact, we also confirmed robust enrichment of cell-type-specific markers derived from neuron- and astrocyte-specific immune-panned transcriptomes reported by Zhang et al. (2014b) (not shown). We went on and applied the same stringent criteria as used for in vitro cultures above to detect readthrough events and identified 50 transcripts with at least 1% readthrough in the TRAP-RF data (Table S4), and these were reproducible across replicates. To confirm these were likely coding, we examined the codon bias of these regions. Protein-coding regions show a conserved preference for particular codons, and comparing the readthrough fragments to an equivalent number of length-matched fragments from transcripts without readthrough revealed a significantly enriched codon bias in the readthrough fragments (p < 0.002) that matched the codon preferences of standard CDSs (not shown), consistent with coding potential. Of the 50 transcripts, 21 were neuron enriched, 19 were astrocyte enriched, and the remaining 10 were non-cell-type specific (Figure 6E). Comparing the in vitro and in vivo results, we found that 13 out of the 18 in vitro candidates had clearly detectable readthrough in vivo (Figure 6F), including Aqp4 and Mdh1.
Finally, we analyzed the 50 readthrough candidates to determine whether any cis-regulatory elements in the nearby sequence mediate this phenomenon. Of the three standard stop codons, we found TGA to be overrepresented in the readthrough candidates (Figure S6C), consistent with prior studies (Jungreis et al., 2011). Similarly, motifs for specific RNA-binding proteins were enriched in readthrough regions as compared to shuffled sequence controls and readthrough-negative controls, suggesting these proteins might regulate specific aspects of readthrough candidates (Figures S6D and S6E). In contrast to prior studies in other systems, however, we saw no bias for CUAG immediately downstream of the stop codon (Loughran et al., 2014). Further, we found that readthrough regions were only moderately, but not significantly, conserved as compared to the analog regions in readthrough-negative transcripts (odds ratio 1.83, p = 0.19) (Figure S6F). Finally, we noticed that readthrough regions contain less free energy than the analogous regions in readthrough-negative transcripts (Figure S6G), suggesting the possibility that in addition to the motifs for specific RNA-binding proteins, stable RNA secondary structures might influence the phenomenon. Overall, our TRAP-RF analysis identified both cell-type-specific and shared readthrough candidates that possess specific sequence features and possibly generate functionally important C-terminally extended protein variants in the brain.
DISCUSSION
Initially identified as viral strategies to diversify protein repertoire (Sangar et al., 1987; Schwartz et al., 1992; Weiner and Weber, 1971), non-canonical translational events were later also reported in eukaryotes, including humans (Bazykin and Kochetov, 2011; Dunn et al., 2013; Ingolia et al., 2011; Jungreis et al., 2011; Loughran et al., 2014). Now we present the first study showing their extensive use in the brain. Specifically, we show that hundreds of neural transcripts use uTISs and dTISs in vitro, and dozens undergo stop codon readthrough in specific cell types of the brain in vivo. Our MS data demonstrate that several of the uTISs and dTISs do give rise to measurable protein products from neurons. Although MS did not identify any readthrough peptides (more below), we ascertained the presence of AQP4X in vivo using a readthrough-specific antibody. Thus, we establish that non-canonical translation contributes to the heterogeneity of the neural proteome.
We also provide two lines of evidence that the products of non-canonical translation are likely to have functional significance in the brain but are not simply abnormal proteins destined for immediate degradation. First, we found that they are regulated: more than one hundred transcripts altered their preference for different TISs in response to KCl stimulation. De novo protein synthesis in the brain is required for not only housekeeping functions but also specialized functions such as long-term potentiation (Kandel, 2001). It is conceivable that the protein isoforms induced by depolarization are involved in carrying out such brain functions. Second, we found that readthrough localizes AQP4 to astrocyte endfeet that enwrap the blood-brain barrier, and the amounts of AQP4 and AQP4X are differentially altered in diverse pathological conditions involving gliosis. These findings on AQP4 and AQP4X might be of importance in the context of diseases such as stroke, in which cerebral edema is often a fatal complication (Rosand and Schwamm, 2001), and infantile Batten disease, in which the blood-brain barrier is disrupted (Saha et al., 2012). Intriguingly, endfoot-polarized AQP4 is required for the efficient removal of amyloid beta from the brain (Iliff et al., 2012; Kress et al., 2014) and is lost in mouse models as well as human patients with Alzheimer’s disease (Park et al., 2014; Yang et al., 2011; Zeppenfeld et al., 2017), suggesting that promoting AQP4X biosynthesis improves the disease outcome. Identification of regulators of Aqp4 readthrough and generation of AQP4X−/− mice will allow the explicit testing of the possible role of AQP4X in these diseases. Overall, we provide compelling evidence that alternative protein isoforms have implications in both normal brain functions and neurological diseases.
Thus far, we have confirmed alternative isoforms for only a subset of proteins by MS, although RF suggested the possibility of hundreds of such isoforms. To some extent, this reflects the fact that peptide-based measures are inherently more challenging and less sensitive than sequencing-based ones. For instance, Menschaert et al. detected only 16 N-terminally extended proteins in mouse embryonic stem cells using MS (Menschaert et al., 2013), although Ingolia et al. had shown the possibility of 570 of such proteins in the same cells using RF (Ingolia et al., 2011). Similarly, Sendoel et al. detected 13 peptides corresponding to uTISs in transformed epidermal cells, although they had identified 215 uTISs using RF (Sendoel et al., 2017). Some of the alternative isoforms might exist in very small amounts or be rapidly degraded after translation, further limiting the ability of MS to detect them. In addition, our MS experiment was conducted on a purer neuronal population cultured from a younger age than the RF studies and hence was insensitive to astrocyte-specific proteins such as AQP4. Another challenge is that it is not entirely clear which amino acid would be included in the peptide chain in lieu of the stop codon, and thus the expected MS spectra of readthrough peptides are inherently unpredictable. Indeed, the peptide found for Aqp4 had a mass-to-charge ratio indicating it included arginine at the stop position. It is also quite possible that many alternative TIS and readthrough events do not produce stable proteins but serve regulatory functions by engaging ribosomes. uTISs specifically have been reported to serve regulatory functions in several transcripts (Calvo et al., 2009), often serving as decoys to suppress the usage of the aTIS in a regulated manner. A prevalence of such sequences would be consistent with the difficulty in detecting protein products, the relatively high frequency of movement of ribosomes from uTISs to aTISs during stimulation, and potentially even significantly lower conservation of NTGs in UTR sequences in general. Evolution across species is posited to be driven more by changes in gene regulation (King and Wilson, 1975), typically interpreted with regard to enhancer usage (Rubinstein and de Souza, 2013). The same logic may be true at the level of regulation of translation initiation, and thus evolution of new uTISs might serve to modulate protein levels across species. Thus, negative results from the MS study should be interpreted cautiously.
Our findings on TISs and readthrough highlight the several potential mechanisms that may warrant future investigation. We found that many alternative TISs are differentially used in response to KCl, which is known to stimulate neurons by phosphorylating CREB protein via an influx of Ca2+ through L-type Ca2+ channels (Macías et al., 2001). Previously, we have shown that KCl stimulation of neural cultures modulates the ribosomal occupancy of specific CDSs (Dalal et al., 2017). Others have shown that KCl stimulation of neuronal cultures enhances TIS by promoting the expression and activity of EIF4E (Moon et al., 2009). What remains to be shown is how exactly the KCl pathway influences the choice of a TIS by ribosomes. It is intriguing to speculate whether it does so by regulating cis elements like RNA secondary structures or phosphorylation and regulation of trans factors such as RNA-binding proteins. Likewise, we found that AQP4 readthrough is of functional significance, and AQP4X level is differentially regulated by gliosis. This, together with the conservation of several read-through peptides in humans (De Bellis et al., 2017; Dunn et al., 2013; Eswarappa et al., 2014; Stiebler et al., 2014), suggests that there are pathways that actively regulate the process. Targeted manipulation of any such pathways may help define the role of stop codon readthrough in the brain or other tissues. Until then, targeting individual transcripts for deep analysis, as was done for Aqp4 (De Bellis et al., 2017), may continue to uncover novel roles for individual products arising from such alternative translation events.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-AQP4, goat | Santa Cruz | SC-9888; RRID:AB_2059853 |
| Anti-AQPX, rabbit | This study | Cell Signaling Technology, 60789 |
| Anti-GFAP, mouse | Biogenex | MU020-UC |
| Anti-V5, mouse | Sigma | V8012; RRID:AB_261888 |
| Anti-cMyc, rabbit | Sigma | C3956; RRID:AB_439680 |
| Anti-cMyc, mouse | Santa Cruz | 9E10; RRID:AB_627268 |
| Anti-PECAM1 | BD Pharmigen | 550274; RRID:AB_393571 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Cycloheximide | Sigma | C1988 |
| Homoharringtonine | LKT Labs | H5750 |
| Critical Commercial Assays | ||
| Dual Luciferase Reporter Assay | Promega | E1960 |
| Deposited Data | ||
| Cell culture RF without HHT and with or without KCl | Dalal et al., 2017 | GEO GSE77076 |
| In vivo TRAP-RF | This study | GSE115483 |
| Cell culture RF with HHT and with or without KCl | This study | http://genetics.wustl.edu/Dougherty_Data/SapkotaEtAl_2018_upload_jdlab/ |
| Mouse brain mass spectrometry dataset | Sharma et al., 2015 | ProteomeXchange Consortium, PXD001250 |
| Synthetic 5’UTR polysome profiling dataset | Sample et al., 2018 | Biorxiv https://www.biorxiv.org/content/early/2018/04/29/310375 |
| Experimental Models: Cell Lines | ||
| DBT glioblastoma cell line | Kumanishi etal., 1973 | N/A |
| Experimental Models: Organisms/Strains | ||
| Snap25::eGFP-RpL10a (B6;FVB-Tg(Snap25-EGFP/Rpl10a)JD362Htz/J) | Dougherty et al., 2012 | https://www.jax.org/strain/30273 |
| Aldh1l1::eGFP-RpL10a (B6;FVB-Tg(Aldh1l1-EGFP/Rpl10a)JD133Htz/J) |
Dougherty et al., 2012; Doyle et al., 2008 |
https://www.jax.org/strain/30248 |
| FVB | Jackson Laboratory | https://www.jax.org/strain/001800 |
| C57BL/6J | Jackson Laboratory | https://www.jax.org/strain/000664 |
| Ppt1−/− (Ppt1tm1Hof) | Gupta et al., 2001 | http://www.informatics.jax.org/allele/MGI:2176390 |
| Recombinant DNA | ||
| pdLUC dual luciferase vector | Fixsen and Howard, 2010 | N/A |
| AAV9-Gfap::cMyc-Aqp4X- | This study | N/A |
| AAV9-Gfap::cMyc-Aqp4X+ | This study | N/A |
| AAV9-Gfap::cMyc-CFP | Sakers et al., 2017 | N/A |
| Software and Algorithms | ||
| R | The R Foundation | https://www.r-project.org/ |
| FastQC (version 0.11.2) | Simon, 2010 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trimmomatic (version 0.32) | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| STAR (version 2.3.1z8) | Dobin et al., 2013 | https://github.com/alexdobin/STAR/ |
| Bowtie2(version 2.2.2) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/index.shtml |
| riboSeqR | Bioconductor | 10.18129/B9.bioc.riboSeqR |
| BEDTools | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| edgeR | Robinson et al., 2010 | 10.18129/B9.bioc.edgeR |
| Specificity Index | Dougherty et al., 2010 | https://cran.r-project.org/web/packages/pSI/index.html |
| MaxQuant software version 1.5.2.10 | http://www.coxdocs.org/doku.php?id=maxquant:start | MaxQuant |
| riboWaltz | Lauria et al., 2018 | https://github.com/LabTranslationalArchitectomics/RiboWaltz |
| WebLogo | Crooks et al., 2004 | http://weblogo.berkeley.edu/logo.cgi |
| ViennaRNA Package 2.0 | Lorenz et al., 2011 | https://www.tbi.univie.ac.at/RNA/ |
| Biostrings | Bioconductor | 10.18129/B9.bioc.Biostrings |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Joseph D. Dougherty (jdougherty@genetics.wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All procedures involving mice conformed to the Washington University institutional animal care and use committee. FVB and C57BL/6J mice were purchased from the Jackson Laboratory. We have previously described Snap25::eGFP-RpL10a and Aldh1l1::eGFP-RpL10a mice (Dougherty et al., 2012; Doyle et al., 2008), and others have described Ppt1−/− mice (Gupta et al., 2001). For immunostaining, tMCAO and viral transduction experiments, three mice (per test group where applicable) were used. Except tMCAO, for which only male mice were used, all mouse experiments included both sexes. Mouse ages were embryonic day {E} 16.5 (LC-MS); postnatal day {P} 0 (in vitro RF); P1 (viral experiment for perivascular proportions of Aqp4 and Aqp4X); E17.5, P0, and P21 (Western blot); P21 (TRAP-RF, Aqp4X immunostaining); 14 weeks (tMCAO); 3 months (needle injury); and 6 months (immunostaining in Ppt1−/−).
Cell Lines and Primary Cultures
DBT glioblastoma cell line, derived from female mice, was used in dual-luciferase and antibody validation experiments has been previously reported (Kumanishi et al., 1973) and was obtained from Dr. Keith M. Rich’s laboratory at Washington University in St. Louis. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 5% fetal calf serum and 100 units/mL each of penicillin and streptomycin and incubated at 37°C and in a humidified atmosphere with 5% CO2.
We have previously described the mouse primary neuron-glia cultures used for RF (Dalal et al., 2017) and primary neuronal cultures used for LC-MS (Prabakaran et al., 2014).
METHOD DETAILS
Culture
Neuron-glia mix cultures from the cortices of full litters of P0 FVB mouse pups were generated as described (Dalal et al., 2017). After 7 days in vitro, cells were treated with 2 mg/ml HHT ((LKT lab) and compared to parallel cultures treated with DMSO (Dalal et al., 2017) (Sigma) for 2 min, and then with 100ug/ml CHX (Sigma) for 7 min at 37°C before being lysed for RF (Ingolia et al., 2011). For examining activity-induced TISs, cells were depolarized with 55mM KCl for 3 h (Bading et al., 1993; Kim et al., 2010) before HHT and CHX treatments. RF was conducted in duplicates.
RF
RF of cultured cells was performed as described (Dalal et al., 2017; Ingolia et al., 2011). Briefly, the cell lysates were treated with RNaseI and subjected to sucrose-cushion ultracentrifugation for pelleting monosomes. mRNA fragments were isolated from the monosomes, size-selected in a polyacrylamide, ligated to cloning linkers, and converted to cDNAs. The cDNAs were then circularized, depleted of rRNAs, PCRed and size-selected and finally deep-sequenced on Illumina Hiseq 2000 (50 bp, single end). Ribosomal profiles of replicate cultures were highly reproducible, with Pearson’s r > 0.96.
TRAP-RF
P21 Snap25::eGFP-RpL10a, Aldh1l1:: eGFP-RpL10a, along with their eGFP-negative littermates were euthanized, and their brains frozen in liquid nitrogen and stored at −80°C until use. Two brains were pooled per sample, and replicate experiments were done. TRAP was performed as described (Heiman et al., 2008) with a few modifications. Briefly, the brains were homogenized in ice in a buffer (20 mM pH 7.4 HEPES, 150 mM KCl, 5 mM MgCl2, 0.5 mM dithiothreitol, 100 mg/ml CHX, Turbo DNase, protease inhibitors, and RNase inhibitors). The lysates were cleared by centrifuging at 2000 xg for 10 min at 4°C and then treated with DHPC (to 30mM, Avanti) and NP-40 (to 1%, Ipgal-ca630, Sigma) for 5 min in ice. Lysates were then further cleared by centrifuging at 20,000 xg for 15 min at 4°C and then mixed with protein L-coated magnetic beads (Invitrogen), previously conjugated with a mix of two monoclonal anti-GFP antibodies (Doyle et al., 2008), and incubated with rotation for 4 h at 4°C. Beads were washed 5 times with a high-salt buffer (20 mM pH7.4 HEPES, 350 mM KCl, 5 mM MgCl2, 1% NP-40, 0.5 mM dithiothreitol, and 100 μg/ml CHX) and then resuspended in normal-salt buffer (150 mM KCl, otherwise as above). To couple to RF, on-bead RNA digestion was performed with RNase I (Invitrogen) for 1 h with end-to-end rotation, followed by washing three times with normal-salt buffer. Small ribosomal subunits with the mRNA fragments were eluted with ribosome dissociation buffer as described (20 mM pH7.3 Tris-HCl, 250 mM NaCl, 0.5% Triton X-100, 50mM EDTA) (Gonzalez et al., 2014). RNA was extracted with phenol-chloroform, quality-tested with Agilent BioAnalyzer, and then subjected to dephosphorylation and subsequent library preparation as described for RF above.
Analysis of Sequence Data
Culture and TRAP-RF sequencing results were quality-tested using FastQC (version 0.11.2) (Simon, 2010), and one culture RF sample was removed due to low read depth. Trimmomatic (version 0.32) was used to trim low-quality bases from the ends of reads and to remove adaptor sequences (Bolger et al., 2014). Only fragments 25–35 nt in length were retained for subsequent analysis. Reads aligning to the mouse rRNA were removed using STAR (version 2.3.1z8) (Dobin et al., 2013). Surviving reads were then aligned, using bowtie2(version 2.2.2) (Langmead and Salzberg, 2012), to the mouse transcriptome (downloaded from Ensembl Release 75) after first removing degenerate sequences from the transcriptome as described (Dunn et al., 2013; Ingolia et al., 2009), retaining both uniquely mapped and multi-mapped reads.
• TIS identification
For TIS analysis on neuron-glia culture RF data, we focused on the 5′-UTR region and proximal 300bp of the CDS and excluded the downstream region where ribosome runoff after HHT treatment was not complete. In order to calculate TIS score, we used riboSeqR package and identified ribosomal P sites, which are known to exist at the 12th, 13th or 14th nts of the footprints of lengths 28–29, 30–31, and 32–33 nts, respectively (Ingolia et al., 2009; Lee et al., 2012). To redress small possible errors in the P site estimation, the P site counts were smoothed by taking an average of the total counts in a 3 nt window, one nt on each side of the initially identified P site. TIS score at every nt position on a transcript was calculated as:
Where RHHT = XHHT / NHHT * 10 and RCHX = XCHX / NCHX * 10. X is the smoothed P site count at a position, and N is the total counts in the 5′UTR and the proximal 300 nt region of the transcript.
The following procedure was used to call TISs
10 candidate peaks were found in each transcript. The first peak was picked based on highest TIS scores. On each side of this peak, the locations were masked when finding the next peaks if: the location is less than k-bp (k = 9) away from the current peak; or TIS scores keep decreasing from the current peak. The next peaks are selected the same way in the unmasked region of the transcript.
Assuming that there is at least one TIS per transcript, the first peak is always called a TIS.
If m (m < 10) of the 10 candidate peaks were already called as TISs, for the next peak, a score is calcuated, where Di is the TIS score at the i-th peak. If Sm + 1 > 0.08, this peak is also called as a TIS. Otherwise, only the previous m TISs are called for this transcript. This step assures we are only focusing on TISs capturing at least ~8% of initiating ribosomes.
Using this algorithm, TISs were called based on the total P site counts in KCl treated and untreated samples. TISs for a transcript in the two samples were considered the same if they were % 3bp away from each other. For each TIS, the P site count rate was calculated as the P site count over the total counts over all TIS positions. A TIS was excluded if the average rate in KCl+ and KCl− samples was < 0.08. Although we started off with 10 candidate peaks, not all of them could be reliably detected across samples; therefore, at this step we kept only the top five that were reliably detectable across most samples. If more than 5 TISs were called for a transcript, only the top 5 TISs with the highest total P site count rates across KCL+ and KCL-samples were kept. If a NUG codon lay within 3bp from the called TIS, the TIS was considered overlapping and hence relocated to the position of that NUG codon.
With above criteria, only transcripts with high P site counts at TIS locations can be used to get reliable TIS calls across samples. Requiring the peak P site counts of ≥ 32 in the considered region of a transcript in the HHT treated group for both KCl+ and KCl−, a total of 426 transcripts were kept for TIS evaluations.
• Readthrough identification
In order to count reads aligning to non-overlapping regions for readthrough analysis in culture and TRAP-RF data, we used the BEDTools intersect command (Quinlan and Hall, 2010). Then, similar to prior work (Dunn et al., 2013), we removed reads aligning to the following positions: 12 nts after the start codon; 15 nts before and 9 nts after the first stop codon, and 15 nts after the second stop codon. This resulted in four remaining transcriptomic regions: 5′UTR, CDS, readthrough region, and distal 3′UTR. Reads were counted in each of these regions using the uniq Unix utility.
Readthrough analysis was performed in R. Transcripts were filtered based on CDS counts as follows: in culture RF, transcripts with ≥ 320 CDS counts across all samples were retained, and in TRAP RF, transcripts with ≥ 128 CDS counts across replicates in either the Aldh1l1 or Snap25 samples were retained. Subsequently, to avoid false positive identification of readthrough events because of alternative splicing, if a gene was annotated as having multiple isoforms with stop codons at different genomic locations, only the transcript with the downstream-most stop codon was retained.
Using edgeR (Robinson et al., 2010), counts were normalized to RPKM, and readthrough rate was defined for each transcript as the ratio of readthrough region RPKM to CDS RPKM. Next, transcripts with ≥ 32 readthrough region counts across samples were retained. In both culture RF and TRAP-RF datasets, surviving transcripts with ≥ 1% readthrough rate in at least 2 samples were retained for readthrough analysis. Genes with ≥ 100% readthrough or with RPKM (distal 3′UTR) ≥ RPKM (readthrough region), for any transcript isoform or in any sample, were labeled false positives and eliminated.
• Identification of cell type-specific transcripts in TRAP-RF data
For quality assessment and filtering of TRAP-RF data, CDS counts were normalized to CPM using the edgeR package. Due to the expression of some glial transcripts in neuronal samples and vice versa, transcripts were classified as either detectable with high confidence in neurons or astrocytes or as nonspecific background, consistent with prior work (Dougherty et al., 2010). Transcripts were classified as neuron-detectable by first calculating the average log2-fold-change (logFC) in expression between Aldh1l1 and Snap25 samples for genes identified as neuronal markers (Dougherty et al., 2012). Among those transcripts surviving initial CDS count filtering, transcripts with logFC greater than this average plus two standard deviations were identified as detectible in neurons with 96% confidence. Astrocyte-detectable transcripts were identified analogously. Remaining transcripts were designated non cell-type specific.
Proteomics
• Sample preparation
Neuronal MS data were obtained similar to the method of Prabakaran et al., 2014 (Prabakaran et al., 2014). In short, E16.5 cortical neurons were cultured for 7 days (3 biological replicates) and depolarized with 55 mM potassium chloride (KCl) for 0, 15, 30, 60, 90, 120 m. For each time point, proteins were extracted in Qproteome mammalian cell lysis buffer (Complete mini and PhosphoSTOP tablets added (Roche)) using a probe sonicator (15sec, 2x). Protein extracts were reduced with 50 mM dithiothreitol (20 min, 56°C), alkylated with iodoacetamide (30 min, RT), diluted with 8M urea in 50mM triethyl ammonium bicarbonate (TEAB) and digested using filter aided sample preparation (FASP Protein Digestion Kit, Expedeon). Digestion was performed overnight 37°C with 2 ng/μl trypsin (Sequencing Grade Modified Trypsin, Promega) in 50 mM TEAB and peptides were subsequently labeled on-filter using isobaric tags (TMT6plex, Thermo Fisher) according to the manufacturer’s instructions. Labeling was quenched using 5% hydroxylamine and peptides were eluted from the filters using 500 mM NaCl and acidified with 2% formic acid. 10 uL of each peptide eluate were pooled, desalted using C18 silica tips (Nestgroup) to assess labeling efficiency and to adjust sample amounts to ensure 1:1 total protein ratios across channels prior to analysis of the remaining samples. Labeled samples were mixed 1:1 and samples were de-salted using Oasis HLB columns (Waters). One half of the sample volume was fractionated using high-pH HPLC fractionation on an Xbridge C18 (3.5mm) 10cm column (Waters) into 55 fractions, the other half by isoelectric focusing on off-gel pH 3–10 Immobiline Dry Strips (GE Healthcare) into 24 fractions. HPLC fractions were combined based on peptide chromatogram intensities into 22 fractions, whereas isoelectric focusing fractions were desalted using C18 silica tips (Nestgroup). Post vacuum drying, fractions were reconstituted in sample buffer (5% formic acid, 5% acetonitrile) for MS analysis.
• MS analysis
Samples were analyzed on a QExactive mass spectrometer (Thermo) coupled to a micro-autosampler AS2 and a nanoflow HPLC pump (Eksigent). Peptides were separated using an in-house packed C18 analytical column (Magic C18 particles, 3 μm, 200Å, Michrom Bioresource) on a linear 120 min gradient starting from 95% buffer A (0.1% (v/v) formic acid in HPLC-H2O) and 5% buffer B (0.2% (v/v) formic acid in acetonitrile) to 35% buffer B. A full mass spectrum with resolution of 70,000 (at mass-to-charge of 200) was acquired in a mass range of 300–1500 mass-to-charge (AGC target 3 × 106, maximum injection time 20 ms). The 10 most intense ions were selected for fragmentation via higher-energy c-trap dissociation (HCD, resolution 17,500, AGC target 2 × 105, maximum injection time 250 ms, isolation window 1.6 mass-to-charge, normalized collision energy 27%).
Raw data were analyzed by MaxQuant software version 1.5.2.10 and peptide lists were searched against the mouse Uniprot protein sequence database (February 2016, reviewed entries appended with common laboratory contaminants [cRAP database, 247 entries]) appended with the alternative translational products using the Andromeda search engine. The following settings were applied: trypsin (specificity set as C-terminal to arginine and lysine) with up to two missed cleavages, mass tolerances set to 20 ppm for the first search and 4.5 ppm for the second search. Methionine oxidation and N-terminal acetylation were chosen as dynamic modifications and carbamidomethylation of cysteine and TMT labeling of peptide N-termini and lysine residues were set as static modifications. The minimum peptide length was set to seven amino acids. False discovery rates (FDR) were set to 1% on peptide and protein levels as determined by reverse database search. Peptide identification was performed with an allowed initial precursor mass deviation up to 7 ppm and an allowed fragment mass deviation of 20 ppm. For all other search parameters, the default settings were used.
In Silico Analysis of TISs and readthrough
• Conservation analysis of uTISs and dTISs and readthrough regions
For computing conservation scores of uTIS per transcript, phastCons format files were downloaded from UCSC Table Browser, querying the coordinates of uTIS codon from the table of phastCons60way. Next, the average score of the three nucleotides was calculated to define the codon conservation score. Each non-uTIS NTGs in the 5′UTR was annotated using a sliding window approach, and then the codon conservation score was extracted using a similar strategy as for uTISs. These codons were further classified into in-frame and out-of-frame based on their location relative to translation start site. To compare uTIS codon score with average score of 5′UTR, average score of the entire 5′UTR given transcripts with uTIS was calculated. dTISs were analyzed similarly, except that the corresponding CDSs were considered in place of the 5′UTRs.
For the readthrough region, phastCons format files were downloaded using stop codon coordinates, and the average of the conservation scores of the nucleotides in the region was calculated.
• Three nucleotide periodicity
Three-nucleotide periodicity was plotted using the riboWaltz R package as described (Lauria et al., 2018).
• Kozak score computation
A Kozak score of each TIS was computed by comparison to a position weight matrix derived from an empirical assay of ribosome recruitment on synthetic sequences (Sample et al., 2018). Briefly, from a pool of 300,000 randomized 5′UTRs driving a reporter expression, the repressive strength of all out-of-frame uTISs was calculated considering all permutations of NNNAUGNN (except when NNN is AUG). Using the 20 most repressive sequences (i.e., competent to recruit ribosomes), a position weight matrix was created, based on which the scores for Kozak sequences in natural 5′ UTRs studied here were calculated using “log2probratio” function in Biostrings package.
• Motif analysis
Analysis of motif enrichment in the readthrough region was done using CISBP-RNA mouse specific database as described (McLeay and Bailey, 2010). As controls, 1080 shuffled sequences derived from the readthrough region or 46 readthrough-negative transcripts were used. Readthrough-negative transcripts were selected to be have robust brain expression and comparable lengths between the first and second stop codons. Position weight matrices were created using WebLogo (Crooks et al., 2004).
• Free energy computation
Minimum free energies of 48 readthrough transcripts and the 46 readthrough-negative transcripts with comparable lengths between the first and second stop codons were calculated using ViennaRNA Package 2.0 as described (Lorenz et al., 2011).
• Codon bias analysis
Sequences of interest were selected using BioMart in R, and relative synonymous codon usage (RSCU) (Sharp et al., 1986) was calculated using the seqinr package for candidate sequences. For readthrough fragments, a codon bias was calculated for each amino acid as RSCU of the most preferred codon – RSCU of the least preferred codon, and a paired t test was used to compare this bias calculated from the readthrough positive fragments and a matched set of readthrough negative fragments (defined as above). For dTIS compared to aTIS, we analyzed codon frequency for 36 nucleotides, starting from the ATG for all sequences and analyzing only the dTIS with highest confidence for coding potential (in frame, starting with NUG). We tested for alteration of codon usage using a Chi-test, with Benjamin Hochberg multiple testing correction.
Dual-luciferase assay
Aqp4 test cassette (the last 15 nts of the CDS + 1st stop + 84 nts before the 2nd stop), a negative control (extra stop added after the 1st stop) or a positive control (1st stop mutated to a sense codon) was cloned between the Renilla and Firefly luciferases of pdLUC, a dual-luciferase vector that expresses Renilla luciferase constitutively but Firefly luciferase only if the cloned construct is read by ribosomes (Fixsen and Howard, 2010). The test and control plasmids were transfected into DBT glioblastoma cell cultures. After 48 hr, the two luciferase activities were quantified using the Dual-Luciferase Assay System and GloMax Luminometer with dual injectors (Promega) following manufacturer’s instructions. Readthrough rate was calculated as described (Grentzmann et al., 1998) and as follows:
Immunofluorescence staining
Brain sections, except for those from mice with MCA occlusion, were collected and processed as follows. Adult mice were euthanized and perfused transcardially with 15 mL ice-cold PBS and then 20 mL 4% ice-cold paraformaldehyde in PBS. Brains were harvested, fixed in 4% ice-cold paraformaldehyde overnight, and cryoprotected with 10%, 20%, and 30% ice-cold sucrose in PBS for 4 h, 4 h, and overnight, respectively. Brains were then frozen in OCT (Sakura Inc), sectioned at 40 mm, and stored at 4°C in PBS+0.01%NaN3 until staining. For staining, floating brain sections were blocked with 5% normal donkey serum plus 0.3% Tritona® X-100 in PBS at room temperature for 1 h, incubated with primary antibody in block at 4°C overnight, washed three times with PBS, incubated with Alexa fluorophore-conjugated secondary antibodies (1:500, Invitrogen) in block at room temperature for 1 h, washed two times with PBS, incubated with 300nM DAPI (Sigma) at room temperature for 5 m, washed two times with PBS, and mounted for confocal imaging (Perkin Elmer).
Brain sections from mice with MCA occlusion were prepared as follows. After transcardial perfusion and overnight fixation as above, the brains were sliced at 1.5 mm, washed with cold PBS for 1 hr and then dehydrated in a series of ethanol concentrations (50%, 70%, 80%, 95%, 100%, 100%, and 100%, each for 1.5 h). After clearing twice with xylene and vacuum-infiltering twice with 58°C paraffin, each time for 1 hr, slices were aligned in molds containing 58°C paraffin and embedded in the same paraffin letting the paraffin cool down. Sections were made at 7 um, collected on charged slides, and dried overnight. For staining, slides were first rehydrated by treating in Xylene (3 × 5 m), 100% ethanol (2 × 10 m), 95% ethanol (1 × 5 m), 80% (1 × 5 m), 70% (1 × 5 m), 50% (1 × 5 m) and distilled water (2 × 5 m). Antigen unmasking was then performed by dipping the slides in 99°C 10 mM sodim citrate buffer and letting the buffer cool down on bench top for 30 m. Slides were then washed 2 × 5 m with PBST, and then blocked and stained as described for floating sections above.
Staining of coverslip cultures were done as described previously (Dalal et al., 2017). Processing and staining of the retinas and kidneys were done as described for floating brain sections above, except that 14 um-thick sections were collected on charged slides. Primary antibodies and dilutions were: mouse anti-V5 (Sigma, V8012, 1:1000), mouse anti-GFAP (Biogenex, MU020-UC, 1:1000), rabbit anti-cMyc (Sigma, C3956, 1:100), mouse anti-cMyc (Santa Cruz, 9E10, 1:100), goat anti-AQP4 (Santa Cruz, SC-9888, 1:100), rat anti-PECAM-1 (BD Pharmigen, 550274, 1:50), and rabbit anti-AQP4X (made in collaboration with Cell Signaling Technology, 60789, 1:1000). The anti-AQP4X was a polyclonal antibody generated by immunizing rabbits with a synthetic peptide corresponding to residues surrounding Asp333 of mouse AQPX and purifying the antibody with protein A and peptide affinity chromatography.
Western blot
Mice were euthanized, and their brains were rapidly homogenized on ice in RIPA buffer (50 mM pH 8.0 Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1mM NaVO4, 1mM NaF, Roche complete protease inhibitor tablet). Lysates were cleared by centrifuging at 16,000 xg at 4°C for 10 m. Protein concentration was measured using a BCA kit (ThermoScientific). An aliquot of 30 mg of protein was boiled for 5 min in an equal volume of 2x Laemmli buffer (BioRad) and electrophoresed in Mini-Protean Precast gels (BioRad) for 1h at 120 V. Proteins were transferred to a polyvinyl membrane using Semi-Dry Transfer Cell (BioRad) for 30 min at 10 V and following manufacturer’s instructions. The blot was then blocked in 5% non-fat dried milk in tris-buffered saline with 0.1% Tween-20 (TBST) for 1 h at room temperature, incubated with anti-AQP4X in block (1:2000, Cell Signaling Technology) overnight at 4°C, washed three times with TBST, incubated with HRP-conjugated secondary antibody in block (1:5000, BioRad) for 1 h at room temperature, washed three times with TBST, and finally developed using Signal-Fire ECL reagent (Cell Signaling Technology) and My ECL imager (ThermoScientific).
Transfection
DBT glioblastoma cell line was grown in 6 well plates in DMEM with 10 fetal bovine serum (Sigma) and 1x penicillin and streptomycin. At 60% confluency, the cells were transfected with 1.5 μg of plasmids using Lipofectamine 2000 (Invitrogen) and following manufacturer’s instructions. Medium was changed 24 h post-transfection, and immunofluorescence staining was done 48 h post-transfection.
AAV preparation and intracranial injection
A cMyc tag was cloned at the 5′ ends of the Aqp4 cDNA containing an extra stop codon beyond the first stop codon (cMycAqp4X-), Aqp4 cDNA with the first stop codon mutated to a sense codon (TGA to TGG, cMycAqp4X+), and CFP cDNA. Modified cDNAs were then cloned into pAAV-GFAP-EGFP after removing the EGFP with AgeI and HindIII. The plasmids were packaged into AAV9 by the Hope Center Viral Vector Core at Washington University. Two uL of the viruses were intracranially injected into P1 (AAV9-Gfap::cMyc-Aqp4X- and AAV9-Gfap::cMyc-Aqp4X+) or P90 (for AAV9-Gfap::cMyc-CFP) mice. After 3 weeks, the mice were processed for floating brain section immunofluorescence.
tMCAO
tMCAO was performed as described previously (Miller et al., 2001). Briefly, 14 week-old C57BL/6 mice were anesthetized with isoflurane, and the left common carotid artery (CCA) was exposed through a midline cervical incision. A 6.0-gauge nylon suture coated with silicone was inserted in the CCA lumen and advanced through the internal carotid artery to the origin of the MCA. Interruption of the blood flow in the MCA territory was confirmed with laser Doppler. After 60 m, the suture was withdrawn, and the reperfusion of the MCA territory was confirmed by inspection with an operating microscope and more distally by laser Doppler. After the procedure, animals recovered in an incubator before returning to home cages. The brains were harvested after 24 h and processed for immunofluorescence staining as mentioned above.
QUANTIFICATION AND STATISTICAL ANALYSIS
P site counts (Figures 1E, 2A, 2B, 3, S1, and S6; Table S1), readthrough rate calculations (Tables S3 and S4), Pearson correlation calculations (Figures 6B, 6C, S2B, S2C, S2E, and S2G) and statistical tests for significance were performed in R. Identification of cell-type-specific transcripts in the TRAP-RF dataset (Figure 6D) was done with the Specificity Index package (Dougherty et al., 2010). For uTIS and dTIS conservation analysis (Figures 2C and 2D) and minimum free energy analysis for readthrough candidates (Figure S6), levels of significance were calculated using Wilcoxon’s rank sum test since the score distributions appeared bimodal. For quantifying immunofluorescence signals (Figures 5 and S5), three mice per condition and six sections per mouse were analyzed, and t test was used. KCl depolarization-mediated changes in TIS usage were tested for significance using either Chi-square test (Figure 3A, across individual transcripts) or paired t test (Figure 3C, pairwise comparisons). Overall difference in Kozak strengths of the three TISs was tested post hoc using Tukey’s HSD test, whereas pairwise differences were tested using t test (Figure S2A). Statistical tests and results are also reported in the text, figure panels and figure legends. Difference was deemed significance when p < 0.05.
DATA AND SOFTWARE AVAILABILITY
Raw and analyzed sequencing data are available at GEO GSE77076 (Cell culture RF without HHT and with or without KCl), GSE115483 (in vivo TRAP-RF) and our lab URL http://genetics.wustl.edu/Dougherty_Data/SapkotaEtAl_2018_upload_jdlab/ (Cell culture RF with HHT and with or without KCl).
Scripts used for sequencing data processing as well as R codes used for downstream analyses are available upon request.
Supplementary Material
Highlights.
Hundreds of brain transcripts show translation initiation in the5′ UTR and CDS
Neuronal activity regulates the choice of translation initiation sites
Dozens of transcripts undergo stop codon readthrough in the brain
Gliosis differentially regulates the amounts of normal AQP4 and readthrough AQP4
ACKNOWLEDGMENTS
We thank Gary Loughran for the dual-luciferase plasmid, the Hope Center Viral Vectors Core at Washington University in St. Louis for viral vector preparation, Mike Vasek for assistance with image analysis, Yating Liu for TIS feature analysis, and members of the J.D.D. laboratory for scientific discussions. This work was supported by the NIH (1R21DA038458, 1R01NS102272), Hope Center, and Children’s Discovery Institute of Washington University in St. Louis (to J.D.D.), McDonnell Center for Cellular and Molecular Neurobiology postdoctoral fellowship (to D.S.), and NIH R01GM112007R (to J.A.S.), R01NS084028 (to J.-M.L.), R01NS043205 (to M.S.S.), and P30CA91842 and UL1TR000448 (to the Genome Technology Resource Center at Washington University in St. Louis). J.D.D. is a NARSAD Independent Investigator of the Brain and Behavior Research Foundation.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and four tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.077.
DECLARATION OF INTERESTS
J.D.D. has received royalties related to TRAP in the past.
REFERENCES
- Bading H, Ginty DD, and Greenberg ME (1993). Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260, 181–186. [DOI] [PubMed] [Google Scholar]
- Bazykin GA, and Kochetov AV (2011). Alternative translation start sites are conserved in eukaryotic genomes. Nucleic Acids Res. 39, 567–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron D, Lapointe C, Bissonnette C, Tremblay G, Motard J, and Roucou X (2013). An out-of-frame overlapping reading frame in the ataxin-1 coding sequence encodes a novel ataxin-1 interacting protein. J. Biol. Chem 288, 21824–21835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bible E, Gupta P, Hofmann SL, and Cooper JD (2004). Regional and cellular neuropathology in the palmitoyl protein thioesterase-1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol. Dis 16, 346–359. [DOI] [PubMed] [Google Scholar]
- Bilguvar K, Tyagi NK, Ozkara C, Tuysuz B, Bakircioglu M, Choi M, Delil S, Caglayan AO, Baranoski JF, Erturk O, et al. (2013). Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc. Natl. Acad. Sci. USA 110, 3489–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caley DW, and Maxwell DS (1970). Development of the blood vessels and extracellular spaces during postnatal maturation of rat cerebral cortex. J. Comp. Neurol 138, 31–47. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Pagliarini DJ, and Mootha VK (2009). Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc. Natl. Acad. Sci. USA 106, 7507–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiurchiù V, Maccarrone M, and Orlacchio A (2014). The role of reticulons in neurodegenerative diseases. Neuromolecular Med. 16, 3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua JJE, Schob C, Rehbein M, Gkogkas CG, Richter D, and Kindler S (2012). Synthesis of two SAPAP3 isoforms from a single mRNA is mediated via alternative translational initiation. Sci. Rep 2, 484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa-Mattioli M, Sossin WS, Klann E, and Sonenberg N (2009). Translational control of long-lasting synaptic plasticity and memory. Neuron 61, 10–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, and Brenner SE (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski M, Bukowy-Bieryllo Z, and Zietkiewicz E (2015). Translational readthrough potential of natural termination codons in eucaryotes—the impact of RNA sequence. RNA Biol. 12, 950–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal JS, Yang C, Sapkota D, Lake AM, O’Brien DR, and Dougherty JD (2017). Quantitative nucleotide level analysis of regulation of translation in response to depolarization of cultured neural cells. Front. Mol. Neurosci 10, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bellis M, Pisani F, Mola MG, Rosito S, Simone L, Buccoliero C, Trojano M, Nicchia GP, Svelto M, and Frigeri A (2017). Translational readthrough generates new astrocyte AQP4 isoforms that modulate supramolecular clustering, glial endfeet localization, and water transport. Glia 65, 790–803. [DOI] [PubMed] [Google Scholar]
- Delmas V, Laoide BM, Masquilier D, de Groot RP, Foulkes NS, and Sassone-Corsi P (1992). Alternative usage of initiation codons in mRNA encoding the cAMP-responsive-element modulator generates regulators with opposite functions. Proc. Natl. Acad. Sci. USA 89, 4226–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JD, Schmidt EF, Nakajima M, and Heintz N (2010). Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res. 38, 4218–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JD, Fomchenko EI, Akuffo AA, Schmidt E, Helmy KY, Bazzoli E, Brennan CW, Holland EC, and Milosevic A (2012). Candidate pathways for promoting differentiation or quiescence of oligodendrocyte progenitor-like cells in glioma. Cancer Res. 72, 4856–4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, et al. (2008). Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135, 749–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JG, Foo CK, Belletier NG, Gavis ER, and Weissman JS (2013). Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. eLife 2, e01179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eswarappa SM, Potdar AA, Koch WJ, Fan Y, Vasu K, Lindner D, Willard B, Graham LM, DiCorleto PE, and Fox PL (2014). Programmed translational readthrough generates antiangiogenic VEGF-Ax. Cell 157, 1605–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fixsen SM, and Howard MT (2010). Processive selenocysteine incorporation during synthesis of eukaryotic selenoproteins. J. Mol. Biol 399, 385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavell SW, and Greenberg ME (2008). Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu. Rev. Neurosci 31, 563–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Quintanilla A, and Miranzo-Navarro D (2016). Extraintestinal manifestations of celiac disease: 33-mer gliadin binding to glutamate receptor GRINA as a new explanation. BioEssays 38, 427–439. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Sims JS, Hornstein N, Mela A, Garcia F, Lei L, Gass DA, Amendolara B, Bruce JN, Canoll P, and Sims PA (2014). Ribosome profiling reveals a cell-type-specific translational landscape in brain tumors. J. Neurosci 34, 10924–10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR (2001). Alternative splicing: increasing diversity in the proteomic world. Trends Genet. 17, 100–107. [DOI] [PubMed] [Google Scholar]
- Green KM, Linsalata AE, and Todd PK (2016). RAN translation—what makes it run? Brain Res. 1647, 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL, and Greenberg ME (2008). From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron 59, 846–860. [DOI] [PubMed] [Google Scholar]
- Grentzmann G, Ingram JA, Kelly PJ, Gesteland RF, and Atkins JF (1998). A dual-luciferase reporter system for studying recoding signals. RNA 4, 479–486. [PMC free article] [PubMed] [Google Scholar]
- Gupta P, Soyombo AA, Atashband A, Wisniewski KE, Shelton JM, Richardson JA, Hammer RE, and Hofmann SL (2001). Disruption of PPT1 or PPT2 causes neuronal ceroid lipofuscinosis in knockout mice. Proc. Natl. Acad. Sci. USA 98, 13566–13571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, et al. (2008). A translational profiling approach for the molecular characterization of CNS cell types. Cell 135, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermey G, Blüthgen N, and Kuhl D (2017). Neuronal activity-regulated alternative mRNA splicing. Int. J. Biochem. Cell Biol 91, 184–193. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, and Takemura R (2005). Molecular motors and mechanisms of directional transport in neurons. Nat. Rev. Neurosci 6, 201–214. [DOI] [PubMed] [Google Scholar]
- Hofhuis J, Schueren F, Nötzel C, Lingner T, Gärtner J, Jahn O, and Thoms S (2016). The functional readthrough extension of malate dehydrogenase reveals a modification of the genetic code. Open Biol. 6, 160246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, et al. (2012). A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci. Transl. Med 4, 147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, and Weissman JS (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Lareau LF, and Weissman JS (2011). Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell 147, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson RJ, Hellen CUT, and Pestova TV (2010). The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol 11, 113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungreis I, Lin MF, Spokony R, Chan CS, Negre N, Victorsen A, White KP, and Kellis M (2011). Evidence of abundant stop codon readthrough in Drosophila and other metazoa. Genome Res. 21, 2096–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER (2001). The molecular biology of memory storage: a dialogue between genes and synapses. Science 294, 1030–1038. [DOI] [PubMed] [Google Scholar]
- Kim T-K, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. (2010). Widespread transcription at neuronal activity-regulated enhancers. Nature 465, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MC, and Wilson AC (1975). Evolution at two levels in humans and chimpanzees. Science 188, 107–116. [DOI] [PubMed] [Google Scholar]
- Kozak M (1999). Initiation of translation in prokaryotes and eukaryotes. Gene 234, 187–208. [DOI] [PubMed] [Google Scholar]
- Kress BT, Iliff JJ, Xia M, Wang M, Wei HS, Zeppenfeld D, Xie L, Kang H, Xu Q, Liew JA, et al. (2014). Impairment of paravascular clearance pathways in the aging brain. Ann. Neurol 76, 845–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumanishi T, Ikuta F, and Yamamoto T (1973). Brain tumors induced by Rous sarcoma virus, Schmidt-Ruppin strain. 3. Morphology of brain tumors induced in adult mice. J. Natl. Cancer Inst 50, 95–109. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria F, Tebaldi T, Bernabò P, Groen EJN, Gillingwater TH, and Viero G (2018). riboWaltz: optimization of ribosome P-site positioning in ribosome profiling data. PLoS Comput. Biol 14, e1006169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Liu B, Lee S, Huang S-X, Shen B, and Qian S-B (2012). Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 109, E2424–E2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi DD, and Darnell RB (2006). Splicing regulation in neurologic disease. Neuron 52, 93–101. [DOI] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Höner Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, and Hofacker IL (2011). ViennaRNA Package 2.0. Algorithms Mol. Biol 6, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughran G, Chou M-Y, Ivanov IP, Jungreis I, Kellis M, Kiran AM, Baranov PV, and Atkins JF (2014). Evidence of efficient stop codon readthrough in four mammalian genes. Nucleic Acids Res. 42, 8928–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macías W, Carlson R, Rajadhyaksha A, Barczak A, and Konradi C (2001). Potassium chloride depolarization mediates CREB phosphorylation in striatal neurons in an NMDA receptor-dependent manner. Brain Res. 890, 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeay RC, and Bailey TL (2010). Motif enrichment analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 11, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menschaert G, Van Criekinge W, Notelaers T, Koch A, Crappé J, Gevaert K, and Van Damme P (2013). Deep proteome coverage based on ribosome profiling aids mass spectrometry-based protein and peptide discovery and provides evidence of alternative translation products and near-cognate translation initiation events. Mol. Cell. Proteomics 12, 1780–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middeldorp J, and Hol EM (2011). GFAP in health and disease. Prog. Neurobiol 93, 421–443. [DOI] [PubMed] [Google Scholar]
- Miller BA, Perez RS, Shah AR, Gonzales ER, Park TS, and Gidday JM (2001). Cerebral protection by hypoxic preconditioning in a murine model of focal ischemia-reperfusion. Neuroreport 12, 1663–1669. [DOI] [PubMed] [Google Scholar]
- Mobasheri A, Marples D, Young IS, Floyd RV, Moskaluk CA, and Frigeri A (2007). Distribution of the AQP4 water channel in normal human tissues: protein and tissue microarrays reveal expression in several new anatomical locations, including the prostate gland and seminal vesicles. Channels (Austin) 1, 29–38. [PubMed] [Google Scholar]
- Moon IS, Cho S-J, Seog D-H, and Walikonis R (2009). Neuronal activation increases the density of eukaryotic translation initiation factor 4E mRNA clusters in dendrites of cultured hippocampal neurons. Exp. Mol. Med 41, 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagelhus EA, and Ottersen OP (2013). Physiological roles of aquaporin-4 in brain. Physiol. Rev 93, 1543–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, Hartl C, Leppa V, Ubieta LT, Huang J, et al. (2016). Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park R, Kook S-Y, Park J-C, and Mook-Jung I (2014). Ab1–42 reduces P-glycoprotein in the blood-brain barrier through RAGE-NF-kB signaling. Cell Death Dis. 5, e1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabakaran S, Hemberg M, Chauhan R, Winter D, Tweedie-Cullen RY, Dittrich C, Hong E, Gunawardena J, Steen H, Kreiman G, and Steen JA (2014). Quantitative profiling of peptides from RNAs classified as noncoding. Nat. Commun 5, 5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesnel-Vallières M, Dargaei Z, Irimia M, Gonatopoulos-Pournatzis T, Ip JY, Wu M, Sterne-Weiler T, Nakagawa S, Woodin MA, Blencowe BJ, and Cordes SP (2016). Misregulation of an activity-dependent splicing network as a common mechanism underlying autism spectrum disorders. Mol. Cell 64, 1023–1034. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bio-conductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosand J, and Schwamm LH (2001). Management of brain edema complicating stroke. J. Intensive Care Med 16, 128–141. [Google Scholar]
- Rubinstein M, and de Souza FSJ (2013). Evolution of transcriptional enhancers and animal diversity. Philos. Trans. R. Soc. Lond. B Biol. Sci 368, 20130017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A, Sarkar C, Singh SP, Zhang Z, Munasinghe J, Peng S, Chandra G, Kong E, and Mukherjee AB (2012). The blood-brain barrier is disrupted in a mouse model of infantile neuronal ceroid lipofuscinosis: amelioration by resveratrol. Hum. Mol. Genet 21, 2233–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakers K, Lake AM, Khazanchi R, Ouwenga R, Vasek MJ, Dani A, and Dougherty JD (2017). Astrocytes locally translate transcripts in their peripheral processes. Proc. Natl. Acad. Sci. USA 114, E3830–E3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sample PJ, Wang B, Reid DW, Presnyak V, McFadyen I, Morris DR, and Seelig G (2018). Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. bioRxiv. 10.1101/310375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangar DV, Newton SE, Rowlands DJ, and Clarke BE (1987). All foot and mouth disease virus serotypes initiate protein synthesis at two separate AUGs. Nucleic Acids Res. 15, 3305–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, and Liu JO (2010). Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol 6, 209–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Felber BK, and Pavlakis GN (1992). Mechanism of translation of monocistronic and multicistronic human immunodeficiency virus type 1 mRNAs. Mol. Cell. Biol 12, 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendoel A, Dunn JG, Rodriguez EH, Naik S, Gomez NC, Hurwitz B, Levorse J, Dill BD, Schramek D, Molina H, et al. (2017). Translation from unconventional 5′ start sites drives tumour initiation. Nature 541, 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma K, Schmitt S, Bergner CG, Tyanova S, Kannaiyan N, Manrique-Hoyos N, Kongi K, Cantuti L, Hanisch U-K, Philips M-A, et al. (2015). Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci 18, 1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM, Tuohy TM, and Mosurski KR (1986). Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 14, 5125–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicard KM, and Fisher M (2009). Animal models of focal brain ischemia. Exp. Transl. Stroke Med 1, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simkin D, Cavanaugh EJ, and Kim D (2008). Control of the single channel conductance of K2P10.1 (TREK-2) by the amino-terminus: role of alternative translation initiation. J. Physiol 586, 5651–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon A (2010). A quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Slavoff SA, Mitchell AJ, Schwaid AG, Cabili MN, Ma J, Levin JZ, Karger AD, Budnik BA, Rinn JL, and Saghatelian A (2013). Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat. Chem. Biol 9, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler RA, Gan Y, Zhang W, Liou AK, Gao Y, Cao G, and Chen J (2010). Heat shock proteins: cellular and molecular mechanisms in the central nervous system. Prog. Neurobiol 92, 184–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiebler AC, Freitag J, Schink KO, Stehlik T, Tillmann BAM, Ast J, and Bölker M (2014). Ribosomal readthrough at a short UGA stop codon context triggers dual localization of metabolic enzymes in fungi and animals. PLoS Genet. 10, e1004685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studtmann K, Olschläger-Schütt J, Buck F, Richter D, Sala C, Bock-mann J, Kindler S, and Kreienkamp H-J (2014). A non-canonical initiation site is required for efficient translation of the dendritically localized Shank1 mRNA. PLoS ONE 9, e88518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Plant LD, Wilkens CM, McCrossan ZA, and Goldstein SAN (2008). Alternative translation initiation in rat brain yields K2P2.1 potassium channels permeable to sodium. Neuron 58, 859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, and Pilpel Y (2010). An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141, 344–354. [DOI] [PubMed] [Google Scholar]
- Vanderperre B, Staskevicius AB, Tremblay G, McCoy M, O’Neill MA, Cashman NR, and Roucou X (2011). An overlapping reading frame in the PRNP gene encodes a novel polypeptide distinct from the prion protein. FASEB J. 25, 2373–2386. [DOI] [PubMed] [Google Scholar]
- Vanderperre B, Lucier J-F, Bissonnette C, Motard J, Tremblay G, Vanderperre S, Wisztorski M, Salzet M, Boisvert F-M, and Roucou X (2013). Direct detection of alternative open reading frames translation products in human significantly expands the proteome. PLoS ONE 8, e70698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan J, and Qian S-B (2014). TISdb: a database for alternative translation initiation in mammalian cells. Nucleic Acids Res. 42, D845–D850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner AM, and Weber K (1971). Natural read-through at the UGA termination signal of Q-b coat protein cistron. Nat. New Biol 234, 206–209. [DOI] [PubMed] [Google Scholar]
- Yang J, Lunde LK, Nuntagij P, Oguchi T, Camassa LMA, Nilsson LNG, Lannfelt L, Xu Y, Amiry-Moghaddam M, Ottersen OP, and Torp R (2011). Loss of astrocyte polarization in the tg-ArcSwe mouse model of Alzheimer’s disease. J. Alzheimers Dis 27, 711–722. [DOI] [PubMed] [Google Scholar]
- Zeppenfeld DM, Simon M, Haswell JD, D’Abreo D, Murchison C, Quinn JF, Grafe MR, Woltjer RL, Kaye J, and Iliff JJ (2017). Association of perivascular localization of aquaporin-4 with cognition and Alzheimer disease in aging brains. JAMA Neurol. 74, 91–99. [DOI] [PubMed] [Google Scholar]
- Zhang M, Cai F, Zhang S, Zhang S, and Song W (2014a). Overexpression of ubiquitin carboxyl-terminal hydrolase L1 (UCHL1) delays Alzheimer’s progression in vivo. Sci. Rep 4, 7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014b). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupanic A, Meplan C, Grellscheid SN, Mathers JC, Kirkwood TBL, Hesketh JE, and Shanley DP (2014). Detecting translational regulation by change point analysis of ribosome profiling data sets. RNA 20, 1507–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and analyzed sequencing data are available at GEO GSE77076 (Cell culture RF without HHT and with or without KCl), GSE115483 (in vivo TRAP-RF) and our lab URL http://genetics.wustl.edu/Dougherty_Data/SapkotaEtAl_2018_upload_jdlab/ (Cell culture RF with HHT and with or without KCl).
Scripts used for sequencing data processing as well as R codes used for downstream analyses are available upon request.