

Abstract
Our goal is to draw a line – hypothetical in its totality but experimentally supported at each individual step – connecting the ribosomal DNA and the phenomenon of transgenerational epigenetic inheritance of induced phenotypes. The reasonableness of this hypothesis is offset by its implication, that many (or most) (or all) of the cases of induced-and-inherited phenotypes that are seen to persist for generations are instead unmapped induced polymorphisms in the ribosomal DNA, and thus are the consequence of the peculiar and enduringly-fascinating genetics of the highly-transcribed repeat DNA structure at that locus.
Keywords: rDNA, Ribosomal DNA, Epigenetics
The Goal of this Article
Herein, we will argue that many cases of transgenerational epigenetic – induced organismal or cellular phenotypes that are stably inherited in the absence of the inducing factor – are due to environmentally-induced variation at the ribosomal RNA gene cluster (the rDNA). This may be understood as the confluence of three properties of the rDNA:
-
(i)
the sensitivity of rDNA copy number to mutation and environmental perturbations,
-
(ii)
the mapping of phenotypes to rDNA copy number variations,
-
(iii)
the general difficulty in molecularly manipulating or analyzing the rDNA as a consequence of its particular molecular-genetic characteristics.
We will discuss support for each of these three properties, then provide a brief example of the hypothesis as a whole; we will start with point (iii).
We will conclude that it is the definition of “transgenerational epigenetic inheritance” itself that stymies understanding of these phenomena because it purposefully and unnecessarily rejects any valid alternative explanations, including the genetics of the rDNA.
The Ribosomal DNA
As a reader of this special issue, you are probably unopposed to considering the specialness of the ribosomal DNA loci. Early concepts of genetic allelism, disproven conceptually by Muller (Muller 1932) and experimentally by many others (e.g., Lewis 1952), considered that genes were polymeric structures that could be tuned – amorphic to hypomorhic to wild-type to hypermorphic – through internal deletions or duplications of internally-identical units. The rDNA remains the last standing such polygene, and it seems noteworthy through casual reading of classical literature how many times the bobbed locus (the genetic manifestation of the rDNA locus or loci in Drosophila melanogaster (Ritossa et al. 1966)) is mentioned as an exception to then-emerging rules of genetics, or as a confusing counter-example of some new phenomenon (Muller 1932; Waddington 1957; Spofford and DeSalle 1991). The unusual behaviors of the rDNA are no less compelling today.
In eukaryotes, the rDNA is generally composed of two different types of clusters of genes: the 35S (or 45S) and the 5S, the former processed into the 5.8S, 18S, and 28S rRNAs, and the latter providing the final of the four rRNA subunits of the ribosome. Beyond just diploidy, each of these types of arrays is functionally redundant, for example the two (X- and Y-linked) 35S arrays of Drosophila are independently capable of providing all of the rRNAs necessary for translation throughout embryogenesis, larval instars, pupation, and adult life. Other eukaryotes possess a wide variation in rDNA cluster numbers (Long and Dawid 1980; Prokopowich et al. 2003) and, by extension, it would be unsurprising if they were all each also functionally redundant. Nucleolar dominance (e.g., Endow 1983; Chen et al. 1998; Pontes et al. 2003; Preuss and Pikaard 2007; Greil and Ahmad 2012) argues that fewer than half are necessary for viability in some organisms. And while little formal proof has been generated in humans, cell lines occasionally lacking clusters or possessing supernumerary ones through aneuploidy or genome rearrangements, and the reduction or loss of rDNA clusters in the numerous Robertsonian Translocations that occur proximal to the rDNA loci (Hurley and Pathak 1977; Cheung et al. 1990), again argue that functional redundancy is the norm and genetic robustness the result.
In addition to this inter-cluster redundancy in trans, each array of rRNA genes is internally redundant in cis, containing sometimes hundreds of copies of the same pre-rRNA gene despite the necessity for far fewer. It is thought that in most tissues in most organisms that a large fraction of genes, around half, are silenced through the formation of heterochromatin on individual rDNA copies (McStay and Grummt 2008; Sanij et al. 2008; Guetg et al. 2010; Grummt and Langst 2013). The adaptive value of the wide-spread or deep evolutionary invention of silencing supernumerary copies is not known, but may have something to do with facile transcriptional regulation or with stability of the large repeat gene array (Kobayashi 2008; Peng and Karpen 2008; Ianni et al. 2017; Paredes et al. 2018). That many gene copies are silenced can hide considerable sequence variation (Kim et al. 2018; Parks et al. 2018), since not all copies of the rDNA need be perfect if potentially toxic mutant rDNA are preferentially inactivated (Eickbush et al. 2008; Guerrero and Maggert 2011; Larson et al. 2012; Wang et al. 2018). Polymorphisms in the intergenic “non-transcribed” spacer, in the regions corresponding to mature rRNAs, in the presence or absence of interrupting retrotransposable elements (Eickbush et al. 1997; Eickbush and Eickbush 2003), can all be hidden by silencing, or may contribute to the choice of which copies are silenced (Guerrero and Maggert 2011).
A variation in enhancer/promoter sequences, which can control rRNA expression level (Grimaldi and Di Nocera 1988; Hayward and Glover 1988; Kuhn et al. 1990), and in retrotransposon (e.g., the R1-R7 elements of arthropods) residency (Kojima and Fujiwara 2003; Averbeck and Eickbush 2005), which could alter 18S/28S/5.8S rRNA stoichiometry, would naturally lead to variation in copy number. Copies may be added or removed without much consequence. Additionally, the ability to form heterochromatin at individual genes allows for the accumulation of even maladaptive sequences without any obvious consequence to translation.
The multiple rDNA arrays, heterochromatin-induced silencing, uncatalogued variation, and copy number polymorphisms together have made it very difficult to determine what is the minimum number of rDNA genes necessary for viability in animals and plants. Initial studies in Drosophila focused on the bobbed phenotype which expresses as cuticular etchings and truncated macrochaete bristles. It was reasoned that about 90 rDNA copies are needed to avoid a lethal phenotype (Tartof 1973; Terracol and Prud’homme 1986; Terracol et al. 1990), and about 120 to avoid the bobbed phenotype altogether (Terracol and Prud’homme 1981; Paredes and Maggert 2009a), but none of these studies had knowledge of the sequences of the rDNA copies, likely unnecessarily counting those that were retrotransposon-laden, and assuming equal contribution of each copy regardless of enhancer/promoter sequence polymorphisms. Thus, making direct correlative links between copy number and any phenotype is still beyond our reach.
In general, correlative observations (from mining human disease databases) or experiments are difficult to interpret simply, as phenotypes that map to the rDNA are hard to confirm using modern molecular-genetic approaches. The base necessity for the rDNA to accommodate the basic cellular function of protein expression makes true amorphic phenotypes impossible to attain. The repetitive structure and common copy number changes makes “gold standard” experiments like transgenesis or rescue unfeasible. Molecular cloning of repeat DNAs is still difficult, and even with new sequencing technologies, low-abundance polymorphisms are often overlooked. In most data-handling pipelines, the rDNA is viewed as an uninteresting contamination, and reads with homology to the rDNA are the first sequences jettisoned from the raw data. And that most organisms have multiple redundant arrays makes even linkage via meiotic mapping, QTL, or GWAS underpowered or impossible. Despite these challenges, some strongly correlative and altogether convincing studies have made strong connections between disparate phenotypes and the rDNA.
Extent and Origins of Copy Number Variation
For organisms with multiple rDNA loci, either as arrays or dispersed individual copies, there is plenty of opportunity to possess a wide range of copy number. In humans with their ten (diploid) 45S rDNA clusters, there are approximately one thousand combinations of rDNA clusters that can be inherited from parents, and thirty that can be transmitted to offspring. As each allele of each array may differ in copy number, wide variation in copy number between individuals is to be expected. Even in organisms with only two rDNA clusters, the variation in copy number can be at least six-fold (Lyckegaard and Clark 1989).
Multiple mechanisms have evolved to increase copy number at individual rDNA arrays. For example, the yeast Saccharomyces cerevisiae expands short rDNA arrays through the joint activity of Replication fork block protein (Fob1) and replication barrier sequences in the rDNA array (Kobayashi et al. 1998; Kobayashi et al. 2001). It has also been shown that some Drosophila rDNA can exhibit meiotic magnification, spontaneously reverting the bobbed phenotype (Hawley and Tartof 1985; Hawley and Marcus 1989). This magnification seems to act through a different mechanism, as Drosophila lacks obvious fob1 homologues and replication-blocking sequences in their rDNA, molecular data suggests an extrachromosomal intermediate (Bianciardi et al. 2012), and importantly meiotic magnification appears to be a property of only some rDNA arrays (Endow 1980; Endow 1982; Komma and Endow 1987) under some conditions (Ashburner et al. 2005). rDNA copy number variation need not rely on such mechanisms, as it may also change in a less-directed way during meiosis as a result of unequal sister chromatid exchange within the array (Tartof 1974; Endow et al. 1984; Stults et al. 2008). In theory, exchange between homologues could also occur, however the data for crossovers in the rDNA (outside of a magnification or mutational context (Ritossa 1973; Rasooly and Robbins 1991)) are scant.
The possibility of exchange between rDNA arrays on heterologous chromosomes (e.g., the X and Y in Drosophila, any two of 13, 14, 15, 20, 21 in humans), where copy numbers may be shuffled between non-allelic autosomal arrays, adds a compelling dimension to the dynamics of rDNA copy number and linkage.
It stands to reason that mechanisms for rDNA expansion are balanced in some way. Unequal sister chromatid exchange in the germ line would of course generate a complementary chromosome with a reduced rDNA array (Tartof 1974; Stults et al. 2008). Recent data show that alteration of insulin/insulin-like signaling can cause reductions to rDNA copy number in individuals and their offspring, as can extreme dietary stress (Aldrich and Maggert 2015; Holland et al. 2016; Danson et al. 2018). Environmental stresses may also lead to wildly variant rDNA copy numbers (Schneeberger and Cullis 1991; Kwan et al. 2016). It appears that while there is much variation within populations, there is a tendency toward some balance, as if excess copies are deleterious and actively lost to maintain an ideal copy number. Observationally, rDNA copy numbers may be broadly varied in populations, but are relatively robust in family lineages.
The sum of these meiotic shufflings, increases, and decreases results in a steady-state variation that, once established, and in the absence of subsequent rare copy number changes, is maintained through normal DNA replication.
But in contrast to meiosis, rDNA copy number stability is not the norm in the mitotic cell lineages within individuals. Famously, the rDNA are lost in mitotically-active Saccharomyces cerevisiae, particularly by the asymmetric retention of extrachromosomal rDNA circles in mother cells (Sinclair and Guarente 1997). A similar phenomenon has been reported as occurring in Drosophila male stem cells, producing sperm with fewer rDNA arrays as the male ages (Lu et al. 2018). While this would be a dramatic source of natural rDNA variation, those results have yet to be repeated; we ourselves have not observed any such changes in our experiments (Paredes and Maggert 2009b; Aldrich and Maggert 2014; Aldrich and Maggert 2015), or in any of the lines that we’ve maintained for the last decade including those that straddle the extreme-bobbed/bobbed-lethal phenotypic thresholds (either in terms of phenotypic enhancement or quantified rDNA copy number) (Paredes and Maggert 2009a). It is clear, however, that rDNA are lost in the normal healthy developing tissues of at least Drosophila melanogaster, Arabidopsis, daisy, and mouse (Cohen et al. 2003; Cohen et al. 2005; Cohen et al. 2008; Xu et al. 2017). The amount lost does not appear to be tightly regulated, instead being a consequence of transcription-dependent expression and repair (Warmerdam et al. 2016), resulting in some cell lineages with relatively slow diminishment of rDNA copy number intermixed with cell lineages with large stepwise losses of contiguous blocks of rDNA genes (Paredes et al. 2011). The resulting heterogeneity must be held in mind because often rDNA copy number determination is done in whole organisms, resulting in an average of these heterogeneous array sizes. Too few studies have surveyed changes in individual tissues, although rDNA copy number is now known in whole organs of mice (Xu et al. 2017). The lack of clear patterns or consistency between individuals indicates that it’s probably also stochastic. Cataloging the tempo of changes through development, during stress, or in mutants have not been undertaken in a metazoan.
Mutations in genes that act to protect rDNA or genes that act to repress rDNA expression (indeed, those activities may be one-and-the-same) increase the rate of loss in the soma (among many others, Schawalder et al. 2003; Peng and Karpen 2007; Grierson et al. 2012; Larson et al. 2012; Zhou J 2012; Aldrich and Maggert 2014; Salim et al. 2017; Xu et al. 2017; Paredes et al. 2018). The resulting extrachromosomal circles and supernumerary nucleoli are cytological signs of the accelerated loss (Peng and Karpen 2007; Paredes and Maggert 2009b). Accelerated loss has also been observed in offspring of even heterozygous mutants of these genes. In one experiment, we exposed a normal rDNA array (on the Y chromosome) to heterozygous mutation in Su(var)205, which encodes the heterochromatin protein HP1. After multiple generations, and after removing the mutation from the genetic background and swapping out all of the other DNA (X chromosome, autosomes, mitochondria), we found we had induced a bobbed phenotype that mapped to the Y-linked rDNA, which we quantified to be a 15% decrease in rDNA copy number (Aldrich and Maggert 2014) (Figure 1A). Similarly, Drosophila laboratory strains bearing heterozygous mutations in the Su(var)3–9 gene, which encodes a histone H3 Lysine-9 methyltransferase, are very-often associated with a bobbed phenotype (Paredes and Maggert 2009b), and we now know this to be causal (Figure 1A). This loss in Su(var)3–9 mutants and the resulting phenotype were also noticed by others (Greil and Ahmad 2012; Larson et al. 2012), and appears to be a common property of mutations in Su(var) genes. We have seen the same effects in two additional genes, the rDNA repressor CTCF (Torrano et al. 2006; van de Nobelen et al. 2010; Guerrero and Maggert 2011), and the BLM DNA repair/replication helicase (Killen et al. 2012; our work in preparation) (Figure 1B).
Figure 1. rDNA copy number is reduced by exposure to mutations that affect rDNA expression.
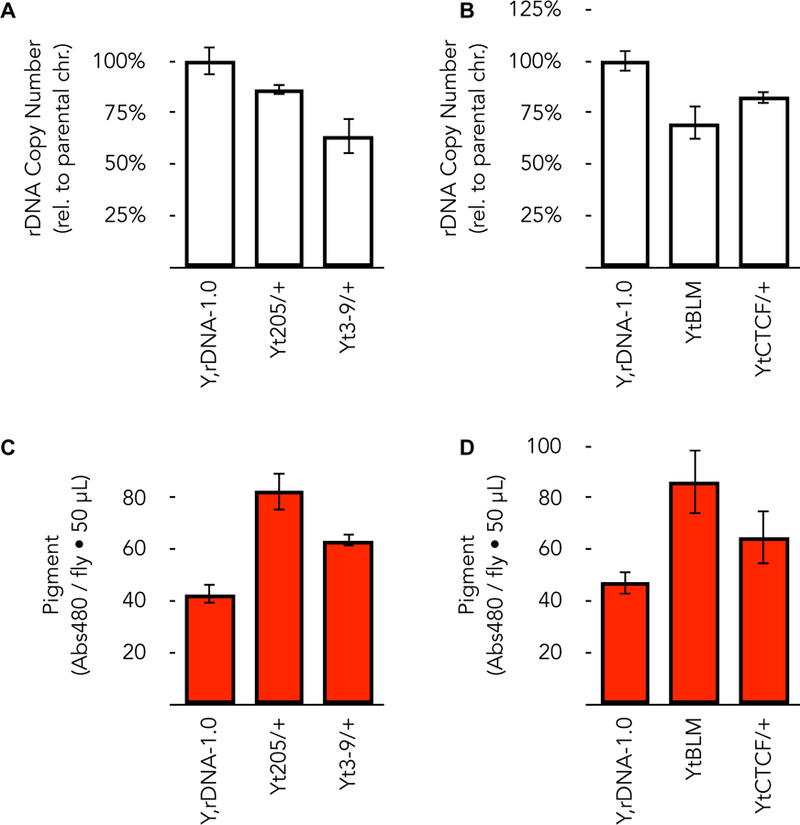
(A) A normal Y chromosome that possesses, by definition, 100% of a normal amount of rDNA (strain rDNA-1.0 is our “standard” Y chromosome that possesses a natural/wild-type amount of rDNA (Paredes and Maggert 2009)) was crossed into a genetic background containing Su(var)2055 or Su(var)3–91 mutations as heterozygotes. After multiple generations, the Y chromosome was genetically isolated by two subsequent backcrosses to isogenized y; bw; e; ey females, replacing all genetic material save for the Y chromosomes. The Y-linked rDNA was made sole-source by crossing males to C(1)DX, y f rDNA0 / Y, BS females and female progeny subjected to real-time PCR to quantify rDNA using copy number of tRNAK-CTT as an internal normalizer. (B) Similar experiment to that in (A) using mutations not previously known to affect variegation, but were discovered to affect rDNA stability (BLM trans-heterozygotes, blmN1/blmD2) or rDNA expression and stability (CTCF∆9 heterozygotes). (C) Males from (A) were crossed to the position effect variegating X-chromosome inversion In(1)wm4 to assess effects on heterochromatin-induced gene silencing in male progeny. Extent of pigment-expressing gene silencing indicates heterochromatin function, with greater amount of pigment indicating greater white+ expression, indicating loss of heterochromatin-induced silencing. Pigment from thirty heads were extracted in acidified ethanol for 24 hours in the dark. (D) similar to (C).
We consider these experiments employing mutations to be revealing of a possible outcome of altered gene activity. Su(var) heterozygosity, and the rDNA loss that results, may be similar to the response of natural populations to conditions that reduce heterochromatin function (Specchia et al. 2010; Gibbons et al. 2015). We work with the proposition that any condition that reduces function of a gene necessary for rDNA stability, for example as has been demonstrated for heat- or dietary-stress, can lead to increased rDNA copy number variation in populations by accelerating loss.
Phenotypes Associated with rDNA Polymorphisms
Copy number variation is common in populations, spanning upwards of 10-fold (Long and Dawid 1980). This variation exists in inbred lab-reared strains of Drosophila, despite presumably uniform selective pressures, suggesting that extra rDNA are without phenotypic consequence (Lyckegaard and Clark 1989). However, a growing number of studies have identified phenotypes that map to the rDNA – either sequence polymorphisms, or copy number, or both – that may be hidden by the relatively easy life of laboratory organisms.
In a dramatic example, Cullis and colleagues found rare variants of flax that grew much taller and with greatly-elaborated inflorescence (Schneeberger and Cullis 1991). These mapped to increases of either the 45S or 5S rRNA genes (Cullis and Cleary 1986). Whether this phenotype is an effect of rDNA dose, or as an aberration to the 5S-45S stoichiometry has not been determined. Dose of the rDNA is an important concern, as some believe that stoichiometry is a critical metric in the genome (Gibbons et al. 2015). Further, key developmental decisions are mediated by rDNA in somatic and germinal stem cells of Drosophila (Zhang et al. 2014; Sriskanthadevan-Pirahas et al. 2018), although no analysis was made concerning the 5S, and so here too it is not known if the effect maps to 35S, one of the 35S-derived rRNAs, the 5S, or the stoichiometry between rRNA subunits. That analysis of the 5S cluster has lagged behind that of the 45S, hampering such detailed work.
The 5S and 35S rDNA of S. cerevisiae are interspersed into one cluster, which assures stoichiometry. Here, expansions and contractions of the cluster affects cell cycle progression through a competition between rDNA-linked origins of replication and other, non-rDNA, origins (Kwan et al. 2013; Foss et al. 2017). Altering the cell cycle timing, the use of origins, and consequent genome instability from replication-coupled defects (Kobayashi 2014; Sanchez et al. 2017) contribute to an altered lifespan in yeast with increased rDNA copy numbers or particular polymorphisms (Kwan et al. 2013). This is supported by the observation connecting the appearance of the nucleolus to longevity in Caenorhabditis elegans, Drosophila, mouse, and human (Tiku et al. 2017), and the changes that occur to rDNA copy number in aging humans (Malinovskaya et al. 2018). The literature on the rDNA and somatic aging is vast and growing (Tiku and Antebi 2018).
Years ago, we created an allelic series of Y-linked rDNA in animals (Paredes and Maggert 2009a). They are superficially indistinguishable, however males bearing the Y chromosomes with shorter arrays starve more quickly when removed from a food source, suffocate more quickly when provided limited oxygen, and consume fat stores more quickly than do their otherwise-isogenic cousins (Figure 2). These data suggest that shorter arrays are associated with increased metabolic rate, although it is also possible that they have a decreased stress resistance (Beiko et al. 2005), or a combination of both. This observation indicates that rDNA polymorphisms may have manifold pleiotropic effects mediated indirectly through metabolic rate and a stress-response to metabolic byproducts. To wit, these shortened rDNA arrays act as genetic enhancers of defective JAK-STAT signaling producing excess lamellocytes (in preparation), a developmental decision known to be controlled by reactive oxygen signaling (Small et al. 2014). The production of reactive oxygen species might contribute to any aging phenotypes correlated with rDNA activity.
Figure 2. rDNA copy number polymorphism express consequential and selectable metabolic phenotypes.
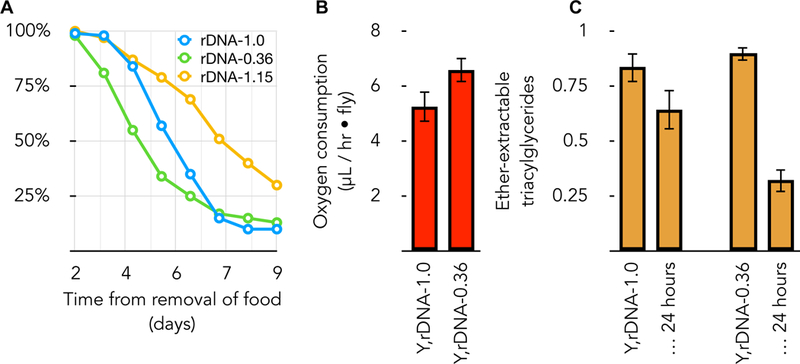
(A) Three Drosophila strains that differ only in their rDNA content exhibit different rates of starvation. Y,rDNA-1.0 is as described in Figure 1. Y,rDNA-0.36 is a derived Y chromosome with the smallest rDNA array we possess (64% of the rDNA removed). Strain Y,rDNA-1.15 is a revertant originally derived from rDNA-1.0 and contains extra rDNA copies. All strains are X, rDNA+ / Y, rDNA-(as indicated), so possess sufficient X-linked rDNA for viability and fertility. X-axis is days after removal from food, Y-axis is fraction of adults surviving at each time point. (B) Normal and smallest rDNA array-bearing flies consume oxygen at different rates. Genotypes are as described for (A). (C) Normal and smallest rDNA array-bearing flies consume ether-extractable triacylglycerides at a different rate. Populations of flies were extracted at the beginning of a starvation experiment, and again at 24 hours after removal from food. Quantification was done after thin-layer chromatographic separation (in 70:30:1 hexane:diethylether:acetic acid) and staining with 8% (w/v) H3PO4 containing 10% (w/v) copper (II) sulfate pentahydrate. Values are reported relative to purchased Triolien standards.
In S. cerevisiae and Drosophila the rDNA has also been linked to silent chromatin and heterochromatin formation, respectively. In yeast, spontaneously suppressed mutants of silenced marker transgenes exhibit rDNA loss, and the suppression maps to the reduced copy number (Michel et al. 2005). In flies, a similar situation is observed, as mutations that affect heterochromatin formation lead to rDNA loss (Peng and Karpen 2007; Aldrich and Maggert 2014), and losses of rDNA lead to suppression of heterochromatin-induced gene silencing (Paredes and Maggert 2009b; Larson et al. 2012; Zhou J 2012). It is likely that even natural somaclonal variation of heterochromatin-induced gene silencing is heavily influenced by the natural stochastic developmental losses of rDNA (Paredes and Maggert 2009b).
Mutations that compromise heterochromatin might be expected to have dramatic but ill-defined pleiotropic effects. With heterochromatin loss would come chromosome mis-segregation, cohesion defects, loss of repeat sequences, DNA damage, telomere defects, and transposon derepression. The consequence of this could range from trivial to lethal, because the underlying defect is the release of potentially-mutagenic transposons or chromosome loss, and so there is no clear pattern from which to draw predictions.
We analyzed the expression of transposons using RT-QPCR in flies that bore either the Y with the full rDNA array, or those that bore a smaller array, as above. All classes increased in expression by 1.1- to 2.5-fold in the flies with fewer rDNA, with the biggest effect on the LTR/IR class, of which some were increased by 10- to 30-fold (Bughio and Maggert, unpublished).
The connection between rDNA copy number and metabolism or heterochromatin-mediated gene silencing is still frustratingly opaque, but if the defects seen in Drosophila and yeast are general, and if similar effects occur in humans, they might reasonably be seen to contribute to pre-disease states.
It is worth noting that phenotypes stemming from rDNA copy number changes need not be a result of changes to rRNA expression or steady-state rRNA levels. In our published experiments, shorter rDNA arrays are not associated with less rRNA (Paredes and Maggert 2009b; Guerrero and Maggert 2011), and we could detect no effects on protein translation. As-yet no experiment has ruled out an adaptive (or maladaptive) role for silent rDNA copies.
Disease Phenotypes – the real and the potential
Hypothetically, fields of cells with different rDNA content may result in fields of cells with different rates of heterochromatin dysfunction, chromosome mis-segregation and transposon mobilization. Those might prefigure pre-disease states associated with genome instability, for instance cancer. The causal relationship between rDNA, heterochromatin, and cancer is at this point compellingly speculative; recent work has demonstrated that laboratory cancer cell lines have less rDNA than non-cancer counterparts and defects common in cancer can induce such changes (Stults et al. 2009; Xu et al. 2017; Udugama et al. 2018). Similarly, we have found that breast cancer tumor tissue possesses less rDNA than the marginal cells adjacent to the tumor (Valori and Maggert, unpublished). Mitotic rDNA loss may prefigure cells with an increased risk of defect leading to disease (e.g., Daroit et al. 2018), and rDNA copy number polymorphisms may predispose individuals to such diseases. Conditions that aggravate the rate of mitotic loss – stress, diet, mutation – would be expected to aggravate the risk to disease (Roche et al. 2017; Salim et al. 2017; Wang and Lemos 2017; Chen et al. 2018; Lindstrom et al. 2018).
Other, less well-investigated, human diseases are beginning to see the groundwork being laid. Ribosomal DNA expression, rDNA loss, nucleolus-mediated stress responses, and heterochromatin defects are now being connected to neurological diseases (Jesse et al. 2017; Maina et al. 2018; Nunez Villacis et al. 2018; Sun et al. 2018) – whether these are confirmed, or are found to be consequential to the disease, remains to be seen.
Heterochromatin defects alone may not explain the entirety of any risk to developing disease, but may contribute along with altered metabolic rate and genetic enhancement of defective signaling pathways. Further, the rDNA organizes the nucleolus. Despite some excellent work, it is not unfair to say that studies of the nucleolus are at their infancy, as the function and purpose of nucleolar residency of the some many-thousand nucleolar proteins has only just begun (Andersen et al. 2005; Pendle et al. 2005; Ahmad et al. 2009; Montacie et al. 2017; Pineiro et al. 2018). There is an expanse of potential implications that altering the regulation, localization or activities these proteins may have on cancer or other pre-disease/disease states.
A Specific Example of the “Epigenetic” Effects of rDNA Loss
Conditions that reduce rDNA copy number might be expected to assimilate induced phenotypes, such as altered metabolic rate or disease risk, into a population (Waddington 1957; Waddington 1959) provided any destabilization of the rDNA in the soma also occurs in the germ line. In the above-described experiment carrying Y-linked rDNA arrays in individuals mutant for Su(var)205 and Su(var)3–9, the heterochromatin was compromised in the soma, and the rDNA lost in both soma and germ line. The rDNA losses caused those Y chromosomes to act as stronger suppressors of variegation (Figure 1C), even after the genetic suppressor-of-variegation mutations had been removed from the genotype. So it would appear that the Y was epigenetically altered by transient “exposure” to the mutation. In fact, this perfectly fits the paradigm of induced transgenerational epigenetic effects (Greer et al. 2011), except we knew to look to rDNA copy number for explanation.
This “transgenerational” effect is not limited to Su(var)s, but appears a common feature of mutations that destabilize the rDNA leading to reduced copy number. Both CTCF and BLM mutants also permanently reduce rDNA copy number on chromosomes (Figure 1B), and those chromosomes are also permanently less potent in their ability to reinforce heterochromatin function (Figure 1D).
The rDNA therefore seems to be a polygene, subject to expansion and reduction, with graded effects on multiple phenotypes. Such acquired changes are present somatically and inherited meiotically, and thus the rDNA locus acts as an inducible and heritable modifier of any genetic variation that maps to the rDNA or its dependents (Mather 1944; Spofford 1976). Discriminating this from “transgenerational epigenetic inheritance” is a matter of terms.
Revisiting “epigenetic”
What makes a thing epigenetic? Conventionally, the term has been co-opted from Waddington’s philosophical treatise on developmental genetics (Waddington 1957) to now mean phenotypes that are established by some perturbation, then maintained in the absence of the inducing event (Berger et al. 2009). Maintenance has come to mean transmission through meiosis or mitosis, which is considered the biggest challenge to this hypothesized ancillary inheritance system. Epigenetic phenotypes appear to be carried by chromatin structure, even though S-phase replication has been thought to erase all chromatin structure as proteins at the the replication fork expose and copy the DNA.
Examples of epigenetics are divided into two broad classes (which others have striven to discriminate (Youngson and Whitelaw 2008; Heard and Martienssen 2014)): those that are stable in mitosis but not meiosis, and those that are transgenerationally transmitted. The former includes genomic imprinting and X chromosome inactivation in female mammals, which last only one generation and are reset in or around meiosis. These are mediated by DNA methylation, and are limited to those organisms that possess such biochemical activities.
The latter is more broad and ill-defined, and includes induced and inherited metabolic changes, chromosome and chromatin properties, fertility, etc., that persist for multiple generations (e.g., Anway et al. 2005; Carone et al. 2010; Greer et al. 2011; Seong et al. 2011; Braunschweig et al. 2012; Manikkam et al. 2012; Padmanabhan et al. 2013; Ost et al. 2014; Remely et al. 2015; Klosin et al. 2017). These so-called “transgenerational epigenetic inheritances” are caused by environmental stress, diet, drugs, and mutations, as reported in human, mouse, and in organisms without DNA methylation (S. cerevisiae, S. pombe, C. elegans, D. melanogaster). These phenomena would be indistinguishable from the induced changes to rDNA copy number we describe above because they are induced by stress or mutation, have some enhancing or suppressing effect on a marker gene or phenotype, and are inherited thenceforth in lineages. In fact the only conceptual difference between rDNA-mediated modification of phenotypes and transgenerational epigenetic inheritance is that the former is known to be affiliated with a discrete locus, and the latter is assumed to not be. As of yet, none of those studies have discounted directed genome damage as their foundational genetic lesion. The one that has mapped the epigenetic effect in Drosophila (Ost et al. 2014) indeed maps it to the rDNA-bearing Y chromosome.
Epigenetic inheritance manifests in most experiments as “heritable changes to a phenotype without a change to DNA sequence” (Berger et al. 2009). This informal description has quietly embedded itself as gate-keeper definition, precluding consideration of any mechanism that can explain transgenerational epigenetic phenotypes. Critically, in almost all cases of “epigenetic” inheritance, the attempt is never made to ascertain whether there are any DNA (especially rDNA) sequence changes – genomes are not sequenced, complementation is not attempted, linkage is not determined. Instead, “epigenetics” simply refuses to entertain alternatives in order to save its initial unsupported assertion. But if induced rDNA polymorphisms can explain some cases of transgenerational epigenetic inheritance, should we not let them?
Acknowledgements
The work was funded by an NIH Director’s Transformative Research Award (1R01GM123640), and support was provided by the UA. Cancer Center Core Grant (P30CA023074). We gratefully acknowledge Drs. Pamela Geyer, C.-Ting Wu, and Harmit Malik for encouragement.
Bibliography
- Ahmad Y, Boisvert FM, Gregor P, Cobley A and Lamond AI, 2009. NOPdb: Nucleolar Proteome Database−−2008 update. Nucleic Acids Res 37: D181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich JC, and Maggert KA, 2014. Simple quantitative PCR approach to reveal naturally occurring and mutation-induced repetitive sequence variation on the Drosophila Y chromosome. PLoS One 9: e109906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich JC, and Maggert KA, 2015. Transgenerational inheritance of diet-induced genome rearrangements in Drosophila. PLoS Genet 11: e1005148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen JS, Lam YW, Leung AK, Ong SE, Lyon CE et al. , 2005. Nucleolar proteome dynamics. Nature 433: 77–83. [DOI] [PubMed] [Google Scholar]
- Anway MD, Cupp AS, Uzumcu M and Skinner MK, 2005. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308: 1466–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Golic KG and Hawley RS, 2005. Drosophila : a laboratory handbook Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y. [Google Scholar]
- Averbeck KT, and Eickbush TH, 2005. Monitoring the mode and tempo of concerted evolution in the Drosophila melanogaster rDNA locus. Genetics 171: 1837–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiko NN, Terekhov SM, Shubaeva NO, Simirnova TD, Ivanova SM et al. , 2005. [Early and late responses to oxidative stress in human dermal fibroblasts of healthy donors and rheumatoid arthritis patients. Relationship between the cell death rate and the genomic dosage of active ribosomal genes]. Mol Biol (Mosk) 39: 264–275. [PubMed] [Google Scholar]
- Berger SL, Kouzarides T, Shiekhattar R and Shilatifard A, 2009. An operational definition of epigenetics. Genes Dev 23: 781–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianciardi A, Boschi M, Swanson EE, Belloni M and Robbins LG, 2012. Ribosomal DNA organization before and after magnification in Drosophila melanogaster. Genetics 191: 703–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig M, Jagannathan V, Gutzwiller A and Bee G, 2012. Investigations on transgenerational epigenetic response down the male line in F2 pigs. PLoS One 7: e30583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carone BR, Fauquier L, Habib N, Shea JM, Hart CE et al. , 2010. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 143: 1084–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Lobb IT, Morin P, Novo SM, Simpson J et al. , 2018. Identification of a novel TIF-IA-NF-kappaB nucleolar stress response pathway. Nucleic Acids Res 46: 6188–6205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Comai L and Pikaard CS, 1998. Gene dosage and stochastic effects determine the severity and direction of uniparental ribosomal RNA gene silencing (nucleolar dominance) in Arabidopsis allopolyploids. Proc Natl Acad Sci U S A 95: 14891–14896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung SW, Sun L and Featherstone T, 1990. Molecular cytogenetic evidence to characterize breakpoint regions in Robertsonian translocations. Cytogenet Cell Genet 54: 97–102. [DOI] [PubMed] [Google Scholar]
- Cohen S, Agmon N, Yacobi K, Mislovati M and Segal D, 2005. Evidence for rolling circle replication of tandem genes in Drosophila. Nucleic Acids Res 33: 4519–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Houben A and Segal D, 2008. Extrachromosomal circular DNA derived from tandemly repeated genomic sequences in plants. Plant J 53: 1027–1034. [DOI] [PubMed] [Google Scholar]
- Cohen S, Yacobi K and Segal D, 2003. Extrachromosomal circular DNA of tandemly repeated genomic sequences in Drosophila. Genome Res 13: 1133–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullis CA, and Cleary W, 1986. Rapidly varying DNA sequences in flax. Canadian Journal of Genetics and Cytology 28: 252–259. [Google Scholar]
- Danson AF, Marzi SJ, Lowe R, Holland ML and Rakyan VK, 2018. Early life diet conditions the molecular response to post-weaning protein restriction in the mouse. BMC Biol 16: 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daroit NB, Salgueiro AP, Maito F, Visioli F and Rados PV, 2018. The use of cytopathology to identify disturbances in oral squamous cell carcinoma at early stage: A case report. Diagn Cytopathol [DOI] [PubMed] [Google Scholar]
- Eickbush DG, and Eickbush TH, 2003. Transcription of endogenous and exogenous R2 elements in the rRNA gene locus of Drosophila melanogaster. Mol Cell Biol 23: 3825–3836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickbush DG, Ye J, Zhang X, Burke WD and Eickbush TH, 2008. Epigenetic regulation of retrotransposons within the nucleolus of Drosophila. Mol Cell Biol 28: 6452–6461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eickbush TH, Burke WD, Eickbush DG and Lathe WC 3rd, 1997. Evolution of R1 and R2 in the rDNA units of the genus Drosophila. Genetica 100: 49–61. [PubMed] [Google Scholar]
- Endow SA, 1980. On ribosomal gene compensation in Drosophila. Cell 22: 149–155. [DOI] [PubMed] [Google Scholar]
- Endow SA, 1982. Molecular characterization of ribosomal genes on the Ybb- chromosome of Drosophila melanogaster. Genetics 102: 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endow SA, 1983. Nucleolar dominance in polytene cells of Drosophila. Proc Natl Acad Sci U S A 80: 4427–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endow SA, Komma DJ and Atwood KC, 1984. Ring chromosomes and rDNA magnification in Drosophila. Genetics 108: 969–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss EJ, Lao U, Dalrymple E, Adrianse RL, Loe T et al. , 2017. SIR2 suppresses replication gaps and genome instability by balancing replication between repetitive and unique sequences. Proc Natl Acad Sci U S A 114: 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons JG, Branco AT, Godinho SA, Yu S and Lemos B, 2015. Concerted copy number variation balances ribosomal DNA dosage in human and mouse genomes. Proc Natl Acad Sci U S A 112: 2485–2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Ucar D, Hauswirth AG, Mancini E et al. , 2011. Transgenerational epigenetic inheritance of longevity in Caenorhabditis elegans. Nature 479: 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greil F, and Ahmad K, 2012. Nucleolar Dominance of the Y Chromosome in Drosophila melanogaster. Genetics [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grierson PM, Lillard K, Behbehani GK, Combs KA, Bhattacharyya S et al. , 2012. BLM helicase facilitates RNA polymerase I-mediated ribosomal RNA transcription. Hum Mol Genet 21: 1172–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi G, and Di Nocera PP, 1988. Multiple repeated units in Drosophila melanogaster ribosomal DNA spacer stimulate rRNA precursor transcription. Proc Natl Acad Sci U S A 85: 5502–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grummt I, and Langst G, 2013. Epigenetic control of RNA polymerase I transcription in mammalian cells. Biochim Biophys Acta 1829: 393–404. [DOI] [PubMed] [Google Scholar]
- Guerrero PA, and Maggert KA, 2011. The CCCTC-binding factor (CTCF) of Drosophila contributes to the regulation of the ribosomal DNA and nucleolar stability. PLoS One 6: e16401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guetg C, Lienemann P, Sirri V, Grummt I, Hernandez-Verdun D et al. , 2010. The NoRC complex mediates the heterochromatin formation and stability of silent rRNA genes and centromeric repeats. EMBO J 29: 2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley RS, and Marcus CH, 1989. Recombinational controls of rDNA redundancy in Drosophila. Annu Rev Genet 23: 87–120. [DOI] [PubMed] [Google Scholar]
- Hawley RS, and Tartof KD, 1985. A two-stage model for the control of rDNA magnification. Genetics 109: 691–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward DC, and Glover DM, 1988. Analysis of the Drosophila rDNA promoter by transient expression. Nucleic Acids Res 16: 4253–4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard E, and Martienssen RA, 2014. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157: 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland ML, Lowe R, Caton PW, Gemma C, Carbajosa G et al. , 2016. Early-life nutrition modulates the epigenetic state of specific rDNA genetic variants in mice. Science [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JE, and Pathak S, 1977. Elimination of nucleolus organizers in a case of 13/14 Robertsonian translocation. Hum Genet 35: 169–173. [DOI] [PubMed] [Google Scholar]
- Ianni A, Hoelper S, Krueger M, Braun T and Bober E, 2017. Sirt7 stabilizes rDNA heterochromatin through recruitment of DNMT1 and Sirt1. Biochem Biophys Res Commun 492: 434–440. [DOI] [PubMed] [Google Scholar]
- Jesse S, Bayer H, Alupei MC, Zugel M, Mulaw M et al. , 2017. Ribosomal transcription is regulated by PGC-1alpha and disturbed in Huntington’s disease. Sci Rep 7: 8513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killen MW, Stults DM, Wilson WA and Pierce AJ, 2012. Escherichia coli RecG functionally suppresses human Bloom syndrome phenotypes. BMC Mol Biol 13: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Dilthey AT, Nagaraja R, Lee HS, Koren S et al. , 2018. Variation in human chromosome 21 ribosomal RNA genes characterized by TAR cloning and long-read sequencing. Nucleic Acids Res 46: 6712–6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klosin A, Casas E, Hidalgo-Carcedo C, Vavouri T and Lehner B, 2017. Transgenerational transmission of environmental information in C. elegans. Science 356: 320–323. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, 2008. A new role of the rDNA and nucleolus in the nucleus--rDNA instability maintains genome integrity. Bioessays 30: 267–272. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, 2014. Ribosomal RNA gene repeats, their stability and cellular senescence. Proceedings of the Japan Academy, Series B 90: 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Heck DJ, Nomura M and Horiuchi T, 1998. Expansion and contraction of ribosomal DNA repeats in Saccharomyces cerevisiae: requirement of replication fork blocking (Fob1) protein and the role of RNA polymerase I. Genes Dev 12: 3821–3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Nomura M and Horiuchi T, 2001. Identification of DNA cis elements essential for expansion of ribosomal DNA repeats in Saccharomyces cerevisiae. Mol Cell Biol 21: 136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima KK, and Fujiwara H, 2003. Evolution of target specificity in R1 clade non-LTR retrotransposons. Mol Biol Evol 20: 351–361. [DOI] [PubMed] [Google Scholar]
- Komma DJ, and Endow SA, 1987. Incomplete Y chromosomes promote magnification in male and female Drosophila. Proc Natl Acad Sci U S A 84: 2382–2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn A, Deppert U and Grummt I, 1990. A 140-base-pair repetitive sequence element in the mouse rRNA gene spacer enhances transcription by RNA polymerase I in a cell-free system. Proc Natl Acad Sci U S A 87: 7527–7531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan EX, Foss EJ, Tsuchiyama S, Alvino GM, Kruglyak L et al. , 2013. A Natural Polymorphism in rDNA Replication Origins Links Origin Activation with Calorie Restriction and Lifespan. PLoS Genet 9: e1003329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan EX, Wang XS, Amemiya HM, Brewer BJ and Raghuraman MK, 2016. rDNA Copy Number Variants Are Frequent Passenger Mutations in Saccharomyces cerevisiae Deletion Collections and de Novo Transformants. G3 (Bethesda) 6: 2829–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson K, Yan SJ, Tsurumi A, Liu J, Zhou J et al. , 2012. Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet 8: e1002473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EB, 1952. The Pseudoallelism of White and Apricot in Drosophila Melanogaster. Proc Natl Acad Sci U S A 38: 953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindstrom MS, Jurada D, Bursac S, Orsolic I, Bartek J et al. , 2018. Nucleolus as an emerging hub in maintenance of genome stability and cancer pathogenesis. Oncogene 37: 2351–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long EO, and Dawid IB, 1980. Repeated genes in eukaryotes. Annu Rev Biochem 49: 727– 764. [DOI] [PubMed] [Google Scholar]
- Lu KL, Nelson JO, Watase GJ, Warsinger-Pepe N and Yamashita YM, 2018. Transgenerational dynamics of rDNA copy number in Drosophila male germline stem cells. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyckegaard EM, and Clark AG, 1989. Ribosomal DNA and Stellate gene copy number variation on the Y chromosome of Drosophila melanogaster. Proc Natl Acad Sci U S A 86: 1944–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maina MB, Bailey LJ, Wagih S, Biasetti L, Pollack SJ et al. , 2018. The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol Commun 6: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinovskaya EM, Ershova ES, Golimbet VE, Porokhovnik LN, Lyapunova NA et al. , 2018. Copy Number of Human Ribosomal Genes With Aging: Unchanged Mean, but Narrowed Range and Decreased Variance in Elderly Group. Front Genet 9: 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manikkam M, Guerrero-Bosagna C, Tracey R, Haque MM and Skinner MK, 2012. Transgenerational actions of environmental compounds on reproductive disease and identification of epigenetic biomarkers of ancestral exposures. PLoS One 7: e31901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mather K, 1944. The genetical activity of heterochromatin. Proceedings of the Royal Society B 132. [Google Scholar]
- McStay B, and Grummt I, 2008. The epigenetics of rRNA genes: from molecular to chromosome biology. Annu Rev Cell Dev Biol 24: 131–157. [DOI] [PubMed] [Google Scholar]
- Michel AH, Kornmann B, Dubrana K and Shore D, 2005. Spontaneous rDNA copy number variation modulates Sir2 levels and epigenetic gene silencing. Genes Dev 19: 1199–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montacie C, Durut N, Opsomer A, Palm D, Comella P et al. , 2017. Nucleolar Proteome Analysis and Proteasomal Activity Assays Reveal a Link between Nucleolus and 26S Proteasome in A. thaliana. Front Plant Sci 8: 1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ, 1932. Further Studies on the Nature and Causes of Gene Mutations. Proceedings of the 6th International Congress of Genetics: 213–255. [Google Scholar]
- Nunez Villacis L, Wong MS, Ferguson LL, Hein N, George AJ et al. , 2018. New Roles for the Nucleolus in Health and Disease. Bioessays 40: e1700233. [DOI] [PubMed] [Google Scholar]
- Ost A, Lempradl A, Casas E, Weigert M, Tiko T et al. , 2014. Paternal diet defines offspring chromatin state and intergenerational obesity. Cell 159: 1352–1364. [DOI] [PubMed] [Google Scholar]
- Padmanabhan N, Jia D, Geary-Joo C, Wu X, Ferguson-Smith AC et al. , 2013. Mutation in folate metabolism causes epigenetic instability and transgenerational effects on development. Cell 155:81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes S, Angulo-Ibanez M, Tasselli L, Carlson SM, Zheng W et al. , 2018. The epigenetic regulator SIRT7 guards against mammalian cellular senescence induced by ribosomal DNA instability. J Biol Chem 293: 11242–11250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes S, Branco AT, Hartl DL, Maggert KA and Lemos B, 2011. Ribosomal DNA deletions modulate genome-wide gene expression: “rDNA-sensitive” genes and natural variation. PLoS Genet 7: e1001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes S, and Maggert KA, 2009a. Expression of I-CreI Endonuclease Generates Deletions Within the rDNA of Drosophila. Genetics 181: 1661–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredes S, and Maggert KA, 2009b. Ribosomal DNA contributes to global chromatin regulation. Proc Natl Acad Sci U S A 106: 17829–17834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks MM, Kurylo CM, Dass RA, Bojmar L, Lyden D et al. , 2018. Variant ribosomal RNA alleles are conserved and exhibit tissue-specific expression. Sci Adv 4: eaao0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendle AF, Clark GP, Boon R, Lewandowska D, Lam YW et al. , 2005. Proteomic analysis of the Arabidopsis nucleolus suggests novel nucleolar functions. Mol Biol Cell 16: 260–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng JC, and Karpen GH, 2007. H3K9 methylation and RNA interference regulate nucleolar organization and repeated DNA stability. Nat Cell Biol 9: 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng JC, and Karpen GH, 2008. Epigenetic regulation of heterochromatic DNA stability. Curr Opin Genet Dev 18: 204–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineiro D, Stoneley M, Ramakrishna M, Alexandrova J, Dezi V et al. , 2018. Identification of the RNA polymerase I-RNA interactome. Nucleic Acids Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontes O, Lawrence RJ, Neves N, Silva M, Lee JH et al. , 2003. Natural variation in nucleolar dominance reveals the relationship between nucleolus organizer chromatin topology and rRNA gene transcription in Arabidopsis. Proc Natl Acad Sci U S A 100: 11418–11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss S, and Pikaard CS, 2007. rRNA gene silencing and nucleolar dominance: insights into a chromosome-scale epigenetic on/off switch. Biochim Biophys Acta 1769: 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prokopowich CD, Gregory TR and Crease TJ, 2003. The correlation between rDNA copy number and genome size in eukaryotes. Genome 46: 48–50. [DOI] [PubMed] [Google Scholar]
- Rasooly RS, and Robbins LG, 1991. Rex and a suppressor of Rex are repeated neomorphic loci in the Drosophila melanogaster ribosomal DNA. Genetics 129: 119–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remely M, Stefanska B, Lovrecic L, Magnet U and Haslberger AG, 2015. Nutriepigenomics: the role of nutrition in epigenetic control of human diseases. Curr Opin Clin Nutr Metab Care 18: 328–333. [DOI] [PubMed] [Google Scholar]
- Ritossa F, 1973. Crossing-over between X AND Y chromosomes during ribosomal DNA magnification in Drosophila melanogaster. Proc Natl Acad Sci U S A 70: 1950–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritossa FM, Atwood KC and Spiegelman S, 1966. A molecular explanation of the bobbed mutants of Drosophila as partial deficiencies of “ribosomal” DNA. Genetics 54: 819–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche B, Arcangioli B and Martienssen R, 2017. New roles for Dicer in the nucleolus and its relevance to cancer. Cell Cycle 16: 1643–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim D, Bradford WD, Freeland A, Cady G, Wang J et al. , 2017. DNA replication stress restricts ribosomal DNA copy number. PLoS Genet 13: e1007006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez JC, Kwan EX, Pohl TJ, Amemiya HM, Raghuraman MK et al. , 2017. Defective replication initiation results in locus specific chromosome breakage and a ribosomal RNA deficiency in yeast. PLoS Genet 13: e1007041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanij E, Poortinga G, Sharkey K, Hung S, Holloway TP et al. , 2008. UBF levels determine the number of active ribosomal RNA genes in mammals. J Cell Biol 183: 1259–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schawalder J, Paric E and Neff NF, 2003. Telomere and ribosomal DNA repeats are chromosomal targets of the bloom syndrome DNA helicase. BMC Cell Biol 4: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger RG, and Cullis CA, 1991. Specific DNA alterations associated with the environmental induction of heritable changes in flax. Genetics 128: 619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong KH, Li D, Shimizu H, Nakamura R and Ishii S, 2011. Inheritance of stress-induced, ATF-2-dependent epigenetic change. Cell 145: 1049–1061. [DOI] [PubMed] [Google Scholar]
- Sinclair DA, and Guarente L, 1997. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell 91: 1033–1042. [DOI] [PubMed] [Google Scholar]
- Small C, Ramroop J, Otazo M, Huang LH, Saleque S et al. , 2014. An unexpected link between notch signaling and ROS in restricting the differentiation of hematopoietic progenitors in Drosophila. Genetics 197: 471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Specchia V, Piacentini L, Tritto P, Fanti L, D’Alessandro R et al. , 2010. Hsp90 prevents phenotypic variation by suppressing the mutagenic activity of transposons. Nature 463: 662–665. [DOI] [PubMed] [Google Scholar]
- Spofford JB, 1976. Position-effect variegation in Drosophila, pp. 955–1019 in The Genetics and Biology of Drosophila, edited by Ashburner M and Novitski E. Academic Press. [Google Scholar]
- Spofford JB, and DeSalle R, 1991. Nucleolus organizer-suppressed position-effect variegation in Drosophila melanogaster. Genet Res 57: 245–255. [DOI] [PubMed] [Google Scholar]
- Sriskanthadevan-Pirahas S, Lee J and Grewal SS, 2018. The EGF/Ras pathway controls growth in Drosophila via ribosomal RNA synthesis. Dev Biol 439: 19–29. [DOI] [PubMed] [Google Scholar]
- Stults DM, Killen MW, Pierce HH and Pierce AJ, 2008. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res 18: 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stults DM, Killen MW, Williamson EP, Hourigan JS, Vargas HD et al. , 2009. Human rRNA gene clusters are recombinational hotspots in cancer. Cancer Res 69: 9096–9104. [DOI] [PubMed] [Google Scholar]
- Sun W, Samimi H, Gamez M, Zare H and Frost B, 2018. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat Neurosci 21: 1038–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartof KD, 1973. Regulation of ribosomal RNA gene multiplicity in Drosophila melanogaster. Genetics 73: 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartof KD, 1974. Unequal mitotic sister chromatid exchange and disproportionate replication as mechanisms regulating ribosomal RNA gene redundancy. Cold Spring Harb Symp Quant Biol 38: 491–500. [DOI] [PubMed] [Google Scholar]
- Terracol R, Iturbide Y and Prud’Homme N, 1990. Partial reversion at the bobbed locus of Drosophila melanogaster. Biol Cell 68: 65–71. [DOI] [PubMed] [Google Scholar]
- Terracol R, and Prud’homme N, 1981. 26S and 18S rRNA synthesis in bobbed mutants of Drosophila melanogaster. Biochimie 63: 451–455. [DOI] [PubMed] [Google Scholar]
- Terracol R, and Prud’homme N, 1986. Differential elimination of rDNA genes in bobbed mutants of Drosophila melanogaster. Mol Cell Biol 6: 1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiku V, and Antebi A, 2018. Nucleolar Function in Lifespan Regulation. Trends Cell Biol 28:662–672. [DOI] [PubMed] [Google Scholar]
- Tiku V, Jain C, Raz Y, Nakamura S, Heestand B et al. , 2017. Small nucleoli are a cellular hallmark of longevity. Nat Commun 8: 16083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrano V, Navascues J, Docquier F, Zhang R, Burke LJ et al. , 2006. Targeting of CTCF to the nucleolus inhibits nucleolar transcription through a poly(ADP-ribosyl)ation-dependent mechanism. J Cell Sci 119: 1746–1759. [DOI] [PubMed] [Google Scholar]
- Udugama M, Sanij E, Voon HPJ, Son J, Hii L et al. , 2018. Ribosomal DNA copy loss and repeat instability in ATRX-mutated cancers. Proc Natl Acad Sci U S A 115: 4737–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Nobelen S, Rosa-Garrido M, Leers J, Heath H, Soochit W et al. , 2010. CTCF regulates the local epigenetic state of ribosomal DNA repeats. Epigenetics Chromatin 3: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH, 1957. The Stretegy of the Genes [Google Scholar]
- Waddington CH, 1959. Canalization of development and genetic assimiliation of acquired characters. Nature 183: 1654–1655. [DOI] [PubMed] [Google Scholar]
- Wang M, and Lemos B, 2017. Ribosomal DNA copy number amplification and loss in human cancers is linked to tumor genetic context, nucleolus activity, and proliferation. PLoS Genet 13: e1006994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wan T, Becher H, Kuderova A, Leitch IJ et al. , 2018. Remarkable variation of ribosomal DNA organization and copy number in gnetophytes, a distinct lineage of gymnosperms. Ann Bot [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warmerdam DO, van den Berg J and Medema RH, 2016. Breaks in the 45S rDNA Lead to Recombination-Mediated Loss of Repeats. Cell Rep 14: 2519–2527. [DOI] [PubMed] [Google Scholar]
- Xu B, Li H, Perry JM, Singh VP, Unruh J et al. , 2017. Ribosomal DNA copy number loss and sequence variation in cancer. PLoS Genet 13: e1006771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngson NA, and Whitelaw E, 2008. Transgenerational epigenetic effects. Annu Rev Genomics Hum Genet 9: 233–257. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Shalaby NA and Buszczak M, 2014. Changes in rRNA transcription influence proliferation and cell fate within a stem cell lineage. Science 343: 298–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J ST, Martinsen L, Lemos B, Eickbush TH, Hartl DL, 2012. Y chromosome mediates ribosomal DNA silencing and modulates the chromatin state in Drosophila. Proc Natl Acad Sci U S A 10.1073/pnas.1207367109. [DOI] [PMC free article] [PubMed] [Google Scholar]