

Abstract
Background:
The pathogenesis of asthma and airway obstruction is the result of an abnormal response to different environmental exposures. The scientific premise of our study was based on the finding that FoxO1 expression is increased in lung macrophages of mice after allergen exposure and human asthmatic patients. Macrophages are capable of switching from one functional phenotype to another, it is important to understand the mechanisms involved in the transformation of macrophages and how their cellular function affects the peribronchial stromal microenvironment.
Methods:
We employed a murine asthma model, in which mice were treated by intranasal insufflation with allergens for 2 to 8 weeks. We used both a pharmacologic approach using a highly specific FoxO1 inhibitor and genetic approaches using FoxO1 knockout mice (FoxO1fl/flLysMcre). Cytokine level in biological fluids were measured by ELISA and the expression of encoding molecules by NanoString assay and qRT-PCR.
Results:
We show that the levels of FoxO1 gene are significantly elevated the in airway macrophages of patients with mild asthma in response to subsegmental bronchial allergen challenge. Transcription factor FoxO1 regulates a pro-asthmatic phenotype of lung macrophages that is involved in the development and progression of chronic allergic airway disease. We have shown that inhibition of FoxO1 induced phenotypic conversion of lung macrophages and down regulates pro-asthmatic and pro-fibrotic gene expression by macrophages, which contribute to airway inflammation and airway remodeling in allergic asthma.
Conclusion:
Targeting FoxO1 with its downstream regulator IRF4 is a novel therapeutic target for controlling allergic inflammation and potentially reversing fibrotic airway remodeling.
Keywords: FoxO1, IRF4, macrophages, asthmatic inflammation, airway remodeling
Graphical Abstract
Forkhead box protein O1 (FoxO1) gene is significantly elevated in the airway macrophages of patients with mild asthma in response to subsegmental bronchial allergen challenge.
Transcription factor FoxO1 regulates a pro-asthmatic phenotype of lung macrophages that is involved in the development and progression of chronic allergic airway disease.
Targeting FoxO1 with its downstream regulator interferon regulatory factor 4 (IRF4) is a novel therapeutic target for controlling allergic inflammation and potentially reversing fibrotic airway remodeling.
INTRODUCTION
Asthma is an inflammatory pulmonary disease that is characterized by immune cells-mediated downstream events, including eosinophilic airway infiltration, mast cell activation, peribronchial goblet cell hyperplasia, and chronic airway remodeling1. Asthma is widely considered to be a “type 2,” immune system response as it is strongly linked to allergic inflammation. However, non-type 2 asthmatic phenotypes, such as obesity- or smoking-associated asthma, are separate category of asthma that may not be accompanied by type 2 inflammation and airway eosinophils. Although type 2 inflammation is a prevalent form of asthma, a substantial subset of asthma has a low or non-type 2 inflammatory response in nature2. Although dendritic cells and allergen-specific CD4+ T cells have a dominant role in the pathogenesis of airway inflammation and hyper-responsiveness, there is a growing awareness that alternatively activated macrophages contribute to the patho-physiological events that result in airway inflammation and remodeling in asthma3–5.
While identification of macrophage phenotype to M1 and M2 provides a strong foundation for understanding their functions, the purist form of the M1-M2 paradigm is an in vitro phenomenon and does not reflect the incomplete polarization that occurs in asthmatic patients. However, it does appear that resident macrophages convert into an M2-like phenotype in the Th2 cytokine-enriched microenvironment of asthma6. In the type 2 cytokine enriched allergic inflammation associated with exposure to allergen, the alternatively (also known as M2) activated macrophages that are seen in various animal models, contribute to Th2 immunity. M2-like macrophages are linked through multiple pathways with Th2 immune mediators that can work synergistically in the promotion of allergic responses4,7. Though the activation of subset-defining transcription factors is well characterized of commitment to T cell lineages, the transcription factors that underlie pro-asthmatic macrophage polarization remain largely undefined. We recently reported involvement of the transcription factor forkhead box proteins, O1 (FoxO1) in regulating alternative macrophages activation5,8 and in this manuscript we examined whether FoxO1 has a role in regulating airway remodeling in sensitized mice in response to allergen challenge.
FoxO1 modulates various cellular responses inlcuding the oxidative stress response, immune homeostasis, cell multiplication, cell death, and metabolism. FoxO1 plays a direct role in regulating inflammation by transcriptional regulation, signal transduction, and partnering with other transcription factors, which mediate its various multifunctional roles9. While FoxO1 has been proposed to be important for functional aspects of Ccr2-dependent M1 adipose tissue macrophage population10, the role of FoxO1 in lung macrophages has not been previously investigated. In this manuscript, we examined whether blocking FoxO1 in macrophages prevents eosinophilic inflammation and reverses chronic allergic aiway changes. Remodeling of structural and functional peribronchial tissues is a hallmark of chronic persistent asthma. In order to develop novel therapeutics against allergic inflammation, it is necessary to understand the etiology of asthmatic airway remodeling. It is believed that persistent airway inflammation promotes dysregulated deposition of extracellular matrix protein and subsequent fibrosis. These series of events are thought to lead to irreversible airway remodeling, and the magnitude of airway remodeling is associated with increased severity and mortality in severe asthmatics11. Recent discoveries indicate that profibrotic alternatively activated macrophages can mediate tissue remodeling during airway inflammation. Lung macrophages are fortified to participate in asthma during chronic airway repair after injury by the production of IL-13, transforming growth factor (TGF)-β, and other pro-fibrotic factors that directly activate fibroblasts12–15. Therefore, influencing lung macrophage function related to the pulmonary fibrotic response is an attractive therapeutic strategy.
Our data show that FoxO1 gene expression is increased in lung macrophages from patients with asthma and in mouse and human lung macrophages in response to in vitro IL-4 treatment. Increasing FoxO1 correlates with expression of interferon regulatory factor 4 (IRF4), which is known to be associated with the M2 macrophage inflammatory phenotype. Furthermore, pharmacologic inhibition of FoxO1 reverses goblet cell hyperplasia when given to chronically allergen sensitized mice and is associated with expression of M2 like gene expression by lung macrophages. In a LysM-cre-driven and a conditional Csf1r-driven FoxO1 knockout mice, we were able to show a marked attenuation of various M2 gene expression, decreased IRF4, and chronic airway changes. Finally, adoptive transfer of lung macrophages isolated from LysM-cre-driven FoxO1 transgenic mice had a marked accentuation of chronic airway changes. These in vivo loss and gain of function experiments, in combination with the literature, indicate that blocking FoxO1 is a possible treatment for prevention of asthma and, based on our data, has the potential to reverse established airway changes.
MATERIAL and METHODS
Detailed methods are described in the supplementary material.
Subsegmental Bronchoprovocation with Allergen Bronchoscopy Protocol
This protocol was approved by the Institutional Review Board of the University of Illinois (Chicago, IL) and an IND was obtained from the FDA for bronchoscopic administration of allergens to volunteers. The details of the protocol were described in our previous publication3. In brief, subjects underwent screening for inclusion and exclusion criteria that included skin prick testing to dust mites, short ragweed, and cockroach allergens and spirometry with bronchodilator reversibility and/or methacholine challenge. Subjects taking daily asthma-controlling medications were excluded. To obtain the prechallenge bronchial sample, BAL was performed at a subsegmental bronchus before allergen challenge. Subsegmental bronchoprovocation with the identified allergen (SBP-AG) was performed in a different subsegment. A starting dose of 10-fold greater than the previously defined skin endpoint titration dose in bioequivalent allergen units (BAU) or weight/volume (wt/vol) of allergen was administered. If no significant airway edema was noted after 10 minutes, the challenge dose of allergen (i.e., 100-fold greater than the previously defined skin endpoint titration dose) was administered to the subsegment. The maximum challenge dose for SBP-AG was 5 mL of a 100 BAU/mL or 1:2,000 wt/vol concentration of allergen. At 48 hrs after the initial bronchosopy, Post-challenege BAL samples were obtained. After BAL fluid was filtered, the mononuclear cell fraction was isolated by Percoll density gradient centrifugation as previously described16. Alveolar macrophages (AM) were further purified from the mononuclear cell fraction by 1 hr of adhesion purification. Additional detail on the methods for the SBP-AG protocol is provided in the online supplement.
Mice
All experiments involving mice were conducted with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the Ohio State University. Generation of myeloid FoxO1 knockout mice, FoxO1fl/fl mice were crossed with LysM-cre mice (Jackson Laboratories, Bar Harbor, ME) to homozygosity (FoxO1fl/flLysMcre)8. Their littermates were used as controls. FoxO1CAfl/fl (R26floxneoΔ256FoxO1) mice5 were bred with LysM-cre mice to homozygozity (FoxO1CAfl/flLysMcre). DNA extraction and genotyping were performed as described previously. Mice at age of 8–12 weeks were used in this study. Csf1r-cre (FVB-Tg(Csf1r-icre)1Jwp/J; Jackson Laboratories, back- crossed for >10 generations into the C57BL/6J background) and crossed with FoxO1fl/fl mice to obtain FoxO1fl/flCsf1rcre mice. Knockout of FoxO1 in myeloid cells was induced by daily subcutaneous injection of 3 μg tamoxifen (Sigma) per mouse for 2 weeks, before allergen treatment17.
Induction of acute/chronic murine asthma model
Triple allergens (DRA) include extracts of dust mite (Dermatophagoides farina), ragweed (Ambrosia artemisiifolia), and Aspergillus fumigates (Greer Laboratories, Lenoir, NC). Aluminum (Inject Alum; Thermo Scientific) was used for adjuvant. Quantities of allergens for intraperitoneal (100 μl) per mouse were used as follows: D. farina (5 μg, 3–35 EU by means of LAL assay), ragweed (50 μg, 5 EU), and Aspergillus fumigates (5 μg, 0.1 EU)18. Quantities of allergens for intranasal injection (30 μl) were used as follows: D. farina (8.3 μg), ragweed (83.4 μg), and Aspergillus fumigates (8.3 μg).
Mice were sensitized with the DRA allergen mixture (60 μg of DRA) on Days 0 and 5 by intraperitoneal injection with alum and then challenged with the DRA mixture (200 μg of DRA) at the same concentration used for sensitization on Days 12, 13, and 14 by intranasal delivery3. The mice were sacrificed on Day 15, and bronchoalveolar lavage (BAL) fluid and lung tissues were collected for further analysis. All sensitized animals had elevated levels of total IgE. Chronic murine asthma model was conducted for mimicking human asthmatic inflammation. Eight-week-old FoxO1fl/flLysMcre and FoxO1fl/fl mice were exposed 100 μg of DRA intranasally, twice per week for up to 10 weeks, without exogenous adjuvant18. After the 10-week exposure period, these mice were rested for 2 weeks before analyses. For each treatment, 8–10 mice were used per group. Timelines of the DRA models are shown in Fig.3A and 4A, respectively.
Fig.3. FoxO1fl/flLsyMcre has attenuated DRA-induced Th2 immunity in lungs of allergic mice.
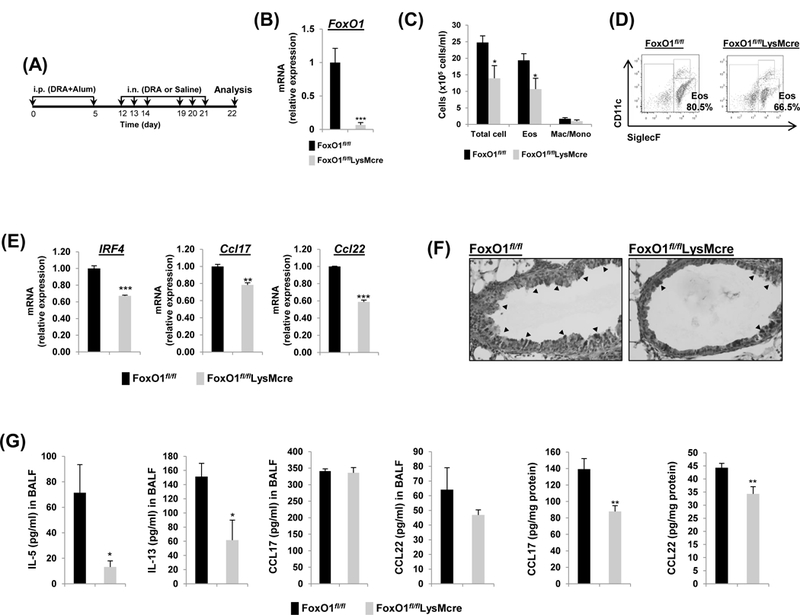
(A) FoxO1fl/fl and FoxO1fl/flLysMcre mice were sensitized and challenged by daily i.n. administration of DRA or saline, 3 days a week for 2 weeks. (B) Relative expression of FoxO1 in sorted AM. FoxO1 mRNA was analyzed by rt-qPCR. (C) Eosinophilic inflammation was attenuated in FoxO1fl/flLysMcre mice. Total cells and eosinophils influx in BAL fluid were counted, analyzed by flow cytometry. (D) Cell staining for markers of eosinophils (CD45+SiglecF+CD11c-) and AM (CD45+SiglecF+CD11c+). (E) Expression of mRNA for IRF4, Ccl17, Ccl22 in DRA-exposed murine AM from FoxO1fl/fl and FoxO1fl/flLysMcre mice. (F) PAS stained (black arrowheads) lung sections from the mice exposed to DRA. (G) IL-5, IL-13, CCL17, CCL22 were quantified with ELISA in BALF and DRA-exposed lung homogenates (N=6–8). Results are shown as mean ± SE. P values were obtained using a t test. *p<0.05, **p<0.01, and ***p<0.001. i.p., intraperitoneal; i.n., intranasal; Eos, eosinophil; Mac/Mono, macrophage/monocyte.
Fig.4. FoxO1 deficiency protects against chronic allergen-induced asthmatic inflammation.
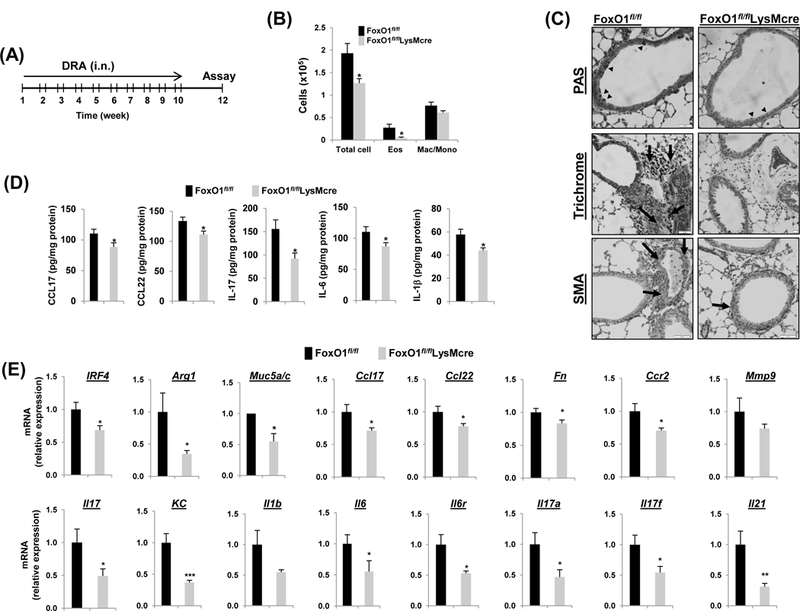
(A) FoxO1fl/fl and FoxO1fl/flLysMcre mice were challenged i.n. with saline or DRA 2 times a week for 10 weeks. End point analysis was performed 2 weeks after the last administration of DRA. (B) The number of BALF inflammatory cells from DRA-challenged FoxO1fl/fl mice were compared with FoxO1fl/flLysMcre mice (N=10). (C) Representative histologic lung sections stained with PAS (black arrowheads), Trichrome (black arrows, peribronchial), and SMA (brown staining) are shown. (D) CCL17, CCL22, IL-17, IL-6, IL-1β cytokines were quantified with ELISA in DRA-exposed lung homogenates (N=6–8). (E) Expression of mRNA for Th2 and Th17 markers in DRA-induced chronic asthmatic lung tissues from FoxO1fl/fl and FoxO1fl/flLysMcre mice. Results are shown as mean ± SE. P values were obtained using a t test. *p<0.05, **p<0.01, ***p<0.001. i.n., intranasal; Eos, eosinophil; Mac/Mono, macrophage/monocyte.
Inhibitor administration
C57BL/6 mice were given DRA 3 days/week i.n. for 2 week. At 3 week, mice were given i.p. AS1842856 (20 mg/kg) or vehicle (DMSO) 5 times for 2 week along with concurrent DRA treatment (4 week total DRA).
Adoptive transfer of macrophages
Murine lung macrophages were isolated from whole lung following collagenase digestion and suspended at a concentration of 3 × 106 cells/ml in PBS18. Donor cells from FoxO1CAfl/flLysMcre or FoxO1CAfl/fl were intranasally (i.n.) injected into a DRA-sensitized recipient FoxO1CAfl/fl mouse at 1 day (D11) before DRA challenge. On Day 15, recipient mice were sacrificed and the allergic asthmatic inflammation was analyzed. The protocol of adoptive transfer of macrophages is shown in Fig.6A.
Fig.6. The adoptive transfer of resident lung macrophage from FoxO1CAfl/flLysMcre has accentuated ability to induce DRA-mediated airway inflammation.
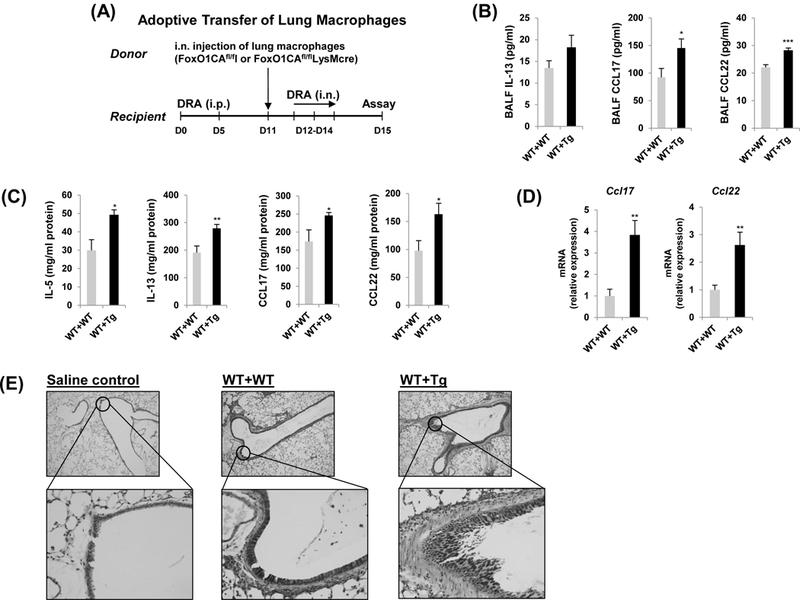
(A) Donor FoxO1CAfl/fl mice were received residential lung macrophages from FoxO1CAfl/fl and FoxO1CAfl/flLysMcre mice intranasally (i.n.), then asthma was induced. Then the mice received transferred macrophages were intranasally challenged with DRA on Days 12, 13, and 14. The allergic inflammation was detected on Day 15. IL-5, IL-13, CCL17, CCL22 cytokines were quantified with ELISA in BALF (B) and lung homogenates (C). (N=6–8) (D) Expression of mRNA for CCL17 and CCL22 in DRA-induced murine lung tissues. (E) Lung lobes were fixed and PAS staining was performed for identification of goblet cells in the epithelium. Mucin within the airway stained pink or red. Results are shown as mean ± SE. P values were obtained using a t test. *p<0.05, **p<0.01, ***p<0.001. i.n., intranasal; i.p., intraperitoneal; WT, wild type; Tg, transgenic.
Flow cytometry
Cells collected from BAL fluid were incubated with Fc blocking anti-mouse CD 16/32 antibody (BD Bioscience) followed by PE-conjugated anti-SiglecF, PerCP Cy5.5 conjugated CD45, PE-Cy7-conjugated CD11c, and APC-conjugated anti-CD11b antibodies. Cells were analyzed on a BD LSR II (BD bioscience) where gating was based on respective unstained cell population and isotype matching control antibodies. The data were analyzed with FlowJo software (TreeStar).
Mouse immunology gene-expression analysis
Mouse alveolar macrophages were purified by cell sorting (SiglecF+CD11c+sidescatterhigh) after 8 weeks of allergen challenge from 10 animals using FACS Aria. RNA was purified using Direct-zol™ RNA Kits (Zymo Research) according to the manufacture’s instruction. Total RNA was hybridized to a custom gene expression CodeSet and analyzed on an nCounter Digital Analyzer (Nanostring Technologies) in the OSU genomics shared resources. Counts were normalized to geometric means of internal spike-in and endogenous controls per Nanostring Technologies’ specifications and averaged background subtraction using Partek Genomics Suite™ 6.6 (Partek Inc., St. Louis, MO). For express of patterns, unsupervised analysis including unsupervised hierarchical clustering and Principal Component Analysis (PCA) were performed (data not shown). For heatmaps, the Euclidian distance between the 2 groups of samples was calculated by the average linkage19.
Statistical analysis
Results are expressed as means ± SEM. Comparison between two groups were performed with Student’s t-test for unpaired variables. Statistical significance is indicated in figure legends. A P <0.05 was considered statistically significant.
RESULTS
Induced FoxO1 expression in pro-asthmatic macrophages.
Whereas our previous report defined a role for alternatively activated M2-like macrophages in contributing the human immune response to allergen3, the current study seeks to extend these data to determine whether FoxO1 has a role in regulating gene expression levels in lung macrophages. To investigate the involvement of FoxO1 in allergic asthma, we first measured FoxO1 expression in AM obtained from pre- and post-allergen challenge in the SBP-AG protocol, a model of allergic airway inflammation, in human volunteers with mild intermittent asthma as previously described16. AMs from subjects with asthma were purified and total RNA analyzed by quantitative RT-PCR. We found that FoxO1 mRNA level was readily expressed in post-allergen challenged AMs compared with pre-challenge AM (Fig.1A). We also observed that FoxO1 expression was increased in AM from normal donor control lung in response to in vitro treatment with IL-4 (Fig.1B) and in AM from patients with asthma (Fig.1C). Consistent with this pattern, western blots of mouse pulmonary macrophages proteins demonstrated that FoxO1 is increased in response to in vitro IL-4 stimulation and in response to in vivo allergen inhalation in the asthma model (Fig.1D). Using intracellular staining of FoxO1 by flow cytometry, we observed a noticeable increase of FoxO1 in AM exposed to IL-4 (Supple Fig.1A, from 22.1% to 44.1%) supporting our hypothesis of a critical role of FoxO1 in pro-asthmatic macrophages.
Fig.1. FoxO1 is a critical component of pro-asthmatic macrophages.
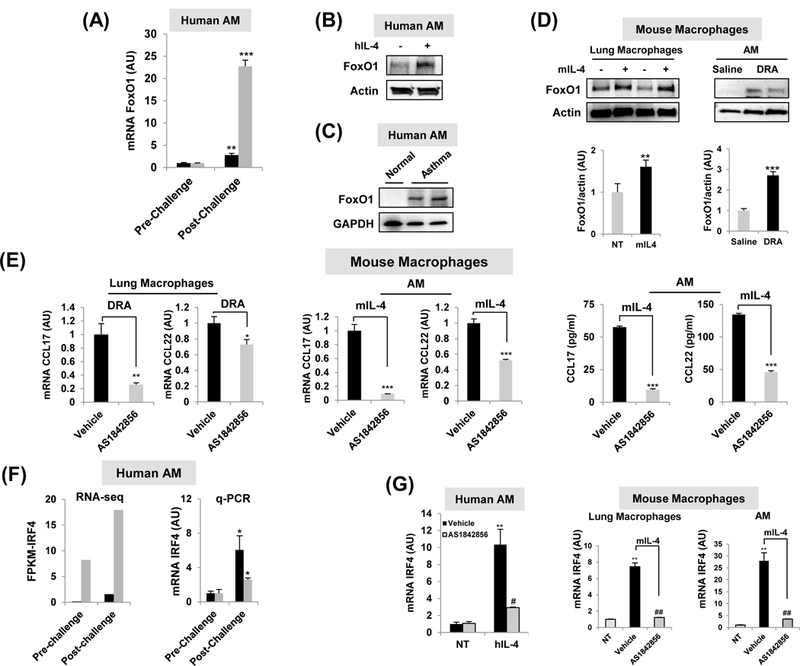
(A) FoxO1 expression comparison from patients with mild asthma enrolled in the subsegmental bronchoprovocation with allergen (SBP-AG) protocol. The post-allergen-challenged AMs were obtained from the allergen-challenged site (experimental site). Total RNA was purified from AM obtained from pre- and post-allergen challenge in the SBP-AG protocol (N=2). (B)In vitro studies demonstrated that Th2 cytokines IL-4 significantly enhanced FoxO1 in healthy human AM. Image is representative of two blots (N=2). (C) AM from a patient with asthma. Representative Western blot image showing FoxO1 observed in asthma patient (N=2). (D)In vitro studies demonstrated that Th2 cytokines IL-4 significantly enhanced FoxO1 in mouse lung macrophages. Western blot analysis of AM from DRA-induced allergic asthma mice. (E) Pretreatment with AS1842856 blunted the upregulation of CCL17 and CCL22 transcript in DRA-challenged mouse lung macrophages and mIL-4-activated AM (N=3–4). CCL17 and CCL22 cytokines were quantified with ELISA in supernatant from mIL-4 treated AM (N=3). (F) IRF4 expression comparison from patients with mild asthma enrolled in the SBP-AG protocol. Normalized mRNA expression levels presented as FPKM values of Irf4 as detected by RNA seq and its validation by q-PCR in AM obtained from pre- and post-allergen challenge in the SBP-AG protocol. (G) Pretreatment with FoxO1 inhibitor blunted the upregulation of IRF4 transcript in IL-4-activated healthy human AM (N=2), mouse lung macrophages (N=4), and sorted AM (N=4). Results are shown as mean ± SE. P values were obtained using a t test. *,#p<0.05, **,##p<0.01, ***,###p<0.001. NT, non-treated; AU, arbitrary unit; FPKM, fragments per kilobase million.
To test the role of FoxO1 signaling in pro-asthmatic phenotype in pulmonary macrophages, we utilized a pharmacological inhibitor of FoxO120. AS1842856, a selective FoxO1 inhibitor markedly suppressed Th2-attracting chemokines CCL17 and CCL22 in pulmonary macrophages exposed to in vivo allergen inhalation and IL-4. It is notable the mRNA level of CCL17 was approximate 11-fold decrease in AS1842856-treated IL-4-stimulated AM (Fig.1E). Moreover, FoxO1 inhibitor treatment substantially decreased CCL17 and CCL22 in IL-4 stimulated AM as compared to their vehicle treated counterparts. These data indicate that FoxO1 is highly expressed in both human and murine pro-asthmatic macrophages suggesting involvement of positive regulation of pro-asthmatic macrophages function.
FoxO1 inhibition regulates pro-asthmatic IRF4 expression in pulmonary macrophages.
Recently published work has demonstrated that FoxO1-IRF4 signaling in macrophages contributes to the enhancement of Th2 immune responses5. RNA-seq analysis in AM obtained from pre- and post-allergen challenge in the SBP-AG protocol revealed that IRF4 transcript was statistically significant (q-value <0.05) (Fig.1F). Out of 29,691 transcripts, IRF4 had one of the higher levels of expression, with a normalized FPKM value. Induction of IRF4 gene was further validated by qPCR. Treatment with a FoxO1 inhibitor attenuated increase in IRF4 mRNA in response to in vitro IL-4 treatment in both human and murine pulmonary macrophages (Fig.1G), and protein (Supple Fig.1B) indicating that FoxO1 regulates IRF4 at a transcriptional level. A luciferase reporter assay showed significantly reduced IRF4 promoter activity to 54% of IL-4 treatment in the pre-treatment of AS1842856 (Supple Fig.1C). In order to confirm this finding, we compared nuclear translocation of IRF4 between WT and FoxO1 knockout BMDMs after IL-4 stimulation. IL-4 enhanced nuclear enrichment of IRF4 in WT BMDM but in FoxO1KO BMDM nuclear IRF4 was not detected under the same stimulation (Supple Fig.1D). Additionally, there was significance that an increase in macrophage-derived Th2 promoting chemokines CCL17 and CCL22 was blunted in myeloid-specific FoxO1 deficient mice (Supple Fig.1E). These results confirm that FoxO1 has been regarded as an upstream regulator of IRF4, which is well known to influence Th2/M2 immune response.
Pharmacological FoxO1 inhibition reverses allergic lung inflammation in DRA-challenged mice.
To assess the therapeutic potential of pharmacological inhibition of FoxO1, we used AS1842856 (A highly specific inhibitor of FoxO1 transcription activity). In a chronic murine asthma model, in which mice were challenged with DRA chronically for 36 days. AS1842856 was introduced by a single daily dosage (20 mg/kg) from day 27 until day 35 and the mice were sacrificed at day 36 (illustrated in Fig.2A), in order to determine whether inhibition of FoxO1 could reverse well established asthmatic lung inflammation. As shown, treatment with the FoxO1 inhibitor significantly decreased the total number of cells and alveolar eosinophils in BAL fluid by almost half (Fig.2B). This was confirmed by flow cytometric analyses that showed that treatment with FoxO1 inhibitor markedly reduced alveolar eosinophils (SiglecF+CD11c-) in the WT mice challenged with allergens compared with vehicle treated control mice (Fig.2C). We also examined lung tissues with Periodic acid-Schiff (PAS) staining that detects mucus glycol conjugates in goblet cells within the asthmatic airway epithelium. As seen in tissue sections (Fig.2D), compared with DRA-challenged groups, AS1842856 reduced the DRA-mediated increases in allergic inflammation and goblet cell hyperplasia compared to the controls. Lastly, BALF level of Th2 cytokines (IL-5, IL-13) and chemokines (CCL17/TARC, CCL22/MDC) were statistically significantly reduced in FoxO1 inhibitor-treated DRA-challenged group compared to control (Fig.2E), indicating an important role for FoxO1 in regulating Th2-mediated airway inflammation by governing inflammatory cell recruitment. Because IL-13 is directly associated with secretion of perostin, a matricellular protein in asthmatic airway remodeling21, we measured periostin release in BALF from DRA-exposed mice. As expected, periostin level was abolished in FoxO1 inhibitor-treated DRA-challenged group when compared to control. These data indicate the necessity of FoxO1 for Th2-mediated airway inflammation and provide important supportive data that globally blocking FoxO1 is an effective pharmacologic intervention strategy that hastens the recovery of fully established allergic airway inflammation and chronic airway remodeling in allergen sensitized mice.
Fig.2. FoxO1 inhibitor reverses DRA-induced allergic lung inflammation.
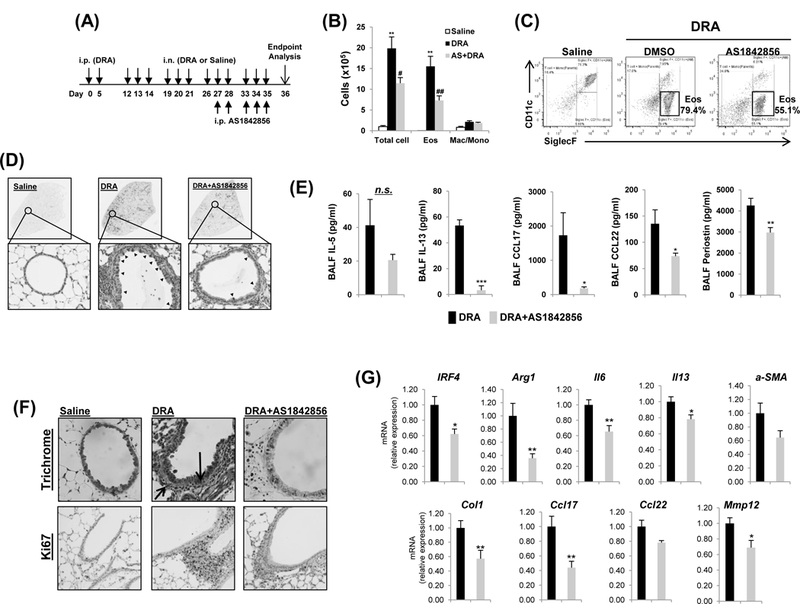
(A) Mice were sensitized and challenged by daily i.n. administration of DRA or saline, 3 days a week for 4 weeks. Prior to allergen challenge, mice were treated with vehicle or 20mg/kg of AS1842856. (B) Eosinophilic inflammation was attenuated in WT mice by pharmacologic inhibition of FoxO1. Total cells and eosinophils influx in BAL fluid were counted, analyzed by flow cytometry. (C) Cell staining for markers of eosinophils (CD45+SiglecF+CD11c-). (D) PAS stained (black arrowheads) lung sections from the mice exposed to DRA. (E) IL-5, IL-13, CCL17, and CCL22 cytokines were quantified with ELISA in BAL fluid (BALF) of AS1842856 pre-treatment group. (N=6–8). (F) Reversible effect of FoxO1 inhibitor on tissue airway remodeling in DRA-induced asthmatic lung. Sections were stained with Masson trichrome (black arrows, peribronchial) and Ki-67 (brown staining) (N=6). (G) The mRNA expression in lung tissues from DRA-induced asthma model (N=4–6). Graphs are plotted as mean ± SE. P values were obtained using a t test. *,#p<0.05, **,##p<0.01, ***p<0.001. AS, AS1842856; i.p., intraperitoneal; i.n., intranasal; Eos, eosinophil; Mac/Mono, macrophage/monocyte; n.s., not significant.
Airway remodeling is a repair process that occurs in response to injury resulting in increased airway obstruction in asthma. Pathways of airway remodeling are activated in lungs of murine asthma model, and there evidence of lung tissue remodeling with a fibrotic component around the airways in trichrome staining. AS1842856 treatment markedly reduced collagen deposition around small bronchi and also reduced proliferation marker protein Ki67-positive cells (Fig.2F). Furthermore, by the qPCR, the expression levels of profibrotic genes were significantly downregulated in the FoxO1 inhibitor-treated mice lung as compared with DRA-challenged mice (Fig.2G). Collectively, these results show that treatment with FoxO1 inhibitor is able to reverse airway fibrosis in a chronic murine model of allergic asthma and thus, is potentially a disease-modifying treatment.
FoxO1 inhibitor has broad immunosuppressive potential in macrophages of mice with DRA-induced asthma.
To elucidate the responses of mouse alveolar macrophages RNA profiles to allergen challenge and candidate genes downstream of FoxO1 involved in the allergic inflammation, immune gene expression was profiled using a NanoString-based immunology panel. We first screened the altered AM RNA levels after allergen challenge and compared them with saline controls. AM from the BAL fluid were purified by cell sorting (CD45+SiglecF+CD11c+sidescatterhigh) after allergen challenge from 10 animals using FACS Aria (Supple Fig.2A). Unsupervised clustering analysis clearly defined a differential Th2/M2 immune gene expression patterns in DRA compared to saline control AM (Supple Fig.2B). Moreover, with only a few exceptions, pretreatment with AS1842856 abrogated these responses, resulting in an expression profile that closely resembled saline controls. We confirmed that the gene expressions of a number of Th2-related genes were reduced in AS1842856-treated AM in response to allergen challenge. Interestingly, genes that are involved in the profibrotic response, such as Tgfb1, Tgfbr1, Tgfbr2, and Fn1 were also dramatically reduced in response to FoxO1 inhibition; suggesting inhibition of FoxO1 could be developed as a treatment to prevent or reverse asthmatic peribronical fibrotic remodeling (Supple Fig.2C).
FoxO1 inhibition in macrophages attenuates allergic airway inflammation.
To assess the functional importance of macrophages FoxO1 in Th2 response, we used LysM-cre driven myeloid-specific FoxO1 knockout mice (FoxO1fl/flLysMcre)5. As shown in Fig.3B, FoxO1 mRNA levels were 14.5-fold higher in alveolar macrophages isolated from FoxO1fl/fl as compared with those from FoxO1fl/flLysMcre. FoxO1fl/flLysMcre mice at 8 weeks of age received multiple intranasal DRA challenges to assess the role FoxO1 deletion on DRA challenged airway inflammation and Th2 responses (Fig.3A). As can be seen in Fig.3C and 3D, the numbers of BAL cells from DRA-challenged FoxO1fl/flLysMcre mice were significantly reduced compared with those from FoxO1fl/fl mice. In sorted AM from BALF after DRA challenge, the expression of FoxO1, CCL17 and CCL22, were significantly downregulated in this FoxO1fl/flLysMcre mice (Fig.3E). Mucus producing goblet cells, shown by PAS staining, were also dramatically reduced in the FoxO1fl/flLysMcre mice than in FoxO1fl/fl mice (Fig.3F). These mice showed a significantly marked deficiency in Th2-cytokine production too in acute asthma model, compared with FoxO1fl/fl mice (Fig.3G). The gene expression levels of characteristic features of asthma from FoxO1fl/flLysMcre mice were decreased in DRA-treated FoxO1fl/fl (Supple Fig.3A). In addition, the expression levels of both IRF4 and Arg1 proteins were much less in FoxO1fl/fl mice asthma group than FoxO1fl/flLysMcre mice in whole lung tissue (Supple Fig.3B). Based on these data, we conclude that FoxO1 depletion in LysM-cre expressing myeloid cells mitigates DRA-induced allergic asthma in mice suggesting a role for macrophage FoxO1 in the pathogenesis of asthma.
FoxO1 deficiency in myeloid cells protects against chronic allergen-induced asthmatic inflammation.
Many asthma patients are repeatedly exposed to multiple inhaled allergens overtime. Thus, we employed a chronic murine asthma model, in which mice were exposed to repeated inhaled allergen, for better understanding the pathologic process of asthma. To investigate whether FoxO1 is required for the development of chronic allergic airway inflammation, we implemented a chronic allergen-exposed FoxO1fl/flLysMcre mice model. In contrast to the acute DRA model, in the chronic model the DRA allergen is administered by nasal insufflation twice weekly for 10 weeks without the use of adjuvant and was followed by a 2 week “resting” period (Fig.4A)3,5,16,18. All sensitized animals had elevated levels of total IgE (Supple Fig.3D). In response to instillation of DRA, significantly decreased eosinophil numbers were detected in the airways of chronic allergen challenged FoxO1fl/flLysMcre mice as compared with FoxO1fl/fl mice (Fig.4B and Supple Fig.3D). Notably, in contrast to the acute model, the levels of infiltrating immune cells were somewhat less in the chronic model. To further examine the chronic condition, we performed histologic examination of lungs from chronic allergen challenged mice and observed that FoxO1fl/flLysMcre mice had much lower numbers of PAS-positive goblet cells, whereas FoxO1fl/fl mice showed a marked increase in PAS-positive cells. Moreover, FoxO1fl/fl mice lungs in addition displayed larger areas of peribronchial connective tissue that were stained positive for collagen, which was reduced in lungs of FoxO1fl/flLysMcre mice (Fig.4C) indicating that FoxO1 in LysM expressing myeloid cells drives peribronchial fibrosis.
In support of this finding, in response to chronic DRA exposure, FoxO1fl/flLysMcre mice had significantly decreased mRNA levels of characteristic Th2 immune response (i.e. Muc5A/C, Irf4, Ccl17) and significantly lower mRNA levels of Th17 response (i.e. Il-6r, Il-17, Il-21, KC) (Fig.4E) than the FoxO1fl/fl mice. Similar to the mRNA data, secreted Th17 cytokines (i.e. Il-17, Il-6, Il-1β) were significantly decreased in BALF from DRA-challenged FoxO1fl/flLysMcre mice compared to the FoxO1fl/fl mice (Fig.4D). Levels of Il-17 mRNA and cytokine were increased in the lungs of chronic DRA-exposed mice compared with saline, and yet acute exposure did not induce Th17 response, which suggests that Th17 has a role in chronic but not acute asthmatic inflammation. As shown in Supple Fig.3C, DRA-challenged FoxO1fl/flLysMcre mice showed a significantly marked decrease in representative Th2/M2-related proteins (IRF4 and Arg1) in whole lung tissue compared with FoxO1fl/fl mice. All together, these data support a conclusion that LysM expressing myeloid cells contribute chronic airway inflammation and asthmatic airway remodeling via a FoxO1 mediated mechanism.
FoxO1fl/flLysMcre mice display reduced activation of the M2 phenotype in AM of mice with chronic allergic airway inflammation.
Using the NanoString assay, we characterized the expression profile of the immune-inflammatory genes in AM (CD45+SiglecF+CD11C+sidescatterhigh) with and without FoxO1. Unsupervised clustering analysis (Fig.5A) clearly defined separate expression patterns in FoxO1fl/fl mice and FoxO1 depleted AM in chronic DRA challenged mice. We found that the expressions of a number of Th2- and profibrotic-response genes were reduced in FoxO1-deficient AM in response to chronic DRA challenge. As shown in Fig.5B and 5C, FoxO1 depletion downregulated immunomodulation factors such as Ccl2, Csf1r, Ccl22, Ccr2, Il10, Il10rb, stat3, stat6 and profibrotic genes including ICAM1, ICAM3, Fn1, Tgfb1, Tgfbi, and upregulated a subset of Th1-like factors such as Csf2, Ifng, Il12b, and Nos2. Interestingly, cytokine receptors that are involved in the Th2 response (IL-10ra and IL-4R) were upregulated in FoxO1fl/fl mice AM but not in FoxO1fl/flLysMcre AM. FoxO1 depleted AM presented low levels of Tgfbr1 and Tgfbr2 mRNA, with 10-fold higher than WT counterpart in response to chronic DRA challenge. Similar results were seen in FoxO1 inhibitor treated DRA-challenged AM in WT mice (Supple Fig.2B), indicating that FoxO1 has a role as a determinant of a profibrotic macrophage transcriptome in response to DRA.
Fig.5. FoxO1 deficient AM express a distinct profile of immune gene in response to DRA.
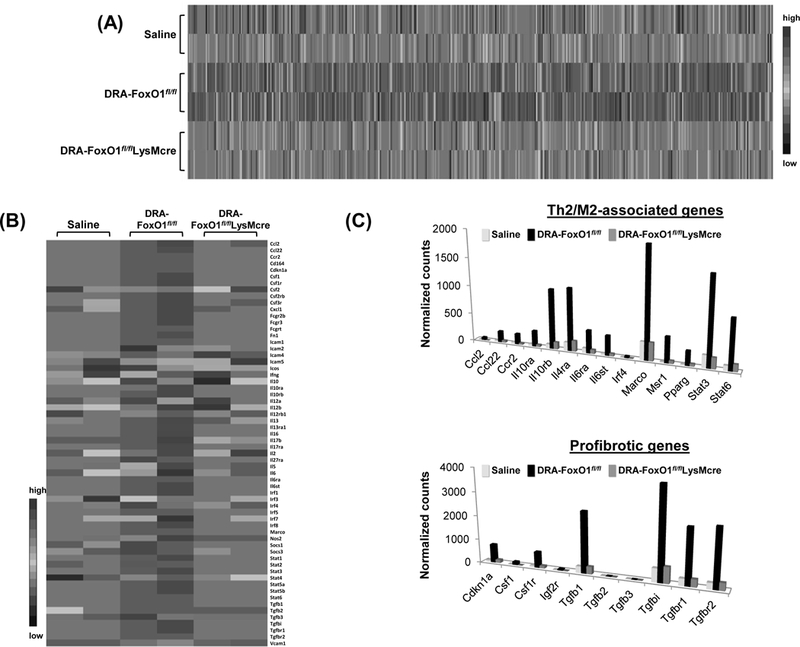
(A) RNA from AM of allergen-challenged mice was subjected to nCounter Nanostring analysis. Gene expression profiles are displayed as a heat map with hierarchical clustering indicated by dendrogram between FoxO1fl/fl and FoxO1fl/flLysMcre. (B) NanoString expression analysis of representative immune genes. (C) Bar graphs show the mean normalized counts of Th2/M2 immune-related and profibrotic mRNAs that are expressed less frequently in FoxO1fl/flLysMcre AM than in FoxO1fl/fl. Data are presented as means ± standard errors of biological duplicates.
Effect of conditional FoxO1 depletion on asthmatic inflammation.
To evaluate if an established asthma phenotype can be mitigated by inactivation of myeloid specific FoxO1, we generated FoxO1 conditional inactivation mice using Cre driven by the colony-stimulating factor-1 receptor promotor (Csf1r), crossed with FoxO1fl/fl (Supple Fig.4A). Inducible ablation of FoxO1 was achieved in cultured BMDMs treated with 4-hydroxytamoxifen (Supple Fig.4B). These FoxO1-inactivated BMDMs did not enhance M2-related IRF4, Fizz1, and Arg1 genes, when compared to control BMDMs (Supple Fig.4C). Cre expression was induced in double transgenic adult mice by applying tamoxifen dissolved in corn oil by subcutaneous injection for 5 consecutive days. In contrast to vehicle treated mice, in vivo injection of tamoxifen substantially down-regulated FoxO1 expression in AM (SiglecF+CD11c+sidescatterhigh) by 50–60% (Supple Fig.4D). Once this was determined, we subjected the mice to the DRA protocol (Supple Fig.4E) to evaluate whether or not tamoxifen-induced FoxO1 depletion was associated with changes in the asthmatic phenotype. Known as, tamoxifen treatment had no significant effect on the asthma phenotype in WT mice22. Interestingly, we found that loss of FoxO1 by tamoxifen caused an additional decline in total BAL cells the percentage of eosinophils (from 60.5% to 48.9%) in BAL was reduced in the FoxO1fl/flCsf1rcre mice. Similarly, Th2 cytokines IL-5 and IL-13 in BALF were significantly reduced in lungs from DRA-challenged FoxO1fl/flCsf1rcre mice compared with that of WT mice (Supple Fig.4F). Consistent with this reduced Th2 inflammation, DRA/Tamoxifen-treated FoxO1fl/flCsf1rcre mice showed decreased Th2/M2 markers (Il-5, Il-13, CCL17, IRF4, and Arg1) in mRNA and protein levels from lungs of tamoxifen/DRA treatment (Supple Fig.4G and 4H).
Adoptive transfer of lung macrophages from FoxO1CAfl/flLysMcre accentuates DRA-induced Th2 response.
To determine whether the specific overexpression of FoxO1 in macrophages would confer to increase Th2 immune response to DRA, lung resident macrophages were isolated from collagenase treated lung tissue of the FoxO1CAfl/fl (WT) and FoxO1CAfl/flLysMcre (Tg) mice. These cells were adoptively transferred to WT recipient mice that subsequently received intranasal DRA challenges to induce airway inflammation as illustrated (Fig.6A). As shown in Fig.6B, levels of IL-5, CCL17, and CCL22 were significantly higher in BALF from WT recipient mice from Tg donor mice (referred WT+Tg in graph) as compared with WT donor mice (WT+WT in graph). Similarly, lung homogenate level of IL-5, IL-13, CCL17, and CCL22 were significantly higher in WT recipient mice from Tg donor mice than in counterpart mice (Fig.6C). Furthermore, the mRNA expression of CCL17 and CCL22, macrophage-derived specific Th2 chemokines in lungs of recipients of WT mice from Tg donor mice were found to be increased compared to that in counterpart mice (Fig.6D). Lung histology showed an induction in inflammatory cell infiltration and goblet cell hyperplasia in recipients of WT mice from Tg donor mice (WT+Tg) as compared with WT donor mice (WT+WT, Fig.6E). Thus, these experiments are consistent with the conclusion that an increase FoxO1 in macrophages results in a marked accentuation of asthmatic airway inflammation.
DISCUSSION
We and others have shown that alternatively activated pro-asthmatic macrophages contribute to Th2 immune allergic response4,7,23. In this study, we have used a murine model of asthma, which uses exposure to three allergens, to determine the contribution of lung macrophages in allergic inflammation in the lung. Our data show that the transcription factor, FoxO1, has a dominant effect on determining the inflammatory phenotype in macrophages that contributes to asthma pathogenesis. The scientific premise of our study was based on the finding that FoxO1 expression was increased in lung macrophages of mice after allergen exposure and human asthmatic patients compared to controls (Fig.1). Of note, our clinical data show that the levels of FoxO1 and IRF4 genes are significantly elevated the in airway macrophages of patients with mild asthma in response to subsegmental bronchial allergen challenge. Our data further show that a highly selective pharmacologic FoxO1 inhibitor reversed eosinophilic lung infiltration in allergen challenged mice. We also employed three independent genetic strategies to confirm these data. We first showed that the Th2 immune response to DRA that are associated airway inflammation, goblet cell hyperplasia, and cytokine/chemokine production in the FoxO1fl/flLysMcre mice was markedly attenuated indicating that LysM-Cre expressing myeloid cells were involved. Next we showed that passive adoptive transfer of lung macrophages LysM-Cre driven FoxO1 transgenic mice had a marked accentuation of chronic airway changes. Finally, we used novel conditional Csf1r-Cre driven FoxO1 deficient mice and confirmed that there was attenuation of various M2 gene expression and chronic airway changes. Our data indicate that both pharmacologic inhibition and genetic ablation of FoxO1 results in an attenuation of IRF4, a transcriptional cofactor, which many other laboratories have linked to the M2 macrophage phenotype24,25. Most remarkably, our data show that blocking FoxO1 in mice that have been repeatedly challenged with allergen inhibits profibrotic gene expression by macrophages and reverses peribronchial fibrosis in established airway remodeling.
Numerous studies have indicated that FoxO1 is a mediator of major upstream inflammatory signals26,27. Functionally, FoxO1 has a direct role in regulating inflammation by signal transduction, transcriptional regulation, and in a combinatorial manner in conjunction with other transcription factors. Interestingly, FoxO1 possess bifunctional role in anti- and pro-inflammatory properties depending on context and binding partners9. Recent literature has reported that the expression of FoxO1 enhances the development of Th9 cells in allergic inflammation28. Notably, IL-9 was shown to be associated with Th2 cells in allergic inflammation models29. Numerous other reports demonstrate the role of FoxO1 as an modulator of inflammation in adpocyte and macrophages that has a downstream impact on effector organs27,30,31. FoxO1 could have a role in the pathogenesis of allergic inflammation in both macrophages and T cells. However, considering the potential mechanism by which FoxO1 contributes to phenotypic plasticity of macrophages, we were interested in the role of pulmonary macrophages subsets in DRA-induced asthmatic inflammation. Pro-asthmatic M2 macrophages phenotypes is mainly arbitrated through specific IRF4 transcription factor pathway32, therefore FoxO1 could regulate a key checkpoint of the IRF4 pathway in macrophages. Our data are consistent with this literature that FoxO1 regulates the macrophage inflammatory phenotype through an IRF4-dependent manner in allergic inflammation and in asthma biology28,33. Mechanistically, FoxO1 is known to bind to IRF4 promoter and mediate transactivation34. Our data adds to the literature by showing that FoxO1 is required for the expression of IRF4 in IL-4-treated human alveolar macrophages, as inhibition of FoxO1 in human cells suppressed IRF4. Furthermore, we have shown that this pathway is “druggable” since we observed that the treatment with a highly selective FoxO1 inhibitor blocks IL-4-mediated IRF4 expression and inhibits M2 macrophages activation. It is highly likely that FoxO1 and IRF4 synergistically enhance the transactivation of Th2 cytokines due to FoxO1-IRF4 protein interaction. Our data, in combination with the literature, suggests that FoxO1 is upstream to IRF4 in the IL-4 mediated immune cell response. We have shown that, FoxO1 depletion in the myeloid cell lineage shifting macrophage activation toward a pro-asthmatic phenotype which negatively regulated the eosinophilic inflammatory reactions in DRA-challenged mice.
Th17 biased-response has been implicated in chronic allergic airway inflammation and is increased in paitents with severe asthma35. Eosinophilia is known to be predominated in acute asthmatic inflammation, while IL-17 and neutrophilic cytokines/chemokines increased in chronic inflammatory response in a classic murine asthma model36. Therefore we measured production of further cytokines IL-17, IL-6, and IL-1β in our chronic murine asthma model and IL-17, IL-17a, IL-17f, IL-21, IL-6, KC, and IL-1β gene expressions. These Th17 cytokines were undetectable in BAL fluid, but were released in lung homogenate. It is clear that Th17 immune response in our model could contribute to airway remodeling by increased goblet cell hyperplasia and airway smooth muscle mass (Fig.4). Our data indicate that FoxO1-LysM conditional deficient mice suppressed the severity of Th17 (IL-17 and IL-6) and Th2 (IRF4 and CCL17) responses, respectively.
The pathogenesis of asthma is mediated by many different cell types in the lungs, including epithelial, smooth muscle, and innate and adaptive immune cells. Recently, it has been reported that there is a pro-asthmatic macrophage phenotype that has prominent features that are consistent with alternatively activated M2 macrophages, activating with IRF4, Arg1, and Fizz16. Accumulating data have already suggested that there is a contribution of pro-asthmatic M2 macrophages to the cell and molecular pathobiology of airway inflammation and even chronic airway remodeling18,37. Alternatively activated macrophages are known to be participated in the repair of the damaged lung, but as our data show M2-like macrophages so are also a source of excessive production of pro-fibrotic factors that can contribute to the chronic airway remodeling that characterizes severe asthma37. Our data indicates that FoxO1 contributes to the maintenance of alternatively activated profibrotic M2 lung macrophages that are involved in the progression of asthmatic airway remodeling that leads to chronic pulmonary disability. Persistent allergic inflammation leads to remodeling responses in the airways of asthmatic patients, which affect millions of people comprehensively and represent some of the leading causes of chronic morbidity. Macrophages produce various mediators that could facilitate structural alteration of asthmatic airway6. For example, macrophage-derived transforming growth factor-β1 (TGF-β1) contributes to repair process synthesizing interstitial fibrillar collagen in myofibroblasts38. Macrophages are also a major source of a secreted glycoprotein, YKL-40, which is elevated in severe asthmatic patients39 and macrophages are a key source of factors that regulate fibroblast growth, differentiation and survival, such as TGF-β, PDGF and several members of the fibroblast growth factor family40,41.
In addition to macrophages, FoxO1 has critical role in regulation of T cell homeostasis and dendritic cells, which are all involved in allergic inflammation. Because dendritic cells are clearly involved in Th2 immune responses, the involvement of FoxO1-deficient dendritic cells cannot be excluded in our asthmatic models. Our previous study indicate that the depletion of macrophages resulted in significant attenuation of DRA-induced asthmatic inflammation by using Macrophage Fas-Induced Apoptosis (MAFIA) transgenic mice16, suggesting a non-redundant role of macrophages in asthmatic inflammation. To further confirm whether FoxO1-overexpressed macrophages are sufficient to confer such aggravating DRA-induced allergic asthma, we performed adoptive-transfer experiments. Using WT mice adoptively transferred with FoxO1CAfl/flLysMcre macrophages, in which FoxO1-mediated macrophages activation was enhanced, we demonstrated that FoxO1-dependent M2-like macrophage activation contributes to asthmatic lung inflammation and airway remodeling, which could be exploited as a potential therapeutic targets for asthma. Chemokines produced by exogenous FoxO1+ pro-asthmatic macrophages such as CCL17 and CCL22 modulate leukocyte trafficking that could enhance the mucus hypersecretion, as determined by using PAS staining to the site of inflammation. By contrast, in our passive adoptive transfer model, we did not see a large difference in the number of eosinophils in the lungs or a difference in the amount of IL-4 or IL-5 in the BAL fluid between WT or FoxO1+ transfer groups suggesting that the Th2 cells were being recruited and activated sufficiently in both settings. We have also demonstrated similar findings in DRA-induced allergic FoxO1CAfl/flLysMcre mice, in which greater abundance of alternatively activated macrophages in the lungs of these mice (unpublished observation SC and JWC). A remaining question is which lung macrophages subset can be assumed to exert asthmatic airway remodeling. Resident lung macrophages comprise two distinct groups: alveolar macrophages (AMs) and interstitial macrophages (IMs). Future study should continue to identify the role of various macrophage subpopulations and their associated phenotypes in type 2 fibrosis accompanying airway remodeling in the lung. This will allow for a better understanding of macrophage activation status, and potentially help identify new markers and their associated functions in pathophysiology of asthmatic airway remodeling. To further determine whether FoxO1 deletion in lung macrophages can relieve DRA-induced airway remodeling in clinically relevant concept, we employed the tamoxifen-inducible myeloid FoxO1 depleted mouse model (FoxO1fl/flCsf1rcre). A key advantage of the Csf1r-iCre mouse is that it permits both cell-specific and temporal control of Cre-mediated recombination. This is an important potential therapeutic concept because prophylactic treatment with a FoxO1 inhibitor would only have a limited application, where as a disease modification which returns a damaged lung to normal would have a very high impact on human health. Our data indicate that loss of functional FoxO1 in lung macrophages results in a reduction in asthmatic airway inflammation. These genetically defined mice will be a powerful tool to study the role of FoxO1-mediated genes in recruited or resident pulmonary macrophages in the context of Th2-driven asthma pathogenesis. Additional investigation into the role for FoxO1 in cell-specific targets using this new conditional mice model will afford valuable insights to define the pathophysiology of asthmatic airway remodeling. Employing genetic approaches to target FoxO1 activity enabled us to perform mechanistic studies without the confounding possible off-target effects of using FoxO1 inhibitor. Future studies are necessary to determine the best FoxO1 inhibitors that are therapeutic without off target deleterious side effects.
In summary, we demonstrated the first time that FoxO1 acts as a critical transcription factor in the development of allergic lung inflammation and airway remodeling through polarizing and inducing the expression of pro-allergic and profibrotic factors. Limitation to the current study includes the relatively small number of human samples, however our results are consistent with the expression of FoxO1 in macrophages is required for the pro-asthmatic phenotype which contributes to aggravating the inflammatory reaction of DRA-induced allergic asthma. Very recent reports have also demonstrated a crucial role of FoxO1 in other type of asthmatic inflammation28,42. Our data, in combination with the literature, suggest that the impact of FoxO1 on the macrophage inflammatory phenotype is transduced through expression of the binding partner, IRF4. Our finding indicates that FoxO1 is a critical target for inhibiting myeloid-directed therapeutic strategies that attenuate allergic inflammatory diseases.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by grants from the National Institutes of Health (R01HL075557, R01HL137224, 5R01HL126852) and American Lung Association Biomedical Research Grant (RG-416620). The authors thank the generous subjects with asthma who volunteered for the SBP-AG protocol and the staff of the Clinical Interface Core at the University of Illinois Center for Clinical and Translational Science (UIC-CCTS) for assistance with patient recruitment, screening, and performing bronchoscopy. We thank Andrew Feldman (Mayo Clinic) for the IRF4-luciferase reporter vector. In addition, we would like to thank analytical flow cytometry core (P30CA016058) and OSU genomics shared resources for their technical assistance.
Footnotes
CONFLICT of INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
- 1.Tjota MY, Williams JW, Lu T, et al. IL-33-dependent induction of allergic lung inflammation by FcgammaRIII signaling. J Clin Invest. 2013;123(5):2287–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Draijer C, Peters-Golden M. Alveolar Macrophages in Allergic Asthma: the Forgotten Cell Awakes. Curr Allergy Asthma Rep. 2017;17(2):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park GY, Lee YG, Berdyshev E, et al. Autotaxin production of lysophosphatidic acid mediates allergic asthmatic inflammation. Am J Respir Crit Care Med. 2013;188(8):928–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Girodet PO, Nguyen D, Mancini JD, et al. Alternative Macrophage Activation Is Increased in Asthma. Am J Respir Cell Mol Biol. 2016;55(4):467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chung S, Lee TJ, Reader BF, et al. FoxO1 regulates allergic asthmatic inflammation through regulating polarization of the macrophage inflammatory phenotype. Oncotarget. 2016;7(14):17532–17546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park GY, Christman JW. Hidden in Plain Sight: The Overlooked Role of Pulmonary Macrophages in the Pathogenesis of Asthma. Am J Respir Cell Mol Biol. 2016;55(4):465–466. [DOI] [PubMed] [Google Scholar]
- 7.Novak N, Peng WM, Bieber T, Akdis C. FcepsilonRI stimulation promotes the differentiation of histamine receptor 1-expressing inflammatory macrophages. Allergy. 2013;68(4):454–461. [DOI] [PubMed] [Google Scholar]
- 8.Chung S, Ranjan R, Lee YG, et al. Distinct role of FoxO1 in M-CSF- and GM-CSF-differentiated macrophages contributes LPS-mediated IL-10: implication in hyperglycemia. J Leukoc Biol. 2015;97(2):327–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sundaresan S, Puthanveetil P. Is FoxO1 the culprit, partner in crime, or a protector in systemic inflammation? Am J Physiol Cell Physiol. 2017;313(2):C239–C241. [DOI] [PubMed] [Google Scholar]
- 10.Kawano Y, Nakae J, Watanabe N, et al. Loss of Pdk1-Foxo1 signaling in myeloid cells predisposes to adipose tissue inflammation and insulin resistance. Diabetes. 2012;61(8):1935–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaza V, Bandi V, Guntupalli KK. Acute severe asthma: recent advances. Curr Opin Pulm Med. 2007;13(1):1–7. [DOI] [PubMed] [Google Scholar]
- 12.Prieto J, Lensmar C, Roquet A, et al. Increased interleukin-13 mRNA expression in bronchoalveolar lavage cells of atopic patients with mild asthma after repeated low-dose allergen provocations. Respir Med. 2000;94(8):806–814. [DOI] [PubMed] [Google Scholar]
- 13.Kolodsick JE, Toews GB, Jakubzick C, et al. Protection from fluorescein isothiocyanate-induced fibrosis in IL-13-deficient, but not IL-4-deficient, mice results from impaired collagen synthesis by fibroblasts. J Immunol. 2004;172(7):4068–4076. [DOI] [PubMed] [Google Scholar]
- 14.Malavia NK, Mih JD, Raub CB, Dinh BT, George SC. IL-13 induces a bronchial epithelial phenotype that is profibrotic. Respir Res. 2008;9:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Murray LA, Argentieri RL, Farrell FX, et al. Hyper-responsiveness of IPF/UIP fibroblasts: interplay between TGFbeta1, IL-13 and CCL2. Int J Biochem Cell Biol. 2008;40(10):2174–2182. [DOI] [PubMed] [Google Scholar]
- 16.Lee YG, Jeong JJ, Nyenhuis S, et al. Recruited Alveolar Macrophages, in Response to Airway Epithelial-Derived Monocyte Chemoattractant Protein 1/CCL2, Regulate Airway Inflammation and Remodeling in Allergic Asthma. Am J Respir Cell Mol Biol. 2015;52(6):772–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qian BZ, Li J, Zhang H, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qian F, Deng J, Lee YG, et al. The transcription factor PU.1 promotes alternative macrophage polarization and asthmatic airway inflammation. J Mol Cell Biol. 2015;7(6):557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song MA, Brasky TM, Marian C, et al. Racial differences in genome-wide methylation profiling and gene expression in breast tissues from healthy women. Epigenetics. 2015;10(12):1177–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan P, Guan H, Xie L, et al. FOXO1 inhibits osteoclastogenesis partially by antagnozing MYC. Sci Rep. 2015;5:16835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corren J, Lemanske RF, Hanania NA, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365(12):1088–1098. [DOI] [PubMed] [Google Scholar]
- 22.Chen M, Hegde A, Choi YH, et al. Genetic Deletion of beta-Arrestin-2 and the Mitigation of Established Airway Hyperresponsiveness in a Murine Asthma Model. Am J Respir Cell Mol Biol. 2015;53(3):346–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li K, Zhang Y, Liang KY, et al. Rheb1 deletion in myeloid cells aggravates OVA-induced allergic inflammation in mice. Sci Rep. 2017;7:42655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Satoh T, Takeuchi O, Vandenbon A, et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol. 2010;11(10):936–944. [DOI] [PubMed] [Google Scholar]
- 25.Huang SC, Smith AM, Everts B, et al. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity. 2016;45(4):817–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown J, Wang H, Suttles J, Graves DT, Martin M. Mammalian target of rapamycin complex 2 (mTORC2) negatively regulates Toll-like receptor 4-mediated inflammatory response via FoxO1. J Biol Chem. 2011;286(52):44295–44305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fan W, Morinaga H, Kim JJ, et al. FoxO1 regulates Tlr4 inflammatory pathway signalling in macrophages. EMBO J. 2010;29(24):4223–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malik S, Sadhu S, Elesela S, et al. Transcription factor Foxo1 is essential for IL-9 induction in T helper cells. Nat Commun. 2017;8(1):815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Veldhoen M, Uyttenhove C, van Snick J, et al. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008;9(12):1341–1346. [DOI] [PubMed] [Google Scholar]
- 30.Siqueira MF, Li J, Chehab L, et al. Impaired wound healing in mouse models of diabetes is mediated by TNF-alpha dysregulation and associated with enhanced activation of forkhead box O1 (FOXO1). Diabetologia. 2010;53(2):378–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.I OS, Zhang W, Wasserman DH, et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat Commun. 2015;6:7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eguchi J, Kong X, Tenta M, Wang X, Kang S, Rosen ED. Interferon regulatory factor 4 regulates obesity-induced inflammation through regulation of adipose tissue macrophage polarization. Diabetes. 2013;62(10):3394–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Penberthy KK, Buckley MW, Arandjelovic S, Ravichandran K. Ex vivo modulation of the Foxo1 phosphorylation state does not lead to dysfunction of T regulatory cells. PLoS One. 2017;12(3):e0173386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eguchi J, Wang X, Yu S, et al. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 2011;13(3):249–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Newcomb DC, Peebles RS Jr, Th17-mediated inflammation in asthma. Curr Opin Immunol. 2013;25(6):755–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao J, Lloyd CM, Noble A. Th17 responses in chronic allergic airway inflammation abrogate regulatory T-cell-mediated tolerance and contribute to airway remodeling. Mucosal Immunol. 2013;6(2):335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moreira AP, Hogaboam CM. Macrophages in allergic asthma: fine-tuning their pro- and anti-inflammatory actions for disease resolution. J Interferon Cytokine Res. 2011;31(6):485–491. [DOI] [PubMed] [Google Scholar]
- 38.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chupp GL, Lee CG, Jarjour N, et al. A chitinase-like protein in the lung and circulation of patients with severe asthma. N Engl J Med. 2007;357(20):2016–2027. [DOI] [PubMed] [Google Scholar]
- 40.Wynn TA, Vannella KM. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity. 2016;44(3):450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gieseck RL 3rd, Wilson MS, Wynn TA . Type 2 immunity in tissue repair and fibrosis. Nat Rev Immunol. 2018;18(1):62–76. [DOI] [PubMed] [Google Scholar]
- 42.Buttrick TS, Wang W, Yung C, et al. Foxo1 Promotes Th9 Cell Differentiation and Airway Allergy. Sci Rep. 2018;8(1):818. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.