

Abstract
Background and Purpose
The expression of the human organic cation transporter‐1 (hOCT1, gene SLC22A1) is reduced in hepatocellular carcinoma (HCC). The molecular bases of this reduction and its relationship with the poor response of HCC to sorafenib were investigated.
Experimental Approach
HCC transcriptomes from 366 samples available at TCGA were analysed. Alternative splicing was determined by RT‐PCR. The role of miRNAs in SLC22A1 downregulation was investigated. Expression of Oct1 was measured in rodent HCC models (spontaneously generated in Fxr ‐/‐ mice and chemically‐induced in rats). hOCT1 was overexpressed in human hepatoma cells (HuH7 and HepG2). Sorafenib and regorafenib uptake was determined by HPLC‐MS/MS.
Key Results
hOCT1 overexpression enhanced sorafenib, but not regorafenib, quinine‐inhibitable uptake by hepatoma cells. In rodent HCC, Oct1 was downregulated, which was accompanied by impaired sorafenib uptake. In mice with s.c.‐implanted HCC, sorafenib inhibited the growth of hOCT1 overexpressing tumours. In human HCC, hOCT1 expression was inversely correlated with SLC22A1 promoter methylation, whereas demethylation with decitabine enhanced hOCT1 expression in hepatoma cells. Increased proportion of aberrant hOCT1 mRNA variants was found in HCC samples. In silico analysis identified six miRNAs as candidates to target hOCT1 mRNA. When overexpressed in HepG2 cells a significant hOCT1 mRNA decay was induced by hsa‐miR‐330 and hsa‐miR‐1468. Analysis of 39 paired tumour/adjacent samples from TCGA revealed that hsa‐mir‐330 was consistently upregulated in HCC.
Conclusion and Implications
Impaired hOCT1 expression/function in HCC, in part due to epigenetic modifications, plays an important role in the poor pharmacological response of this cancer to sorafenib.
Abbreviations
- 2‐AAF
2‐acetaminofluorene
- DAC
decitabine (5‐aza‐2′‐deoxycytidine)
- DEN
diethyl nitrosamine
- HCC
hepatocellular carcinoma
- MOC
mechanisms of chemoresistance
- NT
non‐tumour
- OCT1
organic cation transporter 1
- ORF
open reading frame
- RBPs
RNA binding proteins
- T
tumour
- TCGA
the Cancer Genome Atlas
- TKI
tyrosine kinase inhibitor
- TSS
transcriptional start site
- UTR
untranslated region
Introduction
Hepatocellular carcinoma (HCC), the most frequent type of liver cancer, constitutes one of the three leading causes of cancer‐related death worldwide (Torre et al., 2016). Albeit surgery may be curative in the early stages, primary liver tumours are often silent, which leads to a late diagnosis when surgical resection cannot be recommended (Marin et al., 2010; Kudo, 2017). Although in patients with advanced unresectable HCC, many drugs, such as doxorubicin, cisplatin and tamoxifen, have been assayed; several studies have shown that none of them is efficacious (Llovet and Bruix, 2003). Sorafenib, a multi‐tyrosine kinase inhibitor (TKI), has been approved for the treatment of advanced HCC, showing a beneficial effect by slowing down the tumour progression. However, the impact of sorafenib treatment on overall survival of HCC patients is very limited (Llovet et al., 2008). The main reason for the low efficacy of antitumour drugs in HCC is the pre‐existence and/or rapid acquirement of powerful mechanisms of chemoresistance (MOC) (Marin et al., 2010). Among different types of MOC, an important role is played by processes leading to a reduction in intracellular concentrations of active drugs (Marin et al., 2014). This can be caused by dysregulation of expression of genes encoding uptake/efflux transporters (MOC‐1) and enzymes involved in prodrug activation or metabolic inactivation of drugs (MOC‐2) but also to the appearance of genetic variants, which results in altered function (enhanced or impaired) of proteins involved in MOC‐1 and MOC‐2 (Marin et al., 2012). This is the case of the human organic cation transporter 1 (hOCT1), encoded by the SLC22A1 gene, which plays a major role in the transport of many drugs including metformin (Shikata et al., 2007), platinum derivatives (Zhang et al., 2006), anthracyclines (Koepsell et al., 2007) and TKIs, such as sorafenib (Minematsu and Giacomini, 2011; Herraez et al., 2013). Importantly, the expression of hOCT1 variants markedly determines the biodistribution of these drugs (Lozano et al., 2013). Thus, HCC cells expressing functional hOCT1 have an increased uptake and a better response to sorafenib (Herraez et al., 2013). Unfortunately, deficient hOCT1 expression has been suggested to be a common trait in the multidrug resistance (MDR) phenotype shared by the three major types of liver cancer, that is, HCC, cholangiocarcinoma and hepatoblastoma (Martinez‐Becerra et al., 2012; Herraez et al., 2013). hOCT1 expression is regulated by a complex series of events that take place at both transcriptional and post‐transcriptional levels. Although DNA hypermethylation has been described for SLC22A1 (Schaeffeler et al., 2011) the global mechanisms accounting for attenuated hOCT1 expression and the appearance of aberrant transcripts in HCC have not been fully characterized. The goal of this study was to elucidate the molecular bases accounting for the impaired function of hOCT1 in HCC and their relationship with the poor response of this cancer to sorafenib.
Methods
Human samples and data
Samples from surgically resected HCC and paired adjacent non‐tumour tissue (NT, n = 19) were obtained from the Tumour Biobank of the University Hospital of Salamanca (Salamanca, Spain). None of these patients had received chemotherapy prior to the resection. Information regarding age, gender, tumour stage and underlying diseases of these patients is included in Supporting Information Table S1. The Ethical Committees for clinical research of this institution approved the research protocols, and all patients signed written consents for the use of their samples in biomedical research.
In addition, we analysed gene expression and DNA methylation data from The Cancer Genome Atlas (TCGA) on 366 HCC (Cancer Genome Atlas Research Network. Electronic address and Cancer Genome Atlas Research, 2017, RRID:SCR_003193). We also included in our study the 49 cases out of these 366 samples that had information available regarding paired adjacent NT tissue. Only 39 of them had information on miRNA expression. In gene expression analysis, level 3 RNA‐seq data were quantified in reads per kilobase per million mapped reads (RPKM) and compared between groups using Mann–Whitney two‐tailed unpaired t‐test. In DNA methylation analysis, level 1 Infinium 450k (Illumina) data were extracted and processed using RnBeads package (including removal of poor quality probes, sex chromosome probes and probes mapping to genomic regions containing >2 single nucleotide polymorphisms (SNPs), followed by BMIQ normalization) (Assenov et al., 2014). DNA methylation was quantified using β‐value metric, ranging from 0 to 1 (0 to 100% methylation). Differentially methylated probes between two groups were defined as those with a β‐value difference equal to or greater than 0.2 and a P‐value < 0.05 (Mann–Whitney two‐tailed unpaired t‐test). Correlation between DNA methylation and gene expression levels was assessed by Spearman correlation test.
Animals and experimental groups
Male Wistar rats (150–180 g) were obtained from the University of Salamanca Animal House (RGD Cat# 13508588, RRID:RGD_13 508 588), whereas 8‐week‐old female immunodeficient nude mice (Swiss nu/nu) were purchased from Charles River Laboratories (Barcelona) (MGI Cat# 2668662, RRID:MGI:2668662) and were maintained under pathogen‐free conditions and handled under stringent sterile conditions. Fxr knockout (Fxr −/−) mice B6.129X1(FVB)‐Nr1h4tm1Gonz/J (IMSR Cat# JAX:007214, RRID:IMSR_JAX:007214) were obtained from Jackson Laboratory (Bar Harbor, Maine, USA). Animals were housed under controlled conditions of temperature, humidity and light cycle (12 h/12 h) and fed on standard rodent chow (Panlab) and water ad libitum. Methods were performed in accordance with the relevant guidelines and regulations, and all protocols were approved by the University of Salamanca Bioethical Committee, which are in agreement with requirements for studies using animals established by this journal. Rats and mice were selected because appropriate models of chemically induced and spontaneously generated HCC, respectively, had been well‐characterized previously. The immunodeficient nude mouse is a common model used in xenograft protocols. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010).
Hepatocarcinogenesis was chemically induced in rats as previously described (Monte et al., 1999) using the Solt and Farber protocol (Solt et al., 1977). In brief, diethyl nitrosamine (DEN) was administered on day 1 (200 mg·kg−1 b.w., i.p.), 2‐acetaminofluorene (2‐AAF) was injected twice a week from days 14 to 35 (50 mg·kg−1 b.w., s.c.), and on day 21, the animals underwent 2/3 partial hepatectomy. Experiments to study sorafenib content in tumour (T) and NT liver tissues were carried out 32 weeks after starting the protocol. The rats were anaesthetized with sodium pentobarbital (50 mg·kg−1 b.w., i.p.); a catheter was implanted into the left jugular vein, and, after a resting period of 15 min, sorafenib was administered (i.v.) as a bolus (10 mg·kg−1 b.w.). After 1 h, pieces of T and NT liver tissue were obtained and immediately placed in RNAlater (Thermo Fisher) or immersed in liquid nitrogen and stored at −80°C until used for sorafenib content determination. In Fxr −/− mice, spontaneous development of HCC occurred after 14 months of age (Yang et al., 2007). Sorafenib was injected i.p. (10 mg·kg−1 b.w.), and 1 h later, T and adjacent NT liver tissue samples were obtained and processed as described above for rat tissues. In both models, serum, kidney and muscle samples were obtained to compare sorafenib content.
To study the impact of hOCT1 expression on sorafenib response in vivo, nude mice were anaesthetized with isoflurane, and 107 HepG2 cells stably expressing hOCT1 after transduction with lentiviral vectors or Mock cells (transduced with empty vectors) were suspended in 100 μL Matrigel (BD Biosciences, Madrid, Spain) and s.c. implanted into distant locations of the flanks of each mouse. When the tumours reached 15 mm3, approximately 3 weeks later, the animals were randomly divided into two groups, which received sorafenib (10 mg·kg−1 b.w., i.p.) or the vehicle alone (sterile saline) three times per week. At the end of the experimental period (day 28), the animals were killed with an overdose of pentobarbital, and tumours were measured with a sliding calliper to calculate the volume using the formula (length × width2)/2.
Gene expression studies
Total RNA extraction from tissues and cells was performed using the RNAspinMini kit (GE Healthcare Life Sciences, Barcelona, Spain). RT was carried out using the ‘SuperScript® VILO™ cDNA Synthesis Kit’ (Invitrogen, Thermo Fisher) or the ‘High capacity reverse transcription kit’ (Applied Biosystems, Thermo Fisher). Real‐time quantitative PCR (RT‐qPCR) was performed using AmpliTaq Gold polymerase (Applied Biosystems) in an ABI Prism 7300 Sequence Detection System (Applied Biosystems) and specific primers (Supporting Information Table S2). GAPDH/HPRT1, Gapdh or β‐actin mRNA abundance for human, mouse and rat samples, respectively, was determined for normalization. Steady‐state mRNA levels of these genes under our experimental conditions were previously confirmed. Liver RNA from the appropriate species was used as a calibrator in each reaction. Detection of amplification products was carried out using SYBR Green I. Gene expression values were calculated by the ∆∆Ct method.
Immunoblot and immunofluorescence analyses
Crude suspensions of cell membranes were obtained in 10 mM HEPES‐Tris by use of a freezing/thawing procedure and dilution (1:1) with buffer A (500 mM sucrose, 0.4 mM CaCl2, 20 mM MgCl2, 200 mM KNO3 and 10 mM HEPES‐Tris), followed by centrifugation at 230 000× g. The pellet was suspended in buffer B (250 mM sucrose, 0.2 mM CaCl2, 10 mM MgCl2, 100 mM KNO3 and 10 mM HEPES‐Tris) containing protease inhibitors cocktail (1%). Immunoblotting analyses were carried out in 7.5% SDS‐PAGE, loading 50 μg of protein per lane and transferred onto nitrocellulose membranes (Bio‐Rad, Hercules, CA, USA). Primary antibodies against hOCT1 (1:1000) (Abcam Cat# ab118539, RRID:AB_2754976) and Na+/K+‐ATPase (1:500) (Abcam Cat# ab2871, RRID:AB_303373) were diluted in PBS‐Tween with 3 and 5% milk respectively. Anti‐rabbit (Abcam Cat# ab6721, RRID:AB_955447) and anti‐mouse (Santa Cruz Biotechnology Cat# sc‐516 102, RRID:AB_2687626) IgG HRP‐linked secondary antibodies were diluted 1:2000. Bands of immunoreactive proteins were visualized using enhanced chemiluminescence (ECL; Amersham Pharmacia Biotech, Freiburg, Germany).
To carry out immunofluorescent analysis, cultured cells were fixed in ice‐cold methanol for 3 min. After being blocked with PBS supplemented with 5% FBS for 30 min, the cells were incubated for 1 h with primary antibodies against hOCT1 or Na+/K+‐ATPase. Secondary antibodies [anti‐mouse or anti‐rabbit Alexa 594‐ or Alexa 488‐conjugated antibodies, both from Life Technologies, Thermo Fisher (Carlsbad, CA)] were used as appropriate. The nuclei were stained with DAPI. The images were obtained using a confocal microscope (Leica TCS SP2).
In vitro cytostatic assay
To measure sorafenib and regorafenib antiproliferative activities, monoclonal selected HepG2 and HuH7 cells transduced with lentiviral vectors (pWPI) either empty (Mock) or containing hOCT1 open reading frame (ORF) were seeded in 96‐well plates (≈5 × 103 cells per well). After 24 h, cells were incubated with 5 μM sorafenib (with or without 250 μM quinine) or regorafenib for 6 h. Culture medium was then replaced by a fresh one without sorafenib or regorafenib, and cell viability was determined 66 h later using the formazan test as reported elsewhere (Briz et al., 2000).
Production of cells stably expressing transporters
The ORF of hOCT1 and hOCT3 were amplified from total RNA isolated from healthy liver by RT followed by high‐fidelity PCR using AccuPrime Pfx DNA polymerase (Invitrogen, Thermo Fisher) and specific primers (Supporting Information Table S3). The ORF of hOCT1 or hOCT3 were cloned into the PacI site of the pWPI lentiviral vector. Recombinant lentiviruses were produced in HEK293T cells transfected using a standard polyethyleneimine (PEI) protocol with the transfer vector pWPI‐hOCT1/hOCT3, encoding both hOCT1 or hOCT3 and the eGFP, or simply pWPI (to generate ‘empty vectors’ encoding only eGFP) and the packaging plasmids psPAX2 (RRID:Addgene_12260) and pMD2.G (RRID:Addgene_12259). Viral titres were determined by transduction of HEK293T cells with serial dilutions of the viral solution, and analysis of eGFP‐positive cells was carried out with a FACSCalibur flow cytometer (BD Biosciences, Madrid, Spain). Lentiviral vectors were added to target cells at a multiplicity of infection of 25 in the presence of polybrene (in the case of HepG2), and the transduced cells were incubated for at least 4 days prior to the selection of monoclonal stably expressing cells using the limiting dilution method.
Transport assays
To determine sorafenib and regorafenib uptake by sub‐confluent cultures of HepG2 or HuH7 cells, these were seeded in 6‐well plates (≈75 × 103 and ≈50 × 103 cells per well, respectively), and experiments were performed the day after. Culture medium was removed, and cells were incubated with fresh medium containing 5 μM sorafenib or regorafenib without or with 250 μM quinine for 60 min. Uptake was stopped by rinsing the cultures 4 times with 1 mL of ice‐cold serum‐free culture medium. Cells were then lysed using pure water supplemented with prednisolone (5 μM), and sorafenib or regorafenib concentrations in the lysates were determined. An adaptation of a previously described method (Haouala et al., 2009) for sorafenib measurement by HPLC‐MS/MS was used in a 6420 Triple Quad LC/MS (Agilent Technologies, Santa Clara, CA, USA). Separation was carried out by HPLC using a Zorbax C18 column (30 mm × 2.1 mm, 3.5 μm). The chromatographic conditions were 65:35 methanol/water, both containing 5 mM ammonium acetate and 0.1% formic acid. Flow rate was 0.3 mL·min−1 at 35°C. Positive electrospray ionization was carried out with the following conditions: gas temperature 350°C, gas flow 10 L·min−1, nebulizer 20 psi and capillary voltage 2500 V. In multiple reaction monitoring mode (MRM), the selected m/z transitions were 465.1 m/z to 252.0 and 270.0 m/z for sorafenib and 483.0 m/z to 287.9 and 269.9 m/z for regorafenib. Prednisolone was used as an internal standard (transition 361.0 m/z to 343.0 and 307.0 m/z).
hOCT1 silencing by shRNA or miRNA
The selection of miRNAs potentially involved in the regulation of hOCT1 expression was carried out in two steps: First, an in silico analysis was performed to choose mature miRNAs (hsa‐miR‐X) with a better alignment on hOCT1 pre‐mRNA using bioinformatic prediction tools available at the following websites: miRbase (RRID:SCR_003152), Data Integral Portal DIP analysis (http://ophid.utoronto.ca/mirDIP), EMBL/EBI, miRDB (RRID:SCR_010848), miRTarbase (RRID:SCR_010851) and microrna.org. Then, we used as selection criteria the existence of a correlation between high expression in HCC samples and low hOCT1 mRNA expression from data publicly available at TCGA. Commercial DNA oligonucleotides for shRNA targeting hOCT1 (sh‐hOCT1; positive control) were obtained from Origene. An shRNA targeted against firefly luciferase (Luc2) was used as a negative control (Gonzalez‐Sanchez et al., 2016). Four oligonucleotides per miRNA were annealed to mimic the pre‐miRNA (hsa‐mir‐X) sequence including the loop (Supporting Information Table S3) and cloned into the MluI and ClaI restriction sites of the lentiviral pLVTHM plasmid. Plasmids containing the sequences mentioned above were co‐transfected with psPAX2 and pMD2.G in HEK293T to generate complete lentiviral particles. These viruses were used to transduce HepG2 cells that were then incubated with 5 mM sodium butyrate for 24 h. In all cases, eGFP was used as a reporter to track transduced cells. The effect on the expression of hOCT1 was investigated using RT‐qPCR and immunoblot, carried out as described above, in RNA and crude membrane suspensions, respectively, which were obtained 5 days after lentiviral‐mediated transduction.
Determination of alternative splicing
Based on previous reports of alternatively spliced hOCT1 variants (Herraez et al., 2013), we designed primers annealing in exon 6 (Forward) and exon 11 (Reverse) that are shared by all hOCT1 isoforms (Supporting Information Table S2). Using cDNA of paired HCC T and adjacent NT tissue PCR was carried out with Platinum‐Taq DNA polymerase using 30 cycles of amplification (Thermo Fisher). The presence and size of the PCR products were determined by gel electrophoresis, and then, a semi‐quantitative determination of the amount of each splicing variant was carried out by densitometry of the bands using the LAS‐4000 luminescent image analyser. Additionally, qPCR was performed using specific primers to determine relative abundance of the wild‐type full sequence and total isoforms of hOCT1 mRNA (Supporting Information Table S2).
Data and statistical analyses
To compare two means, Student's paired or unpaired t‐tests were used as appropriate. When indicated, Mann–Whitney two‐tailed t‐test was used. The one‐way ANOVA plus Bonferroni method of multiple‐range testing were used to calculate the statistical significance of differences among groups. Spearman's correlation was applied to datasets containing patient‐matched expression and methylation. Differences were considered significant when P < 0.05. Microsoft Excel (version 15.32, RRID:SCR_016137) and GraphPad (Prism5, RRID:SCR_002798) were used for the statistical analyses.
Chemicals and cell lines
Decitabine (DAC; 5‐aza‐2′‐deoxycytidine), DEN, 2‐AAF, sodium butyrate and quinine were purchased from Sigma‐Aldrich (Madrid, Spain). Sorafenib tosylate was obtained from Santa Cruz Biotechnology (Heidelberg, Germany). Regorafenib was from Selleckchem (Munich, Germany). The purity of all these compounds was ≥97%.
Cell lines from human hepatoma [HepG2, American Type Culture Collection (ATCC) Cat# HB‐8065, RRID:CVCL_0027], HEK cells (HEK293T, ATCC Cat# CRL‐3216, RRID:CVCL_0063) and CHO cells (CHO K1, ATCC, Cat# CCL‐61, RRID:CVCL_0214) were obtained from the ATCC (LGC Standards, Barcelona, Spain). Human hepatoma HuH7 cell line was obtained from the Japanese Collection of Research Bioresources Cell Bank (Japan) (JCRB Cat# JCRB0403, RRID:CVCL_0336). HepG2 cells were cultured in MEM medium and HEK293T and CHO cells in DMEM medium. HuH7 cells were cultured in DMEM medium supplemented with sodium bicarbonate (2.2 g·L−1), HEPES (2.38 g·L−1) and D‐glucose (3.5 g·L−1). All media were supplemented with 10% heat‐inactivated FCS, with penicillin/streptomycin/amphotericin solution B (Thermo Fisher) (HepG2 medium) or with penicillin/streptomycin and glutamax® solutions (Thermo Fisher) (HEK293T, HuH7 and CHO media). The cells were routinely tested to ensure they were mycoplasm free.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Results
Impaired expression of OCT1 in HCC
HCC spontaneously developed in >80% of Fxr −/− mice older than 14 months (Figure 1A). The procedure of chemical induction of HCC in rats was accompanied by ≈50% of mortality that occurred during the exposure to 2‐AAF for 2 weeks after partial hepatectomy. All rats that survived this stage lived until the end of the experimental period (32 weeks) and generated liver tumours (Figure 1B). In both models of HCC, a consistent decrease in the expression of OCT1 orthologues, that is, mOct1 in mice (Figure 1C) and rOct1 in rats (Figure 1D), was found. Moreover, these experimental tools revealed that OCT1 down‐regulation was accompanied by a reduction in the tumour ability to take up sorafenib. Analysis by HPLC‐MS/MS of samples collected 1 h after administration of sorafenib indicated that the concentration of this drug was lower in T than in adjacent NT tissue, in both mice (Figure 1E) and rats (Figure 1F). Values of sorafenib content in kidney and skeletal muscle, determined for comparison, were <30% and <1% of those found in liver respectively (data not shown).
Figure 1.
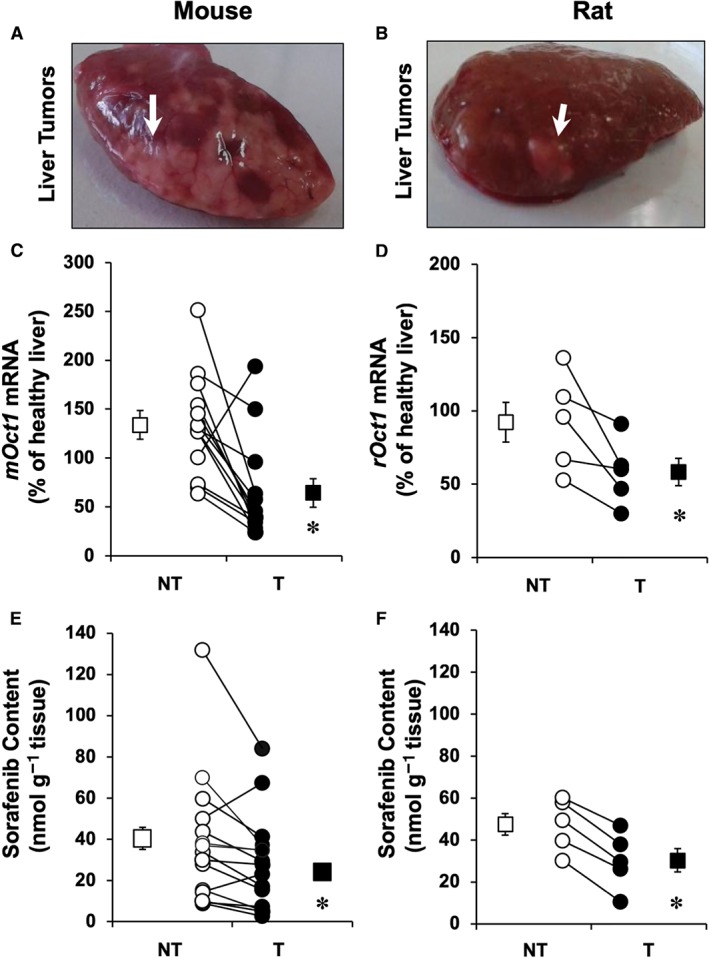
Representative images showing liver tumours (arrows) in (A) mice and (B) rats. Hepatocellular carcinoma was spontaneously generated in aged (≥14 months) Fxr −/− mice (n = 13) and chemically induced (Solt and Farber method) in rats (n = 5). Relative expression (mRNA) of mouse (mOct1) and rat (rOct1) orthologues was determined by RT‐qPCR in T and adjacent NT liver tissue in (C) mice and (D) rats. Sorafenib content in T and NT was measured by HPLC‐MS/MS 60 min after its administration (10 mg·kg−1 b.w.) i.p. in (E) mice or i.v. in (F) rats. Paired samples of tissue and serum were collected. Average values of serum concentrations at 60 min were 13.8 μM (range 2–32 μM) in mice and 2.43 μM (range 1–4 μM) in rats. Results are shown as individual values (circles) and mean ± SEM (squares). *P < 0.05 on comparing T with NT.
Relationship between hOCT1 expression and drug uptake/activity
To investigate the relationship between the ability of hOCT1 to mediate the uptake of sorafenib and regorafenib by HCC cells and hence increase their sensitivity to these drugs, HepG2 and HuH7 cells were transduced using lentiviral vectors either empty (Mock) or containing hOCT1 ORF. Overexpression of this transporter in transduced cells was confirmed by measuring hOCT1 mRNA (Supporting Information Figure S1A). The specificity of the primary antibody used to detect hOCT1 was first evaluated in CHO cells transduced with Mock vectors or vectors containing hOCT1 or hOCT3 ORF (Supporting Information Figure S1B). Immunoblotting of these cells revealed only positive signal in hOCT1, but not hOCT3, transduced cells. Using this anti‐hOCT1 antibody, a weak expression of hOCT1 was detected in HepG2 cells transduced with Mock vectors, whereas enhanced abundance of this protein was detected in hOCT1 transduced cells (Supporting Information Figure S1C). Immunofluorescence analysis showed that the enhanced expression of hOCT1 resulted in a higher level of this protein at the plasma membrane (Supporting Information Figure S2). The uptake of sorafenib was evaluated after the exposure to this drug for 1 h. In cells expressing hOCT1, sorafenib uptake was enhanced, which was prevented by co‐incubation with the hOCT1 inhibitor quinine (Figure 2A). Similar results were obtained using a different cell line (HuH7) (Figure 2B). Regorafenib is a novel TKI that has shown mild survival benefit in HCC patients with poor response or low tolerance to sorafenib treatment (Bruix et al., 2017). Our results suggested that hOCT1 does not play an important role in regorafenib uptake by HCC cells, as regorafenib uptake was not enhanced by hOCT1 expression in HepG2 (Figure 2C) or HuH7 (Figure 2D) cells. Consistently with these results, hOCT1 expression conferred higher sensitivity to HepG2 (Figure 2E) or HuH7 (Figure 2F) cells to sorafenib as shown by cytostatic assays in vitro, without affecting the response of both types of cells to regorafenib (Figure 2E,F). Co‐exposure with quinine prevented not only hOCT1‐mediated sorafenib uptake but also sorafenib‐induced cytotoxicity (Figure 2E,F).
Figure 2.
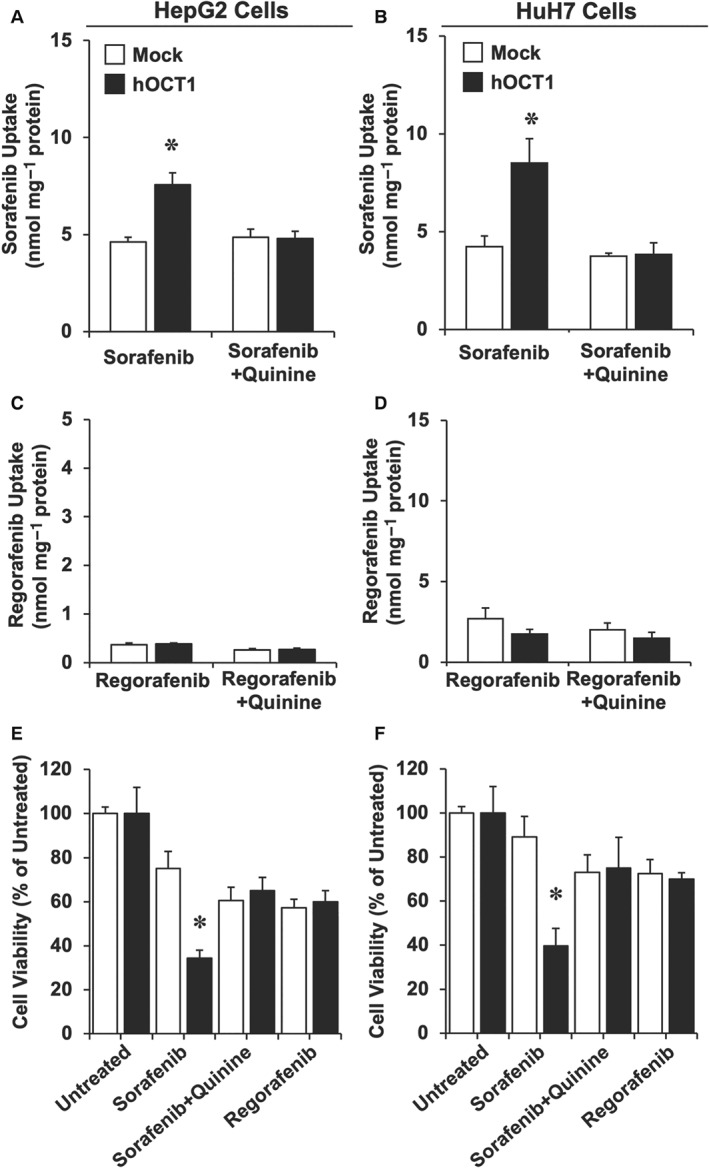
Sorafenib and regorafenib uptake by (A, C) HepG2 and (B, D) HuH7 cells and their (E, F) viability, as determined in cells transduced with empty lentiviral vectors (Mock) or containing the ORF of hOCT1 (hOCT1). Sorafenib and regorafenib were measured by HPLC‐MS/MS in the cells after incubating them with these compounds at 5 μM for 1 h. To determine cytotoxicity, cells were incubated with 5 μM sorafenib (with and without 250 μM quinine) or regorafenib for 6 h, and (E, F) cell viability was measured by MTT test 66 h later. Values are shown as mean ± SEM from five experiments performed in triplicate. *P < 0.05 on comparing hOCT1 versus Mock cells.
Consistent results were obtained in the xenograft model, in which stably transduced HepG2 cells, that overexpressed or not (Mock) hOCT1, were s.c. implanted in two distant locations of the flanks of each mouse. Once tumours were formed (15 mm3), the animals were divided into control group that received only the vehicle (saline) (Figure 3A) and the group that received sorafenib (Figure 3B). Treatment with sorafenib resulted in marked inhibition of tumour growth but only if the tumour was formed by cells overexpressing hOCT1 (Figure 3C).
Figure 3.
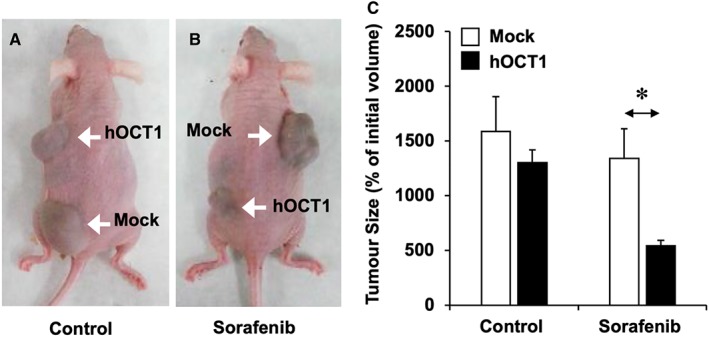
(A–B) Representative images of mice bearing tumours formed after s.c. implantation of ≈107 HepG2 cells transduced with lentiviral vectors either empty (Mock) or containing hOCT1 ORF. Three weeks later, the animals were randomly divided into two groups of five animals each. Control group (A, n = 4 for both types of tumours because one of the animals died during the experimental period) received only the vehicle (saline), whereas the sorafenib group received this drug (10 mg·kg−1 b.w., i.p.) three times per week for 4 weeks (B, n = 5 for both types of tumours). (C) Tumour size was measured at the end of treatment after killing the animals. Values are mean ± SEM. *P < 0.05, upon comparing Mock versus hOCT1 in mice treated with sorafenib.
Reduced hOCT1 expression/function in HCC due to aberrant splicing
In a previous study, the existence in HCC biopsies of several already known and newly identified variants of aberrant splicing of hOCT1 pre‐mRNA had been described (Herraez et al., 2013). This is pharmacologically relevant because these variants encode truncated non‐functional proteins (Herraez et al., 2013). To quantify the importance of aberrant splicing in overall hOCT1 expression/function in HCC, two approaches have been used in paired samples of HCC and adjacent NT tissue: (i) we performed two RT‐qPCR, one specific for the wild type sequence (full‐length mRNA) of hOCT1 (Figure 4A) and the other to determine total hOCT1 mRNA (full length plus all alternative splicing variants) (Figure 4B) and (ii) we evaluated the presence of splicing variants using specific primers to amplify the hOCT1 fragment between exons 6 and 11 (Figure 4C), and the length of the amplified fragment was used to identify the presence of wild type or splicing variants forms of hOCT1 (Figure 4D). Measurement by densitometry of the abundance of splicing variants indicated higher proportion of aberrant forms in HCC than in healthy liver. However, enhanced aberrant splicing was detected in both T and adjacent NT tissue (Figure 4E). It should be noted that, in all these cases, NT was ‘cirrhotic’, not ‘healthy’ liver tissue.
Figure 4.
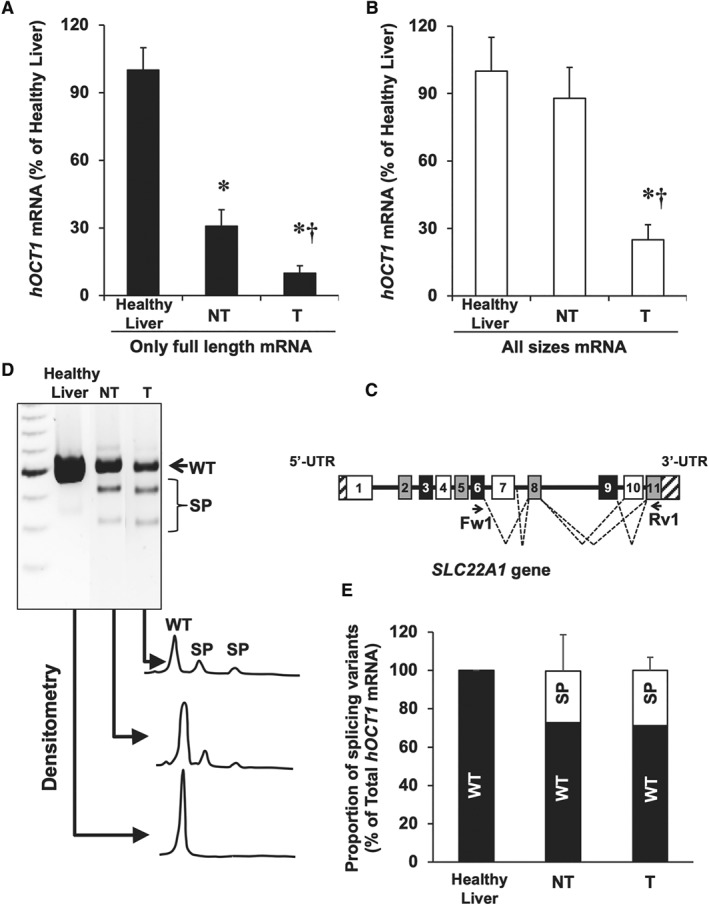
Analysis of hOCT1 mRNA splicing in HCC. Using RT‐qPCR with appropriate primers, the abundance of (A) only the full‐length sequence and (B) total mRNA (full length plus splicing variants) was determined in healthy liver (Control) and in paired samples of T and adjacent NT tissue (n = 19). (C) Structure of exons (boxes), introns (horizontal lines) and UTRs (striped bar) of SLC22A1 pre‐mRNA. Dashed lines indicate exon skipping or intron retention mechanisms of alternative splicing. The location of forward (Fw1) and reverse (Rv1) primers used to detect spliced forms is shown. (D) Representative gel after electrophoresis showing the products of semi‐quantitative RT‐PCR (during the linear phase of the amplification) using cDNA from healthy liver, NT and T samples. (E) Densitometry analysis of relative abundance of wild‐type (WT) versus all splicing variants (SP) of hOCT1. *P < 0.05 as compared with Control; †P < 0.05 as compared with NT.
Role of DNA methylation
Previous studies in smaller cohorts have shown hOCT1 down‐regulation in HCC compared with the expression levels in NT surrounding liver tissue. Here, using data from a larger cohort, this characteristic of the HCC phenotype was confirmed (Figure 5A). As aberrant DNA methylation alters gene expression in many types of cancer (Kelly and Issa, 2017), including hOCT1 down‐regulation in HCC (Schaeffeler et al., 2011), HepG2 and HuH7 cells were incubated with 1 μM of the demethylating agent DAC. The results showed significant stimulation of hOCT1 expression (Figure 5B). Because it has been reported that the effect of DAC can be enhanced by histone deacetylase inhibitors (Shieh et al., 2017), HepG2 and HuH7 cells were exposed to 5 mM sodium butyrate, a previously evaluated non‐toxic concentration (data not shown), for 24 h, and the hOCT1 expression was measured. Although sodium butyrate alone stimulated hOCT1 expression, the combination with DAC did not further increase hOCT1 expression (data not shown). To further evaluate the relationship between hOCT1 expression and DNA methylation of SLC22A1 gene, we have analysed genome‐wide DNA methylation data in 366 T and 49 NT samples. SLC22A1 was significantly hypermethylated at two probes located in TSS1500 and in exon 1 (Figure 5C,E) in HCC compared to adjacent NT samples. Moreover, an inverse correlation between hOCT1 mRNA level and the degree of SLC22A1 gene methylation was found (Figure 5D,F).
Figure 5.
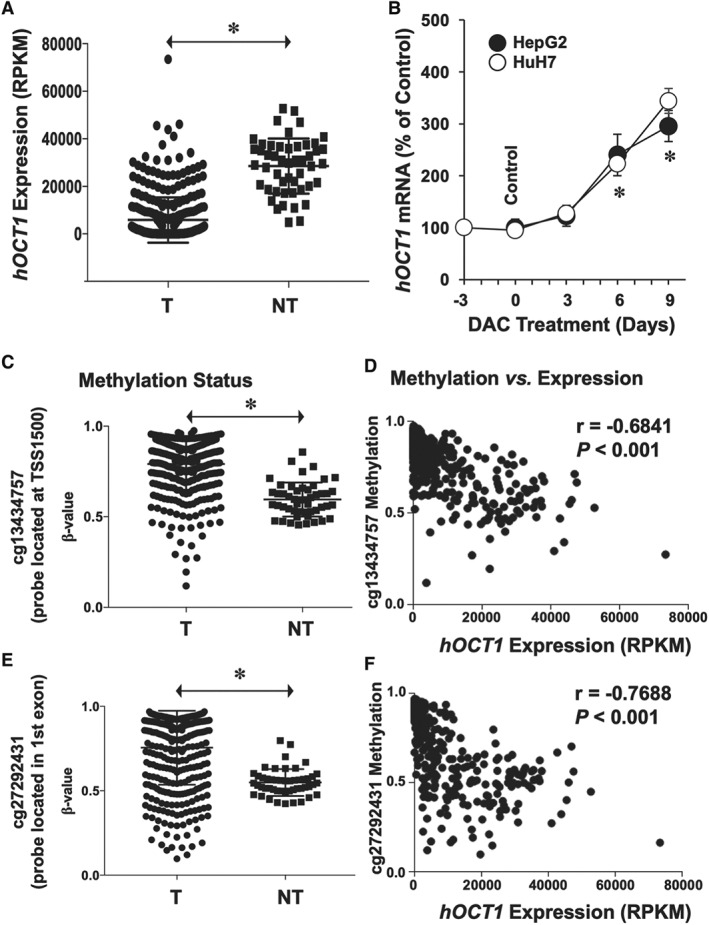
(A) Expression levels of hOCT1 in T and surrounding NT as determined by transcriptome profiling of HCC. Data were obtained from patients (n = 366) included in the publicly available database TCGA. (B) Effect of treating HepG2 and HuH7 cells with 1 μM of the demethylating agent DAC for the indicated time on hOCT1 expression (mRNA levels). Values (mean ± SEM from five different experiments performed in triplicate) are compared with basal hOCT1 expression in HepG2 and HuH7 cells (Control group, before starting the treatment). *P < 0.05 as compared with Control. (C–F) Methylation status of SLC22A1 gene in HCC (n = 366 from TCGA). Genome‐wide DNA methylation profiling uncovered two critical CpG regions whose methylation status was significantly increased in HCC (C: cg13434757 located at TSS1500 and E: cg27292431 located in exon 1). Relationship between expression levels and methylation status of (D) cg13434757 and (F) cg27292431 regions. Mann–Whitney two‐tailed t‐test was used to compare mean values between groups; *P < 0.05. Spearman's correlation was applied to datasets containing patient‐matched expression and methylation. RPKM, reads per kilobase per million mapped reads.
Changes in miRNA landscape
An important role in post‐transcriptional inhibition of gene expression, which markedly affects cancer cells phenotype, is played by miRNAs. To investigate whether this mechanism is involved in HCC‐associated hOCT1 down‐regulation, we carried out in silico studies to select those miRNAs expressed in HCC according to the information available at TCGA database. Selection criteria included expression‐related (Supporting Information Table S4) and structure‐related (Supporting Information Table S5) specific requirements. The five miRNAs that reached the highest score in this selection process were as follows: miR‐141‐5p, miR‐330‐3p, miR‐769‐3p, miR‐1287‐3p and miR‐1468‐3p. Accordingly, hsa‐mir‐141, hsa‐mir‐330, hsa‐mir‐769, hsa‐mir‐1287 and hsa‐mir‐1468 were cloned in lentiviral vectors. We added to this list hsa‐mir‐6806, whose expression in HCC is low according to TCGA; however, it was reported as a predicted miRNA that targets hOCT1 mRNA (Hyrsova et al., 2016). HepG2 cells were first transduced to obtain overexpression of these miRNAs, then treated with 5 mM sodium butyrate for 24 h, which enhanced (2.2‐fold) basal hOCT1 mRNA expression (Supporting Information Figure S3). Short hairpins against Luc2 (sh‐Luc2) (negative control) had no effect on hOCT1 expression (data not shown). In contrast, sh‐hOCT1 markedly reduced hOCT1 mRNA (positive control) (Figure 6A). Among the six miRNAs assayed, three (hsa‐mir‐6806, hsa‐mir‐1287 and hsa‐mir‐769) failed to affect hOCT1 mRNA abundance, whereas hsa‐mir‐141 displayed a moderate effect, and both hsa‐mir‐330 and hsa‐mir‐1468 induced a marked decrease in mRNA levels, which was similar to that induced by sh‐hOCT1 (Figure 6A). Although immunoblot was assayed to confirm, at the protein levels, the silencing ability of these microRNA, the very low basal expression of hOCT1 (Supporting Information Figure S1C) has precluded to accurately measure a further reduction of hOCT1 protein levels (data not shown). To further evaluate the role of these microRNA in HCC, values (from TCGA) of expression in 39 paired samples T tissue and adjacent NT tissue were investigated (Figure 6B). Whereas miR‐141 and miR‐1468 were significantly down‐regulated in T, a consistent and significant increase in the abundance of miR‐330 was found (Figure 6C).
Figure 6.
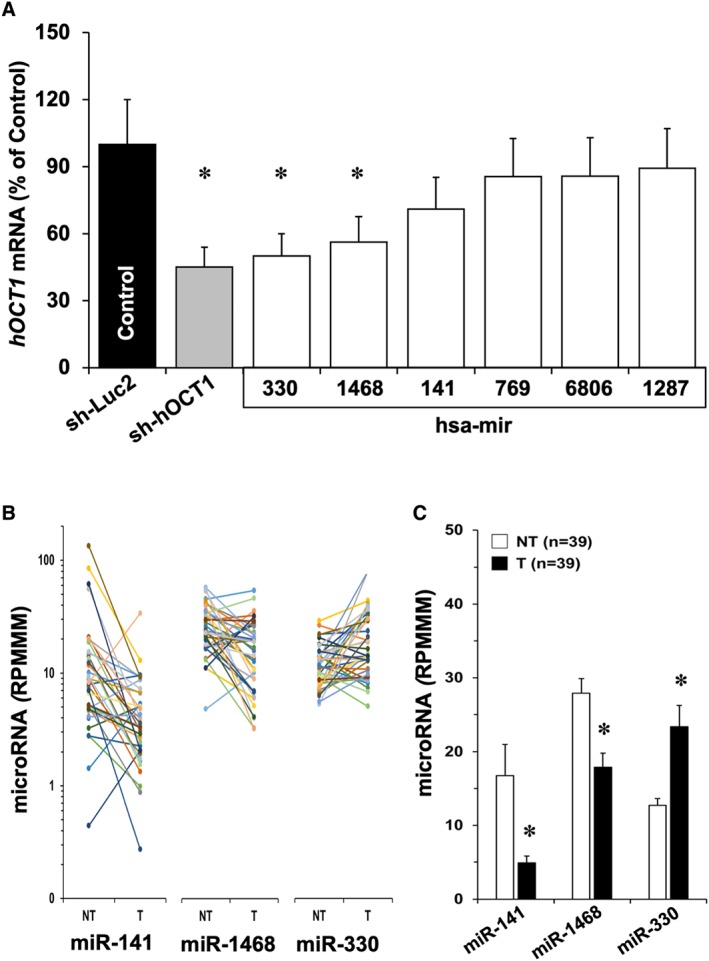
(A) Evaluation of the ability of several miRNAs to inhibit hOCT1 expression in HepG2 cells. The cells were transduced with lentiviral vectors able to induce the expression of sh‐Luc2, used as negative control, or against hOCT1 (sh‐hOCT1), or one of the six miRNAs selected from their expression in HCC and predicted interaction with pre‐mRNA by in silico analysis or reported inhibitory effect on hOCT1 expression. Values are mean ± SEM from five experiments carried out in triplicate. *P < 0.05 on comparing with sh‐Luc2. (B) Individual values of expression levels downloaded from TCGA of miR‐141, miR‐1468 and miR‐330 in 39 paired samples of T and adjacent NT. Results are shown as RPMMM (read per million miRNA mapped). (C) Average values (mean ± SEM) calculated from data shown in B; *P < 0.05 on comparing T and NT by paired t‐test.
Role of mRNA stability and decay
Expression/function of proteins accounting for preserving or reducing mRNA stability are known to be altered in cancer cells (Geissler and Grimson, 2016). Here, we have determined the expression of several important modulators of mRNA stability/degradation (Supporting Information Figure S4). When comparing T with NT tissue, no significant changes were found regarding the expression of proteins that favour mRNA stability (HuR) or degradation (CUGBP, BRF1, BRF2, FBP2 and AUF1). The only significant difference was observed in the expression of tristetraprolin (TTP) expression. However, this change could not be expected to induce hOCT1 mRNA decay, because TTP favours mRNA degradation, and this protein was down‐regulated in HCC (Supporting Information Figure S4).
Discussion
Although targeted therapies based on TKIs were initially very promising when used to treat several types of cancer, this has not been the case for advanced HCC, where TKIs developed until now have offered only modest beneficial effects. Several types of MOC account for this limitation (Marin et al., 2010). Two of them (MOC‐1 and MOC‐2) participate in the reduction in intracellular concentrations of active drugs (Marin et al., 2014). It should be considered that to carry out their pharmacological action, TKIs must interact with the catalytic site of TK receptors, which are located intracellularly. This means that TKIs must be taken up by cancer cells to act as antiproliferative drugs. However, in aqueous solution at physiological pH, these compounds are mainly charged molecules, which determine high polarity and poor ability to freely diffuse across the plasma membrane. This implies that uptake by cancer cells must occur through carrier proteins. Unfortunately, in HCC, the expression of the main transporter accounting for sorafenib uptake – hOCT1, because hOCT3 contribution seems to be minor (Heise et al., 2012) – is markedly reduced. Understanding the mechanism(s) involved in the impaired hOCT1 expression/function is crucial to envision the future development of chemo‐sensitizing strategies. The present study supports the concept that dysregulation of hOCT1 expression is a common trait in MDR phenotype of HCC (Martinez‐Becerra et al., 2012; Herraez et al., 2013). Moreover, the relationship between this characteristic and the reduced ability to take up and respond to sorafenib was demonstrated. This is consistent with the clinical data, suggesting that reduced intratumour abundance of hOCT1 mRNA might be a prognostic biomarker in sorafenib‐based HCC therapy (Grimm et al., 2016). Nevertheless, we have further reported that the presence of hOCT1 at the plasma membrane of cancer cells, as measured by immunohistochemical techniques, rather than the amount of hOCT1 mRNA, is associated with better outcome of patients with advanced HCC following sorafenib therapy (Geier et al., 2017).
Several putative mechanisms were evaluated here as the reason for impaired hOCT1 expression. Alternative splicing is an important regulator mechanism of gene expression. Dysregulation in splicing patterns have been associated with many human diseases, including cancer. Indeed, aberrant splicing of many genes, including SLC22A1 (Herraez et al., 2013), has been reported to be increased in HCC (Tremblay et al., 2016). Changes in the profile of alternative splicing have been suggested to play a role in the high variability existing among individual tumours. In HCC biopsies, we have previously identified the existence of aberrant splicing variants of hOCT1, which are generated following the loss of one or more exons and partial intron retention. These alternative hOCT1 transcripts encode for truncated non‐functional proteins that accumulate inside the cell instead of being targeted to the plasma membrane (Herraez et al., 2013). We have shown here that, in addition to the reduced hOCT1 mRNA abundance in tumour cells as compared to adjacent liver tissue, the proportion of aberrantly spliced isoforms is augmented, which markedly contributes to further impair hOCT1 function. Interestingly, the presence of aberrant splicing is not restricted to T tissue, because this was also found in surrounding cirrhotic tissue, but not in healthy liver. Whether splicing dysregulation is an early event in pre‐malignant lesions or adjacent tissue is affected by changes in the factors controlling pre‐mRNA splicing is an interesting question that deserves further investigation.
Several studies reported DNA methylation to be typically altered in many cancers. This is consistent with previous studies reporting hypermethylation of SLC22A1 promoter as quantified by MALDI‐TOF MS in 22 fresh‐frozen samples of HCC (Schaeffeler et al., 2011). This was inversely correlated with hOCT1 expression (Schaeffeler et al., 2011). The results obtained by analysing Gene Expression Omnibus database confirmed that SLC22A1 is among the genes with lowest expression and highest degree of methylation in HCC (Liu et al., 2017b). Moreover, our experimental evidence provides direct link between DNA methylation and hOCT1 expression. Thus, incubation of hepatoma cells with DAC resulted in enhanced hOCT1 expression. In agreement with these findings, DNA hypomethylating agents, such as DAC, are used clinically in some types of cancer. For instance, in the treatment of acute myeloid leukaemia, when combined with sorafenib, DAC has a synergistic effect, whereas adverse effects are still tolerable for these patients (Muppidi et al., 2015).
The panel of miRNAs expression changes in cancer, which is thought to contribute to alter physiological processes, such as apoptosis or autophagy, leading to carcinogenesis, progression and metastasis (Kim and Kim, 2014), and may also play a role in the impaired regulation of hOCT1 expression. In HCC biopsies, a marked difference in the expression panel of miRNAs between T and NT tissues has been reported (Murakami et al., 2006). Because miRNAs are also important in the prognosis, a set of miRNAs has been proposed to form part of a signature useful to predict the chance of overall survival of HCC patients (Liu et al., 2017a). The development of available bioinformatic tools has enabled in silico prediction of candidate miRNAs that regulate a precise gene or the possible targets of a known miRNA (Peterson et al., 2014). Using this technology, we have selected a few potential miRNA, highly expressed in HCC, as possible regulators of hOCT1 expression. Among them, in vitro assays have validated the strong ability of both hsa‐miR‐330 and hsa‐miR‐1468 to reduce hOCT1 expression; however, analysis of TCGA database revealed that hsa‐mir‐330 but not hsa‐mir‐1468 was up‐regulated in HCC compared with adjacent NT tissue. An association between hsa‐mir‐330 and cancer has already been described. This miRNA seems to play an oncogenic role in human glioblastoma (Qu et al., 2012) and is involved in the inflammatory response (Gu et al., 2017). Recent data by Hu et al. (2017) have also showed that miR‐330 levels were significantly higher in HCC than in the surrounding liver tissues. Interestingly, the high expression of miR‐330 was significantly associated with more aggressive phenotypes of the tumour and shorter overall survival in these patients.
Dysregulated expression/function of RNA binding proteins (RBPs), key regulators of post‐transcriptional events, affects mRNA degradation rate (Sanchez‐Diaz and Penalva, 2006). RBPs interact with the AU rich element in the 3′ untranslated region (3′UTR) leading to mRNA stability regulation. Since in 3′UTR of hOCT1 mRNA there is an AU rich region, RBPs could be involved in enhanced hOCT1 mRNA decay in HCC. The regulatory network formed by these proteins is a complex mechanism that has not been fully explored here. As a preliminary step, we have only determined the expression level of RBPs in T and NT samples. Our results do not support a role of changes in the expression of RBPs in reducing hOCT1 mRNA levels.
In conclusion, lower expression of hOCT1 in HCC, due in part to a high rate of aberrant splicing and epigenetic factors, such as hypermethylation status and miRNA‐mediated (mainly miR‐330) mRNA decay, results in impaired sorafenib uptake and cytotoxic effect. Accordingly, to increase hOCT1 function constitutes an interesting goal to sensitize HCC to the pharmacological effect of sorafenib. This is clinically relevant because the response of HCC to conventional chemotherapy is very poor and that to sorafenib and regorafenib is mediocre and only moderately improve the outcome of these patients. The results obtained in this study support the concept that manipulating the expression/function of uptake transporters in cancer cells may increase the chance of TKIs to reach their intracellular targets and hence enhance their pharmacological efficacy.
Author contributions
E.L., R.I.R.M., R.A.A., O.B. and J.J.G.M. contributed to the study concept and design; R.A.A., M.J.M., O.B., E.L., R.I.R.M. and M.A.S. carried out the experiments; R.A.A., A.G., J.B.A., C.J.O., M.J.M., O.B., E.L., R.I.R.M. and M.A.S. contributed to data acquisition; R.A.A., C.J.O., M.J.M., O.B., E.L. and R.I.R.M. conducted the statistical analysis; J.J.G.M., R.A.A., A.G., J.B.A., J.M.B., M.A.A., M.L.M.C. and R.I.R.M. interpreted the data; J.J.G.M., O.B., R.I.R.M., J.B.A., A.G., J.M.B., M.A.A. and M.L.M.C. obtained funding; J.J.G.M., R.A.A. and E.L. drafted the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Demographic and clinical characteristics of 19 HCC patients analysed.
Table S2 Sequence of oligonucleotide primers used for quantitative PCR.
Table S3 Sequence of forward (F) and reverse (R) oligonucleotide primers used for cloning.
Table S4 Selected miRNA based on expression‐related criteria.
Table S5 Selected miRNA based on structure‐related criteria.
Figure S1 (A) Relative hOCT1 mRNA levels in HepG2 cells transduced with lentiviral vectors either empty (Mock) or containing the hOCT1 coding sequence. Results are mean ± SEM (n = 5). *P < 0.05 on comparing both groups by Student t‐test. (B) Specificity test of the primary antibody used in immunoblotting against hOCT1. CHO cells were transduced with empty vectors (Mock) or vectors containing hOCT1 or hOCT3 coding sequence. Immunoblot was carried out with cell lysates. Gapdh was used for normalization. (C) Immunoblots (n = 5) were then performed in crude preparations of cell membranes obtained from HepG2 cells (Mock and hOCT1). Na+/K+‐ATPase was used as normalizer.
Figure S2 Immunofluorescence detected by confocal microscopy in HepG2 cells transduced with Mock vectors (A‐C) or hOCT1 (D‐F) stained with anti‐hOCT1 (red) and anti‐Na+/K+‐ATPase (green) antibodies. Merge images (C and F) show the nuclei stained with DAPI.
Figure S3 Relative expression (mRNA levels) of hOCT1 in HepG2 cells after incubation without or with 5 mM sodium butyrate for 24 h. Results are mean ± SEM (n = 5). *P < 0.05 on comparing both groups by paired t‐test.
Figure S4 Relative expression (mRNA levels) in biopsies (n = 13) of hepatocellular carcinoma (HCC) of genes involved in favouring mRNA degradation or stability (see table in the inset) as determined by RT‐qPCR in the tumour (T) and paired adjacent non‐tumour (NT) tissue. Results are shown as individual values (circles) and mean ± SEM (squares). *P < 0.05 comparing T with NT; N.S., not significant difference.
Acknowledgements
This study was supported by the Carlos III Institute of Health, Spain [Grants PI15/00179 and PI16/00598, co‐financed by European Regional Development Fund (ERDF)]; Ministry of Science and Innovation, Spain (SAF2013‐40620‐R, SAF2016‐75197‐R and SAF2017‐87301‐R); ‘Asociación Española Contra el Cancer’ (AECC‐2017); ‘Junta de Castilla y León’, Spain (SA015U13 and SA063P17); ‘Fundación Samuel Solórzano Barruso’ (FS/10‐2014, FS/08‐2017 and FS/13‐2017), Spain; ‘Fundación Mutua Madrileña’ (Call 2015), Spain; Fundación Vasca de Innovación e Investigación Sanitarias, Spain (Bioef BIO15/CA/011); Gobierno Vasco‐Departamento de Salud, Spain (2013111114); Basque Foundation for Innovation and Health Research: EiTB Maratoia, Spain (BIO15/CA/016/BD); ‘Proyecto Centro Internacional sobre el Envejecimiento’ (0348‐CIE‐6‐E), Spain. R.A.A. was supported by a pre‐doctoral contract funded by the ‘Junta de Castilla y León’ and the ‘Fondo Social Europeo’, Spain (EDU/828/2014). J.B.A. and C.J.O. were supported by the Danish Cancer Society (R98‐A6446), Novo Nordisk Foundation (14040) and Marie Sklodowska‐Curie Fellowship (Epi‐Target) respectively. E.L. was supported by a post‐doctoral fellowship funded by the University of Salamanca, Spain. M.A.A. was supported by Fundación Bancaria La Caixa, Hepacare Project, Spain.
Al‐Abdulla, R. , Lozano, E. , Macias, R. I. R. , Monte, M. J. , Briz, O. , O'Rourke, C. J. , Serrano, M. A. , Banales, J. M. , Avila, M. A. , Martinez‐Chantar, M. L. , Geier, A. , Andersen, J. B. , and Marin, J. J. G. (2019) Epigenetic events involved in organic cation transporter 1‐dependent impaired response of hepatocellular carcinoma to sorafenib. British Journal of Pharmacology, 176: 787–800. 10.1111/bph.14563.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Catalytic receptors. Br J Pharmacol 174: S225–S271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assenov Y, Muller F, Lutsik P, Walter J, Lengauer T, Bock C (2014). Comprehensive analysis of DNA methylation data with RnBeads. Nat Methods 11: 1138–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briz O, Serrano MA, Macias RI, Marin JJ (2000). Overcoming cisplatin resistance in vitro by a free and liposome‐encapsulated bile acid derivative: BAMET‐R2. Int J Cancer 88: 287–292. [PubMed] [Google Scholar]
- Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G et al (2017). Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 389: 56–66. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Electronic address wbe , Cancer Genome Atlas Research N (2017). Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell 169: 1327–1341 e1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geier A, Macias RI, Bettinger D, Weiss J, Bantel H, Jahn D et al (2017). The lack of the organic cation transporter OCT1 at the plasma membrane of tumor cells precludes a positive response to sorafenib in patients with hepatocellular carcinoma. Oncotarget 8: 15846–15857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler R, Grimson A (2016). A position‐specific 3′UTR sequence that accelerates mRNA decay. RNA Biol 13: 1075–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez‐Sanchez E, Perez MJ, Nytofte NS, Briz O, Monte MJ, Lozano E et al (2016). Protective role of biliverdin against bile acid‐induced oxidative stress in liver cells. Free Radic Biol Med 97: 466–477. [DOI] [PubMed] [Google Scholar]
- Grimm D, Lieb J, Weyer V, Vollmar J, Darstein F, Lautem A et al (2016). Organic cation transporter 1 (OCT1) mRNA expression in hepatocellular carcinoma as a biomarker for sorafenib treatment. BMC Cancer 16: 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu TT, Song L, Chen TY, Wang X, Zhao XJ, Ding XQ et al (2017). Fructose downregulates miR‐330 to induce renal inflammatory response and insulin signaling impairment: attenuation by morin. Mol Nutr Food Res 61: 1–20. [DOI] [PubMed] [Google Scholar]
- Haouala A, Zanolari B, Rochat B, Montemurro M, Zaman K, Duchosal MA et al (2009). Therapeutic drug monitoring of the new targeted anticancer agents imatinib, nilotinib, dasatinib, sunitinib, sorafenib and lapatinib by LC tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci 877: 1982–1996. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise M, Lautem A, Knapstein J, Schattenberg JM, Hoppe‐Lotichius M, Foltys D et al (2012). Downregulation of organic cation transporters OCT1 (SLC22A1) and OCT3 (SLC22A3) in human hepatocellular carcinoma and their prognostic significance. BMC Cancer 12: 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herraez E, Lozano E, Macias RI, Vaquero J, Bujanda L, Banales JM et al (2013). Expression of SLC22A1 variants may affect the response of hepatocellular carcinoma and cholangiocarcinoma to sorafenib. Hepatology 58: 1065–1073. [DOI] [PubMed] [Google Scholar]
- Hu X, Feng Y, Sun L, Qu L, Sun C (2017). Roles of microRNA‐330 and its target gene ING4 in the development of aggressive phenotype in hepatocellular carcinoma cells. Dig Dis Sci 62: 715–722. [DOI] [PubMed] [Google Scholar]
- Hyrsova L, Smutny T, Trejtnar F, Pavek P (2016). Expression of organic cation transporter 1 (OCT1): unique patterns of indirect regulation by nuclear receptors and hepatospecific gene regulation. Drug Metab Rev 48: 139–158. [DOI] [PubMed] [Google Scholar]
- Kelly AD, Issa JJ (2017). The promise of epigenetic therapy: reprogramming the cancer epigenome. Curr Opin Genet Dev 42: 68–77. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KM, Kim SG (2014). Autophagy and microRNA dysregulation in liver diseases. Arch Pharm Res 37: 1097–1116. [DOI] [PubMed] [Google Scholar]
- Koepsell H, Lips K, Volk C (2007). Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res 24: 1227–1251. [DOI] [PubMed] [Google Scholar]
- Kudo M (2017). Systemic therapy for hepatocellular carcinoma: 2017 update. Oncology 93 (Suppl. 1): 135–146. [DOI] [PubMed] [Google Scholar]
- Liu D, Liu P, Cao L, Zhang Q, Chen Y (2017b). Screening the key genes of hepatocellular adenoma via microarray analysis of DNA expression and methylation profiles. Oncol Lett 14: 3975–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Wang H, Fu JD, Liu JY, Yan AG, Guan YY (2017a). A five‐miRNA expression signature predicts survival in hepatocellular carcinoma. APMIS 125: 614–622. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Bruix J (2003). Systematic review of randomized trials for unresectable hepatocellular carcinoma: chemoembolization improves survival. Hepatology 37: 429–442. [DOI] [PubMed] [Google Scholar]
- Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF et al (2008). Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359: 378–390. [DOI] [PubMed] [Google Scholar]
- Lozano E, Herraez E, Briz O, Robledo VS, Hernandez‐Iglesias J, Gonzalez‐Hernandez A et al (2013). Role of the plasma membrane transporter of organic cations OCT1 and its genetic variants in modern liver pharmacology. Biomed Res Int 2013: 692071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin JJ, Briz O, Monte MJ, Blazquez AG, Macias RI (2012). Genetic variants in genes involved in mechanisms of chemoresistance to anticancer drugs. Curr Cancer Drug Targets 12: 402–438. [DOI] [PubMed] [Google Scholar]
- Marin JJ, Monte MJ, Blazquez AG, Macias RI, Serrano MA, Briz O (2014). The role of reduced intracellular concentrations of active drugs in the lack of response to anticancer chemotherapy. Acta Pharmacol Sin 35: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin JJ, Romero MR, Briz O (2010). Molecular bases of liver cancer refractoriness to pharmacological treatment. Curr Med Chem 17: 709–740. [DOI] [PubMed] [Google Scholar]
- Martinez‐Becerra P, Vaquero J, Romero MR, Lozano E, Anadon C, Macias RI et al (2012). No correlation between the expression of FXR and genes involved in multidrug resistance phenotype of primary liver tumors. Mol Pharm 9: 1693–1704. [DOI] [PubMed] [Google Scholar]
- Minematsu T, Giacomini KM (2011). Interactions of tyrosine kinase inhibitors with organic cation transporters and multidrug and toxic compound extrusion proteins. Mol Cancer Ther 10: 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monte MJ, Dominguez S, Palomero MF, Macias RI, Marin JJ (1999). Further evidence of the usefulness of bile acids as molecules for shuttling cytostatic drugs toward liver tumors. J Hepatol 31: 521–528. [DOI] [PubMed] [Google Scholar]
- Muppidi MR, Portwood S, Griffiths EA, Thompson JE, Ford LA, Freyer CW et al (2015). Decitabine and sorafenib therapy in FLT‐3 ITD‐mutant acute myeloid leukemia. Clin Lymphoma Myeloma Leuk 15 (Suppl): S73–S79. [DOI] [PubMed] [Google Scholar]
- Murakami Y, Yasuda T, Saigo K, Urashima T, Toyoda H, Okanoue T et al (2006). Comprehensive analysis of microRNA expression patterns in hepatocellular carcinoma and non‐tumorous tissues. Oncogene 25: 2537–2545. [DOI] [PubMed] [Google Scholar]
- Peterson SM, Thompson JA, Ufkin ML, Sathyanarayana P, Liaw L, Congdon CB (2014). Common features of microRNA target prediction tools. Front Genet 5: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu S, Yao Y, Shang C, Xue Y, Ma J, Li Z et al (2012). MicroRNA‐330 is an oncogenic factor in glioblastoma cells by regulating SH3GL2 gene. PLoS One 7: e46010. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sanchez‐Diaz P, Penalva LO (2006). Post‐transcription meets post‐genomic: the saga of RNA binding proteins in a new era. RNA Biol 3: 101–109. [DOI] [PubMed] [Google Scholar]
- Schaeffeler E, Hellerbrand C, Nies AT, Winter S, Kruck S, Hofmann U et al (2011). DNA methylation is associated with downregulation of the organic cation transporter OCT1 (SLC22A1) in human hepatocellular carcinoma. Genome Med 3: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh JM, Tang YA, Hu FH, Huang WJ, Wang YJ, Jen J et al (2017). A histone deacetylase inhibitor enhances expression of genes inhibiting Wnt pathway and augments activity of DNA demethylation reagent against nonsmall‐cell lung cancer. Int J Cancer 140: 2375–2386. [DOI] [PubMed] [Google Scholar]
- Shikata E, Yamamoto R, Takane H, Shigemasa C, Ikeda T, Otsubo K et al (2007). Human organic cation transporter (OCT1 and OCT2) gene polymorphisms and therapeutic effects of metformin. J Hum Genet 52: 117–122. [DOI] [PubMed] [Google Scholar]
- Solt DB, Medline A, Farber E (1977). Rapid emergence of carcinogen‐induced hyperplastic lesions in a new model for the sequential analysis of liver carcinogenesis. Am J Pathol 88: 595–618. [PMC free article] [PubMed] [Google Scholar]
- Torre LA, Siegel RL, Ward EM, Jemal A (2016). Global cancer incidence and mortality rates and trends – an update. Cancer Epidemiol Biomarkers Prev 25: 16–27. [DOI] [PubMed] [Google Scholar]
- Tremblay MP, Armero VE, Allaire A, Boudreault S, Martenon‐Brodeur C, Durand M et al (2016). Global profiling of alternative RNA splicing events provides insights into molecular differences between various types of hepatocellular carcinoma. BMC Genomics 17: 683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Huang X, Yi T, Yen Y, Moore DD, Huang W (2007). Spontaneous development of liver tumors in the absence of the bile acid receptor farnesoid X receptor. Cancer Res 67: 863–867. [DOI] [PubMed] [Google Scholar]
- Zhang S, Lovejoy KS, Shima JE, Lagpacan LL, Shu Y, Lapuk A et al (2006). Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res 66: 8847–8857. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Demographic and clinical characteristics of 19 HCC patients analysed.
Table S2 Sequence of oligonucleotide primers used for quantitative PCR.
Table S3 Sequence of forward (F) and reverse (R) oligonucleotide primers used for cloning.
Table S4 Selected miRNA based on expression‐related criteria.
Table S5 Selected miRNA based on structure‐related criteria.
Figure S1 (A) Relative hOCT1 mRNA levels in HepG2 cells transduced with lentiviral vectors either empty (Mock) or containing the hOCT1 coding sequence. Results are mean ± SEM (n = 5). *P < 0.05 on comparing both groups by Student t‐test. (B) Specificity test of the primary antibody used in immunoblotting against hOCT1. CHO cells were transduced with empty vectors (Mock) or vectors containing hOCT1 or hOCT3 coding sequence. Immunoblot was carried out with cell lysates. Gapdh was used for normalization. (C) Immunoblots (n = 5) were then performed in crude preparations of cell membranes obtained from HepG2 cells (Mock and hOCT1). Na+/K+‐ATPase was used as normalizer.
Figure S2 Immunofluorescence detected by confocal microscopy in HepG2 cells transduced with Mock vectors (A‐C) or hOCT1 (D‐F) stained with anti‐hOCT1 (red) and anti‐Na+/K+‐ATPase (green) antibodies. Merge images (C and F) show the nuclei stained with DAPI.
Figure S3 Relative expression (mRNA levels) of hOCT1 in HepG2 cells after incubation without or with 5 mM sodium butyrate for 24 h. Results are mean ± SEM (n = 5). *P < 0.05 on comparing both groups by paired t‐test.
Figure S4 Relative expression (mRNA levels) in biopsies (n = 13) of hepatocellular carcinoma (HCC) of genes involved in favouring mRNA degradation or stability (see table in the inset) as determined by RT‐qPCR in the tumour (T) and paired adjacent non‐tumour (NT) tissue. Results are shown as individual values (circles) and mean ± SEM (squares). *P < 0.05 comparing T with NT; N.S., not significant difference.