


Abstract
The DNA binding domain (DBD) of the tumor suppressor p53 is the site of several oncogenic mutations. A subset of these mutations lowers the unfolding temperature of the DBD. Unfolding leads to the exposure of a hydrophobic β-strand and nucleates aggregation which results in pathologies through loss of function and dominant negative/gain of function effects. Inspired by the hypothesis that structural changes that are associated with events initiating unfolding in DBD are likely to present opportunities for inhibition, we investigate the dynamics of the wild type (WT) and some aggregating mutants through extensive all atom explicit solvent MD simulations. Simulations reveal differential conformational sampling between the WT and the mutants of a turn region (S6–S7) that is contiguous to a known aggregation-prone region (APR). The conformational properties of the S6–S7 turn appear to be modulated by a network of interacting residues. We speculate that changes that take place in this network as a result of the mutational stress result in the events that destabilize the DBD and initiate unfolding. These perturbations also result in the emergence of a novel pocket that appears to have druggable characteristics. FDA approved drugs are computationally screened against this pocket.
INTRODUCTION
Cellular trauma triggers the stabilization of the transcription factor/tumor suppressor p53, which in turn regulates downstream genes involved in apoptosis, DNA repair, senescence and metabolism. Any perturbation in the p53 gene network causes abnormalities/disorders in these cellular pathways (1). The gene encoding p53, TP53, is mutated in 50–70% of cancers (2), thus making it a potential therapeutic target, with some molecules already in clinical trials (1,3–5).
The IARC TP53 Mutation Database (6) lists that ∼95% of these oncogenic mutations are located in the DNA binding domain (DBD) (alternatively referred to as the core domain or p53C). The DBD is structurally composed of a β-sandwich, helices and loops (Figure 1) and initiates transcription by binding to DNA promoters in a sequence specific manner (7). The oncogenic mutants have been classified into 2 groups based on their mode of action: the DNA contact mutants and the destabilizing (also referred as structural or aggregating) mutants. The DNA contact mutants lose their ability to interact with specific DNA promoters (loss of function) and sustain local structural changes with negligible perturbations in the thermodynamic stability of the DBD (8). Some contact mutants oligomerize with WT p53 (when the mutation is in one allele) and inactivate the WT allele and also bind to the paralogs p63 and p73, exhibiting ‘dominant negative’ and ‘gain of function’ effects respectively (9,10). In contrast, the destabilizing mutants, which constitute 30% of all the oncogenic mutations in p53, are thermodynamically unstable at physiological temperature and undergo partial unfolding and/or aggregation. Such mutants also trigger the unfolding and co-aggregation of WT p53 (dominant negative effect) and its interactions with the paralogs p63 and p73 (gain of function effect) (11). This weakens the apoptotic propensity and expression of p63 and p73 transcribed genes in cells (12). Destabilizing mutants can be partly or fully functional at sub-physiological temperatures (4,13,14). For instance, the V143A mutant of DBD aggregates at physiological temperatures, but binds DNA non-specifically at sub-physiological temperatures. The aggregation kinetics of a destabilizing mutant Y220C (15,16) suggest that the aggregation competent state results from a slow rate-determining first step followed by rapid polymerization of the unfolded DBD (15). Indeed, even the WT p53 DBD is inherently unstable, unfolds at temperatures slightly above physiological temperatures (∼44°C) and has a low kinetic stability with a half-life of just ∼9 min at physiological temperature (13,17,18) Computational and experimental studies suggest that the strand S9 (residues 251–257) (Figure 1) of the DBD is involved in the nucleation process that leads to aggregation (11,19,20). However, a recent report presents a more complex picture, with multiple regions in DBD demonstrated to mediate aggregation (21).
Figure 1.

DNA binding domain (DBD) with secondary structures labeled (PDB ID- 2XWR_A). Green sphere represents Zn and the residues (Cys176(SG), Cys238(SG), Cys242(SG) and His 179(ND1)) coordinated to Zn are shown in sticks. The aggregation prone region in p53 DBD is present in strand S9 and is colored in magenta. The N and C termini of the domain are labeled. Figure on right shows dominant conformational states of the S6–S7 turn in p53 DBD. WT (blue) and V143A (red) DBD from MD simulations (at 310 K) superimposed to highlight differential conformational sampling of the S6–S7 turn in these two forms of the domain. Atoms, which are labeled on the S6–S7 turn and S9–S10 turn, are used to quantify the conformational sampling of this turn by measuring the distance between them. WT DBD samples a ‘closed’ state whereas V143A DBD samples an ‘open’ state of the S6–S7 turn.
Elegant genetic experiments have identified several second site mutations that could revert the effects of destabilizing mutations and restabilize mutant p53 (22–27). These observations inspired the discovery of ligands that stabilized mutant p53 by inducing it to adopt a WT conformation (3,5,28–31). PRIMA-1, a compound already in clinical trials, is thought to stabilize p53 by alkylating cysteine residues near the L1/S3 region of the DBD (32–34). The same site was also shown to accommodate Stictic acid, which was shown to be more efficient than PRIMA-1 at stabilizing p53 (32). In Y220C, another destabilizing mutant of DBD, a pocket resulting from the Tyr to Cys mutation was successfully targeted by small molecules (29,35–37). A different approach targeted the DNA binding surface of mutant DBD with peptides, speculating that the peptides will act as chaperones and stabilize mutant p53 and subsequently would be competed out in the presence of natural binding partners of p53 (38–40). Some mutants are thought to unfold because they lose zinc (41). A small molecule ZMC1, has been shown to act as a zinc-mettalochaperone, increasing the intracellular concentrations of zinc, and enhancing zinc binding to DBD (42,43). Recently, a peptide was designed to block aggregation by targeting the APR (residues 251–257) and shown to rescue the function of p53 in R248 and R175 mutants of p53 (20). There still remains a compelling need to identify new ways to rescue the function of p53. Although a ‘global suppressor motif’ made up of N235, N239 and S240 has been proposed (44), it was found to rescue only half of the most common p53 oncogenic mutations. This suggests that there likely are different pathways of destabilization of DBD. However, is it possible that there is a common initial event that triggers the unfolding of the DBD? If so, can it offer opportunities that enable a new mode of therapeutic targeting towards the prevention of unfolding of p53?
To begin our investigation, we first query whether the DBD is a good model system for our studies. Amongst the various reports in the literature, there are some that state that the DBD is a good model to recapitulate some properties of full length p53 such as destabilization and unfolding, while other reports suggest that it is necessary to study the dynamics of p53 in the context of a tetrameric unit composed of full length p53 monomers. This contradiction has been pointed out by (45) where it is inferred that the thermodynamics of the process are likely the same irrespective of whether the DBD alone is used or full length p53 is used. Further, (45) also report that misfolding of p53 occurs exclusively in the context of multiple units of DBD. They build a construct of p53 DBD and the tetrameric unit to support that misfolding involves intramolecular DBD–DBD association within a single tetrameric molecule. However, they suggest that this association is promoted by destabilization of DBD (caused by mutation). Indeed, DBD–DBD association alone will not lead to misfolding, otherwise even the WT DBDs would misfold. Hence we hypothesize that the DBD is the site of certain initial events which presumably are enhanced in mutants, resulting in misfolding and subsequently aggregation. One region that has been highlighted as important is the hydrophobic strand S9, which is usually buried, but in the mutants, under local perturbations, is exposed and nucleates aggregation. We apply molecular dynamics simulations to the WT and mutant DBDs to capture these initial destabilizing structural changes/unfolding events. We further aim to mine these changes towards the identification of transient pockets which could be drugged to stabilize p53 in the native conformation and prevent further downstream unfolding and aggregation. Previous computational efforts to understand the dynamics of aggregating mutants of p53 DBD have successfully identified structural perturbations in mutant p53 or even molecules that have resulted in the stabilization of mutant p53 (29,32,46).
We carry out molecular dynamics (MD) simulations of WT and the temperature sensitive V143A mutant of DBD; the latter has been reported to be less thermodynamically stable than the WT by about ∼3.7 kcal/mol (47). We observe that the S6–S7 turn of DBD samples different conformations in the WT from those in the destabilizing mutants, which appears to be associated with the thermodynamic stability of the protein at a particular temperature. This landscape of the S6–S7 turn is also seen in several additional simulations that we carry out (Supplementary Table S1) on four other destabilizing mutants (E258V, R110L, R175H and R248Q) which have been reported to aggregate through strand S9 (11). All the simulations reveal a common structural mechanism as an initiating event towards destabilization and are also characterized by a cryptic pocket which offers the potential to be targeted towards stabilizing the conformations of the mutants towards the WT state.
MATERIALS AND METHODS
Molecular dynamics simulation protocol
Monomers of the WT and mutant p53 DNA Binding Domain (DBD) (residues 94–293) were simulated using the amber ff99SB force field (48). The crystal structure of WT DBD (PDB id 2AHI (49), chain A, 1.85 Å resolution) was used for the modeling studies. This chain has only one missing residue (Ser94) and only 3 residues (Ser95, Lys292, and Gly293) have one missing atom each. The missing coordinates of the atoms/residues of Ser94, Ser95 and Gly293 were grafted from the structure with PDB id 3IGL (50) (1.8 Å resolution) and for Lys292 were taken from the structure with PDB id1GZH (51) (2.6 Å resolution). The Modloop program (52) was used to refine the coordinates of these missing atoms/residues. For creating the mutant structures, the mutation was introduced in the WT DBD using the program PyMOL (53) and the rotamers with minimal clashes were chosen.
The programs PDB2PQR (54,55), MOLPROBITY (56) and WHATIF (57) were used to generate the likely protonation states of amino acids. The final protonation states (Histidines with protonation on ND1 were residues 178, 214, 233, while those with protonation on NE2 were residues 115, 168, 179, 193) were chosen after visual inspection. ACE and NME residues were used to cap the N-terminal and C-terminal residues respectively. Zinc is coordinated to four residues in the L2 and L3 loops, including Cys176(SG), Cys238(SG), Cys242(SG) and His 179(ND1). We used the bonded model of Zinc (58) and covalently bonded it to these four residues as it preserves the tetrahedral coordination which has been reported to be crucial for L2–L3 stability and also for the DBD interactions with DNA (14). The structures were solvated in cubic boxes of TIP3P water molecules (59). The minimum distance between any protein atom and the edge of the water box was 12 Å. The systems were then neutralized by the addition of appropriate numbers of Na+ and Cl- ions.
We used the SANDER module of Amber 12 (and later switched to Amber 14 for speed) to perform minimization and molecular dynamics (MD) simulations. Minimization was carried out using 1000 steps each of steepest descent followed by conjugate gradient methods. Subsequently, the systems were gradually heated to 310 K. This was followed by a 2 ns NPT equilibration and a subsequent production run in the NPT ensemble for 500 ns using a GPU implementation of PMEMD (60). The SHAKE algorithm (61) was used to constrain bonds involving hydrogen atoms. Particle Mesh Ewald method (62) was used to calculate the long range electrostatic interactions beyond a distance of 8 Å. We used the Langevin thermostat to regulate the temperature (63). Across the simulations, the temperature fluctuates around the average (preset/target temperature) with a standard deviation of ∼3 K (data not shown). A weak coupling with a relaxation time of 1 ps is used to maintain constant pressure (1.0 bar) in the simulations. We applied an integration time step of 2 fs; coordinates were saved at an interval of 50 ps. The convergence of the simulations was checked using RMSD plots (Supplementary Figure S13).
Several variants of the DBD simulations (details in Supplementary Table S1) have been carried out. The crystal structures were used as the starting structures for the simulations of thermodynamically stable paralogs of p53 i.e. p63 (PDB id 3QYM, 3.2 Å resolution) and p73 (PDB id 3VD2, 4 Å resolution). The goal, here is not to investigate global unfolding but local conformational changes that could potentially destabilize the DBD and trigger subsequent unfolding.
Umbrella sampling simulations
The free energy associated with the conformational state of the S6–S7 turn of DBD was computed from the Umbrella sampling simulations. The distance measure used to characterize the conformational state of S6–S7 turn (distance between CZ atom of Arg209 and backbone carbonyl O atom of Asp259) is used as a reaction coordinate. A harmonic restraint of 1 kcal/mol/Å2 acts as the biasing potential along this reaction coordinate to considerably enhance the sampling of the phase space. The equilibrium values for distances ranging from 6 to 28 Å were sampled at intervals of 1 Å (thus resulting in 23 windows). Each window of the umbrella sampling MD is run for 50 ns. The distance distributions are used to derive the potential of mean force (PMF) using Alan Grossfield's implementation of weighted histogram analysis method (WHAM) (64). The error bars for the PMF were computed from the umbrella sampling simulations by splitting each 50 ns MD for each window into three windows (20–30, 20–40, 20–50 ns) and computing the PMF for each window, then taking the average PMF and the corresponding error bars (as standard deviation).
Benzene mapping simulations
The difference in the exposure of APR, as measured using SASA, between the simulations of WT and the destabilizing mutants of DBD appeared to be quite small and it was difficult to discriminate between them. This is not surprising given the limitations of currently accessible timescales of simulations. Thus, we turned to fragment mapping simulations (65–67) which are a type of non-canonical MD where a protein is immersed in a water box at a defined concentration of fragment(s). Together with their use in locating novel cryptic pockets (68–75), they have also been used to induce conformational changes (76). The benzene probe is ideally suited to explore the possibility of revealing cryptic regions such as the hydrophobic APR (sequence- ILTIITL). We carried out benzene mapping simulations for the WT and five destabilizing mutants of DBD including V143A, E258V, R110L, R175H and R248Q. A 0.2 M concentration of benzene was used and 10 simulations with random orientations of benzenes around the DBD were carried out for WT and for each mutant. Each system was allowed to evolve for 20 ns resulting in a total simulation time of 200 ns for each system. The benzene density was contoured at two times the density at which no benzenes are visible in the bulk solvent. This arbitrary cut off for displaying benzene densities filters out sites that are either unbound or weakly bound to benzenes.
Conformational differences between WT and mutant DBD: MUTINF
The differential conformational sampling between the WT and destabilizing mutants of DBD were examined by using the MUTINF analysis package. This method uses Kullback–Leibler Divergence (KL-div) and outputs the differential sampling of internal coordinates of residues between two simulations (77). The program splits a trajectory into a user defined number of windows (we used five windows) over which the distribution of internal coordinates is considered for computing KL-div. Splitting the trajectory into multiple small windows eliminates the weak correlations (not biologically meaningful) between the residues. Both the side-chain and backbone dihedrals are included for performing these calculations.
Structure based search of the identified pocket
We identified a pocket that seemed to be common to the initiating events that result in destabilizing the mutant structures. We next wondered whether we could find a ligand that would bind to this pocket and stabilize the conformation of mutant DBD against destabilization. We scanned the 101 609 proteins in the Protein Data Bank (PDB) to search for pockets similar to the pocket identified in our simulations using the CLICK algorithm (78,79). CLICK superimposes pairs of protein structures based on similarity of local structural packing, and thus is capable of aligning structures with dissimilar topologies, conformations, or even molecular types, thus revealing regions/pockets with similar shapes across proteins which otherwise may not share any global sequence/structure homologies (80). The residues Arg82, Glu88, Gln100, His101 (HIE), Asp115, Asp116, Arg117, Phe120, Arg121 and His122 (HID) were identified in our simulations to capture the shape of the pocket identified in the S6–S7 region. These pocket residues are structurally aligned to the dataset proteins by matching cliques with similar features of Cartesian coordinates and solvent accessibility. In this study, Cα and Cβ were chosen as the representative atoms. The root mean square deviation (RMSD) and structure overlap for each alignment between the pocket residues and dataset protein were monitored. In our study, structure overlap (also called equivalent positions) is defined as the percentage of the representative Cα and Cβ atoms of the pocket residues that are within 1.5 Å (RMSD cut-off = 1.5 Å) of the corresponding Cα and Cβ atoms of equivalent/aligned residues in the superimposed structure in the dataset proteins. A structure overlap cut-off of 95% was empirically chosen to determine the most accurate matches.
Testing D207A as a second site suppressor mutation
We identified D207A as a possible mutation that would stabilize the mutants to the WT conformation. Second-site reversion by the D207A mutant was evaluated using a reporter gene assay in p53-null H1299 cells. Details of the experiments are in the supplementary text. Plasmids expressing full length wild-type 53 and inactive mutants with or without the D207A mutation were transfected into H1299 cells and expression of a co-transfected p53 reporter gene measured. The D207A mutation in isolation resulted in slightly reduced p53 transactivation function compared to wild-type (Supplementary Figure S10). The single inactive mutants showed little or negligible activity as expected. The presence of D207A did not lead to second site reversion and rescue of p53 function for any of the mutants in this assay. Expression levels of all the p53 variants tested were comparable as shown in Western blot, indicating no gross destabilization by introduction of the D207A mutation.
Virtual screening of FDA approved drugs
A set of FDA approved drugs was screened against the pocket associated with the S6–S7 turn of the DBD that was seen during the simulations. The 2D structures of the library of the molecules were obtained from the ZINC database (81). The 3D structures of the FDA approved drugs were built using Maestro and minimized using the Macromodel module employing the OPLS-2005 force field (82) in Schrodinger 9.0 (83). All the inhibitors were then prepared with Ligprep that generates low energy tautomers and enumerates realistic protonation states at physiological pH.
The prepared inhibitors were docked into the pocket (we picked one conformation from each of the V143A, R110L R175H, R248Q and E258V mutant MD simulations) using Glide (84). These conformations were chosen such that they are maximally open and yet distinct from each other. A box of size 10 Ǻ × 10 Ǻ × 10 Ǻ for molecular docking centered on the selected pocket residues (Arg174, Gln192, Asp207 and His 214) was used to restrict the search space of each docked ligand. Default Glide settings were used to generate the grids. A rigid receptor and flexible ligands were used during the docking. The docked conformation of each ligand was evaluated using the Glide Extra Precision (XP) scoring function.
RESULTS AND DISCUSSION
Differential conformational sampling of S6–S7 turn in the WT and destabilizing mutants of p53 DBD
The S6–S7 turn (residues 208–213) of the DBD links the S6 and S7 β-strands (Figure 1). Simulations of WT and V143A mutant of DBD at 310K show distinct conformations of the S6–S7 turn (Figure 1) which we quantify by measuring the distance between the CZ atom of Arg209 on the S6–S7 turn and the backbone carbonyl oxygen of Asp259 on the S9–S10 turn. The rationale behind choosing this distance measure is that these atoms interact predominantly in the WT DBD and not in the mutant DBD.
This distance distributions across the simulations of the WT and V143A mutant of DBD (Figure 2A), at 310K (physiological temperature) peak at ∼5–7 Å (we refer to this as the ‘closed’ state of the S6–S7 turn) and ∼23 Å (we refer to this as the ‘open’ state of the S6–S7 turn) respectively. Existing experimental data suggests that WT is stable and retains DNA binding at 310 K while the V143A mutant is unstable and loses DNA binding ability (47,85). We thus formulate a hypothesis that the S6–S7 turn samples two dominant conformations—‘open’ and ‘closed’, that are predominant in the destabilizing (e.g. V143A) mutant(s) and WT DBD respectively, suggesting that the conformational state of the S6–S7 turn relates to the thermodynamic stability of the DBD.
Figure 2.

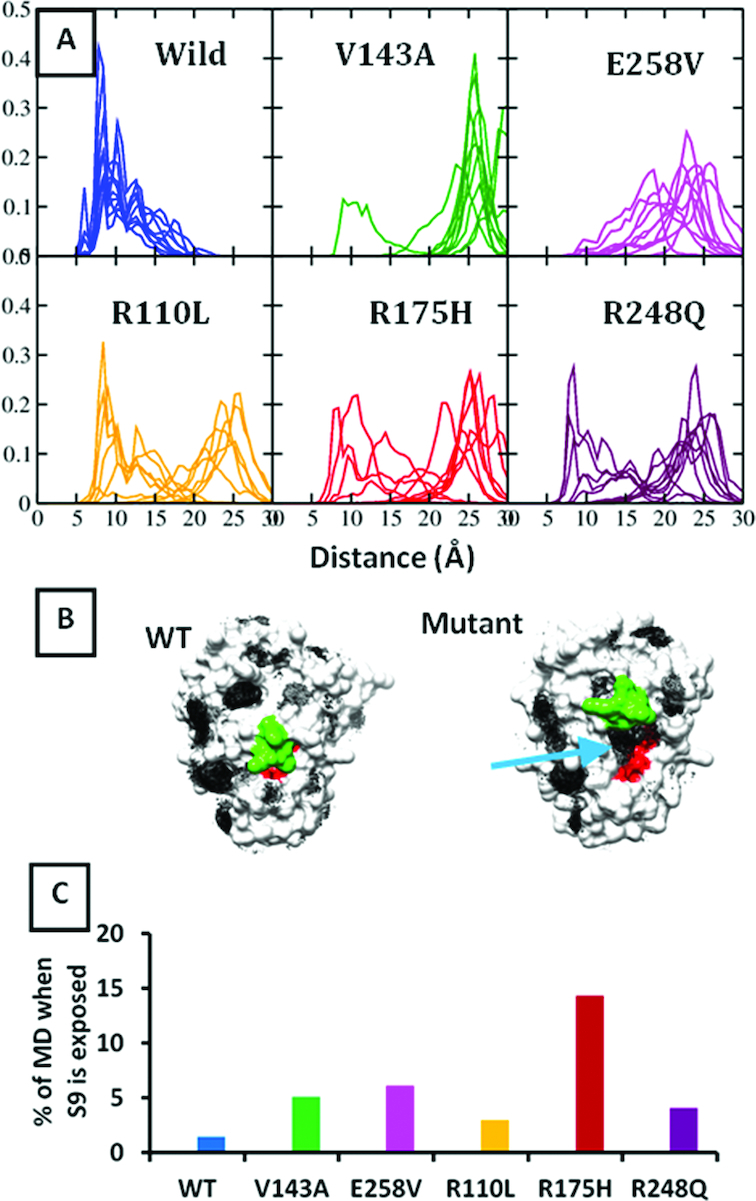
Quantifying the conformational state of S6–S7 turn of p53 DBD across simulations of DBD variants by measuring the distance between CZ atom of Arg209 (S6–S7 turn) and backbone carbonyl oxygen atom of Asp259 (S9–S10 turn). (A) Distance distribution for WT and V143A DBD. Final frame of V143A DBD simulation is ‘back-mutated’ to WT DBD and simulated for 600ns (as shown in the flowchart); (B) Structure of the p53 DBD. Residue positions undergoing a destabilizing mutation (only the ones studied here), are labeled in green boxes while positions of secondary site suppressor mutations are labeled in blue boxes; (C) Distance distribution for secondary site suppressor mutations of V143A DBD and; (D) additional destabilizing mutants of p53 DBD- E258V, R110L, R175H and R248Q. Simulations suggest that a thermodynamically stable DBD (e.g. WT) is likely to sample a ‘closed’ state whereas a destabilized DBD (e.g. the destabilizing mutants) is likely to sample an ‘open’ state.
To examine the robustness of the observation that the V143A mutation is associated with the emergence of the ‘open’ states of the S6–S7 turn, we take the final structure from the MD trajectory of the V143A mutant at 310K and mutate A143 to V143 (corresponding to the WT) and run an independent (reassigned velocities) simulation. It is clear that the conformation of the S6–S7 turn shifts from the ‘open’ towards the ‘closed’ state as the simulation progresses (Figure 2A). The gradual transition from the ‘open’ to the ‘closed’ states could imply the presence of an energy barrier separating these two states (which we subsequently probe using umbrella sampling and is described below).
Brachmann et al. have identified three secondary site suppressor mutations for the V143A mutant of DBD (22) including N239Y, S240N and N268D. Of these, the N239Y and N268D point mutations in the V143A DBD were found to restore p53 activity at 37°C in yeast while the S240N was found to restore p53 activity at 30°C. This agrees well with the human reporter gene assays which show an increased transcriptional activity for the V143A-N239 and V143A-N268D double mutants. Using this data to benchmark our calculations, we simulate the following reversal mutants: V143A-N239Y, V143A-S240N, V143A-N268D at 310 K from the crystal structure (PDB id 2AHI (49), chain A as the starting conformation. We find that the V143A-N239Y and V143A-N268D simulations of DBD largely sample the wild-type like ‘closed’ conformation of the S6–S7 turn while the V143A-S240N simulation of DBD samples the mutant-type like ‘open’ conformation (Figure 2C), which correlates very well with the experimentally known activity/stability of these reversal mutants.
We next ask if the ‘open’ state of the S6–S7 turn could be a general feature of other destabilizing mutants of the DBD. We simulate the dynamics of four other aggregating hot spot mutants of DBD: E258V, R110L, R175H and R248Q (Figure 2D). We specifically chose these mutants since the APR is known to be residues 251–257 (11); the APR for the temperature sensitive V143A DBD is not known. E258V is in the immediate vicinity of the APR, ie strand S9, while R110L is part of the stand S1. R175H (destabilized by ∼3 kcal/mol (13)) and R248Q (destabilized by 2 kcal/mol (13)) are parts of loops L2 and L3 respectively (we could not find any reports of similar experimental data for E258V and R110L). The relative position of these mutations with respect to the aggregation prone region can be seen in Figure 2B. These mutations are present on very different regions of the DBD and are biochemically very diverse: V143A (large to small residue), E258V (anionic to hydrophobic residue), R110L (cationic to hydrophobic residue), R175H (large to small sidechain), R248Q (cationic to uncharged polar residue). The mutations were modeled onto the WT state. Simulations show that these four mutants of the DBD also sample ‘open’ conformations of the S6–S7 turn (Figure 2D). Together these simulations suggest that the ‘open’ conformation of the S6–S7 turn could indeed represent a destabilizing (or destabilized) state of the DBD, while the ‘closed’ conformation of the S6–S7 turn represents the stable WT-like state. Moreover, our observations are also in accord with a recent computational study where it was shown that the R248Q destabilizing mutant of the DBD samples a solvent exposed (‘open’) S6–S7 turn while the WT samples a more buried state (‘closed’) (46).
We next wondered whether the S6–S7 loop dynamics was conserved in the two homologs of p53, ie p63 and p73, both of which are known to be more thermostable (86,87). Previously, we used the distance between the CZ atom of Arg209 on the S6–S7 turn and the backbone carbonyl oxygen of Asp259 on the S9–S10 turn to characterize the ‘open’ and ‘closed’ conformational states of the S6–S7 turn in the DBD. However, we could not use the same metric to characterize the conformational states of the S6–S7 turn in the DBDs of p63 and p73 because of differences in the amino acids at the homologous positions in these paralogs: Arg209 in the p53 corresponds to Pro279 and Pro229 in p63 and p73 respectively. So, we measure the Cα-Cα backbone distance between residues at position 209 and 259 (of p53 DBD) and the corresponding residues in p63 and p73 (Figure 3). This backbone distance distribution is bimodal for the WT p53 DBD as well as for the p63 and p73 DBD whereas it is trimodal for the destabilizing mutants of p53 DBD (V143A, E258V, R110L, R175H and R248Q). We refer to the additional peak that is sampled almost exclusively in the destabilizing mutants as ‘far-open’ conformation (pink shaded area in Figure 3 at a distance of ∼25 Å). MD simulations of the WT p53 DBD, p63 and p73 DBD very rarely sample the ‘far-open’ state.
Figure 3.
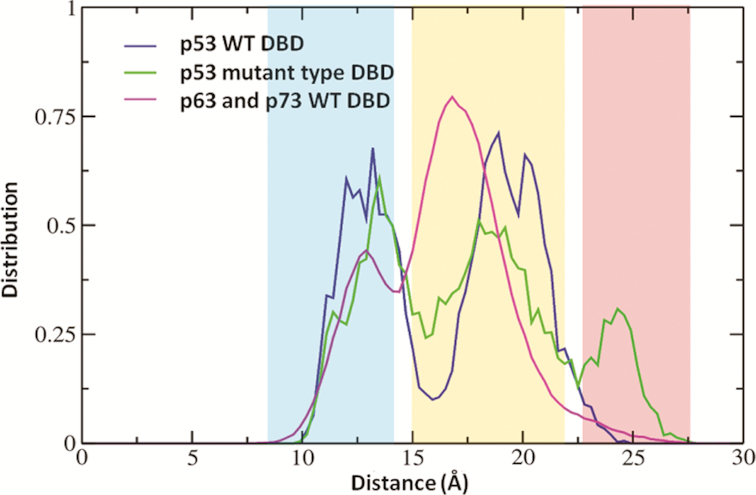
The distribution of the distance between Cα atoms of Arg209 and Asp259, measured across simulations of WT and destabilizing mutants (V143A, E258V, R110L, R175H and R248Q) of p53 DBD as well as of WT p63 and p73 DBD. This distance measure is used to characterize the conformational state of the S6–S7 turn in DBD. Shaded areas in the plot correspond to the conformational states of S6–S7 turn-‘closed’ (blue), ‘open’ (orange), ‘far-open’ (red).
To probe the existence and magnitude of energy barriers characterizing the closed-open transitions, we carried out umbrella sampling simulations using the distance between the CZ of Arg209 and the backbone carbonyl O of Asp259 as the reaction coordinate (this interaction has been key to our definition of the ‘closed’ conformation of the S6–S7 turn). It is clear (Supplementary Figure S1) that the WT DBD is likely to sample a ‘closed’ conformation while V143A is likely to sample an ‘open’ conformation of the S6–S7 turn.
A major cause for skepticism/concern in these simulations is that the Zinc is held with covalent linkages in all our simulations. However, it is known that the R175H mutant loses its ability to coordinate the Zinc ion (43,88). To examine this, we run a control simulation of WT and R175H DBD without the zinc ion (Supplementary Figure S2). In the simulations of WT DBD without zinc ion, we do observe dramatic conformational changes in loops L2 and L3 because they are no longer being held together by the zinc ion. However, the N-terminus samples a crystal like native conformation and the S6–S7 turn samples a ‘closed’ state, as we saw in the simulations with Zinc retained. In the R175H mutant, the N-terminus is displaced from its native position and the S6–S7 turn samples an ‘open’ state. These results suggest that the presence of Zinc in our simulations does not seem to influence either the conformations of the N-terminus of DBD or the conformational landscape of the S6–S7 turn, at least in the timescales of our simulations. Moreover, the findings of (89) about the conformation of S6–S7 turn sampled in the DNA bound state and apo state were shown to be invariant with respect to the electrostatics and van der Waals parameters for the non-bonded zinc model.
Biological/functional implications of the ‘open’ conformation of the S6–S7 turn of the DBD?
The APR residues 251–257 in the S9 ß-strand of the DBD (11), are largely buried in the native fold and must get exposed in order to nucleate aggregation (16). Based on the idea that quantifying the exposure of this APR can presumably help us gauge the extent of destabilization of the DBD, we measure the exposure of the strand S9 using Solvent Accessible Surface Area (SASA) of strand S9. However, SASA did not report noticeable differences in the exposure of this APR in the WT and destabilizing mutants. So we turn to enhanced sampling methods for which we chose benzene mapping, which has been used to identify cryptic hydrophobic pockets in the binding sites of proteins (69,73,76). Benzene mapping simulations (Figure 4) show distinct benzene density near the strand S9 in the destabilizing mutants that contrasts with the minimal benzene density at the strand S9 seen in the WT DBD. It is apparent that the S6–S7 turn, regulates the accessibility of benzenes to the hydrophobic strand S9, by acting as a valve which can ‘open’ and ‘close’. In the ‘open’ state (predominantly observed in the mutants) (Figures 4A and 4B), the S9 strand is accessible to the benzenes while in the ‘closed’ state (which WT samples exclusively), the S9 strand is largely buried and inaccessible to the benzenes. To quantify the exposure of strand S9 using benzene densities, we compute the number of unique benzene moieties within 7 Å of any heavy atom from strand S9 and set an arbitrary cut off of ≥3 benzenes to characterize the exposure of the strand. (Figure 4C) shows the percentage of simulation frames where the strand S9 is exposed. It is thus clear that strand S9 is considerably more exposed in the mutants than in the WT. Higher benzene densities at the APR in the destabilizing mutants capture the experimentally known higher propensity for their APR to be exposed and therefore the enhanced aggregation propensity.
Figure 4.
Results from benzene mapping simulations- (A) Distance distribution between CZ atom of Arg209 (S6–S7 turn) and backbone carbonyl oxygen atom of Asp259 (S9–S10 turn). (B) Benzene densities (black mesh) around WT and a representative mutant p53 DBD. APR is shown in red. S6–S7 turn is shown in green. Blue arrow points to the density near the APR S9 in the mutant. (C) Quantifying exposure of APR S9 by measuring the benzene moieties in its vicinity (detailed protocol in described where the figure is referred to).
We next look at the interactions that stabilize the ‘closed’ and ‘open’ states of the S6–S7 turns (Figure 5). The sidechain of Arg209 makes a hydrogen bond with the backbone carbonyl oxygen of Asp259 and a salt bridge with the side chain of Glu258 in the ‘closed’ state (largely observed in the WT DBD. In the ‘open’ state (largely observed in the destabilizing mutants of DBD), Arg209 is stabilized by a salt bridge with Asp207 instead. Additionally, one of the destabilizing mutants (also simulated here) that is known to aggregate is E258V. This is quite interesting as it suggests that the Arg209-Glu258 salt bridge is lost in this mutant (which is what we observe in the simulations) and that the loss of this interaction might well underlie the loss of stability in the E258V mutant of the DBD. This strongly implies the importance of the Arg209-Glu258 salt bridge interaction in stabilizing the DBD.
Figure 5.

Interactions that stabilize the ‘closed’ (A) and ‘open’ (B) states of the S6–S7 turn in WT and V143A mutant forms of p53 DBD respectively. Residue 143 is shown in red sticks. The ‘closed’ state is populated in simulations of the WT DBD while the ‘open’ state is largely populated in the simulations of destabilizing mutants.
Traditionally, the markers for WT and mutant conformations were the ability of each to uniquely bind to antibodies Pab1620/Pab246 and PAb240/DO11/DO12 respectively (90–93). Supplementary Figure S3 shows the epitopes of these antibodies mapped onto the DBD. We compute the solvent accessible surface area for these epitopes in the MD simulations of the WT and mutant variants of DBD (Supplementary Figures S3 and S4). Unfortunately we do not see any significant differences in the exposure of these epitopes between the WT and mutant forms of the protein. This suggests that the conformational changes and epitope exposures required for antibody recognition occur at timescales that are much longer than those simulated here. Alternatively, it is also possible that other structural modifications are responsible for altered antibody binding. These could include post-translational modifications and/or interactions with other domains that may modulate the structural dynamics or, indeed the initial encounter complex of the DBD with the antibody may itself result in some local induced changes that may result in the exposure of the epitope; for example something similar has been hypothesized for the binding of an antibody to an otherwise buried epitope in EGFR in (94).
A detailed simulation study followed by extensive bioinformatics analysis recently suggested that the ‘closed’ state of S6–S7 turn is presumably a low-energy state of the DBD that binds DNA while the ‘open’ state of the S6–S7 turn of DBD might represent an energetically unfavorable state (89). In that work (89), ‘closed’ and ‘open’ states are referred to as ‘A/D’ and ‘B/C’ respectively. They elegantly relate this state to bind negative regulators of p53 transcriptional activity such as Aurora-A kinase, Ku70. The authors also showed that simulations of the DBD with Ser215 phosphorylation- that is known to destabilize the protein (95–98), result exclusively in the sampling of an ‘open’ state by the S6–S7. We too simulated and confirmed this observation by phosphorylating Ser215 and running 500 ns MD in triplicates. We tried both the protonation states for the phosphorylated Ser215: PO2-OH and PO3, both of which sample an ‘open’ state of the S6–S7 turn (Supplementary Figure S5). Together, these observations strongly associate the ‘open’ state of the S6–S7 turn of DBD with the loss of native conformation and onset of destabilization in the DBD.
We next examine the N-terminus of the DBD as it wraps across the APR or strand S9, which is buried in the WT conformation. It is clear that the N-terminus must be displaced from its native position for the strand S9 to be exposed to solvent (16). Fersht et al. have shown that the extended N-terminus of the DBD (residues 91–293) stabilizes the DBD by ∼2 kcal/mol (99). However, all the simulations reported here have been carried out on residues 94–293 of the DBD. Motivated by this we decided to investigate the consequences on our findings when we use the extended N-terminus in our starting models. We examine this for the WT and two of the most common destabilizing mutants of DBD, i.e. R248Q and R175H. We observe that the N-terminus in the simulations of the mutant DBD is more likely to sample an ‘open’ state as compared to the WT DBD (Supplementary Figure S6). Importantly, while the N-terminus of WT DBD samples a native conformation, it is displaced into a non-native conformation in the mutants, thereby implying the onset of destabilization in the mutants (Figure 6). It is also possible that the initial steps of the unfolding process that we speculate, where the N-terminal region of the DBD peels away from the rest of the surface of the DBD, may adopt disordered states, subsequently resulting in conformational templating, prion-like aggregates, as it engages with other monomers.
Figure 6.

Final frames from the 500ns triplicate simulations of the p53 DBD (residues 91–293) are shown. The N-terminus is shown in ribbon representation and the rest of the protein is shown in surface. It is apparent that the N-terminus in the WT DBD samples a crystal-like native conformation whereas the N-terminus in the destabilizing mutants of the DBD- R175H and R248Q is displaced from its native position into a non-native conformation.
Finally, the umbrella sampling simulation of V143A where the S6–S7 turn is sampled starting from the ‘open’ state towards the WT like ‘close’ state, captures the stabilization of N-terminus and loop L3 (Supplementary Figure S7 and Movie Mov1). As mentioned previously, N-terminus is prone to being displaced into a non-native conformation when the mutant adopts an‘open’ state for the S6–S7 turn. Similarly, sometimes, loop L3 was also observed to adopt a non-native conformation. As the S6–S7 turn deviates from the ‘open’ state towards the ‘close’ state (in the umbrella sampling MD of V143A) other regions of the V143A mutant DBD too appear to assume crystal like native conformations and therefore imply stabilization of the entire DBD as the S6–S7 turn adopts a ‘closed’ conformation.
Together, the analysis strongly suggests that the ‘open’ conformation of the S6–S7 turn of DBD (predominantly observed in the destabilizing mutants) represents a destabilized state of the protein and might be an initial event in the onset of unfolding and aggregation in the DBD.
A network of interacting residues that regulates the conformation of the S6–S7 turn
Simulations of the DBD suggest that the S6–S7 turn assumes ‘open’ and ‘closed’ states in the destabilizing mutants and WT forms respectively. We next proceeded to identify a path, if any, describing the ‘closed’ to ‘open’ state conformational change. For this, we perform Kullback-Leibler divergence (KL-div) calculations on the MD ensembles of the WT and destabilizing mutants of the DBD using the program MUTINF (77,100). KL-div characterizes the differential sampling of dihedral angles of residues between two MD trajectories.
We compare the distribution of internal coordinates of the residues between the MD trajectories of the WT and the destabilizing mutants V143A, E258V, R110L, R175H and R248Q (Figure 7). The residues with a high degree of divergence (between the WT and mutant DBD) are located in loops L2, L3 and strands S6, S7. This correlates well with the available experimental data on the NMR of the destabilizing mutants R248Q and V143A (101).
Figure 7.

Structures of p53 DBD colored according to divergence value derived from Kl-div computations across simulations of WT and mutant DBDs. Mutation sites are shown with a red arrow.
The residues in the Zinc binding site have the largest values of divergence. Experimental data suggesting that the loss of Zinc from the DBD is a major factor for the destabilization and subsequent aggregation is only available for the R175H mutant (43); in any case, the Zinc remains covalently linked in our simulations and hence we cannot comment on this aspect of destabilization.
A tabulation of the divergence calculations for several aggregating mutants (Supplementary Table S2) shows the residues that consistently sample different dihedral angles between the WT and aggregating mutants. Visualization allowed us to identify Arg174, Glu180, Gln192, Asp207, Arg209 and His214 as critical to the ‘opening’ of the S6–S7 turn. Figure 8 and movie Mov2 shows the time series plots for the distance and the dihedral reaction coordinates involving this subset of residues for the V143A DBD. It is quite apparent how the perturbations in one reaction coordinate trigger a downstream change in another. To begin with, residues Arg174–Glu180–Gln192 appear to form an interacting triad. During the course of the simulation, this triad is perturbed (Plot A in Figure 8). The loss of the Gln192–Arg174 interaction is followed by a change in Gln192, His214 and Asp207 χ1 dihedral angles (Plot B in Figure 8). This culminates in the ‘opening’ of the S6–S7 turn as can be seen by the distance between CZ of Arg209 and the backbone carbonyl O of Asp259. Finally, this change displaces the N-terminus (measured by the Val172–Asp259 distance) which then adopts a non-native conformation (Plot C in Figure 8). Recent computational analysis (89) reported the existence of allostery between the DNA binding region (Loop L1) and the S6–S7 turn of the DBD. Our simulations and the KL-div computations are in agreement with their observations and also identify a probable route along which the changes in conformations of residues regulate the conformational state of the S6–S7 turn.
Figure 8.

A change in the interaction pattern of a subset of residues across MD simulations causes ‘opening’ of the S6–S7 strand in the destabilizing mutants. Structure of the V143A DBD is shown on the left. Plots (A), (B) and (C) on the right show how a perturbation in the reaction coordinate (involving the residues labelled in the structure on the left) (dihedral/distance) trigger a change in another reaction coordinate across a 500 ns MD (x-axis). Plot (A) shows that the triad of three interacting residues (R174, E180 and Q192) is destabilized. Plot (B) records the following changes in the side chain dihedrals of the vicinal residues (Q192, D207, H214). Subsequently the S6–S7 strand assumes an ‘open’ conformation (Plot (C)) displacing the N-terminus into a non-native conformation (as shown in the structure on the left).
To test the hypothesis that a stable Arg174–Glu180–Gln192 interaction triad is vital for the ‘closed’ conformation of the S6–S7 turn, we introduced a double mutation (R174A–E180A) in the WT DBD and carried out a 500 ns simulation. The interaction triad is no longer stable and Gln192 loses interactions with residues Ala180 and Ala174. Subsequently, the S6–S7 turn assumes an ‘open’ conformation (Supplementary Figure S8). Furthermore, the loop L3 is observed to assume a non-native conformation in this double mutant of DBD. However, single mutation simulations of R174A and E180A sample a ‘closed’ conformation of the S6–S7 turn (data not shown) thereby suggesting that the loss of Gln192 interactions with both Arg174 and Glu180 might be crucial to triggering the changes leading to the loss of the ‘closed’ conformation.
Several lines of evidence underscore the significance of these residues which are involved in this intricate network of interactions. Arg174 interacts with an N-terminal residue Trp91 and is known to stabilize DBD by ∼2 kcal/mol (99); indeed, a stable and less flexible Arg174 might be crucial to maintaining this interaction with Trp91 (Supplementary Figure S9). A very exciting piece of evidence for the importance of these ‘network-residues’ comes from an examination of the DBD of p53 of the subterranean blind mole rat (Spalax) that leads a hypoxic life and is not prone to cancer. Striking differences between the Spalax and human p53 map to residues 174 and 209, which form the two ends of the interaction network (Figure 8), identified here. Both 174 and 209 are Arg in humans but Lys in Spalax. A double mutant of Arg at these two positions in Spalax p53 is known to be five times more potent in promoting growth arrest (102,103) underscoring the significance of Arg at these two positions. Additionally, Arg174 and Arg209 have been directly implicated in cancer: R174L has been noted in breast, colon, lung and liver cancers, while an Arg209 mutation (to Lys among others) often exists in several cancers like skin, colon and oesophagus. Finally, these network residues are quite conserved across other species (Supplementary Figure S10). Since, these network residues are not directly involved in any intermolecular interactions of p53 DBD (to the best of our knowledge), it is likely that they are critical to the conformational regulation of the S6–S7 turn and hence, to the structural viability of the DBD.
Experimental tests on full length p53
Analysis of the network residues that regulate the S6–S7 turn conformation showed that in the simulations of WT, the side chain of Arg209 interacts with the backbone carbonyl oxygen of Asp259 on the S9–S10 turn, stabilizing the ‘closed’ state. In contrast, Arg209 loses this interaction in the ‘open’ state (and is stabilized by D207). This suggested that disrupting the Arg209-Asp207 interaction (by making the D207A mutant for example) in the mutant DBD might shift the conformation of S6–S7 turn towards the WT-like ‘closed’ state. To examine this, the following double mutants were expressed as full length human p53 and tested for function: V143A-D207A, E258V-D207A, R110L-D207A, R175H-D207A and R248Q-D207A. However all of them failed to show activity suggesting that D207A is not a second site suppressor mutation (Supplementary Figure S11), at least in the context of full length p53. Furthermore, Asp207 is conserved across evolution (Supplementary Figure S10) and is perhaps critical to protein function (in the figure the D207A mutant has lost 30% of its activity) and hence any perturbation is not tolerated.
Identifying a novel druggable pocket in the S6–S7 turn of p53 DBD
The next question we asked is whether a pocket can be identified in the conformational landscape of the mutants which can be stabilized by a ligand, thus preventing unfolding. It is well recognized that stabilizing the destabilizing mutants of p53 is a potential approach to treat cancer. We found a pocket (quantified using a geometric criteria as shown in Figure 9 B) that exists mostly in the mutants where the S6–S7 turn occupies the ‘open’ conformation (Figure 9 and Movie Mov4); it is less populated in the simulations of the WT DBD (Movie Mov3). Supplementary Table S3 shows the occupancy of the pocket in the simulations of the WT and several destabilizing mutants of DBD. The largest occupancy of the open pocket is seen for the R248Q destabilizing mutant. The amino acids lining this pocket are: Arg174, Glu192, Asp207, Asp208, Arg209, Asn210, Thr211, Phe212, Arg213 and His214. The pocket is hydrophobic at the base while the walls of the pocket have charged moieties.
Figure 9.

Characterizing the novel S6–S7 pocket. (A) The final frame of the V143A mutant type DBD is taken as a representative to highlight the S6–S7 pocket. The residues lining the pocket are indicated. (B) Residues used to quantify the pocket are shown in p53 DBD. Two dihedral angles (arrows) and a distance measure (dotted line), shown in the figure on right, are used to define a geometric criteria to characterize the pocket.
We wondered if this pocket is druggable, and can, upon occupation by a suitable ligand, stabilize the DBD close to its functional WT conformation. There are some features in favor of this pocket's potential as a druggable site (Figure 10). For instance, we identified a yet unreported dehydron in the S6–S7 turn (208Asp-213Arg) in the simulations (which is absent in the X-ray crystal structures of DBD). Dehydrons are backbone hydrogen bonds in proteins that are prone to hydration (104), are concentrated at protein-protein/ligand interfaces (105–107) and their presence has proved immensely useful in the structure guided design of inhibitors (108,109). A recent study also implicated the role of dehydrons in thermodynamic stabilities and aggregation tendencies in p53 family members (110). Thus a small molecule could potentially bind to the S6–S7 pocket. In addition, Ф-value analysis (suggestive of the extent of local unfolding) of the DBD (37) suggested that Val216 and Val217, residues that are present at the base of this pocket, exhibit the highest values of Фu (Ф for unfolding) implying significant local unfolding in this region. This further suggests that these two residues (as well as the vicinal residues) may be responsive to compounds designed to stabilize the DBD and inhibit unfolding and aggregation. Thirdly, benzene mapping simulations reveal distinct benzene densities in the pocket for the WT and the mutants. Fourthly, we carried out fragment screening using FTmap (111,112) on one of the mutant structures (where the pocket is maximally open) taken from the final frame of the simulation and identified this pocket as the dominant cluster where a range of fragments are able to dock in high density. Finally, while this pocket is not observable in the crystal structures of DBD, it is partially formed in some of the conformations of NMR structures of WT DBD (PDB ID: 2FEJ). Therefore we proceeded to explore the possibility of finding a ligand that could fit into this pocket.
Figure 10.

Computationally probing the druggability of the S6–S7 pocket. (A) Dehydrons (green dashes) mapped onto the final frame from the simulation of V143A destabilizing mutant of the p53 DBD. Two new dehydrons (208Asp–213Arg) in the S6–S7 turn of the domain are identified and highlighted in circle. (B) Fragment mapping carried out on a mutant p53 DBD conformation taken from the MD simulations (where the pocket is open) using FTMap identified the S6–S7 pocket as the primary pocket with the largest cluster of a range of fragments. (C) Benzene mapping simulations of the mutant p53 DBD identified a cluster of benzenes at the S6–S7 pocket. The S6–S7 turn is colored in green. (D–F) Structure based search of the S6–S7 pocket in p53 DBD identified small molecules that are bound to a similar pocket in beta-lactoglobulin-dodecane trimethylamine, Dodecyl sulfate and [N,N-di(pyridin-2-yl-kappaN)dodecanamide]rhodium respectively. (G) Pocket as observed in the WT NMR structure (PDB ID: 2FEJ).
To do this, we adopted a strategy to scan for shape-similarity in the structural database of proteins. We scanned the entire Protein Data Bank (PDB) to search for similar (structurally and/or chemically) pockets with the hope that one of them might already have been crystallized with a suitable ligand bound. Our pocket is defined by residues Arg174, Glu180, Gln192, His193(HIE), Asp207, Asp208, Arg209, Phe212, Arg213 and His214 (HID). Using the program CLICK (see methods) we identified 3 structures of beta-lactoglobulin (PDB ids 4IB7, 4IBA and 4KII) with similar pockets that had the ligands dodecane trimethylamine, dodecyl sulfate and [N,N-di(pyridin-2-yl-kappaN)dodecanamide]rhodium bound. Identifying three compounds bound to a pocket with similar topology further suggests that the identified S6–S7 pocket may potentially be druggable.
Of the 2798 FDA approved compounds screened against the pockets, ∼100 compounds could not be docked due to lack of topological and chemical complementarity between the S6–S7 pocket and the compound. The top 100 compounds from each of the 5 screens (for the mutants V143A, R110L, R175H, R248Q, E258V) were chosen (based on the glide docking score). The glide docking score (which is also the binding energy) was within 1 kcal/mol for the top 100 compounds. We found that among these top 100 compounds, Supplementary Figure S12, Baclofen was found to bind to all the 5 mutants of DBD. The glide docking score for this compound was in the range of –4.9 to –6.5 kcal/mol.
These observations provide compelling evidence to suggest that the identified pocket in DBD is druggable and can be harnessed to thermodynamically stabilize DBD mutants.
Testable hypothesis to probe loss of function in destabilizing mutants of p53 DBD
Based on the observations from the simulations in this study, the following experiments could be carried out to probe the proposed hypotheses:
In the simulations, an interacting triad formed by Arg174–Glu180–Gln192 is broken, as the S6–S7 loop assumes the ‘open' state in the mutants. Hence the R174A-E180A double mutant should accelerate the transition to the ‘open’ state and result in lowering the melting temperature of the DBD (Supplementary Figure S8). (b) Although we have not carried out any investigations of residue His214 located at the base of the S6–S7 pocket, its importance in modulating the dynamics of S6–S7 turn is compelling given its conservation in all the orthologs of p53 (Supplementary Figure S10), the H214Q mutant has been shown to be more potent at inducing apoptosis than the WT and is regarded to be a ‘super p53′ (113), and additionally, is a Glutamine in the more thermostable paralogs p63 and p73. It is possible that H214Q may be a powerful second site suppressor (we could not find any reports of this). (c) The observation that the ‘open’ state of S6–S7 turn is associated with a pocket that in our simple virtual screen appears to accommodate molecules such as dodecane trimethylamine and/or Baclofen, and recent experimental data on the Ф-value analysis of DBD suggesting that this region is perturbed by compounds that are aimed to stabilize DBD (37), suggests that simple thermal shift experiments could be carried out to probe whether these molecules can stabilize the mutant DBDs and if successful, would open a new avenue to restabilize mutant p53.
CONCLUSIONS
A subset of the mutations in TP53 is known to destabilize the protein, cause unfolding and aggregation and is linked to cancer pathologies. Although, most of these mutations are monoallelic and missense in nature, a dominant negative effect can abrogate the function of the WT allele (9). It is well recognized that stabilizing the mutant p53 to reactivate its WT function can allow us to specifically target cancer/tumor cells (37,114). However, the diverse nature of these mutations in the TP53 gene makes restoring normal levels of native p53 in cells as a strategy to treat cancers very challenging. Finding solutions to this problem requires a mechanistic understanding of the unfolding and aggregation pathway of p53.
The DNA Binding Domain (DBD) of p53 houses most of the high frequency oncogenic mutations, as well as an Aggregation Prone Region (APR), and in several studies, has been shown to be a good model system to recapitulate the behavior of full length p53, including aggregation. Based on this hypothesis, we carried out extensive MD simulations of the WT and numerous destabilizing mutants of the DBD, and observe differences in conformational behavior of the S6–S7 turn between the WT and the destabilizing mutants. Although there is some degree of overlap between the conformations of the S6–S7 turn sampled in the WT and in the mutants, overall the data suggests that the WT is characterized largely by a ‘closed' conformation of the S6–S7 turn while the mutant DBD preferentially adopts an ‘open’ conformation. This observation was tested by several control simulations and additional rigorous free energy calculations, and also was in agreement with some published computational and experimental reports. Upon probing the structural mechanisms underlying these differences, it was found that there were distinct differences between the dynamics of specific amino acids in the WT and mutant DBD; these residues were part of a network of interactions and govern the conformational state of the S6–S7 turn. These residues belong to the loop L2, helix H1 and ß-strands S6 and S7. Thus, we hypothesize a probable mechanism of the loss of function in the destabilizing mutants of the DBD. The Nterminal region of the DBD is largely associated with the rest of the DBD in the WT and begins to peel away from the surface in the mutant. This initial event is associated with the adoption by the S6–S7 turn of an ‘open’ conformational state. Together the incurred destabilization eventually results in unfolding of the DBD and likely aggregation.
This ‘open’ state of the S6–S7 turn resulted in the formation of a pocket in the mutant DBD. Ф-value studies from literature, an analysis of dehydrons in the ensemble structures (performed in this study), ligand mapping simulations, structure based searches, analysis of the reported NMR structures, and a screen of FDA approved drugs provided compelling evidence that the pocket is very likely druggable and functional.
The failure to observe stabilization of one of the mutants through the design of a second site suppressor mutation in an experimental study by our collaborators could either reflect upon the failure of the DBD (at least of this mutant) to recapitulate the complex behavior of full length p53 in the experimental construct or could result from the fact that any mutation at that conserved position is not tolerated. Nevertheless we hope that our observations will inspire experimental groups to test our observations.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Dr Dilraj Lama for critical discussion and useful insights as well as to all the members of the Verma lab for fruitful discussions regarding the data and helpful criticism of the work. M.N. Nguyen would like to thank A*STAR Joint Council Office (JCO) Career Development Award [15302FG145] for support. We thank BMRC-A*STAR for support and NSCC for providing computational support.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Research Scholarship Award by Bioinformatics Institute-NTU/SCE Joint PhD Program (to M.R.P.); A*STAR’s Biomedical Sciences Institute (BMSI). Funding for open access charge: BII, A*STAR.
Conflict of interest statement. Chandra S. Verma and Srinivasaraghavan Kannan are the founder directors of Sinopsee therapeutics, a biotech company developing molecules for therapeutic purposes; the current work has no conflict with the company.
REFERENCES
- 1. Vogelstein B., Lane D., Levine A.J.. Surfing the p53 network. Nature. 2000; 408:307–310. [DOI] [PubMed] [Google Scholar]
- 2. Schuijer M., Berns E.M.. TP53 and ovarian cancer. Hum. Mut. 2003; 21:285–291. [DOI] [PubMed] [Google Scholar]
- 3. Brown C.J., Lain S., Verma C.S., Fersht A.R., Lane D.P.. Awakening guardian angels: drugging the p53 pathway. Nat. Rev. Cancer. 2009; 9:862–873. [DOI] [PubMed] [Google Scholar]
- 4. Bullock A.N., Fersht A.R.. Rescuing the function of mutant p53. Nat. Rev. Cancer. 2001; 1:68–76. [DOI] [PubMed] [Google Scholar]
- 5. Brown C.J., Cheok C.F., Verma C.S., Lane D.P.. Reactivation of p53: from peptides to small molecules. Trends Pharmacol. Sci. 2011; 32:53–62. [DOI] [PubMed] [Google Scholar]
- 6. Olivier M., Eeles R., Hollstein M., Khan M.A., Harris C.C., Hainaut P.. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum. Mut. 2002; 19:607–614. [DOI] [PubMed] [Google Scholar]
- 7. Vyas P., Beno I., Xi Z., Stein Y., Golovenko D., Kessler N., Rotter V., Shakked Z., Haran T.E.. Diverse p53/DNA binding modes expand the repertoire of p53 response elements. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:10624–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Joerger A.C., Fersht A.R.. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 2008; 77:557–582. [DOI] [PubMed] [Google Scholar]
- 9. Brosh R., Rotter V.. When mutants gain new powers: news from the mutant p53 field. Nat. Rev. Cancer. 2009; 9:701–713. [DOI] [PubMed] [Google Scholar]
- 10. Joerger A.C., Rajagopalan S., Natan E., Veprintsev D.B., Robinson C.V., Fersht A.R.. Structural evolution of p53, p63, and p73: implication for heterotetramer formation. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:17705–17710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu J., Reumers J., Couceiro J.R., De Smet F., Gallardo R., Rudyak S., Cornelis A., Rozenski J., Zwolinska A., Marine J.C. et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011; 7:285–295. [DOI] [PubMed] [Google Scholar]
- 12. Gaiddon C., Lokshin M., Ahn J., Zhang T., Prives C.. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001; 21:1874–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bullock A.N., Henckel J., DeDecker B.S., Johnson C.M., Nikolova P.V., Proctor M.R., Lane D.P., Fersht A.R.. Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:14338–14342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bullock A.N., Henckel J., Fersht A.R.. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene. 2000; 19:1245–1256. [DOI] [PubMed] [Google Scholar]
- 15. Wilcken R., Wang G., Boeckler F.M., Fersht A.R.. Kinetic mechanism of p53 oncogenic mutant aggregation and its inhibition. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:13584–13589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang G., Fersht A.R.. First-order rate-determining aggregation mechanism of p53 and its implications. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:13590–13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ang H.C., Joerger A.C., Mayer S., Fersht A.R.. Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J. Biol. Chem. 2006; 281:21934–21941. [DOI] [PubMed] [Google Scholar]
- 18. Friedler A., Veprintsev D.B., Hansson L.O., Fersht A.R.. Kinetic instability of p53 core domain mutants: implications for rescue by small molecules. J. Biol. Chem. 2003; 278:24108–24112. [DOI] [PubMed] [Google Scholar]
- 19. Ghosh S., Ghosh D., Ranganathan S., Anoop A., P S.K., Jha N.N., Padinhateeri R., Maji S.K.. Investigating the intrinsic aggregation potential of evolutionarily conserved segments in p53. Biochemistry. 2014; 53:5995–6010. [DOI] [PubMed] [Google Scholar]
- 20. Soragni A., Janzen D.M., Johnson L.M., Lindgren A.G., Thai-Quynh Nguyen A., Tiourin E., Soriaga A.B., Lu J., Jiang L., Faull K.F. et al. A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell. 2016; 29:90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang G., Fersht A.. Multisite aggregation of p53 and implications for drug rescue. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:2634–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brachmann R.K., Yu K., Eby Y., Pavletich N.P., Boeke J.D.. Genetic selection of intragenic suppressor mutations that reverse the effect of common p53 cancer mutations. EMBO J. 1998; 17:1847–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Joerger A.C., Ang H.C., Veprintsev D.B., Blair C.M., Fersht A.R.. Structures of p53 cancer mutants and mechanism of rescue by second-site suppressor mutations. J. Biol. Chem. 2005; 280:16030–16037. [DOI] [PubMed] [Google Scholar]
- 24. Joerger A.C., Allen M.D., Fersht A.R.. Crystal structure of a superstable mutant of human p53 core domain. Insights into the mechanism of rescuing oncogenic mutations. J. Biol. Chem. 2004; 279:1291–1296. [DOI] [PubMed] [Google Scholar]
- 25. Nikolova P.V., Wong K.B., DeDecker B., Henckel J., Fersht A.R.. Mechanism of rescue of common p53 cancer mutations by second-site suppressor mutations. EMBO J. 2000; 19:370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Merabet A., Houlleberghs H., Maclagan K., Akanho E., Bui T.T., Pagano B., Drake A.F., Fraternali F., Nikolova P.V.. Mutants of the tumour suppressor p53 L1 loop as second-site suppressors for restoring DNA binding to oncogenic p53 mutations: structural and biochemical insights. Biochem. J. 2010; 427:225–236. [DOI] [PubMed] [Google Scholar]
- 27. Otsuka K., Kato S., Kakudo Y., Mashiko S., Shibata H., Ishioka C.. The screening of the second-site suppressor mutations of the common p53 mutants. Int. J. Cancer. 2007; 121:559–566. [DOI] [PubMed] [Google Scholar]
- 28. Foster B.A., Coffey H.A., Morin M.J., Rastinejad F.. Pharmacological rescue of mutant p53 conformation and function. Science. 1999; 286:2507–2510. [DOI] [PubMed] [Google Scholar]
- 29. Joerger A.C., Bauer M.R., Wilcken R., Baud M.G., Harbrecht H., Exner T.E., Boeckler F.M., Spencer J., Fersht A.R.. Exploiting transient protein states for the design of small-molecule stabilizers of mutant p53. Structure. 2015; 23:2246–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Selivanova G., Wiman K.G.. Reactivation of mutant p53: molecular mechanisms and therapeutic potential. Oncogene. 2007; 26:2243–2254. [DOI] [PubMed] [Google Scholar]
- 31. Wiman K.G. Pharmacological reactivation of mutant p53: from protein structure to the cancer patient. Oncogene. 2010; 29:4245–4252. [DOI] [PubMed] [Google Scholar]
- 32. Wassman C.D., Baronio R., Demir O., Wallentine B.D., Chen C.K., Hall L.V., Salehi F., Lin D.W., Chung B.P., Hatfield G.W. et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 2013; 4:1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bykov V.J., Issaeva N., Shilov A., Hultcrantz M., Pugacheva E., Chumakov P., Bergman J., Wiman K.G., Selivanova G.. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 2002; 8:282–288. [DOI] [PubMed] [Google Scholar]
- 34. Bykov V.J., Issaeva N., Selivanova G., Wiman K.G.. Mutant p53-dependent growth suppression distinguishes PRIMA-1 from known anticancer drugs: a statistical analysis of information in the National Cancer Institute database. Carcinogenesis. 2002; 23:2011–2018. [DOI] [PubMed] [Google Scholar]
- 35. Boeckler F.M., Joerger A.C., Jaggi G., Rutherford T.J., Veprintsev D.B., Fersht A.R.. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:10360–10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu X., Wilcken R., Joerger A.C., Chuckowree I.S., Amin J., Spencer J., Fersht A.R.. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013; 41:6034–6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang G., Fersht A.R.. Mechanism of initiation of aggregation of p53 revealed by Phi-value analysis. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:2437–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Friedler A., Hansson L.O., Veprintsev D.B., Freund S.M., Rippin T.M., Nikolova P.V., Proctor M.R., Rudiger S., Fersht A.R.. A peptide that binds and stabilizes p53 core domain: chaperone strategy for rescue of oncogenic mutants. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:937–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Issaeva N., Friedler A., Bozko P., Wiman K.G., Fersht A.R., Selivanova G.. Rescue of mutants of the tumor suppressor p53 in cancer cells by a designed peptide. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:13303–13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tal P, Eizenberger S., Cohen E., Goldfinger N., Pietrokovski S., Oren M., Rotter V.. Cancer therapeutic approach based on conformational stabilization of mutant p53 protein by small peptides. Oncotarget. 2016; 15:11817–11837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Meplan C., Richard M.J., Hainaut P.. Metalloregulation of the tumor suppressor protein p53: zinc mediates the renaturation of p53 after exposure to metal chelators in vitro and in intact cells. Oncogene. 2000; 19:5227–5236. [DOI] [PubMed] [Google Scholar]
- 42. Blanden A.R., Yu X., Wolfe A.J., Gilleran J.A., Augeri D.J., O’Dell R.S., Olson E.C., Kimball S.D., Emge T.J., Movileanu L. et al. Synthetic metallochaperone ZMC1 rescues mutant p53 conformation by transporting zinc into cells as an ionophore. Mol. Pharmacol. 2015; 87:825–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu X., Blanden A.R., Narayanan S., Jayakumar L., Lubin D., Augeri D., Kimball S.D., Loh S.N., Carpizo D.R.. Small molecule restoration of wildtype structure and function of mutant p53 using a novel zinc-metallochaperone based mechanism. Oncotarget. 2014; 5:8879–8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baroni T.E., Wang T., Qian H., Dearth L.R., Truong L.N., Zeng J., Denes A.E., Chen S.W., Brachmann R.K.. A global suppressor motif for p53 cancer mutants. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:4930–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lubin D.J., Butler J.S., Loh S.N.. Folding of tetrameric p53: oligomerization and tumorigenic mutations induce misfolding and loss of function. J. Mol. Biol. 2010; 395:705–716. [DOI] [PubMed] [Google Scholar]
- 46. Ng J.W., Lama D., Lukman S., Lane D.P., Verma C.S., Sim A.Y.. R248Q mutation-beyond p53-DNA binding. Proteins. 2015; 83:2240–2250. [DOI] [PubMed] [Google Scholar]
- 47. Joerger A.C., Ang H.C., Fersht A.R.. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:15056–15061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C.. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006; 65:712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kitayner M., Rozenberg H., Kessler N., Rabinovich D., Shaulov L., Haran T.E., Shakked Z.. Structural basis of DNA recognition by p53 tetramers. Mol. Cell. 2006; 22:741–753. [DOI] [PubMed] [Google Scholar]
- 50. Kitayner M., Rozenberg H., Rohs R., Suad O., Rabinovich D., Honig B., Shakked Z.. Diversity in DNA recognition by p53 revealed by crystal structures with Hoogsteen base pairs. Nat. Struct. Mol. Biol. 2010; 17:423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Derbyshire D.J., Basu B.P., Serpell L.C., Joo W.S., Date T., Iwabuchi K., Doherty A.J.. Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. EMBO J. 2002; 21:3863–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fiser A., Sali A.. ModLoop: automated modeling of loops in protein structures. Bioinformatics. 2003; 19:2500–2501. [DOI] [PubMed] [Google Scholar]
- 53. The PyMOL Molecular Graphics System, Version 2.0. 2010; Schrödinger, LLC. [Google Scholar]
- 54. Dolinsky T.J., Czodrowski P., Li H., Nielsen J.E., Jensen J.H., Klebe G., Baker N.A.. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007; 35:W522–W525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dolinsky T.J., Nielsen J.E., McCammon J.A., Baker N.A.. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004; 32:W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen V.B., Arendall W.B. 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C.. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D, Biol. Crystallogr. 2010; 66:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vriend G. WHAT IF: a molecular modeling and drug design program. J. Mol.Graph. 1990; 8:52–56. [DOI] [PubMed] [Google Scholar]
- 58. Peters M.B., Yang Y., Wang B., Fusti-Molnar L., Weaver M.N., Merz K.M. Jr.. Structural survey of zinc containing proteins and the development of the Zinc AMBER Force Field (ZAFF). J. Chem. Theory Comput. 2010; 6:2935–2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983; 79:926–935. [Google Scholar]
- 60. Gotz A.W., Williamson M.J., Xu D., Poole D., Le Grand S., Walker R.C.. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012; 8:1542–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ryckaert J.-P., Ciccotti G., Berendsen H.J.. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977; 23:327–341. [Google Scholar]
- 62. Darden T., York D., Pedersen L.. Particle mesh Ewald: an N⋅ log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993; 98:10089–10092. [Google Scholar]
- 63. Loncharich R.J., Brooks B.R., Pastor R.W.. Langevin dynamics of peptides: the frictional dependence of isomerization rates of N-acetylalanyl-N'-methylamide. Biopolymers. 1992; 32:523–535. [DOI] [PubMed] [Google Scholar]
- 64. Grossfield A. WHAM: an implementation of the weighted histogram analysis method Version 2.0.7 http://membrane.urmc.rochester.edu/content/wham/.
- 65. Guvench O., MacKerell A.D. Jr.. Computational fragment-based binding site identification by ligand competitive saturation. PLoS Comput. Biol. 2009; 5:e1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Raman E.P., Yu W., Guvench O., Mackerell A.D.. Reproducing crystal binding modes of ligand functional groups using Site-Identification by Ligand Competitive Saturation (SILCS) simulations. J. Chem. Inform. Model. 2011; 51:877–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Foster T.J., MacKerell A.D. Jr., Guvench O.. Balancing target flexibility and target denaturation in computational fragment-based inhibitor discovery. J. Comput. Chem. 2012; 33:1880–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bakan A, Nevins N., Lakdawala A.S., Bahar I.. Druggability assessment of allosteric proteins by dynamics simulations in the presence of probe molecules. J. Chem. Theory Comput. 2012; 8:2435–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lama D., Brown C.J., Lane D.P., Verma C.S.. Gating by tryptophan 73 exposes a cryptic pocket at the Protein-Binding interface of the oncogenic eIF4E protein. Biochemistry. 2015; 54:6535–6544. [DOI] [PubMed] [Google Scholar]
- 70. Lexa K.W., Carlson H. A.. Full protein flexibility is essential for proper hot-spot mapping. J. Am. Chem. Soc. 2011; 133:200–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Prakash P., Hancock J.F., Gorfe A.A.. Binding hotspots on K-ras: consensus ligand binding sites and other reactive regions from probe-based molecular dynamics analysis. Proteins. 2015; 83:898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Seco J., Luque F.J., Barril X.. Binding site detection and druggability index from first principles. J. Med. Chem. 2009; 52:2363–2371. [DOI] [PubMed] [Google Scholar]
- 73. Tan Y.S., Sledz P., Lang S., Stubbs C.J., Spring D.R., Abell C., Best R.B.. Using ligand-mapping simulations to design a ligand selectively targeting a cryptic surface pocket of polo-like kinase 1. Angew. Chem. 2012; 51:10078–10081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Yang C.Y., Wang S.. Hydrophobic binding hot spots of Bcl-xL protein-protein interfaces by cosolvent molecular dynamics simulation. ACS Med. Chem. Lett. 2011; 2:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhu M., De Simone A., Schenk D., Toth G., Dobson C.M., Vendruscolo M.. Identification of small-molecule binding pockets in the soluble monomeric form of the Aβ42 peptide. J. Chem. Phys. 2013; 139:035101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tan Y.S., Spring D.R., Abell C., Verma C.S.. The application of Ligand-Mapping molecular dynamics simulations to the rational design of peptidic modulators of protein-protein interactions. J. Chem. Theory Comput. 2015; 11:3199–3210. [DOI] [PubMed] [Google Scholar]
- 77. McClendon C.L., Hua L., Barreiro A., Jacobson M.P.. Comparing conformational ensembles using the Kullback-Leibler divergence expansion. J. Chem. Theory Comput. 2012; 8:2115–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nguyen M.N., Madhusudhan M.S.. Biological insights from topology independent comparison of protein 3D structures. Nucleic Acids Res. 2011; 39:e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Nguyen M.N., Tan K.P., Madhusudhan M.S.. CLICK–topology-independent comparison of biomolecular 3D structures. Nucleic Acids Res. 2011; 39:W24–W28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nguyen M.N., Verma C.. Rclick: a web server for comparison of RNA 3D structures. Bioinformatics. 2015; 31:966–968. [DOI] [PubMed] [Google Scholar]
- 81. Irwin J.J., Shoichet B.K.. ZINC–a free database of commercially available compounds for virtual screening. J. Chem. Inform. Model. 2005; 45:177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Banks J.L., Beard H.S., Cao Y., Cho A.E., Damm W., Farid R., Felts A.K., Halgren T.A., Mainz D.T., Maple J.R. et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005; 26:1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Schrödinger Release 2017-4: Maestro. 2017; NY: Schrödinger, LLC. [Google Scholar]
- 84. Friesner R.A., Murphy R.B., Repasky M.P., Frye L.L., Greenwood J.R., Halgren T.A., Sanschagrin P.C., Mainz D.T.. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006; 49:6177–6196. [DOI] [PubMed] [Google Scholar]
- 85. Zhang W., Guo X.Y., Hu G.Y., Liu W.B., Shay J.W., Deisseroth A.B.. A temperature-sensitive mutant of human p53. EMBO J. 1994; 13:2535–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cañadillas J.M., Tidow H., Freund S.M., Rutherford T.J., Ang H.C., Fersht A.R.. Solution structure of p53 core domain: structural basis for its instability. Proc. Natl. Acad. Sci. U.S.A. 2006; 14:2109–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Brandt T., Kaar J.L., Fersht A.R., Veprintsev D.B.. Stability of p53 homologs. PLoS One. 2012; 7:e47889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Blanden A.R., Yu X., Loh S.N., Levine A.J., Carpizo D.R.. Reactivating mutant p53 using small molecules as zinc metallochaperones: awakening a sleeping giant in cancer. Drug Discov. Today. 2015; 20:1391–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Lambrughi M., De Gioia L., Gervasio F.L., Lindorff-Larsen K., Nussinov R., Urani C., Bruschi M., Papaleo E.. DNA-binding protects p53 from interactions with cofactors involved in transcription-independent functions. Nucleic Acids Res. 2016; 44:9096–9109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Coomber D.W., Ward R.L.. Isolation of human antibodies against the central DNA binding domain of p53 from an individual with colorectal cancer using antibody phage display. Clin. Cancer Res. 2001; 7:2802–2808. [PubMed] [Google Scholar]
- 91. Fraser J.A., Vojtesek B., Hupp T.R.. A novel p53 phosphorylation site within the MDM2 ubiquitination signal. I. phosphorylation at Ser269 in vivo is linked to inactivation of p53 function. J. Biol. Chem. 2010; 285:37762–37772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Selivanova G., Wiman K.G.. Reactivation of mutant p53: molecular mechanisms and therapeutic potential. Oncogene. 2007; 26:2243–2254. [DOI] [PubMed] [Google Scholar]
- 93. Wang P.L., Sait F., Winter G.. The ‘wildtype’ conformation of p53: epitope mapping using hybrid proteins. Oncogene. 2001; 20:2318–2324. [DOI] [PubMed] [Google Scholar]
- 94. Ng Y.Z., Kannan S., Lane D.P., Fuentes G., Verma C.S.. mAb806 binding to epidermal growth factor receptor: a computational study. Proteins. 2015; 83:153–168. [DOI] [PubMed] [Google Scholar]
- 95. Liu Q., Kaneko S., Yang L., Feldman R.I., Nicosia S.V., Chen J., Cheng J.Q.. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J. Biol. Chem. 2004; 279:52175–52182. [DOI] [PubMed] [Google Scholar]
- 96. Fraser J.A., Madhumalar A., Blackburn E., Bramham J., Walkinshaw M.D., Verma C., Hupp T.R.. A novel p53 phosphorylation site within the MDM2 ubiquitination signal: II. a model in which phosphorylation at SER269 induces a mutant conformation to p53. J. Biol. Chem. 2010; 285:37773–37786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cai Q., Xiao B., Si H., Cervini A., Gao J., Lu J., Upadhyay S.K., Verma S.C., Robertson E.S.. Kaposi's sarcoma herpesvirus upregulates Aurora A expression to promote p53 phosphorylation and ubiquitylation. PLoS Pathog. 2012; 8:e1002566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Warnock L.J., Raines S.A., Milner J.. Aurora A mediates cross-talk between N- and C-terminal post-translational modifications of p53. Cancer Biol. Ther. 2011; 12:1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Natan E., Baloglu C., Pagel K., Freund S.M., Morgner N., Robinson C.V., Fersht A.R., Joerger A.C.. Interaction of the p53 DNA-binding domain with its n-terminal extension modulates the stability of the p53 tetramer. J. Mol. Biol. 2011; 409:358–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. McClendon C.L., Friedland G., Mobley D.L., Amirkhani H., Jacobson M.P.. Quantifying correlations between allosteric sites in thermodynamic ensembles. J. Chem. Theory Comput. 2009; 5:2486–2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Wong K.B., DeDecker B.S., Freund S.M., Proctor M.R., Bycroft M., Fersht A.R.. Hot-spot mutants of p53 core domain evince characteristic local structural changes. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:8438–8442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Ashur-Fabian O., Avivi A., Trakhtenbrot L., Adamsky K., Cohen M., Kajakaro G., Joel A., Amariglio N., Nevo E., Rechavi G.. Evolution of p53 in hypoxia-stressed Spalax mimics human tumor mutation. Proc. Natl Acad. Sci. U.S.A. 2004; 101:12236–12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Pagano B., Jama A., Martinez P., Akanho E., Bui T.T., Drake A.F., Fraternali F., Nikolova P.V.. Structure and stability insights into tumour suppressor p53 evolutionary related proteins. PLoS One. 2013; 8:e76014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Fernandez A., Berry R.S.. Extent of hydrogen-bond protection in folded proteins: a constraint on packing architectures. Biophys. J. 2002; 83:2475–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Fernandez A. Functionality of wrapping defects in soluble proteins: what cannot be kept dry must be conserved. J. Mol. Biol. 2004; 337:477–483. [DOI] [PubMed] [Google Scholar]
- 106. Fernandez A., Scheraga H.A.. Insufficiently dehydrated hydrogen bonds as determinants of protein interactions. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Fernandez A., Scott R.. Dehydron: a structurally encoded signal for protein interaction. Biophys. J. 2003; 85:1914–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Accordino S.R., Rodriguez Fris J.A., Appignanesi G.A.. Wrapping effects within a proposed function-rescue strategy for the Y220C oncogenic mutation of protein p53. PLoS One. 2013; 8:e55123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Fernandez A., Sanguino A., Peng Z., Ozturk E., Chen J., Crespo A., Wulf S., Shavrin A., Qin C., Ma J. et al. An anticancer C-Kit kinase inhibitor is reengineered to make it more active and less cardiotoxic. J. Clin. Invest. 2007; 117:4044–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Cino E.A., Soares I.N., Pedrote M.M., de Oliveira G.A., Silva J.L.. Aggregation tendencies in the p53 family are modulated by backbone hydrogen bonds. Scientific Rep. 2016; 6:32535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Brenke R., Kozakov D., Chuang G.Y., Beglov D., Hall D., Landon M.R., Mattos C., Vajda S.. Fragment-based identification of druggable ‘hot spots’ of proteins using Fourier domain correlation techniques. Bioinformatics. 2009; 25:621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Kozakov D., Grove L.E., Hall D.R., Bohnuud T., Mottarella S.E., Luo L., Xia B., Beglov D., Vajda S.. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015; 10:733–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Kakudo Y., Shibata H., Otsuka K., Kato S., Ishioka C.. Lack of correlation between p53-dependent transcriptional activity and the ability to induce apoptosis among 179 mutant p53s. Cancer Res. 2005; 65:2108–2114. [DOI] [PubMed] [Google Scholar]
- 114. Rangel L.P., Costa D.C., Vieira T.C., Silva J.L.. The aggregation of mutant p53 produces prion-like properties in cancer. Prion. 2014; 8:75–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.