

Abstract
Background
Bardet‐Biedl syndrome (BBS) is an autosomal recessive pleiotropic disorder of the primary cilia that leads to severe visual loss in the teenage years. Approximately 80% of BBS cases are explained by mutations in one of the 21 identified genes. Documented causative mutation types include missense, nonsense, copy number variation (CNV), frameshift deletions or insertions, and splicing variants.
Methods
Whole genome sequencing was performed on a patient affected with BBS for whom no mutations were identified using clinically approved genetic testing of the known genes. Analysis of the WGS was done using internal protocols and publicly available algorithms. The phenotype was defined by retrospective chart review.
Results
We document a female affected with BBS carrying the most common BBS1 mutation (BBS1: Met390Arg) on the maternal allele and an insertion of a ~1.7‐kb retrotransposon in exon 13 on the paternal allele. This retrotransposon insertion was not automatically annotated by the standard variant calling protocols used. This novel variant was identified by visual inspection of the alignment file followed by specific genome analysis with an available algorithm for transposable elements.
Conclusion
This report documents a novel mutation type associated with BBS and highlights the importance of systematically performing transposon detection analysis on WGS data of unsolved cases.
Keywords: Bardet‐Biedl syndrome, BBS, BBS1, ciliopathy, human genome, mutation, repetitive element, SVA, transposable element
1. BRIEF REPORT
Bardet‐Biedl syndrome (BBS:MIM209900) is a complex disease characterized mainly by severe photoreceptor degeneration, truncal obesity, postaxial polydactyly, autism‐like behavior, cognitive impairment, hypogonadism, renal anomalies, among other secondary features (Gerth, Zawadzki, Werner, & Heon, 2008; Habibullah & Mohiuddin, 2009; Heon et al., 2005; Kerr, Bhan, & Heon, 2015; Weihbrecht et al., 2017). BBS is phenotypically and genetically heterogeneous, and demonstrates considerable overlap with other ciliopathies such as Joubert syndrome (JBST) (Beales, Elcioglu, Woolf, Parker, & Flinter, 1999; Billingsley et al., 2010; Branfield Day et al., 2015; Deveault et al., 2011; Gerth et al., 2008; Heon et al., 2016; Kerr et al., 2015). Biallelic mutations have been identified in at least 21 BBS genes (Heon et al., 2016; Khan et al., 2016), all of which are involved in primary cilia structure and/or function (Alvarez‐Satta, Castro‐Sanchez, & Valverde, 2017; Khan et al., 2016). Approximately 80% of the clinically examined BBS cases (Forsythe & Beales, 2015) have been associated with biallelic mutations in one of the 21 BBS genes, of which Bardet‐Biedl syndrome‐1 gene (BBS1: 209,901) is the most frequently mutated (Billingsley, Deveault, & Heon, 2011). Currently, reported mutations in BBS are as follows: missense, nonsense, frameshift, and nonframeshift variants; copy number variation; alternative splicing; and a few descriptions of complex rearrangements (Billingsley et al., 2011; Khan et al., 2016; Weihbrecht et al., 2017). In this study, we report a novel exonic retrotransposon insertion into BBS1, which represents a new type of mutational event for BBS patients and may play a role in the missing heritability of this condition. This study was approved by the Research Ethic Board of the Hospital for Sick Children and met the Tenets of the Declaration of Helsinki.
The proband was a female from a sibship of three, born to nonconsanguineous parents of European origin (Figure 1a). She presented with night blindness, constriction of visual field, and photophobia at the age of nine (Table 1). Distance visual acuity was reduced (right: 20/400, left: 20/600), and severe generalized rod and cone photoreceptor degeneration was documented on an electroretinogram (ERG) (Supporting information Figure S1). This correlated with the observed retinal structural changes (macular atrophic changes and pigmentary retinal changes and vessel attenuation; Figure 1b,c). It was not possible to perform a visual field test due to the developmental delay. However, based on the small residual central island of photoreceptors documented by optical coherence tomography (OCT), it is likely that the fields were very constricted (Figure 1c). Other features included obesity (BMI 37.6 at 13 years old), postaxial polydactyly, fatty infiltration of the liver, elevated lipids, autism, and absence‐type seizures (Table 1). The clinical phenotype was consistent with BBS (Beales et al., 1999; Daniels et al., 2012) and part of the phenotype variability associated with BBS1 mutations (Deveault et al., 2011).
Figure 1.
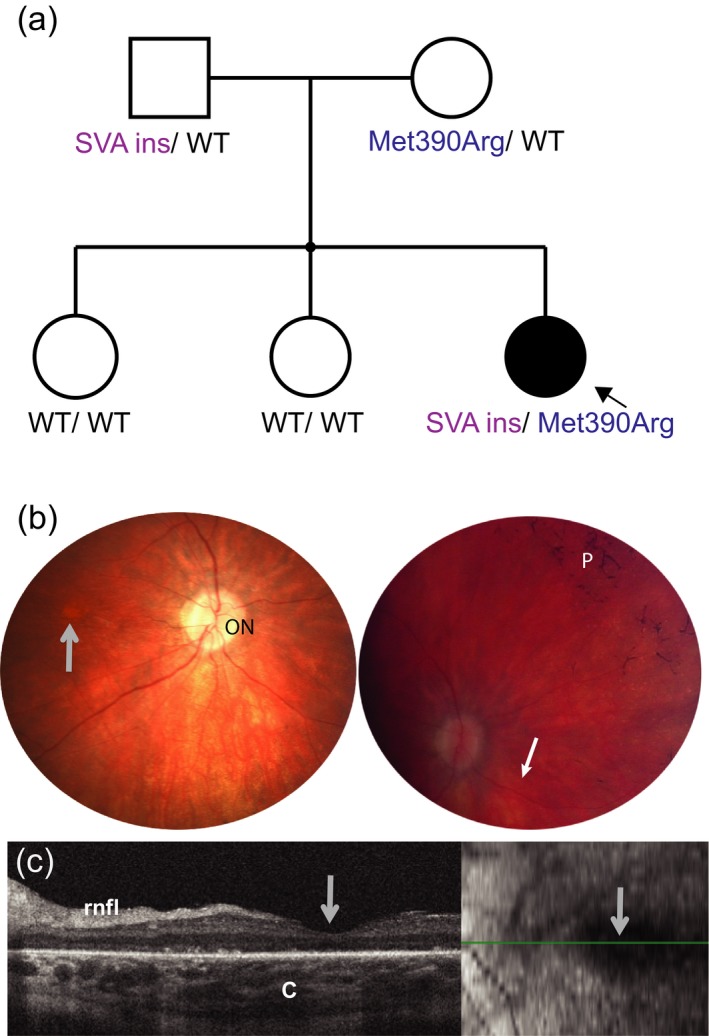
(a) Pedigree of the affected family and BBS1 variant segregation; (b) retinal photography of the right eye centered on the optic nerve (ON, left image) and on the nasal retina (right image). Gray arrow locates the foveal area which shows atrophy (center of the retina), and a blunted reflex. Filled white arrow (right) points toward narrowed (very thin) vessels, p: bone spiculing pigmentary deposits typical of retinal degeneration. (c) Optical coherence tomography (OCT) of the right eye centered on the fovea (gray arrow) shows markedly disruption of outer retinal layers including the photoreceptor outer and inner segments, and the outer nuclear layer. rnfl: retinal nerve fiber layer, c: choroidal layer. Insert to the right shows the area scanned (green line). The quality of the images was limited due to the patient's ability to participate
Table 1.
Phenotype summary of the proband
| Onset symptoms | Age 9–10 years (nyctalopia, visual field constriction) |
|---|---|
| VA (14 years) | 20/400 (right eye), 20/600 (left eye) |
| Anterior segment | Normal |
| Retinal exam (15 years) | Macular atrophy, vessel attenuation, bone spiculing changes |
| ERG amplitude (1 years) | Severe rod‐cone dystrophy |
| Neurological findings (age 13 years) | Absent Seizures, autism |
| Normal Brain MRI | |
| Head circumference: 57 cm | |
| Kidney (age 13 years) | Normal structure and function |
| Liver (age 13 years) | Fatty Infiltration, normal transaminases |
| Lipidsa (age 13 years) | Cholesterol 6.4 mmol/L (⇑) |
| Triglyceride 8.63 mmol/L (⇑) | |
| Heart | Situs Solitus, levocardia |
| Spleen | Mild splenomegaly |
| Digits | Postaxial polydactyly × 3 limbs |
| Weight | BMI 38.9 |
| Menarche | 13 years |
| Developmental | Delayed |
| Other | Recurrent ear infections, strabismus |
Normal lipid values; cholesterol: 0.65–2.5 mmol/L, triglyceride: 0.40–103 mmol/L.
Genetic testing initially done using a CLIA‐approved laboratory performing next‐generation sequencing on 19 BBS genes revealed a heterozygous BBS1 (NM_024649.4) variant: c.1169 T>G, p.Met390Arg, the most common mutation in BBS1. Segregation analysis of the variant by Sanger sequencing confirmed that the mutation was heterozygous in the proband and that the mother was a carrier. No other potentially pathogenic mutation was found in BBS1 or other BBS‐associated genes. No significant copy number variation (CNV) was detected by microarray analysis.
Whole genome sequencing (WGS) was performed, and single‐nucleotide variants (SNVs), insertions/deletions (indels), and CNVs were annotated with our standard in‐house variant filtration pipeline approach (Supporting information Data S1 and Figure S2). The five genes with biallelic variants (three genes with homozygous variants and two genes with compound heterozygous variants) were excluded because these were either platform based errors (ZFPM1, SLCA6), inframe deletion in a polymorphic region (UBXN11), or the variants did not segregate with the disease phenotype (DNAH8, IGSF3) (Supporting information Figure S2). Since BBS best fits the proband's phenotype, a CNV analysis was performed for BBS1 (for which a heterozygous missense mutation was previously identified; Met390Arg), and for two other ciliopathy‐related genes (for which heterozygous variants were also identified in the proband's WGS filtering (Supporting information Figure S2): NPHP4 (NM_015102:exon26:c.3644+1G>C), CEP78 (NM_032171:exon15:c.1801–1G>C)). CNV was not detected for any of the three genes (Supporting information Data S1, Figure S2), but during the visualization of the binary alignment map (BAM) for these three genes, we identified discrepant reads (split reads) in exon 13 of BBS1 only (Figure 2a). The split portion of the forward reads (127 bp) was 98.5% identical to a retrotransposon of the category SINE/VNTR/Alu subtype F (SVAF) in RepeatMasker (http://www.repeatmasker.org). This novel transposable element (TE) insertion on BBS1 had not yet been reported in the database of retrotransposon insertion polymorphisms dbRIP (http://dbrip.brocku.ca/searchRIP.html). The corresponding mate pairs of the split reads were mapped at different parts of the human genome (hg19) where TEs of the similar category had been mapped. The TE insertion (BBS1:NM_02649.4:c.1214–1215ins (1700_1800);1198_1214) was validated using PCR and Sanger sequencing and verified to be paternally inherited (Figure 2b,c, Supporting information Data S1, Figure S3, Table S1). The final sequence (GenBank MH395756, Supporting information Data S2) had higher similarity (95%–97.7%) with multiple SVA types B, D, and mostly F from the human whole genome sequence (hg19 in UCSC—https://genome.ucsc.edu/‐ using RepeatMasker track). The allele carrying the SVA insertion was expressed in patient‐derived lymphoblast cell line; these results showed that BBS1 protein is translated up to AA residue 406, followed by insertion of 46 new AA residues (corresponding to the SVA) before reaching stop codon (Supporting information Figure S4).
Figure 2.
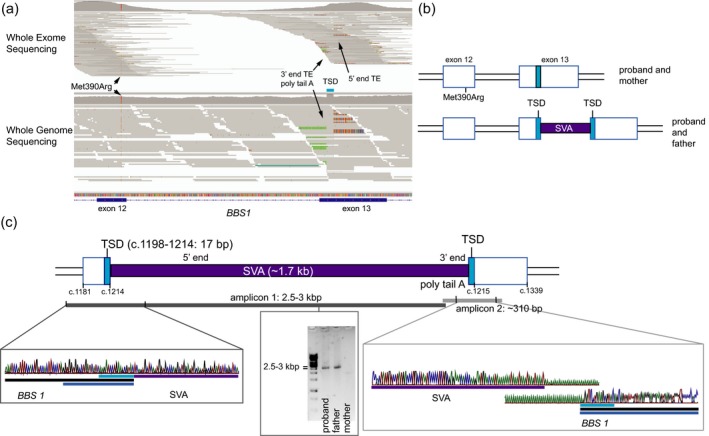
Disease‐causing mutations in the proband with retrotransposon insertion detail. Pathogenic mutations are represented in purple, Target Site Duplication (TSD) is shown in cyan, and exon is represented in blue. (a) Schematic representation of the retrotransposon insertion in exon 13 of BBS1 in Integrative Genome Viewer using the whole exome and the whole genome sequence alignment maps, respectively (Robinson et al., 2011). Nucleotides matching the reference are gray; unmatched A, T, C, and G nucleotides to reference are color coded in green, red, blue, and orange, respectively. (b) Schematic diagram of the two alleles comprising both BBS1 mutations (Met390Arg and the exonic TE insertion in exon 13) observed in the proband and parents. (c) Detailed characterization of the novel SVAF insertion detected in this study and schematic representation of the results used to assemble the mutation map. Each PCR product is represented as a bar overlapping the region they comprise in the mutation map. Chromatograms from Sanger sequencing represent the BBS1‐SVAF junction
The shorter PCR segment amplification (Supporting information Data S1, Figure S3) did not identify this particular SVA insertion in our cohort of 24 genetically unsolved BBS probands. Additionally, the insertion was not found in 2,504 control individuals from 1,000 genomes phase three databases, for which MELT, an automatic TE detection method was applied (Gardner et al., 2017; Rishishwar, Tellez Villa, & Jordan, 2015).
We validated our finding using Mobster (Thung et al., 2014), an algorithm designed to detect transposable element insertions from next‐generation sequencing data (Supporting information Data S1). We then applied this method to five of unsolved BBS probands for which WGS data were available. However, no potentially disease‐causing TEs were identified for them.
We could not automatically detect the SVA insertion from the whole exome sequencing (WES) of the proband using the same protocols applied to WGS, mostly due to the lower number of reads bearing the BBS1 and SVA junction in WES. However, some of those split reads can be observed in the WES reads map (Figure 2a).
2. DISCUSSION
We report for the first time an SVA retrotransposon insertion in a BBS1 exon as a disease‐causing mechanism for BBS.
DNA transposable elements (TE) are mobile DNA elements that occupy nearly half of the human Genome (http://genome.ucsc.edu), yet are poorly explored (Kazazian & Moran, 2017). The aggregate length of these sequences is 40 times greater than protein‐coding exons (Kazazian & Moran, 2017; Lander et al., 2001; Mills, Bennett, Iskow, & Devine, 2007). Active TEs account for around 0.02% of the human genome and belong to the retrotransposon class (Kaer & Speek, 2013; Lander et al., 2001). Retrotransposons can be transcribed into RNA, reverse transcribed into cDNA, which is then reinserted in the genome at a new location (Kaer & Speek, 2013; Lander et al., 2001). It is estimated that one de novo insertion happens every 10–100 live births. The most abundant retrotransposon in the human genome is the Long Interspersed Nuclear Element (LINE; 17%), followed by Alu (11%) and composite SVA repeats (0.2%) (Kaer & Speek, 2013; Lander et al., 2001; Solyom & Kazazian, 2012).
The role of transposable elements in disease has only been minimally explored. However, these can be pathogenic by inserting themselves into coding or regulatory portions of genes, facilitating chromosomal rearrangements, duplications, and deletions, among other mechanisms (Mills et al., 2007). Only a few cases of human retinal degeneration have been reported to be caused by inserted retrotransposons; an Alu insertion on exon 9 of MAK, and intronic Alu insertion in OPA1 (Bujakowska, White, Place, Consugar, & Comander, 2015; Gallus et al., 2010; Tucker et al., 2011). A few cases have also been reported in animals; an L1 insertion in rodent Nr2e3 (Chen, Rattner, & Nathans, 2006), an intronic SINE insertion in canine FAM161A (Downs & Mellersh, 2014), and an intronic insertion in equine TRPM1 (Bellone et al., 2013). Recently, two cases affected with nonBBS ciliopathy phenotypes were associated with retrotransposon insertions; a L1 in exon 7 of CC2D2A and an Alu in exon 16 of ALMS1 (Takenouchi et al., 2017; Taschner et al., 2016). A recent study of BBS cases also found that 40% (6 out of 15) of the CNV deletions detected in their cohort (in BBS1, BBS4, BBS5, and IFT74) were mediated by Alu–Alu recombination (Lindstrand et al., 2016).
The high amount of active TE in the human genome, and the technical difficulties associated with their detection suggests that they are likely an underexplored category of disease‐causing mutational events. Therefore, the implementation of TE detection protocols in the standard pipeline for search of disease‐causing variants can significantly increase diagnostic yield and improve patient care. Traditional methods such as candidate gene sequence and microarray analysis are unsuitable to capture large insertions such as TEs. Large‐scale sequence analysis such as whole exome and whole genome sequencing allows identification of TEs but require specific algorithms for automatic detection (Ewing, 2015; Gardner et al., 2017; Thung et al., 2014), which are not systematically incorporated in the current practice (Takenouchi et al., 2017). The WES is less efficient to WGS in automatic TE detection because some of the genomic segments including the junction between the gene of interest and inserted TE are eliminated at the exome capture step (Tucker et al., 2011). Therefore, standard implementation of Mobster or other TE detection methods should be planned forward for WGS data. Currently, there is only one “population frequency database” of nonreference TEs insertions available, making it difficult to determine the prevalence of this mutational event in disease or in the general population (Gardner et al., 2017; Rishishwar et al., 2015). The incorporation of TE insertion frequencies from the 1,000 genomes, and other additional population databases, will allow for more efficient identification of potentially pathogenic TEs. Further larger‐scale implementation of such detection methods will allow for the discovery of more disease‐causing variants and provide a clearer idea of the role of TEs in the population and disease processes.
CONFLICT OF INTEREST
The authors have no conflict of interest to declare.
Supporting information
ACKNOWLEDGMENTS
We thank the patient family for participating in our study. We also thank Dr. Andrew Paterson for critical discussion and Dr. Rola Ba‐Abbad for technical participation in an earlier phase of the study.
Tavares E, Tang CY, Vig A, et al. Retrotransposon insertion as a novel mutational event in Bardet‐Biedl syndrome. Mol Genet Genomic Med. 2019;7:e521 10.1002/mgg3.521
Funding information
Mira Godard Research Fund; Canadian foundation Fighting Blindness; Career Development Award (Foundation Fighting Blindness, USA); SickKids Ophthalmology Research Fund; Accelerator Grant in Genomic Medicine, McLaughlin Centre, University of Toronto.
REFERENCES
- Alvarez‐Satta, M. , Castro‐Sanchez, S. , & Valverde, D. (2017). Bardet‐Biedl syndrome as a chaperonopathy: Dissecting the major role of chaperonin‐like BBS proteins (BBS6‐BBS10‐BBS12). Frontiers in Molecular Biosciences, 4, 55 10.3389/fmolb.2017.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beales, P. L. , Elcioglu, N. , Woolf, A. S. , Parker, D. , & Flinter, F. A. (1999). New criteria for improved diagnosis of Bardet‐Biedl syndrome: Results of a population survey. Journal of Medical Genetics, 36(6), 437–446. [PMC free article] [PubMed] [Google Scholar]
- Bellone, R. R. , Holl, H. , Setaluri, V. , Devi, S. , Maddodi, N. , Archer, S. , … Brooks, S. A. (2013). Evidence for a retroviral insertion in TRPM1 as the cause of congenital stationary night blindness and leopard complex spotting in the horse. PLoS ONE, 8(10), e78280 10.1371/journal.pone.0078280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley, G. , Deveault, C. , & Heon, E. (2011). BBS mutational analysis: A strategic approach. Ophthalmic Genetics, 32(3), 181–187. 10.3109/13816810.2011.567319. [DOI] [PubMed] [Google Scholar]
- Billingsley, G. , Bin, J. , Fieggen, K. J. , Duncan, J. L. , Gerth, C. , Ogata, K. , … Heon, E. (2010). Mutations in chaperonin‐like BBS genes are a major contributor to disease development in a multiethnic Bardet‐Biedl syndrome patient population. Journal of Medical Genetics, 47(7), 453–463. 10.1136/jmg.2009.073205. [DOI] [PubMed] [Google Scholar]
- Branfield Day, L. , Quammie, C. , Heon, E. , Bhan, A. , Batmanabane, V. , Dai, T. , & Kamath, B. M. (2015). Liver anomalies as a phenotype parameter of Bardet‐Biedl syndrome. Clinical Genetics, 10.1111/cge.12684. [DOI] [PubMed] [Google Scholar]
- Bujakowska, K. M. , White, J. , Place, E. , Consugar, M. , & Comander, J. (2015). Efficient In Silico Identification of a Common Insertion in the MAK Gene which Causes Retinitis Pigmentosa. PLoS ONE, 10(11), e0142614 10.1371/journal.pone.0142614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Rattner, A. , & Nathans, J. (2006). Effects of L1 retrotransposon insertion on transcript processing, localization and accumulation: Lessons from the retinal degeneration 7 mouse and implications for the genomic ecology of L1 elements. Human Molecular Genetics, 15(13), 2146–2156. 10.1093/hmg/ddl138. [DOI] [PubMed] [Google Scholar]
- Daniels, A. B. , Sandberg, M. A. , Chen, J. , Weigel‐DiFranco, C. , Fielding Hejtmancic, J. , & Berson, E. L. (2012). Genotype‐phenotype correlations in Bardet‐Biedl syndrome. Archives of Ophthalmology, 130(7), 901–907. 10.1001/archophthalmol.2012.89. [DOI] [PubMed] [Google Scholar]
- Deveault, C. , Billingsley, G. , Duncan, J. L. , Bin, J. , Theal, R. , Vincent, A. , … Heon, E. (2011). BBS genotype‐phenotype assessment of a multiethnic patient cohort calls for a revision of the disease definition. Human Mutation, 32(6), 610–619. 10.1002/humu.21480. [DOI] [PubMed] [Google Scholar]
- Downs, L. M. , & Mellersh, C. S. (2014). An Intronic SINE insertion in FAM161A that causes exon‐skipping is associated with progressive retinal atrophy in Tibetan Spaniels and Tibetan Terriers. PLoS ONE, 9(4), e93990 10.1371/journal.pone.0093990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing, A. D. (2015). Transposable element detection from whole genome sequence data. Mobile DNA, 6, 24 10.1186/s13100-015-0055-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe, E. , & Beales, P. L. (2015). In Adam M. P., Ardinger H. H., & Pagon R. A. (Eds.), GeneReviews. Internet (https:, www.ncbi.nlm.nih.gov, books, NBK1363, ), (Eds.), Bardet‐Biedl Syndrome. Seattle: University of Washington.
- Gallus, G. N. , Cardaioli, E. , Rufa, A. , Da Pozzo, P. , Bianchi, S. , D'Eramo, C. , … Federico, A. (2010). Alu‐element insertion in an OPA1 intron sequence associated with autosomal dominant optic atrophy. Molcular Vision, 16, 178–183. [PMC free article] [PubMed] [Google Scholar]
- Gardner, E. J. , Lam, V. K. , Harris, D. N. , Chuang, N. T. , Scott, E. C. , Pittard, W. S. , … Devine, S. E. (2017). The Mobile Element Locator Tool (MELT): Population‐scale mobile element discovery and biology. Genome Research, 27(11), 1916–1929. 10.1101/gr.218032.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth, C. , Zawadzki, R. J. , Werner, J. S. , & Heon, E. (2008). Retinal morphology in patients with BBS1 and BBS10 related Bardet‐Biedl Syndrome evaluated by Fourier‐domain optical coherence tomography. Vision Research, 48(3), 392–399. 10.1016/j.visres.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habibullah, M. , & Mohiuddin, A. A. (2009). Bardet‐Biedl syndrome. Mymensingh Medical Journal, 18(1), 80–83. [PubMed] [Google Scholar]
- Heon, E. , Kim, G. , Qin, S. , Garrison, J. E. , Tavares, E. , Vincent, A. , … Sheffield, V. C. (2016). Mutations in C8ORF37 cause Bardet Biedl syndrome (BBS21). Human Molecular Genetics, 25(11), 2283–2294. 10.1093/hmg/ddw096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heon, E. , Westall, C. , Carmi, R. , Elbedour, K. , Panton, C. , Mackeen, L. , … Sheffield, V. C. (2005). Ocular phenotypes of three genetic variants of Bardet‐Biedl syndrome. American Journal of Medical Genetics. Part A, 132A(3), 283–287. 10.1002/ajmg.a.30466. [DOI] [PubMed] [Google Scholar]
- Kaer, K. , & Speek, M. (2013). Retroelements in human disease. Gene, 518(2), 231–241. 10.1016/j.gene.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Kazazian, H. H. Jr , & Moran, J. V. (2017). Mobile DNA in health and disease. New England Journal of Medicine, 377(4), 361–370. 10.1056/NEJMra1510092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr, E. N. , Bhan, A. , & Heon, E. (2015). Exploration of the cognitive, adaptive and behavioral functioning of patients affected with Bardet‐Biedl syndrome. Clinical Genetics, 89(4), 426‐433. 10.1111/cge.12614. [DOI] [PubMed] [Google Scholar]
- Khan, S. A. , Muhammad, N. , Khan, M. A. , Kamal, A. , Rehman, Z. U. , & Khan, S. (2016). Genetics of human Bardet‐Biedl syndrome, an updates. Clinical Genetics, 90(1), 3–15. 10.1111/cge.12737. [DOI] [PubMed] [Google Scholar]
- Lander, E. S. , Linton, L. M. , Birren, B. , Nusbaum, C. , Zody, M. C. , & Baldwin, J. … International Human Genome Sequencing, C (2001). Initial sequencing and analysis of the human genome. Nature, 409(6822), 860–921. 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Lindstrand, A. , Frangakis, S. , Carvalho, C. M. , Richardson, E. B. , McFadden, K. A. , Willer, J. R. , … Katsanis, N. (2016). Copy‐Number variation contributes to the mutational load of bardet‐biedl syndrome. American Journal of Human Genetics, 99(2), 318–336. 10.1016/j.ajhg.2015.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills, R. E. , Bennett, E. A. , Iskow, R. C. , & Devine, S. E. (2007). Which transposable elements are active in the human genome? Trends in Genetics, 23(4), 183–191. 10.1016/j.tig.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Rishishwar, L. , Tellez Villa, C. E. , & Jordan, I. K. (2015). Transposable element polymorphisms recapitulate human evolution. Mobile DNA, 6, 21 10.1186/s13100-015-0052-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, J. T. , Thorvaldsdottir, H. , Winckler, W. , Guttman, M. , Lander, E. S. , Getz, G. , & Mesirov, J. P. (2011). Integrative genomics viewer. Nature Biotechnology, 29(1), 24–26. 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solyom, S. , & Kazazian, H. H. Jr (2012). Mobile elements in the human genome: Implications for disease. Genome Medicine, 4(2), 12 10.1186/gm311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenouchi, T. , Kuchikata, T. , Yoshihashi, H. , Fujiwara, M. , Uehara, T. , Miyama, S. , … Kosaki, K. (2017). Diagnostic use of computational retrotransposon detection: Successful definition of pathogenetic mechanism in a ciliopathy phenotype. American Journal of Medical Genetics. Part A, 173(5), 1353–1357. 10.1002/ajmg.a.38167. [DOI] [PubMed] [Google Scholar]
- Taschner, M. , Weber, K. , Mourao, A. , Vetter, M. , Awasthi, M. , Stiegler, M. , … Lorentzen, E. (2016). Intraflagellar transport proteins 172, 80, 57, 54, 38, and 20 form a stable tubulin‐binding IFT‐B2 complex. EMBO Journal, 35(7), 773–790. 10.15252/embj.201593164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thung, D. T. , de Ligt, J. , Vissers, L. E. , Steehouwer, M. , Kroon, M. , de Vries, P. , … Hehir‐Kwa, J. Y. (2014). Mobster: Accurate detection of mobile element insertions in next generation sequencing data. Genome Biology, 15(10), 488 10.1186/s13059-014-0488-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker, B. A. , Scheetz, T. E. , Mullins, R. F. , DeLuca, A. P. , Hoffmann, J. M. , Johnston, R. M. , … Stone, E. M. (2011). Exome sequencing and analysis of induced pluripotent stem cells identify the cilia‐related gene male germ cell‐associated kinase (MAK) as a cause of retinitis pigmentosa. Proceedings of the National Academy of Sciences United States of America, 108(34), E569–E576. 10.1073/pnas.1108918108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihbrecht, K. , Goar, W. A. , Pak, T. , Garrison, J. E. , DeLuca, A. P. , Stone, E. M. , … Sheffield, V. C. (2017). Keeping an eye on Bardet‐Biedl syndrome: A comprehensive review of the role of Bardet‐Biedl syndrome genes in the eye. Medical Research Archives, 5(9), 1–16. 10.18103/mra.v5i9.1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials