

Abstract
Recipient infusion of donor apoptotic cells is an emerging strategy for inducing robust transplantation tolerance. Daily clearance of billions of self apoptotic cells relies on homeostatic engagement of phagocytic receptors, in particular, receptors of the tyrosine kinase family TAM (Tyro3, Axl and MerTK), to maintain self-tolerance. However, an outstanding question is if allogeneic apoptotic cells trigger the same receptor system for inducing allogeneic tolerance. Here, we employed allogeneic apoptotic splenocytes and discovered that the efferocytic receptor MerTK on recipient phagocytes is a critical mediator for transplantation tolerance induced by this strategy. Our findings indicate that the tolerogenic properties of allogeneic apoptotic splenocytes require MerTK transmission of intracellular signaling to suppress the production of inflammatory cytokine IFN-α. We further demonstrate that MerTK is crucial for subsequent expansion of myeloid derived suppressor cells and for promoting their immunomodulatory function, including maintaining graft-infiltrating CD4+CD25+Foxp3+ regulatory T cells. Consequently, recipient MerTK deficiency resulted in failure of tolerance by donor apoptotic cells, and this failure could be effectively rescued by IFN-α receptor blockade. These findings underscore the importance of the efferocytic receptor MerTK in mediating transplantation tolerance by donor apoptotic cells, and implicate MerTK agonism as a promising target for promoting transplantation tolerance.
1. Introduction
To date, bone marrow chimerism is the only tolerance approach that has reached clinical transplantation(1–5). This approach, however, requires aggressive bone marrow conditioning that has significant toxicities, and additionally carries a formidable risk of long-term graft-versus-host-disease (GVHD). A conceptually different tolerance approach is by delivering apoptotic donor cells to tolerize the recipient immune system in a donor-specific manner(6, 7). Supporting the rationale for this approach is the body’s impressive immunologically quiescent daily clearance of billions of self apoptotic cells that preserves systemic homeostasis(8) through maintenance of a nonphlogistic milieu(9), a process termed efferocytosis. Harnessing the innate potential of efferocyotsis for therapeutic transplantation tolerance takes advantage of pre-evolved immunosuppressive properties of apoptotic cells. While this approach has recently shown remarkable safety and efficacy in a phase I/IIa clinical trial for GVHD prophylaxis in patients receiving HLA-matched allogeneic bone marrow transplantation (10), it has not yet been successfully applied to HLA-mismatched allogeneic organ and/or tissue transplantation for tolerance induction. Because phagocytes are the first point of encounter between host and infused apoptotic donor cells(6), a precise understanding of this interaction will significantly enhance our ability to manipulate this process for therapeutic tolerance induction.
Implicated in the homeostatic clearance of apoptotic cells by host phagocytes are three members of the Receptor Tyrosine Kinase (RTK) family TYRO3, AXL and MER, collectively referred to as TAM RTKs(11). TAM RTKs have two primary functions: (1) to mediate “efferocytosis,” the process of homeostatic phagocytosis of apoptotic cells via actin-mediated signaling(12, 13); and (2) to transmit regulatory signals that modulate post-phagocytic immune responses(14–16). TAM signaling has been linked to tumor escape through the inhibition of immunosurveillance (17), whereas impairments in TAMs have been implicated in autoimmunity and protracted resolution of injuries(11, 14, 16).
The role of TAM RTKs in regulating alloimmunity, however, is entirely unknown. We hypothesized that TAM RTKs represent the first point of encounter between recipient phagocytes and donor apoptotic cells, thus playing a determining and targetable role in the outcome of transplant tolerance by donor apoptotic cells. A promising form of donor apoptotic cells demonstrating robust efficacy for transplant tolerance induction is donor splenocytes treated with the chemical crosslinker 1-ehtyl-3-(3-dimethylaminopropyl) carbodiimide (ECDI-SP)(18). Donor ECDI-SP infused to recipients on day −7 and day +1 (with day 0 being the day of transplant) induce indefinite full MHC-mismatch allograft survival in the complete absence of immunosuppression in murine models of allogeneic islet(18, 19) and heart(20, 21) transplantation. Upon injection, donor ECDI-SP are rapidly phagocytosed by recipient splenic macrophages and dendritic cells, effectively promoting downstream anti-inflammatory cytokine production and tolerogenic effects on donor-specific T cells(19, 21–23).
In this report, we identified one of the three TAM RTKs, MerTK, as the key mediator of the tolerogenic effects of donor ECDI-SP. Our studies reveal that macrophage MerTK mediates efferocytosis of donor ECDI-SP, inhibits a pro-inflammatory TRIF/TBK1 signaling axis that is induced by allogenic ECDI-SPs, consequently inhibits inflammatory cytokine production, and promotes expansion of myeloid-derived suppressor cells (MDSCs) and intra-graft CD4+Foxp3+ regulatory T cells. For the first time, our studies provide a mechanistic link between MerTK and transplantation tolerance by donor apoptotic cells, and establish MerTK as a promising target for manipulation to achieve tolerance in transplantation.
2. Methods
Mice:
Ten-to 12-wk-old male BALB/c, C57BL/6 (B6) mice were purchased from the Jackson Laboratory or Harlan. B6.Mertk−/− mice were previously described(12). B6.2C mice were provided by Dr. Fairchild (Cleveland Clinic). All mice were housed under SPF conditions at Northwestern University and usage approved by IACUC.
Preparation and injection of ECDI-SP:
Donor (BALB/c) splenocytes were treated with ECDI as described(18).
Islet and heart transplantation experiments:
Diabetes induction, islet transplantation, determination of graft rejection were described previously(24). Abdominal heart transplantation was performed as described(20).
Flow cytometry:
The following Abs(clones) were used: Gr1-APC(RB6–8C5), Ly6C-PE-Cyanine7(HK1.4), Ly6G-PE(1A8); CD11b-APC-eFluor780(M1/70), CD11c-APC-eFluor780(N418), CD3e-PE-Cyanine7(145–2C11), CD3e-FITC(145–2C11), CD19-PerCP-Cyanine5.5(eBio1D3), CD4-PE(GK1.5), CD4-PE-Cyanine7(GK1.5), CD8a-APC-eFluor780(53–6.7), CD45-FITC(30-F11), CD25-eFluor450(PC61.5), F4/80-PerCP-Cyanine5.5(BM8), SIGN-R1-APC(eBio22D1), from eBioscience; CD68-FITC(FA-11), CD169-PE-Cyanine7(3D6.112), PD-L1-PerCP-Cyanine5.5(10F.9G2) from BioLegend; MerTK-PE or MerTK-APC(108928), ARG1-PE(Polyclonal) from R&D Systems; Foxp3-APC(3G3) from TONBO and CD3e-V500(500A2) from BD Biosciences.
Immunoblot analysis:
Immunoblots were probed first with primary antibodies to: mouse MerTK(AF591, R&D Systems), phospho-MerTK(PMKT-140AP, FabGennix international, Inc.), Phospho-TBK1/NAK(Ser172, Cell Signaling), and GAPDH(FL-335, Santa Cruz Biotechnology); then developed by respective secondary antibodies: anti-rabbit IgG, HRP-linked Antibody(7074S, Cell Signaling) and donkey anti-goat IgG H&L (HRP)(ab205723, abcam).
Quantitative RT-PCR:
TaqMan primers and probes used were: Ifnα1 Ifnα6 Ifnα5(Mm03030145_gH); Ifng(Mm01168134_m1); Myd88(Mm00440338_m1); Ticam1(Mm00844508_s1); and Gapdh (Mm99999915_g1); all from Applied Biosystems. Gene expression levels were standardized to that of GAPDH. SYBR Green primers used were: Cd209b(qMmuCED0050986, Bio-Rad); Csf1: forward ATG GAC ACC TGA AGG TCC TG and reverse CAT CCA GCT GTT CCT GGT CT; B2m: forward CTG CTA CGT AAC ACA GTT CCA CCC and reverse CAT GAT GCT TGA TCA CAT GTC TCG. Gene expression levels were standardized to that of B2M.
SiRNA knockdown
For transient siRNA in BMDM, Mertk−/− and Mertk+/+ BMDMs were transfected with FlexiTube GeneSolution GS17874 for Myd88, FlexiTube GeneSolution GS106759 for Ticam1 (also called TRIF), or scrambled siRNA (all from Qiagen) with HiPerFect Transfection Reagent (Qiagen) per manufacturer’s instructions. After 48 hrs, allogeneic ECDI-SPs were added. After an additional 24 hrs, BMDMs were harvested for measurement of IFN-α production by qRT-PCR.
T cell proliferation assays:
B6 or B6.2C splenic T cells were purified, labeled with 2.5 μM CFSE (Molecular Probes), plated at 1 × 105 per well as responder cells, and stimulated with 5 × 105 irradiated donor (BALB/c) APCs. In some cases, MACS-sorted G-or M-MDSCs were added at a 1:1 MDSC:T ratio. Additional recombinant mouse IFN-α1(1000U/ml; PBL Assay Science) or anti-IFNαR1(20μg/ml, clone MAR1–5A3; BioXCell) was added to indicated cultures. T cell proliferation was measured by CFSE dilution after 72 or 96 hrs of co-culturing.
In vivo antibody and cytokine treatment:
In experiments in which mice were treated with anti-IFNαR1(clone MAR1–5A3, BioXCell), the antibody was injected i.p. at a dose of 250 μg/mouse on day −7, −6, −5, +1, +2, +3, with day 0 as the day of transplantation. In experiments in which mice were treated with recombinant mouse IFN-α (rIFN-α, BioLegend), rIFN-α was injected s.c. at a dose of 600 U/gm on day −7 and +1, and 400 U/gm on −6, −5, +2 and +3.
Statistical analysis:
Data represented were mean ± SEM. Significance between groups was calculated by Student two-tailed t tests, two-way ANOVA with the Bonferroni post test or the Log-rank (Mantel-Cox) test, as deemed appropriate. A P value ≤ 0.05 was considered significant. All statistical analysis was performed using Prism v5.0 (GraphPad).
3. Results
Recipient MerTK plays an obligatory role in donor ECDI-SP-mediated allograft protection
Based on the known critical role of TAM RTKs in homeostatic clearance of apoptotic cells, we hypothesized that graft protection by donor ECDI-SP infusions seen in Mertk+/+ recipients (20, 21) would be impaired in Mertk−/−(25) recipients. Donor ECDI-SP exerts its tolerogenic effects by controlling the immune response of both the direct and indirect allo-recognition pathways(19). We hypothesize that recipient MerTK deficiency would impair the control of indirect allo-immune response by donor ECDI-SP via impairing its homeostatic clearance, but would not impair its control of direct allo-immune response. We tested our hypothesis in two transplant models: (1) allogeneic islet transplantation which predominantly rely on indirect allo-recognition for rejection(26); (2) allogeneic heart transplantation which rely on both indirect and direct allo-recognition for rejection(20). We injected donor (BALB/c) ECDI-SP to C57BL/6 (B6) Mertk+/+ or Mertk−/− recipients on day −7 and day +1, and transplanted BALB/c islets or hearts on day 0. As shown in Fig. 1A, in allogeneic islet transplantation, while in Mertk+/+ recipients infusion of donor ECDI-SP led to indefinite islet allograft survival, in Mertk−/− recipients this protection was completely lost, implicating a dominant role for recipient MerTK mediating the effect of donor ECDI-SP via the indirect pathway. In allogeneic heart transplant (Fig. 1B), Mertk−/− recipients also exhibited a significant impairment of allograft protection by donor ECDI-SP compared with Mertk+/+ recipients. However, a partial protection by donor ECDI-SP treatment in Mertk−/− recipients was still evident in this model, implicating that recipient MerTK deficiency does not impair the effect of donor ECDI-SP on direct allo-immune response. As a control for donor specificity, infusion of a non-donor third-party ECDI-SP did not protect either islet (Fig. 1A) or heart (Fig. 1B) allograft survive. Collectively, these data indicate that recipient MerTK plays a critical role in allograft protection provided by donor ECDI-SP, an apoptotic cell based tolerance therapy.
Figure 1. Recipient deficiency in MerTK impairs transplantation tolerance.
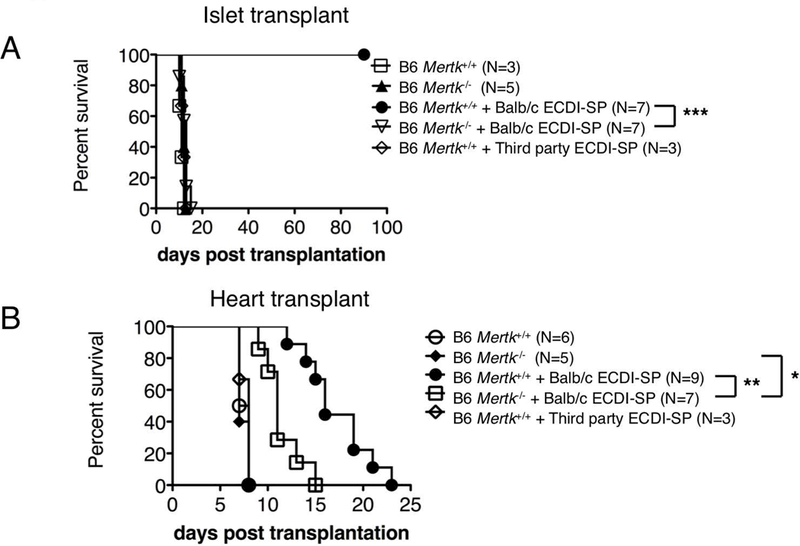
A full MHC-mismatched BALB/c to B6 islet (A) or heart (B) transplant model was used. B6 recipients were either Mertk+/+ or Mertk−/−. Recipients were either untreated or injected with BALB/c or third party (C3H) ECDI-SP on day-7 and day+1. Transplant was performed on day 0. Graft survival was determined as described in Methods. (A-B) Statistical significance was determined using Log-rank (Mantel-Cox) test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
MerTK facilitates efferocytosis of allogeneic ECDI-SP and transmission of tolerogenic regulatory signaling
Recipient splenic macrophages are the initial primary phagocytes interacting with the i.v. injected allogeneic ECDI-SP(19), and MerTK is known to be expressed on prototypical macrophage populations(27, 28). As Mertk−/− recipients exhibited a significant impairment of tolerance by donor ECDI-SP treatment (Fig. 1A and 1B), we first examined if MerTK deficiency had an effect on the number of splenic macrophages at baseline and in response to allogeneic ECDI-SP treatment. Using CD11b+F4/80+CD64+ as a marker for splenic macrophages, we found that at baseline Mertk−/− mice harbor a lower number of splenic macrophages compared with Mertk+/+ mice (Fig. 2A). However, treatment with allogeneic ECDI-SP raised the number of splenic macrophages in Mertk−/− mice to that comparable to Mertk+/+ mice (Fig. 2A). We next examined the expression level of MerTK in splenic macrophages in Mertk+/+ mice following injection of allogeneic ECDI-SP. B6 mice were injected with BALB/c ECDI-SP fluorescently labeled with PKH-67, and spleens were harvested 18hrs later for analysis. As shown in Fig. 2B, upon injection of allogeneic ECDI-SP, splenic CD11b+F4/80+CD64+ macrophages could be discriminated by their status of engulfing the injected allogeneic ECDI-SP (by PKH-67 positivity). Only macrophages that had engulfed allogeneic ECDI-SP (PKH-67+) showed an enhanced MerTK expression compared to baseline (Fig. 2B). To determine if MerTK signaling, in addition to expression, was also enhanced in response to allogeneic ECDI-SP, we co-cultured B6 bone marrow derived macrophages (BMDM) with BALB/c ECDI-SP for 5hrs, and examined phosphorylated MerTK following co-culturing. Indeed, phosphorylation of MerTK (p-MerTK) was significantly enhanced in B6 BMDM co-cultured with BALB/c ECDI-SP (Fig. 2C, lane 1). This enhancement was dependent on ECDI treatment of the allogeneic SP, as live untreated SP did not induce a similar increase of p-MerTK (Fig. 2C, lane 4). As a negative control, BMDMs from Mertk−/− mice did not exhibit measurable p-MerTK under any conditions. Collectively, these data indicate that allogeneic ECDI-SP induce a robust MerTK expression as well as signaling in the engulfing macrophages.
Figure 2. MerTK is activated in response to ECDI-SPs.
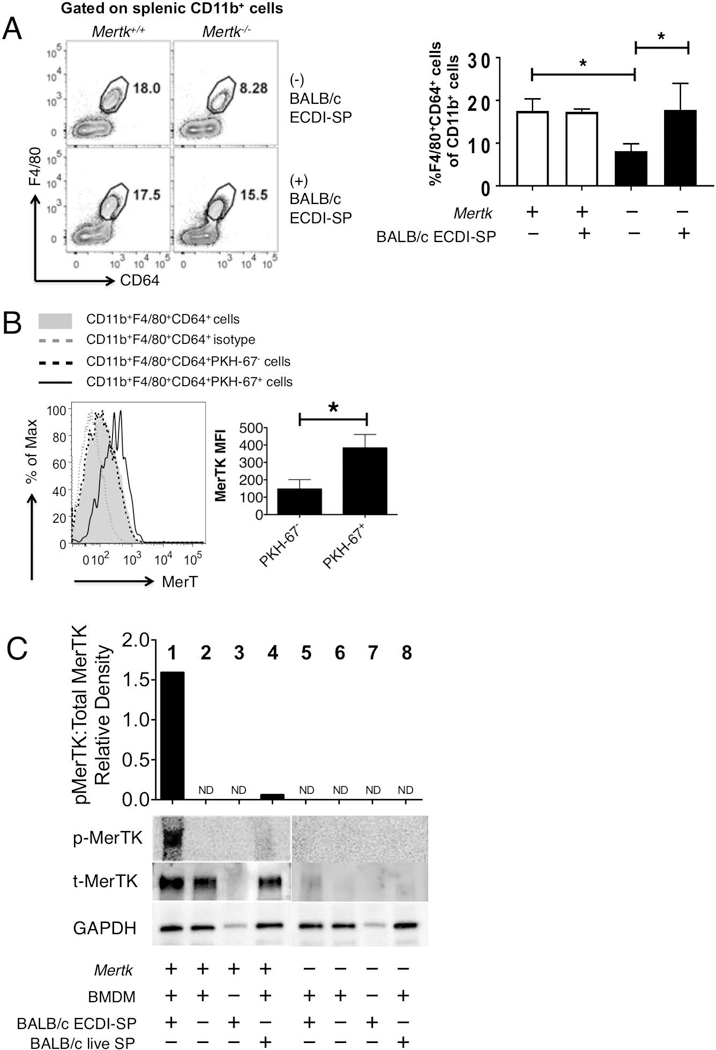
(A) Total splenic macrophages (CD11b+F4/80+CD64+) in Mertk+/+ and Mertk−/− at baseline and 4 days after allogeneic ECDI-SP injection. Left: representative FACS contour plots; Right: bar graph showing average percentages of splenic CD11b+F4/80+CD64+ among total CD11b+ cells in various conditions. Data for (A) were obtained and averaged from three animals per group from one experiment. Statistical significance was determined using t test. *P ≤ 0.05. (B) MerTK expression on Mertk+/+ splenic macrophages engulfing allogeneic ECDI-SP. BALB/c ECDI-SP were labeled with PKH-67 to allow differentiation of splenic macrophages engulfing ECDI-SP from those that had not. Total splenic macrophages (CD11b+F4/80+CD64+) were analyzed. Isotype was used as negative control. Bar graph shows averaged MerTK mean fluorescence intensity (MFI) on PKH-67+ vs. PKH-67− splenic macrophages. Data for (B) were obtained and averaged from a total of six animals per group from three independent experiments. Statistical significance was determined using t test. *P ≤ 0.05. (C) Western blot analyses of phosphorylated MerTK (p-MerTK), total MerTK (t-MerTK) and GAPDH, in BMDM alone, or following 5-hr co-culturing with either BALB/c ECDI-SP or BALB/c live SP. Lanes 1–4: BMDMs from Mertk+/+ mice; lanes 5–8: BMDMs from Mertk−/− mice. BMDMs from Mertk−/− mice were shown as a negative control. Top bar graph shows densitometry quantification of relative density of p-MerTK and t-MerTK. Data shown in (C) is representative of three independent experiments.
Allogeneic ECDI-SPs trigger a unique MerTK dependent anti-inflammatory response
We hypothesized that MerTK plays two critical roles in mediating tolerogenic effects of allogeneic ECDI-SP: (1) to facilitate their uptake; and (2) to trigger a pro-tolerogenic macrophage reprogramming. To test our hypothesis, we co-cultured Mertk+/+ or Mertk−/−BMDM with PKH-67-labelled allogeneic ECDI-SP for 24hrs. First, Mertk−/− macrophages exhibited a reduced ability to internalize allogeneic ECDI-SP (Fig. 3A), measured both by the percentage of BMDM engulfing ECDI-SP (“%PKH+”) as well as by the per cell degree of engulfment (“PKH MFI”). Furthermore, in response to the co-cultured allogeneic ECDI-SP, Mertk−/−BMDM exhibited markedly elevated levels of IFN-α and IFN-γ compared to Mertk+/+ BMDM (Fig. 3B). Similar findings were measured in vivo: allogeneic ECDI-SPs were injected into Mertk+/+ or Mertk−/− recipients. 18hrs later, splenic CD11b+F4/80+CD64+ macrophages were sorted by FACS and analyzed. As shown in Fig. 3C, macrophages from Mertk−/− recipients exhibited a significantly more heightened expression level of IFN-α and IFN-γ by qPCR compared with macrophages from Mertk+/+ recipients following injection of allogeneic ECDI-SP. Thus, in the absence of MerTK, macrophages are induced by allogeneic ECDI-SP to produce higher levels of inflammatory cytokines.
Figure 3. MerTK mediates macrophage uptake of ECDI-SP and inhibits their production of inflammatory cytokines.
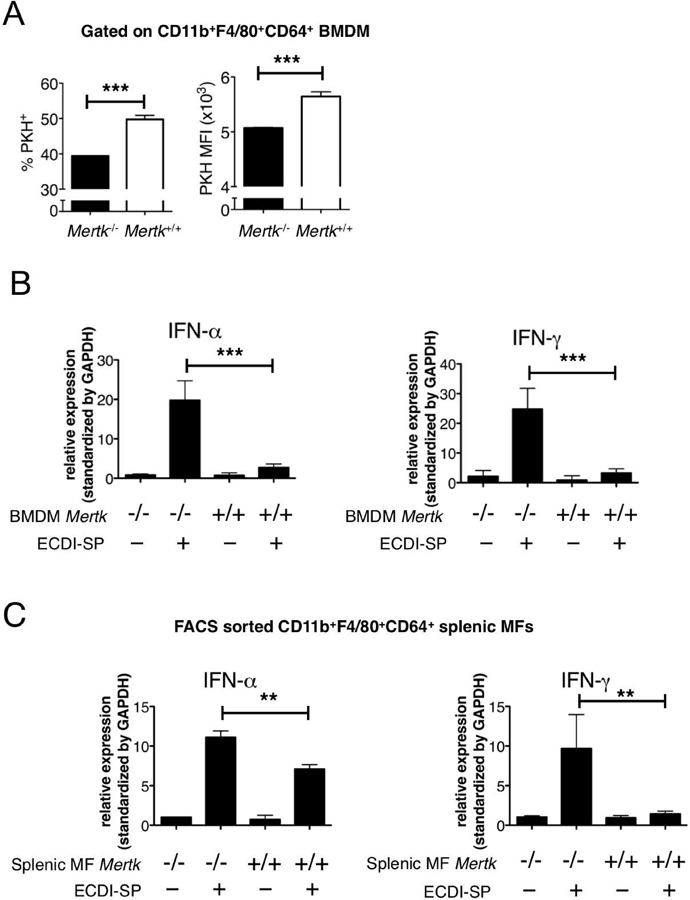
(A) Uptake of PKH-67-labeled BALB/c ECDI-SP by Mertk−/− and Mertk−+/+ BMDM in co-cultures. Left bar graph: %PKH-67+ BMDM after co-culturing; Right bar graph: PKH-67 MFI of PKH-67+ BMDM. Data were obtained and averaged from three independent experiments. (B) In vitro gene expressions by Mertk−/− and Mertk+/+ BMDM, alone or following co-culturing with BALB/c ECDI-SP. Data were obtained and averaged from three independent experiments with triplicates per group per experiment. (C) In vivo gene expressions by FACS sorted splenic CD11b+F4/80+CD64+ macrophages from Mertk−/− and Mertk−+/+ mice, untreated or following injection of BALB/c ECDI-SP. Data were obtained and averaged from three independent experiments with two animals per group per experiment. (A-C) Statistical significance was determined using t test. **P ≤ 0.01, ***P ≤ 0.001.
MerTK inhibits macrophage inflammatory cytokines via inhibition of TRIF/TBK1
We next determined if the enhanced IFN-α production seen in Mertk−/− in response to allogeneic ECDI-SP was due to impaired suppression of TLR signaling by MerTK. We examined this in the same BMDM and allogeneic ECDI-SP co-culture system as in Fig. 3B. We first examined the role of Myd88, a universal adapter protein for all TLRs except TLR3. Knockdown of Myd88 did not significantly inhibit the exaggerated IFN-α production seen in Mertk−/−BMDM (Fig.4A). Next, we examined the role of TRIF, the main adaptor protein for TLR3. Knockdown of TRIF significantly suppressed the exaggerated IFN-α production seen in Mertk−/−BMDM (Fig. 4A). To further interrogate the involvement of the TLR3/TRIF signaling pathway, we measured phosphorylated TBK1/NAK (p-TBK1/NAK), a known TRIF downstream transcription factor, in Mertk+/+ and Mertk−/−BMDM co-cultured with allogeneic ECDI-SP. As shown in Fig. 4B, p-TBK1/NAK was significantly induced by allogeneic ECDI-SP in Mertk−/−, but not Mertk+/+ BMDM. Collectively, these findings suggest that in the absence of MerTK, allogeneic ECDI-SP activate TRIF/TBK1 signaling in macrophages, possibly via TLR3, leading to their enhanced production of the inflammatory cytokine IFN-α.
Figure 4. MerTK inhibits IFN-α production via suppression of TRIF/TBK1 signaling.
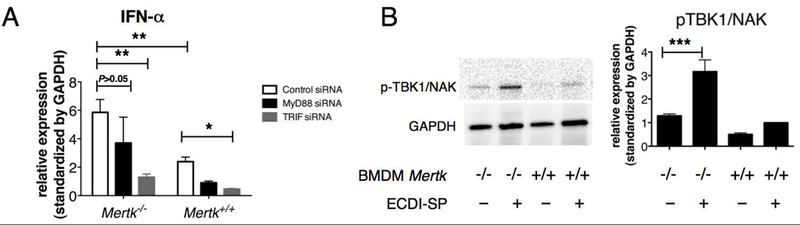
(A) IFN-α gene expression by Mertk−/− or Mertk+/+ BMDM following treatment with control (scrambled), MyD88 or TRIF siRNA prior to co-culturing with BALB/c ECDI-SP. IFN-α gene expression by BMDM was quantified by qPCR. Data were obtained and averaged from five independent experiments with triplicates per group per experiment. Statistical significance was determined by either two-way ANOVA with Bonferroni post-test or one-way ANOVA with Bonferroni post-test. *P ≤ 0.05, **P ≤ 0.01. (B) Western blot analyses of phosphorylated TBK1/NAK (p-TBK1/NAK) and GAPDH, in BMDM alone, or following 24-hr co-culturing with BALB/c ECDI-SP. Bar graph on the right shows p-TBK1/NAK signal standardized to that of GAPDH. Western blot shown is representative of three independent experiments. Bar graph data were obtained and averaged from three independent experiments. Statistical significance was determined using t test. ***P ≤ 0.001.
MerTK is required for optimal expansion of myeloid derived suppressor cells (MDSCs) induced by allogeneic ECDI-SP
In the same co-culture above, we observed that Mertk+/+ BMDM at baseline exhibited a higher level of M-CSF production compared to Mertk−/−BMDM, and that this production was further enhanced by co-culturing with allogeneic ECDI-SP (Fig. 5A). It has been previously reported that the pattern of an enhanced M-CSF together with a down-regulation of inflammatory cytokines is implicated in the expansion of myeloid derived suppressor cells (MDSCs)(29), a cellular event that we have previously reported to be critical for transplantation tolerance induced by donor ECDI-SP (20, 21). Since MerTK on macrophages mediated both suppression of inflammatory cytokines (Fig. 3B–3C) and up-regulation of M-CSF (Fig. 5A) in response to allogeneic ECDI-SP, we next examined if MerTK had an effect on MDSC expansion in response to allogeneic ECDI-SP. Mertk+/+ and Mertk−/− mice were injected with allogeneic ECDI-SP, and 14 days later, MDSCs were enumerated in the spleen. MDSCs were gated as CD11b+Gr1hiLy6G+Ly6Cdim (referred to as granulocytic MDSCs, or G-MDSCs) and CD11b+Gr1dimLy6G-Ly6Chi (referred to as monocytic MDSCs, or M-MDSCs) cells. The gating strategy for MDSCs is shown in Fig. S1. As shown in Fig. 5B, in the spleen of Mertk+/+ mice, total numbers of both G-MDSCs and M-MDSCs were significantly increased following allogeneic ECDI-SP injection. In contrast, this increase was absent in the spleen of Mertk−/− mice. These data suggest that expansion of MDSCs in response to allogeneic ECDI-SP infusion is dependent on MerTK.
Figure 5. MerTK is required for the optimal expansion of MDSCs in response to ECDI-SP.
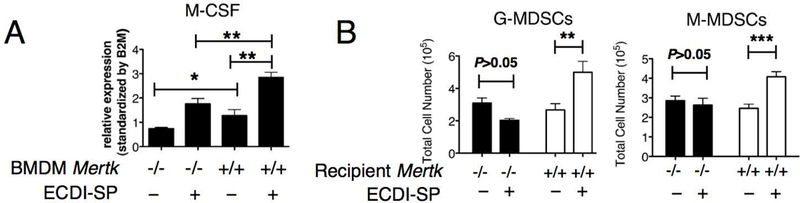
(A) In vitro M-CSF gene expression by Mertk−/− and Mertk+/+ BMDM, alone or following co-culturing with BALB/c ECDI-SP. Data were obtained and averaged from three independent experiments with triplicates per group per experiment. (B) Enumeration of CD11b+Gr1hiLy6G+Ly6Cdim (abbreviated as the G-MDSCs) and CD11b+Gr1dimLy6G-Ly6Chi (abbreviated as the M-MDSCs) cells in the spleen of Mertk−/− and Mertk+/+ mice injected with BALB/c ECDI-SP. Data were obtained and averaged from a total of eight animals per group from three independent experiments. (A-B) Statistical significance was determined using t test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
MDSC function is compromised by an inflammatory milieu in Mertk−/−
In addition to the impaired MDSC expansion seen above (Fig. 5B), we next determined if MDSC function was also compromised by the heightened IFN-α production seen with MerTK deficiency (Fig. 3B–3C).
We first examined the effect of IFN-α on MDSC suppression of donor-stimulated T cell proliferation in vitro. We used 2C CD8 T cell receptor transgenic (TCR tg) T cells that have direct donor specificity in the BALB/c to B6 combination by recognizing a naturally occurring ubiquitous octapeptide presented by Ld molecule, thus proliferating to BALB/c stimulation(30). A 72-hr mixed lymphocyte reaction (MLR) of CFSE-labeled 2C CD8 T cells and BALB/c APCs was set up. M-MDSCs and G-MDSCs were freshly isolated from the spleen of naïve B6 mice and added to the MLR. As shown in Fig. 6A, 2C CD8 T cells proliferated rigorously to BALB/c APC stimulation at baseline. When M-MDSCs were added to the MLR, significant suppression of 2C cell proliferation was observed (Fig. 6A, left histograms). Interestingly, addition of recombinant IFN-α (rIFN-α) completely reversed the suppression of 2C cell proliferation by M-MDSCs, whereas blocking IFN-α receptor signaling by an antagonizing antibody (anti-IFNαR1) further enhanced the suppression of 2C cell proliferation by M-MDSCs (Fig. 6A, left histograms). As neither rIFN-α nor anti-IFNαR1 alone in the absence of M-MDSCs had any direct effect on 2C proliferation (Fig. S2A), we concluded that the observed effects of rIFN-α or anti-IFNαR1 on 2C proliferation was mediated through M-MDSCs. In contrast to M-MDSCs, G-MDSCs did not show detectable suppression of 2C proliferation in this MLR (Fig. 6A, right histograms).
Figure 6. MDSC function is compromised by the inflammatory cytokine IFN-α.
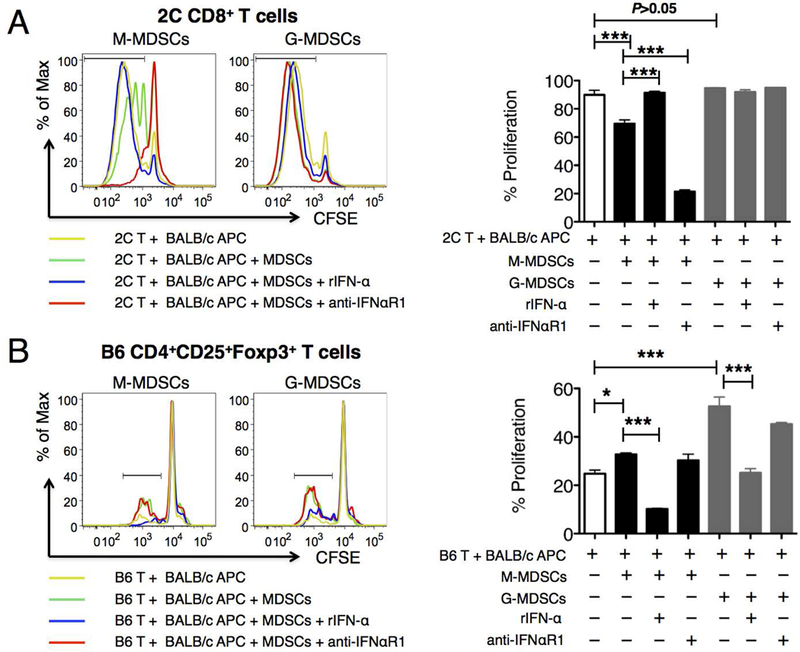
(A) 2C CD8 T cell proliferation to BALB/c stimulation was measured by CFSE dilution in MLRs set up as described in Methods. In some cases, MACS sorted splenic M-or G-MDSCs from B6 mice were added to the MLRs to determine their suppression of 2C T cell proliferation. In additional cases, rIFN-α (1000 U/ml) or anti-IFNaR1 (20 mg/ml) was further added to the cultures to determine their effects on the MDSC-mediated suppression of 2C proliferation. Representative histograms were gated on 2C CD8+ T cells. Percentage of proliferation in the right bar graph was calculated as (2C CD8+ T cells with diluted CFSE)/(total 2C CD8+ T cells). (B) B6 CD4+CD25+Foxp3+ Treg proliferation to BALB/c stimulation was measured by CFSE dilution in MLRs set up as described in Methods. In some cases, MACS sorted splenic M-or G-MDSCs from B6 mice were added to the MLRs to determine their effect on CD4+CD25+Foxp3+ Treg proliferation. In additional cases, rIFN-α or anti-IFNaR1 was further added to the cultures to determine their effects on the MDSC-mediated promotion of CD4+CD25+Foxp3+ Treg proliferation. Representative histograms were gated on CD4+CD25+Foxp3+ Tregs. Percentage of proliferation in the right bar graph was calculated as (CD4+CD25+Foxp3+ Tregs with diluted CFSE)/(total CD4+CD25+Foxp3+ Tregs). Data for (A-B) were obtained and averaged from three independent experiments. Statistical significance was determined by either two-way ANOVA with Bonferroni post-test or one-way ANOVA with Bonferroni post-test. *P ≤ 0.05, ***P ≤ 0.001.
We next examined the effect of IFN-α on MDSC-mediated expansion of allo-reactive CD4+CD25+Foxp3+ regulatory T cells (Tregs) in vitro. To do so, we set up a 96-hr MLR using CFSE-labeled B6 total T cells and BALB/c APCs in the presence or absence of B6 MDSCs, and examined the proliferation of CD4+CD25+Foxp3+ Treg by CFSE dilution. As shown in Fig. 6B, allo-reactive Treg proliferation was enhanced by the presence of M-MDSCs (left histograms) and more pronouncedly by the presence of G-MDSCs (right histograms). Interestingly, rIFN-α again completely reversed the enhanced allo-reactive Treg expansion promoted by either MDSC populations; although blocking IFN-α signaling by anti-IFNαR1 did not further enhance Treg proliferation. Again, as rIFN-α alone in the absence of MDSCs had no direct effect on Treg proliferation (Fig. S2B), we concluded that the observed effects of rIFN-α on Treg proliferation was mediated through MDSCs.
Collectively, these data suggest that heightened IFN-α seen with MerTK deficiency may compromise MDSC function in their suppression of donor-stimulated T cell proliferation and expansion of allo-reactive Tregs.
Graft-infiltrating CD4+CD25+Foxp3+ Tregs are diminished in Mertk−/− recipients
Next, we determined if in vivo Treg expansion in Mertk−/− transplant recipients was also compromised compared with Mertk+/+ recipients. To do so, we used a BALB/c to B6 cardiac transplant model to allow retrieval of sufficient numbers of graft-infiltrating cells for analysis. Mertk+/+ and Mertk−/−B6 mice were treated with BALB/c ECDI-SP and transplanted with a BALB/c cardiac allograft as in Fig. 1B, and the cardiac allografts were examined on post-transplant day 10 for CD4+CD25+Foxp3+ Tregs. As shown in Fig. 7A, rejecting cardiac allografts from either untreated Mertk−/− or untreated Mertk+/+ recipients showed minimal Treg cell infiltration (left dot plots). Donor ECDI-SP infusions significantly increased graft-infiltrating Treg cells in Mertk+/+ but not Mertk−/− recipients (middle dot plots). Interestingly, additional treatment of Mertk−/− recipients with anti-IFNαR1 almost completely restored graft-infiltrating Tregs to the level seen in Mertk+/+ recipients (right dot plots). Consistent with a role of MDSCs in the expansion of graft-infiltrating Tregs(31), we observed that graft-infiltrating G-MDSCs from ECDI-SP treated Mertk+/+ recipients expressed significantly higher levels of Arginase-1 and PD-L1 than those from Mertk−/− recipients (Fig. 7B).
Figure 7. Cardiac allografts from Mertk−/− recipients exhibit a markedly reduced number of intra-graft CD4+CD25+Foxp3+ Tregs and an altered MDSC phenotype.
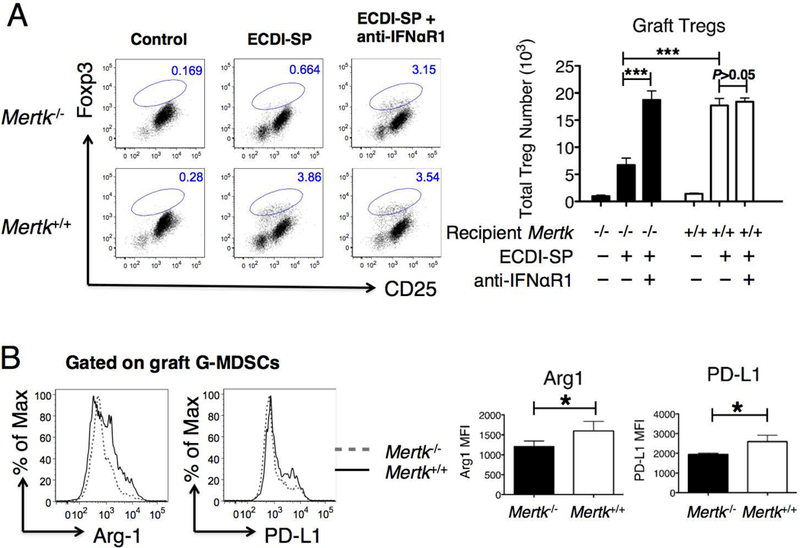
(A) Heart allografts from Mertk−/− or Mertk+/+ recipients were retrieved on post-transplant day 10 and enumerated for CD4+CD25+Foxp3+ Tregs. Grafts from control untreated recipients are shown in the left dot plots. Grafts from recipients treated with donor (BALB/c) ECDI-SP are shown in the middle dot plots. Grafts from recipients further treated with anti-IFNaR1 as described in Methods are shown in the right dot plots. Representative dot blots were all gated on CD4+ cells. The bar graph on the right shows averaged total number of intra-graft Tregs from the various groups. (B) Heart allografts from Mertk−/− or Mertk+/+ recipients treated with BALB/c ECDI-SP were retrieved on post-transplant day 10, and graft-infiltrating G-MDSCs were analyzed for expression levels of Arginase-1 and PD-L1 by FACS. Representative histograms are shown on the left. The bar graph on the right shows the averaged MFI of Arginase-1 or PD-L1 of G-MDSCs in heart allografts from Mertk−/− or Mertk+/+ recipients. Data for (A-B) were obtained and averaged from a total of eight animals per group from three independent experiments. Statistical significance was determined using t test. *P ≤ 0.05, ***P ≤ 0.001.
Efficacy of donor ECDI-SP in Mertk−/− recipients can be rescued by IFN-α blockade
Based on the effect of anti-IFNαR1 in restoring the intragraft Treg accumulation (Fig. 7A), we hypothesized that the same treatment would restore graft protection by donor ECDI-SP in Mertk−/− recipients. Indeed, anti-IFNαR1 treatment of Mertk−/− recipients restored islet allograft tolerance by donor ECDI-SP (Fig. 8A). Similarly, in the allogeneic heart transplant model, anti-IFNαR1 treatment of Mertk−/− recipients also rescued the protective effect on heart allografts by donor ECDI-SP to that seen in Mertk+/+ recipients (Fig. 8B). Reciprocally, we tested whether graft protection by donor ECDI-SP in Mertk+/+ recipients could be abrogated by recipient treatment with rIFN-α. As shown in Fig. 8C, administration of rIFN-α significantly reduced heart allograft survival in donor ECDI-SP-treated Mertk+/+ recipients to that seen in Mertk−/− recipients. Based on these findings, we conclude that IFN-α plays a critical role in the observed impaired tolerance efficacy by donor ECDI-SP in Mertk−/− recipients.
Figure 8. Impaired efficacy of donor ECDI-SP in Mertk−/− recipients is rescued by IFN-α blockade.
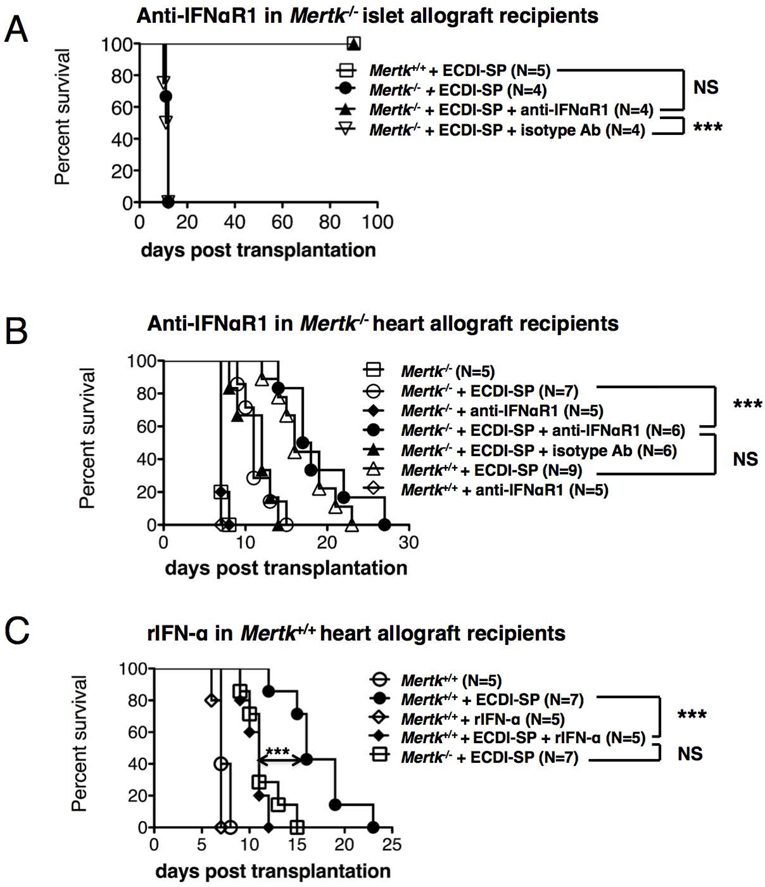
(A) Groups of Mertk−/− or Mertk+/+ recipients were treated with BALB/c ECDI-SP with or without anti-IFNaR1 (or isotype antibody) as indicated, and transplanted with a BALB/c islet allograft. Graft survival was determined as described in Methods. (B) Groups of Mertk−/− or Mertk+/+ recipients were treated with BALB/c ECDI-SP and/or anti-IFNaR1 (or isotype antibody) as indicated, and transplanted with a BALB/c heart allograft. Graft survival was determined as described in Methods. (C) Groups of Mertk−/− or Mertk+/+ recipients were treated with BALB/c ECDI-SP and/or rIFN-α as indicated, and transplanted with a BALB/c heart allograft. Graft survival was similarly determined. (A-C) Statistical significance was determined using Log-rank (Mantel-Cox) test. ***P ≤ 0.001.
4. Discussion
This is the first study to demonstrate a role of the TAM RTK family member MerTK in modulating alloimmunity. In this study, we demonstrate that apoptotic donor ECDI-SP engage host phagocytes via MerTK. MerTK in turn plays an obligatory role in suppressing phagocyte inflammatory cytokine production following their engulfment of allogeneic cells, thus promoting optimal expansion and function of MDSCs as well as graft-infiltrating CD4+CD25+Foxp3+ Tregs. Consequently, recipient MerTK deficiency results in exaggerated inflammatory cytokine production, compromised MDSC expansion and graft-infiltrating Tregs, and ultimately loss of tolerance efficacy by ECDI-SP. We have previously shown that both MDSCs(20, 21) and CD4+CD25+Foxp3+ Tregs(18, 32) play critical roles in mediating tolerance by donor ECDI-SP. The current study now extends our previous understanding by providing a mechanistic link between defective MerTK signaling and the impairment of the MDSC Treg axis needed for mediating transplantation tolerance by this powerful strategy.
We previously reported that donor splenocytes treated with ECDI undergo efficient early apoptosis(23). When infused intravenously, they are readily phagocytized by recipient splenic APCs(19), and induce robust donor-specific tolerance in murine models of allogeneic and xenogeneic transplantation(18, 20, 33). This approach has also demonstrated promising efficacy in a non-human primate model of allogeneic islet transplantation(34), and the same concept has been successfully applied to treating autoimmunity(35) in a clinical trial for multiple sclerosis(36). Independently, a recent phase I/IIa clinical trial using a single infusion of donor early apoptotic cells for prophylaxis of GVHD in 13 patients receiving allogeneic bone marrow transplantation has demonstrated remarkable safety and potential efficacy of this approach in reducing acute GVHD (10). While our original studies utilized a regimen of giving donor cells 7 days prior to and the day of transplantation(18, 19), we have subsequently validated the efficacy of this approach when the first dose of donor cells was given as late as 2 days prior to transplantation(37). This flexibility significantly enhances the clinical feasibility of this approach, particularly when only deceased donor organs are available for transplantation. Future studies, however, will be needed to determine the efficacy of this approach when the timing of the first dose of donor cells is further delayed and/or is given after transplantation.
Quality control of the initial encounter between host phagocytes and the infused donor apoptotic cells to ensure tolerance rather than inflammation can be challenging, particularly because infusion of apoptotic cells could in fact trigger innate inflammatory responses as seen with Mertk−/− macrophages (Fig. 3B and 3C). Our study demonstrates that MerTK is a critical gate keeper of this encounter (Fig. 3B and 3C). This function of MerTK may be mediated through suppression of TLR signaling (Fig. 4A) and subsequently suppression of TBK/NAK signaling (Fig. 4B). It is also possible that TBK/NAK is a direct downstream target of MerTK signaling(38), therefore the enhanced IFN-α expression in MerTK−/− mice is due to a direct dysregulation of TBK/NAK by the lack of MerTK. Regardless, engaging MerTK could be advantageous for precisely controlling the phagocytic process and eliminating variabilities from donor cells. Practically, as bioengineering platforms becomes more mature for manufacturing nanocarriers for tolerogenic donor antigen delivery (37, 39), it is now conceivable to directly assemble MerTK targeting signals to nanocarriers to precisely instruct their interaction with host phagocytes towards tolerance.
Based on current understanding of ligand-receptor interaction and subsequent signal transduction of MerTK(40), there are several strategies for forced augmentation of MerTK signaling. These include: (1) prevention of MerTK cleavage; (2) signaling via MerTK ligands Growth arrest specific factor 6 (Gas6) and Protein S; (3) signaling via activating anti-MerTK antibodies; and (4) signaling directly via the tyrosine kinase domain. The concept of prevention of MerTK cleavage for transplant tolerance can be tested using either genetically mutated MerTK cleavage-resistant mice(41) or inhibitors of protease implicated in MerTK cleavage(42). Forced signaling via MerTK ligands or agonizing antibodies is another attractive approach, as reagents for this approach are now available(43). Additionally, functionalization of nanospheres using specific signaling peptides to activate MerTK signaling represents an attractive and highly controllable approach for manipulating this pathway for transplant tolerance in the future. Lastly, while directly inhibiting the tyrosine kinase activity of MerTK is an approach actively tested in cancer therapies (40), directly activating the kinase domain of MerTK is currently a concept that awaits the development of target-specific small molecule activators of this domain.
Lastly, our study reveals an intrinsic MerTK-mediated mechanism for restraining inflammatory cytokine IFN-α during transplantation. Excessive IFN-α as a risk for allograft rejection has been well recognized when interferon treatment was the first line of therapy for recurrent hepatitis C following liver or kidney transplantation(44). IFN-α increases cell surface HLA antigens, enhances cytokine production and function of NK cells, cytotoxic T cells, and monocytes(45). Our study demonstrates that host phagocytes are an important source of IFN-α, whose production is tightly controlled by MerTK, particularly in their interaction with donor apoptotic cells. IFN-α has been further implicated in destabilizing established transplant tolerance in setting of microbial infections(46, 47). Our study now provides the rationale for testing the role of MerTK in stabilizing tolerance during such infections.
In summary, our study is the first to demonstrate a mechanistic link between MerTK and transplantation tolerance by donor apoptotic cells. It further establishes MerTK and/or downstream IFN-α as promising targets to manipulate for achieving and maintaining stable tolerance in transplantation.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health R01 AI114824 (L.Z., E.T., X.L.), P01 AI112522 (A.D., X.Z., Z.Z., X.L.), HL139812 (X.L., E.T) and R01 HL122309 (E.T.).
Abbreviations:
- ECDI
1-ethyl-3-(3′-dimethylaminopropyl)-carbodiimide
- BMDM
bone marrow derived macrophages
- GVHD
graft-versus-host-disease
- MDSCs
myeloid-derived suppressor cells
- MERTK
Myeloid-Epithelial-Reproductive Receptor Tyrosine Kinase
- RTK
Receptor Tyrosine Kinase
- Tregs
regulatory T cells
- TAM RTKs
RTK family TYRO3, AXL and MER
- SP
Splenocyte
- TAM
(Tyro3, Axl and MerTK)
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to declare as defined by the American Journal of Transplantation.
Supporting Information
Additional Supporting Information may be found online in the supporting information tab for this article.
References
- 1.Scandling JD, Busque S, Dejbakhsh-Jones S, Benike C, Millan MT, Shizuru JA, et al. Tolerance and chimerism after renal and hematopoietic-cell transplantation. N Engl J Med 2008;358(4):362–8. [DOI] [PubMed] [Google Scholar]
- 2.Kawai T, Cosimi AB, Spitzer TR, Tolkoff-Rubin N, Suthanthiran M, Saidman SL, et al. HLA-mismatched renal transplantation without maintenance immunosuppression. N Engl J Med 2008;358(4):353–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leventhal J, Abecassis M, Miller J, Gallon L, Ravindra K, Tollerud DJ, et al. Chimerism and tolerance without GVHD or engraftment syndrome in HLA-mismatched combined kidney and hematopoietic stem cell transplantation. Sci Transl Med 2012;4(124):124ra28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leventhal J, Abecassis M, Miller J, Gallon L, Tollerud D, Elliott MJ, et al. Tolerance induction in HLA disparate living donor kidney transplantation by donor stem cell infusion: durable chimerism predicts outcome. Transplantation 2013;95(1):169–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leventhal JR, Mathew JM, Salomon DR, Kurian SM, Suthanthiran M, Tambur A, et al. Genomic Biomarkers Correlate with HLA-Identical Renal Transplant Tolerance. Journal of the American Society of Nephrology : JASN 2013. [DOI] [PMC free article] [PubMed]
- 6.Morelli AE, and Larregina AT. Concise Review: Mechanisms behind apoptotic cell-based therapies against transplant rejection and graft versus host disease. Stem Cells 2016. [DOI] [PMC free article] [PubMed]
- 7.McCarthy DP, Bryant J, Galvin JP, Miller SD, and Luo X. Tempering allorecognition to induce transplant tolerance with chemically modified apoptotic donor cells. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2015;15(6):1475–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arandjelovic S, and Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol 2015;16(9):907–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Henson PM, and Bratton DL. Antiinflammatory effects of apoptotic cells. J Clin Invest 2013;123(7):2773–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mevorach D, Zuckerman T, Reiner I, Shimoni A, Samuel S, Nagler A, et al. Single infusion of donor mononuclear early apoptotic cells as prophylaxis for graft-versus-host disease in myeloablative HLA-matched allogeneic bone marrow transplantation: a phase I/IIa clinical trial. Biol Blood Marrow Transplant 2014;20(1):58–65. [DOI] [PubMed] [Google Scholar]
- 11.Lemke G, and Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol 2008;8(5):327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001;411(6834):207–11. [DOI] [PubMed] [Google Scholar]
- 13.Lemke G, and Burstyn-Cohen T. TAM receptors and the clearance of apoptotic cells. Ann N Y Acad Sci 2010;1209:23–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu Q, and Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the Tyro 3 family. Science 2001;293(5528):306–11. [DOI] [PubMed] [Google Scholar]
- 15.Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, and Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007;131(6):1124–36. [DOI] [PubMed] [Google Scholar]
- 16.Cai B, Thorp EB, Doran AC, Sansbury BE, Daemen MJ, Dorweiler B, et al. MerTK receptor cleavage promotes plaque necrosis and defective resolution in atherosclerosis. J Clin Invest 2017;127(2):564–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Akalu YT, Rothlin CV, and Ghosh S. TAM receptor tyrosine kinases as emerging targets of innate immune checkpoint blockade for cancer therapy. Immunol Rev 2017;276(1):165–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo X, Pothoven KL, McCarthy D, DeGutes M, Martin A, Getts DR, et al. ECDI-fixed allogeneic splenocytes induce donor-specific tolerance for long-term survival of islet transplants via two distinct mechanisms. Proc Natl Acad Sci U S A 2008;105(38):14527–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kheradmand T, Wang S, Bryant J, Tasch JJ, Lerret N, Pothoven KL, et al. Ethylenecarbodiimide-fixed donor splenocyte infusions differentially target direct and indirect pathways of allorecognition for induction of transplant tolerance. J Immunol 2012;189(2):804–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen G, Kheradmand T, Bryant J, Wang S, Tasch J, Wang JJ, et al. Intragraft CD11b(+) IDO(+) cells mediate cardiac allograft tolerance by ECDI-fixed donor splenocyte infusions. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2012;12(11):2920–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bryant J, Lerret NM, Wang JJ, Kang HK, Tasch J, Zhang Z, et al. Preemptive Donor Apoptotic Cell Infusions Induce IFN-gamma-Producing Myeloid-Derived Suppressor Cells for Cardiac Allograft Protection. J Immunol 2014;192(12):6092–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, et al. Tolerance Induced by Apoptotic Antigen-Coupled Leukocytes Is Induced by PD-L1+ and IL-10-Producing Splenic Macrophages and Maintained by T Regulatory Cells. J Immunol 2011;187(5):2405–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang S, Zhang X, Zhang L, Bryant J, Kheradmand T, Hering BJ, et al. Preemptive Tolerogenic Delivery of Donor Antigens for Permanent Allogeneic Islet Graft Protection. Cell transplantation 2014. [DOI] [PMC free article] [PubMed]
- 24.Luo X, Yang H, Kim IS, Saint-Hilaire F, Thomas DA, De BP, et al. Systemic transforming growth factor-beta1 gene therapy induces Foxp3+ regulatory cells, restores self-tolerance, and facilitates regeneration of beta cell function in overtly diabetic nonobese diabetic mice. Transplantation 2005;79(9):1091–6. [DOI] [PubMed] [Google Scholar]
- 25.Thorp E, Cui D, Schrijvers DM, Kuriakose G, and Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe−/−mice. Arteriosclerosis, thrombosis, and vascular biology 2008;28(8):1421–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Makhlouf L, Yamada A, Ito T, Abdi R, Ansari MJ, Khuong CQ, et al. Allorecognition and effector pathways of islet allograft rejection in normal versus nonobese diabetic mice. J Am Soc Nephrol 2003;14(8):2168–75. [DOI] [PubMed] [Google Scholar]
- 27.Lemke G Biology of the TAM receptors. Cold Spring Harb Perspect Biol 2013;5(11):a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nature immunology 2012;13(11):1118–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Condamine T, and Gabrilovich DI. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol 2011;32(1):19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stegall MD, Elices M, Shepard G, Gup C, Ninova D, Ferguson D, et al. The 2C T-cell transgenic mouse: an in vivo model of allospecific cytotoxic T-cell activation and homing. Transplant Proc 1999;31(1–2):779. [DOI] [PubMed] [Google Scholar]
- 31.Luan Y, Mosheir E, Menon MC, Wilson D, Woytovich C, Ochando J, et al. Monocytic myeloid-derived suppressor cells accumulate in renal transplant patients and mediate CD4(+) Foxp3(+) Treg expansion. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2013;13(12):3123–31. [DOI] [PubMed] [Google Scholar]
- 32.Kang HK, Wang S, Dangi A, Zhang X, Singh A, Zhang L, et al. Differential Role of B Cells and IL-17 Versus IFN-gamma During Early and Late Rejection of Pig Islet Xenografts in Mice. Transplantation 2017;101(8):1801–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang S, Tasch J, Kheradmand T, Ulaszek J, Ely S, Zhang X, et al. Transient B-cell depletion combined with apoptotic donor splenocytes induces xeno-specific T-and B-cell tolerance to islet xenografts. Diabetes 2013;62(9):3143–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lei J, Kim JI, Shi S, Zhang X, Machaidze Z, Lee S, et al. Pilot Study Evaluating Regulatory T Cell-Promoting Immunosuppression and Nonimmunogenic Donor Antigen Delivery in a Nonhuman Primate Islet Allotransplantation Model. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2015;15(10):2739–49. [DOI] [PubMed] [Google Scholar]
- 35.Getts DR, McCarthy DP, and Miller SD. Exploiting apoptosis for therapeutic tolerance induction. J Immunol 2013;191(11):5341–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lutterotti A, Yousef S, Sputtek A, Sturner KH, Stellmann JP, Breiden P, et al. Antigen-specific tolerance by autologous myelin Peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Transl Med 2013;5(188):188ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bryant J, Hlavaty KA, Zhang X, Yap WT, Zhang L, Shea LD, et al. Nanoparticle delivery of donor antigens for transplant tolerance in allogeneic islet transplantation. Biomaterials 2014;35(31):8887–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Linger RM, Keating AK, Earp HS, and Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res 2008;100:35–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tostanoski LH, Chiu YC, Gammon JM, Simon T, Andorko JI, Bromberg JS, et al. Reprogramming the Local Lymph Node Microenvironment Promotes Tolerance that Is Systemic and Antigen Specific. Cell Rep 2016;16(11):2940–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Davra V, Kimani SG, Calianese D, and Birge RB. Ligand Activation of TAM Family Receptors-Implications for Tumor Biology and Therapeutic Response. Cancers (Basel) 2016;8(12). [DOI] [PMC free article] [PubMed]
- 41.Thorp E, Vaisar T, Subramanian M, Mautner L, Blobel C, and Tabas I. Shedding of the Mer tyrosine kinase receptor is mediated by ADAM17 protein through a pathway involving reactive oxygen species, protein kinase Cdelta, and p38 mitogen-activated protein kinase (MAPK). The Journal of biological chemistry 2011;286(38):33335–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Choi JY, Park HJ, Lee YJ, Byun J, Youn YS, Choi JH, et al. Upregulation of Mer receptor tyrosine kinase signaling attenuated lipopolysaccharide-induced lung inflammation. J Pharmacol Exp Ther 2013;344(2):447–58. [DOI] [PubMed] [Google Scholar]
- 43.Zagorska A, Traves PG, Lew ED, Dransfield I, and Lemke G. Diversification of TAM receptor tyrosine kinase function. Nature immunology 2014;15(10):920–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stravitz RT, Shiffman ML, Sanyal AJ, Luketic VA, Sterling RK, Heuman DM, et al. Effects of interferon treatment on liver histology and allograft rejection in patients with recurrent hepatitis C following liver transplantation. Liver Transpl 2004;10(7):850–8. [DOI] [PubMed] [Google Scholar]
- 45.Gessani S, Conti L, Del Corno M, and Belardelli F. Type I interferons as regulators of human antigen presenting cell functions. Toxins (Basel) 2014;6(6):1696–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen L, Wang T, Zhou P, Ma L, Yin D, Shen J, et al. TLR engagement prevents transplantation tolerance. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2006;6(10):2282–91. [DOI] [PubMed] [Google Scholar]
- 47.Wang T, Ahmed EB, Chen L, Xu J, Tao J, Wang CR, et al. Infection with the intracellular bacterium, Listeria monocytogenes, overrides established tolerance in a mouse cardiac allograft model. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2010;10(7):1524–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.