

Abstract
Single missense mutations in the F8 gene encoding the coagulation protein factor VIII give rise predominantly to non-severe hemophilia A. Despite only a single amino acid sequence difference between the replacement, therapeutic factor VIII and the patient’s endogenous factor VIII, therapeutic factor VIII may still be perceived as foreign by the recipient’s immune system and trigger an immune response (inhibitor). Inhibitor formation is a life-long risk for patients with non-severe hemophilia A treated with therapeutic factor VIII, but remains difficult to predict. The aim of this study was to understand whether fortuitous, primary sequence cross-matches between therapeutic factor VIII and proteins in the human proteome are the reason why certain F8 mutations are not associated with inhibitor formation. We predicted which therapeutic factor VIII differences are potentially perceived as foreign by helper T cells – a necessary precursor to inhibitor development – and then scanned potentially immunogenic peptides against more than 100,000 proteins in the proteome. As there are hundreds of disease-causing F8 missense mutations and the human leukocyte antigen gene complex governing peptide presentation to helper T cells is highly polymorphic, these calculations pose a huge combinatorial challenge that we addressed computationally. We found that cross-matches between therapeutic factor VIII and the human proteome are commonplace and have a profound impact on the predicted risk of inhibitor development. Our results emphasize the importance of knowing both the F8 missense mutation and the human leukocyte antigen alleles of a patient with missense mutation hemophilia A if his underlying risk of inhibitor development is to be estimated.
Introduction
Subjects with all severities of hemophilia A are at risk of an alloimmune response (inhibitor formation) against infused, therapeutic FVIII (tFVIII) concentrate. It is well recognized that the more disruptive the F8 mutation, the more severe the hemophilia and the more likely it is that inhibitors will arise.1 Consequently, severe hemophilia A has been the priority for inhibitor-related research, surveillance and intervention over the past decades.2–5 However, it is also clear that only a single amino acid difference between an endogenous F8 genotype and the wild-type tFVIII sequence is sufficient to induce an immune response that results in clinically relevant inhibitors6–8 and that this risk is life-long in the context of non-severe hemophilia A.8
Hemophilia A caused by a missense mutation is typically associated with a less severe bleeding phenotype than that caused by incomplete F8 transcripts. In contrast to boys and men with severe hemophilia A, those living with non-severe hemophilia A are more likely to remain hospital dependent for on-demand tFVIII administration throughout their lives in the event of injury or surgery. The treatment burden for this group is surprisingly high, with 44% of a large London cohort being reported to have received some hemostatic treatment in a 2-year observation window, 79% of whom received tFVIII concentrate.9 Consequently, inhibitor surveillance in non-severe hemophilia A requires adult treaters to be ever vigilant.8 In contrast to the systematic inhibitor screening in patients exposed early to tFVIII for severe hemophilia A,5 inhibitor screening in the setting of non-severe hemophilia A is currently more reactive and sporadic,9 but recognized to be of increasing importance given the aging population of those living with non-severe hemophilia A.10 Inhibitor occurrence in non-severe hemophilia A can be devastating, with neutralization of infused FVIII concentrate and potential cross-reactivity with endogenous FVIII. This cross-reactivity occurs in at least 50% of identified cases11 and results in loss of a patient’s previous non-severe FVIII activity baseline level (FVIII:C) resulting in a worsening bleeding phenotype, often in later decades of life.8 This, in turn, results in increased bleed rates and an increased risk of premature mortality.12 In this context, the early detection of inhibitor occurrence – or, better still, the ability to reliably predict an individual’s risk of developing inhibitors before any have formed – has the potential to influence subsequent clinical decisions in ways that would substantially improve the patient’s outcomes.
The T-cell dependency of inhibitor generation is well described, with confirmed tFVIII-specific CD4+ T-cell responses13–15 and immunoglobulin class switching.16 The activation of CD4+ T cells depends on their interaction with “foreign” peptides – in this case, tFVIII-derived peptides spanning the location of the endogenous F8 missense mutation – presented by major histocompatibility complex (MHC) class II molecules. However, not all such foreign peptides are perceived to be immunologically different from self and, if the difference is undetected, there is presumed negligible risk of an immune response. There are two key mechanisms at work here. Firstly, not all peptides are capable of binding to an individual’s repertoire of MHC molecules and are therefore never presented to T cells. Secondly, not all binding peptides are distinguishable from self-peptides bound to the same MHC molecules; in such cases, T cells that are capable of binding to these MHC:peptide complexes are expected to have been removed from the T-cell repertoire by self-tolerance mechanisms.17
What is unclear, however, is whether an understanding of MHC presentation and self-tolerance can enable us to make useful predictions about the inhibitor risk of patients with missense mutation hemophilia A – for example, by accurately predicting whether individual patients have a negligible risk of developing inhibitors. The aim of this study was to address this point directly. Given that MHC molecules are encoded by genes that are among the most polymorphic in the human genome, and there are several hundred disease-causing F8 missense mutations, the first aim of this study was to predict inhibitor risk based on an analysis of tFVIII peptide presentation by MHC molecules. Such an investigation poses a huge combinatorial challenge – one that is arguably impractical to address using purely in vitro techniques. Building on the approach developed in an earlier study that we undertook using a much smaller dataset,18 we analyzed MHC:peptide complexes associated with 25 common human leukocyte antigen (HLA) class II alleles and 956 distinct F8 missense mutations, requiring over four million peptide-HLA isoform combinations to be evaluated.
However, this preliminary analysis did not take into account the possibility that fortuitous cross-matches between tFVIII-derived peptides and peptides at other locations – both within the FVIII protein sequence itself, and more generally to other proteins in the human proteome – may play a protective role by ensuring that T cells capable of triggering an immune response have been removed from the repertoire by self-tolerance mechanisms. This includes, but is far from limited to, the well-described homology between FVIII and factor V.19 Such human proteome cross-matching is the main focus of this study. Here we demonstrate that cross-matches between tFVIII and other parts of the proteome are commonplace and have a profound impact on the predicted inhibitor risk for individuals living with non-severe hemophilia A.
Methods
Novel peptide-MHC surfaces
In previous work, we developed a methodology for predicting which patients with non-severe hemophilia A are at risk of developing antibodies against tFVIII.18 Specifically, we predicted which F8 missense mutation/HLA isoform combinations would present novel peptide-MHC (pMHC) surfaces to CD4+ T cells, taking into account the reasonable assumption that T cells capable of binding to pMHC surfaces that are formed by endogenous FVIII peptides and presented by the same MHC molecules would have been removed from the T-cell repertoire by central tolerance mechanisms. Novelty arises when a tFVIII-derived peptide is an MHC-binder (most are not) and either (i) the equivalent endogenous FVIII-derived peptide is a non-binder (this may occur if the missense mutation is at an MHC-facing, peptide-anchoring position, as residues at such positions anchor the peptide to the MHC molecule) or (ii) the relevant amino-acid difference is at a T-cell receptor (TCR)-facing position (Figure 1A).
Figure 1.

Schematic diagram showing how side-chain differences may, or may not, lead to novel peptide-MHC surfaces. (A) If a tFVIII peptide spanning the location of the F8 missense mutation is a non-binder, it poses no risk of forming a novel pMHC surface capable of inducing an immune response. Otherwise, one needs to consider the position of the F8 missense mutation within the MHC groove of the tFVIII peptide and the corresponding endogenous peptide. For most HLA class II isoforms, positions 1,4 6 and 9 are MHC-facing, and positions 2, 3, 5, 7 and 8 are TCR-facing. Where the F8 missense mutation is at a downward, MHC-facing position (top row of A, denoted by a diamond), there are two scenarios: both tFVIII and endogenous peptides are binders, implying no risk; or the tFVIII peptide is a binder and the endogenous peptide is a non-binder, implying a potential risk. Where the F8 missense mutation is at an upward, TCR-facing position (bottom row of A, denoted by a diamond) and both peptides are binders, there is a potential risk. (B) Where a tFVIII peptide is associated with a potential risk according to the preceding assessment, a peptide from elsewhere in the human proteome that (i) has the same TCR-facing residues and (ii) is a binder, will militate against this risk, as no novel pMHC surface will be formed. FVIII: factor VIII; tFVIII: therapeutic factor VIII; TCR: T-cell receptor; MHC: major histocompatibility complex; pMHC: peptide-major histocompatibility complex.
Our earlier work focused exclusively on the location of the hemophilia-causing F8 missense mutation and HLA-DR presentation. Here we extend our analysis to include HLA-DP and -DQ presentation and, crucially, to take into account the possibility of fortuitous peptide cross-matches to other locations – both within FVIII itself, and more generally within other proteins in the human proteome (Figure 1B).
Peptide-MHC binding prediction
We used the in silico tool NetMHCII20 to predict the strength with which a given peptide binds to an MHC molecule – specifically, the portable version of NetMHCII 2.2 (Technical University of Denmark, http://www.cbs.dtu.dk/services/NetMHCII/). If a peptide is predicted to bind with a 50% inhibitory concentration (IC50) of <1000 nmol/L – the conventional threshold used to indicate that peptide-MHC class II binding is of biological significance21 – it is a candidate for forming a novel pMHC surface. Deciding whether a given combination of tFVIII peptide and MHC molecule forms a novel pMHC surface in comparison to the corresponding endogenous FVIII-derived peptide requires the considerations outlined in Figure 1 to be made, given: (i) knowledge of the positions of the MHC anchoring pockets for the chosen HLA isoform (generally these are at positions 1, 4, 6 and 9); and (ii) the binding register of the peptide (predicted by NetMHCII), which specifies the stretch of nine consecutive amino-acid residues that form the preferred binding core within the MHC groove. Predictions were made for 25 common HLA-DR, -DP and -DQ isoforms with estimated worldwide population coverages of >70%, >90% and >80%, respectively,22 using UniProt23 FVIII sequence P00451.
For the full set of 25 HLA class II alleles considered for this research, over four million peptide-HLA isoforms combinations were evaluated in order to identify the combinations that are predicted to form a novel pMHC surface – plus a similar order of additional evaluations necessary to identify potential cross-matches to the human proteome. A detailed breakdown of the calculations performed is given in Online Supplementary Table S1.
Scanning the human proteome for cross-matches
Our preliminary risk assessment focused exclusively on the location of the disease-causing F8 missense mutation – that is, we assessed whether one or more peptides would be perceived as foreign, given the binding properties and side-chain orientations of both tFVIII and endogenous FVIII peptides spanning the location of the missense mutation (Figure 1A). The innovative hypothesis explored here is that some pMHC surfaces that were identified as risk-associated by this preliminary approach may not be novel within the broader context of the whole human proteome.
For this research, we compared pMHC surfaces formed by tFVIII peptides spanning the location of the F8 missense mutation with those formed by peptides from the human proteome. Following research showing that the “maximal representation of the ‘immunological self’”17 is made available for tolerance induction in the thymus, we used the complete human proteome from Ensembl24 containing over 100,000 proteins, including alternative isoforms that have an associated protein product.
As previously, we confined our analysis to 15-mers, this being the most common peptide length chosen for MHC class II binding experiments. The canonical proteome was sub-divided into all possible 9-mers. The resultant dataset consists of nearly 38 million 9-mers of which more than 11 million are non-identical.
A summary of the computational pipeline used to identify novel pMHC surfaces is shown in Figure 2.
Figure 2.

Flowchart for the assessment of human proteome cross-matching and missense mutation hemophilia A inhibitor risk. The flowchart shows the process by which the presence, or otherwise, of a novel peptide-MHC surface is determined given a specific combination of endogenous F8 missense mutation and human leukocyte antigen isoform. tFVIII: therapeutic factor VIII; FVIII: factor VIII; TCR: T-cell receptor.
Statistical analysis
We evaluated the accuracy of our method using F8 mutation data downloaded from the largest source of F8 mutation data in the public domain (The European Association for Haemophilia and Allied Disorders. The Factor VIII Gene (F8) Variant Database. http://www.factorviii-db.org. Accessed November 26, 2016). The dataset contained 956 distinct F8 missense mutations at 605 different loci from 3,243 individuals. Ninety of the missense mutations were associated with at least one reported case of inhibitor formation, with a total of 160 individuals (prevalence 4.9%) listed as having inhibitors.
We tested the null hypothesis that our predicted rate and the database reported rate of inhibitor formation are independent. In the absence of HLA-typing information for the patients, we predicted whether a patient with a given missense mutation has a risk of inhibitor formation based on the combined predictions for our chosen set of 25 common HLA class II isoforms. We evaluated different IC50 binding thresholds for tFVIII peptides, on the grounds that binding strength is likely to be an important factor in inhibitor risk.25 However, when considering peptide-MHC binding in the context of self-tolerance prediction (e.g. the binding potential of proteome cross-matches), we maintained a binding threshold of 1000 nM. For a given tFVIII-peptide binding threshold, a patient’s predicted risk of inhibitor formation was deemed to fall within one of the following three categories: “predicted low/negligible risk”, “predicted at risk”, or “unknown risk”. A patient was deemed to be at low/negligible risk if no novel pMHC surfaces were predicted to be formed with any of the 25 HLA isoforms. A patient was deemed to be “at risk” if novel pMHC surfaces were predicted to be formed by all, or all but one, of the HLA isoforms encoded by at least one HLA gene complex – HLA-DRB1, HLA-DRB3/4/5, HLA-DP, and/or HLA-DQ isoforms. (Here the “all, or all but one” condition was designed to rule out the possibility that the patient was a heterozygous individual having a risk-free combination of common HLA isoforms, i.e. two or more low/negligible risk isoforms per gene complex.) The inhibitor risk for patients in neither of the preceding categories was considered “unknown”. Patients with unknown inhibitor risk were omitted from the statistical test.
The statistical test was undertaken using the two-tailed Fisher exact test implemented in the R statistical programming language. The Fisher exact test is necessary because the sample size and background inhibitor rate are both relatively low; in such circumstances, the calculation of exact P values is important. Following standard convention, a P value of <0.05 was used to define statistical significance.
Results
An example of proteome cross-matching
Arg593Cys (R593C) (R612C using Human Genome Variation Society numbering) is a relatively common F8 missense mutation that has been identified as being associated with an “increased” risk of inhibitor formation – for example, 12/106 (11.3%) of R593C individuals in the INSIGHT cohort have been reported as having an inhibitor.8 Taking the common HLA-DR allele HLA-DRB1*01:01 as an example, our analysis of predicted inhibitor risk proceeded as follows.
We began by using NetMHCII20 to predict whether any of the tFVIII 15-mers spanning position 593 are binders. Several such 15-mers were predicted to bind to the MHC molecule associated with HLA-DRB1*01:01, and some had binding cores that span position R593. There were two such cores – IQRFLPNPA and YLTENIQRF – both of which were associated with multiple binding peptides, as shown in Figure 3A.
Figure 3.

An example of information peptide-MHC binding and novel surface: Arg593Cys for HLA-DRB1*0101. (A) NetMHCII predicts there are binding tFVIII 15-mers that have two cores – IQRFLPNPA and YLTENIQRF – containing Arg593 (R593). (B) These two cores form pMHC surfaces that are novel compared to the equivalent surfaces for endogenous FVIII. FVIII: factor VIII; tFVIII: therapeutic factor VIII; MHC: major histocompatibility complex; pMHC: peptide-major histocompatibility complex.
The next step assessed whether either of these cores was associated with predicted pMHC surface novelty compared to their respective endogenous counterparts. Both of the endogenous cores – IQCFLPNPA and YLTENIQCF, respectively [with a Cys (C) in place of the Arg (R) of the equivalent tFVIII peptides] – were associated with predicted binding 15-mers. Hence the question of predicted novelty hinged on the position of R593 within the 9-mer: whether at a position involved in MHC-binding (and hence invisible to a TCR), or at a TCR-facing position. As the binding pockets in the MHC groove for HLA-DRB1*01:01 are at positions 1, 4, 6 and 9, both IQR-FLPNPA (with R593 at TCR-facing position 3) and YLTENIQRF (with R593 at TCR-facing position 8) are associated with the formation of pMHC surfaces that are novel in comparison to those formed by endogenous FVIII, as shown in Figure 3B.
Based on the preceding analysis alone, we would predict that a patient with the R593C mutation and HLA-DR allele HLA-DRB1*01:01 would be at risk of developing inhibitors. However, a tFVIII-derived pMHC surface that is novel with respect to an individual’s endogenous FVIII may not be novel in the wider context of his proteome. To evaluate this possibility of a proteome cross-match that reduces the risk of inhibitor development, we searched for a pattern for each of the 9-mer cores matching at T-cell facing positions 2, 3, 5, 7 and 8. For IQRFLPNPA and YLTENIQRF these patterns are XQRXLXNPX and XLTXNXQRX, respectively, where the letter X matches any amino-acid type. These patterns were scanned against a library containing all 11,272,502 unique 9-mers from the human proteome. In this case, the pattern XQRXLXNPX matched the 9-mer FQRELNNPL in human tubulin polyglutamylase (UniProt25 Q6ZT98), and pattern XLTXNX-QRX matched both the 9-mer GLTENSQRD in dystrobrevin binding protein 1 (dysbindin) (UniProt D6RJC6) and the 9-mer ELTKNAQRA in the uncharacterized human protein C2orf48 (UniProt Q96LS8), as shown in Figure 4A.
Figure 4.

A proteome cross-matching example: Arg593Cys for HLA-DRB1*0101. This example concerns the two tFVIII binding cores from Figure 3, IQRFLPNPA and YLTENIQRF. (A) Both cores cross-match to 9-mers from proteins in the human proteome at TCR-facing positions 2, 3, 5, 7 and 9. (B) Both of the matched 9-mers (one each from tubulin polyglutamylase and C2orf48, but none from dysbindin) are predicted by NetMHCII to form binding cores within 15-mers derived from these proteins. Hence, we conclude that the F8 missense mutation/HLA isoform combination Arg593Cys/HLA-DRB1*0101 is associated with negligible risk of inhibitor formation. FVIII: factor VIII; tFVIII: therapeutic factor VIII; TCR: T-cell receptor; HLA: human leukocyte antigen.
The final step was to check whether a given cross-matching, proteome-derived 9-mer occurred as a binding core for HLA-DRB1*01:01, as only then would we hypothesize tolerance. In this case, NetMHCII predicted that both FQRELNNPL in tubulin polyglutamylase and ELTKNAQRA in C2orf48 form cores within 15-mers with a predicted IC50 <1000 nmol/L, as shown in Figure 4B. Hence, we ultimately predicted that the F8 missense mutation/HLA allele combination R593C/HLA-DRB1*01:01 confers no, or negligible, risk of inhibitor formation owing to fortuitous cross-matches to peptides in the human proteome.
Proteome cross-matches and inhibitor risk stratification
In terms of individual combinations of F8 missense mutation and HLA-DR/DP/DQ isoforms, the impact of proteome cross-matches on predicted risk is shown in a comprehensive heat map (Online Supplementary Figure S1), with a subset of combinations shown in Figure 5. Each individual F8 missense mutation/HLA isoform combination is shown as a single square. An analysis of the full set of data indicates that the percentage of F8 missense mutation/HLA isoform combinations associated with predicted inhibitor risk falls appreciably when proteome cross-matches are taken into account: from 49% to 31% with a binding threshold of 1000 nM; and from 37% to 21% with a binding threshold of 500 nM.
Figure 5.

MHC-binding strengths of F8 peptides predicted to form novel pMHC surfaces with and without proteome scanning. Heatmap showing the predicted occurrence of novel pMHC surfaces and binding strengths for 25 HLA-DR/DP/DQ isoforms (y axis) covering the first 50 missense mutations in the Factor VIII Gene (F8) Variant Database (x axis). Black and gray squares indicate F8 missense mutation/HLA isoform combinations that are not predicted to form a novel pMHC surface. Otherwise the temperature color scale indicates the predicted binding strength of the strongest binding peptide with a novel pMHC surface for each remaining F8 missense mutation/HLA isoform combination. The full heatmap for all missense F8 mutations is given in Online Supplementary Figure S1. (A) MHC-binding strengths of F8 peptides predicted to form novel pMHC surfaces (colored squares), or not (black squares), without proteome scanning. (B) MHC-binding strengths of F8 peptides predicted to form novel pMHC surfaces with proteome scanning. Gray squares indicate F8 missense mutation/HLA isoform combinations that are no longer predicted to form a novel pMHC surface after cross-matches to the proteome are taken into account. MHC: major histocompatibility complex; pMHC: peptide-major histocompatibility complex; HLA: human leukocyte antigen.
These predictions strongly suggest that the risk of inhibitor formation is largely HLA-dependent and, in most cases, cannot be reliably predicted from knowledge of the F8 genotype alone. The proportion of reported F8 missense mutations that we predict to be risk-associated varies considerably between different HLA isoforms. For example, with a binding threshold of 1000 nM, it ranges between 21% (HLA-DQA1*03:01-DQB1*03:02 and HLA-DQA1*04:01-DQB1*04:02) to 86% (HLA-DRB1*01:01) without proteome cross-matches, and between 13% (HLA-DQA1*04:01-DQB1*04:02) and 48% (HLA-DRB1*01:01) with proteome cross-matches. Values for all 25 HLA isoforms and multiple binding thresholds are given in Online Supplementary Table S2. The number of F8 missense mutations associated with no, or negligible, predicted risk (black columns in the heatmap) is only 69 out of 956 (7%).
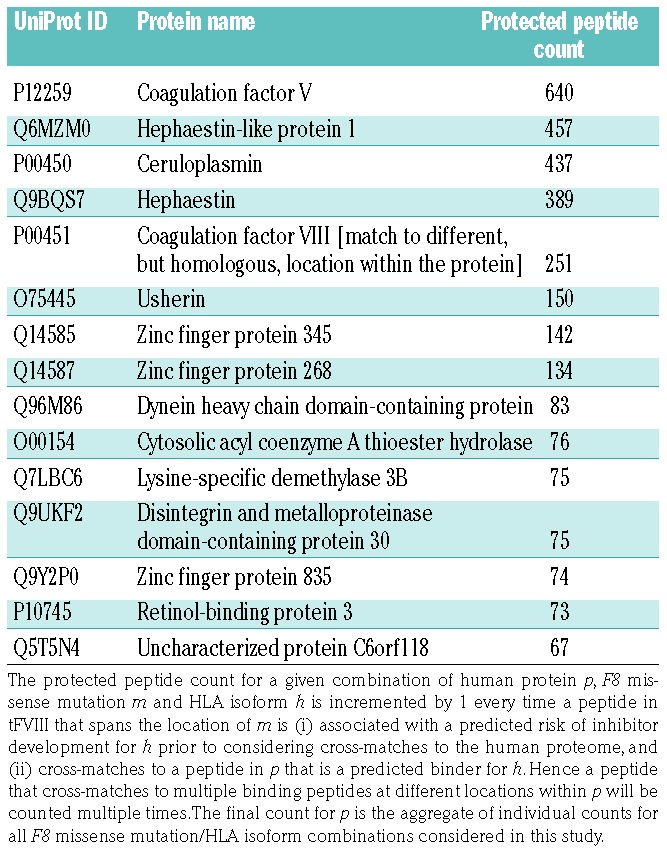
We consider any (human) protein that, in effect, contributes to a reduction in predicted inhibitor risk for one or more F8 missense mutation/HLA isoform combinations to be “protective”. Of the 20,300 proteins in the human proteome, 4,605 are protective. The protein that affords the most protection is factor V, which is recognized as having high homology with FVIII.19 The most protective proteins identified in our analysis are listed in Table 1. It is notable that FVIII and all the top entries in Table 1 (coagulation factor V, hephaestin-like protein 1, ceruloplasmin, and hephaestin) have copper-binding sites.
Table 1.
Human proteins that afford the greatest proteome cross-match protection.
Evaluation of risk prediction accuracy
Given the paucity of published data specifying the HLA profiles of patients with missense mutation hemophilia A, we based the evaluation of how accurate our approach is at predicting potential risk on data in the Factor VIII Gene Variant Database, focusing on F8 missense mutations that we predict to have low or negligible risk with any common HLA isoform.
Arguably the most important performance indicator for our method is the number of false negatives: are there individuals with missense mutation hemophilia A and with data in the Factor VIII Gene Variant Database who we predict to have a zero, or negligible, inhibitor risk but have, in fact, developed inhibitors? The results in Table 2 for our chosen set of 25 HLA alleles show that the number of false negatives (column 3) is very low with conservative cut-offs for peptide-MHC binding affinity. The number of patients predicted to have a zero, or negligible risk of inhibitor development is considerably higher when cross-matches to the proteome are taken into account, but the false negative rate remains comparatively low; these factors taken together contribute to the higher prevalence of statistically significant P values with proteome cross-matching.
Table 2.
Evaluation of zero/negligible inhibitor risk prediction with and without proteome scanning.

Discussion
In this paper, we highlight the potential value of an in silico predictive model of HLA class II antigen presentation as the basis for identifying patients with missense mutation hemophilia A who are at risk of developing inhibitors against tFVIII. Our pipeline incorporates a novel strategy that we term proteome scanning – the identification of fortuitous cross-matches between potentially antigenic tFVIII peptides and peptides arising elsewhere within the human proteome. Such cross-matching peptides are the basis for protection against inhibitor development because of presumed T-cell tolerance mechanisms. Here we focused on the surfaces formed by tFVIII peptides that span the locations of known disease-causing F8 missense mutations and are predicted to bind to the MHC molecules for 25 common HLA class II alleles. Given a conservative binding threshold of 1000 nM, the number of F8 missense mutation/HLA isoform combinations associated with a risk of developing inhibitors was predicted to fall by more than a third – from 49% to 31% – when cross-matches to the proteome are taken into account. These results were shown to be statistically significant with a dataset derived from the Factor VIII Gene Variant Database of patients with missense mutation hemophilia A.
Although our proteome scanning approach reduces the number of patients predicted to be at risk of developing inhibitors, that number remains higher than, albeit closer to, the number of patients that have – at least to date – developed inhibitors. This is inevitable for a model based entirely on a consideration of MHC/TCR interactions, as a range of downstream factors may militate against inhibitor development. These include: the absence of sufficient T cells capable of binding to a given pMHC surface for reasons other than self-tolerance; the lack of co-stimulatory signaling; or the level of exposure to tFVIII being below the threshold necessary for inhibitor formation (only cursory information about a patient’s degree of exposure to tFVIII is available in the Factor VIII Gene Variant Database).
There are a number of ways in which this analysis could be refined. Firstly, we took no account of potential F8 genotype mismatches between tFVIII products (derived from common F8 genotypes H1 and H2 that differ only in the B domain) and rare genotypes H3-8, such as the M2238V found in approximately 23% of black people.26 Nor did we consider the antigenic impact of different linkers used in B-domain-modified tFVIII products. Secondly, proteome scanning was performed against a single reference proteome. It is likely that additional cross-matches will be found if allelic variants are taken into account, adding further to the potential advantages of personalized risk assessment. Scanning against an individual’s own proteome would be the optimal predictive strategy. The impact of proteome variability will be assessed in future work using data from IGSR: The International Genome Sample Resource.27
There are several more challenging issues. Our model of peptide-MHC binding is imperfect, for example: we do not take into account the impact of cathepsin cleavage on the availability of FVIII peptides for MHC class II binding (there are no established computational methods for predicting cleavage by cathepsins, and different sets of cathepsins occur in different professional antigen-presenting cells28); peptide differences at anchoring positions,29 or outside the binding core,30 are known to affect the formation of pMHC surface in specific cases (but the prevalence of such effects is poorly understood); and a given TCR may not be in contact with all TCR-facing residues (but the binding angle and register of individual TCR is currently unpredictable).31
Validating the accuracy of inhibitor risk prediction for patients with non-severe hemophilia is also particularly problematic. In practice, the current, clinical gold standard for inhibitor detection is a functional, clotting-based Bethesda assay. However, heat treatment modifications in the presence of residual FVIII:C (i.e. non-severe hemophilia A) are often omitted, resulting in reduced sensitivity of detection.32 More importantly, a purely “functional” clotting assay does not detect the totality of antibody responses against a protein therapeutic. The absence of a more “neutral” screening assay (e.g. based on enzyme-linked immunosorbence) to pick up any anti-tFVIII antibody response first, and subsequently for the functional assay (Bethesda) to determine its inhibitory potential and clinical relevance, compromises our knowledge of the totality of anti-tFVIII responses in our cohorts of patients. It is also evident that the screening practice for antibody responses in non-severe hemophilia A, in contrast to that for severe hemophilia A, is often opportunistic and passive, further reducing the likelihood of detecting the totality of anti-tFVIII antibody responses by missing the optimal immunological windows for screening after tFVIII exposure.9 Given the life-long risk of inhibitor formation in non-severe hemophilia A, we have concerns that true negatives (i.e. patients confirmed to have a zero risk of inhibitor development) are impossible to identify in a clinical study of non-severe hemophilia A, even when factors such as age and exposure are taken into account.
Notwithstanding these limitations, this study provides compelling evidence of the importance of HLA class II genotyping for analyzing the inhibitor risk of patients with missense mutation hemophilia A. Moreover, we have demonstrated that an innovative computational pipeline incorporating proteome scanning predicts that a large proportion of F8 missense mutation/HLA isoform combinations afford a negligible risk of inhibitor development, with a low error rate when evaluated using the largest available dataset of patients with F8 missense mutations and conservative MHC binding thresholds. This represents an important step forward, as it closes part of the gap between predicted/potential inhibitor risk and observed inhibitor rates. These insights may ultimately contribute to the design of future clinical studies (with HLA typing of missense mutation hemophilia A patients) that are of direct translational relevance.
Supplementary Material
Acknowledgments
DPH received funding from the British Society of Haematology.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/104/3/599
References
- 1.Schwaab R, Brackmann HH, Meyer C, et al. Haemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost. 1995;74(6):1402–1406. [PubMed] [Google Scholar]
- 2.Gouw SC, van der Bom JG, Ljung R, et al. Factor VIII products and inhibitor development in severe hemophilia A. N Engl J Med. 2013;368(3):231–239. [DOI] [PubMed] [Google Scholar]
- 3.Gouw SC, van den Berg HM, Fischer K, et al. Intensity of factor VIII treatment and inhibitor development in children with severe hemophilia A: the RODIN study. Blood. 2013;121(20):4046–4055. [DOI] [PubMed] [Google Scholar]
- 4.Collins PW, Palmer BP, Chalmers EA, et al. Factor VIII brand and the incidence of factor VIII inhibitors in previously untreated UK children with severe hemophilia A, 2000-2011. Blood. 2014;124(23):3389–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collins PW, Chalmers E, Hart DP, et al. Diagnosis and treatment of factor VIII and IX inhibitors in congenital haemophilia: (4th edition). UK Haemophilia Centre Doctors Organization. Br J Haematol. 2013;160(2): 153–170. [DOI] [PubMed] [Google Scholar]
- 6.Fischer K, Iorio A, Lassila R, et al. Inhibitor development in non-severe haemophilia across Europe. Thromb Haemost. 2015;114(4): 670–675. [DOI] [PubMed] [Google Scholar]
- 7.Hay CR. Factor VIII inhibitors in mild and moderate-severity haemophilia A. Haemophilia. 1998;4(4):558–563. [DOI] [PubMed] [Google Scholar]
- 8.Eckhardt CL, van Velzen AS, Peters M, et al. Factor VIII gene (F8) mutation and risk of inhibitor development in nonsevere hemophilia A. Blood. 2013;122(11):1954–1962. [DOI] [PubMed] [Google Scholar]
- 9.Batty P, Austin SK, Khair K, et al. Treatment burden, haemostatic strategies and real world inhibitor screening practice in non-severe haemophilia A. Br J Haematol. 2017;176(5):796–804. [DOI] [PubMed] [Google Scholar]
- 10.Hvas A-M, Poulsen LH. Inhibitor screening in non-severe haemophilia patients; a major challenge. Br J Haematol. 2017;176(5):683–685. [DOI] [PubMed] [Google Scholar]
- 11.van Velzen AS, Eckhardt CL, Hart DP, et al. Inhibitors in nonsevere haemophilia A: outcome and eradication strategies. Thromb Haemost. 2015;114(1):46–55. [DOI] [PubMed] [Google Scholar]
- 12.van Velzen AS, Eckhardt CL, Streefkerk N, et al. The incidence and treatment of bleeding episodes in non-severe haemophilia A patients with inhibitors. Thromb Haemost. 2016;115(3):543–550. [DOI] [PubMed] [Google Scholar]
- 13.James EA, Van Haren SD, Ettinger RA, et al. T-cell responses in two unrelated hemophilia A inhibitor subjects include an epitope at the factor VIII R593C missense site: T-cell responses to a FVIII missense site in hemophilia A. J Thromb Haemost. 2011;9(4):689–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ettinger RA, James EA, Kwok WW, Thompson AR, Pratt KP. HLA-DR-restricted T-cell responses to factor VIII epitopes in a mild haemophilia A family with missense substitution A2201P. Haemophilia. 2010;16(102):44–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.James EA, Kwok WW, Ettinger RA, Thompson AR, Pratt KP. T-cell responses over time in a mild hemophilia A inhibitor subject: epitope identification and transient immunogenicity of the corresponding self-peptide. J Thromb Haemost. 2007;5(12): 2399–2407. [DOI] [PubMed] [Google Scholar]
- 16.Whelan SFJ, Hofbauer CJ, Horling FM, et al. Distinct characteristics of antibody responses against factor VIII in healthy individuals and in different cohorts of hemophilia A patients. Blood. 2013;121(6):1039–1048. [DOI] [PubMed] [Google Scholar]
- 17.Derbinski J, Kyewski B. How thymic antigen presenting cells sample the body’s self-antigens. Curr Opin Immunol. 2010;22(5): 592–600. [DOI] [PubMed] [Google Scholar]
- 18.Shepherd AJ, Skelton S, Sansom CE, Gomez K, Moss DS, Hart DP. A large-scale computational study of inhibitor risk in non-severe haemophilia A. Br J Haematol. 2015;168(3): 413–420. [DOI] [PubMed] [Google Scholar]
- 19.Davidson CJ, Hirt RP, Lal K, et al. Molecular evolution of the vertebrate blood coagulation network. Thromb Haemost. 2003;89(3): 420–428. [PubMed] [Google Scholar]
- 20.Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics. 2009;10(1):296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Southwood S, Sidney J, Kondo A, et al. Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol. 1998;160(7):3363–3373. [PubMed] [Google Scholar]
- 22.Wang P, Sidney J, Kim Y, et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics. 2010;11(1):568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.UniProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 2012;40(D1): D71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yates A, Akanni W, Amode MR, et al. Ensembl 2016. Nucleic Acids Res. 2016;44(D1):D710–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lazarski CA, Chaves FA, Jenks SA, et al. The kinetic stability of MHC class II:peptide complexes is a key parameter that dictates immunodominance. Immunity. 2005;23(1): 29–40. [DOI] [PubMed] [Google Scholar]
- 26.Viel KR, Ameri A, Abshire TC, et al. Inhibitors of factor VIII in black patients with hemophilia. N Engl J Med. 2009;360(16):1618–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.1000 Genomes Project Consortium. Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsing LC, Rudensky AY. The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol Rev. 2005;207(1):229–241. [DOI] [PubMed] [Google Scholar]
- 29.Kersh GJ, Miley MJ, Nelson CA, et al. Structural and functional consequences of altering a peptide MHC anchor residue. J Immunol. 2001;166(5):3345–3354. [DOI] [PubMed] [Google Scholar]
- 30.Deng L, Langley RJ, Brown PH, et al. Structural basis for the recognition of mutant self by a tumor-specific, MHC class II-restricted T cell receptor. Nat Immunol. 2007;8(4):398–408. [DOI] [PubMed] [Google Scholar]
- 31.Rudolph MG, Stanfield RL, Wilson IA. How TCRs bind MHCs, peptides, and coreceptors. Annu Rev Immunol. 2006;24:419–466. [DOI] [PubMed] [Google Scholar]
- 32.Jennings IA, Kitchen D, Kitchen S, et al. Variation in practice for FVIII and FIX inhibitor investigations - results from a UK NEQAS (Blood Coagulation) and UK HCDO multicentre exercise. Res Pract Thromb Haemost. 2017;1(S1):525. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.