

Abstract
The articular cartilage is exquisitely sensitive to mechanical load. Its structure is largely defined by the mechanical environment and destruction in osteoarthritis is the pathophysiological consequence of abnormal mechanics. It is often overlooked that disuse of joints causes profound loss of volume in the articular cartilage, a clinical observation first described in polio patients and stroke victims. Through the 1980s, the results of studies exploiting experimental joint immobilisation supported this. Importantly, this substantial body of work was also the first to describe metabolic changes that resulted in decreased synthesis of matrix molecules, especially sulfated proteoglycans. The molecular mechanisms that underlie disuse atrophy are poorly understood despite the identification of multiple mechanosensing mechanisms in cartilage. Moreover, there has been a tendency to equate cartilage loss with osteoarthritic degeneration. Here, we review the historic literature and clarify the structural, metabolic and clinical features that clearly distinguish cartilage loss due to disuse atrophy and those due to osteoarthritis. We speculate on the molecular sensing pathways in cartilage that may be responsible for cartilage mechanoadaptation.
Keywords: articular cartilage, disuse atrophy, cartilage hypertrophy, mechanotransduction, mechanoadaptation, osteoarthritis
The mechanosensitivity of musculoskeletal tissues is extremely well documented but for the most part this has focused on muscle and bone rather than articular cartilage (Ziaaldini et al. 2017; Galea et al. 2017; Uda et al. 2017). Part of the reason for this lies in the fact that the cartilage is not easily visualised; its thickness is inferred from the gap between the ends of the bone (the joint space) on a plain radiograph. Cartilage has only relatively recently been directly visualised by magnetic resonance imaging (MRI).
Articular cartilage is adapted to withstand mechanical stress
Articular cartilage develops and operates under very high compressive and shear stresses throughout its lifetime, and has only modest purported reparative capability, particularly in the elderly. Cartilage is an extracellular matrix‐rich tissue where only 5–10% of the tissue volume is cell (Hunziker et al. 2002). The principal fibrillar component is type II collagen, forming staggered arrangements of triple helical molecules that are highly cross‐linked and organised in such a way that they lie parallel with the surface layer superficially but perpendicular to it in the deep regions (Fig. 1). The complex organisation of primary and secondary structures provides cartilage with high tensile strength and the orientation of fibres allows mechanical loads to be dissipated over the surface, so as to minimise impact on the focal ‘load‐bearing’ regions of the joint. The tissue holds water by means of fixed negative charges offered by the proteoglycan aggrecan, itself aggregated on hyaluronan chains in the tissue (Dudhia, 2005). Aggrecan has attachment sites for many chondroitin sulfate and keratan sulfate glycosaminoglycan (GAG) chains that lie within the territorial and inter‐territorial parts of the matrix. The hydration allowed by the fixed negative charge of these chains provides the tissue with a swelling pressure enabling absorption of, and resistance to, mechanical shock. The chondrocyte is separated from the territorial matrix by a specialised pericellular matrix (PCM). Together, the chondrocyte and the PCM form the ‘chondron’. Chondrons are themselves orientated relative to local mechanical strain, exhibiting a polarised morphology that varies with tissue depth (Poole, 1997).
Figure 1. Schematic diagram of articular cartilage.
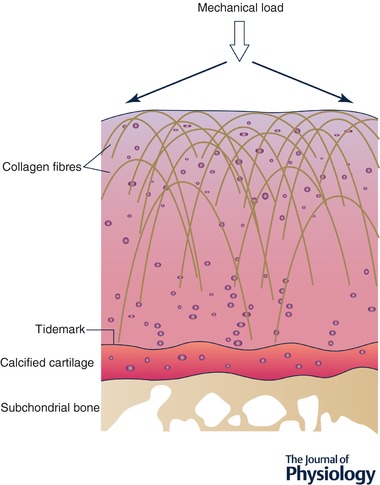
Illustration of tertiary structure of fibrillar collagens in articular cartilage showing parallel arrangement in superficial layer and perpendicular orientation in deeper layers. Arrows show direction of mechanical load and how it is dissipated across the articulating surface.
Loss of proteoglycans by cleavage of the core molecule by aggrecanases occurs in osteoarthritis (OA) and is regarded as a key pathological feature of disease (Glasson et al. 2005). Tissue catabolism is also mediated by other members of the matrix metalloproteinase (MMP) family. MMP13, one of the mammalian collagenases, has a proven role in osteoarthritis (Mitchell et al. 1996), whilst MMP3 (stromelysin) does not appear to drive disease and may be more important in normal tissue turnover (Clements et al. 2003; Troeberg & Nagase, 2012).
Articular chondrocytes are highly mechanosensitive
For articular cartilage to be able to respond to its mechanical environment, it must be able to sense a change in the magnitude and type of mechanical load and respond to this change. Historically, the chondrocyte was regarded as a largely quiescent cell, one that rarely proliferated and was responsible for slow turnover of the matrix. Early studies by Mankin showed that proteoglycan synthesis was moderately active (with a half‐life of around 8 days) (Mankin & Lippiello, 1969), and synthetic activity increased substantially following injury (Meachim, 1963).
Multiple mechanisms by which chondrocytes sense mechanical load have been identified. Cellular mechanosensors include cell surface integrins, stretch and cation‐sensitive ion channels (Ross et al. 2013; Lee et al. 2014; O'Conor et al. 2014; see also review by Drexler et al. 2014). The chondrocyte primary cilium has also been implicated in mechanotransduction, as it has in many other cell types (Wann et al. 2012). Beyond the chondrocyte another important mechanism is release of sequestered heparan sulfate‐bound molecules of the PCM in response to compression or cutting injury (Vincent et al. 2002, 2004; Vincent, 2013) (Fig. 2). The best described of these is fibroblast growth factor 2 (FGF2), which is chondroprotective in vivo in models of osteoarthritis (Chia et al. 2009). Other molecules include connective tissue growth factor (CTGF) which is covalently bound to latent transforming growth factor beta (TGFb). Release of the complex upon cutting injury results in activation of TGFb at the cell surface in a CTGF‐dependent manner (Tang et al. 2018). Activation of a number of intracellular signalling pathways occurs rapidly upon injury including TGFb‐activated kinase 1 (TAK1), β‐catenin, Src, Smads, mitogen activated kinases (ERK, p38 and JNK) and nuclear factor κB (NFκB) (Vincent et al. 2002; Clements et al. 2003, 2011; Gruber et al. 2004; Dell'Accio et al. 2006, 2008; Troeberg & Nagase, 2012; Watt et al. 2013; Ismail et al. 2017). The receptor proximal mechanism of activation for most of these remains unknown.
Figure 2. Schematic diagram of a chondron.
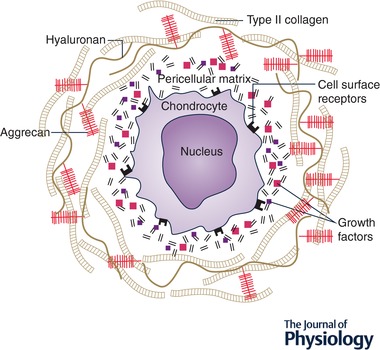
Illustration of the chondrocyte sitting within its pericellular matrix (PCM), together forming the chondron. Note absence of fibrillar collagens and aggrecan in the PCM, but abundance in the adjacent territorial matrix. Multiple heparan sulfate‐bound growth factors are bound within the PCM and released upon tissue compression or injury.
Interestingly, both experimental data and in silico models show that the forces experienced by an individual chondrocyte are largely the same irrespective of the mass of the animal (Simon, 1970). This is most likely explained by ‘isometric’ scaling of the skeleton, where the joint surface area increases in proportion to the mass of the animal. Cartilage thickness does increase with animal size, but exhibits allometric rather than isometric scaling. Indeed, it has been suggested that there is an optimal maximum thickness of cartilage, beyond which perfusion of the chondrocytes deep in the tissue would be compromised (Malda et al. 2013). One might thus hypothesise the existence of an evolutionary ‘mechanostat’ for chondrocytes that has been tuned to an optimal mechanical input. The mechanical input itself, being related to the properties (stiffness) of the surrounding matrix.
Evidence for disuse cartilage atrophy in humans
In 1974, Winford Pool reviewed 200 cases of flaccid paralysis of the lower extremeties. Twenty‐five cases had demonstrable narrowing of the hip joint space by plain radiograph of at least 50% (Pool, 1974). His conclusions at the time were that flaccid paralysis caused significant reduction of all types of load on the joint (compared with spastic paralysis where there is increased static load by action of muscle). He proposed that this reduced the nutrient supply to the cartilage, which is dependent upon pumping action of load‐bearing activity. His findings were supported by those of Anderson and Breidahl who performed a similar study in 71 patients with either flaccid or spastic paralysis due to spinal cord damage. Their results confirmed the strong association between paralysis and joint space narrowing at the hip which was more common in flaccid rather than spastic paralysis (Anderson & Breidahl, 1981). Similar results were obtained when lower limb amputees were examined. Importantly, in this paper they also comment that features of OA were never seen on the amputated side (Benichou & Wirotius, 1982). This is one of the earliest clarifications regarding cartilage atrophy and OA. Despite both conditions leading to thinning of articular cartilage (see Table 1), OA develops as a result of increased mechanical load whereas atrophy results from reduced load.
Table 1.
Causes of thinning of the articular cartilage
| Cause | Mechanism | Reversible? |
|---|---|---|
| Osteoarthritis | Proteolytic degradation of matrix | No, except by regeneration |
| Atrophy | Reduced synthesis of proteoglycan; a mechanoadaptive response to reduced load | Yes, weeks–months |
| Cartilage compression | Water extrusion from matrix due to compression | Yes, minutes–hours |
| Apparent reduction in joint space (‘pseudo thinning’) | Migration of tidemark or meniscal extrusion | Unknown |
The subsequent development in the early 1990s of non‐invasive methodologies to assess the volume and proteoglycan content of articular cartilage by MRI allowed much more sensitive assessments of change with disuse and disease (Recht et al. 1993; Eckstein et al. 1997; Haubner et al. 1997; Lösch et al. 1997). A small number of cross‐sectional and longitudinal studies have been carried out in spinal cord injury patients since then. Cartilage volume loss was seen to decrease by around 6% in the first 6 months from baseline (baseline was on average 4 weeks post‐spinal injury), resulting in a loss of around 25% of the medial tibial thickness by 24 months after injury when compared with normal controls (Vanwanseele et al. 2002, 2003). In individuals who are temporarily immobilised, through ankle fracture, a 6.6% loss of cartilage thickness was measured in the medial tibia over 7 weeks following fracture. Sensitivity of these techniques continues to increase and it is now possible to detect very small strain levels (around 3%) in the tissue that occur on normal weight‐bearing activity (Eckstein et al. 2000; Sutter et al. 2015). No human study to our knowledge has demonstrated recovery of cartilage volume after remobilisation but importantly, moderate programmed exercise was shown to have a positive effect on increasing cartilage GAG content (assessed by delayed gadolinium‐enhanced MRI of cartilage, dGEMRIC) in individuals at risk of OA due to previous meniscal injury (Roos & Dahlberg, 2005).
Disuse atrophy in large animal models
The greatest body of literature on cartilage atrophy stems from in vivo studies, mainly carried out in large animals (dog and rabbit). These studies have done much to provide definitive evidence for the importance of mechanics to joint homeostasis and disuse atrophy, greatly enhancing our understanding of the metabolic processes that lead to the condition. In 1979, Palmoski et al. produced a series of interesting studies in which they immobilised healthy adult dogs using a light cast that fixed the knee joint at 90°. The cast was strapped to the trunk preventing weight bearing or active flexion/extension. These studies revealed that 6 days of immobilisation was sufficient to observe a reduction in proteoglycan staining in the tissue and by 8 weeks there was a 30–50% reduction in cartilage thickness with almost complete loss of proteoglycan. Although the authors did not describe whether the changes occurred in the calcified or non‐calcified cartilage, others have suggested that the calcified cartilage layer (below the tidemark) is more sensitive to atrophic changes (Kiviranta et al. 1987). Interestingly, despite loss of proteoglycan there was an increase in the water content in the tissue, a feature that was confirmed by other similar studies (Palmoski et al. 1979; Behrens et al. 1989), and a change in the proteoglycan produced by the tissue such that it failed to aggregate with hyaluronan in vitro. This aggregation defect suggests a change in the GAG rather than the hyaluronan itself, although a reduction in hyaluronan tissue content has also been described with disuse (Haapala et al. 1996). Hyaluronan synthesis itself was later shown to be mechanosensitive in the synovial joint (Ingram et al. 2009). The hyaluronan aggregation defect was reversed upon voluntary remobilisation (Palmoski et al. 1979) and may be related to restoration of normal hyaluronan synthesis (Müller et al. 1994). Recovery from atrophy has been documented consistently in other studies, although is affected by the severity of atrophy attained and may be incomplete when immature joints are immobilised (Palmoski & Brandt, 1981; Behrens et al. 1989; Kiviranta et al. 1992).
In a follow‐up study in 1981, Palmoski & Brandt showed forced treadmill exercise prevented recovery of the tissue after immobilisation. Newly synthesised proteoglycans measured by 35S incorporation demonstrated that there was a decrease in proteoglycan synthesis upon disuse, consistent with later studies examining aggrecan gene expression (Djurasovic et al. 1998). Proteoglycan synthesis increased upon remobilisation in both the free and forced remobilised groups. The authors concluded that during forced remobilisation there must also be increased proteolytic activity to account for why proteoglycan failed to recover despite increased synthesis (Palmoski & Brandt, 1981). Whether increased proteolysis contributes to atrophic changes in the cartilage has been an area of speculation but to date there is little evidence to support this despite some efforts (Grumbles et al. 1995; Haapala et al. 2001).
The focus on proteoglycan metabolism in these studies is justified. There is significantly less information available to suggest a change in the collagenous matrix in atrophic cartilage apart from some modest reduction in collagen crosslinking which recovers after remobilisation (Haapala et al. 1999). This would be consistent with the known stability of fibrillar collagens and evidence for minimal turnover in human cartilage over the lifetime (Heinemeier et al. 2016). The biomechanical consequences of matrix imbalance in atrophic tissue have been studied, ex vivo, by mechanical testing. These studies collectively show some site‐specific changes in shear and compressive properties of the cartilage, although they show some inconsistencies (Jurvelin et al. 1986b; Leroux et al. 2001).
Disuse atrophy in small animal models
Generally speaking all types of joint immobilisation lead to atrophy although differences in how immobilisation is achieved, e.g. by rigid external fixation or cast immobilisation, and at which sites the effects are measured, does affect the type of outcome measures (Säämänen et al. 1990) and magnitude of change (Behrens et al. 1989). Two studies performed in the last decade have studied immobilisation in rodents. In the first, immobilisation was achieved by spinal cord injury in which the animals drag the affected limb. The study revealed changes in the thickness of cartilage across different regions of the joint at baseline with predicted weight‐bearing regions having higher recorded thickness. Upon spinal cord injury a 30% decrease in cartilage thickness was measured in off‐loaded areas (Moriyama et al. 2008). The second study compared rigid (by externally fixed Kirschner wires) and ‘tail suspension’ immobilisation in mice. Both led to a reduction in cartilage thickness, largely due to a decrease in the calcified cartilage, and were associated with loss of proteoglycan in the non‐calcified layer particularly. Atrophy was slightly enhanced in the rigid fixation group, presumably because the joint was not subjected to movements associated with muscle activity (which are present during tail suspension) (Nomura et al. 2017).
Increased thickness of cartilage upon exercise
If disuse leads to cartilage thinning then it follows that exercise training should result in thicker cartilage, so‐called cartilage hypertrophy. This has been shown in a number of large animal studies and is reflected by changes in the same biochemical metrics as measured in atrophy. Increased synthesis of the guanidine‐extractable proteoglycan was documented in exercised rabbits (Tammi et al. 1983; Säämänen et al. 1988) and in dogs, there was an increase in extractable GAGs that were less able to aggregate with hyaluronan in vitro. These GAGs also had increased ratios of chondroitin‐6 to chondroitin‐4 sulfate (Tammi et al. 1983; Säämänen et al. 1989). Treadmill running of 4 km a day for 15 weeks in dogs was associated with an increase in cartilage thickness (11% in loaded areas) and mechanical stiffness (6%) (Jurvelin et al. 1986a). Treadmill training for up to 40 weeks at 5 h/week was associated with an increase in the non‐calcified cartilage thickness by 19–23% and glycosaminoglycans by 28% (Kiviranta et al. 1988).
Exercise dosage has been examined in rats running on a treadmill. Speeds of 15 m/min with no incline, and 19 m/min with a 5° incline for 5 h per week for 6 weeks were associated with increased cartilage thickness and proteoglycan synthesis compared with non‐exercised control animals (Ni et al. 2013). Similar to studies of atrophy there are site‐specific changes in cartilage that reflect the type of exercise undertaken, e.g. downhill treadmill running (Hamann et al. 2014).
In contrast to exercise‐induced adaptation of muscle and bone in adult human joints, there is much less evidence for cartilage thickness adaptation. While associations exist between rates of cartilage accrual and physical activity during childhood (Jones et al. 2003), studies comparing athletes and inactive cohorts found the variance in cartilage thickness was not clearly attributable to activity levels (Eckstein et al. 2002). However, an association between exercise and joint size (cartilage surface area) did exist, consistent with the isometric relationship described for body mass (or by extrapolation, load) and surface area in other mammals (Malda et al. 2013).
Molecular mechanisms underlying mechanoadaptation in cartilage
The evidence thus far points to cartilage being highly ‘mechanoadaptable’ in contrast to its somewhat ‘inert’ reputation in the post‐developmental context. Early speculation suggested that this was due to altered nutrition of the cartilage due to its dependence on mechanical load to deliver nutrients and oxygen to the chondrocytes. However, the relatively recent appreciation that chondrocytes can sense their mechanical environment and respond differently to distinct mechanical cues, amplitudes and frequencies, suggests that these mechanosensing mechanisms will turn out to be responsible for driving mechanoadaptation. Although, to our knowledge, this has not been directly addressed, careful scrutiny of the literature does provide us with potential clues as to the pathways that may be responsible. For instance, increased cartilage thickness has been described in mice that have been subjected to a transient burst of Wnt signalling (by tamoxifen‐induced transgene activation) (Yuasa et al. 2009). In this case there was an immediate loss of proteoglycan preceding the ‘rebound’ hypertrophy of cartilage. Wnt signalling occurs rapidly upon mechanical injury of cartilage in vitro and a number of Wnt ligands are matrix bound and present in cartilage (Dell'Accio et al. 2006, 2008; Berendsen et al. 2011). Wnts are also involved in joint cavitation during development, a process that is critically dependent upon mechanical force by associated muscle action (Kahn et al. 2009; Shea et al. 2015).
Vincent has also observed an increase in cartilage thickness associated with constitutive activation of Smad2 signalling suggesting that TGFb family members may drive hypertropic cartilage responses (Tang et al. 2018). As TGFb is one of the growth factors present in the pericellular matrix and decreases in cartilage with age (Madej et al. 2016; van Caam et al. 2016), it is tempting to speculate that its release upon tissue compression could be relevant to reduced mechanoadaptation over the life course, a process described in other skeletal tissues (Greig et al. 2011; Brook et al. 2016). Another molecule that is involved in chondrocyte mechanosensing is FGF2. FGF2 is an important mechanosensitive mediator of joint cavitation during development (Kavanagh et al. 2006; Shea et al. 2015) and is also released by cartilage compression (Vincent et al. 2007). However, we found no evidence of a change in cartilage thickness in male or female naive FGF2 knockout mice (Chia et al. 2009). Activation of cell surface mechanoreceptors piezo 1 and Trpv4 in chondrocytes leads to anabolic responses in vitro and chondroprotection in vivo. Moreover, their activity is associated with proteoglycan synthesis, making these interesting molecular candidates for further investigation (Clark et al. 2010; Lee et al. 2014; O'Conor et al. 2014, 2016).
What is the relationship between atrophy and osteoarthritis?
While mechanoadaptation of cartilage has become a somewhat neglected area of study, interest in molecular pathogenesis of osteoarthritis has greatly increased (see Tables 2 and 3 for comparisons). Cartilage loss in OA has completely different molecular and chemical drivers; matrix catabolism is highly protease dependent but is associated with a strong anabolic reaction suggesting an unsuccessful repair response. Increased mechanical load is a critical aetiological factor that is strongly supported by epidemiological studies (Brandt et al. 2009), and by showing that regulation of proteases in vivo upon induction of disease (by surgical joint destabilisation) is abrogated by joint immobilisation (Burleigh et al. 2012). OA is also distinguished from atrophy by loss of integrity of the tissue starting with changes at the articular surface, which spread throughout the tissue as the disease progresses and are accompanied by aberrant bone remodelling. From a molecular and structural point of view, the process of atrophy and OA could not be further apart. The only feature they share is thinning of the cartilage and therefore an apparent decrease in the joint space on plain radiograph.
Table 2.
Clinical distinguishing features between cartilage atrophy and osteoarthritis
| Clinical features | Cartilage atrophy | Osteoarthritis |
|---|---|---|
| Joint space narrowing (JSN) | Yes | Yes |
| History of immobilisation | Yes | No, increased load |
| Pain | No | Yes |
| Synovial hypertrophy/inflammation | No | Yes |
| Bone changes | Local osteoporosis | Subchondral bone sclerosis; osteophytes |
Table 3.
Biochemical changes in atrophic and osteoarthritic cartilage
| Tissue characteristic | Cartilage atrophy | Recovery with ad libitum remobilisation? | Osteoarthritis |
|---|---|---|---|
| Proteoglycan synthesis | Decreased | Yes | Increased |
| Total tissue proteoglycan | Decreased | Yes | Decreased |
| Evidence of proteolysis? | No | N/A | Yes, of proteoglycan and collagen |
| Proteoglycan aggregation defect | Yes | Yes | Probably not |
| Chondroitin‐6 sulfate | Not changed | Reduced relative to chondroitin‐4‐sulfate (Säämänen et al. 1990) | Increased (Ratcliffe et al. 1993) |
| Water content | Increased | Yes | Increased early in disease |
| Collagen cross‐linking | Decreased | Yes | Unknown |
| Appearance | Smooth, shiny, healthy | N/A | Roughened, dull |
But is there a relationship between atrophy and OA? And why does exercise sometimes result in thickening of the cartilage and sometimes OA? The answer likely relates back to the defined ‘mechanostat’ within the tissue. The authors speculate that if mechanical stimuli exceed a given threshold, or fall outside certain parameters, they are perceived as injurious and trigger pathways associated with rapid matrix remodelling and attempted repair. TAK1 activation appears to be critical in this response. TAK1 is a pivotal upstream regulator of inflammatory signalling, is induced by cartilage trauma and drives inflammatory gene regulation and matrix proteolysis (Ismail et al. 2015, 2016). The upstream activator of TAK1 remains elusive and does not appear to be mediated by a soluble factor (Ismail et al. 2017). Exceeding the injury threshold could occur either when the mechanical load is increased, or when the load is moderate but the joint has lost its mechanoprotective mechanisms. Examples of the latter include loss of stability of the joint and weakening of the cartilage matrix by atrophy. This would provide a plausible explanation for why ad libitum remobilisation leads to recovery after disuse but forced remobilisation leads to OA‐like changes in the cartilage (Palmoski & Brandt, 1981). Similarly in joints weakened by papain injection, moderate exercise leads to rapid degeneration of the tissue (Siebelt et al. 2014). A systematic review of experimental daily exercise in animals also suggests a non‐linear dose response with medium loads appearing good for cartilage but high loads precipitating degeneration (Bricca et al. 2017).
We presume that the ‘injury’ threshold changes as the cartilage mechanoadapts and will be different for each individual according to genetics, the mechanical integrity of the tissue, the amount of load usually experienced by the joint and the ‘pre‐tuned’ sensing mechanisms of their chondrocytes. Nonetheless, it is highly likely that mechanical training (graded exercise rather than overnight athleticism) will better protect the cartilage from load‐induced activation of matrix remodelling and subsequent OA. Aiming for thicker cartilage is probably a good start. Whether it is possible to optimise cartilage thickness during skeletal development to prevent OA later in life, remains an important unanswered question.
Conclusions
Mechanoadaptation in cartilage is rapid and reversible, and potentially of a similar scale to that seen in muscle and bone. Comprehensive studies of cartilage atrophy in vivo have been carried out although largely during the ‘pre‐molecular’ era and prior to the discovery of direct chondrocyte mechanosensing mechanisms. Advances in newly available glycobiology and proteomic techniques, in combination with genetic modification in rodents, will add considerable value to future studies. Cartilage atrophy is readily distinguished from osteoarthritis at the clinical, tissue and molecular level but cannot be discerned on a plain radiograph except by noting the absence of bone remodelling. As joint space narrowing is typically used to diagnose OA, it is important to consider atrophy as a differential diagnosis. Harnessing the molecules that drive mechanoadaption in articular cartilage may provide novel strategies to prevent or treat OA.
Additional information
Competing interests
None declared.
Author contributions
Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
We acknowledge salary support from Arthritis Research UK (grant no. 20205) and the Kennedy Trust for Rheumatology Research.
Biographies
Tonia L. Vincent is an academic rheumatologist and Director of the Centre for OA Pathogenesis in Oxford. She works on the molecular response of cartilage to injury and how this relates to the development of osteoarthritis. She did her PhD with Professor Jeremy Saklatvala and has been supported by Wellcome Trust and Arthritis Research UK fellowships. She continues to be clinically active.

Following PhD training under Professor J. Rodney Levick at St George's, University of London, studying the mechanobiology of the synovial joint, Angus Wann moved to Bioengineering at Queen Mary University of London to work with Professor Martin Knight and became fascinated by the primary cilium. He received a fellowship jointly funded by the Kennedy Trust for Rheumatology Research (KTRR) and the Arthritis Research UK Centre for OA Pathogenesis and now runs his own lab focusing on pathophysiological roles for ciliary proteins in musculoskeletal disease.
Edited by: Ole Petersen & Maike Glitsch
References
- Anderson J & Breidahl P (1981). Cartilage atrophy following spinal cord damage. Australas Radiol 25, 98–103. [DOI] [PubMed] [Google Scholar]
- Behrens F, Kraft EL & Oegema TR (1989). Biochemical changes in articular cartilage after joint immobilization by casting or external fixation. J Orthop Res 7, 335–343. [DOI] [PubMed] [Google Scholar]
- Benichou C & Wirotius JM (1982). Articular cartilage atrophy in lower limb amputees. Arthritis Rheum 25, 80–82. [DOI] [PubMed] [Google Scholar]
- Berendsen AD, Fisher LW, Kilts TM, Owens RT, Robey PG, Gutkind JS & Young MF (2011). Modulation of canonical Wnt signaling by the extracellular matrix component biglycan. Proc Natl Acad Sci U S A 108, 17022–17027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt KD, Dieppe P & Radin EL (2009). Commentary: is it useful to subset “primary” osteoarthritis? A critique based on evidence regarding the etiopathogenesis of osteoarthritis. Semin Arthritis Rheum 39, 81–95. [DOI] [PubMed] [Google Scholar]
- Bricca A, Juhl CB, Grodzinsky AJ & Roos EM (2017). Impact of a daily exercise dose on knee joint cartilage – a systematic review and meta‐analysis of randomized controlled trials in healthy animals. Osteoarthritis Cartilage 25, 1223–1237. [DOI] [PubMed] [Google Scholar]
- Brook MS, Wilkinson DJ, Phillips BE, Perez‐Schindler J, Philp A, Smith K & Atherton PJ (2016). Skeletal muscle homeostasis and plasticity in youth and ageing: impact of nutrition and exercise. Acta Physiol (Oxf) 216, 15–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burleigh A, Chanalaris A, Gardiner MD, Driscoll C, Boruc O, Saklatvala J & Vincent TL (2012). Joint immobilization prevents murine osteoarthritis and reveals the highly mechanosensitive nature of protease expression in vivo. Arthritis Rheum 64, 2278–2288. [DOI] [PubMed] [Google Scholar]
- Chia S‐L, Sawaji Y, Burleigh A, McLean C, Inglis J, Saklatvala J & Vincent T (2009). Fibroblast growth factor 2 is an intrinsic chondroprotective agent that suppresses ADAMTS‐5 and delays cartilage degradation in murine osteoarthritis. Arthritis Rheum 60, 2019–2027. [DOI] [PubMed] [Google Scholar]
- Clark AL, Votta BJ, Kumar S, Liedtke W & Guilak F (2010). Chondroprotective role of the osmotically sensitive ion channel transient receptor potential vanilloid 4: age‐ and sex‐dependent progression of osteoarthritis in Trpv4‐deficient mice. Arthritis Rheum 62, 2973–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements KM, Flannelly JK, Tart J, Brockbank SMV, Wardale J, Freeth J, Parker AE & Newham P (2011). Matrix metalloproteinase 17 is necessary for cartilage aggrecan degradation in an inflammatory environment. Ann Rheum Dis 70, 683–689. [DOI] [PubMed] [Google Scholar]
- Clements KM, Price JS, Chambers MG, Visco DM, Poole AR & Mason RM (2003). Gene deletion of either interleukin‐1β, interleukin‐1β‐converting enzyme, inducible nitric oxide synthase, or stromelysin 1 accelerates the development of knee osteoarthritis in mice after surgical transection of the medial collateral ligament and partial medial meniscectomy. Arthritis Rheum 48, 3452–3463. [DOI] [PubMed] [Google Scholar]
- Dell'Accio F, De Bari C, Eltawil NM, Vanhummelen P & Pitzalis C (2008). Identification of the molecular response of articular cartilage to injury, by microarray screening: Wnt‐16 expression and signaling after injury and in osteoarthritis. Arthritis Rheum 58, 1410–1421. [DOI] [PubMed] [Google Scholar]
- Dell'Accio F, De Bari C, El Tawil NMF, Barone F, Mitsiadis TA, O'Dowd J & Pitzalis C (2006). Activation of WNT and BMP signaling in adult human articular cartilage following mechanical injury. Arthritis Res Ther 8, R139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djurasovic M, Aldridge JW, Grumbles R, Rosenwasser MP, Howell D & Ratcliffe A (1998). Knee joint immobilization decreases aggrecan gene expression in the meniscus. Am J Sports Med 26, 460–466. [DOI] [PubMed] [Google Scholar]
- Drexler S, Wann A & Vincent TL (2014). Are cellular mechanosensors potential therapeutic targets in osteoarthritis? Int J Clin Rheumatol 9, 155–167. [Google Scholar]
- Dudhia J (2005). Aggrecan, aging and assembly in articular cartilage. Cell Mol Life Sci 62, 2241–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckstein F, Adam C, Sittek H, Becker C, Milz S, Schulte E, Reiser M & Putz R (1997). Non‐invasive determination of cartilage thickness throughout joint surfaces using magnetic resonance imaging. J Biomech 30, 285–289. [DOI] [PubMed] [Google Scholar]
- Eckstein F, Faber S, Mühlbauer R, Hohe J, Englmeier KH, Reiser M & Putz R (2002). Functional adaptation of human joints to mechanical stimuli. Osteoarthritis Cartilage 10, 44–50. [DOI] [PubMed] [Google Scholar]
- Eckstein F, Lemberger B, Stammberger T, Englmeier KH & Reiser M (2000). Patellar cartilage deformation in vivo after static versus dynamic loading. J Biomech 33, 819–825. [DOI] [PubMed] [Google Scholar]
- Galea GL, Lanyon LE & Price JS (2017). Sclerostin's role in bone's adaptive response to mechanical loading. Bone 96, 38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasson SS, Askew R, Sheppard B, Carito B, Blanchet T, Ma H‐L, Flannery CR, Peluso D, Kanki K, Yang Z, Majumdar MK & Morris EA (2005). Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 434, 644–648. [DOI] [PubMed] [Google Scholar]
- Greig CA, Gray C, Rankin D, Young A, Mann V, Noble B & Atherton PJ (2011). Blunting of adaptive responses to resistance exercise training in women over 75y. Exp Gerontol 46, 884–890. [DOI] [PubMed] [Google Scholar]
- Gruber J, Vincent TL, Hermansson M, Bolton M, Wait R & Saklatvala J (2004). Induction of interleukin‐1 in articular cartilage by explantation and cutting. Arthritis Rheum 50, 2539–2546. [DOI] [PubMed] [Google Scholar]
- Grumbles RM, Howell DS, Howard GA, Roos BA, Setton LA, Mow VC, Ratcliffe A, Müller FJ & Altman RD (1995). Cartilage metalloproteases in disuse atrophy. J Rheumatol Suppl 43, 146–148. [PubMed] [Google Scholar]
- Haapala J, Arokoski JP, Hyttinen MM, Lammi M, Tammi M, Kovanen V, Helminen HJ & Kiviranta I (1999). Remobilization does not fully restore immobilization induced articular cartilage atrophy. Clin Orthop Relat Res 218–229. [PubMed] [Google Scholar]
- Haapala J, Arokoski JP, Rönkkö S, Agren U, Kosma VM, Lohmander LS, Tammi M, Helminen HJ & Kiviranta I (2001). Decline after immobilisation and recovery after remobilisation of synovial fluid IL1, TIMP, and chondroitin sulphate levels in young beagle dogs. Ann Rheum Dis 60, 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapala J, Lammi MJ, Inkinen R, Parkkinen JJ, Agren UM, Arokoski J, Kiviranta I, Helminen HJ & Tammi MI (1996). Coordinated regulation of hyaluronan and aggrecan content in the articular cartilage of immobilized and exercised dogs. J Rheumatol 23, 1586–1593. [PubMed] [Google Scholar]
- Hamann N, Zaucke F, Heilig J, Oberländer KD, Brüggemann G‐P & Niehoff A (2014). Effect of different running modes on the morphological, biochemical, and mechanical properties of articular cartilage. Scand J Med Sci Sports 24, 179–188. [DOI] [PubMed] [Google Scholar]
- Haubner M, Eckstein F, Schnier M, Lösch A, Sittek H, Becker C, Kolem H, Reiser M & Englmeier KH (1997). A non‐invasive technique for 3‐dimensional assessment of articular cartilage thickness based on MRI. Part 2: Validation using CT arthrography. Magn Reson Imaging 15, 805–813. [DOI] [PubMed] [Google Scholar]
- Heinemeier KM, Schjerling P, Heinemeier J, Møller MB, Krogsgaard MR, Grum‐Schwensen T, Petersen MM & Kjaer M (2016). Radiocarbon dating reveals minimal collagen turnover in both healthy and osteoarthritic human cartilage. Sci Transl Med 8, 346ra90. [DOI] [PubMed] [Google Scholar]
- Hunziker EB, Quinn TM & Häuselmann H‐J (2002). Quantitative structural organization of normal adult human articular cartilage. Osteoarthritis Cartilage 10, 564–572. [DOI] [PubMed] [Google Scholar]
- Ingram KR, Wann AKT, Wingate RM, Coleman PJ, McHale N & Levick JR (2009). Signal pathways regulating hyaluronan secretion into static and cycled synovial joints of rabbits. J Physiol 587, 4361–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail HM, Didangelos A, Vincent TL & Saklatvala J (2017). Rapid activation of transforming growth factor β‐activated kinase 1 in chondrocytes by phosphorylation and K63‐linked polyubiquitination upon injury to animal articular cartilage. Arthritis Rheumatol 69, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail HM, Miotla‐Zarebska J, Troeberg L, Tang X, Stott B, Yamamoto K, Nagase H, Fosang AJ, Vincent TL & Saklatvala J (2016). Brief report: JNK‐2 controls aggrecan degradation in murine articular cartilage and the development of experimental osteoarthritis. Arthritis Rheumatol 68, 1165–1171. [DOI] [PubMed] [Google Scholar]
- Ismail HM, Yamamoto K, Vincent TL, Nagase H, Troeberg L & Saklatvala J (2015). Interleukin‐1 acts via the JNK‐2 signaling pathway to induce aggrecan degradation by human chondrocytes. Arthritis Rheumatol 67, 1826–1836. [DOI] [PubMed] [Google Scholar]
- Jones G, Ding C, Glisson M, Hynes K, Ma D & Cicuttini F (2003). Knee articular cartilage development in children: a longitudinal study of the effect of sex, growth, body composition, and physical activity. Pediatr Res 54, 230–236. [DOI] [PubMed] [Google Scholar]
- Jurvelin J, Kiviranta I, Tammi M & Helminen HJ (1986a). Effect of physical exercise on indentation stiffness of articular cartilage in the canine knee. Int J Sports Med 7, 106–110. [DOI] [PubMed] [Google Scholar]
- Jurvelin J, Kiviranta I, Tammi M & Helminen JH (1986b). Softening of canine articular cartilage after immobilization of the knee joint. Clin Orthop Relat Res 246–252. [PubMed] [Google Scholar]
- Kahn J, Shwartz Y, Blitz E, Krief S, Sharir A, Breitel DA, Rattenbach R, Relaix F, Maire P, Rountree RB, Kingsley DM & Zelzer E (2009). Muscle contraction is necessary to maintain joint progenitor cell fate. Dev Cell 16, 734–743. [DOI] [PubMed] [Google Scholar]
- Kavanagh E, Church VL, Osborne AC, Lamb KJ, Archer CW, Francis‐West PH & Pitsillides AA (2006). Differential regulation of GDF‐5 and FGF‐2/4 by immobilisation in ovo exposes distinct roles in joint formation. Dev Dyn 235, 826–834. [DOI] [PubMed] [Google Scholar]
- Kiviranta I, Jurvelin J, Tammi M, Säämänen A‐M & Helminen HJ (1987). Weight bearing controls glycosaminoglycan concentration and articular cartilage thickness in the knee joints of young beagle dogs. Arthritis Rheum 30, 801–809. [DOI] [PubMed] [Google Scholar]
- Kiviranta I, Tammi M, Jurvelin J, Arokoski J, Säämänen A‐M & Helminen HJ (1992). Articular cartilage thickness and glycosaminoglycan distribution in the canine knee joint after strenuous running exercise. Clin Orthop Relat Res 302–308. [PubMed] [Google Scholar]
- Kiviranta I, Tammi M, Jurvelin J, Säämänen A‐M & Helminen HJ (1988). Moderate running exercise augments glycosaminoglycans and thickness of articular cartilage in the knee joint of young beagle dogs. J Orthop Res 6, 188–195. [DOI] [PubMed] [Google Scholar]
- Lee W, Leddy HA, Chen Y, Lee SH, Zelenski NA, McNulty AL, Wu J, Beicker KN, Coles J, Zauscher S, Grandl J, Sachs F, Guilak F & Liedtke WB (2014). Synergy between Piezo1 and Piezo2 channels confers high‐strain mechanosensitivity to articular cartilage. Proc Natl Acad Sci U S A 111, E5114–E5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroux MA, Cheung HS, Bau JL, Wang JY, Howell DS & Setton LA (2001). Altered mechanics and histomorphometry of canine tibial cartilage following joint immobilization. Osteoarthritis Cartilage 9, 633–640. [DOI] [PubMed] [Google Scholar]
- Lösch A, Eckstein F, Haubner M & Englmeier KH (1997). A non‐invasive technique for 3‐dimensional assessment of articular cartilage thickness based on MRI. Part 1: Development of a computational method. Magn Reson Imaging 15, 795–804. [DOI] [PubMed] [Google Scholar]
- Madej W, van Caam A, Blaney Davidson EN, Hannink G, Buma P & van der Kraan PM (2016). Ageing is associated with reduction of mechanically‐induced activation of Smad2/3P signaling in articular cartilage. Osteoarthritis Cartilage 24, 146–157. [DOI] [PubMed] [Google Scholar]
- Malda J, de Grauw JC, Benders KEM, Kik MJL, van de Lest CHA, Creemers LB, Dhert WJA & van Weeren PR (2013). Of mice, men and elephants: the relation between articular cartilage thickness and body mass. PLoS One 8, e57683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankin HJ & Lippiello L (1969). The turnover of adult rabbit articular cartilage. J Bone Joint Surg Am 51, 1591–1600. [PubMed] [Google Scholar]
- Meachim G (1963). The effect of scarification on articular cartilage in the rabbit. J Bone Joint Surg Br 45B, 150–161. [Google Scholar]
- Mitchell PG, Magna HA, Reeves LM, Lopresti‐Morrow LL, Yocum SA, Rosner PJ, Geoghegan KF & Hambor JE (1996). Cloning, expression, and type II collagenolytic activity of matrix metalloproteinase‐13 from human osteoarthritic cartilage. J Clin Invest 97, 761–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama H, Yoshimura O, Kawamata S, Takayanagi K, Kurose T, Kubota A, Hosoda M & Tobimatsu Y (2008). Alteration in articular cartilage of rat knee joints after spinal cord injury. Osteoarthritis Cartilage 16, 392–398. [DOI] [PubMed] [Google Scholar]
- Müller FJ, Setton LA, Manicourt DH, Mow VC, Howell DS & Pita JC (1994). Centrifugal and biochemical comparison of proteoglycan aggregates from articular cartilage in experimental joint disuse and joint instability. J Orthop Res 12, 498–508. [DOI] [PubMed] [Google Scholar]
- Ni G‐X, Liu S‐Y, Lei L, Li Z, Zhou Y‐Z & Zhan L‐Q (2013). Intensity‐dependent effect of treadmill running on knee articular cartilage in a rat model. Biomed Res Int 2013, 172392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura M, Sakitani N, Iwasawa H, Kohara Y, Takano S, Wakimoto Y, Kuroki H & Moriyama H (2017). Thinning of articular cartilage after joint unloading or immobilization. An experimental investigation of the pathogenesis in mice. Osteoarthritis Cartilage 25, 727–736. [DOI] [PubMed] [Google Scholar]
- O'Conor CJ, Leddy HA, Benefield HC, Liedtke WB & Guilak F (2014). TRPV4‐mediated mechanotransduction regulates the metabolic response of chondrocytes to dynamic loading. Proc Natl Acad Sci U S A 111, 1316–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Conor CJ, Ramalingam S, Zelenski NA, Benefield HC, Rigo I, Little D, Wu C‐L, Chen D, Liedtke W, McNulty AL & Guilak F (2016). Cartilage‐specific knockout of the mechanosensory ion channel TRPV4 decreases age‐related osteoarthritis. Sci Rep 6, 29053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmoski M, Perricone E & Brandt KD (1979). Development and reversal of a proteoglycan aggregation defect in normal canine knee cartilage after immobilization. Arthritis Rheum 22, 508–517. [DOI] [PubMed] [Google Scholar]
- Palmoski MJ & Brandt KD (1981). Running inhibits the reversal of atrophic changes in canine knee cartilage after removal of a leg cast. Arthritis Rheum 24, 1329–1337. [DOI] [PubMed] [Google Scholar]
- Pool WH (1974). Cartilage atrophy. Radiology 112, 47–50. [DOI] [PubMed] [Google Scholar]
- Poole CA (1997). Articular cartilage chondrons: form, function and failure. J Anat 191, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe A, Shurety W & Caterson B (1993). The quantitation of a native chondroitin sulfate epitope in synovial fluid lavages and articular cartilage from canine experimental osteoarthritis and disuse atrophy. Arthritis Rheum 36, 543–551. [DOI] [PubMed] [Google Scholar]
- Recht MP, Kramer J, Marcelis S, Pathria MN, Trudell D, Haghighi P, Sartoris DJ & Resnick D (1993). Abnormalities of articular cartilage in the knee: analysis of available MR techniques. Radiology 187, 473–478. [DOI] [PubMed] [Google Scholar]
- Roos EM & Dahlberg L (2005). Positive effects of moderate exercise on glycosaminoglycan content in knee cartilage: a four‐month, randomized, controlled trial in patients at risk of osteoarthritis. Arthritis Rheum 52, 3507–3514. [DOI] [PubMed] [Google Scholar]
- Ross TD, Coon BG, Yun S, Baeyens N, Tanaka K, Ouyang M & Schwartz MA (2013). Integrins in mechanotransduction. Curr Opin Cell Biol 25, 613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Säämänen A‐M, Tammi M, Jurvelin J, Kiviranta I & Helminen HJ (1990). Proteoglycan alterations following immobilization and remobilization in the articular cartilage of young canine knee (stifle) joint. J Orthop Res 8, 863–873. [DOI] [PubMed] [Google Scholar]
- Säämänen A‐M, Tammi M, Kiviranta I & Helminen HJ (1988). Running exercise as a modulator of proteoglycan matrix in the articular cartilage of young rabbits. Int J Sports Med 9, 127–133. [DOI] [PubMed] [Google Scholar]
- Säämänen A‐M, Tammi M, Kiviranta I, Jurvelin J & Helminen HJ (1989). Levels of chondroitin‐6‐sulfate and nonaggregating proteoglycans at articular cartilage contact sites in the knees of young dogs subjected to moderate running exercise. Arthritis Rheum 32, 1282–1292. [DOI] [PubMed] [Google Scholar]
- Shea CA, Rolfe RA & Murphy P (2015). The importance of foetal movement for co‐ordinated cartilage and bone development in utero: clinical consequences and potential for therapy. Bone Joint Res 4, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebelt M, Groen HC, Koelewijn SJ, de Blois E, Sandker M, Waarsing JH, Müller C, van Osch GJVM, de Jong M & Weinans H (2014). Increased physical activity severely induces osteoarthritic changes in knee joints with papain induced sulfate‐glycosaminoglycan depleted cartilage. Arthritis Res Ther 16, R32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon WH (1970). Scale effects in animal joints. I. Articular cartilage thickness and compressive stress. Arthritis Rheum 13, 244–256. [DOI] [PubMed] [Google Scholar]
- Sutter EG, Widmyer MR, Utturkar GM, Spritzer CE, Garrett WE & Defrate LE (2015). In vivo measurement of localized tibiofemoral cartilage strains in response to dynamic activity. Am J Sports Med 43, 370–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tammi M, Säämänen A‐M, Jauhiainen A, Malminen O, Kiviranta I & Helminen H (1983). Proteoglycan alterations in rabbit knee articular cartilage following physical exercise and immobilization. Connect Tissue Res 11, 45–55. [DOI] [PubMed] [Google Scholar]
- Tang X, Muhammad H, McLean C, Miotla-Zarebska J, Flemming J, Didangelos A, Onnerfjord P, Leask A, Saklatvala J & Vincent TL (2018). Connective tissue growth factor contributes to joint homeostasis and osteoarthritis severity by controlling the matrix sequestration and activation of latent TGFb. Ann Rheum Dis. 10.1136/annrheumdis-2018-212964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troeberg L & Nagase H (2012). Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim Biophys Acta 1824, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uda Y, Azab E, Sun N, Shi C & Pajevic PD (2017). Osteocyte mechanobiology. Curr Osteoporos Rep 15, 318–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Caam A, Madej W, Thijsen E, Garcia de Vinuesa A, van den Berg W, Goumans M‐J, ten Dijke P, Blaney‐Davidson E & van der Kraan PM (2016). Expression of TGFβ‐family signalling components in ageing cartilage: Age‐related loss of TGFβ and BMP receptors. Osteoarthritis Cartilage 24, 1235–1245. [DOI] [PubMed] [Google Scholar]
- Vanwanseele B, Eckstein F, Knecht H, Spaepen A & Stüssi E (2003). Longitudinal analysis of cartilage atrophy in the knees of patients with spinal cord injury. Arthritis Rheum 48, 3377–3381. [DOI] [PubMed] [Google Scholar]
- Vanwanseele B, Eckstein F, Knecht H, Stüssi E & Spaepen A (2002). Knee cartilage of spinal cord‐injured patients displays progressive thinning in the absence of normal joint loading and movement. Arthritis Rheum 46, 2073–2078. [DOI] [PubMed] [Google Scholar]
- Vincent T, Hermansson M, Bolton M, Wait R & Saklatvala J (2002). Basic FGF mediates an immediate response of articular cartilage to mechanical injury. Proc Natl Acad Sci U S A 99, 8259–8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent TL (2013). Targeting mechanotransduction pathways in osteoarthritis: a focus on the pericellular matrix. Curr Opin Pharmacol 13, 449–454. [DOI] [PubMed] [Google Scholar]
- Vincent TL, Hermansson MA, Hansen UN, Amis AA & Saklatvala J (2004). Basic fibroblast growth factor mediates transduction of mechanical signals when articular cartilage is loaded. Arthritis Rheum 50, 526–533. [DOI] [PubMed] [Google Scholar]
- Vincent TL, McLean CJ, Full LE, Peston D & Saklatvala J (2007). FGF‐2 is bound to perlecan in the pericellular matrix of articular cartilage, where it acts as a chondrocyte mechanotransducer. Osteoarthritis Cartilage 15, 752–763. [DOI] [PubMed] [Google Scholar]
- Wann AKT, Zuo N, Haycraft CJ, Jensen CG, Poole CA, McGlashan SR & Knight MM (2012). Primary cilia mediate mechanotransduction through control of ATP‐induced Ca2+ signaling in compressed chondrocytes. FASEB J 26, 1663–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt FE, Ismail HM, Didangelos A, Peirce M, Vincent TL, Wait R & Saklatvala J (2013). Src and fibroblast growth factor 2 independently regulate signaling and gene expression induced by experimental injury to intact articular cartilage. Arthritis Rheum 65, 397–407. [DOI] [PubMed] [Google Scholar]
- Yuasa T, Kondo N, Yasuhara R, Shimono K, Mackem S, Pacifici M, Iwamoto M & Enomoto‐Iwamoto M (2009). Transient activation of Wnt/β‐catenin signaling induces abnormal growth plate closure and articular cartilage thickening in postnatal mice. Am J Pathol 175, 1993–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziaaldini MM, Marzetti E, Picca A & Murlasits Z (2017). Biochemical pathways of sarcopenia and their modulation by physical exercise: a narrative review. Front Med (Lausanne) 4, 167. [DOI] [PMC free article] [PubMed] [Google Scholar]