

Abstract
Background
CARD11 encodes a scaffold protein in lymphocytes that links antigen receptor engagement with downstream signaling to NF-κB, JNK, and mTORC1. Germline CARD11 mutations cause several distinct primary immune disorders in humans, including SCID (biallelic null mutations), B cell Expansion with NF-κB and T cell Anergy (BENTA; heterozygous, gain-of-function mutations), and severe atopic disease (loss-of-function, heterozygous, dominant interfering mutations), which has focused attention on CARD11 mutations discovered by whole exome sequencing.
Objectives
To determine the molecular actions of an extended allelic series of CARD11, and to characterize the expanding range of clinical phenotypes associated with heterozygous CARD11 loss-of-function alleles.
Methods
Cell transfections and primary T cell assays were utilized to evaluate signaling and function of CARD11 variants.
Results
Here we report on an expanded cohort of patients harboring novel heterozygous CARD11 mutations that extend beyond atopy to include other immunologic phenotypes not previously associated with CARD11 mutations. In addition to (and sometimes excluding) severe atopy, heterozygous missense and indel mutations in CARD11 presented with immunologic phenotypes similar to those observed in STAT3-LOF, DOCK8 deficiency, common variable immune deficiency (CVID), neutropenia, and immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX)-like syndrome. Pathogenic variants exhibited dominant negative activity, and were largely confined to the CARD or coiled-coil domains of the CARD11 protein.
Conclusion
These results illuminate a broader phenotypic spectrum associated with CARD11 mutations in humans, and underscore the need for functional studies to demonstrate that rare gene variants encountered in expected and unexpected phenotypes must nonetheless be validated for pathogenic activity.
Keywords: CARD11, atopy, atopic dermatitis, dominant negative, primary immunodeficiency, immune dysregulation
Capsule Summary
This description of a large cohort of patients broadens the spectrum of clinical disease phenotypes linked to CARD11 DN mutations, including atopy, cutaneous viral and respiratory infections, hypogammaglobulinemia, autoimmunity, neutropenia, and lymphoma.
Introduction
Hypomorphic mutations in genes whose protein products are critical for immune function provide an opportunity to assess the protein’s roles in the context of a functioning, albeit impaired, immune response. Unlike the unresponsive or absent effector compartments often associated with null mutations that manifest as severe combined immune deficiency (SCID), these mutations preserve sufficient function to allow for development of the particular cell(s) or organ(s) with which it is involved. Studies of such allelic variants in mice and other model organisms have taught us much about immune system development and function. With the wider availability and application of next-generation sequencing technologies, we now have more opportunities to explore the phenomenon of allelic variance and phenotypic heterogeneity in humans. One example can be found in the case of RAG1 – null mutants lead to the absence of T-cells and grossly abnormal thymic development. Hypomorphic mutations lead to functioning T-cells but disruptions of repertoire and thymic development that shed light on immune tolerance, RAG1 protein domain function, and other immune processes (1).
Another example is found in different types of mutations described in CARD11 leading to different phenotypes. CARD11 is a critical 1154 amino acid protein scaffold best known for linking antigen recognition with downstream NF-κB activation in lymphocytes (2, 3). The protein CARD11 (also known as CARMA1, represented by the sequences NP_115791.3 and NM_032415) includes an N-terminal caspase recruitment domain (CARD, 1-110), LATCH (112-130), coiled-coil (CC, 130-449) domains, and a C-terminal membrane-associated guanylate kinase domain (MAGUK, 667-1140) comprised of PDZ, SH3 and GUK domains. Biallelic null mutations of CARD11, in both patients and mouse models, lead to severe T and B-cell immune deficiency (4). Somatic gain-of-function (GOF) CARD11 mutations are commonly found in non-Hodgkin B cell lymphomas, whereas germline GOF mutations give rise to BENTA (B cell Expansion with NF-κB and T cell Anergy) disease in humans (2, 5, 6). Surprisingly, hypomorphic/dominant negative (DN) mutations in both mice and human patients permit sufficient effector function to reveal a strong disposition toward atopic phenotypes, in addition to variable immune deficiency (7–9). Because CARD11 oligomerization is essential for downstream signaling, heterozygous variants can either enhance or dominantly interfere with CARD11 function (10, 11).
Greater access to next-generation sequencing virtually guarantees that variants in a particular gene can now be identified in patients with ever-broadening phenotypes, especially for cases of GOF and hypomorphic loss of function (LOF) mutations (12). Although algorithms can assist in predicting which variants are likely to be benign or deleterious, previously undescribed rare or novel variants in such genes must always be validated for pathogenicity using relevant biological assays. Moreover, in silico prediction methods do not distinguish between variants seen in the heterozygous state and homozygous state, which is critical for CARD11. Furthermore, collection and testing of variants from as many centers as possible can more accurately determine the breadth of disease associated with a given gene, especially given the role that referral bias can play in any given center.
We therefore report our experience with numerous heterozygous mutations in CARD11 in the context of severe familial atopic disease and other immunologic phenotypes not previously associated with CARD11 mutations. The atopy described here is a genetic tendency to develop symptoms of immediate hypersensitivity (e.g. food allergy, allergic rhinitis) or allergic inflammation (e.g. eczema, eosinophilic esophagitis), irrespective of specific allergen sensitization (13). In many cases, rare or novel mutations were uncovered in whole exome/genome sequences (WES/WGS) performed at major referral centers for multiple reasons, particularly in patients without a clear putative genetic diagnosis. Herein we attribute several new DN CARD11 mutations to an expanded list of disease manifestations, and describe assays designed to help differentiate pathogenic vs. non-pathogenic variants in CARD11. Importantly, results of these assays are interpreted within the context of specific genotypic/phenotypic criteria that help to define a differential diagnosis for patients harboring CARD11 DN mutations. In addition to severe atopy, heterozygous missense mutations in CARD11 with DN activity can present with common variable immune deficiency (CVID), neutropenia, cutaneous viral infections, and immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX)-like syndrome. Collectively, our findings define a broader spectrum of immune disease associated with detrimental CARD11 mutations, which are most often confined to specific domains of the CARD11 protein. Nevertheless, our evaluations underscore the idea that rare gene variants found by WES/WGS can be pathogenic even when not matching with reported phenotypes. At the same time, CARD11 mutations associated with an expected phenotype must nonetheless be validated for pathogenic activity using functional studies.
Methods
Patients
Informed consent was obtained from all participating patients and their family members according to protocols approved by Institutional Review Boards and ethics boards at their respective institutions.
Whole exome sequencing (WES) and genetic analysis
WES was performed on the majority of patients described here according to established protocols. For example, Kindreds 6, 19 and 29 were analyzed as follows. Genomic DNA was extracted from peripheral blood cells and Illumina paired-end genomic DNA sample preparation kit (PE-102–1001, Illumina) was used for preparing the libraries followed by exome-enriched library using the Illumina TruSeq exome kit (FC-121–1008, Illumina). Samples were sequenced on an Illumina HiSeq as 100-bp paired-end reads. DNA reads were mapped to the GRCh38 human genome reference using the default parameters of the Burrows-Wheeler Aligner (bio-bwa.sourceforge.net). Single nucleotide substitutions and small insertion deletions were identified and filtered based on quality with SAMtools software package (samtools.sourceforge.net) and annotated with Annovar tool (www.openbioinformatics.org). Filtering of variants for novelty was performed according to minor allele frequency, mouse mutant phenotype in the homologous mouse gene, Mendelian disease associations (OMIM), pathway analysis (gene ontology [GO]), immune system expression (Immgen), CADD, SIFT, and PolyPhen-2 scores (14–16). After filtering and ranking variants, heterozygous CARD11 variants were investigated further.
Kindred 14 (R187P) was initially evaluated separately from the other families. Eight samples were subjected to whole exome sequencing (WES) as described previously (17) (Supplementary Materials and Methods). We selected putative causative variants that had an average read depth >20, a quality score of >200, and were heterozygous in at least one individual. Genetic analysis showed that seven of the samples were related and one sample was not related. The remaining seven samples were from one unaffected individual and six affected individuals. We did a combinatorial search for variants present in the heterozygous state in the six affected individuals and absent in the unaffected individual. We performed multipoint genetic linkage analysis with Superlink-online-SNP (18, 19) assuming a rare disease allele and dominant inheritance. Two variants satisfied the combinatorial condition of being heterozygous in six sampled affected individuals and absent in the one sampled unaffected individual: MICALL2 (p.Q202*) and CARD11 (p.R187P), and were at the same time located in a region (chr7:0.0-4.3 Mbp) consistent with genetic linkage.
PBMC signaling analysis
PBMC were isolated by Ficoll gradient centrifugation. Phosphorylation of p65 and/or S6, and degradation of IkB was assessed by intracellular flow cytometry after short ex vivo stimulations with PMA as previously described (9). To assess S6 phosphorylation under Th0 conditions, total PBMC (106/ml) were seeded in 1 mL RPMI (Gibco) supplemented with 10% FBS, penicillin/streptomycin and L-glutamine in an anti-CD3 antibody (OKT3, 1 μg/mL) coated 48-well plate. Anti-CD28 antibody (L293, 0.2 μg/mL,) and IL-2 (10 ng/mL, Peprotech) were then added to the culture (Th0), and cells were incubated in a 37°C CO 2 chamber. After 24 hours, cells were pelleted by centrifugation, stained with Live/Dead dye (Thermo Fisher), fixed with 1.6% paraformaldehyde in PBS, then centrifuged and permeabilized with absolute methanol. Cells were then stained for flow cytometry with fluorochrome-conjugated antibodies after washes with FACS buffer (PBS with 0.5% BSA). The antibodies (Abs) used for flow cytometry were: CD3 (UCHT1), CD4 (RPA-T4, L200), CD45RA (HI100), phospho-S6 (N7-548) (BD Biosciences). Percentages of phospho-S6+ live CD3+ CD4+ cells, gated on the lymphocyte SSC and CD45RA compartments, are shown in Figure 6.
Figure 6. Clinical diagnostic algorithm for recognizing CARD11 DN patients.
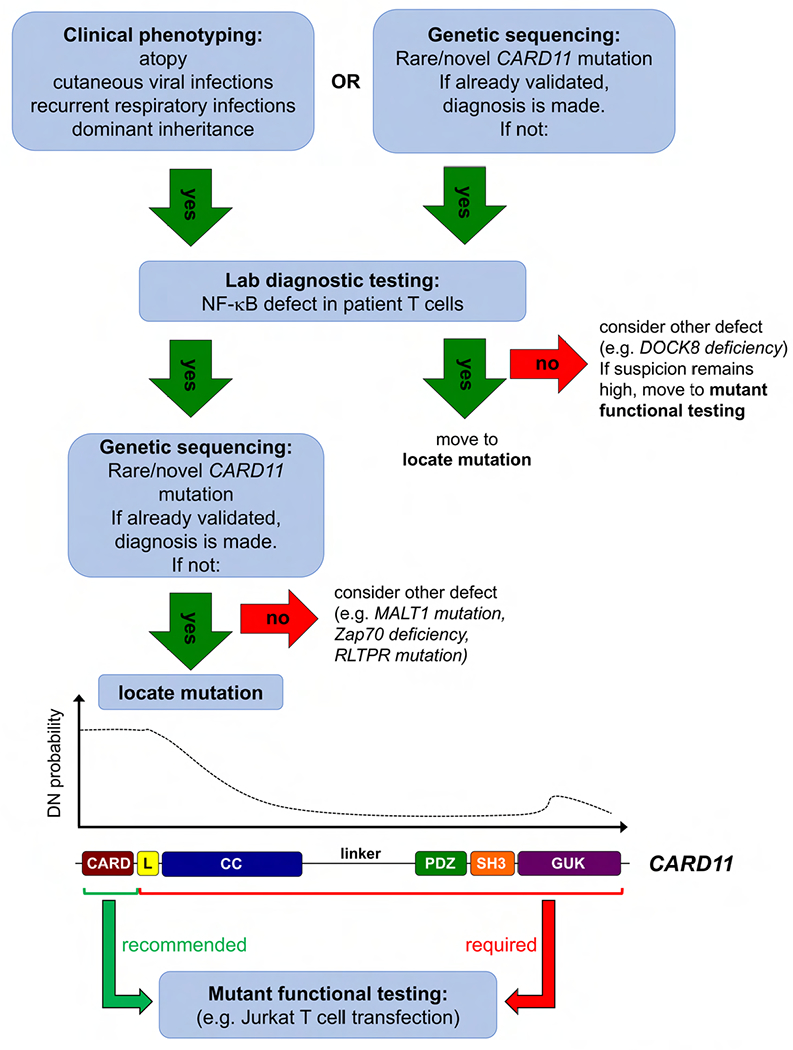
Schematic diagram of an algorithm to be used in identifying and diagnosing potential CARD11 DN patients. Suspected patients typically present with atopy with or without cutaneous viral infections and respiratory infections. Simple lab diagnostic tests to pinpoint TCR-induced NF-κB activation defects in primary patient T cells (e.g. phospho-p65, IκB degradation) are recommended prior to genomic sequencing analysis, or after a CARD11 mutation is uncovered (e.g. via WES). Within these selected patients, a mutation in the CARD domain (or N-terminal CC domain) is highly likely to be DN, whereas mutations in the rest of the protein are more likely benign. Functional testing (e.g. Jurkat transfection assays for NF-κB activation) is recommended for all novel CARD11 variants, especially for those outside the CARD domain.
CARD11 mutant expression plasmids
Modified wild type (WT) and mutant pUNO-CARD11-FLAG plasmids were constructed and purified as previously described (9). Briefly, site-directed mutagenesis was utilized to introduce single nucleotide variants that were putative point mutations into the WT CARD11 construct (Invivogen) using primer-directed linear amplification with Pwo or Pful polymerase (Roche), followed by Dpnl digestion of methylated template DNA (ThermoFisher). All inserted variants were confirmed by Sanger sequencing. All resulting plasmids were purified from DH5α E. Coli (New England Biolabs) using a GenElute HP Plasmid Maxi-Prep Kit (Sigma).
Cell transfection assays
Both WT (clone E6.1, ATCC) and CARD11-deficient Jurkat T cells (referred to as JPM50.6) were cultured and transfected as previously described (9). JPM50.6 cells containing an integrated canonical NF-κB-driven GFP reporter were originally provided by Dr. Xin Lin (MD Anderson Cancer Center). Briefly, 5×106 Jurkat or JPM50.6 T cells were resuspended in 0.4 mL RPMI/10% FBS, placed in 0.4cm cuvettes (Bio-Rad) and electroporated (260 V, 950 μF) with 5-10 μg plasmid DNA (BTX Harvard Apparatus). For JPM50.6, cells were stimulated 24 hours post-transfection with 1 μg/ml anti-CD3/CD28 antibodies and incubated overnight (BD Biosciences). Relative canonical NF-κB activation in JPM50.6 was quantified based on mean fluorescence intensity (MFI) of an integrated -κBGFP reporter using an Accuri C6 flow cytometer (BD Biosciences). To assess S6 phosphorylation in Jurkat transfectants, cells were washed 24 hrs post-transfection in PBS and incubated in PBS/1%BSA for 1 hr at 37°C. Following an additional PBS wash, cells were stimulated with 1 mg/ml anti-CD3/CD28 antibodies (BD Biosciences) for 20 min at 37°C. Activation was stopped by adding ice cold PBS, and cells were pelleted and lysed in 1% NP-40 lysis buffer as previously described (9). Lysates (5–10 μg) were separated on 4-20% Tris-Glycine SDS gels and transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked in 5% milk/TBS/0.1%Tween20 and immunoblotted with the following Abs: anti-phospho-S6 (Ser235/236), anti-S6 (5G10), anti-CARD11 (1D12, Cell Signaling Technology; LS-C368868, LifeSpan Biosciences; OASG00985, Aviva Systems Biology); anti-HA (2-2.2.14, Thermo Fisher), anti-FLAG (M2) and anti-β-actin (AC-15, Sigma). Blots were washed 3× in TBS/0.1%Tween20), incubated in HRP-conjugated secondary Abs (Southern Biotech), and washed again. Bands were visualized by enhanced chemiluminescence (Thermo Fisher) and imaged on a ChemiDoc system (Bio-Rad). Spot densitometric quantification of phosphor-S6 vs. total S6 was performed using ImageLab software (Bio-Rad).
Hierarchical clustering analysis
Hierarchical Clustering with a complete linkage algorithm and an asymmetric binary distance measure (for binary variables) was used to explore the data contained in Online Repository Table 1. Due primarily to a significant proportion of missing data across many of the variables, only a subset of variables was included in the clustering algorithm. Moreover, only subjects with complete data on the chosen subset were included in the hierarchical clustering. Models were also run using different variable sets, distance measures, and clustering algorithms, and led to different sets of clusters. Hence presented results should be considered exploratory and descriptive.
Table 1. Clinical phenotypes for all patients with CARD11 variants.
Kindreds are listed in the order each variant appears within the CARD11 protein.
| Kindred | Mutation | Genotype | Functional defect | # patients | Atopic dermatitis | Asthma | Food allergy | Pneumonia | Bronchiectasis | Molluscum contagiosum | Cutaneous HSV | Warts | Other Phenotypes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | R30G | c.88C>G | DN | 6 | 1 | 3 | 0 | 1 | 0 | 2 | 0 | 0 | bacterial sinusitis/meningitis, visceral leishmaniasis, severe vernal keratoconjunctivitis, florid EoE, severe oral ulcers |
| 2 | R30Q | c.89G>A | DN | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | bullous pemphigoid |
| 3 | R30Q | c.89G>A | DN | 2 | 2 | 1 | 2 | 2 | 0 | 2 | 0 | 2 | viral pneumonia, S. aureus skin infection, flu, constipation |
| 4 | R30W | c.88C>T | DN | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | IPEX-like, FTT |
| 5 | R30W | c.88C>T | DN | 4 | 3 | 4 | 4 | 3 | 1 | 0 | 0 | 0 | recurrent respiratory infections, oral candidiasis, lichen sclerosis of vulva, psoriasis, impetigo |
| 6 | R47H | c.140G>A | DN | 3 | 1 | 1 | 0 | 0 | 0 | 3 | 3 | 3 | neutropenia, LGL |
| 7 | R47H | c.140G>A | DN | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 1 | progressive hypogammaglobulinemia |
| 8 | E57D | c.171 A>C | DN | 2 | 2 | 2 | 1 | 2 | 0 | 1 | 0 | 0 | ulcerative colitis, stroke, peripheral T lymphoma |
| 9 | R72G | c.214C>G | DN | 2 | 2 | 2 | 0 | 1 | 1 | 0 | 0 | 1 | alopecia, joint pain, oral ulcers, pulm TB |
| 10 | R72G | c.214C>G | DN | 1 | 1 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | persistent skin infections (VZV, HPV), EBV viremia, progressive B cell lymphopenia, frequent OM neutropenia |
| 11 | R75Q | c.224G>A | DN | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | neutropenia |
| 12 | L92W | c.275T>G | DN | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | |
| 13 | K143X | c.427A>T | Weak DN | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | ITP, alopecia, EoE |
| 14 | R187P | c.560G>C | DN | 10 | 10 | 5 | 4 | 4 | 2 | 8 | 6 | 5 | alopecia, cutaneous vasculitis, mycosis fungoides, broad nose, retained teeth, shingles |
| 15 | dup183-196 | c.701_713ins T | DN | 3 | 3 | 1 | 0 | 3 | 1 | 2 | 1 | 0 | prom forehead, broad nose, poor dentition, pulm TB, eosinophilic coloproctitis |
| 16 | L194P | c.581T>C | DN | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | 0 | |
| 17 | V195L | c.583G>C | LOF | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | diverticulitis, T2D |
| 18 | K362E | c.1084A>G | LOF | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | healthy |
| 19 | R408H | c.1223G>A | LOF | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | Evan’s syndrome, anhidrosis |
| 20 | P495S | c.C>T | Hypermorphic w/TCR stim | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | IPEX-like, FTT, T1D, alopecia, skin tags |
| 21 | I544L | c.1630A>C | Nil | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | atopic dermatitis |
| 22 | R608H | c.1823G>A | Nil | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | CMV myocarditis, adenoviral hepatitis |
| 23 | V659M | c.1975G>A | Nil | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | recurrent sinopulmonary/skin infections |
| 24 | T670M | c.2009C>T | Nil | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | Seizures, mental retardation, TB, candidiasis, facial dysmorphism |
| 25 | E766D | c2298G>T | Nil | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | severe atopic dermatitis |
| 26 | R848C | c.2542C>T | Hypermorphic w/ TCR stim | 2 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 0 | AHA, ITP, refractory cytopenias, drug-induced lupus, Crohn’s disease |
| 27 | R912Q/D1152N | c.2735G>A, c.3454G>A | Nil | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | late onset recurrent sinopulmonary infections |
| 28 | S923L | c.2768C>T | LOF | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | agammaglobulinemia, giardiasis |
| 29 | R974C | c.2920C>T | Weak DN | 2 | 0 | 1 | 0 | 1 | 0 | 1 | 0 | 0 | neutropenia, mycobacterial disease |
| 30 | R975W | c.2923C>T | DN | 2 | 2 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | |
| 31 | V983M | c.2947G>A | Nil | 2 | 1 | 1 | 1 | 1 | 0 | 0 | 1 | 0 | |
| 32 | E1028K | c.3082G>A | Nil | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | sJIA, MAS, pheochromocytoma, migraines |
| DN | 13 | 44 | 32 | 24 | 14 | 19 | 5 | 23 | 13 | 12 | |||
| % | 73% | 55% | 32% | 43% | 11% | 52% | 30% | 27% | |||||
| Non-DN | 15 | 16 | 6 | 3 | 1 | 7 | 0 | 0 | 1 | 0 | |||
| % | 38% | 19% | 6% | 44% | 0% | 0% | 6% | 0% | |||||
| TOTAL | 28 | 60 | 38 | 27 | 15 | 26 | 5 | 23 | 14 | 12 |
Unsupervised patient clustering
To depict patients with their phenotypic attributes in 2-dimensional space, we used Gower distance transformation (20) (as implemented in the daisy function in the R cluster v2.0.7-1 package (21, 22), which handles datasets with missing data points, followed by tSNE (23) as implemented in the Rtsne v0.13 library (using settings theta=0 and perplexity=10). Next, we divided the patient cohort into three clusters using K-means clustering in R (K=3). The process of using tSNE and K-means to cluster patients was repeated over 10 trials and consensus assignment was used to assign each patient to a final cluster id. To identify phenotypes important for each cluster, we performed attribute selection and logistic regression in Weka v3.8.2 (24). For attribute selection, we used the Fast Correlation-based Feature Search method (25) to select nonredundant attributes. Using the selected attributes, we performed multinomial logistic regression using the SimpleLogistic function (26) with 5-fold cross-validation to build a classifier for assigning patients to one of the three clusters. The features selected along with their predicted coefficients allowed us to assess the importance of phenotypes for each class separately. Based on 10 randomized trials, the classifier demonstrated an overall accuracy of 88.1% with standard deviation (SD) ±11.7%. Sensitivity and specificity for the trials were 98.33% (SD: ±8.42%) and 93.90% (SD: ±11.03%), respectively.
Statistics
For JPM50.6 transfections, two-way ANOVAs with Sidak correction were used to compare GFP MFI between WT and mutant CARD11. pS6/total S6 protein densitometric ratios were normalized to WT values and compared by a Wilcoxon signed-rank test. For primary cell assays, experimental and technical replicates were limited by small numbers of patient samples available. For patients in Kindreds 6, 19 and 29, ratios of pS6 gMFI (stimulated/unstimulated) were compared to healthy controls (matched per experiment) using a Kruskal-Wallis test.
Results
Novel dominant negative CARD11 mutations detected in a broad spectrum of immune disorders
Rare or novel CARD11 mutations were identified by allergy and primary immunodeficiency referral centers in patients with immune-deficient or dysregulatory phenotypes (Table 1). A total of 48 new patients in 27 families, with 26 different heterozygous germline CARD11 variants were referred. Salient patient phenotypes, combined with those already reported (7, 9), are summarized in Tables 1–2. These alleles were then evaluated at centers specialized in CARD11 biology and associated diseases and pathways. First, we investigated whether each variant altered T cell receptor (TCR) signaling. Each CARD11 variant was cloned into an expression construct and transfected in WT Jurkat or CARD11-deficient Jurkat (JPM50.6) T cell lines. As a control, we also tested a variant (p.C150L) identified as a somatic reversion mutant that restored NF-κB signaling in T cells from a CARD11-deficient patient that developed Omenn’s syndrome (27). Upon CD3/CD28 stimulation of transfected JPM50.6 cells, 14/26 mutants demonstrated significantly reduced NF-κB activation, indicating loss of function (LOF) (Figure 1A). Two variants (p.P495S, p.R848C) displayed enhanced NF-κB activation only after stimulation; unlike BENTA-associated mutations, these variants did not induce constitutive NF-κB activity. As all variants were heterozygous, we next tested whether each LOF mutant could dominantly interfere with WT CARD11 signaling when co-expressed at 50:50 ratios. Among 14 LOF mutations, dominant negative (DN) activity (defined as significantly reduced NF-κB activation relative to 100% WT expression) was observed for 10 (Figure 1B). Most of these new DN variants were confined to the CARD domain and proximal coiled-coil (CC) domains, which are critical for both CARD11 oligomerization and BCL10-MALT1 interactions (28, 29). All variants were comparably expressed in transfected cells, suggesting none of the mutations tested affected CARD11 protein translation and stability (Figure 1C and data not shown). Although a faint 15 kD band was noted upon prolonged blot exposure, we could not definitively confirm the presence of a truncated K143X protein by immunoblot using available N-terminus specific CARD11 Abs, which cross-reacted with many non-specific proteins (data not shown). However, the fact that expression of this construct, and not the Q945X truncation mutant (9), exhibits DN activity strongly suggests that it is expressed. Moreover, a K143X CARD11 expression construct engineered to include a C-terminal HA-tag was clearly expressed in JPM50.6 transfectants, and exhibited comparable DN activity compared to the untagged form (Supplemental Figure 1).
Table 2. Clinical summary of CARD11 DN patients.
Summary table of mean age, gender ratio, and relevant phenotypes shared by CARD11 DN patients, with the % of affected patients shown for each category.
| CARD11 DN Patients | |
|---|---|
| Age (mean −/+ SD) | 23.3 −/+ 19.5 |
| Age of disease onset (mean −/+ SD) | 5.2 −/+ 6.7 |
| % Female | 50% |
| Clinical Phenotype | % Patients Affected |
|---|---|
| Atopic disease | 89% |
| Atopic dermatitis | 73% |
| Asthma | 55% |
| Food allergies | 32% |
| Eosinophilic esophagitis | 7% |
| Cutaneous viral infections | 68% |
| Respiratory infections | 68% |
| Autoimmunity | 20% |
| Neutropenia | 14% |
| Oral ulcers | 14% |
| Hypogammaglobulinemia | 11% |
| Lymphoma | 7% |
Figure 1. Jurkat T cell transfection screens for LOF/DN activity of new CARD11 variants.
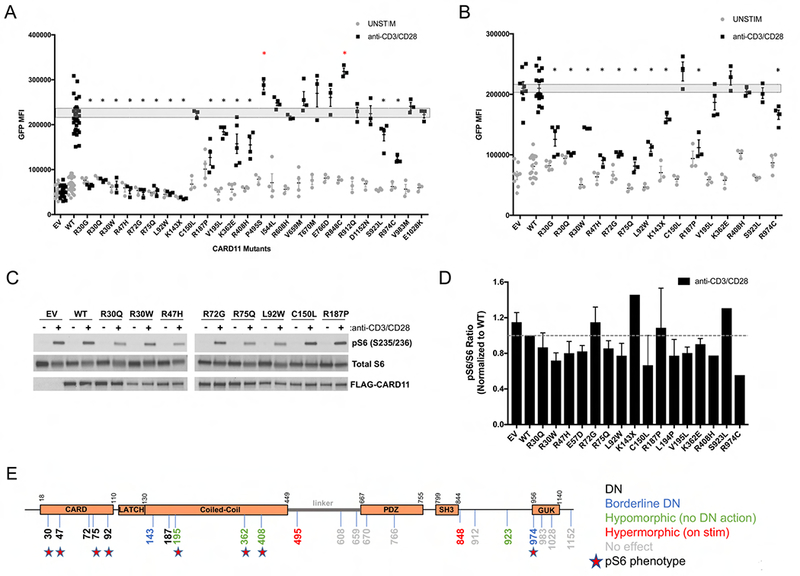
(A) Quantification of NF-κB-induced GFP reporter activity in CARD11-deficient JPM50.6 T cells transfected with empty vector (EV), wild-type (WT), or mutant CARD11 constructs and subsequently stimulated with anti-CD3/CD28 Abs. Data are mean −/+ SEM for ≥3 separate transfections each. (B) Quantification of NF-κB-induced GFP in JPM50.6 cells transfected with equal ratios of WT and mutant CARD11 constructs. Data are mean −/+ SEM for ≥3 separate transfections of each LOF variant; dashed boxes in (A-B) indicate SEM for numerous WT transfections. Asterisks denote statistically significant reductions in GFP MFI versus WT (A) or WT+WT CARD11 (B) after stimulation, indicating DN activity (p<0.05). (C) Immunoblot for phospho-S6, total S6, and FLAG-CARD11 expression in Jurkat T cells transfected with CARD11 constructs −/+ 20 min stimulation with anti-CD3/CD28 Abs. Data are representative of several independent experiments. (D) Spot densitometric quantification of phospho-S6/S6 ratio for each mutant, normalized to WT (dashed line = 1). Data are mean −/+ SD for 2-3 experiments for each variant. (E) Schematic diagram of CARD11 protein including new DN, LOF, hypermorphic and benign (i.e. no effect) variants. Stars indicate mutations with reduced ph-S6 in (D).
CARD11 also affects TCR-induced mTORC1 activation (30, 31). We therefore further tested whether selected CARD11 variants could disrupt TCR-induced mTORC1 activation by measuring ribosomal S6 protein phosphorylation by immunoblotting. For most mutations within the CARD domain, we observed a trend toward decreased phospho-S6 (Figure 1C–D), similar to previously characterized DN mutants (9). These differences did not reach statistical significance, however, due to inherent variation in our assays. mTORC1 signaling is exquisitely sensitive to subtle perturbations in cell viability, culture media contents and stimulation conditions, particularly in Jurkat T cells (K. Hamilton, personal communication). Interestingly, some mutants (e.g. p.R72G, p.R187P) had no appreciable effect on TCR-induced S6 phosphorylation despite impaired NF-κB activation in Jurkat transfectants (Figure 1C–D). Collectively, these transfection results suggest bona fide DN mutations in CARD11, often located in the CARD domain, attenuate TCR-induced NF-κB activation with variable effects on mTORC1 signaling (Figure 1E).
Patient phenotypes with dominant negative CARD11 mutations
We compared a total of 60 patients from 32 kindreds carrying heterozygous CARD11 variants (Table 1). Including those previously published (7, 9), we identified 28 distinct CARD11 alleles, of which 14 are DN as determined by the functional assays described above. All kindred pedigrees with confirmed CARD11 DN variants are shown in Figure 2. All patients were referred with immunological phenotypes for investigation, typically presenting in childhood. We therefore compared the manifestations in those with functional CARD11 DN defects to those with no obvious DN functional defect, even if hypomorphic (Tables 1–2). Severe atopic dermatitis (AD) was present in most patients with DN mutations (32/44; 73%), compared with non-DN variants (5/16, 31%). Other atopic symptoms were noted in patients with DN variants, including asthma (55%) and food allergies (32%), and less frequently, rhinitis and eosinophilic esophagitis (Table 1). Cutaneous viral infections were also more common in patients with DN alleles, including molluscum contagiosum (52%), cutaneous herpes simplex virus type 1 (HSV-1) infection (30%) and warts (27%) (Table 1). We also observed cases of neutropenia, presentations similar to other syndromes associated with elevated IgE and infection (Kindred 14), including IPEX-like syndrome (Kindred 4) in patients with functional DN alleles, but not in the others. Of note, the patient with IPEX-like presentation (including failure to thrive, bloody diarrhea, and severe eczema) carried the identical mutation (p.R30W) to that described recently by Roifman and colleagues (Kindred 5) in a family with severe atopy, infection and mild (late onset) autoimmunity (7). Prominent phenotypes in the collective CARD11 DN patient cohort are summarized in Table 2. In addition to atopic disease (89%), significant viral skin infections (68%) and lung disease (e.g. infections, pneumonia, bronchiectasis) (68%) were the most prominent symptoms shared by patients harboring DN CARD11 mutations (Table 2). Additional phenotypes included autoimmunity (20%)—most commonly alopecia, but also including ITP and bullous pemphigoid. Neutropenia was also observed in five patients (14%, including four with no demonstrable atopic disease), although an autoimmune etiology could not be confirmed. Oral ulcers were also observed (14%), and may be linked to neutropenia. Four patients had lymphoproliferative disease (large granular lymphocytic leukemia (LGL), peripheral T-cell lymphoma, and/or mycosis fungoides). Notably, three patients from two families had little to no atopic disease (one had a mild IgE elevation without clinical manifestations), but did have neutropenia with humoral defects including progressive B-cell lymphopenia, poor class switched B-cell memory, and antibody deficiency. Indeed, low IgM and impaired humoral responses to certain vaccines (e.g. pneumococcal) were observed in several patients. Relevant immunological phenotypes, including total and specific antibody defects and lymphocyte subsets are summarized in Table 3 and Online Repository Table 1.
Figure 2. Pedigrees for new CARD11 DN variants.
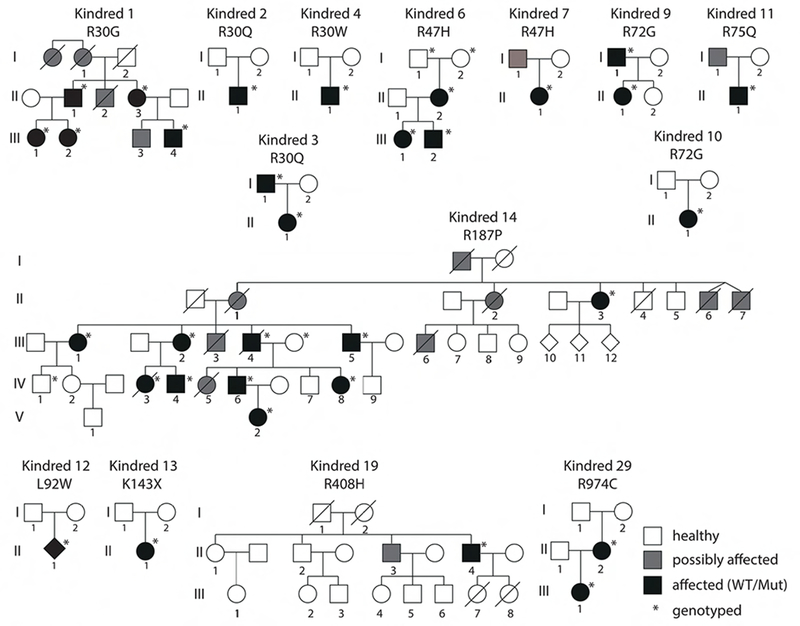
Key indicates healthy (white), affected (symptomatic with confirmed heterozygous DN mutation), and possibly affected (symptomatic, no genotype available). Asterisks denote patients that were definitively genotyped.
Table 3. Immunological phenotypes for patients with CARD11 DN variants.
Descriptors are based on institution-specific reference ranges for each parameter (see Online Repository Table 1). Absent = no cells detected, Low = >10% below reference range minimum, Borderline low = within 10% of reference minimum, High = >10% above reference range maximum. Numbers in parentheses = number of patients with the observed defect/number of patients tested within each kindred.
| Kindred | Mutation | # patients | T cell prolif defect | Spec Ab response defect | Total Ab defect | Total CD4 | Memory CD4 | Total CD8 | Memory CD8 | Total B cells | Low CS/Mem B cells | NK cells | Tregs | IgE | Eosinophils |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | R30G | 6 | no | yes (2/6) | no | normal | ND | normal | ND | normal | normal | normal | ND | high (3/6) | high (2/6) |
| 2 | R30Q | 1 | yes | yes | yes (low IgM) | normal | normal | normal | normal | borderline low | ND | borderline low | ND | high | high |
| 3 | R30Q | 2 | yes (2/2) | yes (2/2) | yes (2/2) | normal | normal | normal | normal | normal | low | normal | ND | high (2/2) | high (1/2) |
| 4 | R30W | 1 | unknown | unknown | unknown | unknown | unknown | unknown | unknown | unknown | unknown | unknown | low | high | ND |
| 5 | R30W | 4 | yes (4/4) | yes (2/4) | yes (2/4) | normal | ND | normal | ND | low (1/4) | ND | normal | ND | high | high (3/4) |
| 6 | R47H | 3 | yes (3/3) | yes (2/2) | yes (1/3) | normal | low (3/3) | normal | low (3/3) | normal | low (3/3) | normal | normal | high 1/3 | normal |
| 7 | R47H | 1 | yes | yes | yes | normal | low | normal | ND | low | low | normal | normal | high | normal |
| 8 | E57D | 2 | yes (1/2) | unknown | yes (1/1) | normal | ND | normal | ND | normal | normal | low (1/2) | normal | high | high |
| 9 | R72G | 2 | yes (1/2) | yes (2/2) | no | normal | ND | normal | ND | low (1/2) | absent | normal | ND | ND | ND |
| 10 | R72G | 1 | no | yes | no | high | normal | high | normal | low | normal | normal | normal | high | high |
| 11 | R75Q | 1 | yes | yes | no | normal | normal | normal | normal (high TEMRA) | low | low | low | low | normal | normal |
| 12 | L92W | 1 | ND | no | yes | normal | normal | normal | normal | normal | normal | normal | normal | high | ND |
| 13 | K143X | 1 | ND | yes | yes | normal | normal | normal | normal | normal | normal | normal | normal | normal | normal |
| 14 | R187P | 10 | ND | yes (2/10) | yes (1/10) | high (3/3) | low (3/3) | low (1/3) | ND | low (1/3) | low (1/3) | low (3/4) | normal (0/3) | high (8/10) | high (8/10) |
| 15 | dup183-196 | 3 | yes (3/3) | no | no | normal | ND | normal | ND | low (1/3) | ND | normal | normal | high | high |
| 16 | L194P | 1 | yes | yes | no | normal | borderline low | normal | borderline low | normal | normal | normal | normal | high | high |
| 29 | R974C | 2 | no | yes (2/2) | IgA (1/2) | normal | normal | normal | normal | normal | normal | normal | normal | normal | normal |
| 30 | R975W | 2 | yes (1/2) | no | no | normal | borderline low (1/2) | normal | normal | normal | normal | borderline low | normal | high | high |
| TOTAL | 44 | 61% (19/31) | 49% (20/41) | 29% (12/42) |
Hierarchical clustering with a complete linkage algorithm and an asymmetric binary distance measure was used to explore the cohort for specific phenotypic patterns for patients (n=36) and phenotypes (n=7) for which phenotypic data were more complete. While the small size of the cohort precludes any assessment of statistical significance, descriptively we saw (a) a cluster of neutropenic patients having low IgM and lower IgE, (b) patients with AD and asthma who had low IgM, (c) patients with AD without asthma who had low IgG and low IgA, and (d) patients with high IgA, normal IgM, lack of neutropenia and IgE levels that tracked with the presence of AD (Supplemental Figure 2A). These clusters did not appear to correlate with specific mutations. To be more inclusive of phenotypes with some missing data, we performed an additional analysis using all phenotypes (n=31) and patients (n=44). Data was transformed into a Gower distance matrix and reduced to 2-dimensions using t-Distributed Stochastic Neighbor Embedding (tSNE) before applying K-means clustering to partition the patients into three clusters. Supplemental Figure 2B summarizes phenotypes characteristic of each cluster. Interestingly, cluster 2 was characterized by lack of AD, skin bacterial infections, pneumonia, and allergic rhinitis, but included patients with neutropenia and abnormal IgM and IgA levels. This cluster included all patients with R30G and R974C, most patients with R47H, and the single patient with R75Q. Again, all phenotypic correlations predicted by these clusters are merely descriptive for this limited patient cohort.
The largest family analyzed included ten patients from the United Kingdom (Kindred 14) who presented with autosomal dominant inheritance of symptoms typically associated with DOCK8 deficiency, including elevated serum-IgE levels with significant atopic disease, recurrent respiratory tract infections, skin abscesses, and recurrent or persisting viral skin infections such as molluscum contagiosum, herpes infections or warts (Table 1, Figure 3). Two patients additionally had mycobacterial complications. Surprisingly, mild skeletal/connective tissue abnormalities characteristic for the STAT3-DN autosomal dominant HIES were also observed in six out of ten affected family members. Patient III.4 died from disseminated mycosis fungoides. Immune phenotyping of patient PBMCs from this family revealed low CD8+ T cell numbers and high CD4+ T cell numbers, with a shift towards the naive compartment and reduction of memory CD4+ T cells. The B cell compartment was normal.
Figure 3. Clinical presentation of patients with DN CARD11 mutations (A) R30Q (Kindred 2) and (B-F) R187P (Kindred 14).
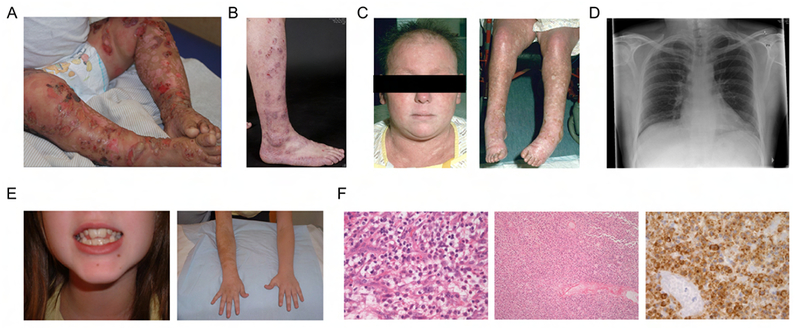
(A) Patient II.1 displaying HSV-1 skin disease. (B) Patient IV.6 displaying severe eczema. (C) Cutaneous vasculitis (both) and alopecia (left) in patient IV.3. (D) Chest radiograph of patient III.5 from 2008 depicting bronchiectasis. (E) Abnormal dentition (left) and brachial hypermelanosis (right) in patient IV.8. (F) Histology findings in patient III.4 with cutaneous T cell lymphoma (mycosis fungoides). Left: viable pleomorphic blasts ×40; middle: necrotic lymph node replaced by lymphoma low power; right: CD2 staining viable and semiviable cells.
Flow cytometric analyses of other patients, when available, often showed B-cell defects including low total/memory B cells in those with specific antibody deficiency, and low IgM in several patients (Table 3). Other assays performed showed diminished mitogen-induced T-cell proliferation in the majority of patients tested, and relatively normal frequencies of regulatory T-cells, as previously reported (Table 3). Detailed clinical and laboratory findings for individual patients, where available, are included in Online Repository Table 1.
Primary cell signaling defects in CARD11 DN patient lymphocytes
In seven of the patients with confirmed DN mutations, primary cells were available for evaluation of NF-κB signaling using short Phorbol 12-myristate 13-acetate (PMA) activation. Hallmarks of TCR-induced canonical NF-κB activation, including p65 phosphorylation and IκBα degradation, were impaired in all samples tested (Figure 4). Impaired upregulation of activation markers at 24 hours and skewed cytokine secretion was also noted in multiple CD4+ T cell patient samples, consistent with previous findings (Supplemental Figures 3–4). Of note, NF-κB activation in lymphocytes from the patient with the V195L substitution—which was LOF but did not show DN activity in our transfection system—was also abnormal. Because haploinsufficiency does not likely lead to direct DN activity (9), the result suggests either that other lesions explain the cellular and clinical phenotype, or that the effects of this mutation on the pathway are not revealed using our transfection system. The variability in pS6 activation in the primary cell assay precludes making a definitive conclusion regarding its utility, though small but reproducible defects were observed in 4 patients tested from Kindreds 6 and 19 (Figure 5A–B). Moreover, we noted a more reproducible diminution of pS6 could be observed in CARD11 DN patient samples activated for 24 hours (Figure 5C).
Figure 4. Defective NF-κB activation in primary T cells from CARD11 DN/LOF patients.
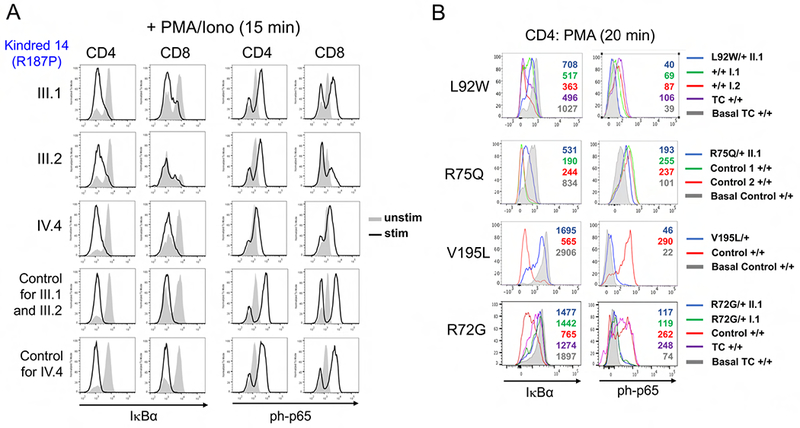
(A) Primary CD4+ and CD8+ T cells from 3 Kindred 14 patients (III.1, III.2, IV.4) and 2 controls were left unstimulated (gray filled histograms) or stimulated for 15 min with PMA + ionomycin (black histograms). I∼B degradation and p65 phosphorylation was detected by intracellular flow cytometry. (B) Total PBMC from affected patients, unaffected family members and controls (labeled at right, + standing for WT) were stimulated for 20 min with PMA or left unstimulated (basal: gray histograms). IκB degradation and p65 phosphorylation was detected in gated CD4+ T cells by intracellular flow cytometry. Geometric mean fluorescence intensity (gMFI) values are listed within each histogram.
Figure 5. Defective S6 phosphorylation in primary T cells from affected CARD11 DN patients after acute and prolonged stimulation.
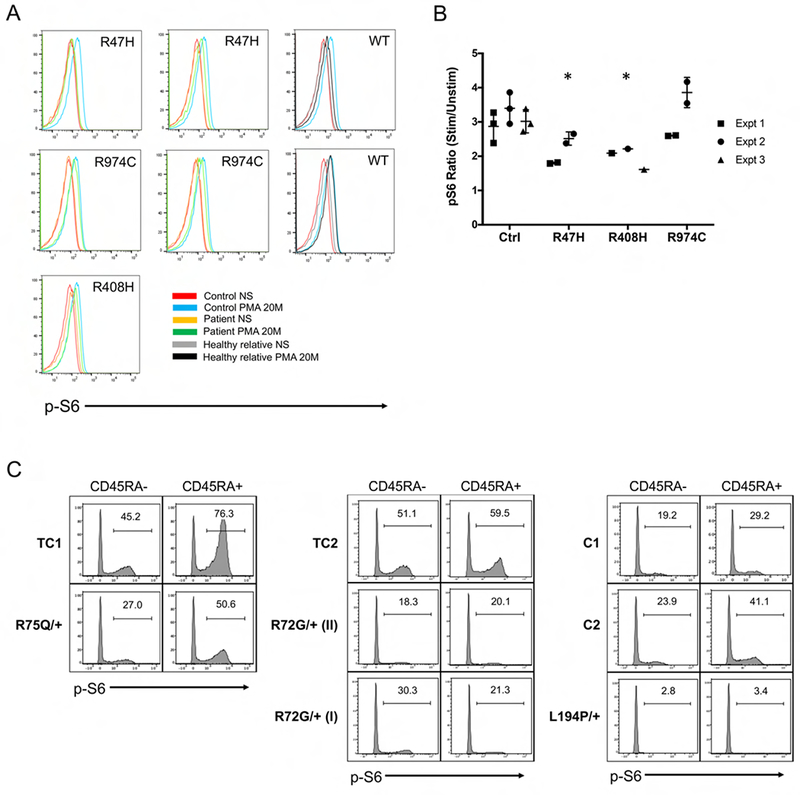
(A) Primary CD4+ T cells from affected patients in Kindreds 6, 19 and 29, healthy relatives and controls were not stimulated (NS) or stimulated with PMA for 20 min. S6 phosphorylation was measured by intracellular flow cytometry. (B) The ratio of pS6 gMFI for stimulated versus unstimulated cells is plotted for each of 3 experiments represented in (A). Asterisks denote statistical significance relative to controls (Kruskal-Wallis test, p<0.05). (C) Total PBMC from affected CARD11 DN patients, controls (C) and travel controls (TC) were cultured under Th0 conditions for 24 hours. S6 phosphorylation was measured in CD4+CD45RA- or RA+ cells gated for high vs. low side scatter (SSC) by intracellular flow cytometry; % pS6+ cells are labeled within each histogram.
Discussion
In this report, we describe multiple new DN CARD11 mutations associated with additional immune-deficient and dysregulatory conditions in human patients. Recent reports identified DN CARD11 mutations in a handful of patients presenting with severe atopic dermatitis and other allergic conditions, with or without additional infections (7, 9). The substantially larger cohort assembled here illuminates a broader phenotypic spectrum of disease tied to CARD11 DN mutations, including frequent sinopulmonary infections, cutaneous viral infections, neutropenia, hypogammaglobulinemia, and lymphoma (Table 2). Atopic disease was the cardinal feature noted in most patients (89%), frequently presenting in childhood as atopic dermatitis, but also including asthma, allergic rhinitis, food allergies, and even eosinophilic esophagitis (Table 2). However, atopy was mild or absent in a number of patients examined in this report, with no measured differences in TCR-driven signaling responses. There were no obvious clinical similarities between non-atopic CARD11 DN patients beyond neutropenia (present in four). It is possible that atopic symptoms improved over time for some older adults for whom detailed clinical history was lacking, similar to previously described patients (9). Furthermore, unrelated patients with the same mutation (e.g. R30Q, R72G) and even family members harboring identical mutations (e.g. p.R47H, p.R187P) demonstrated differences in both the variety and severity of disease symptoms. From our characterization of this expanded patient cohort, we conclude that CARD11 DN mutations exhibit high penetrance and variable expressivity for several phenotypes that can extend beyond atopy. Other genetic variants or polymorphisms could certainly be influencing the heterogeneity and severity of phenotypes observed in certain patients.
Lack of atopic disease in patients with biallelic CARD11 null mutations, associated with SCID, is likely due to the relative lack of lymphocyte effector function (32, 33). However, the absence of atopy observed in certain patients described here with CARD11 DN mutations, all of whom had lived into adulthood without major intervention, is surprising. Interestingly, unsupervised clustering analyses identified a subset of these patients from four kindreds that presented with neutropenia, abnormal Ig levels, and fewer skin/respiratory infections, although the significance of these associations cannot be formally assessed within this limited cohort. We otherwise noted no particular genotype/phenotype correlations, nor were there patterns of other comorbid conditions that suggested substantially different phenotypic manifestations. Phenotypic variation within families suggests that other genetic variants, which may not be pathogenic by themselves, or differences in environmental exposures could influence the phenotype. As more CARD11 variant patients are found, demographic patterns may be possible to establish.
Non-atopic phenotypes associated with bona fide DN mutations shed new light on important biological functions governed by CARD11 signaling in lymphocytes. For example, insufficient (e.g. low IgM) or misdirected humoral responses appear to be a common outcome of attenuated CARD11 signaling, with or without elevated IgE. A number of families presented with more severe humoral defects resembling CVID, which may reflect both intrinsic defects in B cell differentiation and/or poor T cell help. Future studies aimed at elucidating specific abnormalities in class switch recombination and plasma cell differentiation in patient B cells would be helpful. The neutropenia noted in several patients was not associated with obvious bone marrow defects, which could suggest an autoimmune etiology; indeed, CARD11 is primarily expressed in mature lymphocytes. However, given that this is a diagnosis of exclusion, further investigation is warranted.
Cutaneous viral infections (e.g. molluscum, HSV-1, etc.) were also common to several patients. Impaired CD8+ T cell immunosurveillance could be a factor, and may also help to explain tumor development in certain patients. Interestingly, BENTA patients carrying GOF CARD11 mutations often present with molluscum and other viral infections (e.g. EBV), and are at greater risk of lymphoma/leukemia development (5). Unlike many CARD11 DN patients, BENTA patient T cells are only mildly “anergic” and proliferate relatively well in response to robust stimuli, even though IL-2 production is often reduced (6, 34). There is also no evidence of Th2-skewing or atopy in BENTA patients described to date, although this has not been investigated thoroughly. Further studies are needed to elucidate how dysregulated CARD11 variant signaling leads to abnormal CD4+ and CD8+ T cell responses that might predict expressivity of associated phenotypes (e.g. atopy, viral skin infections). Based on our work and the work of others, we suspect alterations in TCR signal strength, metabolic reprogramming, and actin-dependent adhesion and motility could all be contributing factors (13, 35–38).
Our workup of 48 new patients encompassing 25 novel/rare variants further clarifies whether mutations in specific regions of CARD11 are more or less likely to be pathogenic (Figure 6). Notably, missense mutations in the N-terminal CARD domain (aa1-110) are most likely to disrupt NF-κB (and less reliably, mTORC1) signaling, probably by compromising interactions with BCL10 (and by association, MALT1). The N-terminal CC domain (aa130-200) is also a hotspot for pathogenic mutations, although it is still difficult to predict which variants in the CC domain will be LOF or DN; GOF mutations have been identified in the CARD, LATCH, and CC domains (28, 39). In contrast, we have not found DN mutations residing between residues ∼200-970, encompassing the C-terminal portion of the CC domain, the flexible linker, and the PDZ and SH3 domains. In fact, two mutations within this region (p.P495S, p.R848C) significantly boosted TCR-induced NF-κB activity in Jurkat cells; however, more work is required to determine if and how this effect contributes to autoimmune manifestations in these patients. Unlike CARD/CC-associated GOF mutations found in BENTA patients, these mutations did not drive constitutive NF-κB activation in the absence of antigen receptor stimulation. The linker itself (aa449-667) contains an array of redundant repressive and activating elements that govern the complex intramolecular regulation of CARD11 (40, 41), making it unlikely to find single point mutations in the linker that affect CARD11 signaling. Finally, one new (p.R974C) and one previously confirmed DN mutation (p.R975W) were located in the guanylate kinase (GUK) domain), although nearby variants in this region showed no effect on TCR-induced NF-κB (p.V983M, p.E1028K, p.D1152N). In the end, LOF and/or DN activity was not detected in all rare variants from patients referred for a variety of phenotypes, including severe atopy. These results highlight the utility of our simple cell transfection assay in addition to the workup of primary patient cells whenever possible—indeed, as highlighted by the patient with the p.V195L mutation, the observed NF-κB signaling defect detected in primary cells cannot always be ascribed to DN activity. Although defects in mTORC1 signaling are also important, even small perturbations in cell culture conditions make quantification of differences in S6 phosphorylation extremely challenging for both Jurkat and primary T cells. Therefore, we cannot currently recommend pS6 quantification after short term stimulation as a diagnostic assay for CARD11 DN mutations. Our comprehensive allelic series suggests that for CARD11, protein domain architecture might predict functional consequences of specific mutations. However, this too will require broader, more detailed structure-function analyses of the CARD11 protein before predictions could be used for clinical diagnoses.
For clinicians, this study also provides an improved differential diagnosis for immune deficiency and dysregulation linked to CARD11 mutations. An algorithm detailing our strategy for identifying and diagnosing these patients is depicted in Figure 6. Heterozygous CARD11 DN mutations may therefore be suspected and sought - either via analysis of existing exomes or included in other gene sequencing panels - in patients with histories of atopy (especially atopic dermatitis), often in combination with sinopulmonary bacterial infections or cutaneous viral infections (Figure 6), especially when a dominant family history of any of those phenotypes is present. Failure to thrive, neutropenia, autoimmunity (e.g. alopecia), specific antibody deficiency and/or CVID, and even lymphoma may also be noted. Even in patients without atopy, but with a family history of dominant inheritance of any of the other phenotypes above, it may be worth testing for a heterozygous CARD11 DN mutation. A simple lab diagnostic test aimed at uncovering a TCR-induced NF-κB signaling defect in primary T cells is strongly recommended, even before sequencing is performed. Although identification of such mutations may not alter patient care currently, continued evaluation and reporting of new CARD11 variants (perhaps through a registry) will advance our understanding of specific phenotypic associations and hopefully inform future clinical management. Certainly, those with any such phenotypes in whom a rare heterozygous CARD11 mutation is found even by chance should lead to strong suspicion that it is causal, especially if it resides in the CARD domain. Nevertheless, functional tests such as those described here are imperative for definitive diagnosis.
Supplementary Material
Key Messages:
CARD11 DN mutations are associated with a broader spectrum of human disease phenotypes than previously appreciated, extending beyond atopy to include cutaneous viral and respiratory infections, hypogammaglobulinemia, autoimmunity, neutropenia, and lymphoma.
Pathogenic DN mutations are most likely located in the N-terminal CARD and CC domains of CARD11, compromising TCR-induced NF-κB activation
Clinicians should test for causative CARD11 DN mutations in patients that present with an autosomal dominant pattern of atopy, viral skin infections, and/or respiratory infections and exhibit defective TCR-induced NF-κB activation in vitro, with or without impaired TCR-induced S6 phosphorylation.
Acknowledgements
We thank the patients and their families for participating in this research. All patients were enrolled on IRB-approved protocols and provided informed consent. We also thank C. Olsen, C. Lake, K. Voss, R. Chand and H. Pritchett for technical assistance and statistical analysis, and Z. Li in the USUHS Genomics core for primer synthesis and Sanger sequencing support. We thank Dr. Lia Menasce and Dr. Patrick Shenjere, Histopathology Department, Christie Hospital, Manchester, UK for histopathologic data. We thank Drs. James R. Lupski, Richard A. Gibbs, and Zeynep H. Coban Akdemir for providing whole exome sequencing and bioinformatics support in the Baylor-Hopkins Center for Mendelian Genomics (NIH grant UM1HG006542), and T.D Andrews and M. Field for bioinformatics support at John Curtin School of Medical Research. This work was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases, National Library of Medicine, and the German BMBF grants 01E01303 and 01ZX1306F, the DZIF (TTU 07.801), NIAID extramural award R21AI109187, and the SFB1160 (IMPATH), and National Health and Medical Research Council (Australia) Grants 1113577, 1079648.
Abbreviations:
- AHA:
autoimmune hemolytic anemia
- BENTA:
B cell Expansion with NF-κB and T cell Anergy
- CARD:
caspase activation and recruitment domain
- CC:
coiled-coil
- CVID:
common variable immunodeficiency
- DN:
dominant negative
- EoE:
eosinophilic espophagitis
- EBV:
Epstein-Barr virus
- FTT:
failure to thrive
- GOF:
gain-of-function
- HSV-1:
herpes simplex virus 1
- IPEX:
immune dysregulation, polyendocrinopathy, enteropathy, X-linked
- ITP:
idiopathic thrombocytopenic purpura
- JNK:
c-Jun N-terminal kinase
- LGL:
large granular lymphocytic leukemia
- LOF:
loss-of-function
- MAGUK:
membrane-associated guanylate kinase domain
- MAS:
macrophage activation syndrome
- mTORC1:
mechanistic target of rapamycin complex 1
- PMA:
Phorbol 12-myristate 13-acetate
- NF-κB:
nuclear factor kappa B
- SCID:
severe combined immune deficiency
- sJIA:
systematic juvenile idiopathic arthritis
- T1D/T2D:
type 1 / type 2 diabetes
- TCR:
T cell receptor
- WES:
whole exome sequencing
- WGS:
whole genome sequencing
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Notarangelo LD, Kim MS, Walter JE, Lee YN. Human RAG mutations: biochemistry and clinical implications. Nat Rev Immunol. 2016;16(4):234–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Juilland M, Thome M. Role of the CARMA1/BCL10/MALT1 complex in lymphoid malignancies. Curr Opin Hematol. 2016;23(4):402–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meininger I, Krappmann D. Lymphocyte signaling and activation by the CARMA1-BCL10-MALT1 signalosome. Biol Chem. 2016;397(12):1315–33. [DOI] [PubMed] [Google Scholar]
- 4.Turvey SE, Durandy A, Fischer A, Fung SY, Geha RS, Gewies A, et al. The CARD11-BCL10-MALT1 (CBM) signalosome complex: Stepping into the limelight of human primary immunodeficiency. J Allergy Clin Immunol. 2014;134(2):276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arjunaraja S, Angelus P, Su HC, Snow AL. Impaired Control of Epstein-Barr Virus Infection in B-Cell Expansion with NF-κΒ and T-Cell Anergy Disease. Frontiers in Immunology. 2018;9(198). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Snow AL, Xiao W, Stinson JR, Lu W, Chaigne-Delalande B, Zheng L, et al. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med. 2012;209(12):2247–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dadi H, Jones TA, Merico D, Sharfe N, Ovadia A, Schejter Y, et al. Combined immunodeficiency and atopy caused by a dominant negative mutation in caspase activation and recruitment domain family member 11 (CARD11). J Allergy Clin Immunol. 2017. [DOI] [PubMed] [Google Scholar]
- 8.Jun JE, Wilson LE, Vinuesa CG, Lesage S, Blery M, Miosge LA, et al. Identifying the MAGUK protein Carma-1 as a central regulator of humoral immune responses and atopy by genome-wide mouse mutagenesis. Immunity. 2003;18(6):751–62. [DOI] [PubMed] [Google Scholar]
- 9.Ma CA, Stinson JR, Zhang Y, Abbott JK, Weinreich MA, Hauk PJ, et al. Germline hypomorphic CARD11 mutations in severe atopic disease. Nat Genet. 2017;49(8):1192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hara H, Yokosuka T, Hirakawa H, Ishihara C, Yasukawa S, Yamazaki M, et al. Clustering of CARMA1 through SH3-GUK domain interactions is required for its activation of NF-kappaB signalling. Nat Commun. 2015;6:5555. [DOI] [PubMed] [Google Scholar]
- 11.Tanner MJ, Hanel W, Gaffen SL, Lin X. CARMA1 coiled-coil domain is involved in the oligomerization and subcellular localization of CARMA1 and is required for T cell receptor-induced NF-kappaB activation. J Biol Chem. 2007;282(23):17141–7. [DOI] [PubMed] [Google Scholar]
- 12.Meyts I, Bosch B, Bolze A, Boisson B, Itan Y, Belkadi A, et al. Exome and genome sequencing for inborn errors of immunity. J Allergy Clin Immunol. 2016; 138(4):957–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lyons JJ, Milner JD. Primary atopic disorders. J Exp Med. 2018;215(4):1009–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. 2013;Chapter 7:Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81. [DOI] [PubMed] [Google Scholar]
- 17.Schubert D, Klein MC, Hassdenteufel S, Caballero-Oteyza A, Yang L, Proietti M, et al. Plasma cell deficiency in human subjects with heterozygous mutations in Sec61 translocon alpha 1 subunit (SEC61A1). J Allergy Clin Immunol. 2018;141(4): 1427–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silberstein M, Tzemach A, Dovgolevsky N, Fishelson M, Schuster A, Geiger D. Online system for faster multipoint linkage analysis via parallel execution on thousands of personal computers. Am J Hum Genet. 2006;78(6):922–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silberstein M, Weissbrod O, Otten L, Tzemach A, Anisenia A, Shtark O, et al. A system for exact and approximate genetic linkage analysis of SNP data in large pedigrees. Bioinformatics. 2013;29(2):197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gower JC. A general coefficient of similarity and some of its properties. Biometrics. 1971:857–71. [Google Scholar]
- 21.Kaufman L, Rousseeuw PJ. Finding groups in data: an introduction to cluster analysis: John Wiley & Sons; 2009. [Google Scholar]
- 22.Maechler M, Rousseeuw P, Struyf A, Hubert M, Hornik K. cluster: Cluster Analysis Basics and Extensions. R package version 2.0. 1. 2015. 2017. [Google Scholar]
- 23.Van Der Maaten L Accelerating t-SNE using tree-based algorithms. The Journal of Machine Learning Research. 2014;15(1):3221–45. [Google Scholar]
- 24.Witten IH, Frank E, Hall MA, Pal CJ. Data Mining: Practical machine learning tools and techniques: Morgan Kaufmann; 2016. [Google Scholar]
- 25.Yu L, Liu H, editors. Feature selection for high-dimensional data: A fast correlation-based filter solution. Proceedings of the 20th international conference on machine learning (ICML-03); 2003. [Google Scholar]
- 26.Landwehr N, Hall M, Frank E. Logistic model trees. Machine learning. 2005;59(1-2):161–205. [Google Scholar]
- 27.Fuchs S, Rensing-Ehl A, Pannicke U, Lorenz MR, Fisch P, Jeelall Y, et al. Omenn syndrome associated with a functional reversion due to a somatic second-site mutation in CARD11 deficiency. Blood. 2015;126(14):1658–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan W, Schaffer TB, Pomerantz JL. A quantitative signaling screen identifies CARD11 mutations in the CARD and LATCH domains that induce Bcl10 ubiquitination and human lymphoma cell survival. Mol Cell Biol. 2013;33(2):429–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thome M, Charton JE, Pelzer C, Hailfinger S. Antigen receptor signaling to NF-kappaB via CARMA1, BCL10, and MALT1. Cold Spring Harb Perspect Biol. 2010;2(9):a003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamilton KS, Phong B, Corey C, Cheng J, Gorentla B, Zhong X, et al. T cell receptor-dependent activation of mTOR signaling in T cells is mediated by Carma1 and MALT 1, but not Bcl10. Sci Signal. 2014;7(329):ra55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi JH, Sun SC. TCR signaling to NF-kappaB and mTORC1: Expanding roles of the CARMA1 complex. Mol Immunol. 2015;68(2 Pt C):546–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greil J, Rausch T, Giese T, Bandapalli OR, Daniel V, Bekeredjian-Ding I, et al. Whole-exome sequencing links caspase recruitment domain 11 (CARD11) inactivation to severe combined immunodeficiency. J Allergy Clin Immunol. 2013;131(5): 1376–83 e3. [DOI] [PubMed] [Google Scholar]
- 33.Stepensky P, Keller B, Buchta M, Kienzler AK, Elpeleg O, Somech R, et al. Deficiency of caspase recruitment domain family, member 11 (CARD11), causes profound combined immunodeficiency in human subjects. J Allergy Clin Immunol. 2013;131(2):477–85 e1. [DOI] [PubMed] [Google Scholar]
- 34.Brohl AS, Stinson JR, Su HC, Badgett T, Jennings CD, Sukumar G, et al. Germline CARD11 Mutation in a Patient with Severe Congenital B Cell Lymphocytosis. J Clin Immunol. 2015;35(1):32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aronica MA, Mora AL, Mitchell DB, Finn PW, Johnson JE, Sheller JR, et al. Preferential role for NF-kappa B/Rel signaling in the type 1 but not type 2 T cell-dependent immune response in vivo. J Immunol. 1999;163(9):5116–24. [PubMed] [Google Scholar]
- 36.Pollizzi KN, Powell JD. Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat Rev Immunol. 2014;14(7):435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rebeaud F, Hailfinger S, Posevitz-Fejfar A, Tapernoux M, Moser R, Rueda D, et al. The proteolytic activity of the paracaspase MALT1 is key in T cell activation. Nat Immunol. 2008;9(3):272–81. [DOI] [PubMed] [Google Scholar]
- 38.Rueda D, Gaide O, Ho L, Lewkowicz E, Niedergang F, Hailfinger S, et al. Bcl10 controls TCR- and FcgammaR-induced actin polymerization. J Immunol. 2007; 178(7):4373–84. [DOI] [PubMed] [Google Scholar]
- 39.Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319(5870): 1676–9. [DOI] [PubMed] [Google Scholar]
- 40.Jattani RP, Tritapoe JM, Pomerantz JL. Cooperative Control of Caspase Recruitment Domain-containing Protein 11 (CARD11) Signaling by an Unusual Array of Redundant Repressive Elements. J Biol Chem. 2016;291(16):8324–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jattani RP, Tritapoe JM, Pomerantz JL. Intramolecular Interactions and Regulation of Cofactor Binding by the Four Repressive Elements in the Caspase Recruitment Domain-containing Protein 11 (CARD11) Inhibitory Domain. J Biol Chem. 2016;291(16):8338–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.