

Accumulating evidence suggests that the bactericidal activity of some antibiotics may not be directly initiated by target inhibition. The activity of isoniazid (INH), a key first-line bactericidal antituberculosis drug currently known to inhibit mycolic acid synthesis, becomes extremely poor under stress conditions, such as hypoxia and starvation.
KEYWORDS: Mycobacterium tuberculosis, Q203, bedaquiline, electron transport chain, isoniazid, persistence
ABSTRACT
Accumulating evidence suggests that the bactericidal activity of some antibiotics may not be directly initiated by target inhibition. The activity of isoniazid (INH), a key first-line bactericidal antituberculosis drug currently known to inhibit mycolic acid synthesis, becomes extremely poor under stress conditions, such as hypoxia and starvation. This suggests that the target inhibition may not fully explain the bactericidal activity of the drug. Here, we report that INH rapidly increased Mycobacterium bovis BCG cellular ATP levels and enhanced oxygen consumption. The INH-triggered ATP increase and bactericidal activity were strongly compromised by Q203 and bedaquiline, which inhibit mycobacterial cytochrome bc1 and FoF1 ATP synthase, respectively. Moreover, the antioxidant N-acetylcysteine (NAC) but not 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPOL) abrogated the INH-triggered ATP increase and killing. These results reveal a link between the energetic (ATP) perturbation and INH’s killing. Furthermore, the INH-induced energetic perturbation and killing were also abrogated by chemical inhibition of NADH dehydrogenases (NDHs) and succinate dehydrogenases (SDHs), linking INH’s bactericidal activity further to the electron transport chain (ETC) perturbation. This notion was also supported by the observation that INH dissipated mycobacterial membrane potential. Importantly, inhibition of cytochrome bd oxidase significantly reduced cell recovery during INH challenge in a culture settling model, suggesting that the respiratory reprogramming to the cytochrome bd oxidase contributes to the escape of INH killing. This study implicates mycobacterial ETC perturbation through NDHs, SDHs, cytochrome bc1, and FoF1 ATP synthase in INH’s bactericidal activity and pinpoints the participation of the cytochrome bd oxidase in protection against this drug under stress conditions.
INTRODUCTION
Tuberculosis (TB) still represents a severe health threat today, causing in 2016 an estimated 10.4 million cases with morbidity globally (1). TB treatment depends largely on a 6-month chemotherapy regimen, i.e., 2 months of isoniazid (INH), rifampin (RIF), pyrazinamide (PZA), and ethambutol (EMB) and 4 months of INH and RIF (2, 3). However, rapid emergence of drug-resistant TB, such as multidrug-resistant (MDR) strains resistant to both INH and RIF, challenges the TB chemotherapy and calls for development of novel drugs and regimens. A better knowledge of the bactericidal mechanisms of currently available anti-TB drugs would be beneficial to develop better synergistic drug combinations.
INH is a prodrug requiring the mycobacterial catalase-peroxidase (KatG) to form the active INH-NAD adduct. This adduct is known to bind and inhibit the mycobacterial fatty acid synthase II (FAS-II) component enoyl-acyl carrier protein reductase (InhA), which is required for the synthesis of mycolic acids, the core mycobacterial cell wall components (4). Indeed, INH exposure leads to rapid loss of mycolic acids, alteration of cell morphology, and cell death (5, 6). Similar to INH, the second-line drug ethionamide (ETH) is also a prodrug that is activated by the monooxygenase (EthA) to form an ETH-NAD adduct inhibiting InhA (7–9). In agreement with their mode of action, resistance mechanisms are associated with mutations in genes such as inhA (INH and ETH coresistance), katG (INH resistance), and ethA (ETH resistance) (8, 10, 11).
The mycobacterial electron transport chain (ETC) is the focus of current anti-TB research attention, providing new targets for several promising anti-TB agents, including Q203, bedaquiline, and clofazimine (1). Q203 and bedaquiline inhibit cytochrome bc1 and FoF1 ATP synthase, respectively (12–14). Although both Q203 and bedaquiline decrease ATP production in mycobacteria, only bedaquiline exhibits potent bactericidal activity (15–17). The failure of Q203 to kill the bacilli results from the presence of an alternative cytochrome bd oxidase, allowing mycobacteria to reroute electron flow under cytochrome bc1/aa3 inhibition (15, 16). In addition to these branching terminal oxidases, flexibility of the mycobacterial ETC is also demonstrated by the utilization of various dehydrogenases that initiate the ETC, such as proton-pumping type I NADH dehydrogenase (NDH-I), non-proton-pumping NDH-II, and succinate dehydrogenases (SDHs) (18). The relative contributions of these dehydrogenases to a functional ETC in mycobacteria, although not fully understood, could be related to the cell growth status and the available carbon/energy sources (18).
Interestingly, Shoeb et al. observed increased reactive oxygen species (ROS) production in INH-treated mycobacterial cell extracts (19, 20). Moreover, overexpression of the alkyl hydroperoxide reductase C protein (AhpC) conferred INH resistance (21). Conversely, disruption of the superoxide dismutase A (SodA) enhanced INH susceptibility (22). These observations seem to establish a link between INH’s killing and ROS. We presumed that if INH induces ROS, it should also affect the mycobacterial ETC, which is the primary source of ROS (23). Interestingly, mycobacterial mutants with the cytochrome bd assembly disrupted are more susceptible to INH in mice (24). Therefore, it is likely that INH could impact the mycobacterial ETC.
To test this hypothesis, we measured mycobacterial energetics (ATP), oxygen consumption, ROS production, and membrane potential following treatment with several antimycobacterial agents, including INH. We further assessed the effect of ETC inhibitors (Q203, bedaquiline, and various dehydrogenase inhibitors) and antioxidants (e.g., N-acetylcysteine [NAC]) on the INH-induced energetic perturbation and killing to elucidate INH's bactericidal mechanism. These studies provide evidence that INH bactericidal activity involves mycobacterial ETC perturbation.
RESULTS
INH increases mycobacterial ATP rapidly.
We first measured the amount of ATP as an indicator of ETC status. Surprisingly, Mycobacterium bovis BCG treated with the MIC of INH (0.1 µg/ml) (25) showed significantly enhanced ATP levels after 24 h of treatment (Fig. 1A). The ATP increase caused by INH was unexpectedly rapid, occurring already after 1.5 h treatment (Fig. 1B). Since the amount of extracellular ATP exhibited no substantial change, unlike that of total or intracellular ATP (Fig. 1C), we ruled out that the observed ATP increase caused by INH was an artificial effect resulting from cell lysis. Importantly, the level of ATP increase was both time dependent (Fig. 1B) and proportional to the drug’s concentration (Fig. 1D), suggesting that the energetic enhancement might be associated with INH’s killing mechanism.
FIG 1.

INH and ethionamide enhance cellular ATP. (A) M. bovis BCG cultures in DTA medium were treated with the MICs of various drugs for 24 h before ATP determination. #, P < 0.0001 relative to the no-drug control. (B) ATP kinetics after INH treatment at indicated time points. #, P < 0.0001 relative to the corresponding no-drug control. (C) Bacterial cultures were treated with increasing concentrations of INH for 24 h before measuring extracellular (in the culture filtrate) and intracellular ATP. **, ***, and #, P < 0.01, P < 0.001, and P < 0.0001, respectively, relative to the no-drug control. (D) ATP was determined after INH exposure for 24 h and normalized by viability. (E) Bacterial ATP was determined after sonication. ***, P < 0.001. (F) Cells were grown in DTA medium (with or without glucose) and treated with INH for 7 h prior to ATP measurement. Numbers indicate fold increase of ATP. #, P < 0.0001 relative to the no-drug control. These experiments were performed 2 or 3 times (each in triplicate). Representative data are shown. The error bars indicate standard deviations. RLU, relative light units.
Like INH, ETH also elicited a similar ATP response (Fig. 1A), implying that the inhibition of mycolic acid synthesis may be related to the ATP increase. It is important to note that the INH- and ETH-induced ATP enhancement is not a common mycobacterial response to antibiotic stress, since clofazimine and rifampin failed to induce a similar phenotype (Fig. 1A). Interestingly, Shetty and Dick reported, using the same ATP measurement protocol as in this study, an ATP surge for M. bovis BCG treated not only with INH but also with other cell wall inhibitors (e.g., ethambutol, which inhibits arabinogalactan synthesis) (26). This raised the question whether the observed ATP increase could be the result of a cell wall damage-associated artifact (i.e., a damaged cell wall may allow better ATP detection by the commonly applied ATP measurement protocol using the BacTiter-Glo reagent). To clarify this, we compared ATP levels before and after sonication. Sonication was found to significantly elevate the ATP readings (data not shown). In addition to sonication, tetrahydrolipstatin (THL), a drug previously shown to compromise mycobacterial cell wall intactness by reducing phthiocerol dimycocerosate levels (27), also increased the ATP reading, which was abrogated after sonication (see Fig. S1 in the supplemental material). Given these observations, we reassessed the INH-induced ATP change after sonication. We observed that in contrast to THL, INH still elicited a significant ATP increase (Fig. 1E). The enhanced ATP levels in the presence of INH thus cannot be explained simply by increased cell wall permeability.
Enhanced oxygen consumption caused by INH.
Since INH also triggered the ATP level increase in the absence of glycolytic carbon sources (Fig. 1F), we reasoned that the ATP response depends on the mycobacterial ETC but not on glycolysis, which directly generates ATP via two substrate-level phosphorylation reactions (i.e., the conversion of 1,3-bisphosphoglycerate to 3-phosphoglycerate and the conversion of phosphoenolpyruvate to pyruvate [28]). To investigate whether ETC activity was enhanced by INH, we monitored oxygen consumption following INH treatment using methylene blue (15). It is noteworthy that some headspace air was deliberately kept in the glass tube to allow for better drug action. As shown in Fig. 2A, cells treated with INH consumed oxygen significantly faster than those subjected to mock treatment, although INH caused substantial loss of cell viability after 40 h (data not shown). In stark contrast, the oxygen consumption during rifampin treatment was greatly retarded (Fig. 2B), consistent with the cell death (>200-fold less viability relative to the no-drug control at 40 h) and failure of this drug to elevate the ATP (Fig. 1). To further confirm the results obtained with methylene blue, the oxygen consumption was also monitored by cyclic voltammetry using a three-electrode system (see Materials and Methods). The reduction of oxygen at the gold electrode generated a large wave with a peak potential (Ep) at −300 mV versus Ag/AgCl, 3 M KCl (see Fig. S2A, curve 1, in the supplemental material). The intensity of this peak, as indicated by the height of the double-arrowed line in Fig. S2A, reflected the maximal quantity of dissolved oxygen that can be reduced at the electrode surface under the tested conditions. This peak disappeared following bubbling of the medium with nitrogen (data not shown). After incubation with bacteria for 5 h, the height of the oxygen peak was reduced by ∼50% (Fig. S2A, curve 2). In the presence of INH, a greater decrease (∼60%) of the oxygen peak was observed (Fig. S2A, curve 3). In contrast, the oxygen peak was reduced by only ∼40% in the presence of rifampin (Fig. S2A, curve 4). In addition, we noted that the oxygen peak for the INH-treated culture was increased after culture reoxygenation by opening the tube and shaking (Fig. S2B), again supporting the specificity of the peak for oxygen. Using this assay, we also tested whether ethambutol could enhance mycobacterial oxygen consumption. We observed that unlike INH, ethambutol did not augment mycobacterial oxygen consumption (data not shown), suggesting that the ATP increase induced by this drug (26) developed through a mechanism different from that by INH.
FIG 2.

INH enhances oxygen consumption. (A and B) M. bovis BCG cultures were treated with 0.4 µg/ml INH (A) or 0.2 µg/ml rifampin (B), and oxygen consumption was indicated by decolorization of methylene blue (3 µg/ml). (C) Bacterial cultures were treated with 0.4 µg/ml INH (with or without 10 nM Q203), and the oxygen consumption was indicated by methylene blue. Experiments were performed at least 3 times. Representative pictures are shown.
Studies have demonstrated crucial roles for ROS in antibiotic-induced bacterial death (29, 30). The enhanced oxygen consumption, together with ROS generation by INH-treated mycobacterial extracts (19), led us to determine if INH could enhance ROS production in live bacilli. To this end, 2,7-dichlorofluorescin diacetate was used as an ROS probe (31). We measured ROS both before (5 h of treatment) and after (24 h of treatment) INH’s killing was observed (see Fig. S3A in the supplemental material). As a positive control, clofazimine (16 µg/ml) was also included in this assay. Consistent with previous findings (32), clofazimine induced mycobacterial ROS generation at both time points (Fig. S3B). Treatment with INH failed to increase the fluorescence at both time points (Fig. S3B). However, since the culture viability after 24 h of INH treatment was already decreased by >1 log (Fig. S3A), the normalized data showed an elevated ROS level at this time point (Fig. S3C).
Q203 and bedaquiline compromise INH-mediated ATP increase and killing.
To confirm that the INH-induced ATP increase indeed resulted from enhancement of ETC activity, we combined INH with the two mycobacterial ETC-targeting drugs Q203 and bedaquiline, which inhibit cytochrome bc1 and FoF1 ATP synthase, respectively (12–14). As expected, addition of either drug alone resulted in an ATP decrease (Fig. 3A), as reported earlier (15). Interestingly, coadministration of Q203 or bedaquiline significantly compromised the INH-induced ATP enhancement (Fig. 3A), suggesting that the ATP response was dependent on cytochrome bc1 and FoF1 ATP synthase.
FIG 3.

Q203 and bedaquiline abrogate the INH-triggered ATP increase and killing. (A) Normalized ATP levels for M. bovis BCG treated with INH, Q203, or bedaquiline (BDQ). *** and #, P < 0.001 and P < 0.0001, respectively. (B and C) Viability of bacteria determined after 1 (B) and 2 (C) days of treatment with various drugs. *, P < 0.05 relative to the INH group; ***, P < 0.001, relative to viability before treatment for the INH group or to the INH group for the drug combination. ND, not detectable under our plating strategy (<104 CFU/ml). (D and E) Drug kill kinetics following treatment with 0.4 µg/ml INH combined with 10 nM Q203 (D) or 0.25 µg/ml bedaquiline (BDQ) (E). ND, not detectable (<104 CFU/ml). (F) M. tuberculosis H37Rv was treated with 0.4 µg/ml INH and/or 10 nM Q203 for 2 days and viability determined. **, P < 0.01 relative to viability before treatment; #, P < 0.0001 relative to INH group. These experiments were performed 2 or 3 times (each in triplicate). Representative data are shown. The error bars indicate standard deviations.
As expected, INH provoked a concentration-dependent killing (Fig. 3B and C). More strikingly, the drug’s bactericidal activity was significantly abolished by Q203 or bedaquiline (Fig. 3B to E), demonstrating strong antagonistic activities of these ETC-targeting drugs against INH. These results also suggest that INH’s bactericidal activity is largely dependent on a functional cytochrome bc1/aa3 branch and FoF1 ATP synthase. We further assessed the impact of the combination of Q203 and INH on Mycobacterium tuberculosis H37Rv. In agreement with the results obtained with M. bovis BCG, Q203 significantly improved bacterial survival after INH exposure (Fig. 3F). Taken together, these results strongly demonstrate that INH’s bactericidal activity is linked to the enhancement of the ATP level, which is dependent on a functional cytochrome bc1/aa3 branch and FoF1 ATP synthase. We also observed that Q203 but not bedaquiline significantly attenuated the bactericidal activity of both rifampin and moxifloxacin (see Fig. S4 in the supplemental material).
To assess whether the inhibition of ATP increase by the drug combinations correlated with a decreased oxygen consumption rate, we monitored oxygen consumption after treatment with the combination of INH plus Q203. As shown in Fig. 2C, the addition of Q203 efficiently reversed the INH-induced oxygen consumption enhancement, suggesting that the INH-induced ETC perturbation is inhibited by Q203.
Proteomic analysis.
In order to gain insight into metabolic changes associated with INH’s bactericidal activity and Q203-mediated inhibitory effect, we performed proteomic analysis of M. bovis BCG challenged with INH and/or Q203 for 7 h, an incubation sufficient to trigger an enhanced ATP production without significantly decreasing viability (Fig. 1B and S3A).
Relative to the no-drug control, 17 proteins were overexpressed and 22 downregulated in the INH-challenged M. bovis BCG (see Table S1 in the supplemental material). In good agreement with earlier findings (33, 34), INH enhanced the expression of proteins involved in fatty acid beta-oxidation (e.g., two acyl coenzyme A [acyl-CoA] dehydrogenases, FadE23 and FadE24) and mycolic acid synthesis (e.g., acetyl/propionyl-CoA carboxylase [beta subunit] AccD6 and beta-ketoacyl-acyl carrier protein [ACP] synthases KasA and -B), indicative of potential rescuing mechanisms under INH stress.
Strikingly, INH altered the expression of 5 membrane proteins (with 8.9% of total protein hits showing a significant change), indicating that this drug could potentially alter mycobacterial membrane function. Interestingly, INH also triggered a 21-fold overexpression of the Rv0097 protein, a putative taurine catabolism dioxygenase encoded within the operon rv0096-rv0101, which was previously shown to participate in INH’s killing in mice (24). This overexpression could reflect a mycobacterial stress signature and/or a potential killing mechanism of INH. In addition, decreased expression of EphE (a possible epoxide hydrolase) was noted after INH treatment, suggesting that this drug may attenuate the mycobacterial oxidative defense.
The proteomic profile of mycobacteria treated with the Q203/INH combination was compared with the INH-induced proteomic profile (see Table S2 in the supplemental material). A total of 36 and 22 proteins were upregulated and downregulated, respectively, in the Q203/INH-cotreated bacilli. Proteins involved in the synthesis of phthiocerol dimycocerosate (PDIM)/phenolic glycolipid (PGL) (e.g., PpsA and FadD26) and mycolic acid (e.g., AccD6 and KasB) were decreased by the cotreatment. Notably, expression of the Rv0097 protein was decreased by 21-fold in cotreated bacilli, suggesting that Q203 fully abrogates the INH-induced Rv0097 overexpression. Moreover, the downregulation of the membrane protein Rv2203 by INH was also abolished by the addition of Q203. Furthermore, the oxidative stress response regulatory protein OxyS and several stress tolerance proteins (e.g., the iron-regulated universal stress protein TB15.3, the metallothionein MymT, and the bacterioferritin BfrB) were uniformly more abundant in the Q203/INH-cotreated bacilli, indicating that the addition of Q203 enhanced mycobacterial stress tolerance. In addition, we noted increased expression of a putative drug efflux transporter (Rv1410c) in the cotreated bacilli. The possibly enhanced stress tolerance and drug efflux caused by the addition of Q203 may facilitate the development of mycobacterial drug tolerance. Interestingly, the Q203/INH cotreatment upregulated the expression of the Rv3319 protein (SdhB). This protein is a component of mycobacterial SDH2, which may preferentially function as a fumarate reductase instead of an SDH (18). In addition, the Q203/INH-cotreated bacilli also showed enhanced expression of the phosphoenolpyruvate carboxykinase (PckA) catalyzing the interconversion of phosphoenolpyruvate and oxaloacetate (35). Therefore, the reductive tricarboxylic acid cycle depending on PckA and Rv3319 might be activated in the Q203/INH-cotreated bacilli. We also observed an enhanced expression of multiple proteins involved in glutamate metabolism, including l-lysine-epsilon aminotransferase (Lat), glutamate decarboxylase (GadB), and folylpolyglutamate synthase (FolC), in the cotreated bacilli. These metabolic changes, although less related to the current study, may suggest some pathways essential for mycobacterial survival under treatment with Q203 or Q203 plus INH.
NAC abrogates INH-triggered ATP increase and bactericidal activity.
The forgoing results strongly demonstrate that killing by INH involves disruption of the mycobacterial ETC, including energetic perturbation. Although the importance of ROS in antibiotic-induced bacterial death has been documented (29, 30), INH’s bactericidal activity was less likely to be initiated by ROS, as no ROS was induced at early points of drug exposure (Fig. S3). However, ROS may still increase the drug’s effect at later points. To test whether ROS was involved in INH’s killing, we combined INH with two commonly used antioxidants NAC and 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPOL) (16, 32). The effect of these antioxidants on mycobacterial ATP was also determined given the link between energetic perturbation and INH killing. We noted that NAC by itself downregulated ATP levels, and its addition abrogated the INH-induced ATP increase (Fig. 4A). Interestingly, Vilcheze et al. recently observed that addition of cysteine to INH-treated M. tuberculosis enhanced the expression of the dormancy regulon (36). The nonreplicating “dormant” mycobacteria are known to produce smaller amounts of ATP (37). Furthermore, NAC significantly improved mycobacterial survival after INH exposure (Fig. 4B). In sharp contrast, TEMPOL, another ROS-scavenging agent (16, 38), failed to downregulate ATP by itself and did not inhibit the INH-induced ATP increase (Fig. 4C). Accordingly, it conferred no discernible protection against the INH-mediated killing (Fig. 4D). Taken together, these results not only further support the notion that INH’s bactericidal activity involves energetic perturbation but also argue against a major role for ROS in INH-induced killing under the tested conditions.
FIG 4.

NAC but not TEMPOL abolishes the INH-induced ATP increase and killing. (A) M. bovis BCG was treated with 0.4 µg/ml INH (with or without 5 mM NAC) for 24 h prior to ATP measurement. #, P < 0.0001 relative to the no-drug control. (B) Viability was determined after exposure to INH (with or without NAC) for 2 days. **, P < 0.01 relative to the INH group. (C) BCG was treated with 0.4 µg/ml INH (with or without 5 mM TEMPOL) for 24 h prior to ATP measurement. #, P < 0.0001 relative to the no-drug control. (D) Viability was determined after exposure to INH (with or without TEMPOL) for 2 days. These experiments were performed 3 times (each in triplicate). Representative data are shown. The error bars indicate standard deviations.
Chemical inhibition of NDHs and SDHs protects mycobacteria from killing by INH.
The foregoing results strongly demonstrate that INH perturbs the mycobacterial ETC. One possibility is that in the presence of INH, excessive electrons are fed into the ETC via NDHs and SDHs (18). Indeed, under INH stress, bacilli tend to oxidize fatty acids, as suggested by our proteomic and previous transcriptomic data (33). This could lead to overproduction of reducing equivalents (e.g., NADH) and succinates from beta-oxidation, the glyoxylate shunt, and the methylcitrate cycle (39–41), promoting enhanced ETC activity (Fig. 5A). In addition to the beta-oxidation, INH treatment markedly enhanced the expression of the Rv0097 protein (Table S1), a putative taurine dioxygenase catalyzing the production of succinate (Fig. 5A) (42).
FIG 5.

NDH and SDH inhibitors compromise the INH-induced ATP increase and killing. (A) Proposed pathways leading to INH-induced ETC perturbation. Inhibitors targeting the ETC-initiating dehydrogenases are also indicated. (B) M. bovis BCG was treated with various drugs for 24 h before ATP determination. R and T, rotenone and thioridazine, respectively. The concentrations of drugs were used as indicated. *, **, and ***, P < 0.05, P < 0.01, and P < 0.001, respectively, relative to the INH group; #, P < 0.0001 relative to the no-drug control. (C) BCG was challenged with 0.4 µg/ml INH (with or without 200 µM 3-NP, 100 µM rotenone, and 10 µg/ml thioridazine) for 2 days and viability determined. *, P < 0.05; ** and ***, P < 0.001 and P < 0.001, respectively (relative to the INH group). These experiments were performed 3 times (each in triplicate). Representative data are shown. The error bars indicate standard deviations.
To test this hypothesis, we examined the effects of rotenone (an NDH-I inhibitor [37]), thioridazine (an NDH-II inhibitor [37, 43]), and 3-nitropropionic acid (3-NP) (an SDH inhibitor [44]) (Fig. 5A) on the INH-triggered ATP increase and killing. A previous metabolomic analysis showing accumulation of succinate and depletion of downstream tricarboxylic acid cycle metabolites (e.g., fumarate and malate) indicated that 3-NP inhibits mycobacterial SDH (44). Initially, we tested lower inhibitor concentrations based on their MIC values determined in our laboratory and previous studies, i.e., 50 µM rotenone (MIC of >126 µM), 5 µg/ml thioridazine (MIC of 10 µg/ml), and 100 µM 3-NP (44, 45). We noted that the inhibitors, administered either alone or in combinations, failed to kill the bacilli over 2 days of treatment (data not shown). Moreover, their treatment did not rescue the bacilli from INH killing, except that the 3-NP/rotenone/thioridazine triple combination improved bacterial survival by approximately 2.8-fold and 33.2-fold following 1 and 2 days of INH exposure, respectively (data not shown; see Fig. S5 in the supplemental material).
We next tested higher concentration of these inhibitors. We noted that 200 µM 3-NP, like 100 µM, elevated ATP production and failed to inhibit the INH-induced ATP increase and killing (Fig. 5B and C and data not shown). Similarly, 100 µM rotenone or 10 µg/ml thioridazine caused no or only slight inhibition of the INH-induced ATP increase, respectively (Fig. 5B). Accordingly, they conferred no discernible protection against INH’s killing (Fig. 5C). Coadministration of the two NDH inhibitors elicited a stronger inhibition of the ATP increase and slightly but significantly improved bacterial survival under INH treatment (Fig. 5B and C). Notably, the triple combination of these dehydrogenase inhibitors greatly compromised the INH-induced ATP increase and killing (>100-fold) (Fig. 5B and C). It is noteworthy that the triple combination of inhibitors in the absence of INH induced only a negligible decrease (<1 log) in viability under the tested conditions (Fig. 5C). These results further corroborate that the INH bactericidal effect is linked to the ETC perturbation, which is dependent on functional ETC-initiating NDH-I, NDH-II, and SDHs.
INH dissipates mycobacterial membrane potential.
The enhancement of oxygen consumption and the dependency on NDHs, SDHs, the cytochrome bc1/aa3 complex, and FoF1 ATP synthase strongly suggested that INH enhanced mycobacterial ETC activity. We were therefore also interested in how this drug could impact the membrane potential, which is a key factor in mycobacterial energetics (37). The membrane potential was measured using 3,3-diethyloxacarbocyanine iodide [DiOC2(3)] (16, 44). We initially speculated that the INH-induced ETC enhancement could elevate the membrane potential, leading to increased ATP generation.
To validate the membrane potential assay, we used valinomycin as a positive control (37). In our assay, valinomycin treatment for 30 min did not affect mycobacterial membrane potential (data not shown). Unexpectedly, INH exposure moderately decreased the membrane potential, a phenomenon also observed by another group (46). More interestingly, the membrane potential dissipation was observed after 2 h but not 30 min of drug treatment (Fig. 6A and B), reflecting the time-dependent ATP enhancement (Fig. 1B). At 2 h, the INH-induced membrane potential change was not significantly different from that induced by valinomycin (Fig. 6B). We further assessed if Q203 or bedaquiline could abrogate the INH-induced membrane potential dissipation. Interestingly, only bedaquiline, and not Q203, restored the membrane potential (Fig. 6B and C). In contrast to bedaquiline, Q203 by itself reduced the membrane potential (Fig. 6A to C).
FIG 6.

INH dissipates mycobacterial membrane potential in an FoF1 ATP synthase-dependent manner. (A and B) M. bovis BCG was treated with INH and/or Q203 for 30 min (A) and 2 h (B) before measuring membrane potential using DiOC2(3). ** and ***, P < 0.01 and P < 0.001, respectively, relative to the no-drug control. The membrane potential was indicated by the ratio of red to green fluorescence. (C) Membrane potential determined in BCG treated with INH and/or bedaquiline for 2 h. **, P < 0.01 relative to the no-drug control. These experiments were performed 3 times (each in triplicate). Representative data are shown. The error bars indicate standard deviations.
Inhibition of cytochrome bd oxidase reduces cell recovery under INH challenge.
Since chemical inhibition of cytochrome bc1 protected against INH’s killing (Fig. 3), we reasoned that the activation of the cytochrome bd branch prevented INH killing (Fig. 7A). To test this hypothesis, the cytochrome bd inhibitor aurachin D was used (Fig. 7A) (47, 48). We noted that this inhibitor was not bactericidal to M. bovis BCG (data not shown), as reported for M. tuberculosis (47). In particular, the drug-exposed cultures were maintained without agitation during the experiment to allow some cells to settle (cells settling to the bottom of the tubes were expected to be poorly aerated [49] and, consequently, likely to activate the cytochrome bd branch [50]). Since INH’s killing was most prominent after 2 days under our conditions (Fig. 3), we measured the culture viability after 4 and 7 days. As expected, the recovered viability after INH exposure was significantly reduced when aurachin D was simultaneously added (Fig. 7B). We also noted that all the randomly picked colonies recovered from the INH/aurachin D-cotreated cultures could grow in the presence of 0.2 µg/ml INH, suggesting that they were all INH-resistant mutants. In sharp contrast, only between 20% and 73% of colonies recovered from the INH-treated cultures, depending on the experiment, could grow. The failure of some colonies to grow in the presence of the relatively low concentration of INH suggested that they were not resistant mutants but instead survived INH’s challenge by settling and cytochrome bd activation (Fig. 7A). Therefore, these results further support that INH’s killing depends on the cytochrome bc1/aa3 branch and suggest a role for cytochrome bd oxidase in the mycobacterial escape of INH killing (Fig. 7A).
FIG 7.
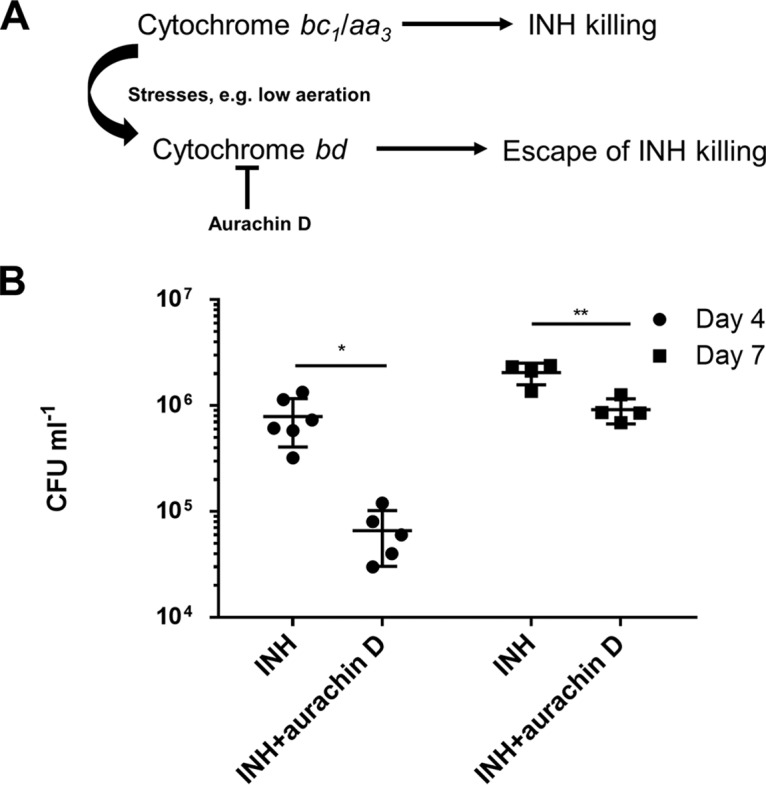
Cytochrome bd oxidase inhibition reduces cell recovery in a culture settling model. (A) Dependence of INH’s bactericidal activity on the cytochrome bc1/aa3 supercomplex and escape of INH killing through the activation of cytochrome bd oxidase under stresses such as low aeration. (B) M. bovis BCG (107 CFU/ml) was treated with 0.4 µg/ml INH (with or without 18 µg/ml aurachin D). The cultures were maintained without agitation to allow for cell settling until viability determination at the indicated time points. *and **, P < 0.05 and P < 0.01, respectively. These experiments were performed 3 times (each in triplicate). Results from one representative experiment are shown. The error bars indicate standard deviations.
DISCUSSION
The mechanism of action of INH is known to be linked to inhibition of the FAS-II component InhA, impeding mycolic acid production (4, 10). Indeed, the INH-treated bacilli lost their ability to produce mycolic acids (5), accumulated FAS-I end product (C26:0), and underwent cell lysis (6). However, under stress conditions, such as low aeration (51), nutrition limitation (52), and acid stress (53), the bacilli become unsusceptible to INH killing. Here, we report a previously underestimated killing mechanism of INH involving mycobacterial ETC perturbation, as demonstrated by the increased ATP and oxygen consumption. The INH-induced ATP surge was reported during the preparation of this article (26).
The INH-triggered increase in ATP may be explained in several ways, e.g., reduced consumption, enhanced production, or both. It is well known that lipid synthesis consumes a large amount of ATP. Every two-carbon elongation of a precursor lipid requires the consumption of one molecule of ATP (consumed at the conversion of acetyl-CoA to malonyl-CoA) (54). Therefore, the INH-induced mycolic acid synthesis inhibition (4, 10) is expected to reduce the ATP consumption. The contribution of reduced consumption to the observed ATP increase, although not investigated in this study, is supported by the finding that even in the presence of an FoF1 ATP synthase inhibitor or ETC-inhibiting dehydrogenase inhibitors, INH still induced a slight ATP increase (Fig. 3A and Fig. 5B). In addition to reduced consumption, the enhanced ATP production through oxidative phosphorylation should also play a pivotal role given that the oxygen consumption was markedly enhanced by INH. This notion is further supported by the observation that chemical inhibition of the ETC at various sites largely compromised the ATP increase and killing activity. INH challenge led to a decrease in FAS-I-derived C16:0 fatty acids (6) and enhanced the expression of proteins involved in fatty acid beta-oxidation, suggesting that the bacilli tend to oxidize fatty acids under INH stress. In addition, INH-treated mycobacteria also showed an important overexpression of a putative taurine dioxygenase (Rv0097) catalyzing the production of succinate (42). These changes may result in enhanced production of reducing equivalents (e.g., NADH) and succinate feeding the ETC (Fig. 5A), leading to augmented oxygen consumption and ATP production.
It was recently shown that a group of mycobacterial cell wall assembly inhibitors, including INH and ethambutol, all increased the ATP level (26), thereby linking the ATP increase to a general cell envelope stress response. However, we showed here that the phthiocerol dimycocerosate (an important mycobacterial cell wall component)-depleting THL (27) failed to increase the ATP level, thus ruling out the general cell envelope stress response as the mechanism for the INH-induced ATP increase. Furthermore, unlike INH, ethambutol did not enhance oxygen consumption (data not shown). Hence, we reason that only INH and not ethambutol can enhance mycobacterial ETC to elevate ATP production.
Interestingly, we also noted membrane potential dissipation after INH treatment, a phenotype observed previously by another group (46). The INH-induced membrane potential change was restored by bedaquiline, an FoF1 ATP synthase proton pump inhibitor (13), suggesting that a functional FoF1 ATP synthase is required for this phenotype. We hypothesize that INH may boost the proton inflow through the proton pump of the FoF1 ATP synthase, leading to the observed membrane potential dissipation and contributing to enhanced ATP production. It can be further speculated that the ETC enhancement may be employed by the bacilli to compensate for the loss of membrane potential by pumping protons across the membrane, which may further exacerbate the ATP surge. However, a recent study demonstrated that bedaquiline can also function as an H+/K+ exchanger and is able to move K+ to generate membrane potential (55). Therefore, bedaquiline can restore membrane potential through its ionophoric activity but not through FoF1 ATP synthase inhibition. Differentiation between these possibilities warrants further investigation. On the other hand, the membrane potential dissipation itself may not be the direct cause of bacterial death, since Q203 or INH plus Q203 also decreased membrane potential in our study. Q203 inhibits the proton-pumping cytochrome bc1/aa3 supercomplex (12, 15). It was shown earlier that this drug did not affect mycobacterial membrane potential in M. tuberculosis (16). This discrepancy may be explained by the use of a different mycobacterial species, a much smaller drug concentration, and a distinct experimental technique in our assay.
The enhanced ATP levels could be linked to INH’s killing because drugs that abrogate the ATP response, including Q203, bedaquiline, NAC, and dehydrogenase inhibitors, substantially attenuated the INH’s bactericidal effect. The loss of INH’s activity in the presence of ETC inhibitors also pinpoints the requirement of ETC-initiating dehydrogenases, the cytochrome bc1/aa3 branch, and FoF1 ATP synthase for INH’s bactericidal activity. This notion is supported by the finding that a defective NADH dehydrogenase conferred INH resistance in M. bovis BCG (56). Since bedaquiline was shown to transiently activate the mycobacterial dormancy regulon (17), one might argue that the bedaquiline-induced loss of INH activity is the result of the nonreplicating “dormant” state. In addition to INH, nonreplicating mycobacteria are also tolerant to other anti-TB drugs such as rifampin and moxifloxacin (37, 51). As bedaquiline failed to prevent rifampin or moxifloxacin killing (see Fig. S4 in the supplemental material), it is less likely that bedaquiline induces a nonreplicating “dormant” state. On the other hand, although Q203 also compromised killing by rifampin and moxifloxacin (Fig. S4), it did not seem to activate the dormancy regulon as suggested by our proteomic data (Sheng Zeng et al, unpublished results). Therefore, we believe that Q203 and bedaquiline exert their protection by inhibiting the INH-induced ATP increase but not by triggering a nonreplicating “dormant” state. In addition, an enhanced NADH/NAD ratio can also compromise INH’s activity (56). Given that the cellular NADH/NAD ratio is not significantly altered in the initial 2 days of Q203 or bedaquiline exposure (16), redox alteration does not seem to contribute to the antagonistic effect of Q203 and bedaquiline observed at 1 and 2 days of treatment. However, another study observed an elevated NADH/NAD ratio after 24 h of treatment with bedaquiline (17), suggesting that the effect of bedaquiline may partially result from the redox alteration in addition to the inhibition of energetic alteration.
This study also unveils that activation of the alternative cytochrome bd oxidase protects from INH killing (Fig. 7A). Exposure to Q203 results in ETC reprogramming to cytochrome bd oxidase (12, 15, 16). Similar respiratory reprogramming can be triggered by other stresses, e.g., hypoxia and bedaquiline treatment, all of which can attenuate INH killing as Q203 does (50, 51, 57). Several studies have investigated the link between INH susceptibility and the cytochrome bd oxidase. Moosa et al. observed that an M. tuberculosis cytochrome bd mutant was not more sensitive to growth inhibition by INH under normal culture conditions (58). In contrast, disruption of cydC, which is required for cytochrome bd oxidase assembly, significantly enhanced INH’s killing in mice (24). We reason that unlike under normal culture conditions, host-derived stresses can lead to the activation of the cytochrome bd oxidase and the consequent escape from INH killing (Fig. 7A) (57). That is, the role of the cytochrome bd oxidase is important mainly under stress conditions. Given that the expression of the cytochrome bd oxidase is regulated by low aeration (59), we tested directly the role of the cytochrome bd oxidase using the inhibitor aurachin D (47) in a settling culture model (49). Interestingly, inhibition of cytochrome bd oxidase (47) significantly reduced cell recovery during INH challenge. This result not only further supports an important role for cytochrome bd oxidase in the protection against INH killing but also demonstrates the potential value of cytochrome bd oxidase inhibitors for future studies. Based on these results, the previously observed induction of cytochrome bd oxidase by INH (50) possibly reflects a mycobacterial rescuing response to counteract the drug’s killing.
A major concern of this study is the use of chemical inhibitors which may have unknown off-target effects. For instance, the above-mentioned effect conferred by aurachin D may be explained by unrecognized effects other than the cytochrome bd oxidase inhibition. Similar experiments with mycobacterial cytochrome bd oxidase mutants should be beneficial in distinguishing between these possibilities. However, we argue that the consistent results obtained with multiple chemicals targeting different sites of the ETC (i.e., dehydrogenases, cytochrome bc1, cytochrome bd, and FoF1 ATP synthase) can largely minimize the influence of the possible off-target effects given that one drug’s off-target effect should be unique from that of another. In addition to the use of chemical inhibitors, Vilcheze et al. showed recently that the addition of cysteine or NAC potentiated INH’s efficacy by preventing the generation of persisters and drug-resistant populations (36). This potentiating effect was more evident after 7 days of drug treatment. In our study, we focused only on early points (i.e., 1 and 2 days) during INH treatment. Therefore, the protection of NAC against INH’s killing we observed here is not contradictory to the previous finding (36). In fact, the early protecting role of NAC can be explained not only by its inhibition of ATP increase (Fig. 4A) but also by the observation that addition of cysteine to INH-treated M. tuberculosis induced DosR regulon expression (36). Interestingly, the cysteine or NAC is proposed to potentiate INH’s killing by enhancing oxygen consumption (36), which supports our finding that INH’s killing involves ETC perturbation.
Although Q203 was not the focus of our study, we believe it is important to point out that this bacteriostatic drug significantly compromised the bactericidal activity of rifampin and moxifloxacin, in addition to INH. A previous study with Escherichia coli and Staphylococcus aureus pinpointed that many bacteriostatic antibiotics decreased bacterial oxygen consumption, whereas bactericidal antibiotics accelerated oxygen consumption (60). More importantly, the bacteriostatic antibiotic-triggered respiration inhibition significantly attenuated the killing by bactericidal drugs (60). Our finding with Q203 and INH seems to fit this notion. As a bacteriostatic drug, Q203 was found to greatly inhibit the INH-induced oxygen consumption. However, unlike INH, rifampin did not seem to enhance the oxygen consumption rate. Therefore, the protection conferred by Q203 may also derive from mechanisms unrelated to respiration, such as cytochrome bd oxidase activation (16, 50). Regardless of the mechanism, it seems that Q203, unlike bedaquiline, confers a general drug tolerance phenotype. Drug tolerance is characterized by an increase in the minimum duration for killing (MDK) for 99% of bacterial population (MDK99) (61). Thus, to ascertain whether Q203 induces drug tolerance, further assays are required to measure the effect of Q203 on MDK99 for INH, rifampin, and moxifloxacin.
In summary, this study links INH’s killing mechanism to mycobacterial ETC perturbation. INH’s bactericidal activity is largely dependent on ETC-initiating dehydrogenases, the cytochrome bc1/aa3 supercomplex, and FoF1 ATP synthase. Conversely, the activation of cytochrome bd oxidase protects from INH’s killing. This in vitro study also suggests that cytochrome bd inhibitors (e.g., aurachin D) potentiate INH’s killing, whereas Q203, bedaquiline, and possibly other ETC inhibitors can inhibit the activity of INH and potentially also ethionamide. These drug interactions should be further investigated in vivo.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
The Mycobacterium bovis BCG wild-type (WT) strain (62) and M. tuberculosis H37Rv were maintained in Dubos medium supplemented with 10% (vol/vol) Dubos medium albumin (DTA medium). The DTA medium contains 0.75% glucose and 0.02% Tween 80 as carbon and energy sources. In some experiments, modified DTA medium without glucose was used.
ATP determination.
Bacterial ATP was determined using the BacTiter-Glo microbial cell viability assay kit (Promega). Briefly, 20 µl of M. bovis BCG was mixed with an equal volume of BacTiter-Glo reagent for 5 min in the dark. Luminescence was recorded using the Lumat LB 9507 instrument (Berthold). In some experiments, bacterial cultures were passed through a 0.2-µm filter and ATP in the culture filtrates was measured. To quantify intracellular ATP, bacteria were pelleted and resuspended in fresh DTA medium before ATP assessment. To facilitate cell lysis prior to ATP measurement, the bacterial culture was repeatedly sonicated using a Bioruptor UCD-200. The sonicated cultures were plated for determining viability to check the efficiency of sonication.
Viability determination.
Mycobacterial cultures were grown in DTA medium to log phase and diluted to an optical density at 600 nm (OD600) of 0.1 (approximately 107 CFU/ml) prior to drug treatment. The viability before and after drug exposure was measured by plating dilutions on 7H11 agar. To minimize any carryover effect, the cultures were diluted at least 100-fold, and only 10 µl of the dilutions was plated on agar plates (i.e., the limit of detection of 104 CFU/ml).
Oxygen consumption assay.
M. bovis BCG were diluted to 107 CFU/ml and dispensed into glass tubes with screw caps. The same volume of headspace air was kept in the tubes. INH, rifampin, ethambutol, and Q203 were added to final concentrations of 0.4 µg/ml, 0.2 µg/ml, 4 µg/ml, and 10 nM, respectively. Methylene blue (3 µg/ml) was used as an oxygen indicator (15). Tightly capped tubes were incubated with shaking for 30 and 40 h. At 40 h, the tubes were also opened for viability measurement.
In addition, cyclic voltammetry was applied to monitor the amount of dissolved oxygen. The potential changed from +100 mV to −600 mV and was subsequently reversed to the initial potential at a scan rate of 50 mV/s. A gold disk (3-mm diameter) served the working electrode, Ag/AgCl–3 M KCl as the reference electrode, and a platinum wire as the auxiliary electrode. Tightly capped tubes (with or without drugs) were incubated with shaking for 5 h. The tubes were then opened, and the three electrodes were immediately dipped in the culture for cyclic voltammetry assay. The experiments were performed 3 times.
ROS determination.
Cells were loaded with 10 µM 2,7-dichlorofluorescin diacetate (Sigma) for 2 h and then treated with INH or clofazimine for 5 and 24 h before ROS determination. The treated culture was transferred to a black-wall, flat, and clear-bottom 96-well plate to record fluorescence (excitation at 485 nm [20-nm filter] and emission at 530 nm [20-nm filter]) (31). ROS data were normalized by viability after 24 h of drug treatment.
Membrane potential measurement.
Mycobacterial cultures were treated with 10 µg/ml valinomycin, 0.4 µg/ml INH, 10 nM Q203, or 0.25 µg/ml bedaquiline for 30 min and 2 h and then incubated with 3,3′-diethyloxacarbocyanine iodide [DiOC2(3)] (15 µM). The culture was washed and resuspended in fresh DTA medium. As described previously (37, 44), green fluorescence and red fluorescence were recorded at 488 nm/530 nm and 488 nm/610 nm, respectively. Since a larger membrane potential results in stronger shift from green to red fluorescence, membrane potentials were indicated as the ratio of red to green fluorescence.
Proteomic analysis.
M. bovis BCG was treated with 0.4 µg/ml INH (with or without 10 nM Q203) for 7 h and harvested for proteomic analysis. Protein extraction was described previously (63), and the detailed proteomic procedure can be found in the supplemental material. Protein hits with a P value of <0.05 and a fold change of <0.8 or >1.2 were further analyzed. Protein function classifications were based on Mycobrowser (https://mycobrowser.epfl.ch/), the NCBI Conserved Domains search tool (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi), and UniProt (https://www.uniprot.org/).
Drug treatment in a settling culture model.
M. bovis BCG grown in DTA medium was diluted to an OD600 of 0.1 (approximately 107 CFU ml−1) and treated with 0.4 µg/ml INH (with or without 18 µg/ml aurachin D [47]). The tubes were maintained without agitation to allow for settling, and the viability was determined after 4 and 7 days of treatment. Subsequently, 20 randomly picked colonies recovered from the drug-treated cultures were subgrown in DTA medium with 0.2 µg/ml INH. Growth was visually checked for 10 days.
Statistical analysis.
Graph preparation and statistics were performed using GraphPad Prism 6.0. The unpaired t test was used to analyze statistical significance. A P value of <0.05 was considered statistically significant. *, **, ***, and # indicate P values of <0.05, <0.01, <0.001, and <0.0001, respectively.
Supplementary Material
ACKNOWLEDGMENTS
Sheng Zeng received a fellowship from the China Scholarship Council and Les Amis des Instituts Pasteur à Bruxelles. This study was sponsored by Les Amis des Instituts Pasteur à Bruxelles and the Belgian Fund for Scientific Research (Grand Equipment-F.R.S-FNRS). The bioprofiling platform used for proteomic analysis was supported by the European Regional Development Fund and the Walloon Region, Belgium.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01841-18.
REFERENCES
- 1.WHO. 2017. Global tuberculosis report 2017. WHO, Geneva, Switzerland. [Google Scholar]
- 2.Zumla A, Nahid P, Cole ST. 2013. Advances in the development of new tuberculosis drugs and treatment regimens. Nat Rev Drug Discov 12:388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 3.Al-Saeedi M, Al-Hajoj S. 2017. Diversity and evolution of drug resistance mechanisms in Mycobacterium tuberculosis. Infect Drug Resist 10:333–342. doi: 10.2147/IDR.S144446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rawat R, Whitty A, Tonge PJ. 2003. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc Natl Acad Sci U S A 100:13881–13886. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takayama K, Wang L, David HL. 1972. Effect of isoniazid on the in vivo mycolic acid synthesis, cell growth, and viability of Mycobacterium tuberculosis. Antimicrob Agents Chemother 2:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vilcheze C, Morbidoni HR, Weisbrod TR, Iwamoto H, Kuo M, Sacchettini JC, Jacobs WR Jr. 2000. Inactivation of the inhA-encoded fatty acid synthase II (FASII) enoyl-acyl carrier protein reductase induces accumulation of the FASI end products and cell lysis of Mycobacterium smegmatis. J Bacteriol 182:4059–4067. doi: 10.1128/JB.182.14.4059-4067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vannelli TA, Dykman A, Ortiz de Montellano PR. 2002. The antituberculosis drug ethionamide is activated by a flavoprotein monooxygenase. J Biol Chem 277:12824–12829. doi: 10.1074/jbc.M110751200. [DOI] [PubMed] [Google Scholar]
- 8.DeBarber AE, Mdluli K, Bosman M, Bekker LG, Barry CE III. 2000. Ethionamide activation and sensitivity in multidrug-resistant Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 97:9677–9682. doi: 10.1073/pnas.97.17.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baulard AR, Betts JC, Engohang-Ndong J, Quan S, McAdam RA, Brennan PJ, Locht C, Besra GS. 2000. Activation of the pro-drug ethionamide is regulated in mycobacteria. J Biol Chem 275:28326–28331. doi: 10.1074/jbc.M003744200. [DOI] [PubMed] [Google Scholar]
- 10.Banerjee A, Dubnau E, Quemard A, Balasubramanian V, Um KS, Wilson T, Collins D, de Lisle G, Jacobs WR Jr. 1994. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 263:227–230. doi: 10.1126/science.8284673. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Y, Heym B, Allen B, Young D, Cole S. 1992. The catalase-peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 358:591–593. doi: 10.1038/358591a0. [DOI] [PubMed] [Google Scholar]
- 12.Pethe K, Bifani P, Jang J, Kang S, Park S, Ahn S, Jiricek J, Jung J, Jeon HK, Cechetto J, Christophe T, Lee H, Kempf M, Jackson M, Lenaerts AJ, Pham H, Jones V, Seo MJ, Kim YM, Seo M, Seo JJ, Park D, Ko Y, Choi I, Kim R, Kim SY, Lim SBin, Yim S-A, Nam J, Kang H, Kwon H, Oh C-T, Cho Y, Jang Y, Kim J, Chua A, Tan BH, Nanjundappa MB, Rao SPS, Barnes WS, Wintjens R, Walker JR, Alonso S, Lee S, Kim J, Oh S, Oh T, Nehrbass U, Han S-J, No Z, Lee J, Brodin P, Cho S-N, Nam K, Kim J. 2013. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat Med 19:1157–1160. doi: 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- 13.Andries K, Verhasselt P, Guillemont J, Gohlmann HW, Neefs JM, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 14.Koul A, Dendouga N, Vergauwen K, Molenberghs B, Vranckx L, Willebrords R, Ristic Z, Lill H, Dorange I, Guillemont J, Bald D, Andries K. 2007. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat Chem Biol 3:323–324. doi: 10.1038/nchembio884. [DOI] [PubMed] [Google Scholar]
- 15.Kalia NP, Hasenoehrl EJ, Ab Rahman NB, Koh VH, Ang MLT, Sajorda DR, Hards K, Gruber G, Alonso S, Cook GM, Berney M, Pethe K. 2017. Exploiting the synthetic lethality between terminal respiratory oxidases to kill Mycobacterium tuberculosis and clear host infection. Proc Natl Acad Sci U S A 114:7426–7431. doi: 10.1073/pnas.1706139114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamprecht DA, Finin PM, Rahman MA, Cumming BM, Russell SL, Jonnala SR, Adamson JH, Steyn AJ. 2016. Turning the respiratory flexibility of Mycobacterium tuberculosis against itself. Nat Commun 7:12393. doi: 10.1038/ncomms12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koul A, Vranckx L, Dhar N, Gohlmann HW, Ozdemir E, Neefs JM, Schulz M, Lu P, Mortz E, McKinney JD, Andries K, Bald D. 2014. Delayed bactericidal response of Mycobacterium tuberculosis to bedaquiline involves remodelling of bacterial metabolism. Nat Commun 5:3369. doi: 10.1038/ncomms4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cook GM, Hards K, Vilcheze C, Hartman T, Berney M. 2014. Energetics of respiration and oxidative phosphorylation in mycobacteria. Microbiol Spectr 2:MGM2-0015-2013. doi: 10.1128/microbiolspec.MGM2-0015-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shoeb HA, Bowman BU Jr, Ottolenghi AC, Merola AJ. 1985. Evidence for the generation of active oxygen by isoniazid treatment of extracts of Mycobacterium tuberculosis H37Ra. Antimicrob Agents Chemother 27:404–407. doi: 10.1128/AAC.27.3.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shoeb HA, Bowman BU Jr, Ottolenghi AC, Merola AJ. 1985. Enzymatic and nonenzymatic superoxide-generating reactions of isoniazid. Antimicrob Agents Chemother 27:408–412. doi: 10.1128/AAC.27.3.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilson TM, Collins DM. 1996. ahpC, a gene involved in isoniazid resistance of the Mycobacterium tuberculosis complex. Mol Microbiol 19:1025–1034. doi: 10.1046/j.1365-2958.1996.449980.x. [DOI] [PubMed] [Google Scholar]
- 22.Dussurget O, Rodriguez M, Smith I. 1998. Protective role of the Mycobacterium smegmatis IdeR against reactive oxygen species and isoniazid toxicity. Tuber Lung Dis 79:99–106. doi: 10.1054/tuld.1998.0011. [DOI] [PubMed] [Google Scholar]
- 23.Lanciano P, Khalfaoui-Hassani B, Selamoglu N, Ghelli A, Rugolo M, Daldal F. 2013. Molecular mechanisms of superoxide production by complex III: a bacterial versus human mitochondrial comparative case study. Biochim Biophys Acta 1827:1332–1339. doi: 10.1016/j.bbabio.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dhar N, McKinney JD. 2010. Mycobacterium tuberculosis persistence mutants identified by screening in isoniazid-treated mice. Proc Natl Acad Sci U S A 107:12275–12280. doi: 10.1073/pnas.1003219107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soetaert K, Rens C, Wang X-M, De Bruyn J, Lanéelle M-A, Laval F, Lemassu A, Daffé M, Bifani P, Fontaine V, Lefèvre P. 2015. Increased vancomycin susceptibility in mycobacteria: a new approach to identify synergistic activity against multidrug-resistant mycobacteria. Antimicrob Agents Chemother 59:5057–5060. doi: 10.1128/AAC.04856-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shetty A, Dick T. 2018. Mycobacterial cell wall synthesis inhibitors cause lethal ATP burst. Front Microbiol 9:1898. doi: 10.3389/fmicb.2018.01898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rens C, Laval F, Daffe M, Denis O, Frita R, Baulard A, Wattiez R, Lefevre P, Fontaine V. 2016. Effects of lipid-lowering drugs on vancomycin susceptibility of mycobacteria. Antimicrob Agents Chemother 60:6193–6199. doi: 10.1128/AAC.00872-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taillefer M, Sparling R. 2016. Glycolysis as the central Core Of Fermentation. Adv Biochem Eng Biotechnol 156:55–77. doi: 10.1007/10_2015_5003. [DOI] [PubMed] [Google Scholar]
- 29.Brynildsen MP, Winkler JA, Spina CS, MacDonald IC, Collins JJ. 2013. Potentiating antibacterial activity by predictably enhancing endogenous microbial ROS production. Nat Biotechnol 31:160–165. doi: 10.1038/nbt.2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang X, Zhao X. 2009. Contribution of oxidative damage to antimicrobial lethality. Antimicrob Agents Chemother 53:1395–1402. doi: 10.1128/AAC.01087-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howell Wescott HA, Roberts DM, Allebach CL, Kokoczka R, Parish T. 2017. Imidazoles induce reactive oxygen species in Mycobacterium tuberculosis which is not associated with cell death. ACS Omega 2:41–51. doi: 10.1021/acsomega.6b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yano T, Kassovska-Bratinova S, Teh JS, Winkler J, Sullivan K, Isaacs A, Schechter NM, Rubin H. 2011. Reduction of clofazimine by mycobacterial type 2 NADH:quinone oxidoreductase: a pathway for the generation of bactericidal levels of reactive oxygen species. J Biol Chem 286:10276–10287. doi: 10.1074/jbc.M110.200501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson M, DeRisi J, Kristensen HH, Imboden P, Rane S, Brown PO, Schoolnik GK. 1999. Exploring drug-induced alterations in gene expression in Mycobacterium tuberculosis by microarray hybridization. Proc Natl Acad Sci U S A 96:12833–12838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karakousis PC, Williams EP, Bishai WR. 2008. Altered expression of isoniazid-regulated genes in drug-treated dormant Mycobacterium tuberculosis. J Antimicrob Chemother 61:323–331. doi: 10.1093/jac/dkm485. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe S, Zimmermann M, Goodwin MB, Sauer U, Barry CE, Boshoff HI. 2011. Fumarate reductase activity maintains an energized membrane in anaerobic Mycobacterium tuberculosis. PLoS Pathog 7:e1002287. doi: 10.1371/journal.ppat.1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vilcheze C, Hartman T, Weinrick B, Jain P, Weisbrod TR, Leung LW, Freundlich JS, Jacobs WR Jr. 2017. Enhanced respiration prevents drug tolerance and drug resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 114:4495–4500. doi: 10.1073/pnas.1704376114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rao SP, Alonso S, Rand L, Dick T, Pethe K. 2008. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 105:11945–11950. doi: 10.1073/pnas.0711697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilcox CS, Pearlman A. 2008. Chemistry and antihypertensive effects of tempol and other nitroxides. Pharmacol Rev 60:418–469. doi: 10.1124/pr.108.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eoh H, Rhee KY. 2014. Methylcitrate cycle defines the bactericidal essentiality of isocitrate lyase for survival of Mycobacterium tuberculosis on fatty acids. Proc Natl Acad Sci U S A 111:4976–4981. doi: 10.1073/pnas.1400390111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Savvi S, Warner DF, Kana BD, McKinney JD, Mizrahi V, Dawes SS. 2008. Functional characterization of a vitamin B12-dependent methylmalonyl pathway in Mycobacterium tuberculosis: implications for propionate metabolism during growth on fatty acids. J Bacteriol 190:3886–3895. doi: 10.1128/JB.01767-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams KJ, Boshoff HI, Krishnan N, Gonzales J, Schnappinger D, Robertson BD. 2011. The Mycobacterium tuberculosis beta-oxidation genes echA5 and fadB3 are dispensable for growth in vitro and in vivo. Tuberculosis (Edinb) 91:549–555. doi: 10.1016/j.tube.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eichhorn E, van der Ploeg JR, Kertesz MA, Leisinger T. 1997. Characterization of alpha-ketoglutarate-dependent taurine dioxygenase from Escherichia coli. J Biol Chem 272:23031–23036. [DOI] [PubMed] [Google Scholar]
- 43.Weinstein EA, Yano T, Li LS, Avarbock D, Avarbock A, Helm D, McColm AA, Duncan K, Lonsdale JT, Rubin H. 2005. Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc Natl Acad Sci U S A 102:4548–4553. doi: 10.1073/pnas.0500469102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eoh H, Rhee KY. 2013. Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 110:6554–6559. doi: 10.1073/pnas.1219375110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pecsi I, Hards K, Ekanayaka N, Berney M, Hartman T, Jacobs WR Jr, Cook GM. 2014. Essentiality of succinate dehydrogenase in Mycobacterium smegmatis and its role in the generation of the membrane potential under hypoxia. mBio 5:e01093-14. doi: 10.1128/mBio.01093-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moreira W, Aziz DB, Dick T. 2016. Boromycin kills mycobacterial persisters without detectable resistance. Front Microbiol 7:199. doi: 10.3389/fmicb.2016.00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu P, Asseri AH, Kremer M, Maaskant J, Ummels R, Lill H, Bald D. 2018. The anti-mycobacterial activity of the cytochrome bcc inhibitor Q203 can be enhanced by small-molecule inhibition of cytochrome bd. Sci Rep 8:2625. doi: 10.1038/s41598-018-20989-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu P, Heineke MH, Koul A, Andries K, Cook GM, Lill H, van Spanning R, Bald D. 2015. The cytochrome bd-type quinol oxidase is important for survival of Mycobacterium smegmatis under peroxide and antibiotic-induced stress. Sci Rep 5:10333. doi: 10.1038/srep10333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wayne LG. 1977. Synchronized replication of Mycobacterium tuberculosis. Infect Immun 17:528–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boot M, Jim KK, Liu T, Commandeur S, Lu P, Verboom T, Lill H, Bitter W, Bald D. 2017. A fluorescence-based reporter for monitoring expression of mycobacterial cytochrome bd in response to antibacterials and during infection. Sci Rep 7:10665. doi: 10.1038/s41598-017-10944-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wayne LG, Hayes LG. 1996. An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect Immun 64:2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Betts JC, Lukey PT, Robb LC, McAdam RA, Duncan K. 2002. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol Microbiol 43:717–731. [DOI] [PubMed] [Google Scholar]
- 53.Baker JJ, Abramovitch RB. 2018. Genetic and metabolic regulation of Mycobacterium tuberculosis acid growth arrest. Sci Rep 8:4168. doi: 10.1038/s41598-018-22343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brose SA, Marquardt AL, Golovko MY. 2014. Fatty acid biosynthesis from glutamate and glutamine is specifically induced in neuronal cells under hypoxia. J Neurochem 129:400–412. doi: 10.1111/jnc.12617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hards K, McMillan DGG, Schurig-Briccio LA, Gennis RB, Lill H, Bald D, Cook GM. 2018. Ionophoric effects of the antitubercular drug bedaquiline. Proc Natl Acad Sci U S A 115:7326–7331. doi: 10.1073/pnas.1803723115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vilcheze C, Weisbrod TR, Chen B, Kremer L, Hazbon MH, Wang F, Alland D, Sacchettini JC, Jacobs WR Jr. 2005. Altered NADH/NAD+ ratio mediates coresistance to isoniazid and ethionamide in mycobacteria. Antimicrob Agents Chemother 49:708–720. doi: 10.1128/AAC.49.2.708-720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shi L, Sohaskey CD, Kana BD, Dawes S, North RJ, Mizrahi V, Gennaro ML. 2005. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc Natl Acad Sci U S A 102:15629–15634. doi: 10.1073/pnas.0507850102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moosa A, Lamprecht DA, Arora K, Barry CE III, Boshoff HIM, Ioerger TR, Steyn AJC, Mizrahi V, Warner DF. 2017. Susceptibility of Mycobacterium tuberculosis cytochrome bd oxidase mutants to compounds targeting the terminal respiratory oxidase, cytochrome c. Antimicrob Agents Chemother 61:e01338-17. doi: 10.1128/AAC.01338-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kana BD, Weinstein EA, Avarbock D, Dawes SS, Rubin H, Mizrahi V. 2001. Characterization of the cydAB-encoded cytochrome bd oxidase from Mycobacterium smegmatis. J Bacteriol 183:7076–7086. doi: 10.1128/JB.183.24.7076-7086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lobritz MA, Belenky P, Porter CB, Gutierrez A, Yang JH, Schwarz EG, Dwyer DJ, Khalil AS, Collins JJ. 2015. Antibiotic efficacy is linked to bacterial cellular respiration. Proc Natl Acad Sci U S A 112:8173–8180. doi: 10.1073/pnas.1509743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brauner A, Fridman O, Gefen O, Balaban NQ. 2016. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat Rev Microbiol 14:320–330. doi: 10.1038/nrmicro.2016.34. [DOI] [PubMed] [Google Scholar]
- 62.Wang XM, Lu C, Soetaert K, S'Heeren C, Peirs P, Laneelle MA, Lefevre P, Bifani P, Content J, Daffe M, Huygen K, De Bruyn J, Wattiez R. 2011. Biochemical and immunological characterization of a cpn60.1 knockout mutant of Mycobacterium bovis BCG. Microbiology 157:1205–1219. doi: 10.1099/mic.0.045120-0. [DOI] [PubMed] [Google Scholar]
- 63.Deschoenmaeker F, Bayon-Vicente G, Sachdeva N, Depraetere O, Cabrera Pino JC, Leroy B, Muylaert K, Wattiez R. 2017. Impact of different nitrogen sources on the growth of Arthrospira sp. PCC 8005 under batch and continuous cultivation—a biochemical, transcriptomic and proteomic profile. Bioresour Technol 237:78–88. doi: 10.1016/j.biortech.2017.03.145. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.