

Abstract
Ribosome stalling triggers quality control pathways targeting the mRNA (NGD: no‐go decay) and the nascent polypeptide (RQC: ribosome‐associated quality control). RQC requires Hel2‐dependent uS10 ubiquitination and the RQT complex in yeast. Here, we report that Hel2‐dependent uS10 ubiquitination and Slh1/Rqt2 are crucial for RQC and NGD induction within a di‐ribosome (disome) unit, which consists of the leading stalled ribosome and the following colliding ribosome. Hel2 preferentially ubiquitinated a disome over a monosome on a quality control inducing reporter mRNA in an in vitro translation reaction. Cryo‐EM analysis of the disome unit revealed a distinct structural arrangement suitable for recognition and modification by Hel2. The absence of the RQT complex or uS10 ubiquitination resulted in the elimination of NGD within the disome unit. Instead, we observed Hel2‐mediated cleavages upstream of the disome, governed by initial Not4‐mediated monoubiquitination of eS7 and followed by Hel2‐mediated K63‐linked polyubiquitination. We propose that Hel2‐mediated ribosome ubiquitination is required both for canonical NGD (NGDRQC +) and RQC coupled to the disome and that RQC‐uncoupled NGD outside the disome (NGDRQC −) can occur in a Not4‐dependent manner.
Keywords: no‐go mRNA decay, ribosome collision, ribosome quality control, RQT complex, ubiquitination
Subject Categories: Post-translational Modifications, Proteolysis & Proteomics; Protein Biosynthesis & Quality Control; Structural Biology
Introduction
Cells have various quality control systems, which selectively degrade aberrant mRNA and defective proteins to ensure precise expression of genome‐encoded information. Ribosome stalling during elongation results in degradation of both the mRNA and the arrested nascent protein by mRNA surveillance pathways and the ribosome‐associated quality control (RQC) systems (Bengtson & Joazeiro, 2010; Becker et al, 2012; Brandman et al, 2012; Defenouillere et al, 2013). Specific mRNA decay pathways (non‐stop decay, NSD; nonsense‐mediated decay, NMD) are triggered by the lack of a stop codon and the presence of a premature a stop codon, respectively. In contrast, the no‐go decay (NGD) mRNA surveillance pathway occurs when the ribosome is road blocked, for example, by stable RNA secondary structures, rare codons or stretches of consecutive positively charged amino acids in the nascent chain (Doma & Parker, 2006; Chen et al, 2010; van den Elzen et al, 2010; Kobayashi et al, 2010; Kuroha et al, 2010). NGD is initiated by endonucleolytic cleavage of mRNA proximal to the ribosomal stalling site (Doma & Parker, 2006). This cleavage results in the production of 5′ NGD and 3′ NGD‐mRNA intermediates, which are further degraded by the Xrn1 exoribonuclease and the exosome, respectively (Doma & Parker, 2006). In this regard, certain combinations of rare codons coding for arginine (CGN; where N=G, C or A) were shown to be very potent for ribosomal stalling (Gamble et al, 2016). Notably, endonucleolytic cleavage events have been reported even in a more general context outside NGD in a study by Mourelatos and co‐workers, showing that endogenous human mRNAs undergo repeated co‐translational and ribosome‐phased endonucleolytic cuts at the exit site of the mRNA ribosome channel (Ibrahim et al, 2018).
Importantly, the potentially toxic protein products of stalled mRNAs have to be degraded. Via the ribosome‐associated quality control (RQC) pathway, a truncated nascent protein is degraded and the stalled ribosome is recycled. Dissociation of the stalled 80S ribosome into 40S and 60S subunits is a crucial step in this process, with the resulting 60S subunit still bearing a peptidyl‐tRNA. This complex is recognized by the Rqc2 and Ltn1 proteins: while Rqc2 stabilizes Ltn1 binding and adds a random sequence of alanines and threonines to the C‐terminus of the nascent peptide (CAT‐tails), the E3 ubiquitin ligase Ltn1 ubiquitinates the arrested peptide, thereby marking it for subsequent proteasomal degradation (Bengtson & Joazeiro, 2010; Brandman et al, 2012; Shao & Hegde, 2014; Hilal & Spahn, 2015; Shen et al, 2015).
In yeast, the RQC pathway is triggered by ubiquitination of the ribosomal protein uS10 at specific lysine residues by the E3 ubiquitin ligase Hel2 (also known as Rqt1, derived from RQC‐trigger; ZNF598 in mammals) and the E2 enzyme Ubc4 (Matsuo et al, 2017). Moreover, initiating the RQC pathway requires an additional protein complex, the so‐called RQC‐trigger (RQT) complex (Matsuo et al, 2017). It is composed of the RNA helicase family protein Slh1/Rqt2, the ubiquitin‐binding protein Cue3/Rqt3 and yKR023W/Rqt4 (Matsuo et al, 2017; Sitron et al, 2017). The RQT complex was found to associate with the ribosome and Hel2, and the ubiquitin‐binding activity of Cue3/Rqt3 and ATPase activity of Slh1/Rqt2 were shown to be crucial to trigger RQC (Matsuo et al, 2017). Recent studies in the mammalian system demonstrated that ZNF598 ubiquitinates the ribosomal proteins eS10 at lysines K138 and K139 and uS10 at K4 and K8. This ubiquitination triggered the RQC response on ribosomes stalled on a poly‐lysine encoding mRNA reporter, indicating that the role of ribosome ubiquitination in quality control is conserved (Garzia et al, 2017; Juszkiewicz & Hegde, 2017; Sundaramoorthy et al, 2017).
Both the NGD and the RQC require common factors and biochemical events suggesting a coupling of these two quality control pathways (Shoemaker & Green, 2012). Both are initiated by translation arrest and are dependent on the 40S subunit‐associated Asc1 (RACK1) in yeast (Kuroha et al, 2010; Ikeuchi & Inada, 2016). Moreover, Hel2‐mediated K63‐linked polyubiquitination has been implicated in RQC after stalling at polybasic amino acid sequences and tandem CGA codons in yeast (Saito et al, 2015). Furthermore, Simms et al recently reported that ribosomal collisions represent the trigger for NGD‐induced mRNA cleavage and proposed that colliding ribosomes induce robust ubiquitination of uS3 by Hel2 However, prior to our study, there was no evidence that K63‐linked polyubiquitination of uS3 or other ribosomal proteins by Hel2 plays a role in NGD, and the exact biochemical and structural interdependencies between NGD and RQC induced by translation arrest remained unclear.
In this study, we demonstrate that NGD and RQC are coupled and that both pathways respond to ribosome collision. We focused on a di‐ribosome (disome) unit consisting of the stalled ribosome (here referred to as the leading ribosome) and the following colliding ribosome. This minimal ribosome collision unit is required to couple NGD and RQC via Hel2. We show that endonucleolytic cleavage of a NGD reporter mRNA occurs at sites within this disome unit. The mRNA cleavage is dependent on Hel2‐mediated K63‐linked polyubiquitination of uS10 as well as on the activity of the RQT component Slh1/Rqt2, showing that NGD and RQC are coupled via this ubiquitination event (here referred to as the NGDRQC+). Furthermore, we show that disomes are preferred as targets for Hel2‐mediated uS10 ubiquitination over monosomes. In addition, we could dissect the NGD pathway into two interdependent branches. We identified a mutant of Hel2 that fails to trigger RQC and only initiates an alternative NGD. In this alternative NGD pathway, endonucleolytic mRNA cleavages occur upstream of the stalled disome (here referred to as the NGDRQC−). We further show that these cleavages require K63‐linked polyubiquitination of ribosomal protein eS7. This polyubiquitination happens in a two‐step mechanism, where the E3 ligase Not4 first monoubiquitinates eS7 which is followed by Hel2‐mediated polyubiquitination. Taken together, we propose a dual role of Hel2 leading to two distinct NGD pathways, which require specific ubiquitination events on the stalled disome.
To get a structural understanding of how ribosome collisions could provide a platform for signalling to mRNA and protein quality control pathways, we determined the Cryo‐EM structure of a disome as an NGD substrate. This structure shows a defined disome arrangement in which the leading ribosome is stalled in the classical POST‐translocation state with an empty A‐site and occupied P‐ and E‐sites. The second ribosome is in a hybrid state with A/P and P/E tRNAs, apparently locked in an incomplete translocation step. The interface between the leading and the colliding ribosomes is mainly formed by the small 40S subunit and to lesser extent also the large 60S subunit. Importantly, the 40S inter‐ribosomal contact interface brings all proteins targeted by Hel2 during quality control in close proximity. Moreover, both Asc1 (RACK1 in humans) molecules are in direct contact forming one of the inter‐ribosomal interaction sites. We suggest that this defined interaction between a stalled leading ribosome and a colliding ribosome generates a unique composite surface for molecular recognition of translational stalling. It may represent the ideal substrate for Hel2, thereby specifically recognizing a prolonged translation stall to initiate RQC and NGDRQC+ by its E3 ubiquitin ligase activity.
Results
Hel2‐mediated K63‐linked polyubiquitination is required for both NGD and RQC
In our previous study, we demonstrated that Hel2 is essential for ubiquitination of the small subunit ribosomal protein uS10, which leads to the degradation of the arrested peptide after translation arrest via RQC in yeast (Matsuo et al, 2017). To investigate the relation between the two quality control pathways induced by ribosome stalling, NGD and RQC, we now investigated the role of Hel2 in the endoribonucleolytic cleavage of mRNA, a triggering step of the NGD quality control using several stalling reporter mRNAs (Fig EV1A). The main reporter contains twelve consecutive CGN codons (N = A/G/C) coding for arginine (R(CGN)12). Notably, this sequence contains codon pairs (CGA‐CGA) which have been described to efficiently cause ribosomal stalling in yeast (Gamble et al, 2016; Matsuo et al, 2017). To estimate the efficiency of NGD, we used the production of intermediate endonucleolytic mRNA cleavage products (Fig 1A). The 5′ NGD‐mRNA intermediate (5′ NGD‐IM) represents the primary product, which is rapidly degraded by the cytoplasmic exosome. Therefore, the 5′ NGD intermediates derived from various arrest‐inducing sequences can be detected in mutants lacking the exosomal co‐factor Ski2 (ski2∆).
Figure EV1. Hel2 and Ubc4 were involved in NGD but not NSD and NMD.
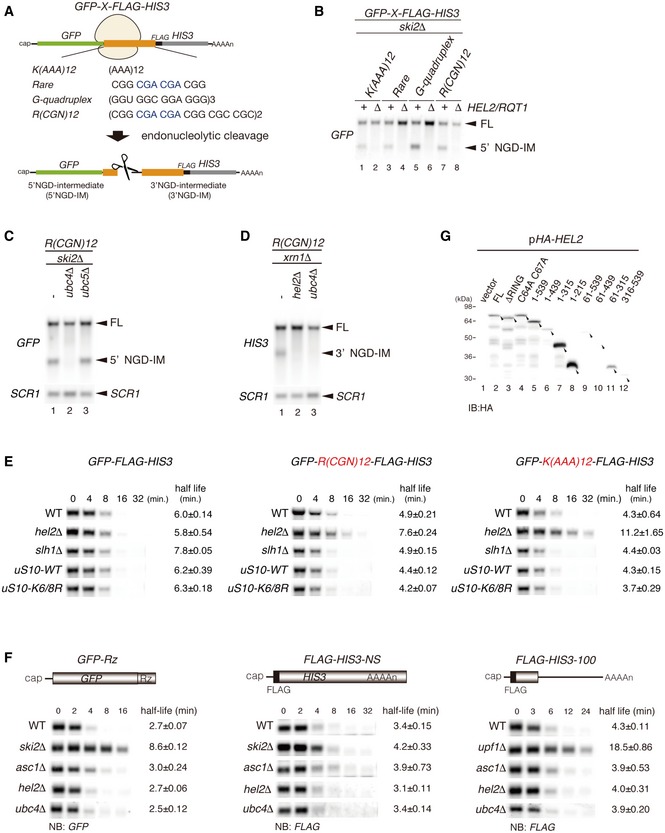
- Schematic drawing of reporter mRNAs containing various arrest‐inducing sequences. The box indicates GFP and HIS3 open reading frames (ORFs), and the black line indicates the untranslated region (UTR). Ribosome stalling takes place during translation of the indicated arrest sequences (shown in red) and induces an endonucleolytic cleavage to produce two fragments, the 5′ NGD intermediate (5′ NGD‐IM) and 3′ NGD intermediate (3′ NGD‐IM).
- Northern blot analysis showing that Hel2 is required for endonucleolytic cleavages by R(CGN)12 arrest‐inducing sequences. The GFP‐R(CGN) 12 ‐HIS3 mRNA (FL) and the 5′ NGD‐IM were detected in the indicated strains using a DIG‐labelled GFP probe. SCR1 coding for RNA in the signal recognition particle (SRP) was used as a loading control.
- Northern blot analysis showing that Ubc4 but not Ubc5 is required for endonucleolytic cleavages by R(CGN)12 arrest‐inducing sequences. The GFP‐R(CGN) 12 ‐HIS3 mRNA (FL) and the 5′ NGD‐IM were detected in the indicated strains with the DIG‐labelled GFP probe.
- Northern blotting showing that Hel2 and Ubc4 are required for endonucleolytic cleavages by R(CGN)12 arrest‐inducing sequences. The GFP‐R(CGN) 12 ‐HIS3 mRNA (FL) and the 3′ NGD‐IM were detected in the indicated strains with the DIG‐labelled HIS3 probe.
- Northern blotting showing that Hel2 reduces the half‐life of reporter mRNA containing R(CGN) 12 or K(AAA) 12 arrest sequences. The stability of GFP‐K(AAA) 12 ‐FLAG‐HIS3 and GFP‐R(CGN) 12 ‐FLAG‐HIS3 mRNAs was determined. The GFP‐K(AAA) 12 ‐FLAG‐HIS3 mRNA was stable in hel2 mutant cells than that in wild‐type cells (t 1/2 = 11.2 min for hel2Δ versus t 1/2 = 4.3 min for wild type). The GFP‐R(CGN) 12 ‐FLAG‐HIS3 mRNA was also stable in hel2 mutant cells than that in wild‐type cells (t 1/2 = 7.6 min for hel2 versus t 1/2 = 4.9 min for wild type). In contrast, the stability of GFP‐K(AAA) 12 ‐FLAG‐HIS3 mRNA was essentially the same in hel2 mutant cells (t 1/2 = 6.0 min) and in wild‐type cells (t 1/2 = 5.8 min). This stabilization of the reporter mRNAs that are substrate for NGD in hel2 mutant cells strongly supports the crucial role of Hel2 in NGD. On the other hand, mRNA stability did not change in slh1∆ and uS10‐K6/8R mutant cells.
- Hel2 is dispensable for NSD and NMD. The GFP‐Rz mRNA is a truncated poly(A) tail‐less non‐stop mRNA that is produced by self‐cleavage of hammerhead ribozyme (Rz) and efficiently degraded in the NSD pathway (Tsuboi et al, 2012). The FLAG‐HIS3‐NS mRNA is a poly(A) tail containing nonstop mRNA and subjected to NSD (van Hoof et al, 2002). The FLAG‐his3‐100 mRNA contains premature termination codon and degraded by NMD quality control (Kuroha et al, 2009). The relative levels of reporter mRNAs in the indicated mutant cells were determined using a DIG‐labeled GFP (GFP‐Rz) or 5′ DIG‐labeled‐LNA‐FLAG (FLAG‐HIS3‐NS or FLAG‐his3‐100) probes. The relative levels of each mRNA were normalized to the mRNA level in time 0, which was assigned a value of 100, and SCR RNA levels were used as a loading control for the RNA samples.
- Western blot analysis to check the expression levels of HA‐tagged Hel2 deletion mutant proteins as schematically displayed in Fig 1B using an anti‐HA antibody.
Figure 1. Hel2‐mediated K63‐linked polyubiquitination is crucial for NGD and RQC.
- Schematic drawing of the R(CGN) 12 reporter mRNA including the two quality control pathways induced by the R(CGN)12 translation arrest sequence. Ribosome stalling occurs during translation of the R(CGN)12 arrest sequence (shown in orange) and induces RQC and NGD. In the RQC pathway, the stalled ribosome is dissociated into subunits, and peptidyl‐tRNA remaining on the 60S subunit is ubiquitinated by Ltn1 (shown in pink) and degraded by the proteasome. In the NGD pathway, an endonucleolytic cleavage produces two fragments, the 5′ NGD intermediate (5′ NGD‐IM) and 3′ NGD intermediate (3′ NGD‐IM). The green and thin grey lines indicate GFP and HIS3 open reading frames (ORFs), and the black line indicates an untranslated region (UTR).
- Schematic drawing of the truncated Hel2 mutant proteins. Activities in RQC or NGD induced by the R(CGN)12 sequence are indicated.
- Western blot showing that Hel2(1–315) is defective in RQC but not in NGD. The arrest products derived from the R(CGN)12 reporter in ltn1Δ cells expressing truncated Hel2 mutant protein were detected with an anti‐GFP antibody.
- Northern blot showing the 5′ NGD‐IM derived from the R(CGN)12 reporter in ski2Δ cells expressing the indicated Hel2 mutant proteins. 5′ NGD‐IMs were detected with a DIG‐labelled GFP probe.
- Primer extension mapping of 5′ ends of 3′ NGD intermediates in Hel2‐WT or Hel2(1–315) mutant cells at nucleotide resolution. The primer extension samples were analysed using 5% TBE‐Urea‐PAGE and detected by fluorescence. Non‐specific reverse transcription (ReTr) products are indicated by asterisks.
- Dissection of NGDRQC+ and NGDRQC−: the carboxyl‐terminal region of Hel2 is required for both NGD and RQC which is likely triggered on a disome unit (pale yellow). It contains the primarily stalled, leading ribosome followed the colliding ribosome. For NGDRQC+, cleavages occur on mRNA covered by the disome, whereby the leading ribosome undergoes RQC. In the mutant Hel2 lacking the C‐terminus, an alternative NGD pathway takes place (NGDRQC−). Here, cleavages occur on mRNA covered by the ribosomes following the disome unit (blue) and the leading ribosome is not affected by RQC (grey).
While the 5′ NGD‐IM in ski2∆ mutant cells could be readily detected, it was absent in hel2∆ski2∆ cells (Fig EV1B) and ubc4∆ski2∆ cells (Fig EV1C). This indicates that Hel2 and its E2 ubiquitin‐conjugating enzyme Ubc4 are indeed required for endonucleolytic mRNA cleavage in various arrest‐inducing sequences and subsequent mRNA decay. Consistently, 3′ NGD‐IMs, which are degraded by the 5′–3′ exonuclease Xrn1, were also not detected in either hel2∆xrn1∆ or ubc4∆xrn1∆ cells (Fig EV1D). Moreover, the full‐length reporter mRNAs containing R(CGN)12 and K(AAA)12 arrest sequences were stabilized in hel2∆ mutant cells (Fig EV1E). Notably, we observed that the half‐lives of the NGD reporters in the hel2Δ mutant cells were longer than in wild‐type cells. This confirms the crucial role of Hel2 in NGD. In contrast to the observed Hel2 dependency of these analysed NGD scenarios, we further confirmed that Hel2 and Ubc4 were not involved in either NSD or NMD (Fig EV1F). Taken together, we show that Hel2 is not only required for degradation of the aberrant peptide by the RQC system, but also for the decay of aberrant mRNA by the NGD pathway.
To obtain a more detailed insight into which parts of Hel2 are involved in NGD and RQC, we introduced a series of deletions in Hel2 (Fig 1B). Hel2 contains an N‐terminal RING domain (61–109), three C2H2‐type ZnF domains (222–307) and a proline‐rich motif at the C‐terminus (Fig 1B). Our deletions comprised N‐terminal deletions including or lacking the RING domain (61–109), which is essential for target ubiquitination, and several C‐terminal deletions of the presumably functionally important ZnF domains. After checking the expression levels of mutant Hel2 proteins (Fig EV1G), we determined the levels of 5′ NGD‐IMs in the ski2∆ background and the protein arrest products in the ltn1∆ background to estimate the efficiencies of NGD and RQC. Both RQC and NGD were effective in the cells expressing the Hel2(1–539) and Hel2(61–539) mutant proteins (Fig 1C, lanes 9–10 and 17–18; Fig 1D, lanes 5 and 9) indicating that the ultimate N‐ and C‐termini of Hel2 are not required for its function. However, neither the 5′ NGD‐IM nor the peptide arrest products accumulated in the hel2 mutants carrying deletions of the RING domain (ΔRING) or alanine substitution of the conserved zinc finger (ZnF) cysteine residues (C64A, C67A) within the RING domain indicating a loss of function.
Interestingly, we found two mutants, Hel2(1–315) and Hel2(61–315), which were only defective in inducing RQC but not NGD. Moreover, this singular function in NGD seemed to be inhibited by the Hel2 region comprising residues 316–439. In fact, these mutants enabled us to investigate a potential NGD‐specific function of Hel2 (Fig 1C, lanes 13–14 and 21–22; Fig 1D, lanes 7 and 11). Intriguingly, the length of 5′ NGD intermediates produced by Hel2(1–315)‐associated endoribonucleolytic cleavages was altered and the cleavage sites shifted upstream compared to the normal condition (Fig EV2). To determine the precise endonucleolytic mRNA cleavage sites induced by Hel2 and Hel2(1–315) mutant protein, we mapped the 5′ ends of 3′ NGD‐IMs derived from the R(CGN) 12 reporter mRNA in the xrn1∆ background by primer extension experiments. The 5′ ends of 3′ NGD‐IMs were determined with a fluorescence‐labelled primer that hybridized to the region of the FLAG‐encoding mRNA sequence (Fig 1A and E). Thereby, we could map four Hel2‐dependent cleavage sites (X1–X4; Fig 1E, lanes 6–7). Our previous study demonstrated that the ribosome is stalled mainly at positions R2(CGA) and R3(CGA) of the R(CGN)12 sequence in P‐ and A‐sites, respectively, and subjected to RQC (Matsuo et al, 2017), indicating that the X1 cleavage site is located in the P‐site of the stalled ribosome. The X2–X4 cleavage sites were approximately 26–36 nt upstream from the X1 cleavage site (X2–X4 in red arrowheads, in Fig 1E and F). Interestingly, in cells expressing the Hel2(1–315) mutant, X1 cleavage was minor and X2–X4 cleavages were almost absent, suggesting that the 316–639 region of Hel2 is important for these cleavage events (Fig 1E, lane 9). To our surprise, this mutant produced alternative major endonucleolytic cleavage sites instead, which occurred more than 45‐51 nt upstream of the X1 cleavage site (blue arrowheads, lane 9 in Fig 1E). This indicates that the X1–X4 cleavage sites are not preferentially used in the Hel2 knockout or the Hel2(1–315) mutant cells and that the cleavage sites could be protected by the stalled ribosomes in RQC‐deficient cells. The leading ribosome would be stalled covering the X1 site, and the colliding ribosome would thereby cover X2–X4.
Figure EV2. Alteration of mRNA cleavage sites in Hel2(1–315) expressing cells.
-
A, BNorthern blot showing that the length of 5′ NGD intermediate was altered in Hel2(1–315) expressing cells and slh1∆ cells. The full‐length GFP‐R(CGN) 12 ‐FLAG‐HIS3 mRNA and 5′ NGD intermediates (5′ NGD‐IM) or 3′ NGD intermediates (3′ NGD‐IM) were detected in the indicated mutant cells with expression of Hel2 wild‐type or 1–315 mutant from plasmid by Northern blotting with DIG‐labelled probes. 5′ NGD intermediates were detected by DIG‐labelled GFP probe in (A), and 3′ NGD intermediates were detected by the DIG‐labelled HIS3 probe in (B). SCR1 is used as loading control. FL; full length. Note an upstream shift of NGD cleavage sites in lanes A3‐4 and B3‐4.
We have previously described an interesting phenotype of sensitivity to the anisomycin translation elongation inhibitor in RQC‐deficient cells (Matsuo et al, 2017). Intriguingly, Hel2 mutants that were unable to trigger RQC (1–315, 61–315, 1–439 and 61–439) remained sensitive to anisomycin, whereas RQC‐competent Hel2 mutants (1–539, 61–539) were not susceptible to this drug (Appendix Fig S1). Anisomycin binds in the ribosomal A‐site and most likely prevents tRNA accommodation, which leads to stalling and ribosome collisions. Thus, it could trap ribosomes stalled in a state which occurs during RQC (presumably a disome) and increase RQC turnover demand.
Based on these results, we hypothesized that at least two adjacent ribosomes as a disome unit, rather than a single‐stalled 80S ribosome, would serve as a minimal control hub on which RQC and NGD are induced after translation arrest. In this scenario, ribosomal collision and subsequent oligoribosome formation could be recognized as a signal of translational arrest, which is in complete agreement with earlier concepts and data by Simms and colleagues (Simms et al, 2017). Further supporting this idea, the observed cleavage pattern occurring in the presence of the RQC‐deficient Hel2(1–315) mutant, with a putative disome unit protecting the canonical cleavage sites (X1–X4), precisely positioned the alternative cleavage sites between following colliding ribosomes (Fig 1F). Together, this allowed us to dissect NGD of mRNA occupied by ribosomes into two branches: the first branch occurs on the primary stalled disome unit with NGD coupled to the RQC response and is dependent on the Hel2 C‐terminal and RING domains (NGDRQC+). The second branch affects the ribosomes colliding with the disome, with NGD uncoupled from RQC, and is only dependent on the Hel2 RING domain (NGDRQC−, Fig 1F).
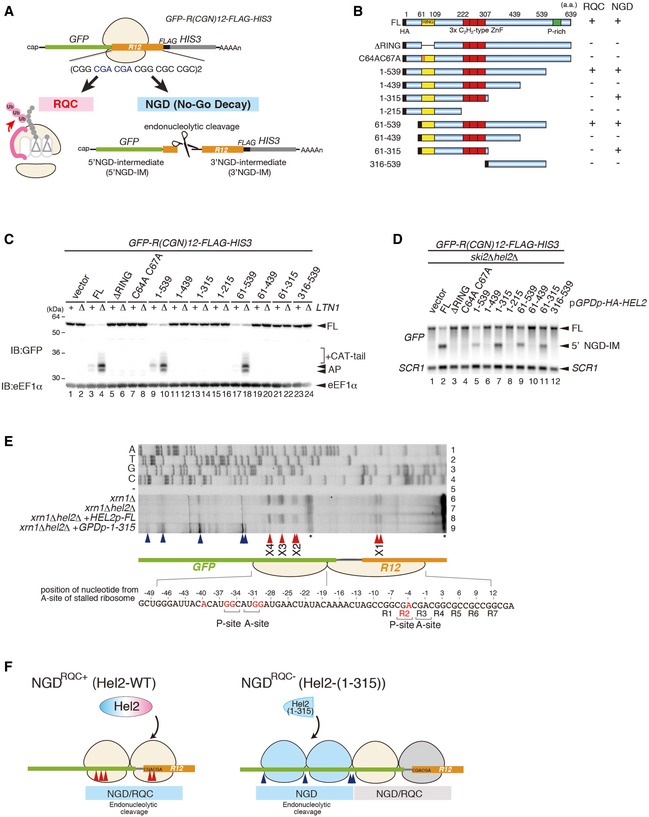
Both uS10 ubiquitination and Slh1/Rqt2 are required for NGDRQC+
We further characterized the role of Hel2 in ribosomal protein ubiquitination by first checking which mode of ubiquitination is employed for NGD. Notably, the role of K63‐linked polyubiquitination has been implicated in translation arrest and RQC before (Saito et al, 2015). To examine whether K63‐linked ubiquitin chains are also critical for NGD, we used ub‐K63R mutant cell strain in which all endogenous ubiquitin‐encoding genes are modified. As a result, only the K63R mutant ubiquitin is expressed. The level of 5′ NGD‐IM derived from the R(CGN) 12 reporter mRNA was measured from an isogenic UB‐WT strain with ski2 deletion, and from the ubiquitin mutant ski2∆ub‐K63R cells. The level of 5′ NGD‐IM was significantly diminished in ski2∆ub‐K63R mutant cells (Fig 2A), indicating that endonucleolytic cleavage of mRNA in NGD required K63‐linked ubiquitination. We also re‐investigated the role of K63‐linked ubiquitination in RQC. The arrest products derived from the R(CGN) 12 reporter are substrates for Ltn1‐dependent degradation, and the level of the arrest product in ltn1∆ mutant cells is an indicator of RQC efficiency. The arrest product was detected in ltn1∆UB‐WT cells, but almost entirely diminished in ltn1∆ub‐K63R mutant cells (Fig 2B). These results indicate that K63‐linked polyubiquitination is required for both NGD and RQC quality control pathways.
Figure 2. Both uS10 ubiquitination and Rqt2 are required for NGDRQC+ .
-
ANorthern blot analysis demonstrating that K63‐linked ubiquitination was required for an endonucleolytic cleavage by R(CGN)12. The ski2ΔUB‐WT and ski2Δub‐K63R cells were transformed with the R(CGN) 12 reporter, and total RNA samples were separated by 2% agarose/MOPS gel. 5′ NGD‐IMs in the cells were detected as in Fig 1D.
-
BWestern blot showing that K63‐linked ubiquitination was required for RQC. Protein samples from UB‐WT, UB‐WT ltn1Δ, ub‐K63R and ub‐K63R ltn1Δ cells expressing the GFP‐R(CGN) 12 ‐HIS3 reporter were subjected to Western blot analysis using an anti‐GFP antibody to detect the arrest products. Note the accumulation of RQC‐specific CAT‐tails in lane 2.
-
CWestern blot analysis demonstrating polyubiquitination of uS10: affinity‐tagged Hel2‐Flag‐TEV‐Protein A (FTP) was co‐purified with ribosomes harbouring HA‐tagged wild‐type uS10 (uS10‐3HA ribosomes) or uS10 mutated in its ubiquitination sites (K6/8R). Western blots of whole protein extracts were performed using an anti‐HA antibody to detect polyubiquitinated uS10‐3HA.
-
DWestern blot analysis showing that uS10‐polyubiquitination in Hel2‐bound ribosomes was mainly K63‐linked: affinity‐tagged Hel2 (Hel2‐ProteinA‐TEV‐His6; PTH‐Hel2) was co‐purified with ribosomes from a strain expressing wild‐type ubiquitin (UB‐WT) or K63R mutant ubiquitin (ub‐K63R) and HA‐tagged uS10 (uS10‐3HA). Western blot analysis of whole protein extracts was performed using anti‐HA antibody.
-
EWestern blot of in vitro polyubiquitination assays of uS10. Reactions were performed with the indicated components including His‐tagged ubiquitin and several ubiquitin mutants. Polyubiquitinated HA‐tagged uS10 (uS10‐3HA) was detected by Western blot analysis using an anti‐HA antibody.
-
F–HNorthern blot analysis of NGD cleavage sites in the absence of uS10 ubiquitination or RQT complex components: the full‐length GFP‐R(CGN) 12 ‐FLAG‐HIS3 mRNA and 5′ NGD intermediates (5′ NGD‐IM) or 3′ NGD intermediates (3′ NGD‐IM) were detected in the indicated mutant cells by Northern blotting with DIG‐labelled probes. 5′ NGD intermediates were detected by DIG‐labelled GFP probe, and 3′ NGD intermediates were detected by the DIG‐labelled HIS3 probe. SCR1 was used as loading control. FL = full length. Note the upstream shift of NGD cleavage sites in lanes F4, G4, H6 and H8.
-
IMapping of 5′ ends of 3′ NGD intermediates as demonstrated in Fig 1E. Non‐specific reverse transcription (ReTr) products are indicated by asterisks. Note that both uS10 ubiquitination and Slh1/Rqt2 were required for an endonucleolytic cleavage within the road‐blocked ribosome (X1–X4).
-
JModel for RQC‐coupled (NGDRQC+) and RQC‐uncoupled (NGDRQC−) endonucleolytic cleavages in the R(CGN)12 reporter mRNA. Endonucleolytic cleavage sites are depicted corresponding to panel (I).
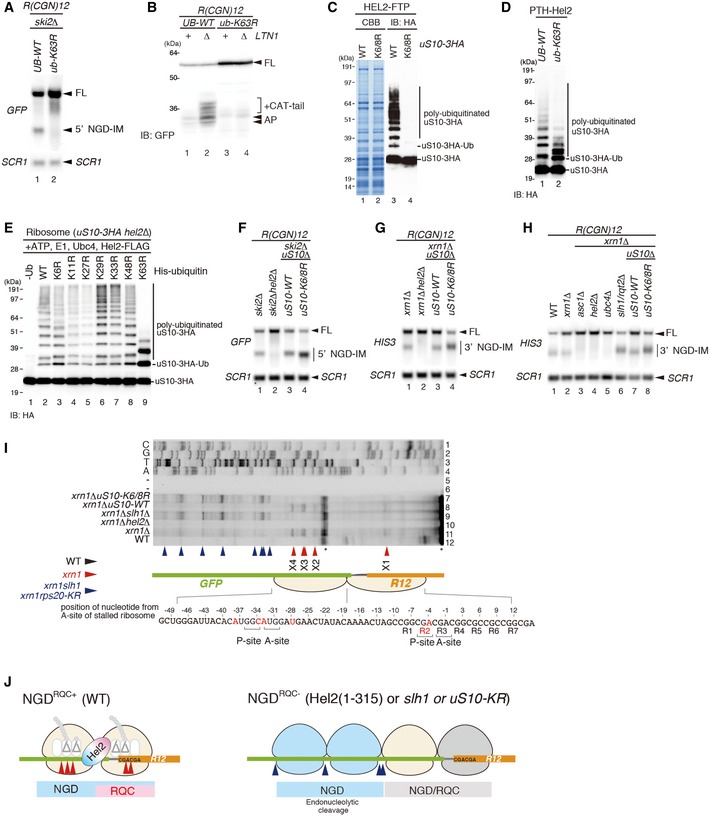
Ribosomal proteins uS10 and uS3 were identified as substrates for Hel2‐dependent ubiquitination at lysines K6 and K8 (uS10) and K212 (uS3) (Matsuo et al, 2017). Therefore, we investigated the roles of Hel2‐dependent uS10 and uS3 ubiquitination in NGD. Although the role of uS3 polyubiquitination has been implicated in NGD before (Simms et al, 2017), the ubiquitination of uS3 by Hel2 was dispensable for endonucleolytic cleavages induced by the R(CGN) 12 sequence (Appendix Fig S2A and B). Therefore, we focused on the role of uS10 in NGD and determined the efficiency of Hel2‐mediated polyubiquitination of uS10. Western blot of the purified Hel2‐bound ribosomes from yeast cells expressing C‐terminally FLAG‐TEV‐Protein A (FTP)‐tagged Hel2 and C‐terminally HA‐tagged uS10 revealed that uS10 was polyubiquitinated in the uS10‐WT cells but not in uS10‐K6R K8R mutant cells (Fig 2C). More than tri‐ubiquitinated forms of uS10 were significantly decreased in ub‐K63R mutant cells, indicating that the polyubiquitination of uS10 by Hel2 is mainly K63‐linked (Fig 2D). We confirmed that this reduction in ub‐K63R mutant cells was not due to a reduced Hel2 level (Appendix Fig S2C). We then reconstituted Hel2‐mediated polyubiquitination of uS10 in an in vitro ubiquitination assay using purified Hel2, Ubc4, uS10‐3HA‐tagged ribosomes, E1, ATP and ubiquitin. In addition, we used ubiquitin mutants bearing lysine to arginine substitutions (KR) that cannot form corresponding polyubiquitin chains. These assays confirmed that Hel2‐mediated polyubiquitination of uS10 is mainly K63‐linked (Fig 2E), and K6 or K8 residues of uS10 represent the target ubiquitination sites (Appendix Fig S2D).
We next examined whether NGD requires Hel2‐mediated ubiquitination of uS10. Northern blot analysis revealed that the length of 5′ NGD‐IM in ski2∆uS10‐K6/8R mutant cells (Fig 2F, lane 4) was shorter than in ski2∆uS10‐WT mutant cells (Fig 2F, lane 3). Consistently, the length of 3′ NGD‐IM in the xrn1∆uS10‐K6/8R and xrn1∆slh1/rqt2∆ mutant cells was longer than in xrn1∆uS10‐WT mutant cells (Fig 2G and H), suggesting that the cleavage sites were shifted upstream in these mutants in the same manner as in the Hel2(1–315) mutant (Fig EV2). The mapping of the 5′ ends of 3′ NGD‐IMs derived from the R(CGN) 12 reporter mRNA by primer extension revealed that the X1–X4 cleavage sites were eliminated in xrn1∆hel2∆ and xrn1∆uS10‐K6/8R mutant cells (Fig 2I). A similar cleavage pattern was observed using a knockout of Slh1/Rqt2 (xrn1∆slh1/rqt2∆ cells). As observed before with the Hel2(1–315) mutant, the endonucleolytic cleavages efficiently took place upstream of the putative disome in uS10‐K6/8R and slh1/rqt2∆ mutant cells (blue arrowheads, lanes 7 and 9 in Fig 2I). Together, these data suggested that the endonucleolytic cleavages in NGDRQC+‐mRNA protected by the colliding ribosomes require the Hel2 C‐terminus, the Hel2‐RING domain‐mediated ubiquitination of uS10 and the Slh1/Rqt2 protein (Fig 2J).
uS10 is efficiently ubiquitinated in the stalling and colliding ribosomes
To assess the effect of ribosome collision on the uS10 ubiquitination, we performed in vitro translation and monitored uS10 ubiquitination. We generated a translation extract from ski2∆uS10‐3HA cells and programmed it with a mRNA containing the previously reported stalling dicodon CGA‐CCG (Gamble et al, 2016; Matsuo et al, 2017) and a sequence coding for an N‐terminal tag for affinity purification (Fig 3A). This dicodon triggered both RQC and NGDRQC+ when tested in vivo (Matsuo et al, 2017, Fig 3B and C), and peptidyl‐tRNA, the indicator for the stalled ribosome, was clearly observed after in vitro translation (Fig 3D). The stalled ribosome–nascent chain complexes (RNCs) were affinity‐purified via the N‐terminal His‐tag on the nascent chain and separated by sucrose density gradient centrifugation. As expected, we observed mono‐ and disomes in the gradient. Consistent with our model, uS10 was more efficiently ubiquitinated in the disome fraction compared to the monosome fraction (Fig 3E). This suggests that the disome as a minimal ribosome collision unit is preferred as a substrate for Hel2‐dependent uS10 ubiquitination over stalled monosomes.
Figure 3. uS10 in colliding ribosome is efficiently ubiquitinated.
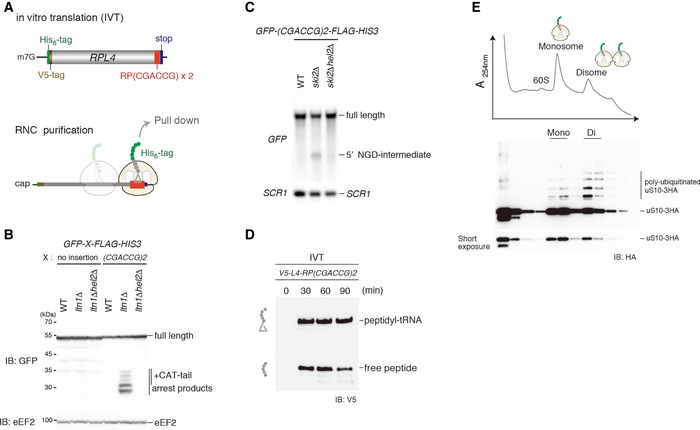
-
ATop: schematic drawing of the (CGA‐CCG) dicodon reporter mRNA used for in vitro translation experiments. Bottom: scheme outlining the principle of ribosome–nascent chain (RNC) purification. Stalled and colliding ribosomes are pulled down via an affinity tag on the nascent chain.
-
B, CBoth RQC and NGDRQC+ are triggered by a (CGA‐CCG) dicodon containing arrest sequence in vivo. (B) Western blot showing the arrest products derived from the GFP‐X‐FLAG‐HIS3 reporter. Translation products were detected by Western blot using an anti‐GFP antibody. (C) Northern blot for the 5′ NGD‐IM derived from the (CGA‐CCG) reporter in ski2Δ cells. 5′ NGD‐IMs were detected with a DIG‐labelled GFP probe. SCR1 was used as a load control.
-
DWestern blot of test translations using the (CGA‐CCG) dicodon stalling mRNA reporter shown in (A). The mRNA reporter was added to a yeast in vitro translation extract obtained from a ski2ΔuS10‐3HA strain. Expression of the translation products (free His‐ and V5‐tagged truncated uL4 protein and the same protein attached to tRNA) was visualized with an anti‐V5 antibody.
-
ESucrose gradient fractions (top) and Western blot analysis (bottom) of the (CGA‐CCG) dicodon‐stalled RNC samples. After the translation reaction, the RNCs were affinity‐purified as indicated in (C). The eluate was loaded on a 10–50% sucrose gradient and fractionated. Each collected fraction was analysed using anti‐HA antibody to detect uS10‐HA. Note that disomes are preferentially polyubiquitinated over monosomes.
Structural analysis of an NGD and RQC substrate disome unit
On the basis of the concept that collision of at least two ribosomes is required to trigger initiation of RQC and NGDRQC+, we asked whether such a ribosome collision would lead to a clearly defined and composite structural interface that could serve as a hub for molecular recognition by Hel2. To that end, we used cryo‐electron microscopy (cryo‐EM) to obtain structural information of a disome as a minimal collision unit formed on a bona fide RQC and NGDRQC+ inducing mRNA substrate. We scaled up the cell‐free translation reaction using the CGA‐CCG stalling reporter mRNA as described above and harvested the disome peak for cryo‐EM analysis (Appendix Fig S3). As expected from previous datasets (Schmidt et al, 2016), translating ribosome populations represent various translational states with respect to tRNA occupancy and intersubunit rotation. To account for this heterogeneity and to separate intact disomes from dissociated ones (80S monosomes), several 3D classification steps were performed resulting in a set of densities representing stably associated disomes. All disomes turned out to have a very similar overall orientation with their small subunits oriented towards each other in order to interact in an apparently somewhat flexible manner. The most stable disome structure adopted the most compact overall conformation permitting a 5.3 Å average resolution and docking of molecular models (Fig 4A and B; Appendix Fig S4, S5A and S6; Schmidt et al, 2016; Ben‐Shem et al, 2010). The two ribosomes interacted via proteins and rRNA of their small subunits and were bridged by a clearly visible mRNA density (Fig 4). In this structure, we identified the ribosome in the POST‐state with P‐ and E‐site tRNAs as the primary stalled ribosome. This was possible by structural analysis of the monosome fraction from the same sucrose gradient, which revealed that the vast majority of stalled primary 80S monosomes (before any collision) is adopting this particular state (Appendix Fig S5B). In contrast, the colliding ribosome was adjacent to the mRNA exit site of the leading ribosome and was stalled in the rotated state with hybrid A/P and P/E tRNAs exclusively (Fig 4C). The presence of these hybrid states and incomplete translocation of mRNA and tRNAs is consistent with the colliding ribosome being incapable of translocating further down on the mRNA after engaging the primary stalled ribosome as a roadblock.
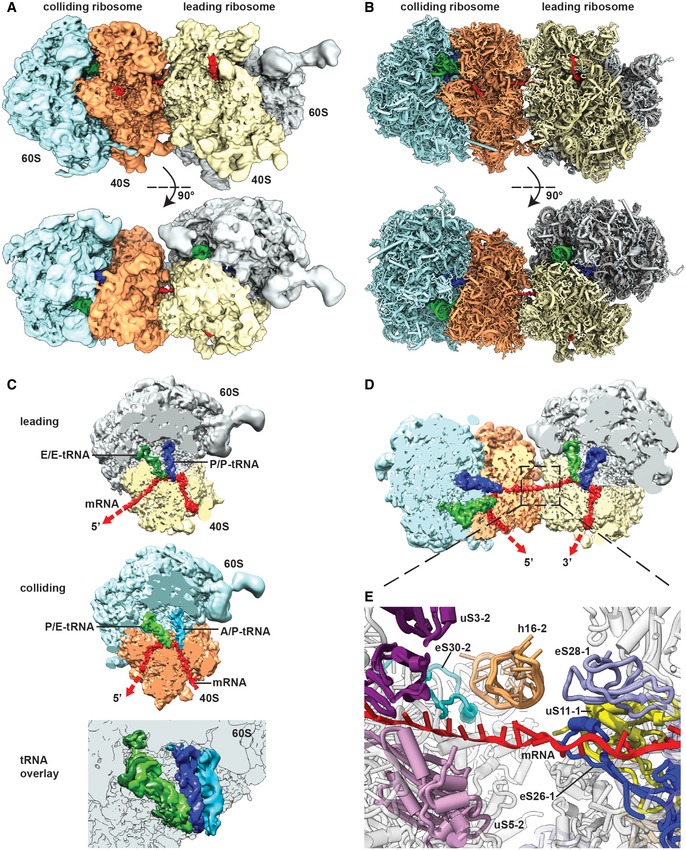
Figure 4. Cryo‐EM structure and molecular model of a stalled disome.
-
A, BSide view and top view of the Cryo‐EM reconstruction (A) and molecular model (B) of the disome stalled on a (CGA‐CCG) reporter mRNA. The first ribosome in the disome stalled on the dicodon mRNA is referred to as the leading ribosome and the second as the colliding ribosome.
-
CCut top views on the leading and colliding ribosome. The leading ribosome is in a non‐rotated POST‐state containing tRNAs in P/P and E/E states and an empty A‐site, whereas the colliding ribosome is in a rotated state containing hybrid A/P and P‐/E‐site tRNAs. An overlay is shown omitting the 40S subunit for clarity.
-
DCut top view on the disome with focus on the mRNA density highlighted in red.
-
EZoom on the mRNA at the interface of the leading and colliding ribosome. The mRNA exits the leading ribosome close to ribosomal proteins uS11, eS26 and eS28 and enters the colliding ribosome near h16, uS3, uS5 and eS30.
We observed a density for the entire mRNA within the disome and could model 60 nucleotides (nt) in total (Figs 4D and E, and EV3A). Both the leading and the colliding ribosome each cover approximately 30 nt long stretches of the template mRNA. At the mRNA exit of the leading ribosome and the mRNA entry of the colliding ribosome, the mRNA is represented by a tube‐like density. The mRNA path straightens at the mRNA exit (leading ribosome; nt 31–35) and entry sites (colliding ribosome; nt 25–30) (Fig EV3A) and is confined between the two ribosomes in a narrow channel formed by eS26, eS28 and uS11 (leading ribosome) and rRNA helix h16, uS2, uS3 and eS30 (colliding ribosome) (Fig 4E). Notably, this mRNA appears rather inaccessible for medium‐sized proteins such as endonucleases. This might explain why NGDRQC− occurs outside this compact structural when the RQC pathway is impaired. Moreover, it is tempting to speculate that Rqt factors such as Hel2 and the ATP‐dependent helicase Slh1/Rqt2 are required for a rearrangement or dissociation of the disome structure in order to efficiently trigger RQC and NGDRQC+.
Figure EV3. Conformational rearrangements in the disome.
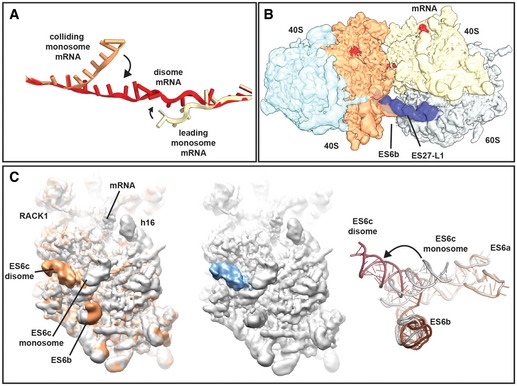
- mRNA was modelled into the refined maps of the hybrid A/P‐P/E tRNA containing monosome and the P/P tRNA containing monosome with ES27 in the L1 conformation (see Appendix Figs S4 and S6). These ribosomes are not involved in disome formation and show density for mRNA in covering approximately 30 nucleotides in the canonical path. In the disome, the mRNA deviates from the canonical path at the exit site of 5′ mRNA and at the entry site of the 3′‐mRNA in the colliding ribosome. In the disome, the corresponding nucleotides are stretching out to connect leading and colliding ribosome.
- Superposition of the disome map shown as in Fig 4A with the isolated ES27 density from the refine monosome sub‐class representing the P/P tRNA containing ribosome with ES27 in the L1 position (blue). Note that ES27 would clash with the 60S‐40S disome bridge involving ES6b.
- Left panel: superposition of the refined colliding ribosome from the disome (yellow orange) with the hybrid A/P‐P/E tRNA containing monosome (grey; see Appendix Figs S4 and S6). The view focuses on ES6. Middle panel: superposition of hybrid A/P‐P/E tRNA containing monosome with the difference map. Note that the only significant difference is in ES6c. Right panel: superposition of molecular models for ES6 in a disome (dark red/brown) and in a monosome (grey).
The large contact interface between the two 80S ribosomes is formed such that the mRNA exit site of the leading ribosome and the entry site of the colliding ribosome are in close proximity. The interface involves mainly the small but to a lesser extent also the large subunit and involves many eukaryote‐specific ribosomal proteins and rRNA expansion segments. The interface can be subdivided into four major contacts regions (Fig 5A), which are (i) a 40S head‐to‐head contact between RACK1 and the C‐terminal tail of uS3 of the leading ribosome with RACK1 and uS10 in the colliding ribosome (Fig 5B), (ii) a 40S body‐to‐body contact formed by eS27 and eS7 of leading ribosome with expansion segment ES6c of the colliding ribosome (Fig 5C), (iii) a 40S platform‐to‐platform contact via eS17 with uS2 and eS21 of the colliding ribosome (Fig 5D) and (iv) dual 60S‐40S contacts between eL27 of the leading with ES6b of the colliding ribosome, as well as ES31L with the C‐terminal helix of eS4 (Fig 5E). Notably, major conformational changes occur in rRNA expansion segments (ES) upon disome formation in both ribosomes compared to the 80S reconstructions derived from the individual 80S ribosomes (Appendix Fig S4). ES27L is a flexible rRNA expansion segment capable of adopting two main conformations in the yeast 80S, one facing towards the peptide exit tunnel (ES27 exit) and the other one facing towards the flexible L1 stalk (ES27‐L1, (Beckmann et al, 2001). In the leading ribosome, we observed ES27L exclusively in the exit position, most likely because ES27L in the L1 position would directly clash with the tip of ES6b of the colliding ribosome forming the 60S‐40S contact site (Fig EV3B). In addition, in the second ribosome, the rRNA expansion segment ES6c rearranges upon disome formation to accommodate the disome interface. Compared to the monosome (Appendix Fig S4), ES6c rearranges at the 3‐way junction connecting ES6a, ES6b and ES6c, resulting in an approximately 90‐degree rotation of ES6c towards the disome interface (Fig EV3C).
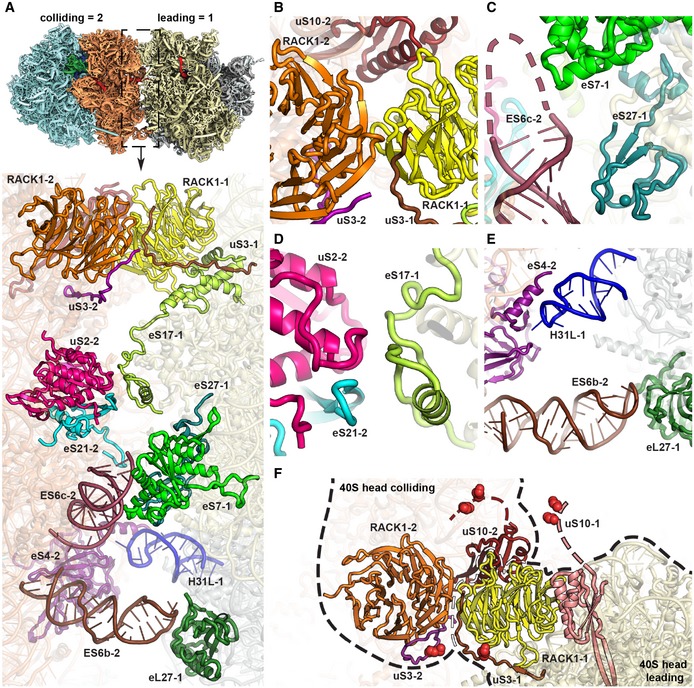
Figure 5. The disome interface and location of ubiquitination sites.
-
AOverview of the contact interface. The thumbnail on top indicates the orientation.
-
B–EZoom views on the 40S head‐to‐head contact site (B), the 40S platform‐to‐platform contact site (C), the 40S body‐to‐body contact site (D) and the 60S‐to‐40S contact site (E). In (D), nucleotides missing in the model at the tip of ES6c are indicated by a dashed line.
-
FLocation of uS3 (K212) and uS10 (K6 and K8) ubiquitination sites. All ubiquitination sites are in the close spatial vicinity highlighted with red spheres. RACK1 proteins from both the leading and the colliding ribosome form the major head‐to‐head contact bringing together both C‐terminal tails of uS3s and N‐termini of uS10s. For the uS10 ubiquitination sites, only an approximate position is shown as indicated by dashed lines since the uS10 N‐termini containing K6 and K8 are invisible in the structure most likely due to flexibility.
Most interestingly, the observed disome assembly on this RQC and NGDRQC+ inducing reporter mRNA created a unique composite surface generated by both the leading and the colliding ribosome, which may serve as a molecular recognition unit for Hel2 (Fig 5F). In agreement with this idea, the composite surface brings important RQT players together in a relatively small area: the RACK1 proteins of the two small subunits, which have been shown to be required for RQC and NGDRQC+, directly interact and stabilize the disome. In addition, the Hel2 targets uS3 and uS10 are also in close vicinity. This suggests that colliding disome formation could indeed provide a distinct composite structural platform for molecular recognition of stalling by Hel2, resulting in subsequent modification and trigger of RQC and NGDRQC+.
Hel2 forms K63‐linked polyubiquitin chains on the Not4 monoubiquitinated eS7A
As shown above, the NGD response can be subdivided by the location of cleavages, occurring within the disome‐covered mRNA or on the mRNA upstream of it. We also found that this second NGDRQC− branch requires the Hel2 RING domain, but is only favoured in the absence of either the C‐terminal domain of Hel2, the entire Slh1/Rqt2 protein or ubiquitination of uS10. To that end, we speculated that for NGDRQC− taking place upstream of the disome, ubiquitination of a different ribosomal protein may play a role. Therefore, we screened a series of 3xHA‐tagged 40S ribosomal proteins by Western blot analysis and identified eS7 as a novel target of Hel2‐mediated ubiquitination (Appendix Fig S7A). Notably, eS7A was previously shown to be a target of the E3 ligase Not4, a component of the Ccr4‐NOT complex (Panasenko & Collart, 2012). We observed that Hel2 overexpression increased polyubiquitination of C‐terminal HA‐tagged eS7A and eS7B as well as of uS10 and uS3 as shown before (Fig 6A). Moreover, an eS7 mutant lacking five lysine residues (K72, K76, K83, K84 and K101) was no longer ubiquitinated by Not4 (Panasenko & Collart, 2012). To further dissect the roles of Not4 and Hel2 ligases in the ubiquitination of eS7A, we constructed eS7A∆eS7B∆, hel2∆eS7A∆eS7B∆ and not4∆eS7A∆eS7B∆ deletion mutant cells harbouring plasmids expressing C‐terminal HA‐tagged wild‐type eS7A (WT) or eS7A‐4KR (4KR) in which four lysine residues (K72, K76, K83 and K84) were substituted with arginine residues (Fig 6B). Interestingly, we found that eS7A was detected in a monoubiquitinated form in the absence of Hel2 (Fig 6C, lane 3), suggesting that Hel2 may facilitate the polyubiquitination of the eS7A, after monoubiquitination by Not4. The mono‐ and polyubiquitinated forms of eS7A were detected in the co‐purified ribosomes with PTH (Protein A‐TEV protease site‐His6)‐tagged Hel2 from eS7A‐WT cells (Fig 6D, lane 4), but disappeared in not4∆eS7A‐HA and eS7A‐4KR‐HA mutant cells (Fig 6D, lanes 5–6).
Figure 6. Hel2 polyubiquitinates the monoubiquitinated eS7A by Not4.
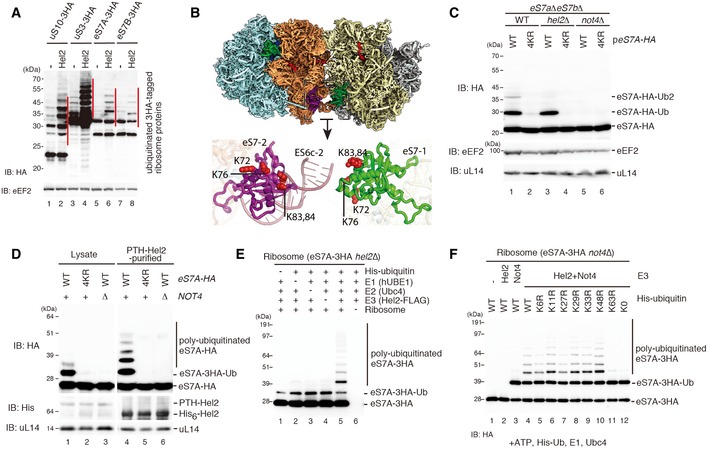
- Western blot analysis after overexpression of Hel2 in strains expressing HA‐tagged ribosomal proteins using an anti‐HA antibody. Note the increase in the polyubiquitinated eS7 as well as uS10 and uS3.
- Structural overview of the ubiquitination sites on eS7. The general overview on the disome interface is depicted according to Fig 4A. Individual ubiquitination sites are highlighted as red spheres in the detailed cartoon model representation.
- Western blot analysis showing the role of Hel2 and Not4 in eS7A ubiquitination: both copies of eS7 (eS7A and eS7B) were deleted and transformed with a plasmid containing HA‐tagged eS7A (eS7‐WT) or eS7A mutated in the four potential ubiquitination sites (4KR). Whole protein extracts we obtained from eS7AΔeS7BΔ cells or cells with an additional deletion in E3 ligases Hel2 and Not4 (hel2Δ and not4Δ). Note that the four mutated lysine residues were responsible for Not4‐dependent monoubiquitination.
- Western blot showing that Not4 was required for Hel2‐mediated polyubiquitination of eS7 in Hel2‐bound ribosomal complexes: cells expressing HA‐tagged eS7A or the eS7‐4KR mutant, as well as PTH‐tagged Hel2 in eS7AΔeS7BΔ and eS7AΔeS7BΔnot4Δ background, were probed for eS7‐ubiquitination. Either cell lysates or affinity‐purified Hel2‐ribosome complexes were used. The levels of the ubiquitinated eS7A in the lysates and in the affinity‐purified samples with PTH‐Hel2 were determined by Western blot analysis using an anti‐HA antibody to detect eS7.
- Western blotting of in vitro ubiquitination assays of eS7A. The reactions were performed with the indicated components including His‐tagged ubiquitin. Ribosomes were purified from Hel2Δ cells expressing HA‐tagged eS7A. Polyubiquitinated HA‐tagged eS7A (eS7A‐3HA) was detected by Western blot analysis using an anti‐HA antibody. We observed Hel2‐mediated polyubiquitination of the monoubiquitinated eS7A.
- Western blot analysis of eS7A in vitro ubiquitination assays showing that Hel2‐mediated polyubiquitination of eS7 was mainly K63‐linked and required Not4‐dependent monoubiquitination: these assays were performed similarly as described in (E) except yeast strains were lacking Not4 (not4Δ). Reactions were performed with the indicated components including His‐tagged ubiquitin and several ubiquitin mutants, and eS7A‐ubiquitination was monitored using an anti‐HA antibody.
To test whether Hel2 polyubiquitinates the Not4‐monoubiquitinated eS7A, we performed an in vitro ubiquitination assay with purified Hel2, Ubc4 and ribosomes from hel2∆eS7A‐3HA mutant cells. Strikingly, the purified eS7A‐3HA ribosomes were found in the monoubiquitinated form, and Hel2 was able to polyubiquitinate monoubiquitinated eS7A (Fig 6E, lane 5). We then affinity‐purified Not4‐FLAG protein together with several components of the Ccr4‐NOT complex (Appendix Fig S7B) and applied it to ribosomes purified from not4∆eS7A‐3HA or not4∆eS7B‐3HA cells. Not4 proved to ubiquitinate both eS7A and eS7B on the ribosome (Appendix Fig S7C). Moreover, Hel2 failed to ubiquitinate eS7A on ribosomes purified from not4∆eS7A‐3HA mutant cells (Fig 6E, lane 2). Instead, Hel2 polyubiquitination of eS7A was entirely dependent on the Not4‐mediated monoubiquitination of eS7 (Fig 6F, lanes 4–12). These results indicate that Hel2 serves as a ubiquitin chain elongation factor for K63‐linked ubiquitination of eS7 and that Not4‐mediated monoubiquitination of eS7A is a prerequisite for this process.
Not4‐dependent ubiquitination is required for NGDRQC− outside the disome
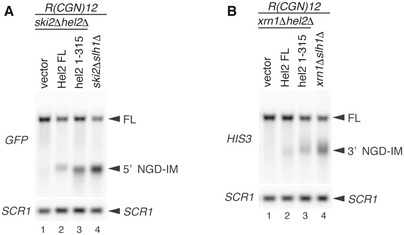
We then examined whether Not4‐dependent ubiquitination of eS7A is required for NGDRQC− in RQC‐defective slh1/rqt2∆ or uS10‐K6/8R mutant cells. The shorter 5′ NGD‐IM derived from the R(CGN)12 reporter was slightly decreased in the ski2∆slh1∆not4∆ cells compared to the ski2∆slh1∆ cells (1.3‐fold decrease; Fig 7A, lanes 3 and 5), and the shorter 5′ NGD‐IMs were also reduced in the ski2∆not4∆uS10‐K6/8R cells compared to that in ski2∆uS10‐K6/8R cells (6.7‐fold increase; Fig 7B, lanes 4 and 3, respectively). These results indicate that Not4 plays an important role in NGDRQC−. We then determined the levels and size of 5′ NGD‐IMs derived from R(CGN) 12 reporter in ski2∆eS7A∆eS7B∆ or ski2∆slh1∆eS7A∆eS7B∆ mutant cells harbouring plasmids expressing either eS7A wild type (WT) or eS7A‐4KR mutant (4KR). The shorter 5′ NGD‐IM in slh1∆eS7A‐WT was produced by NGDRQC−, and its level was higher than the one from NGDRQC+ in eS7A‐WT cells (1.7‐fold increase; Fig 7C, lanes 2 and 1, respectively). Importantly, the level of the shorter 5′ NGD‐IM in the slh1∆eS7A‐4KR mutant was decreased compared to slh1∆eS7A‐WT (3.1‐fold decrease; Fig 7C, lanes 4 and 2, respectively), confirming that Not4‐mediated ubiquitination of eS7A plays a crucial role in the endonucleolytic cleavage. In conclusion, our results lead us to propose that the endonucleolytic cleavages in NGDRQC− outside the disome require step‐wise ubiquitination of eS7A by E3 ligases Not4 and Hel2.
Figure 7. Not4‐dependent ubiquitination of eS7 is crucial for NGDRQC− .
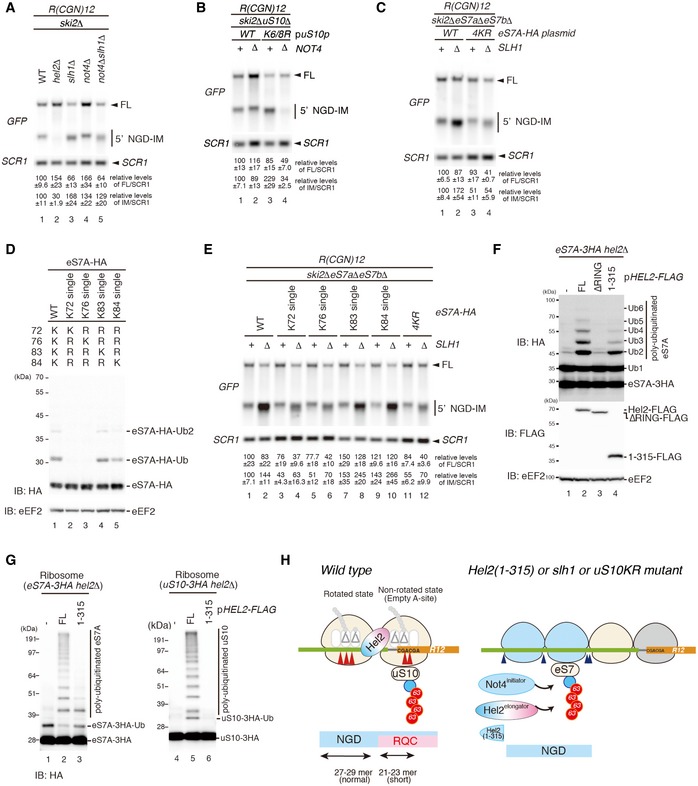
-
A–CNorthern blots probing for the 5′ NGD‐IM in mutant cells expressing the R(CGN)12 reporter. (A) 5′ NGD‐IM detection in not4Δski2Δ, hel2Δski2Δ, slh1Δski2Δ and not4Δslh1Δski2Δ. Quantification of full length and the 5′ NGD‐IM relative to the loading control (SCR1) is given below. Note the size difference of 5′ NGD‐IMs resulting from NGDRQC+ and NGDRQC− and that the shorter 5′ NGD intermediate (representing intermediates of the NGDRQC− pathway) was reduced in not4Δslh1Δski2Δ mutant cells. (B) Northern blot as in (A) except uS10‐WTski2Δ or uS10‐K6/8Rski2Δ mutant cells with or without a deletion of Not4 (not4Δ) was used. Note that the shorter 5′ NGD intermediates are also reduced in ski2Δnot4ΔuS10‐K6/8R cells. (C) Cells expressing HA‐tagged eS7A or the eS7A‐4KR mutant in a ski2ΔeS7AΔeS7BΔ or ski2ΔeS7AΔeS7BΔslh1Δ backgrounds were probed for 5′ NGD intermediates by Northern blotting. Note that the four mutated lysine residues in eS7 were responsible for NGDRQC− in the R(CGN)12 reporter.
-
DWestern blot analysis showing that K83 and K84 of eS7A are main target sites for Hel2 and Not4‐mediated ubiquitination: similarly, as in Fig 6C, eS7AΔeS7BΔ mutant cells were used and wild‐type and single mutants of HA‐tagged eS7A were expressed. Ubiquitinated eS7‐HA was monitored using an anti‐HA antibody.
-
ENorthern Blot analysis probing for the levels of 5′ NGD‐IM derived from the R(CGN)12 reporter expressed in eS7AΔeS7BΔski2Δ cells and HA‐tagged eS7 or eS7 mutants. We observed that ubiquitination at K83 or K84 of eS7A is mainly responsible for NGDRQC−.
-
FThe levels of polyubiquitinated eS7A were increased by the overproduction of wild‐type Hel2 (FL) but neither Hel2ΔRING nor Hel2(1–315) mutants. eS7A‐3HAhel2Δ mutant cells harbouring the indicated plasmids expressing wild‐type Hel2 (FL), Hel2ΔRING or Hel2(1–315) mutant proteins were harvested. Protein samples were analysed by Western blotting with an anti‐HA (top panel) or anti‐FLAG antibody (bottom panel).
-
GHel2(1–315) polyubiquitinates eS7A but not uS10. Western blotting after an in vitro ubiquitination assay using the Hel2(1–315) mutant and purified ribosomes containing HA‐tagged uS10.
-
HModel for quality control pathways induced by R(CGN)12‐mediated translation arrest. If the RQC pathway is intact, the leading ribosome that is stalled by the arrest sequence undergoes RQC. The uS10 ubiquitination and ATPase activity of Slh1/Rqt2‐dependent subunit dissociation induce the endonucleolytic cleavages in the sequence covered by the first and the second ribosome (disome). In the absence of uS10 ubiquitination and Rqt2, RQC in the first ribosome, as well as NGD in the disome, is eliminated. Instead, RQC‐uncoupled NGDRQC− takes place upstream of the disome. The carboxyl‐terminal region of Hel2 is crucial to induce RQC thereby NGD in the disome (NGDRQC+), but dispensable for RQC‐uncoupled NGD (NGDRQC−).
To identify the exact sites of modification, we determined the levels of ubiquitinated eS7A in the eS7A∆eS7B∆ cells expressing eS7A‐HA (WT) or eS7A‐HA mutants with single K72, K76, K83 or K84 residues in four putative ubiquitination sites (eS7A‐K72, K76, K83 and K84 single). We found that the levels of the ubiquitinated eS7A in eS7A‐K83 single or eS7A‐K84 single mutants were almost the same as that in the WT (Fig 7D, lanes 1, 4 and 5). The ubiquitination of eS7A was nearly eliminated in eS7A‐K72 or eS7A‐K76 single mutant cells (Fig 7D, lanes 2–3). Subsequently, we investigated the efficiencies of NGDRQC− in eS7A single mutants. The levels of 5′ NGD‐IM in eS7A‐K83 single or eS7A‐K84 single mutant cells were slightly increased than that in eS7A‐WT cells, indicating that the ubiquitination of K83 or K84 residues of eS7A is sufficient for NGDRQC− (Fig 7E, lanes 1–2 and 7–10). The levels of 5′ NGD‐IM were decreased in eS7A‐K72 or eS7A‐K76 single mutant cells as well as eS7A‐4KR mutant cells, indicating that the level of ubiquitinated eS7A correlates with the efficiency of NGDRQC− (Fig 7D and E, lanes 3–6, 11–12). Taken together, we propose that step‐wise ubiquitination of eS7A at residues K83 or K84, first Not4‐mediated monoubiquitination followed by Hel2‐mediated K63‐linked polyubiquitination, plays a crucial role in NGDRQC− (Fig 7H).
Given that the Hel2(1–315) mutant protein has an ability to induce NGDRQC− but not RQC, we suspected that the Hel2(1–315) mutant protein ubiquitinates the monoubiquitinated eS7 but not uS10. As expected, eS7A was polyubiquitinated by the overexpression of FLAG‐tagged full‐length Hel2(FL) or Hel2(1–315), but not by Hel2ΔRING (Fig 7F, lanes 2–4). In vitro ubiquitination reactions revealed that full‐length Hel2(FL) polyubiquitinated both eS7A and uS10 (Fig 7G, lanes 2 and 5). However, the truncated Hel2(1–315) mutant polyubiquitinated only eS7A but not uS10 (Fig 7G, lanes 3 and 6). Additionally, we confirmed that Not4‐dependent mRNA cleavages in NGDRQC− were not restored by expressing the Hel2(1–315) mutant (Appendix Fig S8D). However, this phenotype could only be confirmed by using the overexpression of Ski2‐E445Q as a dominant negative Ski2 mutant (Appendix Fig S8A–C; Ikeuchi & Inada, 2016), instead of a ski2 deletion background because of putative synthetic lethality induced in the ski2∆hel2∆not4∆ strain. Based on these results, we propose that also in vivo the Hel2(1–315) mutant protein polyubiquitinates the monoubiquitinated eS7 but not uS10, thereby rendering RQC and NGDRQC+ defective on the disome unit. At the same time, it remains functional enough to evoke the alternative NGDRQC− pathway.
To assess the physiological relevance of the two NGD pathways, RQC‐coupled NGD (NGDRQC+) and Not4‐dependent NGD (NGDRQC−) taking place upstream of the disome, we examined the growth and the anisomycin sensitivity of not4∆ and eS7A‐4KR mutant cells. As shown above, anisomycin sensitivity can be used as an indicator to distinguish NGDRQC+ from NGDRQC−. Deletion of Not4 and eS7A‐4KR mutation caused a significant growth defect compared to the wild‐type cells (Fig EV4A). Despite the severe growth defect, anisomycin did not further affect the growth of not4∆ and eS7A‐4KR mutant cells (Fig EV4A), indicating that Not4 and ubiquitination of eS7A are not directly associated with RQC. Intriguingly, knocking out both NGDRQC+ and NGDRQC− using a not4∆hel2∆ double mutation displayed a synthetic growth defect (Fig EV4B). This further indicates that Not4 may be involved in the Hel2‐independent mRNA quality control pathway and play a crucial role in the absence of Hel2‐mediated quality controls. We also found that not4∆xrn1∆ double deletion mutations conferred synthetic lethality (Fig EV4C), suggesting that the Hel2‐independent mRNA quality control pathway might be required for cell growth when Xrn1‐mediated decay is defective.
Figure EV4. Genetic interactions between NOT4 and quality control factors.
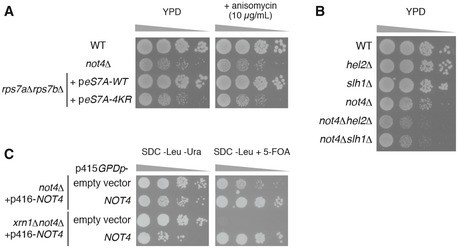
- Spot assay showing that severe growth defect of not4∆ and eS7A‐4KR mutant cells irrespective of the presence of anisomycin. Indicated yeast cells were cultured with liquid YPD media, and 10‐fold serial dilutions of the cells were grown on YPD with or without 10 μg/ml anisomycin.
- Spot assay showing that not4∆hel2∆ and not4∆slh1∆ exhibited synthetic growth defect. Indicated yeast cells were cultured with liquid YPD media, and then, 10‐fold serial dilutions of the cells were grown on YPD.
- Synthetic lethality of xrn1∆not4∆ cells. Spot assay of not4∆ and xrn1∆not4∆ yeast cells harbouring p416GPDp‐FLAG‐NOT4 transformed with p415GPD empty vector or p415GPDp‐FLAG‐NOT4. Transformed cells on the SDC‐Leu medium were cultured with liquid SDC‐Leu media for 1 day, and 10‐fold serial dilutions of the cells were grown on SDC‐Leu‐Ura plate or SDC‐Leu plate with 0.5 g/l 5‐fluoroorotic acid (5‐FOA).
Discussion
One of the major questions in translational quality control is how a stalled ribosome is recognized and how this leads to further steps resulting in degradation of the aberrant mRNA and the arrested nascent protein. Recent work has demonstrated that ribosome collision is a critical event triggering NGD, during which endonucleolytic cleavages in polysomes occur (Simms et al, 2017). In this work, we characterized the role of the E3 ligase Hel2 in this process. Previous studies already demonstrated its function in the ubiquitination of uS10 to trigger RQC in ribosomes stalled on an R(CGN)12 reporter containing rare codon repeats (Matsuo et al, 2017). Western blot analysis with cells expressing a Myc‐tagged Ub‐K48R mutant suggested that Hel2‐dependent ubiquitination of uS10 results in K48‐linked di‐ubiquitination. However, its function was still unclear. Here, we demonstrate that Hel2 is also capable of mediating a second major type of polyubiquitination, via K63 linkage. We show that Hel2 polyubiquitinates uS10 to trigger both endonucleolytic cleavage and RQC in the NGD reporter, thus acting as a master regulator for both pathways. We further identified eS7 as a novel target for Hel2 on stalled ribosomes. We could show that Hel2 polyubiquitinates eS7 and that this linkage is also involving ubiquitin K63. This event, however, is strictly dependent on initial monoubiquitination of eS7 by Not4. Through our mutational analysis of Hel2, we could dissect the NGD pathway into two branches. The first branch was coupled to RQC (NGDRQC+) and lead to cleavage events in the mRNA covered by the first two stalled ribosomes (disome unit). The second branch was uncoupled from RQC (NGDRQC−) and resulted in upstream cleavage events on the mRNA outside of the disome unit and potentially covered by following ribosomes. The exact mechanisms of these cleavage events will be the subject of future research, in particular regarding the identification of the enigmatic endonuclease responsible for this observed activity. We showed that efficient NGDRQC+ and also RQC both require the RING domain as well as the C‐terminal half of Hel2. A mutant Hel2 lacking residues 316–639 was unable to polyubiquitinate uS10 and failed to induce both NGDRQC+ and RQC on mRNA covered by the disome unit. However, the Hel2(1–315) mutant efficiently induced NGDRQC− upstream of the disome unit via K63‐linked eS7 polyubiquitination. While this upstream‐acting NGDRQC− could easily be considered as “alternative” or “non‐canonical” pathway active only under RQC‐deficient conditions, it is important to notice that its products were readily detectable, despite only at low levels, in a wild‐type background (xrn1Δ) as seen in Fig 1E lane 6 and Fig 2I lane 11. Moreover, as demonstrated in Fig EV1E, the mRNA half‐lives of K(AAA)12 and R(CGN)12 reporters were not affected under the RQC‐deficient uS10 point mutant and Slh1 deletion conditions. This showed that NGD could be as efficient in the alternative NGDRQC− pathway as in the “canonical” NGDRQC+ pathway, which strongly argues in favour of its physiological relevance. We therefore propose that the NGDRQC− pathway may not only provide redundancy in the critical physiological process of NGD, but also constitutively contribute to its overall efficiency. This is further supported by results presented in Fig EV4B, where we showed genetic interaction between Not4 and Hel2 or Slh1, and the necessity of at least one working NGD pathway for cell viability. Taken together, these results suggest that there might be a coordinated action by Not4 and Hel2 on the stalled oligoribosome to trigger subsequent mRNA cleavage events. Here, the distinct structure formed by colliding ribosomes (e.g. a disome, a trisome or a tetrasome) may serve as a molecular recognition platform to guide Hel2 to its targets and establish chain specificity for ubiquitin linkage.
A disome represents the minimal and thus most simple example for colliding ribosomes, yet it might represent a primary unit that has to form to initiate downstream quality control pathways that are initiated by Hel2 alone or together with Not4. The disome structure gives a first hint at how these E3 ligases could act as master regulators and play their roles in ubiquitination of uS10 and eS7. First, the disome architecture involves a direct interaction of two ribosomal RACK1 proteins, which may explain the importance of RACK1 for RQC and NGDRQC+ induction in vivo (Kuroha et al, 2010; Brandman et al, 2012; Wolf & Grayhack, 2015; Ikeuchi & Inada, 2016; Juszkiewicz & Hegde, 2017; Matsuo et al, 2017; Sundaramoorthy et al, 2017). Here, a unique composite interface is formed that may be recognized by Hel2 to subsequently modify the nearby targets uS3 and uS10. Second, we note that eS7 and the ZnF‐protein eS27 from the leading ribosome are involved in the body‐to‐body disome contact with ES6b, and also, eS7 of the colliding ribosome is in close vicinity to this contact site. Thus, the structural environment around this contact site might serve as a hub for both Not4 and Hel2, to engage their targets. However, it is unclear which of the ribosomes is polyubiquitinated and where exactly Hel2 binds. Our previous study showed that affinity‐purified Hel2‐ribosome complexes mainly yielded 80S ribosomes in the rotated state (Matsuo et al, 2017), suggesting that Hel2 may preferentially bind colliding rotated ribosomes as a primary contact site (Fig EV5). This contact might be established by its carboxyl‐terminal region, which we found to be required for uS10 ubiquitination in the leading ribosome and subsequent RQC. A potential binding candidate might be eS27, which could interact with one or more of the ZnF domains of Hel2 (222–307). However, this will remain speculative until at least an additional density of Hel2 is observed. Since we did not detect any Hel2 density in our sample, we cannot rule out the possibility that it also recognizes higher order interfaces of trisomes or tetrasomes, which would also be characteristic for efficient stalling. Nonetheless, during the revision of our manuscript, a study by the Hegde and Ramakrishnan groups (Juszkiewicz et al, 2018) was published, confirming that formation of a collided rabbit disome unit with a defined structure indeed serves as a platform for molecular recognition by the mammalian homologue of Hel2.
Figure EV5. Comparison of ribosomal states in Hel2 pulldown and CGA‐CCG‐stalled monosomes (80S).
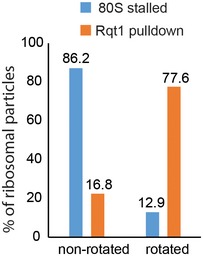
Hel2 preferentially interacted with rotated A/P‐P/E ribosomes (77.6%). Ribosomes in this state are almost not present in the case of leading stalled 80S where 86.2% ribosomal particles are in the P/P E/E non‐rotated state. Therefore, Hel2 seems to preferentially interact with colliding stalled ribosomes.
The question remains unanswered how chain specificity for Hel2‐mediated K63‐linked polyubiquitination is established. Chain specificity is generally reached in concert with an E2‐conjugating enzyme. When paired with RING domain ubiquitin ligases, E2 ubiquitin‐conjugating enzymes define the linkage type during polyubiquitin chain synthesis by positioning the linkage‐defining lysine of the acceptor ubiquitin. Given the analogy with the best‐studied yeast E2 enzyme pair forming K63‐linked polyubiquitin chain Ubc13‐Mms2 (Eddins et al, 2006), we suspect that Hel2 synthesizes polyubiquitin chains by transferring the thioester‐bound donor ubiquitin from E2 (Ubc4) onto an acceptor ubiquitin that is non‐covalently bound to the putative ZnF domain(s) of Hel2. The zinc finger domain (ZnF) is arguably the largest class of ubiquitin‐binding domains (Hurley et al, 2006; Sato et al, 2011), which has been shown to function in many ubiquitin‐related processes including ubiquitin chain editing (Bosanac et al, 2010) and deubiquitination (Reyes‐Turcu et al, 2006). In the light of these findings, we suggest that the ZnF of Hel2(222–307) interacts with the acceptor ubiquitin that is conjugating with the substrate. Deletion analysis suggests that residues 315–439 of Hel2 inhibit the activity of Hel2(1–315) in the polyubiquitination of eS7. We suspect that the interaction of this putative auto‐inhibitory domain with the stalled polysome allows the zinc finger domains to bind to an acceptor ubiquitin. Taken together, we suggest putative domain functions of Hel2 as follows: the RING domain (61–109) recruits a donor ubiquitin‐conjugating Ubc4, ZnF domains (222–307) interact with an acceptor ubiquitin, and a putative auto‐inhibitory domain (315–439) regulates the polyubiquitination of eS7 or uS10 via interaction with stalled poly‐ribosomes in specific states.
We previously reported that the polyubiquitination of uS10 is K48‐linked (Matsuo et al, 2017). In this study, we demonstrated the K63‐linked polyubiquitination activity of Hel2, indicating that Hel2 forms both K48‐linked and K63‐linked polyubiquitin chains on uS10 at residues K6 or K8. Other E3 ligases have also been described to connect ubiquitin chains via multiple linkage types. For instance, the well‐established E3 ligase Parkin is able to connect ubiquitin chains through K11, K48 and K63 (reviewed in Yau & Rape, 2016). The K63‐linked polyubiquitination of uS10 by Hel2 is required for NGD and RQC; however, the function of K48‐linked polyubiquitinations remains unknown and should be addressed in future studies. The polyubiquitination of eS7 requires monoubiquitination by Not4, so we suspect that also the stable yeast Ccr4‐Not complex (Collart & Panasenko, 2012) could interact with stalled poly‐ribosomes. However, it is not clear how and when Not4 recognizes and ubiquitinates the ribosome and how the activity of Not4 can be related to ribosome collisions. We speculate that other component(s) of the Ccr4‐Not complex may serve as selectors of the target ribosome(s) directing Not4 to its sites of action, such as eS7 in the case of NGDRQC−. Further studies will be needed to uncover the regulatory relevance of sequential ubiquitination of eS7 in the determination of mRNA fate.
Materials and Methods
Yeast strains
Saccharomyces cerevisiae yeast strains used in this study are W303‐1a and its derivatives, and are listed in Appendix Table S1. Gene disruption strains and C‐terminally 3xHA‐ or Flag‐TEV‐Protein A (FTP)‐tagged strains were constructed by established homologous recombination strategies using PCR‐amplified selection marker genes with cassette sequences (kanMX4, hphMX4, natMX4, natNT2 or HIS3MX6) (Janke et al, 2004; Longtine et al, 1998). To construct shuffle strains of essential ribosomal protein genes (uS10, uS3, eS7AeS7B), ribosomal protein gene of parent strain, harbouring plasmid expressing the ribosomal protein with URA3 selection marker gene, was disrupted by the PCR‐based method. To construct mutant strains of ribosomal protein genes (uS10‐K6/8R, uS3‐K212R, eS7A‐4KR, etc.), the shuffle strain transformed with plasmid expressing mutant ribosomal protein products was grown on SDC plate containing 0.5 mg/ml of 5‐fluoroorotic acid (5‐FOA) and isolated URA3 absent strain.
Plasmids
Plasmids used in this study are listed in Appendix Table S2. DNA cloning was performed with PCR amplification by using gene‐specific primers and PrimeSTAR HS DNA polymerase (Takara Bio) and by using In‐Fusion HD Cloning Kit (Takara Bio) or T4 DNA ligase (NEB). uS10‐K6/8R mutant was constructed by PCR using primers having corresponding mutations. uS3 and eS7A point mutants were constructed by site‐directed mutagenesis by PCR using primer set harbouring point mutation. All cloned DNAs amplified by PCR were verified by sequencing.
Yeast culture and media
All yeast cells were grown in YPD or synthetic complete (SC) medium with 2% glucose at 30°C, and harvested cells at log phase (OD600 of 0.5–0.8), unless otherwise noted. For polysome analysis, yeast cells grown at 30°C until OD600 of 0.8 were immediately cooled in an ice bucket and treated for 5 min with 0.1 mg/ml of cycloheximide (Nacalai Tesque Cat# 06741‐04) before harvesting and harvested by quick centrifuge. To determine half‐lives of mRNAs, yeast cells harbouring pGAL1p‐GFP‐X‐FLAG‐HIS3‐CYC1t (X = no insertion, R(CGN) 12, K(AAA)12) were grown with SC 2% galactose medium until log phase (OD600 = ~0.5) and harvested cells by centrifuge and discard media. Immediately after that, cell pellet was resuspended with SC 2% glucose medium pre‐incubated at 30°C to shut‐off transcription from GAL1 promoter, and 1.5 ml of yeast cell culture was harvested at time points (2, 4, 8, 16, 32 min or 3, 6, 12, 24 min).
RNA isolation
Total RNA samples were used for Northern blotting. Yeast cells harbouring reporter plasmids were cultured in 15 ml scale, 10 ml of them were harvested by centrifuge, and yeast pellet collected in 1.5‐ml tube was frozen by liquid nitrogen. RNA was extracted by acid phenol–chloroform extraction method as follow. Yeast pellet was resuspended with 300 μl of RNA buffer (Tris–HCl pH 7.5, 300 mM NaCl, 10 mM EDTA, 1% SDS with DEPC‐treated Milli‐Q water, room temperature) and put it on ice before phenol addition. Then, 300 μl of water‐saturated phenol was added and mixed them by vortex for 10 s. Mixture was incubated at 65°C for 5 min., vortexed for 10 s again and put them on ice for 5 min. After centrifugation at 16,000 × g for 5 min, RT, the upper layer was transferred to a new 1.5‐ml RNase‐free tube. 300 μl of water‐saturated phenol was added to the water layer in 1.5‐ml tube, and above operation was performed again. After that, 300 μl of water‐saturated phenol/chloroform (1:1) mixture was added to water layer in 1.5‐ml tube, mixed them by vortex for 10 s and centrifuged the tube at 16,000 × g for 5 min, RT. Obtained water layer was mixed with 300 μl of water‐saturated phenol/chloroform/isoamyl alcohol (25:24:1), mixed by vortex for 10 s and centrifuged the tube at 16,000 × g for 5 min, RT. Finally, gained water layer was subjected to ethanol precipitation and RNA pellet was dissolved with DEPC‐treated water.
RNA electrophoresis and northern blotting
2.5 mg of total RNA in 6 μl was mixed with 24 μl of glyoxal mix (600 μl of DMSO, 200 μl of deionized 40% glyoxal, 120 μl of 10× MOPS buffer (200 mM MOPS, 50 mM NaOAc, 10 mM EDTA, pH 7.0), 62.5 μl of 80% glycerol and 17.5 μl of DEPC‐treated water in 1 ml) and 3 μl of RNA loading buffer (50% glycerol, 10 mM EDTA pH 8.0, 0.05% bromophenol blue, 0.05% xylene cyanol) and incubated at 74°C for 10 min followed by on ice for 10 min. RNA sample was resolved on 1.2 or 2% agarose gel with 1× MOPS buffer by electrophoresis at 200 V for 40 or 50 min, respectively, followed by capillary transfer to Hybond‐N+ membrane (GE Healthcare) with 20xSSC (3M NaCl, 300 mM trisodium citrate dihydrate) for 20 h. RNA was cross‐linked on membrane by CL‐1000 ultraviolet cross‐linker (UVP) at 120 mJ/cm2. Membrane was incubated with DIG Easy Hyb Granules (Roche Cat# 11796895001) for 1 h in a hybridization oven at 50°C. DIG‐labelled GFP, HIS3 or SCR1 probe prepared by PCR DIG Probe Synthesis Kit (Roche Cat# 11636090910) or DIG‐labelled FLAG oligonucleotide (Fasmac) was added and incubated for over 14 h. Membrane was washed with wash buffer I (2× SSC, 0.1% SDS) for 15 min, two times, in a hybridization oven at 50°C, followed by additional wash with wash buffer II (0.1× SSC, 0.1% SDS) for 15 min in a hybridization oven at 50°C. Then, membrane was washed with 1× maleic acid buffer (100 mM maleic acid, 150 mM NaCl, pH 7.0, adjusted by NaOH) for 10 min, RT, and incubated with Blocking Reagent (Roche Cat# 11096176001) for 30 min. Anti‐Digoxigenin‐AP, Fab fragments (Roche Cat# 11093274910), was added to Blocking Reagent and further incubation for 1 h. After that, membrane was washed with wash buffer III (1× maleic acid buffer, 0.3% Tween 20) for 10 min., three times, and equilibrated by equilibration buffer (100 mM Tris–HCl, 100 mM NaCl, pH 9.5). To detect RNA, membrane was reacted with CDP‐Star (Roche Cat# 11759051001) for 10 min, and chemiluminescence was detected by LAS4000 mini (GE Healthcare).
Primer extension
Primer extension was performed to determine 5′ ends of 3′ NGD intermediates as endonucleolytic cleavage sites in NGD. Isolated total RNA was further treated with equal volume of chloroform–isoamyl alcohol (24:1) and collected water layer to new 1.5 ml RNase‐free tube, followed by ethanol precipitation. 30 mg of total RNA was subjected to reverse transcription (RT) by using SuperScript IV Reverse Transcriptase (Invitrogen Cat# 18090010) with 5′‐IRDye700‐labelled primer (LI‐COR) complementary to FLAG nucleotide sequence (5′‐CTTGTCATCGTCGTCCTTGTAGTC‐3′). Equal amount of chloroform was added to reaction and mix it by vortex. Water layer was collected after centrifuge, followed by ethanol precipitation with one‐fifth amount of 10 M NH4OAc, twice and half amount of ethanol and 1 μl of glycogen. Obtained cDNA pellet was dissolved in 6 μl of deionized formamide with 5 mg/ml blue dextran. cDNA sample was linearized at 70°C for 3 min followed by on ice for 3 min and resolved on 5% polyacrylamide‐TBE‐urea sequencing gel by electrophoresis at 1,000 V for 135 min. Fluorescence of IRDye700 was detected by FLA‐9000 (Fujifilm). The size of the RT product was determined compared to a sequencing ladder of corresponding reporter plasmid DNA prepared by using the same primer and Thermo Sequenase Cycle Sequencing Kit (USB Cat# 78500 1 KT).
Total protein sample preparation for SDS–PAGE and Western blotting
Total protein samples were used for Western blotting. Total protein samples for Western blotting were prepared by trichloroacetic acid (TCA) precipitation method as follow. 10 ml of exponentially grown yeast culture were harvested at OD600 of 0.5–0.8. Yeast cell pellet in 1.5‐ml tube (stand on ice) was resuspended with 500 μl of ice‐cold TCA buffer (20 mM Tris–HCl pH 8.0, 50 mM NH4OAc, 2 mM EDTA, 1 mM PMSF) and added 500 μl of 0.5 mm dia. zirconia/silica beads (BioSpec) and 500 μl of ice‐cold 20% TCA followed by thorough vortex for 30 s each, three times at 4°C. Supernatant was collected to new 1.5‐ml tube, and beads were further washed with 500 μl of ice‐cold TCA buffer followed by collection of supernatant to the 1.5‐ml tube. After centrifuge at 18,000 × g, 4°C for 10 min and removing supernatant completely, pellet was dissolved with TCA sample buffer (120 mM Tris, 3.5% SDS, 14% glycerol, 8 mM EDTA, 120 mM DTT and 0.01% BPB; added 150 μl/10 ml culture at OD600 of 0.6). For SDS–PAGE followed by Western blotting except for detection of ubiquitinated proteins, total protein samples were incubated at 100°C for 10 min followed by centrifuge at 16,000 × g for 10 min. For ubiquitinated proteins, total protein samples were incubated at 88°C for 10 min.
Protein purification
Hel2‐ribosome complex
Hel2‐ribosome complex was purified by Hel2‐FTP or PTH‐Hel2 from 3 l of YPD or SC 2% glucose culture at OD600 of 0.8, as previously described (Matsuo et al, 2017). Particularly, yeast cells were ground by mortar and pestle with liquid nitrogen and resulting yeast powder was collected in 50‐ml tube. Powder was resuspended by ice‐cold LB100 (50 mM Tris–HCl pH 7.5, 100 mM NaCl, 10 mM MgCl2, 0.0075% NP‐40, 2 mM 2‐mercaptoethanol, 1 mM PMSF) containing 1 pill/10 ml of complete‐mini EDTA free (Roche Cat# 11836170001) and centrifuged at 15,000 × g, 4°C for 3 min followed by thorough centrifuge of supernatant at 40,000 × g, 4°C for 30 min to obtain clear lysate. Lysate was incubated at 4°C with 50 μl of pre‐equilibrated IgG (Sigma‐Aldrich Cat# I5006)‐conjugated Dynabeads (Thermo Fisher Scientific Cat# 14302D) for 1 h, and beads were washed with LB100 for five times. Hel2‐ribosome complex was eluted by TEV protease at 4°C for 2 h from IgG‐conjugated Dynabeads, and eluate was precipitated with TCA. Pellet was dissolved with 1× Laemmli Sample Buffer (50 mM Tris–HCl pH 6.8, 2% SDS, 10% glycerol, 25 mM DTT, 0.01% BPB). Samples were subjected to PAGE at neutral pH condition followed by CBB staining or Western blotting.
Ribosome for in vitro ubiquitination assay
Ribosome was purified from 1 l of yeast cell culture of SC 2% glucose by one‐step affinity purification method using uS5‐FLAG or uL23‐FLAG expressing plasmid, as described previously (Inada et al, 2002). To detect ubiquitinated ribosomal proteins by PAGE followed by Western blotting after in vitro ubiquitination reaction, C‐terminally 3xHA‐tagged strains were used for purification. It is notable that, to be free of ubiquitinated ribosomal proteins in purified ribosome, ribosome was purified from Hel2∆ or not4∆ mutant background. Grinded yeast pellet was resuspended in ice‐cold LB300 (50 mM Tris–HCl pH 7.5, 300 mM NaCl, 10 mM MgCl2, 0.01% NP‐40, 1 mM DTT, 1 mM PMSF) containing 1 pill/10 ml of complete‐mini EDTA free, centrifuged at 10,000 × g, 4°C for 10 min followed by thorough centrifuge of supernatant at 40,000 × g, 4°C for 30 min to obtain clear lysate. To purify ribosome with FLAG‐tagged protein, lysate was incubated at 4°C with 100 μl of pre‐equilibrated anti‐DYKDDDDK tag antibody beads (Wako Cat# 016‐22784) for 1 h. After wash steps by batch with LB300 for seven times, LB200 for one time and LB100 w/o detergent for three times, ribosome was eluted from beads by 100 μl of Elution Buffer (50 mM Tris–HCl pH 7.5, 300 mM NaCl, 10 mM MgCl2, 0.01% NP‐40, 1 mM DTT, 1 mM PMSF, 250 μg/ml FLAG peptide (GenScript)) at 4°C for 1 h. Eluted ribosomes were confirmed by PAGE and CBB stain, and were used for in vitro ubiquitination assay.
Hel2‐FLAG
Hel2‐FLAG was purified from 1 l of SC 2% glucose culture of yeast cell harbouring pGPDp‐HEL2‐FLAG‐CYC1t. Grinded yeast pellet was resuspended in ice‐cold LB500 (50 mM Tris–HCl pH 7.5, 500 mM NaCl, 10 mM MgCl2, 0.01% NP‐40, 1 mM DTT, 1 mM PMSF) containing 1 pill/10 ml of complete‐mini EDTA free, centrifuged at 10,000 × g, 4°C for 10 min followed by thorough centrifuge of supernatant at 40,000 × g, 4°C for 30 min to obtain clear lysate. To purify Hel2‐FLAG, lysate was incubated at 4°C with 100 μl of pre‐equilibrated anti‐DYKDDDDK tag antibody beads for 1 h. After wash steps by batch with LB500 for five times, LB400 for one time, LB500 for one time, LB200 for one time and LB100 w/o detergent for three times, Hel2‐FLAG was eluted from beads by 100 μl of Elution Buffer at 4°C for 1 h.
Not4 with Ccr4‐NOT
Not4 with Ccr4‐NOT complex (partially at least) was purified by tandem affinity purification using C‐terminal FLAG‐TEV‐Protein A (FTP) tag from 2 l of SC 2% glucose culture of yeast cell harbouring p414GPDp‐NOT4‐FTP‐ADH1t. Grinded yeast pellet was resuspended in ice‐cold LB500 (50 mM Tris–HCl pH 7.5, 500 mM NaCl, 10 mM MgCl2, 0.01% NP‐40, 1 mM DTT, 1 mM PMSF) containing 1 pill/10 ml of complete‐mini EDTA free, centrifuged at 10,000 × g, 4°C for 5 min followed by thorough centrifuge of supernatant at 40,000 × g, 4°C for 30 min to obtain clear lysate. First, lysate was incubated at 4°C with 500 μl of pre‐equilibrated IgG Sepharose 6 Fast Flow (GE Healthcare Cat# 17‐0969‐01) for 1 h. After wash steps by batch with LB500 for three times, LB400 for one time, LB300 for one time, LB200 for one time and LB100 for one time, Not4 was eluted by TEV protease at 4°C for 2 h from beads. Second, TEV elution was incubated with 100 μl of pre‐equilibrated anti‐DYKDDDDK tag antibody beads for 1 h. After binding, beads were washed with LB100 for three times and LB100 w/o detergent for three times. Not4 was eluted from beads by 100 μl of Elution Buffer at 4°C for 1 h.
Ubc4
Recombinant Ubc4 was purified as GST‐Ubc4 from E. coli Rosetta‐gami 2 (DE3) harbouring pGEX‐Ubc4 and was eluted as no tagged Ubc4 by PreScission Protease (GE Healthcare Cat# 27084301). Lysate of E coli cells harbouring pGEX‐Ubc4 was incubated with Glutathione Sepharose 4B (GE Healthcare Cat# 17‐756‐05) followed by wash with LB500 for four times, LB400 for one time, LB300 for one time, LB200 for one time and LB100 w/o detergent for three times. Ubc4 moiety was eluted from beads using PreScission Protease at 4°C for 16 h.
In vitro ubiquitination assay
Ubiquitination reactions in reaction buffer (50 mM Tris–HCl pH 7.5, 100 mM NaCl, 10 mM MgCl2, 1 mM DTT) contained 50 μM His‐ubiquitin (wild type or mutants; UBPBio), 100 nM UBE1 (UBPBio Cat# B1100), 500 nM Ubc4, 300 nM Hel2‐FLAG and/or 200 nM Not4, 2 A260 unit of ribosome and energy regenerating source (1 mM ATP, 10 mM creatine phosphate and 20 μg/ml creatine kinase). Initially, reaction was mixed at room temperature except E3 ligase and ribosome, then ribosome and E3 was added and incubated at 28°C for 90 min. To stop ubiquitination reaction, 4× SDS sample buffer was added to the reaction tube. Samples were incubated at 77°C for 10 min to proceed to PAGE at neutral pH condition followed by Western blotting.
Polysome analysis
Cycloheximide (CHX)‐treated cells were grinded by mortar and pestle with liquid nitrogen and dissolved to lysis buffer (20 mM HEPES‐KOH pH 7.5, 100 mM KOAc, 2 mM Mg(OAc)2, 1 mM DTT, 1 mM PMSF, 0.1 mg/ml CHX and 1 pill/10 ml complete‐mini EDTA free). After centrifuge at 10,000 × g, 4°C for 10 min followed by thorough centrifuge of supernatant at 21,500 × g, 4°C for 30 min, 50 A260 × ml (2 mg RNA amount) of supernatant was layered on sucrose density gradient (10‐50% sucrose in 10 mM Tris–acetate, pH 7.4, 70 mM NH4OAc and 4 mM Mg(OAc)2) prepared in 25 × 89 mm polyallomer tubes (Beckman Coulter) using a Gradient Master (BioComp). After ultracentrifugation at 13,1000 × g, 4°C for 3 h in a P28S rotor and himac CP‐70MX (Hitachi Koki), gradient was fractionated by fractionator (BioComp) and 900 μl of each fraction was collected and precipitated by 10% TCA. Pellet was dissolved to 45 μl of TCA sample buffer (120 mM Tris, 3.5% SDS, 14% glycerol, 8 mM EDTA, 120 mM DTT and 0.01% BPB). Samples were incubated at 88°C for 10 min to proceed to 10% SDS–PAGE followed by Western blotting.
Electrophoresis and Western blotting
Protein samples were separated by SDS–PAGE (for total TCA‐precipitated samples and polysome analysis samples) or PAGE at neutral pH condition (pH 6.8, for purified protein samples and in vitro reaction samples) and were analysed by CBB staining or were transferred on PVDF membrane (Immobilon‐P, Millipore). After blocking with 5% skim milk in PBST, membrane was incubated with primary antibodies for 1 h at room temperature followed by washing with PBST for three times and further incubation with horseradish peroxidase (HRP)‐conjugated secondary antibodies for 1 h at room temperature. Antibodies used in this study are anti‐GFP (1:5,000; Santa Cruz Biotechnology, Cat# sc‐9996, clone B‐2), anti‐Rpl23 (uL14) (1:1,000; Abcam, Cat# ab112587), anti‐FLAG (1:5,000; Sigma‐Aldrich, Cat# F1804, clone M2), anti‐Penta‐His (1:1,000; Qiagen, Cat# 34660), anti‐V5 (1:5,000; Bio‐Rad, Cat# MCA1360, clone SV5‐Pk1), anti‐mouse (1:5,000; GE Healthcare, Cat# NA931V), anti‐rabbit (1:5,000; GE Healthcare, Cat# NA934V), anti‐eEF1α (1:20,000; Lab stock) and anti‐eEF2 (1:25,000; Lab Stock). In case of HA‐tagged protein detection, membrane was incubated with HRP‐conjugated antibodies: anti‐HA (1:10,000; Roche, Cat# 12013819001, clone 3F10). After washing with PBST for three times, chemiluminescence was detected by LAS4000 mini (GE Healthcare). Antibodies used in this study were listed in Key Resource Table.
Spot assay to confirm the synthetic lethality
Yeast not4∆ and xrn1∆not4∆ cells harbouring p416GPDp‐FLAG‐NOT4 with p415GPD empty vector or p415GPDp‐FLAG‐NOT4 were grown on liquid SDC‐Leu media for 1 day. To provide an opportunity to displace p416 plasmids, yeast cells were pre‐grown on the SDC‐Leu medium with uracil for 3 days before growth in liquid media. Then, cell cultures were diluted to OD600 = 0.3 for basal spot, and 10‐fold serial dilutions were prepared. 10 μl droplet of the serial dilutions was grown on SDC‐Leu‐Ura or SDC‐Leu containing 0.5 g/l 5‐fluoroorotic acid (5‐FOA) plate at 30°C for 2–3 days.
Preparation of the CGA‐CCG‐stalled mono‐ and disomes
We generated a reporter containing an mRNA stalling sequence with the codons (CGA‐CCG)2. This sequence was placed downstream of a sequence coding for TEV‐cleavable N‐terminal His‐ and HA tags and the first 86 amino acids of truncated uL4. His‐HA‐uL4‐(CGA‐CCG) 2 mRNA was produced using the mMessage mMachine Kit (Thermo Fisher) and used in a yeast cell‐free translation extract from ski2Δ cells. This yeast translation extract was prepared, and in vitro translation was performed essentially as described before (Waters & Blobel, 1986). The cells were grown in YPD medium to OD600 of 1.5–2.0. Spheroplasts were prepared from harvested and washed cells using 10 mM DTT for 15 min at room temperature and 2.08 mg zymolyase per 1 g of cell pellet for 75 min in 1 M sorbitol at 30°C. Spheroplasts were then washed and lysed in a Dounce homogenizer as described (Waters & Blobel, 1986) before using lysis buffer comprising 20 mM Hepes pH 7.5, 100 mM KOAc, 2 mM Mg(OAc)2, 10% glycerol, 1 mM DTT, 0.5 mM PMSF and complete EDTA‐free protease inhibitors (GE Healthcare). The S100 fraction of lysate supernatant was passed through PD10 column (GE Healthcare) and used for in vitro translation. In vitro translation was performed at 17°C for 75 min using great excess of template mRNA (38 μg per 415 μl of extract) to prevent degradation of resulting stalled ribosomes by endogenous response factors.
(CGA‐CCG)‐dicodon‐stalled RNCs were affinity‐purified using the His‐tag on the nascent polypeptide chain and magnetic anti‐His‐IgG‐beads. After in vitro translation at 17°C for 75 min, the extract was applied to Dynabeads™ (Invitrogen) for His‐Tag isolation and pulldown for 15 min at 4°C. The beads were washed with a buffer containing 50 mM HEPES/KOH, pH 7.5, 100 mM KOAc, 25 mM Mg(OAc)2, 250 mM sucrose, 0.1% Nikkol and 5 mM β‐Mercaptoethanol and eluted in the same buffer containing 300 mM imidazole. The elution was applied to a 10‐50% sucrose gradient wash buffer, and ribosomal fractions were separated by centrifugation for 3 h at 285,000 g at 4°C in a SW40 rotor. For gradient fractionation, a Piston Gradient FractionatorTM (BIOCOMP®) was used. The monoribosome and disome fractions were collected, applied onto 400 μl of sucrose cushion buffer and spun at 100,000 rpm for 45 min at 4°C in a TLA110 rotor. The ribosomal pellets were resuspended carefully on ice in 25 μl of grid buffer (20 mM HEPES/KOH, pH 7.2, 50 mM KOAc, 5 mM Mg(OAc)2, 125 mM sucrose, 0.05% Nikkol, 1 mM DTT and 0.01 U/μl SUPERase‐INTM (Invitrogen®, #AM2696)).
Cryo‐EM
Freshly prepared samples (stalled monosomes and disomes) were applied to 2 nm pre‐coated Quantifoil R3/3 holey carbon support grids and vitrified. Data were collected at Titan Krios TEM (Thermo Fisher) equipped with a Falcon II direct detector at 300 keV under low‐dose conditions of about 25 e‐/Å2 for 10 frames in total and defocus range of −1.3 to −2.8 μm. Magnification settings resulted in a pixel size of 1.084 Å/pixel. Original image stacks were summed and corrected for drift and beam‐induced motion at the micrograph level by using MotionCor2 (Zheng et al, 2017). The contrast transfer function (CTF) estimation and resolution range of each micrograph were performed with Gctf (Zhang, 2016).
Data processing
Both CGA‐CCG‐stalled monosome and disome data sets were processed using standard procedures with programs GAUTOMATCH (http://www.mrc-lmb.cam.ac.uk/kzhang/) for particle picking and RELION (Kimanius et al, 2016).
Data processing of the CGA‐CCG‐stalled disome sample
The disome data set was initially processed using a 80S monosome as a template for particle picking and alignment. A total of 452,903 particles were selected after 2D classification. Initial refinement and 3D classification were performed at a pixel size of 3.61 Å/pixel using a box of 400 pixel (rescaled to 120 pixels) and a mask diameter of 300 Å. The refined map was 3D classified into six classes. One class (class 4) showed a programmed ribosome in the non‐rotated POST‐state occupied with P/P‐ and E/E‐site tRNAs. Another class (class 1) showed rotated state ribosomes with hybrid tRNAs. In both classes, extra density could be observed close to the mRNA exit site (in the non‐rotated one) or the mRNA entry site (in the rotated one). Another class (class 6) contained mainly P‐site tRNA and rRNA expansion segment ES27 mainly in a position facing the L1 stalk (L1 position) and contained no extra densities at the mRNA entry and exit sites. Other classes were either low populated (class 2) or noisy (classes 3 and 5) and were not further processed.
Reconstruction individual monosomes
Class 1 (rotated 80S ribosomes with hybrid state tRNAs), class 4 (non‐rotated 80S ribosomes with P‐ and E‐site tRNAs) and class 6 (non‐rotated 80S ribosomes with P‐site tRNA and ES27 in L1 position) were further subclassified. Class 4 (61,350 particles) was subsorted into three classes, of which one class (15,739 particles) showed a defined extra density for a second ribosome. This class was individually refined to 3.8 Å after post‐processing using a box size of 400 pixels and a mask diameter of 360 Å. Class 1 (126,626 particles) was subsorted into six classes of which one class (class 3; 16,651 particles) showed a defined extra density for a second ribosome. Other classes showed either less well‐defined density (classes 1, 2 and 5) or no density (class 4; 27,632 particles) for the second ribosome. All classes showed rotated ribosomes with tRNAs in the hybrid A/P and P/E states, except for class 6 with A/A and P/E tRNAs.
The disome class (class 3) was individually refined to 3.9 Å after post‐processing using a box size of 400 pixels and a mask diameter of 360 Å. The rotated monosome class with hybrid A/P and P/E tRNAs (class 4) was refined to 3.6 Å after post‐processing using a box size of 400 pixels and a mask diameter of 300 Å. Apart from the presence of a second ribosome, the major difference between these two rotated 80S classes occurs in the conformation on 18S rRNA expansion segment ES6c. This rRNA segment rotates by approximately 90 degrees upon disome formation to form an interface contact. Class 6 (91,034 particles) representing monosomes in the POST‐state with mainly P/P tRNA was subsorted into four classes of which one class (class 1; 22,192 particles) showed ES27 clearly occupying the L1 position whereby L1 was in the “out” position. Other classes showed mixed ES27 and L1 positions. The clean class 1 was refined to 3.7 Å after post‐processing using a box size of 400 pixels and a mask diameter of 300 Å. Notably, ES27 in the L1 position is incompatible with stable disome formation since it occupies the position of ESb of the second ribosome.
Reconstruction of the disome
As mentioned above, class 4 containing the POST‐state ribosomes was further subclassified into three classes. One class (15,739 particles) showed a defined extra density for a second ribosome. The respective particles we first re‐extracted with a box size of 500 pixels (rescaled to 120 pixels) and refined, revealing features of a 40S subunit adjacent to the mRNA exit. After a second re‐extraction with a box size of 700 Å (rescaled to 186 pixels) followed by a refinement allowing large shifts (30 pixels with an offset of five pixels), we obtained the entire disome. Notably, the second ribosome appeared in the rotated state with A/P and P/E tRNAs.
The disome map was used as a new template for particle picking in GAUTOMATCH. Particles were extracted with a box of 700 pixels and rescaled to 104 pixels for 2D classification. This yielded in several classes clearly showing the shape of a stably formed disome and 107,872 particles were selected, initially refined (rescaled box of 186 pixels) and 3D classified into five classes. One class (class 4) showed a defined arrangement of the disome, whereas the other four classes showed less clear features in the interface (classes 2 and 3) and/or one ribosome poorly resolved (classes 1 and 5). Particles representing stable disomes (27,719 particles) were refined to an overall resolution of 5.3 Å after post‐processing and filtered according to local resolution using a negative b‐factor of 50.
Reconstruction of CGA‐CCG‐stalled monosome sample
For the CGA‐CCG‐stalled monosome, only a small data set was collected to check for tRNA states. Approximately 100,000 particles were obtained after 2D classification. Refinement and 3D classification were performed on rescaled images (pixel size 3.61 Å/pixel). The particles were classified into eight classes, of which four (86.2%) contained tRNAs in the P/P and E/E sites (empty A‐site) and only one class with tRNAs in the hybrid sites.
Model building
To generate a molecular model for the disome, existing models for translating yeast ribosomes (Schmidt et al, 2016, pdb‐ID 5MC6; Svidritskiy et al, 2014, pdb‐ID 3j77) and crystal structures of the yeast ribosome (Ben‐Shem et al, 2010, 4V88) were used and 3JBV (Zhang et al, 2015) served as a template for the A/P hybrid tRNA. First, individual subunits and tRNAs were fitted as rigid bodies into the densities. In the disome interface, rRNA expansion segments ES6b and ES6c were remodelled in COOT (Brown et al, 2015). The protein components in the disome interface required only minor adjustments. Cryo‐EM structures and models were displayed with UCSF Chimera (Pettersen et al, 2004) ChimeraX (Goddard et al, 2018) or PYMOL (Version 2.0 Schrödinger, LLC).
Author contributions
Genetic and biochemical experiments were performed by KI, TS, YM and YS under the supervision of KT and TI. PT prepared samples for Cryo‐EM and PT and TB processed the data. JC built the molecular models, and PT and TB analysed the Cryo‐EM data. TI, TB, RB, KI and PT wrote the manuscript. TI and RB primarily conceived the idea and designed the experiments.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
The authors would like to thank Elisabeth Heckel for help with sample preparation and Dr. Joseph Bartho for critical reading of the manuscript. This study was supported by a Grant‐in‐Aid for Scientific Research (KAKENHI) from the MEXT|Japan Society for the Promotion of Science (grant numbers 26116003 to TI) and by Research Grants in the Natural Sciences from the Takeda Foundation (to TI) and the German Research Council (FOR1805 and BE1814/15‐1 to R.B). R.B. acknowledges support by the Center for Integrated Protein Science Munich (CiPS‐M), the Graduate School of Quantitative Biosciences Munich (QBM).
The EMBO Journal (2019) 38: e100276
See also: LL Yan & HS Zaher (March 2019)
Contributor Information
Roland Beckmann, Email: beckmann@genzentrum.lmu.de.
Toshifumi Inada, Email: tinada@m.tohoku.ac.jp.
Data availability
The Cryo‐EM structure reported here has been deposited in the Protein Data Bank under accession code 6I7O and in the Electron Microscopy Data Bank under accession code EMD‐4427.
References
- Becker T, Franckenberg S, Wickles S, Shoemaker CJ, Anger AM, Armache JP, Sieber H, Ungewickell C, Berninghausen O, Daberkow I, Karcher A, Thomm M, Hopfner KP, Green R, Beckmann R (2012) Structural basis of highly conserved ribosome recycling in eukaryotes and archaea. Nature 482: 501–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann R, Spahn CM, Eswar N, Helmers J, Penczek PA, Sali A, Frank J, Blobel G (2001) Architecture of the protein‐conducting channel associated with the translating 80S ribosome. Cell 107: 361–372 [DOI] [PubMed] [Google Scholar]
- Bengtson MH, Joazeiro CA (2010) Role of a ribosome‐associated E3 ubiquitin ligase in protein quality control. Nature 467: 470–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐Shem A, Jenner L, Yusupova G, Yusupov M (2010) Crystal structure of the eukaryotic ribosome. Science 330: 1203–1209 [DOI] [PubMed] [Google Scholar]
- Bosanac I, Wertz IE, Pan B, Yu C, Kusam S, Lam C, Phu L, Phung Q, Maurer B, Arnott D, Kirkpatrick DS, Dixit VM, Hymowitz SG (2010) Ubiquitin binding to A20 ZnF4 is required for modulation of NF‐kappaB signaling. Mol Cell 40: 548–557 [DOI] [PubMed] [Google Scholar]
- Brandman O, Stewart‐Ornstein J, Wong D, Larson A, Williams CC, Li GW, Zhou S, King D, Shen PS, Weibezahn J, Dunn JG, Rouskin S, Inada T, Frost A, Weissman JS (2012) A ribosome‐bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 151: 1042–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Long F, Nicholls RA, Toots J, Emsley P, Murshudov G (2015) Tools for macromolecular model building and refinement into electron cryo‐microscopy reconstructions. Acta Crystallogr D Biol Crystallogr 71: 136–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Muhlrad D, Hauryliuk V, Cheng Z, Lim MK, Shyp V, Parker R, Song H (2010) Structure of the Dom34‐Hbs1 complex and implications for no‐go decay. Nat Struct Mol Biol 17: 1233–1240 [DOI] [PubMed] [Google Scholar]
- Collart MA, Panasenko OO (2012) The Ccr4–not complex. Gene 492: 42–53 [DOI] [PubMed] [Google Scholar]
- Defenouillere Q, Yao Y, Mouaikel J, Namane A, Galopier A, Decourty L, Doyen A, Malabat C, Saveanu C, Jacquier A, Fromont‐Racine M (2013) Cdc48‐associated complex bound to 60S particles is required for the clearance of aberrant translation products. Proc Natl Acad Sci USA 110: 5046–5051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doma MK, Parker R (2006) Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440: 561–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddins MJ, Carlile CM, Gomez KM, Pickart CM, Wolberger C (2006) Mms2‐Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage‐specific polyubiquitin chain formation. Nat Struct Mol Biol 13: 915–920 [DOI] [PubMed] [Google Scholar]
- van den Elzen AM, Henri J, Lazar N, Gas ME, Durand D, Lacroute F, Nicaise M, van Tilbeurgh H, Seraphin B, Graille M (2010) Dissection of Dom34‐Hbs1 reveals independent functions in two RNA quality control pathways. Nat Struct Mol Biol 17: 1446–1452 [DOI] [PubMed] [Google Scholar]
- Gamble CE, Brule CE, Dean KM, Fields S, Grayhack EJ (2016) Adjacent codons act in concert to modulate translation efficiency in yeast. Cell 166: 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garzia A, Jafarnejad SM, Meyer C, Chapat C, Gogakos T, Morozov P, Amiri M, Shapiro M, Molina H, Tuschl T, Sonenberg N (2017) The E3 ubiquitin ligase and RNA‐binding protein ZNF598 orchestrates ribosome quality control of premature polyadenylated mRNAs. Nat Commun 8: 16056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE (2018) UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci 27: 14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilal T, Spahn CM (2015) Ribosome rescue and protein quality control in concert. Mol Cell 57: 389–390 [DOI] [PubMed] [Google Scholar]
- van Hoof A, Frischmeyer PA, Dietz HC, Parker R (2002) Exosome‐mediated recognition and degradation of mRNAs lacking a termination codon. Science 295: 2262–2264 [DOI] [PubMed] [Google Scholar]
- Hurley JH, Lee S, Prag G (2006) Ubiquitin‐binding domains. Biochem J 399: 361–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada T, Winstall E, Tarun SZ Jr, Yates JR 3rd, Schieltz D, Sachs AB (2002) One‐step affinity purification of the yeast ribosome and its associated proteins and mRNAs. RNA 8: 948–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim F, Maragkakis M, Alexiou P, Mourelatos Z (2018) Ribothrypsis, a novel process of canonical mRNA decay, mediates ribosome‐phased mRNA endonucleolysis. Nat Struct Mol Biol 25: 302–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi K, Inada T (2016) Ribosome‐associated Asc1/RACK1 is required for endonucleolytic cleavage induced by stalled ribosome at the 3′ end of nonstop mRNA. Sci Rep 6: 28234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno‐Borchart A, Doenges G, Schwob E, Schiebel E, Knop M (2004) A versatile toolbox for PCR‐based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21: 947–962 [DOI] [PubMed] [Google Scholar]
- Juszkiewicz S, Hegde RS (2017) Initiation of quality control during poly(A) translation requires site‐specific ribosome ubiquitination. Mol Cell 65: 743–750 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juszkiewicz S, Chandrasekaran V, Lin Z, Kraatz S, Ramakrishnan V, Hegde RS (2018) ZNF598 is a quality control sensor of collided ribosomes. Mol Cell 72: 469–481 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimanius D, Forsberg BO, Scheres SH, Lindahl E (2016) Accelerated cryo‐EM structure determination with parallelisation using GPUs in RELION‐2. Elife 5: e18722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Kikuno I, Kuroha K, Saito K, Ito K, Ishitani R, Inada T, Nureki O (2010) Structural basis for mRNA surveillance by archaeal Pelota and GTP‐bound EF1alpha complex. Proc Natl Acad Sci USA 107: 17575–17579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroha K, Tatematsu T, Inada T (2009) Upf1 stimulates degradation of the product derived from aberrant messenger RNA containing a specific nonsense mutation by the proteasome. EMBO Rep 10: 1265–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroha K, Akamatsu M, Dimitrova L, Ito T, Kato Y, Shirahige K, Inada T (2010) Receptor for activated C kinase 1 stimulates nascent polypeptide‐dependent translation arrest. EMBO Rep 11: 956–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Matsuo Y, Ikeuchi K, Saeki Y, Iwasaki S, Schmidt TC, Udagawa T, Sato F, Tsuchiya H, Becker T, Tanaka K, Ingolia N, Beckmann R, Inada T (2017) Ubiquitination of stalled ribosome triggers ribosome‐associated quality control. Nature Commun 8: 159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panasenko OO, Collart MA (2012) Presence of Not5 and ubiquitinated Rps7A in polysome fractions depends upon the Not4 E3 ligase. Mol Microbiol 83: 640–653 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Reyes‐Turcu FE, Horton JR, Mullally JE, Heroux A, Cheng X, Wilkinson KD (2006) The ubiquitin binding domain ZnF UBP recognizes the C‐terminal diglycine motif of unanchored ubiquitin. Cell 124: 1197–1208 [DOI] [PubMed] [Google Scholar]
- Saito K, Horikawa W, Ito K (2015) Inhibiting K63 polyubiquitination abolishes no‐go type stalled translation surveillance in Saccharomyces cerevisiae . PLoS Genet 11: e1005197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y, Fujita H, Yoshikawa A, Yamashita M, Yamagata A, Kaiser SE, Iwai K, Fukai S (2011) Specific recognition of linear ubiquitin chains by the Npl4 zinc finger (NZF) domain of the HOIL‐1L subunit of the linear ubiquitin chain assembly complex. Proc Natl Acad Sci USA 108: 20520–20525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Kowalinski E, Shanmuganathan V, Defenouillere Q, Braunger K, Heuer A, Pech M, Namane A, Berninghausen O, Fromont‐Racine M, Jacquier A, Conti E, Becker T, Beckmann R (2016) The cryo‐EM structure of a ribosome‐Ski2‐Ski3‐Ski8 helicase complex. Science 354: 1431–1433 [DOI] [PubMed] [Google Scholar]
- Shao S, Hegde RS (2014) Reconstitution of a minimal ribosome‐associated ubiquitination pathway with purified factors. Mol Cell 55: 880–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen PS, Park J, Qin Y, Li X, Parsawar K, Larson MH, Cox J, Cheng Y, Lambowitz AM, Weissman JS, Brandman O, Frost A (2015) Protein synthesis. Rqc2p and 60S ribosomal subunits mediate mRNA‐independent elongation of nascent chains. Science 347: 75–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Green R (2012) Translation drives mRNA quality control. Nat Struct Mol Biol 19: 594–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms CL, Yan LL, Zaher HS (2017) Ribosome collision is critical for quality control during no‐go decay. Mol Cell 68: 361–373 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitron CS, Park JH, Brandman O (2017) Asc1, Hel2, and Slh1 couple translation arrest to nascent chain degradation. RNA 23: 798–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaramoorthy E, Leonard M, Mak R, Liao J, Fulzele A, Bennett EJ (2017) ZNF598 and RACK1 regulate mammalian ribosome‐associated quality control function by mediating regulatory 40S ribosomal ubiquitylation. Mol Cell 65: 751–760 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svidritskiy E, Brilot AF, Koh CS, Grigorieff N, Korostelev AA (2014) Structures of yeast 80S ribosome‐tRNA complexes in the rotated and nonrotated conformations. Structure 22: 1210–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi T, Kuroha K, Kudo K, Makino S, Inoue E, Kashima I, Inada T (2012) Dom34:hbs1 plays a general role in quality‐control systems by dissociation of a stalled ribosome at the 3′ end of aberrant mRNA. Mol Cell 46: 518–529 [DOI] [PubMed] [Google Scholar]
- Waters MG, Blobel G (1986) Secretory protein translocation in a yeast cell‐free system can occur posttranslationally and requires ATP hydrolysis. J Cell Biol 102: 1543–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AS, Grayhack EJ (2015) Asc1, homolog of human RACK1, prevents frameshifting in yeast by ribosomes stalled at CGA codon repeats. RNA 21: 935–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau R, Rape M (2016) The increasing complexity of the ubiquitin code. Nat Cell Biol 18: 579–586 [DOI] [PubMed] [Google Scholar]
- Zhang J, Pan X, Yan K, Sun S, Gao N, Sui SF (2015) Mechanisms of ribosome stalling by SecM at multiple elongation steps. Elife 4: e09684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016) Gctf: real‐time CTF determination and correction. J Struct Biol 193: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA (2017) MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 14: 331–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The Cryo‐EM structure reported here has been deposited in the Protein Data Bank under accession code 6I7O and in the Electron Microscopy Data Bank under accession code EMD‐4427.