

Abstract
The ribosomal DNA (rDNA) is the most evolutionarily conserved segment of the genome and gives origin to the nucleolus, an energy intensive nuclear organelle and major hub influencing myriad molecular processes from cellular metabolism to epigenetic states of the genome. The rDNA/nucleolus has been directly and mechanistically implicated in aging and longevity in organisms as diverse as yeasts, Drosophila, and humans. The rDNA is also a significant target of DNA methylation that silences supernumerary rDNA units and regulates nucleolar activity. Here, we introduce an age clock built exclusively with CpG methylation within the rDNA. The ribosomal clock is sufficient to accurately estimate individual age within species, is responsive to genetic and environmental interventions that modulate life-span, and operates across species as distant as humans, mice, and dogs. Further analyses revealed a significant excess of age-associated hypermethylation in the rDNA relative to other segments of the genome, and which forms the basis of the rDNA clock. Our observations identified an evolutionarily conserved marker of aging that is easily ascertained, grounded on nucleolar biology, and could serve as a universal marker to gauge individual age and response to interventions in humans as well as laboratory and wild organisms across a wide diversity of species.
The ultraconserved ribosomal DNA (rDNA) is essential for life and directly relevant to aging, a universal phenotype exhibited by organisms as diverse as yeasts, plants, worms, flies, and humans. The rDNA locus is an ideal candidate to harbor fundamental and evolutionarily conserved aging mechanisms as well as yield widely applicable markers of aging. In eukaryotes, the array originates the nucleolus, the site of ribosomal RNA (rRNA) transcription and major hub influencing molecular processes from cellular metabolism to genome stability and genome-wide gene expression (Pederson 1998; Murayama et al. 2008; Ide et al. 2010; Németh and Längst 2011; Grummt 2013). Nucleolar size and activity have been mechanistically implicated in aging and longevity (Buchwalter and Hetzer 2017; Tiku et al. 2017). Yet ascertaining nucleolar function through easily measurable and scalable markers has been a challenge. Furthermore, the rDNA array has defied sequencing and assembly technologies and has remained missing from genome assemblies of even the best-studied organisms such as human and flies. Thus, epigenetic states of the rDNA have not been ascertained in most epigenomic studies to date, and the segment is missing from commercial array platforms that are widely used to ascertain DNA methylation in human population and epidemiological studies.
Aging is accompanied by dramatic changes in myriad biological attributes across molecular, cellular, and organismal levels (López-Otín et al. 2013). Various genomic and epigenomic modifications, for instance, are gradually altered during the aging process (Fraga and Esteller 2007). Tracing these alterations with widely available technologies provide opportunities to understand the aging process and assess individual age. Telomere loss, for instance, impacts cellular fitness through an undisputable mechanism and is straightforwardly ascertained. However, despite ease of measure and a biological mechanism linking telomere loss with aging, the efficacy of telomere length as an aging biomarker has been equivocal (Mather et al. 2011). On the other hand, CpG sites across the genome can increase or decrease their methylation levels during aging (Garagnani et al. 2012; Heyn et al. 2012). This feature leads to methylation age predictors that can successfully estimate chronological age in humans and mice (Hannum et al. 2013; Horvath 2013; Petkovich et al. 2017; Stubbs et al. 2017; Thompson et al. 2017; Wagner 2017). These models were built from thousands of CpGs scattered across the genome and have proven useful in ascertaining individual age within a species. However, the significance of these methylation clock sites has remained unknown, and the mechanism linking DNA methylation changes with age is unclear. These sites, moreover, differed between studies of the same species and were not conserved between humans and mice. Finally, DNA methylation age studies did not include information about the rDNA array.
The 45S rDNA is a significant target of the DNA methylation machinery that silences supernumerary rDNA units and regulates nucleolar activity (Santoro and Grummt 2001; Espada et al. 2007; McStay and Grummt 2008). Each unit of the tens to hundreds of repeats harbors more than 1500 CpGs, or more than 10 CpGs per 100 nt (Supplemental Fig. S1). Given the close connection between rDNA and nucleolus with aging, we hypothesized that rDNA methylation (rDNAm) might have a uniquely strong association with age and enable the construction of a universally applicable marker of aging that is grounded on nucleolar biology (rDNAm age). To address the issues, we combined several data sets with age information to ascertain the methylation landscape of the rDNA and its dynamics during aging in mice, canids, and humans.
Results
The methylation landscape of the rDNA during aging
We started by examining a recently published data set of whole blood reduced representative bisulfite sequencing (RRBS) from C57BL/6 mice at ages ranging from 0.67 to 35 mo (16 age stages) (Supplemental Table S1; Petkovich et al. 2017). We first confirmed that >99% of rDNA reads are accurately mapped (Supplemental Fig. S2) and that batch effects are not influential (Supplemental Fig. S2G,H; Methods). To address the question of how methylation of individual CpG sites changes during aging, we correlated each of 928 informative CpGs (depth ≥ 50 in >90% samples) with age. We observed that 620 sites (66.8%) uniformly distributed along the transcribed and promoter regions of the rDNA displayed statistically significant positive correlation with age (Spearman's correlation, ρage > 0, FDR < 0.01) (Fig. 1A). Methylation of CpG 7044 located 10 nt downstream from the mature 5.8S sequence displayed the strongest association with age (ρage = 0.78, P < 2.2 × 10−16) (Fig. 1B). The age-associated hypermethylation is consistent for both strands of the rDNA sequence (Supplemental Fig. S2I). Further inspection showed that age has no impact on the sequencing depth of a sample and that our read depth cutoff is sufficient to accurately estimate ρage (Supplemental Fig. S3). These results are consistent with previous qualitative observations that rDNA is more methylated in aged mice and rats (Swisshelm et al. 1990; Oakes et al. 2003). Conversely, a recent study did not observe a clear pattern of age association for about 5 CpGs in the rDNA promoter of humans and rats (D'Aquila et al. 2017). We analyzed two additional data sets to further validate the age association of rDNA methylation in mice (Hahn et al. 2017; Stubbs et al. 2017). Both data sets confirmed that CpGs with higher ρage are also more likely to be hypermethylated in old mice, despite differences in tissue types and strains (Spearman's ρ ≥ 0.24, P ≤ 6.38 × 10−9) (Supplemental Fig. S4).
Figure 1.
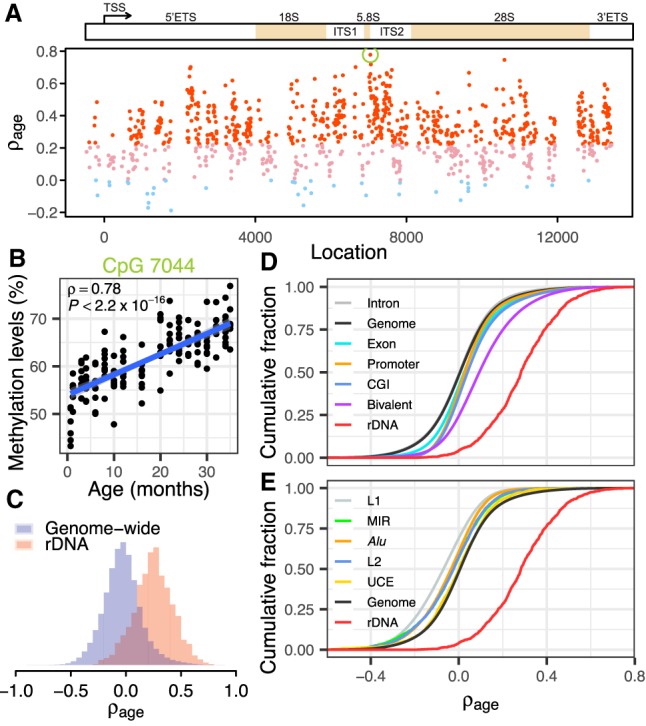
Ribosomal DNA methylation is strongly associated with age. (A) Age-associated hypermethylation of rDNA CpGs in mice. Spearman's correlation coefficients with age (ρage) for CpGs along the rDNA sequence. Red dots indicate CpGs with significant positive correlation with age (ρage > 0, FDR < 0.01); pink and light blue dots denote nonsignificant CpGs (FDR > 0.01) with positive and negative coefficients, respectively. The green circle indicates CpG 7044. (B) Scatterplot shows the correlation between CpG 7044 methylation and age (ρ = 0.78, P < 2.2 × 10−16). (C) The distribution of ρage for CpGs within the rDNA and across the whole genome (Wilcoxon rank-sum test, P < 2.2 × 10−16). (D,E) Cumulative distribution of correlation coefficients for various genomic elements. CpGs within CpG islands (CGIs) and bivalent chromatin have significantly higher ρage than the genome-wide background, although both of them are significantly lower than rDNA CpGs (P < 2.2 × 10−16). Repetitive elements, including L1, L2, Alu (B elements), and mammalian-wide interspersed repeats (MIR), tend to be hypomethylated during aging (P < 2.2 × 10−16) relative to the genome-wide background. All features on D, except for intron, show a shift to the right of the genome-wide background. All features on E, except for the rDNA, show a shift to the left of the genome-wide background. Also see Supplemental Figure S5.
As a comparison, we assessed genome-wide patterns of DNA methylation changes during aging. In contrast to the strong age-associated hypermethylation of the rDNA, we found a slight bias toward loss of DNA methylation with age for CpGs elsewhere in the genome. The proportion of CpGs with positive correlation (ρ > 0.2) across the genome is markedly lower than that in the rDNA (9.14% genome-wide versus 71.8% in rDNA) (Fig. 1C). A closer examination of different classes of genomic segments showed a similar and rather neutral distribution of age association for CpGs within introns, exons, promoters, and ultraconserved elements (UCEs) (Fig. 1D,E). Although CpG islands (CGIs) and bivalent chromatin (regions marked by both H3K27me3 and H3K4me3 modifications) displayed moderate shifts toward hypermethylation, none of them are as pronounced as rDNA. On the other hand, repetitive elements including L1, L2, Alu (B elements), and mammalian-wide interspersed repeats (MIRs) tend to be hypomethylated during aging. We also scanned the mouse genome with windows containing a similar number of informative CpGs as the rDNA, but all of them displayed a much smaller mean ρage than the rDNA (Supplemental Fig. S5). Similarly, a limited fraction of the 15-kb segments (<3%, average of 51 CpGs per segment) displayed mean ρage that was equal to or higher than the mean ρage of rDNA (Supplemental Fig. S5). Collectively, these observations establish the rDNA as a hot spot for age-associated DNA methylation.
CpGs from the rDNA are sufficient to accurately predict age within species
Genomic screens for DNA methylation markers of age across non-rDNA segments identified 353 and 90 CpG sites with the ability to predict chronological age in the human and mouse genomes (Hannum et al. 2013; Horvath 2013; Petkovich et al. 2017; Stubbs et al. 2017). These sites, however, are scattered along the genome and are neither functionally related to one another nor evolutionarily conserved across species. In view of the well-established mechanistic link between rDNA/nucleolus activity and aging as well as the uniquely strong association of rDNA methylation with age, we hypothesized that rDNA CpGs might be sufficient to predict chronological age. To address this, we randomly divided the mouse data set into two subsets of equal size and applied an elastic-net regression model to the 816 CpGs with depth ≥ 50 in all the samples of the Petkovich set (Petkovich et al. 2017). The procedure yielded two models each using one subset for training and the other subset for testing. Each model identified a group of CpGs (or clock sites), estimated site-specific weights, and yielded a predicted age (or rDNAm age) in the training and testing sets (Fig. 2A,B). Due to the stochastic nature of the sample splitting and feature selection steps, we repeated the entire procedure 10,000 times (20,000 models in total). We also used two metrics to ascertain fit: the median absolute error (MAE) between the predicted and observed ages (in units of months) and the correlation between rDNAm and chronological age. Examination of the MAEs and correlations between rDNAm age and chronological age of the test subsets showed strong overall fit (median Spearman's ρ = 0.92 and median MAE = 3.15 mo; highest correlation ρ = 0.98; lowest MAE = 1.63 mo) (Fig. 2C,D). Overall, 736 unique (of the 816) CpGs were selected in these models (with occurrence between 1 and 19,957 models) corresponding to 786,095 total events (considering one CpG occurrence in one model as an event). From the 736 CpGs, the 200 most frequent CpGs accounted for 95.3% of all the models (Supplemental Fig. S6). The 22 most frequent CpGs accounted for 43.2% of all events, and all of these 22 CpGs were selected in more than half of all models. The best-fitted model (lowest MAE) used 72 CpGs located across the rDNA gene region (Fig. 2E; Supplemental Table S2).
Figure 2.
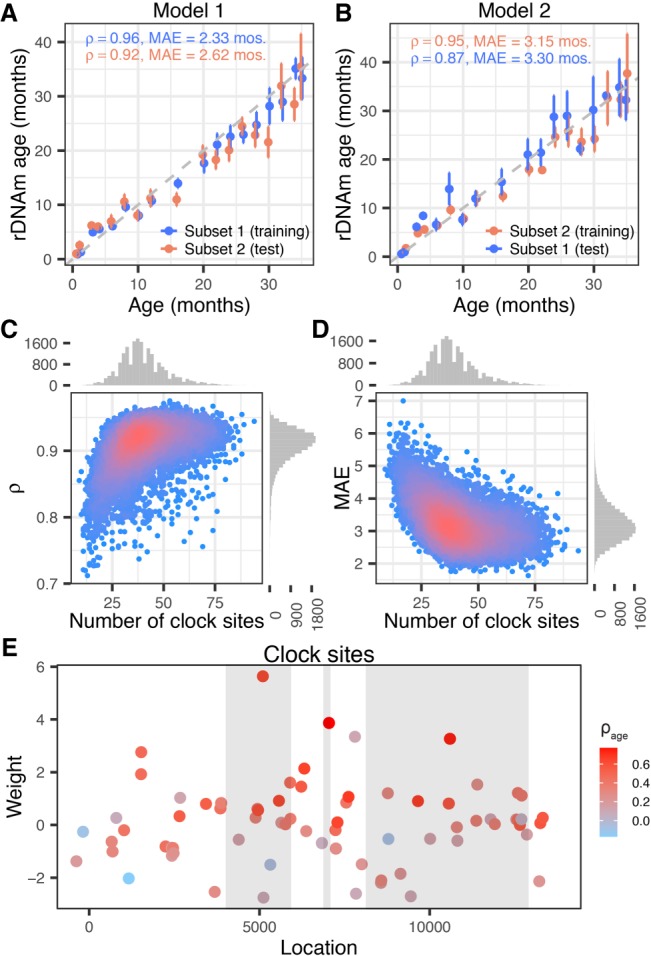
Building the rDNAm age clock. (A,B) Example of two rDNA methylation clock models: (A) Model 1; (B) Model 2. Note that the training and testing subsets are reversed in the two models. (C,D) Performance of 20,000 models trained and tested on randomly split subsets of the mice data set. (C) Correlation coefficients (ρ) between the predicted age (i.e., rDNAm age) and chronological age of the test subsets were plotted against the number of clock CpGs of each model. (D) The median absolute errors (MAEs) of the rDNAm ages were plotted against the number of clock CpGs of each model. (E) Location and weights of the 72 clock sites identified by the best-fitted model. The three gray blocks represent the 18S, 5.8S, and 28S components (from left to right). The color coding represents the strength of age association in each site.
The rDNAm clock is responsive to interventions that modulate life-span
We next examined whether the rDNAm clock is responsive to genetic and environmental interventions that are known to modulate life-span. The best-fitted model was used in this analysis. Calorie restriction (CR) has long been reported to extend life-span and retard aging. For the C57BL/6 mice subjected to CR starting at 14 wk old, we observed lower rDNAm age compared to their ad libitum (AL) controls (one-tailed t-test of the differences between rDNAm age and chronological age, P = 1.17 × 10−9) (Fig. 3A). The CR effect remained obvious when instead examining the B6D2F1 strain mice (P = 0.002) (Supplemental Fig. S7A). Consistently, the slow-aging full-body growth hormone receptor knockout (Ghr KO) mice showed significant reduction in rDNAm age (P = 0.013) (Supplemental Fig. S7B) compared to wild-type controls. Moreover, we also observed significantly lower rDNAm ages for induced pluripotent stem cell (iPSC) lines relative to their kidney and lung fibroblasts progenitors (P ≤ 0.039) (Fig. 3B), which indicates that the iPSC lines have rDNA methylation that more closely resembles those in tissues of young mice. We further examined the change of methylation for the intervention groups. Indeed, both CR and Ghr KO mice showed significant decrease in rDNA methylation (Supplemental Fig. S8), especially for CpG sites with strong age-associations (Fig. 3C; Supplemental Fig. S9) compared to their respective controls. Such pattern also holds for old CR mice compared to their corresponding AL ones from the Hahn data set (Supplemental Figs. S8, S9; Hahn et al. 2017) and for iPSC lines compared to their relative fibroblasts (Fig. 3D,E; Supplemental Fig. S8). Overall, these results indicate that genetic and environmental interventions known to influence longevity can impact rDNA methylation and modulate the rDNAm clock in a coherent manner.
Figure 3.

rDNA methylation and the rDNAm clock are responsive to interventions that modulate life-span. (A) C57BL/6 mice subjected to calorie restriction (CR) (starting at 14 wk old) displayed lower rDNAm age than mice fed ad libitum (AL) (one-tailed t-test of the differences between rDNAm age and chronological age, P = 1.17 × 10−9). CR mice were at four age stages (10, 18, 23, and 27 mo old, each with five samples). The theoretical line is shown in dashed gray. The black and blue lines show the regression line for the relationship between chronological and biological age of control and CR mice, respectively. (B) Derived iPSC cell lines have significantly lower rDNAm ages than their progenitor kidney and lung fibroblasts: (*) P ≤ 0.039; three samples in each group. (C–E) Correlations between the ρage of each of the 816 rDNA CpGs used to train the rDNAm clock and their change in methylation caused by interventions. (C) C57BL/6 strain 27-mo-old CR mice were considered (versus ad libitum 26-mo-old mice, i.e., the ones with the closest ages). (D,E) Derived iPSC cell lines relative to fibroblast progenitors. All samples are from the Petkovich set (Petkovich et al. 2017).
The rDNAm clock is evolutionarily conserved and can be applied across species
The rDNA is the most evolutionarily conserved segment of the genome and essential for ribosome biogenesis and function across all domains of life. Using a highly stringent cutoff for homology, we observed that >70% of human CpGs in the 18S and 5.8S are detected in species as divergent as zebrafish (Fig. 4A; Methods). Hence, we hypothesized that the rDNA clock might be evolutionary conserved and asked whether a rDNAm clock trained in one species could be used to gauge age in a distantly related species. To address the issue, we examined a canid data set with 80 blood RRBS methylation samples collected from dogs and wolves (0.4–14 yr old) (Janowitz Koch et al. 2016). We built 10,000 ribosomal age models with the mouse cohort using 88 mouse-canid homologous rDNA CpGs in both mouse and canid sets. This relatively small number of informative homologous CpGs is primarily due to the random enrichment nature of the RRBS technology used in both data sets, rather than the lack of sequence similarity between the two species. Application of mice rDNAm clock models to canids yielded reasonable correlation between rDNAm age and chronological age despite more than 80 million yr of divergence between the two taxa (median ρ = 0.48) (Fig. 4B). On the other hand, clock models built in mice with 7600 homologous CpGs identified between mice and canids and residing outside the rDNA array yielded meager correlations (median ρ = 0.22) (Fig. 4B). Reversing the training and testing sets (i.e., using canids for training and mice for testing), confirmed the much better fit of models built using homologous CpGs from the rDNA relative to those built with homologous CpGs in other regions of the genome (median ρ, 0.62 versus 0.26) (Fig. 4B). Supplemental Fig. S10 shows the application of the two best-fitted models. In agreement with the better performance of rDNA CpGs for estimating age across species, we also observed that the age association of rDNA CpGs in the two species is evolutionarily conserved and displays significant correlation between mice and canids (ρ = 0.49, P = 1.88 × 10−6) (Fig. 4C). On the other hand, homologous non-rDNA CpGs display a much weaker conservation of age association (ρ = 0.09, P = 7.01 × 10−16) (Fig. 4D).
Figure 4.
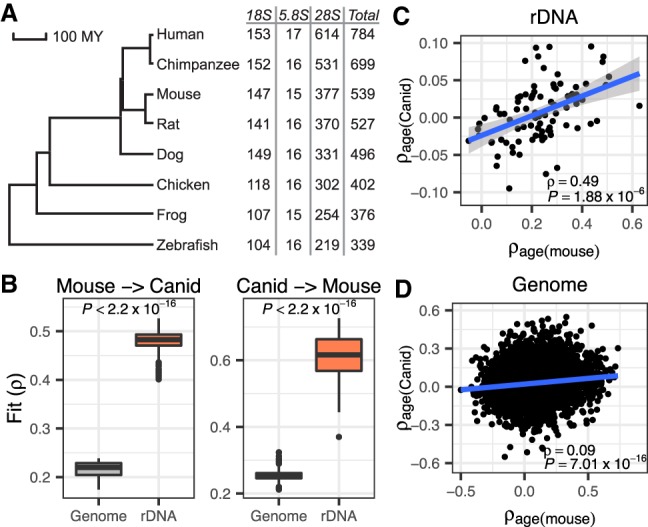
The ribosomal rDNAm clock is conserved between distantly related taxa. (A) Phylogenetic tree of seven vertebrate species with the number of homologous CpGs in each species (relative to human). (B) Interspecific mouse-canid clock models built using homologous CpGs in the rDNA yielded significantly better performance than those built using homologous genome-wide CpGs. Here, the model is trained in one species and applied to the other species. The fit is measured as the correlation coefficient (ρ) between rDNAm age and chronological age in the testing species. (C) Homologous CpGs from the rDNA show conserved age association (ρage) between mice and canids (ρ = 0.49, P = 1.88 × 10−6). (D) Homologous CpGs elsewhere in the genome show age association that is weakly correlated between mice and canid (ρ = 0.09, P = 7.01 × 10−16).
Finally, we applied the mouse rDNAm clock model to human samples. Although rDNA states are not specifically measured in any of the hybridization platforms widely used in human population studies, WGBS data can be used to retrieve rDNA states. We first examined two cell lines (the B lymphocyte derived cell GM12878 from a healthy adult and the embryo stem cell H1). In agreement with a conserved clock, a mouse model built exclusively using mouse-human homologous CpGs yielded much younger rDNAm age for the embryo stem cell than the adult LCL (Supplemental Fig. S11A). To verify this, we applied the mouse model to the skin samples of six adult individuals. As a result, we observed a strong positive correlation between rDNAm age and chronological age (Supplemental Fig. S11B) (ρ = 0.79, P = 0.0041); all skin samples had a predicted age larger than that of the stem cell (Supplemental Fig. S11B). Consistent with the performance of the rDNAm model, CpGs with higher ρage in mice are also more likely to be more hypermethylated in the human adult LCL compared to the human embryo stem cell (Supplemental Fig. S11C), and in the human skin samples of older individuals compared to the skin samples of younger ones (Supplemental Fig. S11D). These observations reveal a conserved ribosomal clock that operates similarly even across distantly related mammalian species, such as mice, humans, and canids.
Discussion
Our study underscores the fundamental role of the ultraconserved rDNA in aging and its ability to serve as a universal predictor of individual age that can be efficiently calibrated for a diversity of species and also be applied across species. The observations are compatible with the well-documented impact of rDNA on aging that is exerted via the manifold consequences of the rDNA on nucleolar activity, cellular metabolism, heterochromatin maintenance, and genome integrity (Murayama et al. 2008; Ide et al. 2010; Larson et al. 2012; Grummt 2013). The data also agree with the observation of significant overrepresentation of ribosomal protein genes among candidates whose expression is associated with age in human, mouse, and Drosophila studies (Zahn et al. 2007; Carlson et al. 2015; Peters et al. 2015). The data presented here further reveal the unique potential of using rDNA CpGs to build methylation age clocks that can be used interspecifically. Collectively, the ribosomal clock likely reflects conserved functions of the nucleolus during aging, can serve as a universal marker of aging, and could be deployed to aging and population studies in natural settings and wild organisms across a variety of species.
Evolutionarily conserved and mechanistic indicators of age represent a critical part of aging research. A satisfying marker of age should (1) be evolutionarily conserved, (2) have a mechanistic relationship with age, (3) be useful in predicting chronological age under control conditions, and (4) be responsive to interventions that are known to decrease or increase longevity. The rDNA array is an excellent candidate to fulfill these criteria. First, the element is ultraconserved and functions consistently across the tree of life from prokaryotes to eukaryotes. Second, the direct role of the rDNA in aging has been unequivocally demonstrated through mechanistic studies. Third, the data show that rDNA methylation is sufficient to predict chronological age and that rDNAm clocks can be applied across species. Finally, rDNA methylation and the rDNAm clock are responsive to calorie restriction and other interventions.
The rDNAm clock reached comparable accuracy to other models calibrated in mouse using sites across the whole genome (Petkovich et al. 2017; Stubbs et al. 2017; Wang et al. 2017). Specifically, the correlation coefficients between predicted and chronological ages for the Petkovich and Wang models were around 0.95 and 0.91, respectively (Petkovich et al. 2017; Wang et al. 2017). For comparison, the median correlation coefficient between predicted and chronological age in the 20,000 rDNAm clocks is ρ = 0.92, and the model with the best correlation displayed a ρ = 0.98. The Stubbs multitissue clock reported a MAE of 3.33 wk (Stubbs et al. 2017). This MAE corresponds to an 8.1% error relative to the oldest mice (MAE of 3.33 wk/age of oldest mice of 41 wk), which is slightly higher than our estimate of 4.7% error for the rDNA clock (MAE of 1.63 mo/age of oldest mice of 35 mo). The performance of the rDNAm clock possibly emerges due to the significant enrichment of rDNA CpGs associated with age, which is much less dramatically observed in other genomic elements and genomic regions.
Noteworthy, the rDNA array has defied sequencing and assembly technologies. The result of this is that the segment remains missing from genome assemblies even in the best-studied organisms such as mouse and human. Thus, the rDNA is not typically incorporated into genomic studies and is similarly missing from prior analysis of age-associated DNA methylation. For instance, in humans, two genomic screens for DNA methylation markers of age did not include the rDNA but identified 353 CpG and 71 CpG sites scattered along the genome with a superb ability to predict chronological age (Hannum et al. 2013; Horvath 2013). The sites, however, are usually located within genes that have no mechanistic link with aging. Hence, skepticism regarding these 353 or 71 nonoverlapping sites has remained partially due to the lack of a functional relationship among the clock sites in each model and the lack of overlap among sites in both models. Similarly, recent methylation age clocks built in mice successfully identified methylation clock sites in mice but again did not include the rDNA array. The efforts led to the identification of some 90 disparate sites that, as in humans, were scattered at seemingly random positions across the genome (Petkovich et al. 2017; Stubbs et al. 2017). There was also no relationship between clock sites in humans and clock sites in mice: Neither the sites nor the nearby genes were conserved between human and mouse, and the clock from one organism could not be applied to the other organism. Although these clock sites are a useful tool they lack a functional relationship with one another, and their identification has not led to a mechanistic understanding of the underlying molecular processes that they were indexing. Our data reconcile observations from transcriptome studies that identified ribosomal genes among the most age-associated transcripts (Zahn et al. 2007; Carlson et al. 2015; Peters et al. 2015) and raise the prospects that methylation clocks might reflect changes in nucleolar biology during aging (Buchwalter and Hetzer 2017; Tiku et al. 2017).
The age-associated rDNA hypermethylation is supported by the alleviation of such pattern in iPSC lines and in CR and Ghr KO mice. The reversal to a more hypomethylated state and near-zero rDNAm age in iPSC lines might also suggest the importance of low rDNA methylation levels in order to maintain the multipotent state. Laron syndrome, which is marked by dwarfism and other characteristic biochemical features, is primarily caused by mutations in the GHR gene that lead to growth hormone resistance (Laron 2004). Here, we observed that Ghr mutant mice displayed hypomethylated rDNA and decelerated rDNAm age; further studies in humans could be relevant to link the clinical outcomes of Laron syndrome to rDNA states. In addition to aging, rDNA hypermethylation has also been observed in young mice whose mothers suffered from poor nutrition (Holland et al. 2016), in the hippocampus of individuals who died by suicide (Carvalho et al. 2010), and linked to diseases including breast carcinoma (Yan et al. 2000; Bacalini et al. 2014) and myelodysplastic syndromes (Raval et al. 2012). The multiple implications of rDNA methylation likely reflect the versatile roles of ribosomal biogenesis and nucleolar activity across various biological processes.
Although the rDNA is universal across eukaryotes, DNA methylation is not. Several unicellular (e.g., some yeasts) and multicellular organisms (e.g., worms) entirely lack CpG methylation or show it at nearly undetectable levels. This is also the case of Drosophila, one of the most widely used complex multicellular model organisms, that either lacks CpG DNA methylation or presents it at an extremely low level (Urieli-Shoval et al. 1982; Goll et al. 2006; Capuano et al. 2014). Yet, the rDNA has been implicated in Drosophila and yeast aging (Larson et al. 2012). Similarly, the most evolutionarily conserved responses to aging (i.e., slower aging upon calorie restriction) are also well-documented in Drosophila and yeast, despite absent or low CpG methylation. Although it is evident that the DNAm clock could not be suitable for Drosophila and yeast, the mechanistic link between the rDNA and aging in Drosophila or yeast needs to be reconciled with the absent or limited role of DNA methylation in these organisms (Kennedy et al. 1997; Capuano et al. 2014). One possibility is that Drosophila and yeast might use unique mechanisms to modulate nucleolar function during aging. This raises the possibility that even more universal age-associated indexes of nucleolar function might exist beyond rDNA methylation, as captured in recent studies of nucleolar size and function during aging (Buchwalter and Hetzer 2017; Tiku et al. 2017).
Overall, it is notable that CpGs identified in this small ∼13 kb segment of the genome is as good a predictor of age as methylation changes monitored across the entire genome. From a practical perspective, clocks based on non-rDNA sites are harder to calibrate because the sites are scattered across the genome and harder to generalize because the sites are not evolutionarily conserved. We envision that rDNAm clocks could be uniquely useful to ascertain age in natural populations for which information on chronological age is not available or is too scarce to enable the calibration of species-specific methylation aging models. The rDNAm clock is not only efficiently developed and calibrated within species with chronological data, such as human and mouse, but it could also be efficiently applied to a diversity of species and nonmodel organisms in the wild, those which lack chronological data.
Methods
rDNA sequences
The consensus 45S rDNA sequences of human, mouse, rat, chicken, and frog were from GenBank (accession numbers: U13369.1, BK000964.3, NR_046239.1, KT445934.2, and X02995.1, respectively). To obtain the rDNA sequences of chimpanzee, canid, and zebrafish, we used BLAT (https://genome.ucsc.edu/cgi-bin/hgBlat) (Kent 2002) to map human rDNA against their genome assemblies (panTro5, canFam3, and danRer11). Specifically, the ChrUn_NW_015976995v1 contig in chimpanzee (18S: 7807–9675, 5.8S: 6583–6739, and 28S: 283–5419; minus strand), the ChrUn_JH373485 contig in canid (18S: 22624–24492, 5.8S: 20599–20755, and 28S: 14596–18348; minus strand), and Chr 5: 820,041–826,807 in zebrafish (18S: 824921–826807, 5.8S: 824487–824644, and 28S: 820041–824135; minus strand) were found with the highest similarities and selected. The downloaded sequences of human and mouse were further modified. Specifically, we moved the promoter (the last 500 bp in the GenBank accession) to be upstream of the transcribed regions (making a contiguous segment with promoter, 5′-ETS, 18S, ITS1, 5.8S, ITS2, 28S, and 3′-ETS).
Identifying homologous CpG sites between species
For the rDNA, we only considered the three mature rRNA regions (18S, 5.8S, and 28S). For each region, we used ClustalW (http://www.genome.jp/tools-bin/clustalw) (Thompson et al. 1994) to align the sequences of pairs of species, and the homologous CpG sites were identified by applying the Perl module Bio::AlignIO. To remove potential error due to misalignment, we further filtered the sites by requiring the two flanking nucleotides (immediately upstream of and downstream from each CpG) also being identical for the considered species. To obtain genome-wide homologous CpGs, we applied the UCSC liftOver chain file, which contains alignment information between genomic assemblies, and the pslMap tool (http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/) to convert the coordinates between two species. We again examined the local context of each CpG and similarly required the two flanking nucleotides to be identical.
Genomic regions
Mouse genes from Ensembl (Aken et al. 2017) release 90 were used to identify exon, intron, and promoter regions. For each gene, the region from 1000 bp upstream of to 500 bp downstream from the transcription start site was considered its promoter. The location of CpG islands, and repetitive elements including Alu (B elements), L1, L2, and MIR, were downloaded from UCSC Table Browser (under CpG islands and RepeatMasker tracks, respectively) (Smit et al. 2016; Tyner et al. 2017). The processed chromatin peaks of H3K27me3 and H3K4me3 modifications of megakaryocyte cells were from the Gene Expression Omnibus (GEO) database (accession numbers: GSM946523 and GSM946527). We converted the peaks from mm9 to mm10 using the UCSC liftOver tool (https://genome.ucsc.edu/cgi-bin/hgLiftOver). Information of ultraconserved elements (UCE) were downloaded from http://ccg.vital-it.ch/UCNEbase (Dimitrieva and Bucher 2013). CpGs along the entire genome and in each of these classes were individually correlated with age (Spearman's rank correlation) using the Petkovich data set (Petkovich et al. 2017). Differences of correlation coefficients among classes were determined with Wilcoxon rank-sum tests.
Description of sequencing data
Our analyses of rDNA methylation rely on whole-genome or reduced representative bisulfite sequencing (WGBS or RRBS) data sets. Methylation arrays that are widely used, for example, the Illumina Infinium BeadChips (450K and 27K), do not currently contain probes that ascertain methylation states of the rDNA loci. The compiled data sets are described below and in Supplemental Table S1. The Petkovich data set (Petkovich et al. 2017) includes 255 samples: (1) 153 samples of C57BL/6 mice from 18 age stages ranging from 0.67 to 35 mo, and 10 B6D2F1 strain mice with two age stages; (2) 20 samples of C57BL/6 and 12 B6D2F1 mice subjected to calorie restriction; (3) 25 samples from two slow-aging mouse models (whole-body growth hormone receptor knockout (Ghr KO) and the snell dwarf, and their respective wild-types controls); and (4) six fibroblasts of lung and kidney (three from each) from 10-wk-old mice, and the six iPSC lines derived from them. Except for fibroblasts and iPSC cells, whole blood was used for RRBS in all the other samples. The Stubbs data set (Stubbs et al. 2017) includes 62 RRBS samples from four tissues (cortex, heart, liver, and lung) of C57BL/6-BABR mice at four age stages (1, 14, 27, and 41 wk). The Hahn data set (Hahn et al. 2017) includes liver WGBS of C3B6F1 female mice that were 5 and 26 mo old, each with three AL and three CR samples (CR started at 12 wk old). The canid data set (Janowitz Koch et al. 2016) includes 45 dogs (0.4 to 14 yr old) and 35 wolves (0.5 to 7 yr old) of both sexes. The WGBS of human skin samples include sun-exposed and sun-protected epidermal tissues from six adult donors (Vandiver et al. 2015). The sun-exposed sample of the 83-yr-old donor was identified as an outlier. The WGBS of human embryo stem cells (H1), and B lymphocyte cells (GM12878) were downloaded from the ENCODE portal (https://www.encodeproject.org) (Sloan et al. 2016). GM12878 was derived from a mother with unknown age and then immortalized.
Processing sequencing data sets
After evaluating sequencing quality using FastQC v0.11.5 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/), we used Trim Galore! v0.4.4 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) to trim the 3′ adaptors as well as low quality bases (BAQ < 20). The “--rrbs” option was used for RRBS reads to remove the filled-in bases. We then used Bismark v0.16.3 (Krueger and Andrews 2011), which invokes Bowtie 2 v2.3.1 (Langmead and Salzberg 2012) to map the bisulfite sequencing reads onto the rDNA reference sequences of each species (with default parameters). The methylated and unmethylated reads were counted using the “bismark_methylation_extractor” script (with optional parameters “-p --no_overlap --CX --bedGraph --gzip --cytosine_report”). For the canid data set we mapped the reads to the ChrUn_JH373485 contig. To examine whether reads derived from other genomic regions were incorrectly mapped onto the rDNA reference, we realigned the rDNA mapped reads from the mouse data sets onto the mouse genome, with the modified BK000964.3 sequence included. We observed that >99% of the reads can be specifically realigned onto BK000964.3 as well as a homologous segment on Chromosome 17 (Supplemental Fig. S2A–C), which can be masked. Overall, protocol mapping choices were made to maximize computational efficiency. Estimates of rDNA methylation were not sensitive to whether the rDNA mapping step was conducted separately or in conjunction with genome mapping. For instance, mapping the RRBS reads of samples from the Petkovich data set (Petkovich et al. 2017) to the rDNA or to the genome (with rDNA) yielded virtually identical estimates of rDNA methylation (Spearman's correlation, median = 0.99, range: 0.96–0.99). Thus, rDNA methylation is consistently estimated regardless of whether one maps reads to the rDNA only or to the assembly. For the human data sets, we similarly remapped human rDNA reads onto a customized version of hg19, which included the modified U13369.1 but with the scaffold GL000220.1 removed. We used hg19 because the assembled locations of 45S rDNA are known in this assembly (scaffold GL000220.1 and the segment Chr 21: 9,815,921–9,838,532), whereas the rDNA is more scattered and less well presented on GRCh38. We observed that >98% of human rDNA reads can be specifically realigned onto U13369.1 and the above segment on Chromosome 21 (Supplemental Fig. S2D,E). For the canid data set, we observed ∼94.68% (ranging from 92.49% to 98.27%) rDNA reads specifically remapped on to ChrUn_JH373485 (Supplemental Fig. S2F). The relatively lower specificity for the canid rDNA reads is likely because of the much less well-assembled genome with several contigs containing rDNA fragments. Overall, the analyses support that almost all of the rDNA mapped reads are indeed from the rDNA sequence. We also separately mapped the reads to mouse (mm10) or canid genomes (canFam3) using Bismark to obtain the methylation of genomic CpGs.
Examination of batch effects
Petkovich et al. (2017) explored batch factors and suggested no perceptible effects on the methylation of genome-wide CpGs. Here, we further examined whether batch effects can be observed for rDNA CpGs. This data set includes three confounding variables: adaptor numbers, library numbers, and flow cells. Because library numbers are almost linearly correlated with flow cells, we instead only considered adaptor numbers and library numbers. We first adopted the linear mixed-effects model method from Petkovich et al. (2017). That is, in a linear mixed-effects model, age and methylation level (of each CpG site) are the response and fixed independent variable, and the confounding factors are random effects. The coefficient of each CpG is then compared with that from a simple linear model, in which age and methylation are the response and independent variable. Indeed, the two coefficients are highly correlated (Spearman's ρ = 0.94, P < 2.2 × 10−16) (Supplemental Fig. S2G). Moreover, as has been suggested (Petkovich et al. 2017), there is certain redundant anti-correlation between library number (flow cell) with age. We also observed that younger mice tend to have larger library numbers, but those mice older than 10 mo had a rather random distribution (ρ = −0.07, P = 0.5). We then only used mice older than 10 mo to estimate the correlation of each CpG with age, and the newly calculated coefficients were compared with those calculated using mice from all age stages. As a result, we observed a very strong correlation between these two coefficients (ρ = 0.83, P < 2.2 × 10−16) (Supplemental Fig. S2H), only with the newly calculated ones having slightly smaller values (possibly due to the reduced sample size and age range). Together, these analyses suggested inconsequential, if any, batch effects on the results.
Building the rDNA age clock models
The rDNAm age clock models were built by using the elastic-net regression algorithm implemented in the glmnet library (Friedman et al. 2010) in R (R Core Team 2017). This method applies multivariate linear regression with the predict and response variables being the methylation levels of CpGs and the logarithm transformed age, respectively. In addition, the model exerts extra constraint on the coefficients of predict variables by adding penalty to the coefficients using the combination of lasso and ridge regulation methods. Specifically, for the set of n mice and p CpG sites, the model finds the set of coefficients, β, that can minimize the following term:
| (1) |
Here, xij is the methylation level of ith mouse at jth CpG; and yi is the log transformed age of ith mouse. Moreover, λ > 0 is a tuning parameter that regulates the overall penalty against the coefficients, and 0 < α < 1 represents a compromise between ridge (α = 0) and lasso (α = 1). In the modeling process, α was set to 0.5 (Horvath 2013; Petkovich et al. 2017), and λ was chosen through 10-fold cross-validation following the one-standard-error rule, for example, the value one standard error larger than the one that minimizes the mean cross-validated error.
For the models applied within the mouse samples, we first randomly split the 153 control-fed C57BL/6 mice samples into two equal subsets and built two models each using one subset for training and the other one for testing. The feature selection nature of the method makes it possible to pick a subset of CpGs to build the model (the rest have coefficients of 0). However, repeating the training process using even the same samples is likely to yield different combinations of CpGs, since the number of input CpGs are much larger than the sample size. To account for such stochasticity, we iterated the division-training-testing procedure for 10,000 times to see how well the method works on average. As in previous methylation clocks (Horvath 2013; Petkovich et al. 2017), the best-fitted model contains some CpGs with low weight or low ρage. It is not entirely clear why including these CpGs with low age correlation helps the clock accuracy (Supplemental Table S3), but they have emerged in all models.
To build models that apply interspecifically, we selected homologous CpGs that have enough reads mapped (≥6 for genomic CpGs and ≥50 for rDNA CpGs) in all samples of both species. The training and testing were processed similarly as within species. Noticing the vast differences in life-span and developmental pace for distinct species, the correlation between rDNAm age and chronological age could be more suitable to evaluate the performance of the model than MAE. When necessary, we scaled the predicted values to obtain the rDNAm ages in the testing species. Specifically, we used the following formula:
| (2) |
Here, P is the predicted value of a sample; and Q0.05 is the 5% quantile of P. Moreover, s is the scale factor:
| (3) |
with Q0.05 being the 95% quantile of P, and Agemax and Agemin being the maximum and minimum chronological ages of the testing samples.
Supplementary Material
Acknowledgments
We thank several members of the Lemos laboratory for their comments and feedback during development of this work and three anonymous reviewers for their comments on an early draft of the manuscript. Earlier aging research in the Lemos laboratory was supported by an Ellison Foundation New Scholars in Aging Award, a Smith Family Award for Excellence in Biomedical Research, and a Star Family Challenge. Work in the Lemos laboratory has been partially supported by a Jere Mead Fellowship, NIGMS grant R01GM122088, and NIEHS grants R01ES027981 and P30ES000002. The authors received no specific funding to conduct this work.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.241745.118.
References
- Aken BL, Achuthan P, Akanni W, Amode MR, Bernsdorff F, Bhai J, Billis K, Carvalho-Silva D, Cummins C, Clapham P, et al. 2017. Ensembl 2017. Nucleic Acids Res 45: D635–D642. 10.1093/nar/gkw1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacalini MG, Pacilli A, Giuliani C, Penzo M, Treré D, Pirazzini C, Salvioli S, Franceschi C, Montanaro L, Garagnani P. 2014. The nucleolar size is associated to the methylation status of ribosomal DNA in breast carcinomas. BMC Cancer 14: 361 10.1186/1471-2407-14-361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchwalter A, Hetzer MW. 2017. Nucleolar expansion and elevated protein translation in premature aging. Nat Commun 8: 328 10.1038/s41467-017-00322-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capuano F, Mülleder M, Kok R, Blom HJ, Ralser M. 2014. Cytosine DNA methylation is found in Drosophila melanogaster but absent in Saccharomyces cerevisiae, Schizosaccharomyces pombe, and other yeast species. Anal Chem 86: 3697–3702. 10.1021/ac500447w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson KA, Gardner K, Pashaj A, Carlson DJ, Yu F, Eudy JD, Zhang C, Harshman LG. 2015. Genome-wide gene expression in relation to age in large laboratory cohorts of Drosophila melanogaster. Genet Res Int 2015: 835624 10.1155/2015/835624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho A, Polanco C, Lima-Brito J, Guedes-Pinto H. 2010. Differential rRNA genes expression in hexaploid wheat related to NOR methylation. Plant Mol Biol Rep 28: 403–412. 10.1007/s11105-009-0165-5 [DOI] [Google Scholar]
- D'Aquila P, Montesanto A, Mandalà M, Garasto S, Mari V, Corsonello A, Bellizzi D, Passarino G. 2017. Methylation of the ribosomal RNA gene promoter is associated with aging and age-related decline. Aging Cell 16: 966–975. 10.1111/acel.12603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrieva S, Bucher P. 2013. UCNEbase—a database of ultraconserved non-coding elements and genomic regulatory blocks. Nucleic Acids Res 41(Database issue): D101–D109. 10.1093/nar/gks1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espada J, Ballestar E, Santoro R, Fraga MF, Villar-Garea A, Németh A, Lopez-Serra L, Ropero S, Aranda A, Orozco H, et al. 2007. Epigenetic disruption of ribosomal RNA genes and nucleolar architecture in DNA methyltransferase 1 (Dnmt1) deficient cells. Nucleic Acids Res 35: 2191–2198. 10.1093/nar/gkm118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Esteller M. 2007. Epigenetics and aging: the targets and the marks. Trends Genet 23: 413–418. 10.1016/j.tig.2007.05.008 [DOI] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R. 2010. Regularization paths for generalized linear models via coordinate descent. J Stat Softw 33: 1–22. 10.18637/jss.v033.i01 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garagnani P, Bacalini MG, Pirazzini C, Gori D, Giuliani C, Mari D, Di Blasio AM, Gentilini D, Vitale G, Collino S, et al. 2012. Methylation of ELOVL2 gene as a new epigenetic marker of age. Aging Cell 11: 1132–1134. 10.1111/acel.12005 [DOI] [PubMed] [Google Scholar]
- Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X, Golic KG, Jacobsen SE, Bestor TH. 2006. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311: 395–398. 10.1126/science.1120976 [DOI] [PubMed] [Google Scholar]
- Grummt I. 2013. The nucleolus—guardian of cellular homeostasis and genome integrity. Chromosoma 122: 487–497. 10.1007/s00412-013-0430-0 [DOI] [PubMed] [Google Scholar]
- Hahn O, Grönke S, Stubbs TM, Ficz G, Hendrich O, Krueger F, Andrews S, Zhang Q, Wakelam MJ, Beyer A, et al. 2017. Dietary restriction protects from age-associated DNA methylation and induces epigenetic reprogramming of lipid metabolism. Genome Biol 18: 56 10.1186/s13059-017-1187-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. 2013. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell 49: 359–367. 10.1016/j.molcel.2012.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ, et al. 2012. Distinct DNA methylomes of newborns and centenarians. Proc Natl Acad Sci 109: 10522–10527. 10.1073/pnas.1120658109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland ML, Lowe R, Caton PW, Gemma C, Carbajosa G, Danson AF, Carpenter AA, Loche E, Ozanne SE, Rakyan VK. 2016. Early-life nutrition modulates the epigenetic state of specific rDNA genetic variants in mice. Science 353: 495–498. 10.1126/science.aaf7040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S. 2013. DNA methylation age of human tissues and cell types. Genome Biol 14: R115 10.1186/gb-2013-14-10-r115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide S, Miyazaki T, Maki H, Kobayashi T. 2010. Abundance of ribosomal RNA gene copies maintains genome integrity. Science 327: 693–696. 10.1126/science.1179044 [DOI] [PubMed] [Google Scholar]
- Janowitz Koch I, Clark MM, Thompson MJ, Deere-Machemer KA, Wang J, Duarte L, Gnanadesikan GE, McCoy EL, Rubbi L, Stahler DR, et al. 2016. The concerted impact of domestication and transposon insertions on methylation patterns between dogs and grey wolves. Mol Ecol 25: 1838–1855. 10.1111/mec.13480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, Gotta M, Sinclair DA, Mills K, McNabb DS, Murthy M, Pak SM, Laroche T, Gasser SM, Guarente L. 1997. Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell 89: 381–391. 10.1016/S0092-8674(00)80219-6 [DOI] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Res 12: 656–664. 10.1101/gr.229202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27: 1571–1572. 10.1093/bioinformatics/btr167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laron Z. 2004. Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958–2003. J Clin Endocrinol Metab 89: 1031–1044. 10.1210/jc.2003-031033 [DOI] [PubMed] [Google Scholar]
- Larson K, Yan SJ, Tsurumi A, Liu J, Zhou J, Gaur K, Guo D, Eickbush TH, Li WX. 2012. Heterochromatin formation promotes longevity and represses ribosomal RNA synthesis. PLoS Genet 8: e1002473 10.1371/journal.pgen.1002473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. 2013. The hallmarks of aging. Cell 153: 1194–1217. 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mather KA, Jorm AF, Parslow RA, Christensen H. 2011. Is telomere length a biomarker of aging? A review. J Gerontol A Biol Sci Med Sci 66: 202–213. 10.1093/gerona/glq180 [DOI] [PubMed] [Google Scholar]
- McStay B, Grummt I. 2008. The epigenetics of rRNA genes: from molecular to chromosome biology. Annu Rev Cell Dev Biol 24: 131–157. 10.1146/annurev.cellbio.24.110707.175259 [DOI] [PubMed] [Google Scholar]
- Murayama A, Ohmori K, Fujimura A, Minami H, Yasuzawa-Tanaka K, Kuroda T, Oie S, Daitoku H, Okuwaki M, Nagata K, et al. 2008. Epigenetic control of rDNA loci in response to intracellular energy status. Cell 133: 627–639. 10.1016/j.cell.2008.03.030 [DOI] [PubMed] [Google Scholar]
- Németh A, Längst G. 2011. Genome organization in and around the nucleolus. Trends Genet 27: 149–156. 10.1016/j.tig.2011.01.002 [DOI] [PubMed] [Google Scholar]
- Oakes CC, Smiraglia DJ, Plass C, Trasler JM, Robaire B. 2003. Aging results in hypermethylation of ribosomal DNA in sperm and liver of male rats. Proc Natl Acad Sci 100: 1775–1780. 10.1073/pnas.0437971100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson T. 1998. The plurifunctional nucleolus. Nucleic Acids Res 26: 3871–3876. 10.1093/nar/26.17.3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters MJ, Joehanes R, Pilling LC, Schurmann C, Conneely KN, Powell J, Reinmaa E, Sutphin GL, Zhernakova A, Schramm K, et al. 2015. The transcriptional landscape of age in human peripheral blood. Nat Commun 6: 8570 10.1038/ncomms9570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkovich DA, Podolskiy DI, Lobanov AV, Lee SG, Miller RA, Gladyshev VN. 2017. Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab 25: 954–960.e6. 10.1016/j.cmet.2017.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Raval A, Sridhar KJ, Patel S, Turnbull BB, Greenberg PL, Mitchell BS. 2012. Reduced rRNA expression and increased rDNA promoter methylation in CD34+ cells of patients with myelodysplastic syndromes. Blood 120: 4812–4818. 10.1182/blood-2012-04-423111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro R, Grummt I. 2001. Molecular mechanisms mediating methylation-dependent silencing of ribosomal gene transcription. Mol Cell 8: 719–725. 10.1016/S1097-2765(01)00317-3 [DOI] [PubMed] [Google Scholar]
- Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, Gabdank I, Narayanan AK, Ho M, Lee BT, et al. 2016. ENCODE data at the ENCODE portal. Nucleic Acids Res 44: D726–732. 10.1093/nar/gkv1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 2016. RepeatMasker Open-4.0 (2013–2015). http://www.repeatmasker.org.
- Stubbs TM, Bonder MJ, Stark AK, Krueger F, BI Ageing Clock Team, von Meyenn F, Stegle O, Reik W. 2017. Multi-tissue DNA methylation age predictor in mouse. Genome Biol 18: 68 10.1186/s13059-017-1203-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swisshelm K, Disteche CM, Thorvaldsen J, Nelson A, Salk D. 1990. Age-related increase in methylation of ribosomal genes and inactivation of chromosome-specific rRNA gene clusters in mouse. Mutat Res 237: 131–146. 10.1016/0921-8734(90)90019-N [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680. 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MJ, vonHoldt B, Horvath S, Pellegrini M. 2017. An epigenetic aging clock for dogs and wolves. Aging (Albany NY) 9: 1055–1068. 10.18632/aging.101211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiku V, Jain C, Raz Y, Nakamura S, Heestand B, Liu W, Späth M, Suchiman HED, Müller RU, Slagboom PE, et al. 2017. Small nucleoli are a cellular hallmark of longevity. Nat Commun 8: 16083 10.1038/ncomms16083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner C, Barber GP, Casper J, Clawson H, Diekhans M, Eisenhart C, Fischer CM, Gibson D, Gonzalez JN, Guruvadoo L, et al. 2017. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res 45: D626–D634. 10.1093/nar/gkw1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urieli-Shoval S, Gruenbaum Y, Sedat J, Razin A. 1982. The absence of detectable methylated bases in Drosophila melanogaster DNA. FEBS Lett 146: 148–152. 10.1016/0014-5793(82)80723-0 [DOI] [PubMed] [Google Scholar]
- Vandiver AR, Irizarry RA, Hansen KD, Garza LA, Runarsson A, Li X, Chien AL, Wang TS, Leung SG, Kang S, et al. 2015. Age and sun exposure-related widespread genomic blocks of hypomethylation in nonmalignant skin. Genome Biol 16: 80 10.1186/s13059-015-0644-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner W. 2017. Epigenetic aging clocks in mice and men. Genome Biol 18: 107 10.1186/s13059-017-1245-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Tsui B, Kreisberg JF, Robertson NA, Gross AM, Yu MK, Carter H, Brown-Borg HM, Adams PD, Ideker T. 2017. Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol 18: 57 10.1186/s13059-017-1186-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan PS, Rodriguez FJ, Laux DE, Perry MR, Standiford SB, Huang TH. 2000. Hypermethylation of ribosomal DNA in human breast carcinoma. Br J Cancer 82: 514–517. 10.1054/bjoc.1999.0955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahn JM, Poosala S, Owen AB, Ingram DK, Lustig A, Carter A, Weeraratna AT, Taub DD, Gorospe M, Mazan-Mamczarz K, et al. 2007. AGEMAP: a gene expression database for aging in mice. PLoS Genet 3: e201 10.1371/journal.pgen.0030201 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.