

Abstract
The homologous recombination repair (HRR) pathway repairs DNA double-strand breaks in an error-free manner. Mutations in HRR genes can result in increased mutation rate and genomic rearrangements, and are associated with numerous genetic disorders and cancer. Despite intensive research, the HRR pathway is not yet fully mapped. Phylogenetic profiling analysis, which detects functional linkage between genes using coevolution, is a powerful approach to identify factors in many pathways. Nevertheless, phylogenetic profiling has limited predictive power when analyzing pathways with complex evolutionary dynamics such as the HRR. To map novel HRR genes systematically, we developed clade phylogenetic profiling (CladePP). CladePP detects local coevolution across hundreds of genomes and points to the evolutionary scale (e.g., mammals, vertebrates, animals, plants) at which coevolution occurred. We found that multiscale coevolution analysis is significantly more biologically relevant and sensitive to detect gene function. By using CladePP, we identified dozens of unrecognized genes that coevolved with the HRR pathway, either globally across all eukaryotes or locally in different clades. We validated eight genes in functional biological assays to have a role in DNA repair at both the cellular and organismal levels. These genes are expected to play a role in the HRR pathway and might lead to a better understanding of missing heredity in HRR-associated cancers (e.g., heredity breast and ovarian cancer). Our platform presents an innovative approach to predict gene function, identify novel factors related to different diseases and pathways, and characterize gene evolution.
Hereditary breast and ovarian cancer (HBOC) is an autosomal dominant cancer susceptibility syndrome, commonly associated with inherited mutations in more than 25 reported genes (Nielsen et al. 2016). Many of these genes belong to the homologous recombination repair (HRR) pathway, which is critical in faithfully repairing cytotoxic DNA double-strand break (DSB) lesions. Commonly, mutations in HRR genes observed in HBOC reduced the ability of the cell to repair DSBs and resulted in a distinguishable pattern of single-base substitutions, termed a genomic signature 3 (Alexandrov et al. 2013; Nik-Zainal et al. 2016). This signature has been used clinically as an indication of mutations in HRR genes such as BRCA1, BRCA2, PALB2, RAD50, and RAD51C (Polak et al. 2017). Polak et al. (2017) showed that signature 3 tumors are variable at the level of base substitutions, and patients can be ranked based on their high to low signature 3. Furthermore, in ∼40% of cancer-derived genomes with a strong signature 3 and in ∼80% of the genomes with a medium signature 3, no pathogenic mutation in any known HRR gene has been detected (Hartmann and Lindor 2016; Polak et al. 2017) despite the observed defect in the HRR. The current notion is that many HBOC cases and their associated signature 3 result from either a combined effect of several gene variants (Hartmann and Lindor 2016) and/or very low frequency mutations, which are distributed across many genes that associate, regulate, or interact with the HRR. These hypotheses point toward a gap in understanding of the HRR pathway and its link to signature 3. This knowledge gap has significant clinical implications as PARP1 inhibitors have recently been approved for treatment of cancer patients with HRR malfunction (Kim et al. 2015). Accordingly, identifying new HRR genes is important for improving diagnostics, opening new therapeutic strategies (Bartz et al. 2006; Lord et al. 2008), and identifying targets for drug development.
Because the HRR pathway is essential across the tree of life and many of its factors show complex evolutionary patterns, we monitored the HRR evolution across multiple eukaryotic species and unbiasedly identified novel HRR factors based on similar evolutionary patterns. The standard phylogenetic profile (PP) methods characterize the evolution of a gene as a pattern of presence or absence of the gene orthologs in a set of genomes (Pellegrini et al. 1999) and search for genes with similar patterns. The underlying assumption is that the proteins encoded by genes may have several biological functions, belong to different pathways, have various functions in different clades, and undergo multiple events of speciation, drift, gene loss, and gene duplication across evolution. If, despite all these possibilities, two or more proteins (or genes) share a similar phylogenetic profiling, they are probably coupled functionally. PP has been used successfully to predict gene function (Eisen and Wu 2002; Enault et al. 2004; Jiang 2008), protein–protein interactions (Sun et al. 2005; Kim and Subramaniam 2006), protein subcellular localization (Marcotte et al. 2000; Pagliarini et al. 2008), cellular organelle location (Avidor-Reiss et al. 2004; Hodges et al. 2012), and gene annotation (Merchant et al. 2007). Nevertheless, a pattern of presence or absence is sometimes too crude to describe evolution, mainly in closely related species in which a protein is rarely completely lost.
Previously we have shown that we can accurately identify coevolution by taking into account minute changes in gene evolution using the normalized phylogenetic profiling (NPP) method (Schwartz et al. 2013; Tabach et al. 2013a,b; Sadreyev et al. 2015). NPP uses a continuous scale of conservation instead of a defining cutoff for the protein being lost or retained and normalizes gene conservation relative to the expected conservation of other genes between the species. We applied NPP to reveal genetic interactions and novel genes in the RNA interference pathway, in RNA methylation, and in different human diseases including cancer (Schwartz et al. 2013; Tabach et al. 2013a,b; Sadreyev et al. 2015; Findlay et al. 2018; Malcov-Brog et al. 2018; Nordlinger et al. 2018; Omar et al. 2018).
In general, PP methods look for global coevolution across the entire tree of life, but a major question is: What happens when the PP is correlated only in part of the tree of life? As the number of sequenced genomes dramatically increases, the question of which species to analyze becomes more central, and a fresh look on species choice is required. Here we consider that the functions of certain sets of proteins became coupled or uncoupled at a defined point in evolution (“locally”) in a specific clade. Hence coevolution between such a set of proteins will be evident in the clade(s) in which they are functionally coupled. This event might happen once or multiple times and is expected to vary for different genes and pathways. Therefore, we hypothesize that correlated PPs should be considered as evidence for a functional relationship between genes even if the correlation is present only in specific clades and not in others. This behavior is expected to be particularly important in complex pathways such as the HRR, which interacts with multiple processes and has genes that are conserved from bacteria to humans (such as the RAD51 family), whereas others are relatively new in evolution (e.g., BRCA1). Moreover, the phenomenon of moonlighting proteins, where genes acquire novel functions, is well documented in HRR (Cabello-Lobato et al. 2017; Kolinjivadi et al. 2017). Moonlighting is an example of a process that causes a deviation in the evolutionary path of a gene between different branches of the evolutionary tree.
Results
HRR genes are coevolved
To follow the coevolution of the HRR pathway across the tree of life, we generated a NPP (Tabach et al. 2013a,b) of all human genes across 578 eukaryotes (Supplemental Table S1) and performed hierarchal clustering on the different NPPs. In parallel, we defined a gold standard list of 79 well-established HRR genes (Supplemental Table S2), including BRCA1, BRCA2, and RAD51, and focused our subsequent analyses on these gold standard genes. We found that the NPPs of the gold standard HRR genes cluster with each other in a statistically significant manner (P = 10−4) (see Supplemental Methods). Furthermore, the clustering reflects the functional stages in the HRR pathway (Fig. 1A; Supplemental Fig. S1). For example, RAD51, RAD51B, RAD51C, RAD51D, DMC1, XRCC2, and XRCC3, which are all members of the RecA/RAD51 family (Lin et al. 2006), cluster together. A second cluster contained RAD9A and RAD1, both members of the 9-1-1 complex, as well as ATM and ATR, which are known to interact with the 9-1-1 complex (Warmerdam et al. 2009; Broustas and Lieberman 2012). Similarly, FANCD2 and FANCI, both members of the related Fanconi anemia (FA) pathway that can heterodimerize (Walden and Deans 2014), were also found in our analyses to coevolve (Fig. 1B). The gold standard HRR genes cluster with genes that have not yet been reported as HRR factors, suggesting that these genes may interact, contribute, or belong to this pathway.
Figure 1.

Evolution of HRR proteins. (A) Normalized phylogenetic profiles (NPPs) of all human protein coding genes after hierarchical clustering and dendrogram leaf order optimization. Each row represents the NPP of a single gene across 578 eukaryotes ordered by their phylogenetic distance from Homo sapiens. The colors in the heat map indicate the relative degree of conservation between a human protein and its ortholog in a certain species (column). When zero, this means that the ortholog is conserved at the average conservation level of orthologs in the species, relative to human; negative values mean less conserved than average, and positive values mean more conserved than average (the values are in Z-scores) (for further details, see Tabach et al. 2013a). White indicates poor conservation, and dark blue indicates highly conserved genes (blue). The bars on the right side represent clusters enriched for known HRR genes, in which the score represents the fraction of known HRR genes in each cluster (Supplemental Methods), and with an FDR-adjusted P-value indicating the significance of the enrichment (hypergeometric test). The colors of the bars indicate the functional module within the HRR pathway (see legend above heat map and in Supplemental Fig. S1). (B) Examples for HRR genes clustered together. Note that only the known HRR genes in each cluster are shown. (C) Detailed view of three couples of genes that coevolved in different clades. (Top) BRCA1 and PALB2 are locally coevolved in animals but not in plants, fungi, and protists. (Middle) RAD52 and FANCA are locally coevolved only in animals. (Bottom) BLM and WRN are locally coevolved in fungi and protists but not in animals or plants. The clades are indicated by colored bars with the Pearson correlation coefficient of the two PPs within the respective clade. The y-axis indicates the NPP score as in A. Red rectangles show regions of coevolution.
Our analyses found that out of all human protein-coding genes, the PP of BRCA1 (associated with 30% of the solved HBOC cases) (Castéra et al. 2014) exhibited the strongest correlation with the PP of PALB2, present in 6% of the solved HBOC cases (Castéra et al. 2014). However, a closer examination revealed that although the NPPs of BRCA1 and PALB2 are strongly correlated when all eukaryotes are considered (R = 0.77, Pearson correlation) (Fig. 1C, top), indicating global coevolution, the actual coevolution was manifested only in animals (R = 0.83) and without any local coevolution in fungi, plants, or protists. This reflects the dramatic evolutionary changes in these genes from single-celled organisms to animals.
The coevolution of two additional functionally related genes showed divergence in different clades. RAD52 and FANCA are important in the single-strand annealing pathway (Palovcak et al. 2017) and are locally coevolved in mammalians (R = 0.63) and invertebrates but not in other clades (Fig. 1C, middle).
As in many cases, functionally related genes are not always coevolved (for several reasons), and thus, we examined whether coevolution in different parts of the tree of life can point to functional associations in the HRR. For example, BLM and WRN, which are both functionally associated in humans and probably across animals, do not show significant coevolution in either animals or plants (like many other functionally related genes). However, BLM and WRN display high local coevolution in fungi (R = 0.93) and protists (R = 0.87), which hints at their functional interactions (Fig. 1C, bottom). These results show that coevolution can be clade specific and that clade specificity may vary between different sets of coevolved genes.
In addition, we tested if clade analysis could identify coevolution undetected across all genomes. We focused on two genes, RAD51 and BLM, as both are important factors in the HRR pathway and play a role in cell cycle regulation. For each of these genes, we identified the 50 genes that most coevolved with them across each of the different clades (eukaryotes, animals, vertebrates, mammalians, plants, fungi, and protists) (Supplemental Table S3). We counted how many genes among the 50 coevolved genes belong either to DNA repair or cell cycle (Fig. 2A; Supplemental Fig. S2). This analysis revealed that DNA repair and cell cycle genes significantly coevolved with BLM and RAD51 in multiple clades (three clades for RAD51 and five out of seven to BLM). Although there was some overlap (Supplemental Fig. S2) among the DNA repair and cell cycle genes between the clades, several of these genes coevolved in a clade-specific manner. Why certain genes coevolved in specific clades and not in others remains a mystery, but these results clearly reflect the functional evolution of RAD51 and BLM in the DNA repair and the cell cycle progression pathways.
Figure 2.
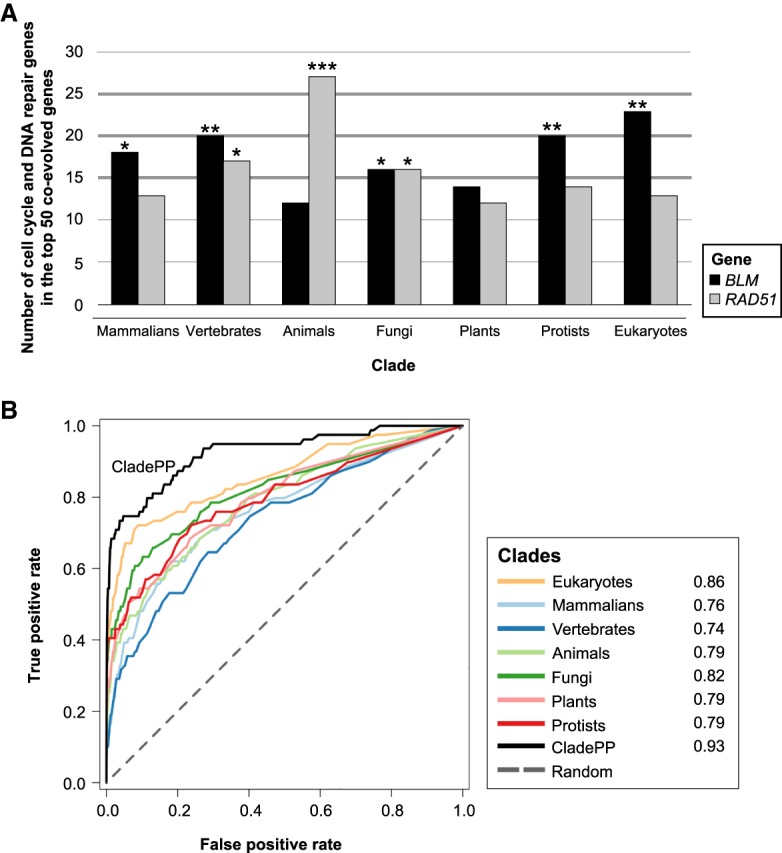
Coevolution of HRR genes is clade-specific. (A) Number of cell cycle and DNA repair genes among the top 50 genes coevolved with BLM and RAD51 in seven different clades. The P-values were calculated using hypergeometric tests. (B) ROC curve of CladePP compared to normalized phylogenetic profiling on individual clades or all eukaryotes, with the HRR gold standard genes used as positives. Numbers in the legend indicate area under the curve (AUC) for each clade. (***) P < 10−5, (**) P < 10−3, (*) P < 0.05.
A method for multiclade phylogenetic profiling
To better map coevolution and identify coevolution that exists only locally, we developed a method to identify systematically genes that significantly coevolve with HRR factors in mammals, vertebrates, animals, plants, protists, or fungi, or across all 578 sequenced eukaryote species. Briefly, we hierarchically clustered the NPP matrix of each clade separately and computed for each gene a score based on how tightly it clustered with at least two of the 79 known HRR gold standard genes in each clade. This method, which we term clade phylogenetic profiling (CladePP), assigns to each gene the maximal score calculated for the clade in which it most strongly coevolves with the gold standard (Methods; Supplemental Methods). As expected, we found that the HRR genes significantly coevolved with each other in the different clades; in particular, many HRR genes locally coevolved in a subset of the clades (Supplemental Fig. S3). For example, we observed that FANCD2 and FANCI coevolved in all eukaryotes and plants but with different partners in each clade. The helicases WRN, BLM, RECQL4, and RECQL5 also clustered together in several clades and coevolved with RECQL in all these clades (Supplemental Fig. S3).
To confirm that the associations predicted by our method were significant, we performed a control analysis in which we ran the algorithm against 10,000 randomized groups of 79 genes (the same size as the gold standard HRR gene list) and tested if they significantly coevolved (Supplemental Methods). The coevolution scores for these random groups were clearly lower compared with the gold standard HRR genes (Supplemental Fig. S4A,B). Moreover, receiver operator characteristic (ROC) analyses showed that our analyses across each clade had sensitive and specific predictions (AUC = 0.93) and outperformed predictions relying on individual clades or relying on all species analysis (Fig. 2B; Supplemental Fig. S4C). The true positives for these analyses were defined as the gold standard.
CladePP analyses yielded 108 genes that significantly coevolved (Q-value ≤ 0.05) with the gold standard genes. These included 41 out of the 79 HRR gold standard genes that tightly coevolved and 67 additional genes that had not previously been implicated in the HRR pathway (Supplemental Table S4). Pathway enrichment analysis of the 67 predicted genes (using GeneAnalytics) (Ben-Ari Fuchs et al. 2016) ranked DNA repair (P < 10−40), DNA recombination (P < 10−23), DSB repair (P < 10−11), and cell cycle (P < 10−10) as the predominant common pathways in the 67 genes.
Validating novel HRR factors
To verify whether the candidate genes identified by our CladePP analysis are associated with the HRR pathway, we validated a spectrum of genes that coevolved with HRR genes in a clade-specific manner. We performed two complementary analyses: the effect of knockdown of these genes on embryonic lethality and brood-size sensitivity to irradiation in Caenorhabditis elegans, and the direct repeat (DR)-GFP assay in human cells.
Effect on brood size in C. elegans
We used the genetically tractable C. elegans to monitor dysfunction of the HRR pathway on the organism level. HRR malfunction is frequently reflected in gametogenesis defects such as embryonic lethal and reduced brood size. A defective HRR pathway results in germline radiation sensitivity (Lemmens et al. 2013). We tested the effect of moderate exposure to ionizing radiation (50 Gy) on early larval (L) stage 4 worms following RNAi treatment. We tested 18 HRR candidates (Supplemental Table S5), out of which 16 showed potential HRR sensitivity. Knockdown of 10 genes caused complete embryonic lethality, and six genes caused a significant reduction in the worm brood size (Fig. 4, below). We calculated the significance of our results using the worm phenotype ontology with the phenotypes “reduced brood size” and “embryonic lethal.” P-values for the hypergeometric test were >3 × 10−7 and >1 × 10−28, respectively, suggesting that the proportion of genes showing phenotypes to the number of genes tested is significant. This assay provides a first indication of the relevance of the genes to the HRR pathway; however, embryonic lethality as well as brood size changes may indicate damage to several different pathways in addition to HRR. For a more specific phenotype, we turned to the DR–GFP system, which measures HRR activity directly.
Figure 4.
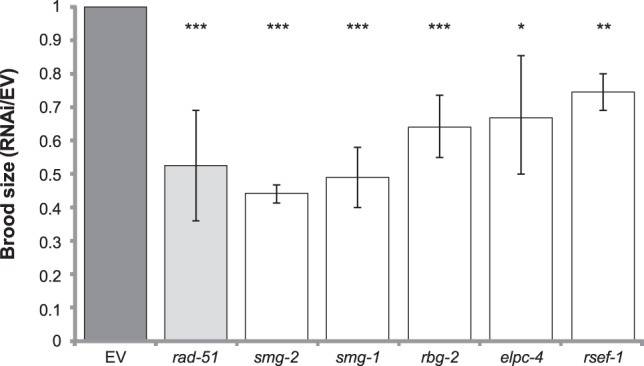
Screen for germline radiation sensitivity in C. elegans. L1 worms were subjected to RNAi of the indicated genes by feeding. Adults were exposed to 50 Gy. After 2 h of recovery, 15 worms were replated with one worm per plate, and 15 nonirradiated worms were plated as controls. After 36 h, the number of eggs and larvae were recorded. The number of progeny is presented relative to worms fed empty-vector containing bacteria. rad-51, a known HRR gene in C. elegans, served as a positive control.
The DR-GFP assay in human cell lines
We monitored the HRR efficiency of a representative sample of these genes in human cells using the well-established DR-GFP assay. It consists of (1) a full-length GFP gene in which an I-SceI restriction site has been introduced such that it disrupts the open reading frame (ORF) of the gene (2) and a truncated GFP version with the correct ORF (iGFP) placed downstream from the full-length GFP gene. Expression of the I-SceI enzyme leads to the generation of a localized DSB in the mutated GFP gene and its subsequent repair by HRR using the iGFP as a template, which can be monitored by the appearance of a GFP signal and quantified by flow cytometry. We chose genes that coevolved in a variety of clades with HRR genes, and analyzed altogether 11 genes in human cells (Fig. 5, below). From these orthogonal validation approaches, we identified eight genes that caused both gametogenesis impairment in C. elegans and a significant reduction in HRR efficiency in two cell lines. Below are some examples of these genes.
Figure 5.

(Top) The DR-GFP reporter assay. SceGFP is a modified version of the GFP gene, which contains an I-Sce site and an in-frame premature stop codon that can be removed by HR-mediated repair of the I-SceI–induced DSB using the internal GFP fragment (iGFP) placed downstream from the SceGFP cassette. (Middle) The experimental setup used to monitor HRR in the HeLa DR-GFP cell line. (Bottom) The percentage of GFP+ cells for each experimental condition (siRNA +/− I-SceI) was measured by flow cytometry (FACS) following the experimental procedure detailed in the top right panel and normalized to the siCtrl + I-SceI conditions, in two human cell lines: HeLa and U2OS. (***) P < 0.0001, (**) P < 0.001, (*) P < 0.01, (ns) nonsignificant.
Examples of validated genes
The first CladePP-based candidate is SMG1, which displayed a strong coevolution pattern with ATR in protists (Fig. 3). SMG1 interacts with multiple gold standard proteins, including ATM, MRE11, CHEK1, and CHEK2 (Szklarczyk et al. 2015). Furthermore, SMG1 is phosphorylated in response to induction of DSBs (Matsuoka et al. 2007), pointing toward a role in DNA repair. We screened this candidate for its involvement in the HRR pathway using C. elegans. Knockdown of smg-1 in C. elegans causes a reduction in brood size, which is a common phenotype of gonadal sensitivity to irradiation (Fig. 4). To further validate that SMG1 is relevant for the HRR pathway, we used the DR-GFP assay. Following knockdown of SMG1 by RNAi, we observed a significant reduction in HRR activity in the cervical HeLa DR-GFP reporter cell line (Fig. 5).
Figure 3.
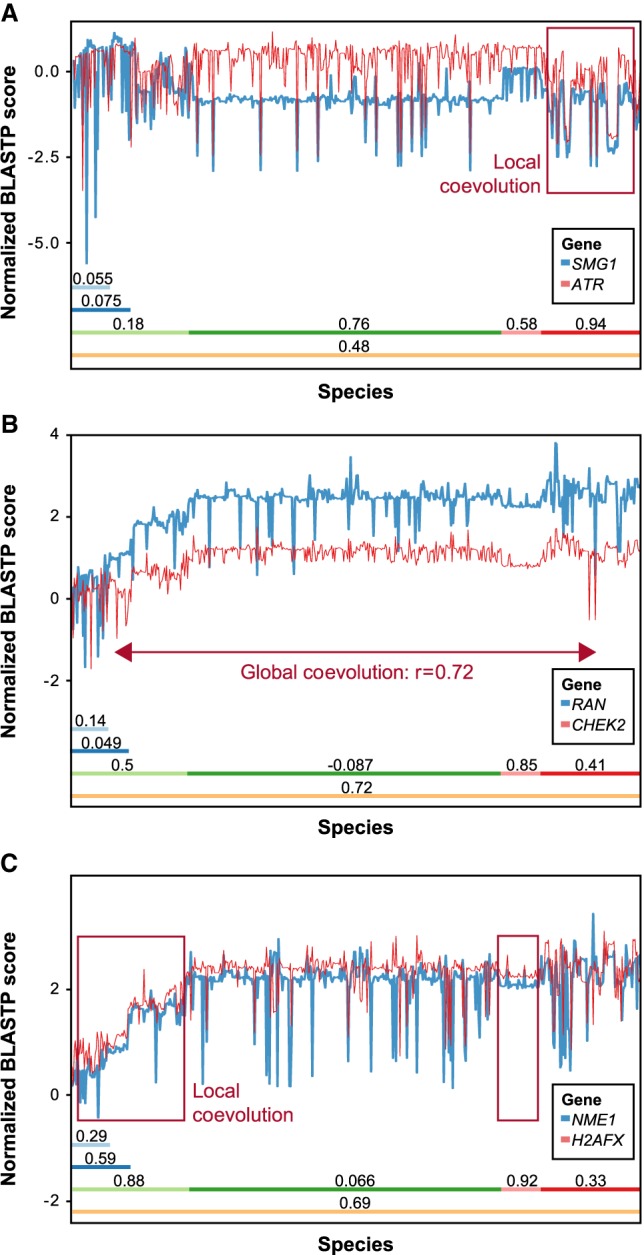
NPPs of SMG1, RAN, and NME1. HRR genes show coevolution within certain clades: (A) SMG1 coevolved with ATR in protists and fungi; (B) RAN coevolved with CHEK2 in plants and across all 578 eukaryotes; and (C) NME1 highly coevolved with H2AFX in plants and animals.
The second gene is RAB3GAP2, which causes Warburg micro syndrome and Martsolf syndrome (Borck et al. 2011). This protein forms the RAB3 GTPase-activating complex with RAB3GAP1 and may be involved in neuronal development. Here we show that depletion of the RAB3GAP2 ortholog rbg-2 in C. elegans correlated with hypersensitivity to irradiation. Similarly, RAB3GAP2 knockdown by RNAi in the HeLa DR-GFP cell line resulted in a strong reduction of HRR activity, pointing toward a role of RAB3GAP2 in HRR (Fig. 5).
RAN is globally coevolved with CHEK2 and with strong local coevolution in plants and animals (Fig. 3). It also came up in a screen in which it reduced HRR in a HeLa DR-GFP assay (Slabicki et al. 2010). Other previous screens found that knockdown of RAN caused retention of H2AFX phosphorylation (Paulsen et al. 2009) and impaired RAD51 foci formation (Herr et al. 2015). The RAN homolog in worms is ran-1, which is known to be embryonic lethal. In both of the human cell lines U2OS and HeLa, RAN siRNA showed a significant reduction in HRR activity.
NME1 is coevolved with H2AFX in animals and plants (Fig. 3). siRNA of the gene also caused increased PARP inhibitor sensitivity in a screen (Lord et al. 2008). NME1 knockdown had a 50% reduction in HRR activity in our experiments in HeLa cells and an even more pronounced reduction in U2OS cells.
Taken together, we validated multiple novel genes as HRR effectors. Overall, we tested 18 genes from the CladePP using the DR-GFP assay and C. elegans. Eight of these genes were validated in both systems (Table 1). These data validate our novel phylogenetic approach, add new genes to the HRR pathway, and suggest that the spectrum of HRR factors is much greater than currently known.
Table 1.
Summary of results of biological validation for novel HRR genes

Discussion
The exponential increase in the availability of fully sequenced eukaryotic genomes revolutionizes our ability to study coevolution and predict protein function using phylogenetic profiling. Although phylogenetic profiling analysis has been used extensively, it was mainly successful in identifying relatively simple metabolic pathways. Most existing PP methods focus on global coevolution that occurs across all analyzed species. In this light, PP has so far been successful in detecting genes that participate in widely conserved systems such as mitochondrial or ciliary genes. In addition, PPs are represented as binary vectors, implying that a protein is completely either present or absent between species. This assumption is unrealistic as genes often show partial conservation, and the level of conservation has a biological significance. In the context of HRR, this is exemplified by RAD52, whose N-terminal region is strongly conserved across eukaryotes, whereas the C-terminal region is poorly conserved. This differential conservation also has important functional implications (Hanamshet et al. 2016). Also, when evolution is considered between closely related species, complete loss of functionally related proteins is a rare event. In such cases, PP analysis should focus on more subtle changes.
Here we resolved these problems by developing a novel phylogenetic profiling approach. We examined coevolution globally by considering the PPs of all human protein-coding coding genes across 578 eukaryotes and locally in six distinct clades: mammalians, vertebrates, animals, fungi, plants, and protists. Our work constitutes a significant conceptual advancement in that we assessed each gene in the clade in which it is most strongly coevolved with known HRR genes, rather than considering all genes across the same set of species. Hence this better uses the information embedded in the unique evolutionary signature of each gene. We applied CladePP to the study of HRR pathway, identified new HRR genes, and experimentally validated multiple candidate genes. The CladePP method is highly scalable, and we anticipate that the method will be applied in the future to the study of additional complex cellular pathways and networks.
We validated our results in two biological systems: C. elegans and human cell lines. Our unbiased approach was even able to identify genes that had a mild effect on the HRR and, as such, are harder to detect. This is even more relevant to the HRR pathway, which has been extensively studied, and thus, it is reasonable to assume that genes with a remarkable effect would probably have been recognized by now. As expected, many of our genes have additional roles in the cells, but their effect on HRR is pronounced. It will be of great value to further elucidate each gene's role in HRR across multiple clades and examine the consistence in their HRR function across evolution.
Methods
HRR gold standard list
We compiled a list, based on an extensive literature review, of 79 recognized HRR genes that affect either DNA damage response or are a part of the HRR pathway (Supplemental Table S2; O'Driscoll and Jeggo 2006; Torres-Rosell et al. 2007; Li and Heyer 2008; San Filippo et al. 2008; Jackson and Bartek 2009; Mladenov and Iliakis 2011; Chapman et al. 2012; Escribano-Diaz et al. 2013; Aparicio et al. 2014; Walden and Deans 2014). Included are genes from the closely related FA pathway and genes such as TP53BP1, which functions in other pathways and yet is known to regulate HRR (Moldovan and D'Andrea 2009; Escribano-Diaz et al. 2013).
Detecting genes coevolved with RAD51 and BLM in different clades
We extracted the subset of columns of the NPP matrix corresponding to each clade. In each clade, we computed the Pearson correlation of the PPs of all genes with the PP of the gene of interest (BLM or RAD51) and retained the top 50 genes. The list of coevolved genes from each clade was then intersected with the list of human genes in the GO biological process terms “strand invasion”; “DNA synthesis involved in DNA repair”; “negative regulation of mitotic cell cycle, embryonic”; “strand displacement”; “mitotic recombination”; “meiotic DNA recombinase assembly”; “response to ionizing radiation”; “reciprocal meiotic recombination”; “regulation of cell proliferation”; “DNA repair”; and “negative regulation of apoptotic process.”
P-values for the intersections were computed using a hypergeometic test, and the P-values for each of the genes were adjusted using the Bonferroni method. We note that the intersection stage was only used for this analysis and that no prior knowledge was used for the rest of the analysis.
Clade phylogenetic profiling
Generating the normalized PPs matrix
To detect genes that coevolved with known HRR genes, we first generated a matrix of PPs of all human protein coding genes as previously described (Tabach et al. 2013a). Specifically, we downloaded the genomes of 578 eukaryotes (Supplemental Table S1) from Ensembl release 83 (Yates et al. 2016) and Ensembl genomes release 30 (Kersey et al. 2016). We then selected from the Homo sapiens genome all genes (ENSG entries) with the gene biotype attribute “protein coding” and a length of at least 40 amino acids. In cases in which a single ENSG entry corresponded to multiple ENSP entries, the ENSP entry with the longest sequence was used. For each gene, we searched for the most similar protein sequence in each of the 578 genomes using BLASTP, obtaining a matrix BS in which BSi,j is the BLASTP bit score of the ith human protein coding gene in the jth genome. Genes that obtained a BLASTP score of 80 or less when aligned against themselves were excluded. To reduce noise, BLASTP scores smaller then a threshold t were floored to t, where t = 24.6, namely, the bitscore value that corresponds to an E-value of 0.05. Genes whose bitscore did not exceed t in at least five genomes were excluded.
When quantitatively comparing the degree of conservation of all human protein coding genes across phylogenetically diverse genomes, two biases are expected:
Bias owing to length. The BLASTP score of an alignment between a pair of proteins linearly increases with the number of positions in the alignment that contain pairs of identical (or similar) amino acids. Consequently, given two pairs of proteins with comparable degrees of sequence similarity, the pair composed of longer proteins will attain a higher score, biasing the analysis toward longer proteins. To correct this bias, we normalized the BLASTP score BSi,j by the BLASTP score of the ith human protein when aligned against itself (BSi,human), obtaining the matrix .
Bias owing to phylogenetic distance. In general, the greater the time span that has passed since the last common ancestor of two genomes, the more differences the genomes accumulate, and the probability to find two proteins in the two genomes with a given BLASTP score s or greater diminishes. To account for this, we transformed each column (representing a species\genome) of the LPP matrix into Z-scores by subtracting from each column its mean μj and dividing the column by its standard deviation σj, yielding , thus attaining for each gene i and genome j a measure for the degree of conservation of gene i relative to the conservation of all other human genes in genome j.
Identifying genes that are coevolved in a specific clade
To identify genes that are coevolved with known HRR genes in the context of a specific clade, we examined six subsets of the complete NPP matrix corresponding to the following clades: mammalians (42 species), vertebrates (the 42 mammalian species and 21 nonmammalian vertebrates), animals (the 63 vertebrates and 57 invertebrates), fungi (315 species), plants (39 species), and protists (104 species), as well as the entire set of eukaryotes (578 species). We decomposed each of these NPP matrices into hierarchical clusters using the hclust function in R (R Core Team 2018), with complete linkage using the distance function , with cor(i,j) being the Pearson correlation coefficient between the PPs of the ith and jth genes. This distance function is minimal when the PPs of the two genes are strongly correlated (cor(i,j) = 1), and maximal when they are anti-correlated (cor(i,j) = −1).
To estimate the extent to which each of the resultant clusters is associated with the HRR pathway, we used our gold standard set of 79 HRR genes and measured for each human protein coding gene how much of its PP clustered with those of the gold standard genes. To do so, we first converted each of the hierarchical clustering objects into a dendrogram. The dendrogram was then traversed recursively, and each cluster a was assigned the cluster ratio score , where G(a) is the set of genes assigned to cluster a and GS is the HRR gold standard. Thus, this score is equal to the fraction of genes in cluster a that belongs to the gold standard. To avoid scenarios in which a candidate gene receives a high score owing to it being clustered with a single gold standard gene, we excluded from this analysis clusters those that contained less than three genes as well as clusters that contained less than two gold standard genes.
Finally, we estimated the extent to which each human protein coding gene g was coevolved with known HRR genes using a two-step maximization process:
-
1.
Intra-clade score maximization. Because of the nature of hierarchical clustering, a dendrogram d contains multiple nested clusters that contain the gene g. Given a dendrogram d(s) derived from the hierarchical clustering of the NPP matrix of clade s, we assign for gene g the maximal clade ratio score MCRS (g, s) = maxc∈d(s):g∈G(c)CR(c), which is the maximal cluster ratio among all clusters in d(s) that contain g.
-
2.
Interclade maximization. Having maximized the ratio score of g in each of the seven clades separately, we then assigned for each gene g the maximal ratio score (MRS): S(g, s) = MCRS (c).
Estimating the statistical significance of the MRS
To validate the significance of the MRS, we simulated 10,000 random sets of genes in the size of the HRR gold standard (79 genes). For each simulated gene set Gsim_i, we computed the MRS for all genes replacing the expression for the cluster ratio score with . This yielded 10,000 vectors of MRSs MRS_[j, sim 1 ],MRS_[j, sim 2 ]…,MRS_[j, s m n], where MRS [j,sim i] is the MRS of the jth gene under the ith simulated gene set. We then estimated the P-value for each gene g the empirical P-value .
Finally, to account for the large number of hypotheses tested, we substituted the P-values with q-values.
Statistical test for the significance of the HRR gold standard being clustered together
To test if the PPs of the HRR gold standard cluster more tightly than expected by chance, we first hierarchically clustered the PPs of all gold standard genes across all 578 species. We then computed the cophenetic distance matrix of the resultant hierarchical clustering dendrogram. For each gene g in the gold standard, we found the cophenetic distance to the gold standard gene nearest to g in the dendrogram. We used the mean of these distances as a summary statistic. We then calculated the mean distance for 10,000 random sets of genes in the same size as the gold standard. The fraction of random sets whose mean distance was lower or equal to the mean distance of the gold standard was used to estimate the P-value.
Software availability
The source code of CladePP is available as Supplemental Code and at GitHub (https://github.com/dolevrahat/CladePP).
Supplementary Material
Acknowledgments
We thank Ayelet Arbel-Eden, Noam Shomron, and Daphna Weissglas for valuable discussions regarding different outputs of this research. This work has been supported by The Israel Cancer Association grant no. 0394837, Melanoma Research Association grant no. 402792, and Israel Innovation Authority grant no. 63374 for Y.T.; National Institutes of Health grant P30 DK04056 for R.S.; and Israel Science Foundation grant no. 1985/13 and Sharett Fund Grant for A.Z. A.O. is the Canada Research Chair (Tier 2) in Genome Stability and Hematological Malignancies. Work in the A.O. laboratory was supported by a Transition Grant from the Cole Foundation and an internal Operating Fund from the Sir Mortimer B. Davis Foundation of the Jewish General Hospital.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.241414.118.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio T, Baer R, Gautier J. 2014. DNA double-strand break repair pathway choice and cancer. DNA Repair 19: 169–175. 10.1016/j.dnarep.2014.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, Zuker CS. 2004. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell 117: 527–539. 10.1016/S0092-8674(04)00412-X [DOI] [PubMed] [Google Scholar]
- Bartz SR, Zhang Z, Burchard J, Imakura M, Martin M, Palmieri A, Needham R, Guo J, Gordon M, Chung N, et al. 2006. Small interfering RNA screens reveal enhanced cisplatin cytotoxicity in tumor cells having both BRCA network and TP53 disruptions. Mol Cell Biol 26: 9377–9386. 10.1128/MCB.01229-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Fuchs S, Lieder I, Stelzer G, Mazor Y, Buzhor E, Kaplan S, Bogoch Y, Plaschkes I, Shitrit A, Rappaport N, et al. 2016. GeneAnalytics: an integrative gene set analysis tool for next generation sequencing, RNAseq and microarray data. OMICS 20: 139–151. 10.1089/omi.2015.0168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borck G, Wunram H, Steiert A, Volk AE, Körber F, Roters S, Herkenrath P, Wollnik B, Morris-Rosendahl DJ, Kubisch C. 2011. A homozygous RAB3GAP2 mutation causes Warburg Micro syndrome. Hum Genet 129: 45–50. 10.1007/s00439-010-0896-2 [DOI] [PubMed] [Google Scholar]
- Broustas CG, Lieberman HB. 2012. Contributions of Rad9 to tumorigenesis. J Cell Biochem 113: 742–751. 10.1002/jcb.23424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabello-Lobato MJ, Wang S, Schmidt CK. 2017. SAMHD1 sheds moonlight on DNA double-strand break repair. Trends Genet 33: 895–897. 10.1016/j.tig.2017.09.007 [DOI] [PubMed] [Google Scholar]
- Castéra L, Krieger S, Rousselin A, Legros A, Baumann JJ, Bruet O, Brault B, Fouillet R, Goardon N, Letac O, et al. 2014. Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur J Hum Genet 22: 1305–1313. 10.1038/ejhg.2014.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Taylor MR, Boulton SJ. 2012. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47: 497–510. 10.1016/j.molcel.2012.07.029 [DOI] [PubMed] [Google Scholar]
- Eisen JA, Wu M. 2002. Phylogenetic analysis and gene functional predictions: phylogenomics in action. Theor Popul Biol 61: 481–487. 10.1006/tpbi.2002.1594 [DOI] [PubMed] [Google Scholar]
- Enault F, Suhre K, Poirot O, Abergel C, Claverie JM. 2004. Phydbac2: improved inference of gene function using interactive phylogenomic profiling and chromosomal location analysis. Nucleic Acids Res 32: W336–W339. 10.1093/nar/gkh365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkáč J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. 2013. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 49: 872–883. 10.1016/j.molcel.2013.01.001 [DOI] [PubMed] [Google Scholar]
- Findlay S, Heath J, Luo VM, Malina A, Morin T, Coulombe Y, Djerir B, Li Z, Samiei A, Simo-Cheyou E, et al. 2018. SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J 37: e100158 10.15252/embj.2018100158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanamshet K, Mazina OM, Mazin AV. 2016. Reappearance from obscurity: mammalian Rad52 in homologous recombination. Genes 7: 63 10.3390/genes7090063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann LC, Lindor NM. 2016. The role of risk-reducing surgery in hereditary breast and ovarian cancer. N Engl J Med 374: 454–468. 10.1056/NEJMra1503523 [DOI] [PubMed] [Google Scholar]
- Herr P, Lundin C, Evers B, Ebner D, Bauerschmidt C, Kingham G, Palmai-Pallag T, Mortusewicz O, Frings O, Sonnhammer E, et al. 2015. A genome-wide IR-induced RAD51 foci RNAi screen identifies CDC73 involved in chromatin remodeling for DNA repair. Cell Discov 1: 15034 10.1038/celldisc.2015.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges ME, Wickstead B, Gull K, Langdale JA. 2012. The evolution of land plant cilia. New Phytol 195: 526–540. 10.1111/j.1469-8137.2012.04197.x [DOI] [PubMed] [Google Scholar]
- Jackson SP, Bartek J. 2009. The DNA-damage response in human biology and disease. Nature 461: 1071–1078. 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Z. 2008. Protein function predictions based on the phylogenetic profile method. Crit Rev Biotechnol 28: 233–238. 10.1080/07388550802512633 [DOI] [PubMed] [Google Scholar]
- Kersey PJ, Allen JE, Armean I, Boddu S, Bolt BJ, Carvalho-Silva D, Christensen M, Davis P, Falin LJ, Grabmueller C, et al. 2016. Ensembl Genomes 2016: more genomes, more complexity. Nucleic Acids Res 44: D574–D580. 10.1093/nar/gkv1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Subramaniam S. 2006. Locally defined protein phylogenetic profiles reveal previously missed protein interactions and functional relationships. Proteins 62: 1115–1124. 10.1002/prot.20830 [DOI] [PubMed] [Google Scholar]
- Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R, et al. 2015. FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res 21: 4257–4261. 10.1158/1078-0432.CCR-15-0887 [DOI] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, de Antoni A, Técher H, Baldi G, Costanzo V. 2017. Moonlighting at replication forks – a new life for homologous recombination proteins BRCA1, BRCA2 and RAD51. FEBS Lett 591: 1083–1100. 10.1002/1873-3468.12556 [DOI] [PubMed] [Google Scholar]
- Lemmens BB, Johnson NM, Tijsterman M. 2013. COM-1 promotes homologous recombination during Caenorhabditis elegans meiosis by antagonizing Ku-mediated non-homologous end joining. PLoS Genet 9: e1003276 10.1371/journal.pgen.1003276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Heyer WD. 2008. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 18: 99–113. 10.1038/cr.2008.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Kong H, Nei M, Ma H. 2006. Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer. Proc Natl Acad Sci 103: 10328–10333. 10.1073/pnas.0604232103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord CJ, McDonald S, Swift S, Turner NC, Ashworth A. 2008. A high-throughput RNA interference screen for DNA repair determinants of PARP inhibitor sensitivity. DNA Repair 7: 2010–2019. 10.1016/j.dnarep.2008.08.014 [DOI] [PubMed] [Google Scholar]
- Malcov-Brog H, Alpert A, Golan T, Parikh S, Nordlinger A, Netti F, Sheinboim D, Dror I, Thomas L, Cosson C, et al. 2018. UV-protection timer controls linkage between stress and pigmentation skin protection systems. Mol Cell 72: 444–456.e7. 10.1016/j.molcel.2018.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte EM, Xenarios I, van Der Bliek AM, Eisenberg D. 2000. Localizing proteins in the cell from their phylogenetic profiles. Proc Natl Acad Sci 97: 12115–12120. 10.1073/pnas.220399497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. 2007. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316: 1160–1166. 10.1126/science.1140321 [DOI] [PubMed] [Google Scholar]
- Merchant SS, Prochnik SE, Vallon O, Harris EH, Karpowicz SJ, Witman GB, Terry A, Salamov A, Fritz-Laylin LK, Marechal-Drouard L, et al. 2007. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318: 245–250. 10.1126/science.1143609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mladenov E, Iliakis G. 2011. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. Mutat Res 711: 61–72. 10.1016/j.mrfmmm.2011.02.005 [DOI] [PubMed] [Google Scholar]
- Moldovan GL, D'Andrea AD. 2009. How the Fanconi Anemia pathway guards the genome. Annu Rev Genet 43: 223–249. 10.1146/annurev-genet-102108-134222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen FC, van Overeem Hansen T, Sørensen CS. 2016. Hereditary breast and ovarian cancer: new genes in confined pathways. Nat Rev Cancer 16: 599–612. 10.1038/nrc.2016.72 [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, et al. 2016. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534: 47–54. 10.1038/nature17676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordlinger A, Dror S, Elkahloun A, Del Rio J, Stubbs E, Golan T, Malcov H, Pricket TD, Cronin JC, Parikh S, et al. 2018. Mutated MITF-E87R in melanoma enhances tumor progression via S100A4. J Invest Dermatol 138: 2216–2223. 10.1016/j.jid.2018.03.1524 [DOI] [PubMed] [Google Scholar]
- O'Driscoll M, Jeggo PA. 2006. The role of double-strand break repair: insights from human genetics. Nat Rev Genet 7: 45–54. 10.1038/nrg1746 [DOI] [PubMed] [Google Scholar]
- Omar I, Guterman-Ram G, Rahat D, Tabach Y, Berger M, Levaot N. 2018. Schlafen2 mutation in mice causes an osteopetrotic phenotype due to a decrease in the number of osteoclast progenitors. Sci Rep 8: 13005 10.1038/s41598-018-31428-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. 2008. A mitochondrial protein compendium elucidates complex I disease biology. Cell 134: 112–123. 10.1016/j.cell.2008.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palovcak A, Liu W, Yuan F, Zhang Y. 2017. Maintenance of genome stability by Fanconi anemia proteins. Cell Biosci 7: 8 10.1186/s13578-016-0134-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen RD, Soni DV, Wollman R, Hahn AT, Yee MC, Guan A, Hesley JA, Miller SC, Cromwell EF, Solow-Cordero DE, et al. 2009. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol Cell 35: 228–239. 10.1016/j.molcel.2009.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. 1999. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci 96: 4285–4288. 10.1073/pnas.96.8.4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polak P, Kim J, Braunstein LZ, Karlic R, Haradhavala NJ, Tiao G, Rosebrock D, Livitz D, Kubler K, Mouw KW, et al. 2017. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat Genet 49: 1476–1486. 10.1038/ng.3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2018. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Sadreyev IR, Ji F, Cohen E, Ruvkun G, Tabach Y. 2015. PhyloGene server for identification and visualization of co-evolving proteins using normalized phylogenetic profiles. Nucleic Acids Res 43: W154–W159. 10.1093/nar/gkv452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. 2008. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77: 229–257. 10.1146/annurev.biochem.77.061306.125255 [DOI] [PubMed] [Google Scholar]
- Schwartz S, Agarwala SD, Mumbach MR, Jovanovic M, Mertins P, Shishkin A, Tabach Y, Mikkelsen TS, Satija R, Ruvkun G, et al. 2013. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell 155: 1409–1421. 10.1016/j.cell.2013.10.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slabicki M, Theis M, Krastev DB, Samsonov S, Mundwiller E, Junqueira M, Paszkowski-Rogacz M, Teyra J, Heninger AK, Poser I, et al. 2010. A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol 8: e1000408 10.1371/journal.pbio.1000408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Xu J, Liu Z, Liu Q, Zhao A, Shi T, Li Y. 2005. Refined phylogenetic profiles method for predicting protein–protein interactions. Bioinformatics 21: 3409–3415. 10.1093/bioinformatics/bti532 [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. 2015. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 43: D447–D452. 10.1093/nar/gku1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabach Y, Billi AC, Hayes GD, Newman MA, Zuk O, Gabel H, Kamath R, Yacoby K, Chapman B, Garcia SM, et al. 2013a. Identification of small RNA pathway genes using patterns of phylogenetic conservation and divergence. Nature 493: 694–698. 10.1038/nature11779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabach Y, Golan T, Hernández-Hernández A, Messer AR, Fukuda T, Kouznetsova A, Liu JG, Lilienthal I, Levy C, Ruvkun G. 2013b. Human disease locus discovery and mapping to molecular pathways through phylogenetic profiling. Mol Syst Biol 9: 692 10.1038/msb.2013.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Rosell J, Sunjevaric I, De Piccoli G, Sacher M, Eckert-Boulet N, Reid R, Jentsch S, Rothstein R, Aragón L, Lisby M. 2007. The Smc5–Smc6 complex and SUMO modification of Rad52 regulates recombinational repair at the ribosomal gene locus. Nat Cell Biol 9: 923–931. 10.1038/ncb1619 [DOI] [PubMed] [Google Scholar]
- Walden H, Deans AJ. 2014. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys 43: 257–278. 10.1146/annurev-biophys-051013-022737 [DOI] [PubMed] [Google Scholar]
- Warmerdam DO, Freire R, Kanaar R, Smits VA. 2009. Cell cycle-dependent processing of DNA lesions controls localization of Rad9 to sites of genotoxic stress. Cell Cycle 8: 1765–1774. 10.4161/cc.8.11.8721 [DOI] [PubMed] [Google Scholar]
- Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, et al. 2016. Ensembl 2016. Nucleic Acids Res 44: D710–D716. 10.1093/nar/gkv1157 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.