

Abstract
Background
Shp1, a tyrosine-phosphatase-1 containing the Src-homology 2 (SH2) domain, is involved in inflammatory and immune reactions, where it regulates diverse signalling pathways, usually by limiting cell responses through dephosphorylation of target molecules. Moreover, Shp1 regulates actin dynamics. One Shp1 target is Src, which controls many cellular functions including actin dynamics. Src has been previously shown to be activated by a signalling cascade initiated by the cytosolic-phospholipase A2 (cPLA2) metabolite glycerophosphoinositol 4-phosphate (GroPIns4P), which enhances actin polymerisation and motility. While the signalling cascade downstream Src has been fully defined, the mechanism by which GroPIns4P activates Src remains unknown.
Methods
Affinity chromatography, mass spectrometry and co-immunoprecipitation studies were employed to identify the GroPIns4P-interactors; among these Shp1 was selected for further analysis. The specific Shp1 residues interacting with GroPIns4P were revealed by NMR and validated by site-directed mutagenesis and biophysical methods such as circular dichroism, isothermal calorimetry, fluorescence spectroscopy, surface plasmon resonance and computational modelling. Morphological and motility assays were performed in NIH3T3 fibroblasts.
Results
We find that Shp1 is the direct cellular target of GroPIns4P. GroPIns4P directly binds to the Shp1-SH2 domain region (with the crucial residues being Ser 118, Arg 138 and Ser 140) and thereby promotes the association between Shp1 and Src, and the dephosphorylation of the Src-inhibitory phosphotyrosine in position 530, resulting in Src activation. As a consequence, fibroblast cells exposed to GroPIns4P show significantly enhanced wound healing capability, indicating that GroPIns4P has a stimulatory role to activate fibroblast migration. GroPIns4P is produced by cPLA2 upon stimulation by diverse receptors, including the EGF receptor. Indeed, endogenously-produced GroPIns4P was shown to mediate the EGF-induced cell motility.
Conclusions
This study identifies a so-far undescribed mechanism of Shp1/Src modulation that promotes cell motility and that is dependent on the cPLA2 metabolite GroPIns4P. We show that GroPIns4P is required for EGF-induced fibroblast migration and that it is part of a cPLA2/GroPIns4P/Shp1/Src cascade that might have broad implications for studies of immune-inflammatory response and cancer.

Electronic supplementary material
The online version of this article (10.1186/s12964-019-0329-3) contains supplementary material, which is available to authorized users.
Keywords: Shp1, EGF, SH2 domain, Glycerophosphoinositols, Phosphoinositides, Actin polymerisation, Membrane ruffles, Cell motility
Background
Glycerophosphoinositols are ubiquitous cellular metabolites derived from the deacylation of the membrane phosphoinositides by receptor-activated cytosolic phospholipase A2α (cPLA2α) [1, 2]. Receptors activating the cPLA2α/glycerophosphoinositol pathway include adrenergic, purinergic and tyrosine kinase receptors such as epidermal growth factor (EGF) or insulin [3–5]. Also lysophosphatidic-acid receptor in fibroblasts [6], Fc receptor in macrophages [7], lipopolysaccharide-TLR4 in human monocytes [8] lead to glycerophosphoinositol production by activating cPLA2α. The glycerophosphoinositol formation is particularly abundant in hematopoietic cells where these compounds are involved in multiple signalling pathways in the context of inflammation and immune reactions [9], generally exerting inhibitory effects [7, 8]. In addition, a major effect of glycerophosphoinositol 4-phosphate (GroPIns4P; the phosphorylated form of glycerophosphoinositol, GroPIns) is the regulation of the actin cytoskeleton dynamics in fibroblasts and immune cells [10–12].
We have previously characterised in depth the activation of the actin cytoskeleton by GroPIns4P, and reported that this metabolite induces membrane ruffling by triggering a phosphorylation cascade initiated by the Src kinase (and, possibly, by the two isoforms of Src) [11]. In NIH3T3 fibroblasts, exogenously added GroPIns4P, acting through Src, led to the phosphorylation – and hence activation – of PLCγ, thereby causing an increase in intracellular Ca2+ levels and the activation of calcium-calmodulin kinase II (CaMKII). In turn, CaMKII induced the activation of the GTPase Rac1 at the plasma membrane by engaging its GTP exchange factor (GEF), Tiam1, finally leading to ruffle formation [11]. A similar pathway controlling actin dynamics was observed in Jurkat T-cells, in which exogenously-added GroPIns4P promotes actin polymerisation by activating the Lck kinase, and inducing the phosphorylation of the GDP/GTP exchanger Vav and subsequent activation of the GTPase Rac, resulting in increased cell motility [9, 12]. This GroPIns4P-dependent Lck activation potentiates the transactivation of the TCR by the SDF1α-stimulated CXCR4 receptor thus enhancing T-cell chemotaxic response [12]. These effects of GroPIns4P might play a role in the immune response by mediating the recruitment of T-cells toward the injured site [9, 12].
Despite the many studies on the numerous and important biological activities of the glycerophosphoinositols [13, 14], the molecular target/s of these metabolites have not yet been identified leaving a major gap in our understanding of their cellular activities.
In this study, we have attempted the isolation of the direct interactors/receptors of the glycerophosphoinositols by pull-down assay coupled with liquid chromatography-tandem mass-spectrometry analysis. Among the molecules identified, we focused on the protein tyrosine-phosphatase 1 (Shp1) because of its well-known role in Src activation and cytoskeleton organisation [15, 16]. Shp1 is a member of the SH2-domain-containing family of non-membrane protein-tyrosine phosphatases expressed in most cells but particularly abundant in hematopoietic cells [17, 18]. It has been implicated in the negative regulation of various receptor-mediated pathways such as the cytokine and chemokine-receptors, T- and B-cell receptors as well as growth factor receptors [15, 16]. Mice deficient in Shp1 (motheaten or viable motheaten) are affected by several immunological, inflammatory, and haematological abnormalities [19, 20]. Studies of hematopoietic cells from these mice indicate that Shp1 plays also an important role in the regulation of the SDF1α-induced signalling pathway [21]. In addition, Shp1 has been shown to have a positive role in regulating the actin cytoskeleton and in activating the Src kinase [15, 22]. Here Shp1 acts by interacting with the Src phosphotyrosine residues and dephosphorylates the inhibitory Tyr530 leading to Src activation [15].
These effects appear to be strikingly reminiscent of those reported for the GroPIns4P-dependent signalling cascade leading to the control of the actin cytoskeleton [10–12]. Prompted by these similarities we examined the interaction between GroPIns4P and Shp1, focussing in particular on the Src activation of the actin cytoskeleton dynamics. We find that GroPIns4P binds to Shp1, through its C-terminal SH2 domain. This binding then leads to enhanced interaction between Shp1 and Src and to Shp1-dependent dephosphorylation and activation of Src kinase which, in turn, results in the induction of actin-dependent ruffling and increased fibroblast cell motility. As these effects are part of the motogenic, pro-invasion activity typically induced by growth factor receptors, we examined whether the GroPIns4P/Shp1 cascade might be required for such activity. We report evidence indicating that this is indeed the case.
These observations represent a breakthrough in our understanding of the physio-pathological roles of both the glycerophosphoinositols and of Shp1. They indicate that these molecules play a role in receptor-activated pathways that lead to the activation of PLA2, followed by generation of GroPIns4P and the activation of Shp1, with important consequences on cell motility. Given the potent activation of PLA2 in several cells involved in the primary immune response, the GroPIns4P/Shp1 cascade is likely to play a role also in inflammatory reactions.
Methods
Antibodies, cDNAs and reagents
NIH3T3 and Raw264.7 cells were obtained from the American Type Culture Collection (ATCC, USA). Dulbecco’s Modified Eagle’s Medium (DMEM), OptiMEM, foetal bovine serum (FBS), calf serum (CS), penicillin, streptomycin, trypsin-EDTA, L-glutamine and the Lipofectamine LTX/plus reagent were obtained from Gibco BRL (Grand Island, NY, USA). Wild-type Shp1 and Shp1-C455S were kindly provided by Dr. Axel Ullrich (Max Planck Institute of Biochemistry, Martinsried, Germany). Src construct was kindly provided by S. Gutkind (NIH, Bethesda, MD, USA).
For the antibodies: anti-p-Src (phosphorylated on tyrosine 530, p-Tyr530), anti-p-Src (phosphorylated on tyrosine 418, p-Tyr418), anti-c-Src, anti-p-PLCγ1 (phosphorylated on tyrosine 783, p-Tyr783) and anti-PLCγ1 antibodies were obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA); the rabbit polyclonal anti-Shp1 and goat polyclonal anti-cPLA2α were obtained from Santa Cruz (Santa Cruz, CA, USA); and the mouse monoclonal anti-Src [clone 327] was obtained from Abcam (Cambridge, UK). Alexa Fluor 488-phalloidin and the Alexa 488- and Alexa 546-conjugated goat anti-rabbit and anti-mouse antibodies were obtained from Molecular Probes (Oregon, USA).
GroPIns4P and GroPIns4P-Bio were obtained from Echelon Biosciences (Salt Lake City, UT, USA). Inositol 4-phosphate (Ins4P) was obtained from Cayman chemicals (Ann Arbor, MI, USA). EGF and PDGF were obtained from Sigma-Aldrich (St. Louis, MO, USA). Protein tyrosine-phosphatase (PTPase) inhibitor I (TPI-1), Shp1/2 PTPase inhibitor (NSC-87877), cPLA2α inhibitor (pyrrolidine) and goat polyclonal HRP-conjugated anti-rabbit and anti-mouse antibodies were obtained from Calbiochem (CA, USA). Dynabeads MyOne Streptavidin C1 beads were obtained from Invitrogen/Gibco. Protein-A Sepharose beads, Protein-G Sepharose beads and Glutathione Sepharose 4B matrix were obtained from Amersham Pharmacia Biotech (NJ, USA). Benzamidine Sepharose 4 Fast Flow and the sensor Chip SA were obtained from GE Healthcare (Germany). All other reagents were of the highest purities from standard commercial sources.
Plasmid construction
To generate the CFP-Shp1 construct, PCR primers were designed to add an EcoRI site to the 5′-end and a XhoI site to the 3′-end of the human Shp1 variant 2 sequence. The PCR product was cut with EcoRI and XhoI and inserted in the correct reading frame into pECFP-C3 (BD Biosciences) cut with the same two enzymes. The same procedure was used to clone Src cDNA into the pEYFP-N1 vector (BD Biosciences). To generate the Shp1-Flag construct, PCR primers were designed to add an EcoRI site to the 5′-end and a XbaI site to the 3′-end of the human Shp1 variant 2 sequence. The PCR product was cut with EcoRI and XbaI and cloned into p3X Flag CMV-14 (Sigma) cut with the same enzymes. The Shp1-GFP construct was generated by cloning the human Shp1 variant 2 sequence, amplified by PCR, into the pEGFP-N3 vector (BD Biosciences).
The primers were designed to add an EcoRI site to the 5′-end and a SacII site, preceded by A to keep Shp1 in the correct frame, to the 3′-end of Shp1. The 6xHis-tagged fusion proteins used in this study were derived by sub-cloning the following cDNA- or PCR-amplified fragments into pETM11 containing a multiple cloning site inserted into the NcoI/NotI sites: the C-terminal truncated form of human Shp1 cDNA (His-Shp1; amino acids 1–529), Shp1 NH2- and COOH-terminal SH2 domains [His-SH2 (N + C); amino acids 1–221] and Shp1 catalytic domain (His-PTPase; amino acids 245–529). The Shp1 S118A/R138E/S140A triple mutant was generated by two sequential site-directed mutagenesis reactions. The primers used for the mutagenesis reaction to generate the Shp1 R138E/S140A mutant were 5′-GGACGTTTCTTGTGGAGGAGGCCCTCAGCCAGCCTGG-3′ and 5′-CCAGGCTGGCTGAGGGCCTCCTCCACAAGAAACGTCC-3′. The primers used for the mutagenesis reaction to generate the Shp1 S118A mutant were 5′-GGTACCATGGCCACATGGCTGGCGGGCAGGCAG-3′ and 5′-CTGCCTGCCCGCCAGCCATGTGGCCATGGTACC-3′. All recombinant plasmids were verified by DNA sequencing.
Cell culture, transfection and treatments
NIH3T3 and Raw 264.7 cells were cultured in DMEM supplemented with 2 mM L-glutamine, 50 U/mL penicillin, 50 mg/mL streptomycin, and 10% CS or heat-inactivated FBS (30 min at 55 °C), respectively. NIH3T3 cells were transfected with different plasmids using the Lipofectamine LTX/plus reagent according to the manufacturer’s instructions. For immunofluorescence experiments, NIH3T3 cells were seeded onto glass coverslips in 24-well plates at a concentration suitable for 70% confluence without transfection or 50% confluence in the case of Lipofectamine-based cell transfection. In the latter, approximately 24 h after seeding, NIH3T3 cells were transiently transfected with different plasmids with the Lipofectamine LTX/plus reagent following the manufacturer’s instructions. Before treatments, NIH3T3 cells were serum-starved in DMEM with 2 mM glutamine, 50 U/mL penicillin and streptomycin for 16–20 h. The cells were then treated with different stimuli and/or inhibitors, as indicated in the text and/or Figure legends. Addition of exogenous GroPIns4P was at 50 μM (unless otherwise indicated), a concentration eliciting an intracellular concentration of about 1.5 μM, as calculated from the Nernst equation (with T = 300 K, z = − 3, and Veq = − 30 mV, an average value for cultured, non-excitable cells).
GroPIns4P-bio pull-down assay
Raw 264.7 cells maintained in the appropriate growth medium were detached using PBS-EDTA, centrifuged at 300×g, washed three times with PBS and re-suspended in lysis buffer (20 mM Tris, pH 7.4, 150 mM KCl, 5 mM MgCl2, 1 mM DTT, 5 mM EGTA, 1% Triton X-100) supplemented with a protease inhibitor cocktail (Complete Mini EDTA-free, Roche). The cell lysate was kept on a rotating wheel for 30 min at 4 °C. The lysate was then centrifuged at 13,000×g for 10 min. The supernatant obtained from the centrifugation was recovered, brought to a 0.2% (w/v) final concentration in Triton X-100, and dialysed twice against 1000 volumes PBS at 4 °C. The dialysis step was performed to remove endogenous GroPIns4P. Ten milligrams of cell extract for each sample was pre-cleared on 1 mg of uncoupled streptavidin-conjugated paramagnetic beads for 4 h at 4 °C on a rotating wheel.
Later, the lysates were recovered and incubated for 16 h at 4 °C with 1 mg of streptavidin-conjugated beads that were previously incubated with 2.5 nmoles of GroPIns4P-Bio or biotin in binding buffer (50 mM Tris-HCl, pH 7.6, 50 mM KCl, 10 mM EDTA) supplemented with the protease inhibitor cocktail. Following incubation, the unbound materials were separated by a magnetic particle concentrator, and the beads were washed five times with 2 ml of binding buffer. GroPIns4P-bound proteins were specifically eluted with 5 mM GroPIns4P, which corresponded to a 100-fold excess compared to the number of moles of GroPIns4P immobilised on the beads. The elution was performed for 30 min at 4 °C on a rotating wheel. Following the incubation, the proteins eluted by GroPIns4P were recovered using a magnetic particle concentrator, and the beads were re-suspended in 100 μL of SDS sample buffer. Both fractions were eluted by specific displacement, and the SDS sample buffer was analysed by 10% SDS/PAGE. The gel was then stained with GelCode Blue Stain Reagent (according to the manufacturer’s instructions). The bands were analysed by LC-MS/MS. For GroPIns4P-Bio pull-down assays with purified Shp1, 1 μg of purified Shp1 was incubated for 16 h at 4 °C with 1 mg of streptavidin-conjugated paramagnetic beads in the presence of 2.5 nmoles of biotin or GroPIns4P-Bio in binding buffer plus protease inhibitors. Following this incubation, the unbound material and beads were washed with binding buffer. The beads with bound protein were boiled in 100 μL of SDS sample buffer, separated by 10% SDS-PAGE, and transferred onto nitrocellulose membranes for western blotting.
Expression and purification of recombinant proteins
pETM11 constructs encoding His-tagged Shp1 fragments were transformed into E. coli BL21(DE3) bacteria. The transformed bacteria were grown to an OD600 of 0.6, and expression of recombinant proteins was induced by the addition of IPTG (0.1 mM). After overnight incubation at 20 °C, the cells were harvested by centrifugation at 6000 rpm for 10 min and rinsed twice with PBS. The pellet was re-suspended in lysis buffer (25 mM Tris-HCl, pH 7.5, 150 mM NaCl, 10 mM β- mercaptoethanol, 20 mM imidazole) containing protease inhibitor cocktail as described above, and lysozyme, MgCl2 and DNase I were added at final concentrations of 0.5 mg/mL, 5 mM and 0.1 mg/mL, respectively. The suspension was incubated at 4 °C for 30 min and sonicated on ice 8 times for 15 s. Subsequently Triton X-100 was added to a final concentration of 1% w/v, and the mixture was incubated for 15 min at 4 °C. The bacterial lysate was then centrifuged at 22,000 rpm for 30 min at 4 °C. The supernatant was applied to a Ni-NTA-agarose column that had been previously equilibrated in lysis buffer for 3 h at 4 °C. After elution with lysis buffer containing 250 mM imidazole, His-Shp1 was dialysed twice against 1000 volumes of PBS and stored in aliquots at − 80 °C.
Fluorescence spectroscopy
The absorption spectra of Shp1 were recorded using a Varian Cary 1 spectrophotometer. Steady-state fluorescence emission measurements were performed with a K2 fluorometer (ISS, Champaign, IL, USA) equipped with a two-cell temperature-controlled sample holder. To selectively excite Trp residues, the excitation wavelength was set at 295 nm with a slit width of 1 nm. To avoid the inner-filter effect, all steady-state fluorescence measurements were performed on Shp1 with optical densities at 295 nm less than 0.1 OD. All measurements were made at 25 °C with the temperature of the samples determined in the cuvette with an accuracy of ±0.2 °C. For each experiment, the Shp1 concentration was fixed at 1.2 μM, while GroPIns4P was varied from 0.0 to 5.0 μM. Thus, a 500 μL solution containing 1.2 μM Shp1 dissolved in PBS pH 7.4 was titrated by successive additions of GroPIns4P (dissolved in PBS buffer, pH 7.4). After each addition, the samples were incubated for 2 min and fluorescence emission spectra were recorded between 310 and 410 nm in triplicate. To exclude the influence of sample dilution, fluorescence titration experiments were performed by adding the same amount of PBS buffer in the total volume (500 μL).
Surface plasmon resonance
The instrument used for surface plasmon resonance was a BIAcore 2000 (BIAcore AB, Uppsala, Sweden). Carboxymethylated dextran pre-immobilised with streptavidin chip (SA chip) was first cleaned with three consecutive 1-min injections of a solution of 1 M NaCl in 50 mM NaOH before the immobilisation procedure. Five minutes after the cleaning process, when the sensorgram reached a stable baseline, GroPIns4P-Bio was diluted in running buffer (HBS-EP buffer: 10 mM Hepes, 150 mM NaCl, 5 mM EDTA 0.005% Surfactant P20, pH 7.4) (GL/min. To obtain the maximum immobilisation level of GroPIns4P-Bio on the surface, multiple injections were performed.
Following GroPIns4P-Bio immobilisation, SPR binding analysis was performed in the concentration range of 0.0 to 5.0 μM of Shp1. The Shp1 stock solution was diluted in 100 μL of HBS-EP buffer at the specific concentrations. The binding flow was fixed to 20 μL/min, and the time of injection was 3 min. After each injection of protein, the binding surface was washed with a buffer containing 2.0 mM glycine/HCl, pH 3.0. A blank flow cell was used as the reference surface to eliminate the bulk effect. Data management and calculation of the kinetics parameters were performed using the BIAevaluation 3.1 software (BIAcore AB, Uppsala, Sweden).
For the competition experiments, SPR assays were performed at a fixed concentration of Shp1 (1.2 μM) in the presence of increasing concentrations of GroPIns4P (0.0 to 5.0 μM). Both Shp1 and GroPIns4P were dissolved in HBS-EP buffer and incubated for 10 min before injection onto the SA sensor chip. The flow was fixed to 20 μL/min, and the time of injection was 3 min. A solution of 2.0 mM glycine/HCl, pH 3.0, was used in the regeneration step to remove the analyte bond before reusing the sensor chip.
Isothermal titration calorimetry (ITC)
ITC measurements were performed by using a Nano-ITC III (TA instruments, New Castle, DE, USA) with a reaction cell volume of 1 ml kept at 25 °C. A protein solution of 10 μM was titrated into the calorimetric cell with a solution of 50–100 μM of the ligand GroPIns4P (Ins4P) in the syringe. A sequence of 16 injections of 15 μL volume was programmed with a stirring speed of 250 rpm at 500 s intervals. Protein samples were prepared with protein solutions at 25 °C in 20 mM Tris buffer pH 7.4 with 50 mM NaCl and 1 mM TCEP. To minimise the differences in buffer composition and pH between cell and syringe, both ligand and protein solutions were prepared with the same batch of buffer. The heat of dilution of the GroPIns4P into the buffer solution was measured in a separate experiment and appropriate corrections were made. The heat evolved after each ligand injection was obtained from the integral of the calorimetric signal. Raw data were integrated, corrected for non-specific heats and normalized for concentration. The equilibrium dissociation constant (KD), binding enthalpy (ΔbH) and binding stoichiometry (n) were obtained by nonlinear regression of the experimental data using a model of independent binding sites, by means of the NanoAnalyze software. Experiments were performed in duplicate.
Nuclear magnetic resonance spectroscopy
A suite of triple resonance experiments (Additional file 1 Table S1) was used for assignment of the 1H, 15N and 13C backbone resonances of the Shp1 cSH2 domain. All experiments were acquired on a 700 MHz Bruker Avance instrument equipped with a TCI cryoprobe. Assignment of the cSH2 domain backbone resonances was complete with the exception of the resonances of S109 and G175. 15N cSH2 (200 μM) was titrated with GroPIns4P, and a series of 1H–15N-HSQC spectra were recorded at different cSH2:GroPIns4P ratios up to a 1:5 ratio. Distinct chemical shift changes were observed in the 1H–15N-HSQC spectra upon addition of GroPIns4P, according to a fast exchange regime. The 1HN and 15N chemical shift perturbations were estimated by using the following equation [23]:
Where ΔHN and ΔN are the differences between the 1H and 15N chemical shifts of free cSH2 and cSH2 in the presence of GroPIns4P, respectively.
Docking calculations
Docking calculations were performed with HADDOCK2.2 implemented in the WeNMR/West-Life GRID-enabled web portal (www.wenmr.eu). The docking calculations were driven by ambiguous interaction restraints between all solvent-exposed residues involved in the intermolecular interactions [24]. The docking calculations were performed using the cSH2 domain (stretch 110–213) of the crystal structure of human tyrosine phosphatase Shp1 (PDB code: 2B3O) [25]. The active residues of the Shp1 cSH2 domain were defined as residues those showing a chemical shift perturbation upon GroPIns4P binding and with at least 50% solvent accessibility. The residues S118, G120, E139, S140 and S142 were defined as active. Passive residues were defined as residues close in space to the active residues and with at least 50% solvent accessibility. The residue solvent accessibility was calculated with the program NACCESS. In the initial rigid body docking calculation phase, 5000 structures of the complex were generated, and the best 400 in terms of total intermolecular energy were further submitted to further calculations and to a final refinement in water. The final 400 structures were then clustered using a cutoff of 5.0 Å of the Root-Mean-Square Deviation (RMSD). Sixteen clusters were obtained and ranked according to their HADDOCK score. Among them, 5 clusters exhibited acceptable values in terms of their energetic and scoring functions (i.e. having an HADDOCK score lower than − 66) (Additional file 1 Table S2).
Immunofluorescence analysis for membrane ruffling
For ruffle assessment after treatments, immunofluorescence labelling and morphological analysis of the cells were performed as previously described in a double-blind fashion [11]. In brief, the assessment was based on a null score (zero) for the absence of the feature, followed by a score of one or two according to the level of response of each individual cell, where one indicate that ruffling was confined to one area of the cell (< 25% of the cell circumference), and two indicate that two or more discrete areas of the cell contained ruffles. The cell phenotypes were quantified by counting 150 cells in each sample. The data were then expressed as percentage scores (±SD) for each treatment response compared with its respective control. Scoring of the control cells was performed in the same way to evaluate the presence of the features of interest in untreated cells.
Wound-healing assay
To perform the wound-healing assay, NIH3T3 cells were seeded in 6-well tissue culture plates to a final density of 3 × 105 cells/well and maintained at 37 °C and 5% CO2 for 24 h until confluence before being starved overnight in DMEM containing 1% bovine serum. The cell monolayer was wounded by scratching with a 10 μL standard sterile pipette tip. The scratched monolayer was rinsed twice with DMEM containing 1% bovine serum to remove cell debris and incubated with or without 50 μM GroPIns4P. Phase contrast images were taken after 24 h, and the size of the wound area was determined automatically using image-processing techniques for an accurate, unbiased process. In detail, the input image was processed by applying a Sobel edge detector [26], obtaining a gradient image. A threshold process [27] was then performed on the gradient image to obtain the foreground corresponding to the wound area. Particularly, all of the pixels of the gradient image with values less than the threshold θ were assumed to be pixels of the foreground (experimentally, θ is set to 4). Finally, procedures based on morphological operations were used to remove small regions and to fill holes of the wound area to complete detection and analysis [28].
Western blotting and immunoprecipitation
NIH3T3 cells were washed with ice-cold PBS and lysed on ice in buffer containing 20 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton-X100, 5 mM Na3VO4, 30 mM β-glycerophosphate and 10 mM NaF supplemented with the protease inhibitor cocktail and processed for SDS-PAGE. Westen blotting was performed with the relevant antibodies. For immunoprecipitation experiments, 2 μg of an anti-Shp1 antibody or 5 μg of an anti-Src antibody were added to 1 mg of NIH3T3 cell lysates at 4 °C and incubated overnight. The samples were then incubated with A or G protein agarose beads for 1 h at 4 °C. Western blotting was performed from the washed and denatured complexes. The extent of Src co-immunoprecipitated was evaluated using an NIH imaging system and normalised for the amount of total Shp1 immunoprecipitated.
In-vitro dephosphorylation of Src
Src was immunoprecipitated from NIH3T3 cells as described above. The immunoprecipitates were washed three times with lysis buffer, three times with lysis buffer without phosphatase inhibitors, and twice with phosphatase buffer (100 mM Na-Hepes, pH 7.4, 150 mM NaCl, 1 mM EDTA, and 10 mM DTT). The immunoprecipitates were then split equally into different tubes for treatment with Shp1 or buffer only. The treatment consisted of the addition of 2 μg of Shp1 to the immunoprecipitates in the absence or presence of 50 μM GroPIns4P and an incubation at 37 °C for different times. The reactions were terminated by the addition of concentrated SDS-sample buffer. The samples were then resolved by SDS-PAGE, and the level of Tyr530 phosphorylation was determined by western blotting and normalised for the total amount of immunoprecipitated Src.
Acceptor photobleaching measurements
Acceptor photobleaching (apFRET) experiments were carried out with Leica SP5 confocal microscope, as follows: for CFP (donor) fluorescence detection, excitation at 458 nm and fluorescence detected at 470–490 nm. For YFP bleaching, a 514 nm argon laser was used. apFRET was measured by increase in CFP fluorescence intensity before (IDA) and after (ID) YFP photobleaching. To ensure reproducibility and reliability of CFP fluorescence measurements in the absence of acceptor (ID), YFP was photobleached to 10% of its initial fluorescence. The FRET efficiency was calculated as E = (ID − IDA)/ ID .
Results
Identification of Shp1 as a GroPIns4P-interacting protein
To identify protein interactors of GroPIns4P we used a pull-down assay followed by liquid chromatography coupled to tandem mass spectrometry (LC/MS-MS) to identify the collected proteins. This procedure yielded 30 potential GroPIns4P-binding proteins (see Table S3 for full list and relative scores). Functional annotation of these proteins allowed their classification under five major clusters: cellular motility and membrane trafficking components; stress and folding proteins; metabolic enzymes; proteins associated with DNA and RNA processing; and cell-cycle regulation proteins.
We next searched these clusters for proteins that could be related to the activities of GroPIns4P on actin dynamics. The cellular motility and membrane trafficking cluster included the tyrosine-phosphatase Shp1, a known regulator of the Src kinase [15, 22]. GroPIns4P induces actin ruffle formation in fibroblasts through a signalling cascade that begins with Src activation; however, no direct interaction occurs between GroPIns4P and Src [11]. We thus hypothesised that Shp1 might represent the origin of this GroPIns4P- and Src-dependent pathway. Based on this hypothesis, we focussed on Shp1 and explored its possible role in GroPIns4P-induced activation of Src and actin cytoskeleton remodelling.
We then asked whether Shp1 is a direct target of GroPIns4P, by in vitro pull-down assays using purified recombinant Shp1 with GroPIns4P-Bio-bound beads (Fig. 1a). Recombinant Shp1 was specifically pulled-down by GroPIns4P-Bio-bound beads (but not by control Biotin-bound beads), indicating that GroPIns4P binds directly to Shp1 (Fig. 1a).
Fig. 1.

Direct binding of GroPIns4P to Shp1. a Representative pull-down of streptavidin-conjugated beads using Biotin or biotinylated GroPIns4P (GroPIns4P-Bio) with either Shp11–529 (His-Shp1), the N-terminal SH2-domain mutant (His-SH2 (N + C)) or the catalytic-domain mutant (His-PTPase) of Shp1. Unbound and eluted (beads) proteins were analysed by western blotting using an anti-Shp1 antibody. Molecular weights (kDa) are indicated on the left of each panel. b Schematic domain structure illustrating the amino acid sequences of the Shp1 mutants used in the pull-down. c Dose-response effect of GroPIns4P on Shp1 fluorescence emission (ΔF) at 332 nm (fluorescence emission spectra of Shp1 upon addition of the indicated μM concentrations of GroPIns4P are shown in supplementary Additional file 1 Figure S1). d Kinetic interaction parameters calculated by surface plasmon resonance (SPR) analysis. Binding of Shp1 on a GroPIns4P-Bio-functionalised chip was analysed SPR as a function of time and analysed using the Langmuir fit. e SPR competition assay. Sensorgram showing Shp1 (1.2 μM) binding to immobilised GroPIns4P-Bio in the absence and presence of increasing concentrations of GroPIns4P. The GroPIns4P concentrations are indicated below the panel. f Data from SPR competition assay (RU = resonance units). Data are representative of three independent experiments, each performed in triplicate
GroPIns4P directly binds to the SH2 domain of Shp1
In determining which region of Shp1 is involved in the interaction with GroPIns4P, we noted that Shp1 (which is composed of 597 amino acids) contains two SH2 domains connected by a short linker at the N-terminus, followed by a catalytic domain (the PTPase domain) and short C-terminal tail. The C-terminal tail is highly disordered and affects protein stability; thus, a tailless-form of Shp1 (residues 1–529) has previously been used [25] and it is used also in this study.
For our experiments, we constructed two His-tagged Shp1 truncated fragments: the N-terminal portion composed of the two SH2 domains [His-SH2 (N + C)], and the central catalytic domain (His-PTPase) (Fig. 1b). These purified recombinant Shp1 truncation mutants, as well as the Shp1 tailless 1–529 form, were used in pull-down assays with GroPIns4P-Bio. As shown in Fig. 1a, the His-SH2 (N + C) fragment fully retained the ability to bind to GroPIns4P-Bio, whereas the catalytic region of the enzyme (His-PTPase) was devoid of GroPIns4P-Bio-binding ability (Fig. 1a). These observations indicate that GroPIns4P binds to one or both SH2 domains of Shp1.
This direct interaction was further analysed by steady-state tryptophan (Trp) fluorescence emission spectroscopy using purified Shp1. We exploited the fact that there are nine Trp residues throughout Shp1, three of which fall in the His-SH2 (N + C) fragment. The addition of a ligand, GroPIns4P in this case, is expected to cause Trp fluorescence quenching if GroPIns4P binds sufficiently close to a Trp residue. When Shp1 was incubated with different concentrations of GroPIns4P, the Shp1 signals in the Trp emission spectra decreased as a function of the GroPIns4P concentration, with maximal quenching of the initial emission of 18% at 5 μM GroPIns4P and an apparent EC50 of approximately 0.6 μM (Fig. 1c and S1a). These results indicate direct saturable binding of GroPIns4P to Shp1. Notably, the concentrations used in our experiments (Fig. 1c) are comfortably within the range of the GroPIns4P levels evaluated in several cell types as in the low micromolar range [3, 5, 29].
By way of confirmation, surface plasmon resonance (SPR) was employed to measure the binding of increasing concentrations of Shp1 to GroPIns4P-Bio immobilised on a sensor chip. Shp1-GroPIns4P binding was clearly measurable and dose-dependent, with an apparent KD of about 13 μM (Figs. 1d and S1b) [30]. This value appeared one order of magnitude higher than that obtained in the Trp-fluorescence quenching experiments, and it might be due to the different availability to binding of the immobilised and chemically-modified GroPIns4P-Bio vs the free compound.
To further analyse this aspect, we first determined the Shp1 concentration that was able to saturate the binding sites on the GroPIns4P-Bio-functionalised chip (1.2 μM under our experimental conditions). Then, to determine the binding affinity between GroPIns4P and Shp1, we performed SPR competitive-binding experiments using GroPIns4P-Bio immobilised on a sensor chip, the saturating concentration of Shp1 and increasing concentrations of non-biotinylated, “free” GroPIns4P to compete for binding to the sensor chip. Free-GroPIns4P competed with the binding of Shp1 to the GroPIns4P-Bio-functionalised chip with an EC50 of approximately 0.8 μM (Figs. 1e-f), in line with the results obtained with the Shp1 Trp fluorescence analysis (Fig. 1c).
Finally isothermal titration calorimetry (ITC) was employed to evaluate the binding of GroPIns4P to Shp1. The titration of Shp1 with GroPIns4P resulted in a binding stoichiometry of 1:1 and a KD of 0.3 μM (Additional file 1 Figure S2). At the same time we evaluated by ITC also the binding of Ins4P, a degradation product of GroPIns4P; no binding was resolved for Ins4P, in line with observations we have previously reported indicating that Ins4P has no effect on actin cytoskeleton organisation under our experimental procedures [10].
Altogether, these results indicate that GroPIns4P binds Shp1 in its SH2-domain portion, and that the affinity of the binding is in the low μM range (0.3–0.8 μM), which is within the cellular physiological concentration range of this metabolite.
Structural features of the GroPIns4P binding site in the C-terminal SH2 domain of Shp1
Since the portion of Shp1 necessary and sufficient for its interaction with GroPIns4P was mapped within the two SH2 domains, we further defined the interaction of this compound with Shp1 by nuclear magnetic resonance (NMR) spectroscopy. The purified C-terminal SH2 domain (cSH2) was selected for these studies, since its stability and solubility enable NMR characterisation in solution.
The 1H-15N HSQC spectrum acquired for the 15N-labelled cSH2 domain showed well-dispersed amide signals, typical of a folded protein [31]. Titration of cSH2 with GroPIns4P, up to a cSH2:GroPIns4P ratio of 1:5, induced chemical shift changes that increased with the GroPIns4P concentration, thus confirming the binding of GroPIns4P to this domain (Figs. 2a-b). This behaviour also indicated that the GroPIns4P-bound and free forms of the cSH2 domain exchange with one another at a rate faster than the resonance frequency differences in the two species. The exchange rate can be set at values higher than 2 × 103 s− 1. The amino acid residues affected by GroPIns4P binding clustered in a well-defined region of the Shp1 protein (PDB code: 2B3O) [25] (Fig. 2b), which includes segments 117MSGG120 and 139ESLSQ143 (Figs. 2c-d) belonging to its pY-binding pocket [32]. This result is in line with previous studies suggesting that some SH2 domains bind lipids in their pY pockets [33, 34]. We next modelled the structure of the complex between the cSH2 domain and GroPIns4P, using HADDOCK2.2 docking calculations driven by chemical shift perturbation data. Five clusters of structural models exhibited acceptable values for the energetic and scoring functions (Additional file 1 Table S2). Comparison between the lowest energy structures of these clusters revealed that only cluster 2 showed an orientation of GroPIns4P bound to the Shp1 cSH2 domain (Figs. 2e-f) that was in full agreement with the experimental NMR data. In this structural model, the glycerol moiety of the GroPIns4P is embedded between the N-terminal region of Shp1 and the loop connecting its β1 and β2 strands, allowing the phosphate group of GroPIns4P to establish contacts with the side chain of Ser118 and Arg138 (β1) (Fig. 2g). Moreover, the inositol group is inserted in the pY-binding pocket, and the 4′ phosphate group of GroPIns4P interacts with the side chains of Ser140, Ser142 and Lys170 (β3) (Fig. 2h).
Fig. 2.

Identification of the Shp1-cSH2 domain residues involved in GroPIns4P binding. a, b Overlay of the 1H-15N Heteronuclear Single-Quantum Coherence (HSQC) spectra of the 15N-labeled cSH2 domain in the absence and presence of different amount of GroPIns4P. Protein alone (black) and in the presence of GroPIns4P at the ratios of 1:1 (blue), 1:2 (green), 1:3 (magenta), 1:4 (brown), 1:5 (red) are reported. c Normalised weighted average chemical shift differences (Δav/Δmax) between the GroPIns4P-bound and free forms of the Shp1 cSH2 domain plotted against the residue number for the amide proton and nitrogen resonances. The horizontal bold line at 0.3 indicates the average value plus one standard deviation. d Chemical shift perturbation mapped onto the cSH2 structure of Shp1 (PDB code: 2B3O). The amino acid residues affected by GroPIns4P binding (Δav/Δmax ≥ 0.3) are depicted in red. e Chemical structure of GroPIns4P. f Ribbon representation of the complex, in which the cSH2 domain is in blue and GroPIns4P in red. Residues involved in the interaction are presented as sticks and are labelled. g Close view of S118 and R138 of the cSH2 domain that interact with the phosphate group of GroPIns4P. h Close view of S140, S142 and K170 of the cSH2 domain that interact with the 4′-phosphate group of GroPIns4P. Similar experiments on the nSH2 domain of Shp1 were hampered by the poor stability of the isolated fragment. i Functional validation of the Shp1 S118A/R138E/S140A mutant. Dose-response effect of GroPIns4P on Shp1 S118A/R138E/S140A mutant fluorescence emission (ΔF) at 332 nm (fluorescence emission spectra of Shp1 upon addition of the indicated μM concentrations of GroPIns4P are shown in supplementary Additional file 1 Figure S3)
Based on this modelling, we produced a panel of proteins with mutations in the above amino acid residues, with the aim of investigating the relevance of the GroPIns4P binding site identified on Shp1-cSH2 in the context of the full-length protein. In each case, correct folding of the mutated proteins was verified by Circular Dichroism spectroscopy, and the mutated proteins were then assayed by Trp fluorescence emission spectroscopy for their ability to bind GroPIns4P. As predicted, the combination of S118A/R138E/S140A mutations significantly impaired GroPIns4P binding, as the binding efficiency of the mutated protein decreased by up to 50% (Figs. 2i and S3). Accordingly, the binding isotherm of Shp1 S118A/R138E/S140A mutant, obtained under the same conditions of the wild-type, was also reduced indicating a significantly lower affinity toward GroPIns4P (Additional file 1 Figure S2).
These data thus indicate that the Ser118, Arg138 and Ser140 residues contribute to the interaction of GroPIns4P with full-length Shp1.
GroPIns4P induces Src activation in NIH3T3 cells
Next, we analysed the involvement of Shp1 in GroPIns4P-induced Src activation in NIH3T3 fibroblasts, the model system where the GroPIns4P/ Src-dependent formation of actin ruffles was first reported [11]. Src is regulated by opposing phosphorylation and dephosphorylation events. Phosphorylation by the C-terminal Src kinase (CSK) at Tyr530, promotes the intramolecular interaction of the C-terminal catalytic domain of Src with the SH2 and SH3 domains and blocks the kinase in an inactive conformation. Dephosphorylation of Tyr530 leads to the release of this block and the opening up of the molecule to assume an active state [22]. Shp1 has previously been shown to bind and dephosphorylate Src at Tyr530, thus inducing activation of the kinase [15].
We therefore examined whether this mechanism could account for GroPIns4P-induced Src activation in NIH3T3 cells. It should be noted here that although glycerophosphoinositols are hydrophilic molecules, they can cross the plasma membrane through specific permeases acting as bidirectional transporters in mammals [35], that are orthologues of the GIT1 transporter identified in yeast [36]. Cells were treated with 50 μM GroPIns4P for 2 to 5 min (see Methods for details) and lysates analysed using western blotting with an antibody that recognises the phosphorylated Tyr530 of Src. The Src inhibitory Tyr530 phosphorylation markedly decreased in response to treatment with GroPIns4P (Fig. 3a). Under these conditions, also the activating Tyr418 phosphorylation was increased and both modifications increased the kinase activity (Fig. 3a; and [11]). Indeed, in further support of this Src activation, we evaluated the phosphorylation of PLCγ on Tyr783, which is a Src substrate that is relevant for the signalling cascade initiated by GroPIns4P, in that it triggers the release of intracellular Ca2+ leading to actin remodelling [11]. In parallel with Src modifications, GroPIns4P treatment induced an increase in phosphorylation of PLCγ (Fig. 3a), thus confirming Src activation.
Fig. 3.

GroPIns4P induces Src dephosphorylation. a Representative western blots using anti-phosphotyrosine 530 in Src (pTyr530-Src), anti-phosphotyrosine 418 in Src (pTyr418-Src) and anti-phosphotyrosine 783 in PLCγ (pTyr783-PLCγ) specific antibodies in serum-starved NIH3T3 cells non-transfected or over-expressing the dominant negative Shp1-C455S mutant and treated with 50 μM GroPIns4P for the indicated times (see the top of the two panels). Total Src and total PLC were used as loading controls. Data are representative of three independent experiments. Molecular weight standards (kDa) are indicated on the left of each panel. b Representative immunoprecipitated Src fraction (IP: Src) from NIH3T3 cell lysates washed and incubated with purified recombinant Shp1 for 10 min at 37 °C in the absence (−) or presence (+) of 50 μM GroPIns4P (as indicated). The top panel shows western blots with an anti-phosphotyrosine antibody (pTyr530-Src) to reveal the specific phosphorylation of Tyr-530 in Src. The blot was then re-probed with an anti-Src antibody for immunoprecipitated proteins (bottom panel). Molecular weight standards (kDa) are indicated on the left of each panel. c Quantification of Src phosphorylation in samples treated with GroPIns4P (as in b) by the ImageJ analysis software. Data (GroPIns4P) are expressed as percentages of untreated sample (untreated) of the means (±SD) of three independent experiments, each of which was performed in duplicate (n = 6). **P < 0.02 (Student’s t-test)
GroPIns4P-induced Src activation in cells is mediated by Shp1
We next examined Src Tyr530 and PLCγ Tyr783 phosphorylations in NIH3T3 cells that over-expressed an inhibitory recombinant point-mutant of Shp1 (Shp1-C455S mutant), to establish whether Shp1 is required for GroPIns4P-induced Src activity. Mutation of the Cys residue to Ser at position 455 of the phosphatase domain results in a Shp1 catalytically inactive mutant that also functions as dominant-negative [37–39]. In NIH3T3 cells overexpressing Shp1 inactive mutant, GroPIns4P failed to induce Src dephosphorylation. In fact, the pTyr530-Src levels appeared to increase (Fig. 3a). GroPIns4P may induce the binding of this mutant to Src and protect the phosphorylated Tyr530 residue from the action of other phosphatases, causing increased phosphorylation of Tyr530. Under these conditions and in line with the hypothesis, no increase in the Src activating Tyr418 or PLCγ tyrosine phosphorylation was detected upon addition of GroPIns4P (Fig. 3a). Altogether, these observations indicate that GroPIns4P-induced Src activation requires and is mediated by Shp1 through dephosphorylation of Src at Tyr530.
We next examined whether GroPIns4P directly regulates Shp1 phosphatase activity against Src using isolated proteins. Phosphorylated Src was immunopurified from NIH3T3 cell lysates using a monoclonal antibody specific for the central portion of the protein, in order not to affect the phosphorylated C-terminal tail. Immunopurified Src was thus used as a substrate in in vitro phosphatase assays of recombinant Shp1 in the absence or presence of 50 μM GroPIns4P, and the phosphorylation of Src at Tyr530 was monitored by western blotting. As shown in Figs. 3b and c, GroPIns4P stimulated Shp1 activity and induced Src dephosphorylation, in line with the data obtained in intact cells (Fig. 3a).
GroPIns4P promotes the association between Shp1 and Src in NIH3T3 cells
Based on our results indicating that GroPIns4P binds to the cSH2 domain region of Shp1, we reasoned that the interaction between GroPIns4P and Shp1 might facilitate the association of Shp1 with, and ultimately dephosphorylation of, Src. To evaluate this possibility, we investigated whether Shp1 and Src physically associate with one another in a GroPIns4P-dependent manner, by immunoprecipitating lysates of NIH3T3 cells over-expressing Shp1 or a Shp1-C455S mutant with an anti-Shp1 antibody. Shp1 and Src co-immunoprecipitated as expected, and GroPIns4P increased their interaction by about 1.6-fold, compared with the control (Figs. 4a-b).
Fig. 4.

GroPIns4P favours the association between Shp1 and Src. a, c, e Interaction between Src and Shp1 wild type (a, Shp WT), Shp1-C455S (c, Shp1-C455S) or Shp1-S118A/R138E/S140A mutant (e, Shp1-S118A/R138E/S140A) was examined by immunoprecipitation (IP) with an anti-Shp1 antibody in serum-starved NIH3T3 cells over-expressing Shp1 WT, Shp1-C455S or Shp1-S118A/R138E/S140A mutant untreated (−) or treated (+) with 50 μM GroPIns4P for 5 min (as indicated). The expression levels of Shp1 WT, Shp1-C455S, Shp1-S118A/R138E/S140A and Src, examined in total lysates (input) indicate comparable amounts of proteins. Molecular weight standards (kDa) are indicated on the right of each panel. b, d, f Quantification of co-immunoprecipitated Src with Shp1 WT (b), Shp1-C455S (d) or Shp1-S118A/R138E/S140A mutant (f) using ImageJ analysis software. Data (GroPIns4P) are expressed as percentages of untreated sample (untreated) and as the means (±SD) of at least three independent experiments (Shp1 WT n = 4; Shp1-C455S n = 4; Shp1-S118A/R138E/S140A n = 3). *P < 0.05 (Student’s t-test)
The binding of Shp1 with its substrates is known to be transient [40], and mutation of Cys455 of Shp1 within the catalytic pocket yields an enzyme in which the phosphatase activity is blocked and the affinity for the substrates is unaffected, thus maintaining binding [40]. Such mutants are called “substrate-trapping mutants” and have been used to identify in vivo substrates for several phosphatases. In cells over-expressing the Shp1-C455S mutant we observed that GroPIns4P caused a 2.2-fold increase in the interaction between Shp1-C455S and Src (Figs. 4c-d), an increase that was higher than that observed for Src binding to the Shp1 wild-type form (see Figs. 4a-b). These results are consistent with the concept that GroPIns4P causes an increase in the association of the substrate Src with its phosphatase Shp1 and that this association results in an increase in Src dephosphorylation and activity.
Next, to further define the molecular details of GroPIns4P-induced regulation of Shp1 activity, we evaluated whether the residues identified in the cSH2 domain that are crucial for the docking of GroPIns4P are also crucial for its biological activity. Therefore, we transfected NIH3T3 cells with Shp1 wild-type or Shp1 S118A/R138E/S140A mutant proteins, and analysed the association of Shp1 with Src under basal conditions or upon GroPIns4P treatment of NIH3T3 cells (as described above). We first observed that the binding of Src to the Shp1 S118A/R138E/S140A mutant was comparable to its binding with Shp1wt (Figs. 4e-f). Upon addition of GroPIns4P, no change in Shp1-Src complex formation was observed, indicating that the docking of GroPIns4P to the Ser118, Arg138 and Ser140 residues is required for the GroPIns4P-induced increase in the interaction of Shp1 with its substrate Src.
Taken together, our data indicate that GroPIns4P modulates the interaction between the phosphatase Shp1 and substrate Src and reveal three residues to be crucial for the binding of GroPIns4P to Shp1.
Finally, to analyse the Shp1-Src interaction within intact cells, fluorescence resonance energy transfer (FRET) experiments were performed using a cyan fluorescent protein (CFP)-labelled construct of Shp1 (Shp1-CFP) and yellow fluorescent protein (YFP)-conjugated Src (Src-YFP). FRET signal was observed only when cells were exposed to GroPIns4P and not in untreated control cells (Figs. 5a-b). The statistical FRET signal analysis of Shp1-Src interaction under GroPIns4P treatment revealed an average efficiency of 9% (Fig. 5b). We further examined the distribution of this FRET over the GroPIns4P-treated cell and we showed that “hot spots” signals occur within the lamellar regions where the efficiency was over 20% (Figs. 5c-d).
Fig. 5.

GroPIns4P promotes the association of Shp1 and Src within the lamellar region. a Representative confocal microscopy images of serum-starved NIH3T3 cells co-transfected with Shp1-CFP (green) and Src-YFP (red) for 24 h and then untreated (−) or treated with 50 μM GroPIns4P (GroPIns4P) for 5 min and subjected to Acceptor Photobleaching apFRET analysis. b Quantification of the FRET efficiency over the cells treated as in a. c Representative confocal microscopy images of serum-starved NIH3T3 cells co-transfected with Shp1-CFP (green) and Src-YFP (red) for 24 h and then treated with 50 μM GroPIns4P for 5 min before apFRET analysis. The dashed rectangle in the lamellar region (close to the plasma membrane) indicates where the FRET efficiency was analysed. d Representative colour-coded apFRET efficiency of the lamellar region (selected in a) where the colour-scale quantifies the degree of protein-protein interaction in each of the indicated regions of interest (ROIs 1 to 5, white circles; the FRET efficiency for each ROI is reported in the table on the right). Data are expressed as the means (±SD) (n = 15 cells/condition). ***P < 0.005 (Student’s t-test). Scale bars, 10 μm
Collectively, these data indicate that GroPIns4P activates Src in vitro by inducing Src dephosphorylation at Tyr530, and that this reaction is facilitated by an enhanced interaction between Shp1 and Src (Figs. 4 and 5).
Shp1 is necessary for GroPIns4P-dependent actin ruffle formation and cell motility
Finally, we evaluated the involvement of Shp1 in the GroPIns4P-dependent induction of actin ruffles in NIH3T3 cells, which would be expected to occur based on the GroPIns4P-Src-actin ruffling cascade described here and in previous reports [10, 11]. To this end, we first used two well-characterised phosphatase inhibitors to reduce Shp1-phosphatase activity and then evaluated the effects on ruffle formation [41, 42].
A 2.5–3-fold increase in peripheral ruffles was clearly induced upon treatment with GroPIns4P (versus the control; Figs. 6a-b), which is comparable to our previous results [10, 11](see Methods for ruffle quantification). However, pre-treatment with either TPI-1 or NSC-87877 inhibitors strongly impair the ability of cells to assemble peripheral ruffles in response to GroPIns4P, suggesting the requirement of the Shp1 enzymatic activity to mediate localised GroPIns4P signalling. That said, the specificity of these inhibitors is limited, as they are known to target other phosphatases [42]. Thus, to specifically inhibit Shp1 activity, we over-expressed the dominant negative mutant Shp1-C455S [37, 38]. As expected, in the presence of the Shp1-C455S mutant, the addition of GroPIns4P did not lead to membrane ruffle formation, as previously observed with phosphatase/Shp1 chemical inhibitors (Figs. 6c-d). Thus, the tyrosine-phosphatase Shp1 is essential for GroPIns4P-dependent formation of membrane ruffles in NIH3T3 cells. The treatment with PDGF (10 ng/ml; used here as a positive control) produced an increase in ruffles of 3.5–4-fold the control that was not affected by Shp1-C455S mutant overexpression (Figs. 6c-d); this indicates that PDGF signalling in NIH3T3 cells diverges from that of GroPIns4P.
Fig. 6.
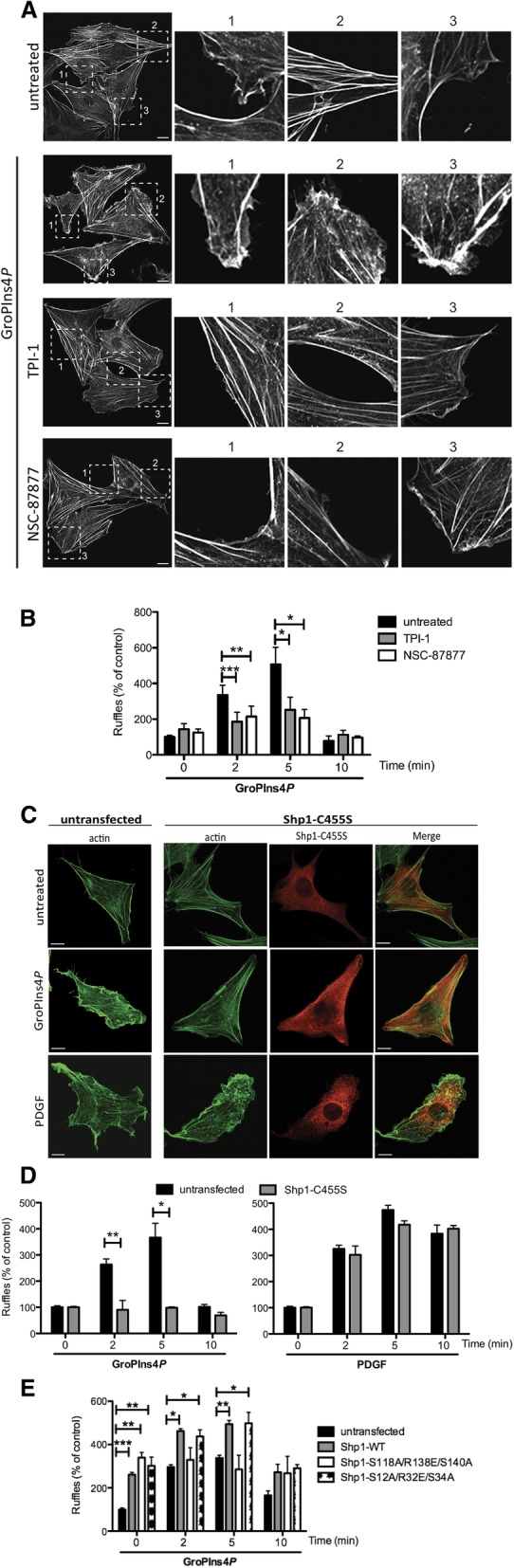
Shp1 directly mediates the GroPIns4P-induced actin ruffle formation. a Representative confocal microscopy images of serum-starved NIH3T3 cells untreated (untreated) or treated with 50 μM of GroPIns4P for 5 min alone (GroPIns4P) or in presence of the Shp1 inhibitors TPI-1 (TPI-1 + GroPIns4P) or NSC-87877 (NSC-87877 + GroPIns4P). The cells were fixed and processed for immunofluorescence analysis with FITC-labelled phalloidin. Zoom 1, 2 and 3: higher magnification images of the membrane area. b Quantification of actin ruffle formation (as percentage of untreated cells) of cells treated as in a (see the Methods). c Representative confocal microscopy images of NIH3T3 cells untransfected or transfected with the Shp1-C455S mutant for 8 h, starved for 24 h and then untreated (untreated) or treated with 50 μM GroPIns4P (GroPIns4P) or 10 ng/ml PDGF (PDGF) for 5 min. The cells were fixed and stained with an anti-Shp1 antibody and FITC-labelled phalloidin. d Quantification of actin ruffle formation (as the percentage of untreated cells) of cells treated as above. e Quantification of actin ruffle formation (as the percentage of untreated cells) of cells transfected with Shp1-WT, Shp1-S118A/R138E/S140A and Shp1-S12A/R32E/S34A mutants for 8 h, starved for 24 h and then treated with 50 μM GroPIns4P for the indicated times. Data are expressed as the means (±SD) of at least three independent experiments. ***P < 0.001; **P < 0.02; *P < 0.05 (Student’s t-test) calculated for each treatment versus untreated samples (untrasfected). Scale bars, 10 μm
To further investigate the molecular basis of this GroPIns4P-induced effect on actin, we expressed the Shp1 S118A/R138E/S140A mutant, which cannot bind GroPIns4P in NIH3T3 cells and analysed peripheral ruffles upon GroPIns4P treatment. Over-expression of this mutant did not significantly affect membrane-ruffling formation compared with the wild-type protein, thus supporting the conclusion that these mutations do not directly impact Shp1 function (Fig. 6e). By contrast, the effect of GroPIns4P on ruffle formation was significantly inhibited, confirming that the Shp1 Ser118, Arg138 and Ser140 residues are crucial for GroPIns4P-mediated interaction between Shp1 and Src and the consequent induction of actin ruffles in NIH3T3 cells.
Based on the above results, we decided to investigate whether the second N-terminal SH2 domain (nSH2) is as crucial for the GroPIns4P activity as the cSH2 domain. We identified a sequence homologous to the S118/R138/S140 stretch of the cSH2 domain in the nSH2 domain of Shp1 at the positions S12/R32/S34. Mutation of these residues yielded a protein (Shp1 S12A/R32E/S34A mutant) that, when over-expressed in the NIH3T3 fibroblasts, responded to GroPIns4P and resulted in the induction of actin ruffles (Fig. 6e). These data indicate that the cSH2 domain of Shp1 is the crucial site initiating the cascade of the GroPIns4P-dependent actin ruffle formation. This conclusion is in line with the data obtained by different biophysical approaches pointing at a single GroPIns4P binding site on Shp1 (Additional file 1 Figure S1 and Figure S2).
The GroPIns4P-increased cell motility and migration during wound healing involves Shp1
Membrane ruffles are often found at the leading edge of motile cells and are involved in cell spreading [43–45]. Hence, due to its ability to induce a local perturbation of the actin cytoskeleton, we analysed the role of GroPIns4P in cell motility by performing wound-healing assays on confluent monolayers of NIH3T3 cells.
Cells exposed to 50 μM GroPIns4P showed a significantly improved wound-healing capability compared with the controls: GroPIns4P-treated cells started to migrate into the scratch area earlier than control cells, and wound closure was almost complete at 30 h. Quantifications performed at 6 h after scratching showed that wound closure was increased by 65% compared to the control (Figs. 7a, d), indicating a stimulatory role of GroPIns4P on fibroblast migration. Pre-treatment of NIH3T3 cells with either TPI-1 or NSC-87877 inhibitors (25 μM and 100 μM, respectively) completely abrogated GroPIns4P-stimulated wound closure (Figs. 7b-d), suggesting that Shp1 is required for GroPIns4P-induced cell motility in NIH3T3 cells. These data thus indicate that GroPIns4P in fibroblasts induces actin ruffles and cell motility and that Shp1 activity is necessary to mediate this GroPIns4P action.
Fig. 7.

GroPIns4P binding to Shp1 facilitates and mediates EGF-induced wound closure in NIH3T3 cell monolayers. a Representative phase-contrast microscopy images after 6 h from scratching in serum-starved NIH3T3 cells untreated (untreated) or treated with 50 μM of GroPIns4P alone (a) or in presence of TPI-1 (b) or NSC-87877 Shp1 inhibitors (c), as indicated. d Quantification of wound healing as the percentage of the untreated control in cells treated as in a-c. e, f Cells without or with pre-incubation with the cPLA2α inhibitor, TPI-1, or NSC-87877 inhibitors (as indicated) were stimulated with 10 ng/ml EGF, 50 μM GroPIns4P or 10 μM arachidonic acid for 6 h. Quantifications of wound healing are presented as the percentage of the untreated controls. The differences between the untreated controls are not statistically significant. Data are expressed as the means (±SE) of at least three independent experiments. ***P < 0.001; **P < 0.02; *P < 0.05 (Student’s t-test) calculated for each treatment versus untreated samples (control). The cPLA2α inhibitor was used at 0.5 μM, but it was effective over a range of concentrations between 0.1 and 2 μM. Arachidonic acid was used at both 10 and 50 μM, with similar results
The EGF-receptor-dependent motogenic activity involves the stimulation of the cPLA2α-GroPIns4P pathway
Previous studies have indicated that glycerophosphoinositols can be produced by the hormonal activation of cPLA2α [2] and that specific increases in GroPIns4P levels are observed in NIH3T3 and Swiss3T3 fibroblasts upon exposure to epidermal growth factor (EGF) [3, 6]. Building on these and the current results, we carried out experiments to test the possibility that the GroPIns4P/Shp1 pathway might be involved in the EGF-induced cell migration.
NIH3T3 fibroblasts were serum-starved overnight and a wound-healing assay was conducted in the absence and presence of EGF (10 ng/ml). EGF stimulation induced a significant increase in cell migration; when cells were treated with Shp1 inhibitors this effect was reduced by 50% (with 25 μM TPI-1) and almost 100% (with 100 μM NSC-87877), indicating that Shp1 is required for EGF-induced cell motility.
To evaluate the role of the cPLA2α pathway and thus of GroPIns4P production in the motogenic effect of EGF, we repeated the above experiments in the presence of a specific cPLA2α inhibitor [46]. Pre-treatment of serum-starved NIH3T3 with 0.5 μM cPLA2α inhibitor strongly reduced EGF-stimulation of cell motility (Figs. 7e-f).
It has been reported that EGF-induced cytoskeleton organisation and cell motility also involve the activation of 5-lipoxygenase and formation of the arachidonic acid-derived leukotrienes [47, 48]. Considering that the inhibitors used in this study could also affect the formation of other metabolites of the arachidonic acid cascade, we directly investigated the role of arachidonic acid in the EGF-induced cell motility. In line with our previous report indicating that leukotriene E4 could induce stress fibres but not ruffle formation in fibroblasts [10], we observed that arachidonic acid (the leukotriene precursor; 10–50 μM) did not affect EGF-induced cell motility under the experimental conditions used in this study (Fig. 7f). When EGF-stimulated fibroblasts were treated as described above with the cPLA2α inhibitor, the lack of effect of the growth factor could be rescued by the addition of GroPIns4P, but not by the addition of arachidonic acid that instead, had a minor inhibitory effect when combined with the PLA2 inhibitor (this decrease was not further investigated since it was out of the scope of the present study; Fig. 7f). These data are consistent with our conclusion that the cPLA2α metabolite mediating the EGF-induced cell motility is mainly GroPIns4P.
Thus, the current and previous evidence collectively indicates that the motogenic activity initiated by EGF involves the cPLA2α/GroPIns4P/Shp1 pathway.
Discussion
The main finding in this study is that the phosphatase Shp1 is an intracellular receptor of the phosphoinositide-derived cell mediator GroPIns4P. To our knowledge, this is the first receptor to be identified for one of these bioactive phosphoinositide metabolites. In addition, we present evidence that the GroPIns4P-controlled Shp1 activity is part of the signalling pathway activated by the EGF receptor to control cell motility. We propose that GroPIns4P endogenously-formed by EGF receptor-activated cPLA2α, interacts with Shp1, starting the Src-dependent signalling cascade that promotes ruffle formation and stimulation of cell motility [10, 11].
The definition of the Shp1 cSH2 domain as the specific GroPIns4P docking site represents a novel aspect of this study. Not all SH2 domains are recognised by GroPIns4P, as shown in the case of the Src SH2 domains where no direct binding of GroPIns4P could be observed [11]. This suggests that the specificity of the binding to Shp1, or other binding domains, can be due to specific structural and charge properties in the individual SH2 domains that affect their direct interaction with ligand such as GroPIns4P, or other phosphate moiety from proteins or lipids.
A SH2 domain ‘signature’ FLVR sequence characterises most SH2-domains, in which the highly conserved Arg is involved in the formation of an electrostatic interaction with the phosphate moiety of the ligands and thus plays a crucial role in phospho-Tyr binding [49]. Using SH2-mutants of Shp1, we show that the Arg residue at position 138 (in the conserved FLVR motif) is critical for GroPIns4P binding and for physiological action, since a lack of effect of GroPIns4P was observed in fibroblasts overexpressing the Shp1 S118A/R138E/S140A mutant. Considering that the SH2 domains play a role in the targeting of proteins to tyrosine-phosphorylated sites, thus allowing localised assembly and activation of signalling effectors, we propose that the binding of GroPIns4P to Shp1-SH2 domains is a means to recruit and compartmentalise Shp1 to a specific cell compartment, or to displace Shp1 when bound to membrane receptors/lipids, thus influencing the recognition of and binding to specific soluble protein substrates. Accordingly, we have shown that GroPIns4P enhances the association of Shp1 and Src in NIH3T3 fibroblasts within the lamella region next to the plasma membrane, but not at the plasma membrane (Fig. 5).
The possibility to modulate Shp1 activity by GroPIns4P is of great pharmacological interest considering the diverse physiological and pathological conditions involving this phosphatase. Deletion of Shp1 is associated with a variety of pathologies including cancer and autoimmune diseases, such as allergic asthma, rheumatoid arthritis and multiple sclerosis [50]. Shp1 is a known negative regulator of cell cycle and of inflammatory cascades, including MAPKs, NF-KB and PI3K-Akt pathways [51]. Similarly, Shp1 directly targets the oncogenic Jak/STAT signalling pathway and thus contributes to apoptosis and tumor suppression in various cancer types, where an abolished or diminished Shp1 expression has been reported [52]. In this context, activating the Shp1 phosphatase activity has been demonstrated to be a promising therapeutic approach to induce apoptosis in cancer cells. Accordingly, the use of GroPIns4P may be a plausible approach to target this enzyme in cancer or in other appropriate contexts, with the aim to restore or stimulate its phosphatase activity.
An important notion proposed in this study is that the action of GroPIns4P on the actin cytoskeleton may mediate the EGF effects on cell migration. EGF is known to promote PLA2 activation [53–55] through several independent signalling pathways [56], that may thus lead to GroPIns4P [6] and arachidonic acid formation. This is the case for the MEK-dependent phosphorylation cascade that has been involved in both the activation of PLA2 [57] and formation of GroPIns4P [6]. Similarly, the activation of PLCγ and cytosolic Ca2+ increase lead to PLA2 activation [58]. Within these diverse signalling pathways the GroPIns4P formed by EGF-dependent stimulation [3, 6] may represent the resulting common messenger mediating the EGF-dependent activation of cell motility.
Indeed, here we demonstrate that the EGF-dependent stimulation of cell motility is inhibited under conditions that reduce the formation of GroPIns4P. Other mediators are certainly involved in the EGF action; however, the virtually overimposable effects on Rac activation and ruffle formation produced by both EGF [59–61] and exogenously-added GroPIns4P [11] suggest that the endogenous formation of GroPIns4P plays a significant role in the motogenic component of the EGF signalling. In this scenario, GroPIns4P could cooperate with other EGF-regulated pathways to coordinate actin remodelling and cell migration.
Another evidence linking EGF-induced cPLA2α activation and GroPIns4P production is related to their cell localisation. EGF is known to induce the translocation of cPLA2α from the cytosol to the plasma membrane [53, 55]. In NIH3T3 cells pre-treated with a cPLA2α inhibitor, cPLA2α translocation in response to EGF stimulation was partially blocked; thus, cPLA2α remained primarily cytosolic (Additional file 1 Figure S4). The lack of localisation of cPLA2α at the leading edge of fibroblasts could prevent the local production of GroPIns4P, thus negatively affecting actin dynamics and cell movement (Fig. 7). In this way both the catalytic activity and localisation of cPLA2α would result relevant for the EGF-dependent cell motility mediated by GroPIns4P.
Arachidonic acid, the other likely product of PLA2 activation, does not appear to affect cell motility under the experimental conditions reported, however it has been shown to promote the transcription of genes relevant in cell motility (at concentrations between 5 and 15 μM) [62, 63]. Thus PLA2 metabolites might control motility directly by acting on the Shp1/Src pathway through GroPIns4P, and at the transcriptional level through arachidonic acid. Finally, and as previously reported [10], GroPIns4P cannot exert its effect upstream of either PI3K or PLA2, enzymes that are also involved in the EGF-induced ruffle formation: their inhibition did not affect the ability of GroPIns4P to induce membrane ruffles, further supporting the relevance of the GroPIns4P/Shp1/Src pathway.
At the same time, it should be considered that Src can also be activated by direct binding to the EGF-receptor initiating diverse signalling cascades involving kinases such as MAP, P38 and PI3, that in turn lead to the remodelling of focal adhesions and cell migration through the phosphorylation of specific downstream substrates [64, 65]. These represent pathways that may be active in parallel or in alternative to the PLA2/GroPIns4P/Shp1-dependent Src activation reported in this study and that may represent either a redundancy of signals or, most probably, a response to diverse cell conditions.
In addition to EGF, other growth factors and ligands, such as insulin, lysophosphatidic acid or ATP, modulate the endogenous GroPIns4P level by activating cPLA2α [6], while the level of GroPIns are modulated, for example, by the Fc receptor-dependent stimulation of cPLA2α in macrophages [7], or the Ret/PTC oncogene-dependent PLA2 activation in human thyroid cancer cells [66]. The formation of the glycerophosphoinositols in normal and pathological conditions might thus represent a widespread mechanism of regulation of signalling pathways leading to cell motility. GroPIns4P is also likely to play an important role in other cell responses such as the immuno-inflammatory response [9], depending on the cell type involved or on specific physiopathological conditions. With the elucidation of the interaction of Shp1 with GroPIns4P all these previous studies can be re-evaluated in the light of the known cellular roles of this specific receptor. Similarly, and possibly more importantly, the numerous pathological conditions that have been related to the inhibition of Shp1, could now be directly modulated by the use of the exogenous administration of GroPIns4P as reported in this study. GroPIns4P represents an ideal lead compound for developing therapeutic approaches requiring the activation of Shp1. The present findings therefore add to the concept that GroPIns4P and GroPIns can be pharmacologically exploited as natural, non-toxic compounds of potential use in proliferative and immuno-inflammatory diseases (patent US9351983 B2 “Use of glycerophosphoinositols for the treatment of septic shock”).
Conclusions
With this study we show that the phosphatase Shp1 is the first-identified cellular receptor of the bioactive metabolite GroPIns4P, and delineate a signalling pathway involved in the regulation of cell motility that is initiated by the activation of the EGF-receptor and followed by the activation of cPLA2α, GroPIns4P formation and Shp1-direct interaction with, and activation of, Src. This in turn initiates a phosphorylation cascade leading to actin polymerisation, hence cell motility (Fig. 8). These data point also at the pharmacological use of GroPIns4P as a new tool to modulate Shp1 and Src activity, with several implications for various physiological and pathological conditions.
Fig. 8.

Schematic representation of the signalling cascade involved in the formation of GroPIns4P in NIH3T3 cells. In NIH3T3 cells, activation of the EGF receptor (EGFR) can lead to activation of cPLA2α through the involvement of MEK kinase. cPLA2α activation results in hydrolysis of PtdIns4P and release of GroPIns4P, which binds to Shp1 and promotes its association with Src. Once activated Src triggers a signalling cascade culminating with the formation of Tiam/Rac complex at the plasma membrane and consequent formation membrane ruffles with stimulation of cell motility. See text for details
Additional file
Figure S1. Direct binding of GroPIns4P to Shp1. Figure S2. Binding of GroPIns4P to the Shp1 WT and S118A/R138E/S140A mutant evaluated by isothermal titration calorimetry (ITC). Figure S3. Binding of GroPIns4P to the Shp1 S118A/R138E/S140A mutant. Figure S4. Localisation of endogenous cPLA2α in NIH3T3 cells. Table S1. Acquisition parameters for NMR experiments performed on the cSH2 domain of Shp1. Table S2. Docking calculations. Table S3. List of proteins identified from proteomic analysis. (DOCX 2706 kb)
Acknowledgments
The authors would like to thank all colleagues who kindly provided antibodies and reagents (as listed under ‘Reagents’); N. Dathan and M. Santoro for preparation of Shp1 mutants; F. Battista for performing part of the ITC experiments; G. Grimaldi for help in the preparation of the figures; A. Beccari for discussion; the BioImaging Facility at the Institute of Protein Biochemistry and the Laboratory of Ultrastructure at the Italian Institute of Technology of Naples, for support in imaging microscopy, data processing and analysis.
Funding
This research was supported by the Italian Association for Cancer Research (to D.C. IG10341 and IG18776; to A.L. 20786), PON01–00862 from MIUR, PNR-CNR Aging Program 2012–2014, Flagship project Nanomax, “FaReBio di Qualità”, OcKey POR Campania FSE 2007/2013, PRONAT project, the SATIN POR project 2014–2020 and the Italian MIUR Cluster project Medintech (CNT01_00177_962865). A.V. and M.V. received Fellowships from the Italian Foundation for Cancer Research (FIRC, Milan, Italy).
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information files.
Abbreviations
- CaMKII
Calcium-calmodulin kinase II
- cPLA2
Cytosolic-phospholipase A2
- EGF
Epidermal growth factor
- FRET
Fluorescence resonance energy transfer
- GEF
GTP exchange factor
- GroPIns4P
Glycerophosphoinositol 4-phosphate
- Ins4P
Inositol 4-phosphate
- NMR
Nuclear magnetic resonance
- PDGF
Platelet-derived growth factor
- SH2
Src-homology 2
- SPR
Surface plasmon resonance
Authors’ contributions
AV designed, performed analysed the experiments, and co-wrote the manuscript; MP, VM, AV, DS, MV, FF, NB, AF performed the experiments; SM, LB, SD’A, MLM, MF, PDV, PP, CI, AL, CV analysed and discussed the data; D.C. conceived and supervised the project, discussed and analysed the data and co-wrote the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Alessia Varone, Phone: +39-081-6132273, Email: a.varone@ibp.cnr.it.
Stefania Mariggiò, Email: s.mariggio@ibp.cnr.it.
Manpreet Patheja, Email: m.patheja@ibp.cnr.it.
Vincenzo Maione, Email: maione@cerm.unifi.it.
Antonio Varriale, Email: antonio.varriale@isa.cnr.it.
Mariangela Vessichelli, Email: mariangela.vessichelli@gmail.com.
Daniela Spano, Email: d.spano@ibp.cnr.it.
Fabio Formiggini, Email: fabio.formiggini@iit.it.
Matteo Lo Monte, Email: m.lomonte@ibp.cnr.it.
Nadia Brancati, Email: nadbra@na.icar.cnr.it.
Maria Frucci, Email: maria.frucci@na.icar.cnr.it.
Pompea Del Vecchio, Email: pompea.delvecchio@unina.it.
Sabato D’Auria, Email: direttore@isa.cnr.it.
Angela Flagiello, Email: flagiello@ceinge.unina.it.
Clara Iannuzzi, Email: clara.iannuzzi@unicampania.it.
Alberto Luini, Email: a.luini@ibp.cnr.it.
Piero Pucci, Email: pucci@unina.it.
Lucia Banci, Email: banci@cerm.unifi.it.
Carmen Valente, Email: c.valente@ibp.cnr.it.
Daniela Corda, Phone: +39-081-6132273, Email: d.corda@ibp.cnr.it.
References
- 1.Valitutti S, Cucchi P, Colletta G, Di Filippo C, Corda D. Transformation by the k-ras oncogene correlates with increases in phospholipase A2 activity, glycerophosphoinositol production and phosphoinositide synthesis in thyroid cells. Cell Signal. 1991;3:321–332. doi: 10.1016/0898-6568(91)90061-X. [DOI] [PubMed] [Google Scholar]
- 2.Mariggio S, Sebastia J, Filippi BM, Iurisci C, Volonte C, Amadio S, De Falco V, Santoro M, Corda D. A novel pathway of cell growth regulation mediated by a PLA2alpha-derived phosphoinositide metabolite. The FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2006;20:2567–2569. doi: 10.1096/fj.05-5397fje. [DOI] [PubMed] [Google Scholar]
- 3.Corda D, Falasca M. Glycerophosphoinositols as potential markers of ras-induced transformation and novel second messengers. Anticancer Res. 1996;16:1341–1350. [PubMed] [Google Scholar]
- 4.Falasca M, Marino M, Carvelli A, Iurisci C, Leoni S, Corda D. Changes in the levels of glycerophosphoinositols during differentiation of hepatic and neuronal cells. European journal of biochemistry / FEBS. 1996;241:386–392. doi: 10.1111/j.1432-1033.1996.00386.x. [DOI] [PubMed] [Google Scholar]
- 5.Corda D, Iurisci C, Berrie CP. Biological activities and metabolism of the lysophosphoinositides and glycerophosphoinositols. Biochim Biophys Acta. 2002;1582:52–69. doi: 10.1016/S1388-1981(02)00137-3. [DOI] [PubMed] [Google Scholar]
- 6.Falasca M, Carvelli A, Iurisci C, Qiu RG, Symons MH, Corda D. Fast receptor-induced formation of glycerophosphoinositol-4-phosphate, a putative novel intracellular messenger in the Ras pathway. Mol Biol Cell. 1997;8:443–453. doi: 10.1091/mbc.8.3.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zizza P, Iurisci C, Bonazzi M, Cossart P, Leslie CC, Corda D, Mariggio S. Phospholipase A2 IValpha regulates phagocytosis independent of its enzymatic activity. J Biol Chem. 2012. [DOI] [PMC free article] [PubMed]
- 8.Vessichelli M, Mariggio S, Varone A, Zizza P, Di Santo A, Amore C, Dell'Elba G, Cutignano A, Fontana A, Cacciapuoti C, et al. The natural phosphoinositide derivative glycerophosphoinositol inhibits the lipopolysaccharide-induced inflammatory and thrombotic responses. J Biol Chem. 2017;292:12828–12841. doi: 10.1074/jbc.M116.773861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patrussi L, Mariggio S, Corda D, Baldari CT. The glycerophosphoinositols: from lipid metabolites to modulators of T-cell signaling. Front Immunol. 2013;4:213. doi: 10.3389/fimmu.2013.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mancini R, Piccolo E, Mariggio S, Filippi BM, Iurisci C, Pertile P, Berrie CP, Corda D. Reorganization of actin cytoskeleton by the phosphoinositide metabolite glycerophosphoinositol 4-phosphate. Mol Biol Cell. 2003;14:503–515. doi: 10.1091/mbc.e02-04-0179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filippi BM, Mariggio S, Pulvirenti T, Corda D. SRC-dependent signalling regulates actin ruffle formation induced by glycerophosphoinositol 4-phosphate. Biochim Biophys Acta. 2008;1783:2311–2322. doi: 10.1016/j.bbamcr.2008.07.021. [DOI] [PubMed] [Google Scholar]
- 12.Patrussi L, Mariggio S, Paccani SR, Capitani N, Zizza P, Corda D, Baldari CT. Glycerophosphoinositol-4-phosphate enhances SDF-1alpha-stimulated T-cell chemotaxis through PTK-dependent activation of Vav. Cell Signal. 2007;19:2351–2360. doi: 10.1016/j.cellsig.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 13.Corda D, Zizza P, Varone A, Filippi BM, Mariggio S. The glycerophosphoinositols: cellular metabolism and biological functions. Cell Mol Life Sci. 2009;66:3449–3467. doi: 10.1007/s00018-009-0113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Corda D, Zizza P, Varone A, Bruzik KS, Mariggio S. The glycerophosphoinositols and their cellular functions. Biochem Soc Trans. 2012;40:101–107. doi: 10.1042/BST20110679. [DOI] [PubMed] [Google Scholar]
- 15.Somani AK, Bignon JS, Mills GB, Siminovitch KA, Branch DR. Src kinase activity is regulated by the SHP-1 protein-tyrosine phosphatase. J Biol Chem. 1997;272:21113–21119. doi: 10.1074/jbc.272.34.21113. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Somani AK, Siminovitch KA. Roles of the SHP-1 tyrosine phosphatase in the negative regulation of cell signalling. Semin Immunol. 2000;12:361–378. doi: 10.1006/smim.2000.0223. [DOI] [PubMed] [Google Scholar]
- 17.Banville D, Stocco R, Shen SH. Human protein tyrosine phosphatase 1C (PTPN6) gene structure: alternate promoter usage and exon skipping generate multiple transcripts. Genomics. 1995;27:165–173. doi: 10.1006/geno.1995.1020. [DOI] [PubMed] [Google Scholar]
- 18.Tsui HW, Hasselblatt K, Martin A, Mok SC, Tsui FW. Molecular mechanisms underlying SHP-1 gene expression. European journal of biochemistry / FEBS. 2002;269:3057–3064. doi: 10.1046/j.1432-1033.2002.02986.x. [DOI] [PubMed] [Google Scholar]
- 19.Shultz LD, Schweitzer PA, Rajan TV, Yi T, Ihle JN, Matthews RJ, Thomas ML, Beier DR. Mutations at the murine motheaten locus are within the hematopoietic cell protein-tyrosine phosphatase (Hcph) gene. Cell. 1993;73:1445–1454. doi: 10.1016/0092-8674(93)90369-2. [DOI] [PubMed] [Google Scholar]
- 20.Shultz LD, Rajan TV, Greiner DL. Severe defects in immunity and hematopoiesis caused by SHP-1 protein-tyrosine-phosphatase deficiency. Trends Biotechnol. 1997;15:302–307. doi: 10.1016/S0167-7799(97)01060-3. [DOI] [PubMed] [Google Scholar]
- 21.Li YF, Xu S, Ou X, Lam KP. Shp1 signalling is required to establish the long-lived bone marrow plasma cell pool. Nat Commun. 2014;5:4273. doi: 10.1038/ncomms5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roskoski R., Jr Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun. 2005;331:1–14. doi: 10.1016/j.bbrc.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 23.Garrett DS, Seok YJ, Peterkofsky A, Clore GM, Gronenborn AM. Identification by NMR of the binding surface for the histidine-containing phosphocarrier protein HPr on the N-terminal domain of enzyme I of the Escherichia coli phosphotransferase system. Biochemistry. 1997;36:4393–4398. doi: 10.1021/bi970221q. [DOI] [PubMed] [Google Scholar]
- 24.Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc. 2003;125:1731–1737. doi: 10.1021/ja026939x. [DOI] [PubMed] [Google Scholar]
- 25.Yang J, Liu L, He D, Song X, Liang X, Zhao ZJ, Zhou GW. Crystal structure of human protein-tyrosine phosphatase SHP-1. J Biol Chem. 2003;278:6516–6520. doi: 10.1074/jbc.M210430200. [DOI] [PubMed] [Google Scholar]
- 26.Sobel I., Duda R., Hart P., J. W: Sobel-Feldman Operator. 2014.
- 27.Brancati N, Frucci M, Sanniti di Baja G. Image segmentation via iterative histogram thresholding and morphological features analysis. In: Campilho A, Kamel M, editors. Image analysis and recognition. Póvoa de Varzim: International Conference Image Analysis and Recognition; 2008. p. 132–41.
- 28.Soille P. Morphological image analysis: principles and applications. 2013. [Google Scholar]
- 29.Iacovelli L, Falasca M, Valitutti S, D'Arcangelo D, Corda D. Glycerophosphoinositol 4-phosphate, a putative endogenous inhibitor of adenylylcyclase. J Biol Chem. 1993;268:20402–20407. [PubMed] [Google Scholar]
- 30.O'Shannessy DJ, Winzor DJ. Interpretation of deviations from pseudo-first-order kinetic behavior in the characterization of ligand binding by biosensor technology. Anal Biochem. 1996;236:275–283. doi: 10.1006/abio.1996.0167. [DOI] [PubMed] [Google Scholar]
- 31.Dyson HJ, Wright PE. Unfolded proteins and protein folding studied by NMR. Chem Rev. 2004;104:3607–3622. doi: 10.1021/cr030403s. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Z, Shen K, Lu W, Cole PA. The role of C-terminal tyrosine phosphorylation in the regulation of SHP-1 explored via expressed protein ligation. J Biol Chem. 2003;278:4668–4674. doi: 10.1074/jbc.M210028200. [DOI] [PubMed] [Google Scholar]
- 33.Rameh LE, Chen CS, Cantley LC. Phosphatidylinositol (3,4,5)P3 interacts with SH2 domains and modulates PI 3-kinase association with tyrosine-phosphorylated proteins. Cell. 1995;83:821–830. doi: 10.1016/0092-8674(95)90195-7. [DOI] [PubMed] [Google Scholar]
- 34.Tokonzaba E, Capelluto DG, Kutateladze TG, Overduin M. Phosphoinositide, phosphopeptide and pyridone interactions of the Abl SH2 domain. Chem Biol Drug Des. 2006;67:230–237. doi: 10.1111/j.1747-0285.2006.00361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mariggio S, Iurisci C, Sebastia J, Patton-Vogt J, Corda D. Molecular characterization of a glycerophosphoinositol transporter in mammalian cells. FEBS Lett. 2006;580:6789–6796. doi: 10.1016/j.febslet.2006.11.039. [DOI] [PubMed] [Google Scholar]
- 36.Patton-Vogt JL, Henry SA. GIT1, a gene encoding a novel transporter for glycerophosphoinositol in Saccharomyces cerevisiae. Genetics. 1998;149:1707–1715. doi: 10.1093/genetics/149.4.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Su L, Zhao Z, Bouchard P, Banville D, Fischer EH, Krebs EG, Shen SH. Positive effect of overexpressed protein-tyrosine phosphatase PTP1C on mitogen-activated signaling in 293 cells. J Biol Chem. 1996;271:10385–10390. doi: 10.1074/jbc.271.17.10385. [DOI] [PubMed] [Google Scholar]
- 38.You M, Zhao Z. Positive effects of SH2 domain-containing tyrosine phosphatase SHP-1 on epidermal growth factor- and interferon-gamma-stimulated activation of STAT transcription factors in HeLa cells. J Biol Chem. 1997;272:23376–23381. doi: 10.1074/jbc.272.37.23376. [DOI] [PubMed] [Google Scholar]
- 39.Xie ZH, Zhang J, Siraganian RP. Positive regulation of c-Jun N-terminal kinase and TNF-alpha production but not histamine release by SHP-1 in RBL-2H3 mast cells. J Immunol. 2000;164:1521–1528. doi: 10.4049/jimmunol.164.3.1521. [DOI] [PubMed] [Google Scholar]
- 40.Tabernero L, Aricescu AR, Jones EY, Szedlacsek SE. Protein tyrosine phosphatases: structure-function relationships. FEBS J. 2008;275:867–882. doi: 10.1111/j.1742-4658.2008.06251.x. [DOI] [PubMed] [Google Scholar]
- 41.Kundu S, Fan K, Cao M, Lindner DJ, Zhao ZJ, Borden E, Yi T. Novel SHP-1 inhibitors tyrosine phosphatase inhibitor-1 and analogs with preclinical anti-tumor activities as tolerated oral agents. J Immunol. 2010;184:6529–6536. doi: 10.4049/jimmunol.0903562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen L, Sung SS, Yip ML, Lawrence HR, Ren Y, Guida WC, Sebti SM, Lawrence NJ, Wu J. Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol Pharmacol. 2006;70:562–570. doi: 10.1124/mol.106.025536. [DOI] [PubMed] [Google Scholar]
- 43.Abercrombie M, Ambrose EJ. Interference microscope studies of cell contacts in tissue culture. Exp Cell Res. 1958;15:332–345. doi: 10.1016/0014-4827(58)90034-X. [DOI] [PubMed] [Google Scholar]
- 44.Abercrombie M, Heaysman JE, Pegrum SM. The locomotion of fibroblasts in culture. II. "RRuffling". Exp Cell Res. 1970;60:437–444. doi: 10.1016/0014-4827(70)90537-9. [DOI] [PubMed] [Google Scholar]
- 45.Small JV, Stradal T, Vignal E, Rottner K. The lamellipodium: where motility begins. Trends Cell Biol. 2002;12:112–120. doi: 10.1016/S0962-8924(01)02237-1. [DOI] [PubMed] [Google Scholar]
- 46.Seno K, Okuno T, Nishi K, Murakami Y, Watanabe F, Matsuura T, Wada M, Fujii Y, Yamada M, Ogawa T, et al. Pyrrolidine inhibitors of human cytosolic phospholipase a(2) J Med Chem. 2000;43:1041–1044. doi: 10.1021/jm9905155. [DOI] [PubMed] [Google Scholar]
- 47.Peppelenbosch MP, Qiu RG, de Vries-Smits AM, Tertoolen LG, de Laat SW, McCormick F, Hall A, Symons MH, Bos JL. Rac mediates growth factor-induced arachidonic acid release. Cell. 1995;81:849–856. doi: 10.1016/0092-8674(95)90005-5. [DOI] [PubMed] [Google Scholar]
- 48.Magi S, Takemoto Y, Kobayashi H, Kasamatsu M, Akita T, Tanaka A, Takano K, Tashiro E, Igarashi Y, Imoto M. 5-lipoxygenase and cysteinyl leukotriene receptor 1 regulate epidermal growth factor-induced cell migration through Tiam1 upregulation and Rac1 activation. Cancer Sci. 2014;105:290–296. doi: 10.1111/cas.12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mayer BJ. The discovery of modular binding domains: building blocks of cell signalling. Nat Rev Mol Cell Biol. 2015;16:691–698. doi: 10.1038/nrm4068. [DOI] [PubMed] [Google Scholar]
- 50.Sharma Y, Bashir S, Bhardwaj P, Ahmad A, Khan F. Protein tyrosine phosphatase SHP-1: resurgence as new drug target for human autoimmune disorders. Immunol Res. 2016;64:804–819. doi: 10.1007/s12026-016-8805-y. [DOI] [PubMed] [Google Scholar]
- 51.Sharma Y, Ahmad A, Bashir S, Elahi A, Khan F. Implication of protein tyrosine phosphatase SHP-1 in cancer-related signaling pathways. Future Oncol. 2016;12:1287–1298. doi: 10.2217/fon-2015-0057. [DOI] [PubMed] [Google Scholar]
- 52.Huang TT, Su JC, Liu CY, Shiau CW, Chen KF. Alteration of SHP-1/p-STAT3 signaling: a potential target for anticancer therapy. Int J Mol Sci. 2017;18. [DOI] [PMC free article] [PubMed]
- 53.Schalkwijk CG, Spaargaren M, Defize LH, Verkleij AJ, van den Bosch H, Boonstra J. Epidermal growth factor (EGF) induces serine phosphorylation-dependent activation and calcium-dependent translocation of the cytosolic phospholipase A2. European journal of biochemistry / FEBS. 1995;231:593–601. doi: 10.1111/j.1432-1033.1995.tb20737.x. [DOI] [PubMed] [Google Scholar]
- 54.Schalkwijk CG, van der Heijden MA, Bunt G, Maas R, Tertoolen LG, Van Bergen en Henegouwen PM, Verkleij AJ, van den Bosch H, Boonstra J. Maximal epidermal growth-factor-induced cytosolic phospholipase A2 activation in vivo requires phosphorylation followed by an increased intracellular calcium concentration. The Biochemical journal. 1996;313(Pt 1):91–96. doi: 10.1042/bj3130091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bunt G, de Wit J, van den Bosch H, Verkleij AJ, Boonstra J. Ultrastructural localization of cPLA2 in unstimulated and EGF/A23187-stimulated fibroblasts. J Cell Sci. 1997;110(Pt 19):2449–2459. doi: 10.1242/jcs.110.19.2449. [DOI] [PubMed] [Google Scholar]
- 56.Jones S, Rappoport JZ. Interdependent epidermal growth factor receptor signalling and trafficking. Int J Biochem Cell Biol. 2014;51:23–28. doi: 10.1016/j.biocel.2014.03.014. [DOI] [PubMed] [Google Scholar]
- 57.Matsuzawa Y, Kiuchi Y, Toyomura K, Matsumoto I, Nakamura H, Fujino H, Murayama T, Kawashima T. Activation of cytosolic phospholipase A2alpha by epidermal growth factor (EGF) and phorbol ester in HeLa cells: different effects of inhibitors for EGF receptor, protein kinase C, Src, and C-Raf. J Pharmacol Sci. 2009;111:182–192. doi: 10.1254/jphs.09201FP. [DOI] [PubMed] [Google Scholar]
- 58.Balsinde J, Balboa MA, Insel PA, Dennis EA. Regulation and inhibition of phospholipase A2. Annu Rev Pharmacol Toxicol. 1999;39:175–189. doi: 10.1146/annurev.pharmtox.39.1.175. [DOI] [PubMed] [Google Scholar]
- 59.Li S, Wang Q, Wang Y, Chen X, Wang Z. PLC-gamma1 and Rac1 coregulate EGF-induced cytoskeleton remodeling and cell migration. Mol Endocrinol. 2009;23:901–913. doi: 10.1210/me.2008-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nogami M, Yamazaki M, Watanabe H, Okabayashi Y, Kido Y, Kasuga M, Sasaki T, Maehama T, Kanaho Y. Requirement of autophosphorylated tyrosine 992 of EGF receptor and its docking protein phospholipase C gamma 1 for membrane ruffle formation. FEBS Lett. 2003;536:71–76. doi: 10.1016/S0014-5793(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 61.Dittmar T, Husemann A, Schewe Y, Nofer JR, Niggemann B, Zanker KS, Brandt BH. Induction of cancer cell migration by epidermal growth factor is initiated by specific phosphorylation of tyrosine 1248 of c-erbB-2 receptor via EGFR. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2002;16:1823–1825. doi: 10.1096/fj.02-0096fje. [DOI] [PubMed] [Google Scholar]
- 62.Oh SY, Lee SJ, Jung YH, Lee HJ, Han HJ. Arachidonic acid promotes skin wound healing through induction of human MSC migration by MT3-MMP-mediated fibronectin degradation. Cell Death Dis. 2015;6:e1750. doi: 10.1038/cddis.2015.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stockton RA, Jacobson BS. Modulation of cell-substrate adhesion by arachidonic acid: lipoxygenase regulates cell spreading and ERK1/2-inducible cyclooxygenase regulates cell migration in NIH-3T3 fibroblasts. Mol Biol Cell. 2001;12:1937–1956. doi: 10.1091/mbc.12.7.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen Z, Oh D, Dubey AK, Yao M, Yang B, Groves JT, Sheetz M. EGFR family and Src family kinase interactions: mechanics matters? Curr Opin Cell Biol. 2018;51:97–102. doi: 10.1016/j.ceb.2017.12.003. [DOI] [PubMed] [Google Scholar]
- 65.Zhang F, Yang H, Pan Z, Wang Z, Wolosin JM, Gjorstrup P, Reinach PS. Dependence of resolvin-induced increases in corneal epithelial cell migration on EGF receptor transactivation. Invest Ophthalmol Vis Sci. 2010;51:5601–5609. doi: 10.1167/iovs.09-4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mariggio S, Filippi BM, Iurisci C, Dragani LK, De Falco V, Santoro M, Corda D. Cytosolic phospholipase A2 alpha regulates cell growth in RET/PTC-transformed thyroid cells. Cancer Res. 2007;67:11769–11778. doi: 10.1158/0008-5472.CAN-07-1997. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Direct binding of GroPIns4P to Shp1. Figure S2. Binding of GroPIns4P to the Shp1 WT and S118A/R138E/S140A mutant evaluated by isothermal titration calorimetry (ITC). Figure S3. Binding of GroPIns4P to the Shp1 S118A/R138E/S140A mutant. Figure S4. Localisation of endogenous cPLA2α in NIH3T3 cells. Table S1. Acquisition parameters for NMR experiments performed on the cSH2 domain of Shp1. Table S2. Docking calculations. Table S3. List of proteins identified from proteomic analysis. (DOCX 2706 kb)
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information files.