

Abstract
The enormous rate accelerations observed for many enzyme catalysts are due to strong stabilizing interactions between the protein and reaction transition state. The defining property of these catalysts is their specificity for binding the transition state with a much higher affinity than substrate. Experimental results are presented which show that the phosphodianion-binding energy of phosphate monoester substrates is used to drive conversion of their protein catalysts from flexible and entropically rich ground states to stiff and catalytically active Michaelis complexes. These results are generalized to other enzyme-catalyzed reactions. The existence of many enzymes in flexible, entropically rich, and inactive ground states provides a mechanism for utilization of ligand-binding energy to mold these catalysts into stiff and active forms. This reduces the substrate-binding energy expressed at the Michaelis complex, while enabling the full and specific expression of large transition-state binding energies. Evidence is presented that the complexity of enzyme conformational changes increases with increases in the enzymatic rate acceleration. The requirement that a large fraction of the total substrate-binding energy be utilized to drive conformational changes of floppy enzymes is proposed to favor the selection and evolution of protein folds with multiple flexible unstructured loops, such as the TIM-barrel fold. The effect of protein motions on the kinetic parameters for enzymes that undergo ligand-driven conformational changes is considered. The results of computational studies to model the complex ligand-driven conformational change in catalysis by triosephosphate isomerase are presented.
Introduction
Bioorganic chemists have understood for more than 50 years that the first step toward determining the mechanism for enzymatic catalysis of polar reactions, such as proton transfer and nucleophilic substitution at carbon, is to determine the mechanisms for catalysis of these reactions by molecules that model the active-site amino acid side chains.1,2 The results from studies on catalysis by these models generally show that enzymes follow one of the reaction mechanisms observed in solution.3,4 However, the synthetic enzyme models fail to capture the large rate accelerations observed for enzyme catalysts.
Why do rate accelerations for catalysis by synthetic enzyme models fall short of those by enzymes? Answers can be found through a consideration of what has been selected for during enzyme evolution. The high conservation of the structure of glycolytic enzymes,5 present in all forms of life, over the past several billion years provides strong evidence that evolution has eliminated non-essential elements of enzyme structure. This suggests that regions distant from the active sites of glycolytic enzymes are essential for efficient function because of interactions between the active site and remote protein side chains. These are not through-space electrostatic interactions, which fall off rapidly with increasing separation from the active site.6 Rather, the interactions are thought to be associated with protein motions that extend from the active site to other parts of the catalyst—hence, the intense interest in establishing links between enzyme catalytic function, enzyme conformational changes, and the dynamics of these conformational changes.7−12
Lock-and-Key or Induced Fit?
The lock-and-key analogy postulated in 1894 by Emil Fischer compares the substrate to a key that must be the correct size and shape to fit into the stiff enzyme and undergo the catalyzed reaction.13 This analogy is supported by the rigid structures of enzyme–ligand complexes from X-ray crystallographic analyses. These structures are routinely used in high-level calculations of activation barriers for formation of enzyme-bound transition states that are in good agreement with the experimental activation barriers.14−19 This suggests that the rigid structures capture the full catalytic power of many enzymes.
By contrast, the induced-fit model postulated by Daniel Koshland in 195820 asserts that binding interactions between flexible enzymes and their substrates are utilized to mold enzyme active sites into structures that are complementary to the reaction transition state. There are abundant examples of such ligand-driven conformational changes,9,21,22 several of which will be discussed in this Perspective. The coexistence of lock-and-key and induced-fit models represents two assessments of enzyme catalysis. In fact, stiffness and flexibility are complementary protein properties that are required to obtain the extraordinary catalytic efficiency of many enzymes. This Perspective presents evidence that the catalytic events for the turnover of enzyme-bound substrate to product occur at stiff protein active sites, and it describes the imperatives for the evolution of enzymes with flexible structures in their unliganded form that undergo large ligand-driven protein conformational changes to an active stiff form.
Reactive Michaelis Complexes Are Stiff
Many results are consistent with the conclusion that the structures for reactive Michaelis complexes of enzyme catalysts are stiff and allow for minimal protein motions away from highly organized forms. As noted above, enzyme-ligand complexes from X-ray crystallographic analyses serve as good starting points for calculations that model the experimental activation barrier for turnover at enzyme active sites,14,15 so that the stiffness of reactive enzyme–substrate complexes is similar to that for crystalline enzymes. The empirical valence bond (EVB) computational methods developed by Arieh Warshel strongly emphasize the modeling of electrostatic interactions.17,23,24 The success of these methods at reproducing the activation barriers for enzymatic reactions is consistent with the primacy of electrostatic interactions in transition-state stabilization and with Warshel’s strongly held conviction that optimal electrostatic stabilization is achieved by preorganization of active-site side chains into a stiff catalytic conformation.25−29 The results of a recent study on the directed evolution of a designed Kemp eliminase provide evidence for the requirement for the precision in placement of catalytic side chains in order to obtain robust catalysis.30 These models and proposals are modern reformulations of Fisher’s lock-and-key model.
Antibodies are also stiff and show affinities for ligands comparable to that of some less proficient enzymes for their transition states. Antibodies have been produced that catalyze chemical reactions, but with smaller rate accelerations than for the most proficient enzymes.31−33 This suggests that protein stiffness alone will not produce the largest enzymatic rate accelerations, but must be combined with protein flexibility to obtain well-rounded and efficient catalysts.
Specificity in Transition-State Binding
The failure to capture the full catalytic rate accelerations of enzymes in synthetic models,34,35 catalytic antibodies,31−33 or in designed protein catalysts36 has driven studies to eliminate gaps in our understanding of enzymatic catalysis.37−42 Watching events as an outsider engaged in studies on organic reaction mechanisms in aqueous solution, I became infected with the ambition to expand our understanding of enzyme catalysis. I was intrigued by William P. Jencks’s proposal that the most important difference between catalysis by enzymes and that by small molecules is that only enzymes have evolved mechanisms for the utilization of substrate-binding energy in the specific stabilization of the transition states for catalyzed reactions.43 These mechanisms remained poorly characterized 30 years after Jencks’s classic 1975 review.43
The difficulty in rationalizing the specificity shown by enzymes in binding their transition states with a higher affinity than substrate is highlighted by the difference between the modest 8 kcal/mol stabilization of the ground-state complex (Kd = 10–6 M) to orotidine 5′-monophosphate (OMP) and the large 31 kcal/mol stabilization of the transition state (Kd⧧ = 10–23 M) for OMP decarboxylase-catalyzed (OMPDC) decarboxylation to form uridine monophosphate (UMP) through a UMP carbanion reaction intermediate (Schemes 1 and 2).44,45 It is as though a switch is turned on at OMPDC as the transition state is approached, which releases the full substrate-binding energy from interactions with both the reacting portions of the substrate and the non-reacting portions such as the phosphodianion and ribosyl hydroxyls.46,47 When there is no such switch, such as for the binding of biotin to avidin with a binding energy of −20 kcal/mol,48 binding is effectively irreversible, and the biotin–avidin complex has a lifetime of 200 days.49,50
Scheme 1. OMPDC-Catalyzed Decarboxylation of OMP through a UMP Carbanion Reaction Intermediate.

Scheme 2. OMPDC-Catalyzed Decarboxylation, with an 8 kcal/mol Stabilization of the Ground-State Complex (Kd = 10–6 M) and a 31 kcal/mol Stabilization of the Rate-Determining Transition State (Kd⧧ = 10–23 M)44,45.

In taking up the challenge to characterize these protein/ligand switches, I hoped to add one missing link to our understanding of enzyme catalysis, while connecting or discarding disparate proposals about how enzymes work. Our studies on the specificity of enzymes for binding their transition states with a higher affinity than substrate have had the unforeseen consequence of identifying a strong imperative for the evolution of enzymes that are flexible in their unliganded form and undergo ligand-driven conformational changes to stiff and active catalysts.
Utilization of Dianion-Binding Energy for Enzyme Activation
Five enzymes which catalyze reactions of substrates that contain a non-reacting phosphate monoester handle have been shown to utilize binding interactions with the phosphite dianion substrate piece to specifically stabilize the transition state for enzyme-catalyzed reactions of phosphodianion-truncated substrates.51−53 In a representative case phosphite dianion shows a ca. 2 kcal/mol binding affinity for free enzyme and provides an 8 kcal/mol stabilization of the transition state for OMPDC-catalyzed decarboxylation of the truncated substrate 1-β-d-erythrofuranosyl-5-fluoroorotate (EO). This gives rise to an 80 000-fold larger second-order rate constant for decarboxylation of EO by the binary E·HPi complex (HPi = phosphite dianion) compared with decarboxylation by E alone (Scheme 3).46 We also reported HPi activation of triosephosphate isomerase (TIM)54 and of glycerol phosphate dehydrogenase (GPDH)55 for catalysis of proton-transfer and hydride-transfer reactions, respectively, of the small phosphodianion-truncated substrate glycolaldehyde (GA, Figure 1). A similar HPi activation of truncated substrate was observed in studies on phosphoglucomutase53 and on 1-deoxy-d-xylulose-5-phosphate reductoisomerase.52,56 Our studies on dianion activation of OMPDC, TIM, and GPDH have been described in several reviews57−61 that focus on the mechanism for dianion activation of enzyme-catalyzed decarboxylation, proton-transfer, and hydride-transfer reactions. I look outwardly in this Perspective and consider whether the architectural elements that enable enzyme activation by dianions are propagated widely in enzymes that catalyze polar reactions in water.
Scheme 3. Phosphite Dianion-Activated, OMPDC-Catalyzed Decarboxylation of a Phosphodianion-Truncated Substrate through a Carbanion Reaction Intermediate.

Figure 1.

Dianion-activated proton- and hydride-transfer reactions catalyzed by TIM (reactions on the left) and by GPDH (reactions on the right). TIM catalyzes isomerization of the whole substrate glyceraldehyde 3-phosphate (GAP) to form dihydroxyacetone phosphate (DHAP)62 and exchange between deuterium in D2O and the α-carbonyl hydrogen of the substrate piece glycolaldehyde (GA) that is activated by the second piece of phosphite dianion (HPi = HPO32–).54,63 GPDH catalyzes reduction of the whole substrate DHAP by NADH to form l-glycerol 3-phosphate (G3P) and reduction of GA by NADH to form ethylene glycol (EG) that is activated by HPi.55 In each case phosphite dianion binds weakly to free enzyme, while the transition state for the reactions of the whole substrate is stabilized by 11–12 kcal/mol by interactions with the substrate phosphodianion, and the transition state for reaction of the truncated substrate is stabilized by 6–8 kcal/mol by interactions with the phosphite dianion.51
A Role for Protein Conformational Changes in Enzyme Catalysis
Our rationale for parallel studies on TIM, OMPDC, and GPDH follows from studies by Jeremy Knowles on TIM,64,65 which show that this enzyme meets two criteria for perfection in achieving efficient catalysis of a reaction in glycolysis.66 The catalytic strategies first realized by TIM more 3 billion years ago5,67 may extend beyond the chemistry of the catalyzed proton-transfer reactions and include perfection of the mechanism for enzyme activation by dianions. This prompted the hypothesis that the proliferation of the TIM barrel protein fold to 10% of all proteins (including OMPDC)68−71 was favored by structural elements that enable dianion and other types of enzyme activation.
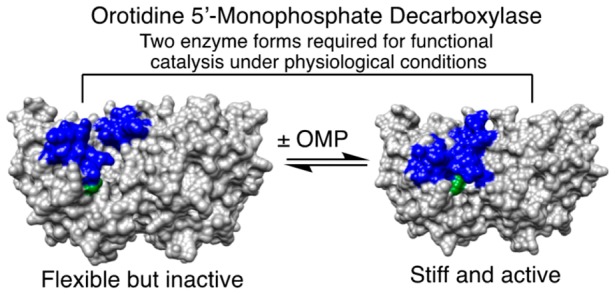
The structures for unliganded and liganded forms of OMPDC, TIM, and GPDH are shown in Figure 2, with the phosphodianion gripper loops shaded blue and a side-chain cation shaded green. Each enzyme undergoes a large conformational change upon substrate binding that is driven by interactions between the protein and substrate phosphodianion (shaded red) or the phosphite dianion piece. Each enzyme is inactive in the open form because of the poor positioning of catalytic side chains at the enzyme active site. In each case, the ligand-driven enzyme conformational change to form the active closed enzyme is the switch that turns on the expression of the full transition-state binding energy.
Figure 2.

Surface structures for TIM (top), GPDH (middle), and OMPDC (bottom). The binding energy of the ligand phosphodianion is utilized to immobilize these loops, in driving the conformational changes to the stiff and catalytically active closed structures shown on the right. The ligand phosphodianion at the closed enzymes is shaded red, and the side-chain cations, which interact with the phosphodianion, are shaded green. Key: Top structures; TIM from Trypanosoma brucei brucei (open form, PDB entry 3TIM; closed form with 3-phosphoglycerate bound, PDB entry 1IIH). The phosphodianion gripper loop (residues 165–177) is shaded blue, and the side chain from K12 is shaded green. Not shown is loop 7 (residues 208–216), whose side chains Y208 and S211 move as the planes defined by the peptide bonds from G209 and G210 undergo 90° and 180° rotations, respectively.74 Middle structures; GPDH from human liver (open form, PDB entry 1X0V; closed form with NAD and DHAP bound, PDB entry 1WPQ). The phosphodianion gripper loop (residues 292–297) is shaded blue, and the side chain from R269 is shaded green. The side chain of Q295 interacts with the substrate phosphodianion through the intervening side chain of R269.75 Bottom structures; OMPDC from Saccharomyces cerevisiae (open form, PDB entry 1DQW; closed form with 6-hydroxyuridine 5′-monophosphate bound, PDB entry 1DQX). The phosphodianion gripper loop (residues 202–220) is shaded blue, and the side chain from R235 is shaded green. The pyrimidine umbrella loop (residues 151–165) is also shaded blue. The blue loops interact at the closed form of OMPDC through a hydrogen bond between the side chains of S154 and Q215.76,77
Figure 2 shows that the dianion-binding energy for OMPDC, TIM, and GPDH is utilized to drive protein conformational changes, which activate these enzymes for catalysis of decarboxylation, proton transfer, and hydride transfer, respectively. This activation is described by the model in Scheme 4.43,61Scheme 4 holds for enzymes that exist mainly in an inactive open form (EO) that is in equilibrium with an active but conformationally unstable (ΔGC > 0) closed form (EC), which shows a much higher affinity than EO for binding to the phosphodianion of whole substrate or to the phosphite dianion piece. Equation 1 in Scheme 4 shows that the observed substrate-binding energy ΔGobsd is then equal to the sum of the intrinsic substrate-binding energy ΔGint plus ΔGC.43,61 The absolute value of ΔGint is greater than ΔGobsd, because of the binding energy ΔGC required to drive the enzyme conformational change from EO to EC. This binding energy is used, partly or entirely, to drive desolvation of active-site side chains at EO and to hold the flexible unliganded protein catalyst in the stiff conformation that is required for the observation of high enzymatic activity.72,73
Scheme 4. Relationship between the Observed and Intrinsic Substrate-Binding Energy, When Binding Drives a Conformational Change from EO to EC.

Connections
The existence of unliganded enzymes in inactive, flexible, and entropically rich open forms provide a mechanism for the utilization of large intrinsic dianion-binding energies to drive conformational changes to stiff, closed, and entropically depleted active enzymes. There are many connections between the common mechanisms for dianion activation of OMPDC, TIM, and GPDH that are relevant to more general observations on enzyme-catalyzed reactions.
(1) The imperatives for the existence of unliganded enzymes in stable open forms deserves scrutiny.43,78 Why do not these enzymes exist in the stiff and catalytically active closed form, thereby eliminating expenditure of substrate-binding energy to create a stiff enzyme? There is a two-part answer to this question. First, Wolfenden noted that the existence of enzymes in an open form with the active site accessible to solvent is required when the substrate is ultimately bound at a protein cage EC that would occlude ligand (Figure 2).73,79 Second, efficient catalysis is facilitated by a sizable difference in the stability of EO and EC whenever the substrate-binding energy required to obtain the total transition-state stabilization is large, because part of this binding energy must then be expended during ligand binding to avoid effectively irreversible ligand association.43,61
(2) The conformational change from EO to EC (Scheme 4) is not limited to the closure of flexible loops over substrate (Figure 2). Others examples include the “oyster-like” clamping motion of protein domains over diaminopimelate (DAP) bound to DAP epimerase,80 the closure of the capping lid domains over substrate observed for members of the enolase81,82 and haloalkane dehalogenase superfamilies,83,84 and the changes in the shape of flexible binding pockets observed upon ligand binding.9 The common feature of these ligand-driven protein enzyme conformational changes is that each activates the enzyme for catalysis, as shown in Scheme 4.
(3) The phosphodianion is one of several non-reacting substrate fragments whose binding energy is utilized to drive enzyme-activating protein conformational changes. Others include the coenzyme A fragment of acetyl CoA,85,86 the ADP-ribose fragment of NAD/NADH,87 the pyrophosphate and tripolyphosphate fragments of ADP and ATP, respectively,42 and fragments that interact with the capping domains of members of the enolase81,82 and haloalkane dehalogenase superfamilies.83,84
(4) TIM barrel proteins undergo rapid conformational changes from movement of 16 enzyme loops. These loops provide a flexible unliganded enzyme and their interactions with bound substrates are used to mold TIM into a stiff and active Michaelis complex. The rapid exploration of many different ground-state conformations during loop movement at TIM-barrel proteins provides access to a large suite of protein conformations, in comparison to the single conformation for a stiff unliganded protein. Each of these conformations is a potential starting point for the evolution of a new enzyme activity. Natural selection of the active conformations has given rise to proteins with a large number of enzymatic activities.88,89
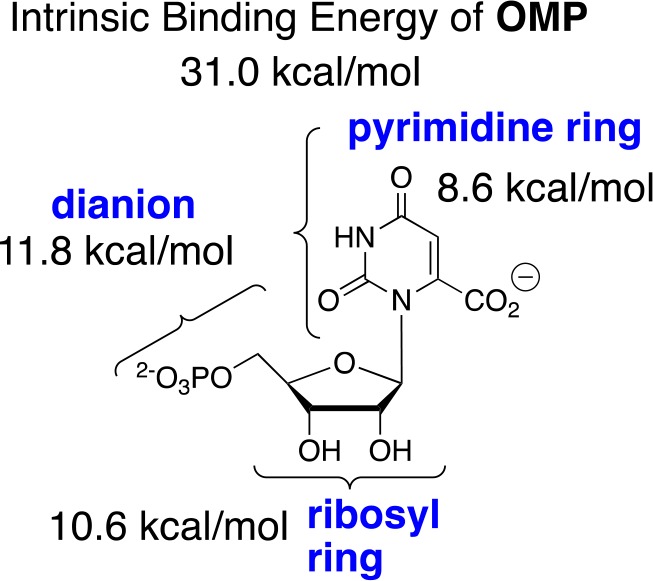
(5) There is evidence for a correlation between the increasing complexity of ligand-driven enzyme conformational changes and increasing total transition-state stabilization. This reflects the increasing number of side-chain interactions that must develop in creating a caged substrate complex with the necessary large transition-state stabilization. For example, the very large 31 kcal/mol total binding energy of OMPDC for the decarboxylation transition state is partitioned between interactions with the phosphodianion, ribosyl, and substrate fragments (Scheme 5).47,90 The interactions of the protein that develop with both the phosphodianion and ribosyl hydroxyls are utilized to drive a complex conformational change that activates OMPDC for catalysis at the pyrimidine ring (Figure 1).47,91
Scheme 5. Partitioning of the Total 31 kcal/mol Intrinsic Binding for OMPDC-Catalyzed Decarboxylation into the Binding Energy for Three Substrate Fragments47.
(6) At the other extreme small enzymatic rate accelerations are associated with small or the absence of ligand-driven conformational changes. Non-enzymatic hydration of CO2 occurs over a period of minutes in water. The rate of the carbonic anhydrase-catalyzed hydration of CO2 is limited by a fast proton-transfer reaction between solvent and enzyme.92 The rate-determining step is thought to involve rapid rotation of the side chain of His-64, which shuttles protons between solvent and the enzyme active site.93 The enzyme 3-oxo-Δ5-steroid isomerase (KSI) catalyzes double-bond migration at a relatively strong carbon acid substrate (pKa = 13, Scheme 6).94 The rate enhancement for KSI is small95 compared to the flexible enzymes triosephosphate isomerase96 and diaminopimelate racemase, which catalyze deprotonation of much more weakly acidic carbon acid substrates.3,80 It is achieved at an active site situated in a shallow cleft on the protein surface,73 which interacts with only a single face of the steroid substrate whose binding induces only a small protein conformational change.97−99 Additional work is needed to extend these observations, which suggest a correlation between enzymatic rate accelerations44 and the magnitude of the substrate-driven conformational change.
Scheme 6. Isomerization Reaction Catalyzed by 3-Oxo-Δ5-Steroid Isomerase (KSI).

(7) The binding pockets of OMPDC, TIM, and GPDH are divided into dianion activation and catalytic sites. The dianion-binding interactions at the activation site trigger protein conformational changes that prime the enzyme for catalysis at the catalytic site.51 Similar principals should govern the operation of these dianion activation sites and traditional allosteric regulation sites, which regulate enzyme activity by binding an effector molecule at a site different from the active site.21,100 It is not known which type of effector site appeared first during evolution. For example, pressure might have been applied first toward the evolution of effector-type sites that optimized the total activity of primordial forms of TIM, OMPDC, and GPDH, through the utilization of the substrate phosphodianion-binding energy. These are cryptic dianion activation sites that also utilize the binding energy of phosphite, sulfate, thiosulfate, and related dianions for activation of the enzyme-catalyzed reactions phosphodianion-truncated substrates.51 They are potential starting points for the evolution of allosteric regulation sites.
(8) OMPDC, TIM, and GPDH use protein–dianion interactions to drive large enzyme conformational changes, which lock their substrates into active protein cages that provide strong stabilization of the transition state for the respective catalyzed reactions.73,101 Another model has been proposed for enzyme-catalyzed hydride-transfer reactions where the substrate-binding energy is used to stabilize a tunneling-ready state that promotes quantum-mechanical (QM) tunneling of the transferred hydron through the energy barrier.38,102,103 The small values for primary deuterium isotope effects (kH/kD = 2.4–3.1) that we have determined for numerous wild-type and mutant GPDH-catalyzed hydride-transfer reactions from NADH/NADD to DHAP or GA (Figure 1) show that there can be only incidental QM tunneling of the transferred hydride through the energy barrier104,105 and no more than a small reduction in the effective barrier height from tunneling.18,106 If this analysis is correct, then there is no imperative for GPDH to utilize the dianion-binding energy for stabilization of a tunneling-ready state.104,105,107
(9) The model from Scheme 4 provides a mechanism for phosphite dianion activation of several enzymes that catalyze polar reactions in water. The model may be generalized to enzymes that catalyze the formation of unstable radical intermediates, for which slow ligand-driven conformational changes to form protein radical cages of defined structure are observed.108,109 Radical cage formation provides for selectivity in the binding of non-reacting substrate fragments at the transition state for enzyme-catalyzed radical formation, while the structured protein cage directs the reaction of reactive and non-selective radical intermediates toward the physiological product(s).
Effect of Protein Motions on Enzyme Turnover
The time scales for protein motions range from femtoseconds for bond vibrations to milliseconds for the large protein conformational changes illustrated by Figure 2.110 It may be difficult for researchers engaged in studies that probe for links between enzymatic rate accelerations and protein dynamics to conclude that there are few important links. However, this possibility should be considered when there are no clear imperatives for coupling protein motions to formation of an enzymatic transition state. For example, if stabilization of the enzymatic transition state by static protein–ligand interactions is sufficient to account for the entire enzymatic rate acceleration, then there may be no requirement for assistance from coupled protein motions.
In many cases loop and side-chain protein motions at entropically rich unliganded enzymes (EO, Scheme 7) exist so that binding energy will be expended for their elimination, thereby providing for specificity in transition-state binding. In these cases the results of biophysical studies on protein dynamics may not be relevant to the explanation for the enzymatic rate acceleration.111 Now the only protein motions clearly relevant to the rate acceleration are those associated with the creation and breakdown of EC·S during the steps for kc, k–c, and k′–c in Scheme 7. These motions may affect the reaction rate, if they occur together with conversion of enzyme-bound substrate to product in a single reaction stage. However, there are no imperatives for such a coupled-concerted reaction mechanism112 and little or no experimental evidence to support this coupling for catalysis by TIM or OMPDC.
Scheme 7. Stepwise Substrate Binding (Kd) Followed by a Protein Conformational Change (kc).

When the protein conformational change is uncoupled from the active-site chemistry (kchem, Scheme 7) the protein motions that control the rate constant kc for this conformational change will only limit the value of the kinetic parameter kcat/Km, when kc is rate determining for turnover at low substrate concentrations (k–c < kchem, Scheme 7).113,114 These motions will only limit the value of kcat when they are rate-determining for reactions at saturating [S] (k′–c < kchem).113,114 The open and closed forms of TIM have been distinguished in solid-state NMR,41,115,116 solution NMR,117 and laser-induced temperature jump fluorescence spectroscopy studies.40 The results from studies on the conversion of EO·S to EC·S provide evidence that closure of flexible loop 6 over the substrate GAP is partly rate determining for kcat/Km and that opening of this loop to release product DHAP is partly rate determining for kcat for TIM-catalyzed isomerization of GAP (Figure 1).40,41,116
The rate of binding of OMP to OMPDC to form EC·S partly limits the value of kcat/Km = 1.1 × 107 M–1 s–1, and the rate of release of product from EC·P partly limits the value of kcat = 16 s–1 for yeast OMPDC-catalyzed decarboxylation of (S = OMP, Scheme 7).118 5-Fluororotidine 5′-monophosphate (S = FOMP, Scheme 7) is ca. 500-fold more reactive toward OMPDC-catalyzed decarboxylation than OMP.113 This large difference in the reactivity of OMP and FOMP is not strongly expressed at the transition states for wild-type OMPDC-catalyzed decarboxylation at low [FOMP] (kcat/Km = 1.2 × 107 M–1 s–1) or at high [FOMP] (kcat = 95 s–1), so that chemistry is not rate-determining for this OMPDC-catalyzed decarboxylation. The values of kcat/Km for wild-type OMPDC-catalyzed decarboxylation of FOMP do not show the linear dependence on solvent viscosity expected for a cleanly diffusion-controlled reaction.119−121 This provides strong evidence that kcat/Km for OMPDC-catalyzed decarboxylation of FOMP is limited by the values of kc for the enzyme conformational change (Scheme 7).113,114 There is good evidence that the rate constant kcat for decarboxylation of FOMP catalyzed by wild-type and several mutant enzymes is limited by k′–c for the enzyme conformational change.113
Lessons from Computational Studies
The difference in the calculated activation barriers ΔG⧧ to kcat and to kcat/Km for an enzymatic reaction provides the substrate-binding energy expressed at the Michaelis complex. This difference may then be compared with the calculated total transition-state binding energy to obtain an estimate for the enzyme specificity in transition-state binding. However, current computational methods are directed toward obtaining the activation barriers to kcat and do not provide the barriers to kcat/Km, presumably because this barrier cannot be accurately modeled by existing computational methods. I am not aware of computational methods that routinely model the substrate-binding energy as the difference between the energy of (E + S) in solution and at the Michaelis complex (ES) for enzymes that undergo large ligand-driven conformational changes. For example, molecular docking methods serve as tools for identifying ligand-binding sites by gauging the strength of protein–ligand interactions,122−125 but do not model the barriers to protein conformational changes. Finally, there have been few computational studies to evaluate proposals that ligand binding is accompanied by the induction of strain into the ligand, which is then relieved at the transition state for the enzymatic reaction.19,43,126−129
One consequence of the lack of computational methods that provide reliable substrate-binding energies is that it is not possible to examine enzyme specificity in binding the reaction transition state by comparing calculated ground-state and transition-state binding energies. The difficulties in interpreting the results of computational studies relevant to this issue are illustrated by calculations on the Michaelis complex between OMP and OMPDC from Methanothermobacter thermautotrophicus. These calculations were concluded to support the conclusion that “the enzyme conformation is more distorted in the reactant state than in the transition state”.129 This distortion energy was proposed to be released by protein conformational relaxation at the transition state, providing a significant contribution to the enzymatic rate acceleration.129,130 However, this analysis failed to note that enzyme or substrate strain, which is induced by formation of the Michaelis complex and then relieved at the reaction transition state, cannot contribute to a reduction in the activation barrier to kcat/Km for OMPDC or for any other enzyme,43 because the Gibbs free energy added to the system in forming the “strained” substrate complex must then be subtracted on formation of the “unstrained” product complex. In other words, binding energy used to induce strain into the substrate or enzyme is not related to the mechanism for transition-state stabilization, but rather ensures specificity in transition-state binding.61
Triosephosphate Isomerase
The results of computational studies on TIM to model the barriers to kcat for reactions of whole substrate and the substrate pieces catalyzed by wild-type and mutant enzymes have been combined with experimental results to provide insight into the role of the dianion-driven conformational change in catalysis.14,15 These studies represent a first step toward modeling the activation of TIM by the dianion-driven conformational change.
TIM-Catalyzed Reaction of the Substrate Pieces
Experimental studies on wild-type and mutant TIM-catalyzed reactions of the whole substrate GAP and the substrate pieces [GA + HPi] show that the two reaction transition states are stabilized by essentially the same interactions with several side chains of the protein catalyst.131,132 This provides strong evidence that these protein–dianion interactions for whole substrate and for substrate pieces are utilized to hold the protein in the active closed conformation. The result predicts that the “stiff” closed conformation of TIM determined by X-ray crystallographic analyses will show the same activation barrier for deprotonation of the whole substrate GAP, of the substrate pieces GA·HPi′, and of GA alone (Scheme 8).
Scheme 8. Proton Transfer from TIM-Bound Carbon Acids to the Carboxylate Side Chain of E165.

This prediction from experiments was confirmed by the results of empirical valence bond (EVB) calculations,14 which give similar activation barriers (Scheme 8) for the TIM-catalyzed deprotonation of GAP [(ΔG⧧)GAP = 12.9 ± 0.8 kcal/mol], for deprotonation of the substrate piece GA [(ΔG⧧)GA = 15.0 ± 2.4 kcal/mol], and for deprotonation of the pieces GA·HPi [(ΔG⧧)GA·HPi = 15.5 ± 3.5 kcal/mol].14 We concluded that the closed form of TIM created by protein–dianion binding interactions is competent to carry out fast deprotonation of the carbon acid whole substrate or the substrate piece GA. The effect of the enzyme-bound dianion on ΔG⧧ for reaction of the active closed enzyme is small (≤2.6 kcal/mol), in comparison to the larger 12 and 5.8 kcal/mol intrinsic phosphodianion and phosphite dianion-binding energy that is utilized in stabilization of the transition states for TIM-catalyzed deprotonation of GAP and GA·HPi, respectively. This analysis provides support for the conclusion that once dianion-binding energy has been used to hold TIM in the active closed conformation, the dianion behaves as a spectator during the proton-transfer reaction.132
I170A and L230A Mutations
The activating conformational change of TIM positions the highly conserved hydrophobic side chains from I170 and L230 [numbering for yeast enzyme] over the carboxylate side chain of the active-site base E165.133 We proposed that this conformational change activates TIM for carbon deprotonation by increasing the basicity of the E165 side chain toward deprotonation of carbon, and then we examined this proposal in studies on I172A, L232A, and I172A/L232A mutants of TIM from Trypanosoma brucei brucei (TbbTIM, numbering displaced two units from the yeast enzyme).134−136 The X-ray crystal structures of complexes for wild-type and the three mutant TIMs with the enediolate analogue 2-phosphoglycolate (PGA) are essentially superimposable, except that the space(s) created by truncation of the hydrophobic side chain(s) at the mutant enzymes are occupied by water molecules that lie ca. 3.5 Å distant from the carboxylate side chain of Glu165.134 This occlusion of water from the active site by these hydrophobic side chains is consistent with an enhancement of the ground-state basicity of E165 at the Michaelis complex to wild-type TIM.137
We were unable to fully rationalize the complex effects of mutations at I170 and L230 on the kinetic parameters for TIM-catalyzed deprotonation of GAP and DHAP.134−136 The interpretation of our experimental results was clarified by EVB calculations, which accurately model the effect of I170A and L230A mutations on the barriers to deprotonation of GAP and DHAP bound to TIM.15Figure 3 shows the reaction free energy profiles for deprotonation of DHAP by TIM to form enediolate reaction intermediates.15,62,138 The computed activation barriers for conversion of the Michaelis complexes to the respective transition states are in good agreement with the activation barriers from experiment. The computed effects of mutations on the thermodynamic barrier to substrate deprotonation to form the enediolate intermediate (ΔΔGocalc, Figure 3) were combined with their effects on the stability of the Michaelis complex (ΔΔlog Km ≈ ΔΔlog Kd) from experiment to give the effect of the mutations on the stability of the complexes to the enediolate reaction intermediates relative to free enyzme.15,134−136 This analysis (Figure 3) focused on the complex interplay of ground- and transition-state effects in catalysis by TIM.
Figure 3.
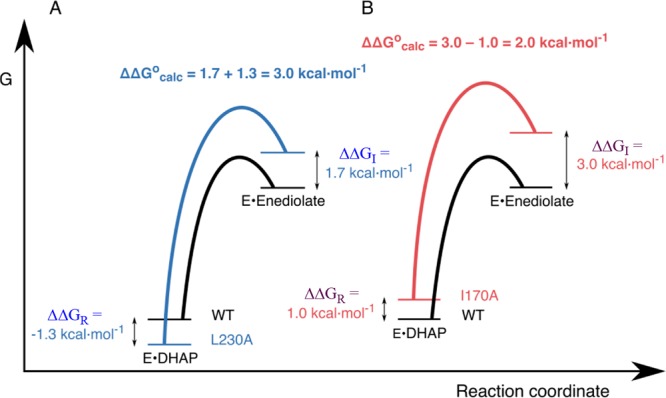
Free energy profiles for deprotonation of enzyme-bound DHAP catalyzed by wild-type and mutant TIMs, which combine results from experimental and computational studies. The diagrams show the effect of these mutations on the stability of the Michaelis complex (ΔΔGR) relative to free TIM determined by experiment, and on the stability of the enediolate intermediate relative to the Michaelis complex (ΔΔGocalc) determined by EVB calculations.15 The effect of these mutations on the stability of the enediolate intermediate relative to free TIM (ΔΔGI) is equal to [(ΔΔGocalc + ΔΔGR]. (A) Profiles for wild-type TIM and the L230A mutant. (B) Profiles for wild-type TIM and the I170A mutant. Reprinted with permission from ref (15). Copyright 2017 American Chemical Society.
The L230A mutation results in a 9-fold decrease in Km ≈ Kd for DHAP, which corresponds to ΔΔGR = −1.3 kcal/mol for Figure 3. This is consistent with the utilization of the binding energy of DHAP to drive desolvation of E165 at wild-type TIM, and with a stabilizing interaction between the side-chain carboxylate and the water molecule that moves into the space created by L230A mutation. A water molecule also enters the space created by the I170A mutation, but this is associated with an increase in Km (destabilization of the Michaelis complex, Figure 3) instead of the decrease in Km observed for the L230A mutation. This effect on ground-state stability cannot be modeled by EVB calculations and is still not understood. However, the EVB calculations do reproduce the effects of the mutations on the activation barriers ΔG⧧ determined be experiment.
The computational results define a linear free energy relationship (LFER, slope = 0.8) between the kinetic (ΔG⧧) and thermodynamic (ΔG°) reaction barriers to formation of the enediolate intermediates of wild-type and mutant TIM-catalyzed deprotonation of DHAP.15 This LFER provides strong support for the conclusions that the I170 and L230 side chains act to minimize the thermodynamic barrier to substrate deprotonation, and that 80% of this effect on reaction driving force is expressed at the transition state for substrate deprotonation.15
The prime imperative for efficient catalysis by TIM is to reduce the large thermodynamic barrier for deprotonation of the carbon acid substrate (pKa = 18 in water)96 to form the enediolate intermediate.58 The value of log Keq for substrate deprotonation at TIM (Scheme 9) is equal to the difference between p(Ka)CH and p(Ka)COOH, where the p(Ka)COOH is similar to the highly perturbed pKa > 10 determined for deprotonation of the carboxylic acid side chain at the complex to the enediolate analogue phosphoglycolate (PGA, Scheme 10).139 There is a good correlation for wild-type and several mutants of TIM between the decrease in log kcat/Km for TIM-catalyzed isomerization of GAP and the decrease in the pKa for deprotonation of the complex between TIM and PGA (Scheme 10). This correlation provides direct evidence that the decrease in the strong side-chain basicity at wild-type TIM is directly linked to the reduction in the catalytic activity of these mutant enzymes.139
Scheme 9. Deprotonation of TIM-Bound Substrate (Keq) and Competing Pathways for Proton Transfer through Solvent Water [(Ka)CH/(Ka)COOH].

Scheme 10. Proton Transfer from the Hydrogen-Bonded TIM·PGA Complex to Water.

Summary and Speculation
Efficient enzymatic catalysis requires a strong stabilization of the enzyme-bound transition state by the protein catalyst, and a switch to activate the expression of this transition-state binding energy following the weak and reversible binding of substrate. These protein switches are often associated with the expenditure of substrate-binding energy to drive a change in enzyme conformation from the stable, flexible, and inactive open enzyme EO (Scheme 4) featured in Koshland’s induced-fit model to the stiff, closed, and active enzyme EC featured in Fisher’s lock-and-key model. The evolution of enzymes that exist in both a flexible unliganded form that shows a weak affinity for the substrate and a stiff liganded form that shows a strong affinity for the transition state has occurred in order to avoid the tight and irreversible binding of substrate. These coexisting flexible and stiff forms for single enzymes favor efficient catalysis at physiological reaction conditions. They comprise two halves that together complete the whole catalyst in enabling the extraordinary operational proficiency of many enzymes.
Experimental and computational protocols for obtaining proteins with enzyme-like activity have focused on optimizing transition-state stabilization from catalysis by stiff proteins.140−143 If these designed proteins were to mimic the very tight transition-state binding observed for some enzymes, then they might suffer the defect of tight and irreversible binding of the substrate and/or product. To the best of my knowledge there have been no efforts to engineer enzyme-activating ligand-driven conformational changes of the type discussed in this Perspective. This may be a requirement to obtain the impressive catalytic efficiency observed for enzymes such as TIM, OMPDC, glycerol 3-phosphate dehydrogenase, and phosphoglucomutase.
Acknowledgments
I acknowledge the National Institutes of Health Grants GM39754 and GM116921 for generous support of my work. I thank Perry Frey for helpful comments.
The author declares no competing financial interest.
References
- Bruice T. C.; Benkovic S. J.. Bioorganic Reaction Mechanisms; W. A. Benjamin: New York, 1966; Vols. 1 and 2. [Google Scholar]
- Jencks W. P.Catalysis in Chemistry and Enzymology; McGraw Hill: New York, 1969. [Google Scholar]
- Richard J. P.; Amyes T. L. Proton transfer at carbon. Curr. Opin. Chem. Biol. 2001, 5, 626–633. 10.1016/S1367-5931(01)00258-7. [DOI] [PubMed] [Google Scholar]
- Nagorski R. W.; Richard J. P. Mechanistic Imperatives for Aldose-Ketose Isomerization in Water: Specific, General Base- and Metal Ion-Catalyzed Isomerization of Glyceraldehyde with Proton and Hydride Transfer. J. Am. Chem. Soc. 2001, 123, 794–802. 10.1021/ja003433a. [DOI] [PubMed] [Google Scholar]
- Fothergill-Gilmore L. A.; Michels P. A. M. Evolution of Glycolysis. Prog. Biophys. Mol. Biol. 1993, 59, 105–235. 10.1016/0079-6107(93)90001-Z. [DOI] [PubMed] [Google Scholar]
- Hine J.Structural Effects on Equilibria in Organic Chemistry; Wiley: New York, 1975; pp 29–54. [Google Scholar]
- Åqvist J.; Kazemi M.; Isaksen G. V.; Brandsdal B. O. Entropy and Enzyme Catalysis. Acc. Chem. Res. 2017, 50, 199–207. 10.1021/acs.accounts.6b00321. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Bora R. P. Perspective: Defining and quantifying the role of dynamics in enzyme catalysis. J. Chem. Phys. 2016, 144, 180901/1–180901/17. 10.1063/1.4947037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stank A.; Kokh D. B.; Fuller J. C.; Wade R. C. Protein Binding Pocket Dynamics. Acc. Chem. Res. 2016, 49, 809–815. 10.1021/acs.accounts.5b00516. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Liddle J. C.; Pardi A.; Ahn N. G. Dynamics of Protein Kinases: Insights from Nuclear Magnetic Resonance. Acc. Chem. Res. 2015, 48, 1106–1114. 10.1021/acs.accounts.5b00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoian P.; Liu C. T.; Hammes-Schiffer S.; Benkovic S. Perspectives on Electrostatics and Conformational Motions in Enzyme Catalysis. Acc. Chem. Res. 2015, 48, 482–489. 10.1021/ar500390e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S.; Klinman J. Emerging Concepts about the Role of Protein Motion in Enzyme Catalysis. Acc. Chem. Res. 2015, 48, 899–899. 10.1021/acs.accounts.5b00113. [DOI] [PubMed] [Google Scholar]
- Fischer E. EinfluD der Konfiguration auf die Wirkung der Enzyme. Ber. Dtsch. Chem. Ges. 1894, 27, 2985. 10.1002/cber.18940270364. [DOI] [Google Scholar]
- Kulkarni Y. S.; Liao Q.; Byléhn F.; Amyes T. L.; Richard J. P.; Kamerlin S. C. L. Role of Ligand-Driven Conformational Changes in Enzyme Catalysis: Modeling the Reactivity of the Catalytic Cage of Triosephosphate Isomerase. J. Am. Chem. Soc. 2018, 140, 3854–3857. 10.1021/jacs.8b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni Y. S.; Liao Q.; Petrović D.; Krüger D. M.; Strodel B.; Amyes T. L.; Richard J. P.; Kamerlin S. C. L. Enzyme Architecture: Modeling the Operation of a Hydrophobic Clamp in Catalysis by Triosephosphate Isomerase. J. Am. Chem. Soc. 2017, 139, 10514–10525. 10.1021/jacs.7b05576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardi-Kilshtain A.; Doron D.; Major D. T. Quantum and Classical Simulations of Orotidine Monophosphate Decarboxylase: Support for a Direct Decarboxylation Mechanism. Biochemistry 2013, 52, 4382–4390. 10.1021/bi400190v. [DOI] [PubMed] [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Parson W. W. Modeling electrostatic effects in proteins. Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764, 1647–1676. 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- Truhlar D. G.; Gao J.; Alhambra C.; Garcia-Viloca M.; Corchado J.; Sánchez M. L.; Villà J. The incorporation of quantum effects in enzyme kinetics modeling. Acc. Chem. Res. 2002, 35, 341–349. 10.1021/ar0100226. [DOI] [PubMed] [Google Scholar]
- Wu N.; Mo Y.; Gao J.; Pai E. F. Electrostatic stress in catalysis: structure and mechanism of the enzyme orotidine monophosphate decarboxylase. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 2017–2022. 10.1073/pnas.050417797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland D. E. Jr. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. U. S. A. 1958, 44, 98–104. 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland D. E. The joys and vicissitudes of protein science. Protein Sci. 1993, 2, 1364–1368. 10.1002/pro.5560020821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt A. D.; Di Cera E. Conformational Selection or Induced Fit? A Critical Appraisal of the Kinetic Mechanism. Biochemistry 2012, 51, 5894–5902. 10.1021/bi3006913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamerlin S. C. L.; Warshel A. The empirical valence bond model: theory and applications. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2011, 1, 30–45. 10.1002/wcms.10. [DOI] [Google Scholar]
- Warshel A.; Weiss R. M. An empirical valence bond approach for comparing reactions in solutions and in enzymes. J. Am. Chem. Soc. 1980, 102, 6218–6226. 10.1021/ja00540a008. [DOI] [Google Scholar]
- Adamczyk A. J.; Cao J.; Kamerlin S. C. L.; Warshel A. Catalysis by dihydrofolate reductase and other enzymes arises from electrostatic preorganization, not conformational motions. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 14115–14120. 10.1073/pnas.1111252108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamerlin S. C. L.; Sharma P. K.; Chu Z. T.; Warshel A. Ketosteroid isomerase provides further support for the idea that enzymes work by electrostatic preorganization. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4075–4080. 10.1073/pnas.0914579107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Xiang Y.; Liu H.; Olsson M. H. M. Electrostatic Basis for Enzyme Catalysis. Chem. Rev. 2006, 106, 3210–3235. 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- Warshel A. Electrostatic Origin of the Catalytic Power of Enzymes and the Role of Preorganized Active Sites. J. Biol. Chem. 1998, 273, 27035–27038. 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- Cannon W. R.; Benkovic S. J. Solvation, Reorganization Energy, and Biological Catalysis. J. Biol. Chem. 1998, 273, 26257–26260. 10.1074/jbc.273.41.26257. [DOI] [PubMed] [Google Scholar]
- Blomberg R.; Kries H.; Pinkas D. M.; Mittl P. R. E.; Grütter M. G.; Privett H. K.; Mayo S. L.; Hilvert D. Precision is essential for efficient catalysis in an evolved Kemp eliminase. Nature 2013, 503, 418–421. 10.1038/nature12623. [DOI] [PubMed] [Google Scholar]
- Debler E. W.; Muller R.; Hilvert D.; Wilson I. A. An aspartate and a water molecule mediate efficient acid-base catalysis in a tailored antibody pocket. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 18539–18544. 10.1073/pnas.0902700106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janda K. D.; Benkovic S. J.; Lerner R. A. Catalytic antibodies with lipase activity and R or S substrate selectivity. Science 1989, 244, 437–440. 10.1126/science.2717936. [DOI] [PubMed] [Google Scholar]
- Tanaka F. Catalytic Antibodies as Designer Proteases and Esterases. Chem. Rev. 2002, 102, 4885–4906. 10.1021/cr010180a. [DOI] [PubMed] [Google Scholar]
- Yang M.-Y.; Iranzo O.; Richard J. P.; Morrow J. R. Solvent Deuterium Isotope Effects on Phosphodiester Cleavage Catalyzed by an Extraordinarily Active Zn(II) Complex. J. Am. Chem. Soc. 2005, 127, 1064–1065. 10.1021/ja044617i. [DOI] [PubMed] [Google Scholar]
- Iranzo O.; Kovalevsky A. Y.; Morrow J. R.; Richard J. P. Physical and Kinetic Analysis of the Cooperative Role of Metal Ions in Catalysis of Phosphodiester Cleavage by a Dinuclear Zn(II) Complex. J. Am. Chem. Soc. 2003, 125, 1988–1993. 10.1021/ja027728v. [DOI] [PubMed] [Google Scholar]
- Zeymer C.; Zschoche R.; Hilvert D. Optimization of Enzyme Mechanism along the Evolutionary Trajectory of a Computationally Designed (Retro-)Aldolase. J. Am. Chem. Soc. 2017, 139, 12541–12549. 10.1021/jacs.7b05796. [DOI] [PubMed] [Google Scholar]
- Schramm V. L.; Schwartz S. D. Promoting Vibrations and the Function of Enzymes. Emerging Theoretical and Experimental Convergence. Biochemistry 2018, 57, 3299–3308. 10.1021/acs.biochem.8b00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinman J. P.; Kohen A. Hydrogen Tunneling Links Protein Dynamics to Enzyme Catalysis. Annu. Rev. Biochem. 2013, 82, 471–496. 10.1146/annurev-biochem-051710-133623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer A. G. III. Enzyme Dynamics from NMR Spectroscopy. Acc. Chem. Res. 2015, 48, 457–465. 10.1021/ar500340a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desamero R.; Rozovsky S.; Zhadin N.; McDermott A.; Callender R. Active Site Loop Motion in Triosephosphate Isomerase: T-Jump Relaxation Spectroscopy of Thermal Activation. Biochemistry 2003, 42, 2941–2951. 10.1021/bi026994i. [DOI] [PubMed] [Google Scholar]
- Rozovsky S.; Jogl G.; Tong L.; McDermott A. E. Solution-state NMR investigations of triosephosphate isomerase active site loop motion: Ligand release in relation to active site loop dynamics. J. Mol. Biol. 2001, 310, 271–280. 10.1006/jmbi.2001.4673. [DOI] [PubMed] [Google Scholar]
- Kerns S. J.; Agafonov R. V.; Cho Y.-J.; Pontiggia F.; Otten R.; Pachov D. V.; Kutter S.; Phung L. A.; Murphy P. N.; Thai V.; Alber T.; Hagan M. F.; Kern D. The energy landscape of adenylate kinase during catalysis. Nat. Struct. Mol. Biol. 2015, 22, 124–131. 10.1038/nsmb.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jencks W. P. Binding energy, specificity, and enzymic catalysis: the Circe effect. Adv. Enzymol. Relat. Areas Mol. Biol. 2006, 43, 219–410. 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- Wolfenden R.; Snider M. J. The Depth of Chemical Time and the Power of Enzymes as Catalysts. Acc. Chem. Res. 2001, 34, 938–945. 10.1021/ar000058i. [DOI] [PubMed] [Google Scholar]
- Radzicka A.; Wolfenden R. A proficient enzyme. Science 1995, 267, 90–93. 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P.; Tait J. J. Activation of orotidine 5′-monophosphate decarboxylase by phosphite dianion: The whole substrate is the sum of two parts. J. Am. Chem. Soc. 2005, 127, 15708–15709. 10.1021/ja055493s. [DOI] [PubMed] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. Enzyme Architecture: Erection of Active Orotidine 5′-Monophosphate Decarboxylase by Substrate-Induced Conformational Changes. J. Am. Chem. Soc. 2017, 139, 16048–16051. 10.1021/jacs.7b08897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green N. M. Adv. Protein Chem. 1975, 29, 85–133. 10.1016/S0065-3233(08)60411-8. [DOI] [PubMed] [Google Scholar]
- Green N. M.; Toms E. J. The properties of subunits of avidin coupled to Sepharose. Biochem. J. 1973, 133, 687–698. 10.1042/bj1330687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green N. M. Avidin. 1. The use of [14C]biotin for kinetic studies and for assay. Biochem. J. 1963, 89, 585–591. 10.1042/bj0890585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Zhai X.; Morgan K. T.; Reinhardt C. J.; Amyes T. L.; Richard J. P. The Activating Oxydianion Binding Domain for Enzyme-Catalyzed Proton Transfer, Hydride Transfer and Decarboxylation: Specificity and Enzyme Architecture. J. Am. Chem. Soc. 2015, 137, 1372–1382. 10.1021/ja5123842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodar S. A.; Allen C. L.; Gulick A. M.; Murkin A. S. The Role of Phosphate in a Multistep Enzymatic Reaction: Reactions of the Substrate and Intermediate in Pieces. J. Am. Chem. Soc. 2015, 137, 2748–2756. 10.1021/ja512911f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray W. J. Jr.; Long J. W.; Owens J. D. An analysis of the substrate-induced rate effect in the phosphoglucomutase system. Biochemistry 1976, 15, 4006–4017. 10.1021/bi00663a015. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. Enzymatic catalysis of proton transfer at carbon: activation of triosephosphate isomerase by phosphite dianion. Biochemistry 2007, 46, 5841–5854. 10.1021/bi700409b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang W.-Y.; Amyes T. L.; Richard J. P. A Substrate in Pieces: Allosteric Activation of Glycerol 3-Phosphate Dehydrogenase (NAD+) by Phosphite Dianion. Biochemistry 2008, 47, 4575–4582. 10.1021/bi8001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodar S. A.; Murkin A. S. DXP Reductoisomerase: Reaction of the Substrate in Pieces Reveals a Catalytic Role for the Nonreacting Phosphodianion Group. Biochemistry 2013, 52, 2302–2308. 10.1021/bi400092n. [DOI] [PubMed] [Google Scholar]
- Amyes T. L.; Malabanan M. M.; Zhai X.; Reyes A. C.; Richard J. P. Enzyme activation through the utilization of intrinsic dianion binding energy. Protein Eng., Des. Sel. 2017, 30, 159–168. 10.1093/protein/gzw064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai X.; Malabanan M. M.; Amyes T. L.; Richard J. P. Mechanistic imperatives for deprotonation of carbon catalyzed by triosephosphate isomerase: enzyme activation by phosphite dianion. J. Phys. Org. Chem. 2014, 27, 269–276. 10.1002/poc.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P. Enzymatic Rate Enhancements: A Review and Perspective. Biochemistry 2013, 52, 2009–2011. 10.1021/bi3017119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow J. R.; Amyes T. L.; Richard J. P. Phosphate Binding Energy and Catalysis by Small and Large Molecules. Acc. Chem. Res. 2008, 41, 539–548. 10.1021/ar7002013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amyes T. L.; Richard J. P. Specificity in transition state binding: The Pauling model revisited. Biochemistry 2013, 52, 2021–2035. 10.1021/bi301491r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P. A Paradigm for Enzyme-Catalyzed Proton Transfer at Carbon: Triosephosphate Isomerase. Biochemistry 2012, 51, 2652–2661. 10.1021/bi300195b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go M. K.; Amyes T. L.; Richard J. P. Hydron Transfer Catalyzed by Triosephosphate Isomerase. Products of the Direct and Phosphite-Activated Isomerization of [1-13C]-Glycolaldehyde in D2O. Biochemistry 2009, 48, 5769–5778. 10.1021/bi900636c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles J. R. To build an enzyme. Philos. Trans. R. Soc. London, Ser. B 1991, 332, 115–121. 10.1098/rstb.1991.0039. [DOI] [PubMed] [Google Scholar]
- Knowles J. R. Enzyme catalysis: not different, just better. Nature 1991, 350, 121–124. 10.1038/350121a0. [DOI] [PubMed] [Google Scholar]
- Knowles J. R.; Albery W. J. Perfection in enzyme catalysis: the energetics of triosephosphate isomerase. Acc. Chem. Res. 1977, 10, 105–111. 10.1021/ar50112a001. [DOI] [Google Scholar]
- Feng D. F.; Cho G.; Doolittle R. F. Determining divergence times with a protein clock: update and reevaluation. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 13028–13033. 10.1073/pnas.94.24.13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterner R.; Hocker B. Catalytic versatility, stability, and evolution of the (β/α)8-barrel enzyme fold. Chem. Rev. 2005, 105, 4038–4055. 10.1021/cr030191z. [DOI] [PubMed] [Google Scholar]
- Copley R. R.; Bork P. Homology among (βα)8 barrels: implications for the evolution of metabolic pathways. J. Mol. Biol. 2000, 303, 627–641. 10.1006/jmbi.2000.4152. [DOI] [PubMed] [Google Scholar]
- Nagano N.; Orengo C. A.; Thornton J. M. One Fold with Many Functions: The Evolutionary Relationships between TIM Barrel Families Based on their Sequences, Structures and Functions. J. Mol. Biol. 2002, 321, 741–765. 10.1016/S0022-2836(02)00649-6. [DOI] [PubMed] [Google Scholar]
- Goldman A. D.; Beatty J. T.; Landweber L. F. The TIM Barrel Architecture Facilitated the Early Evolution of Protein-Mediated Metabolism. J. Mol. Evol. 2016, 82, 17–26. 10.1007/s00239-015-9722-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. A role for flexible loops in enzyme catalysis. Curr. Opin. Struct. Biol. 2010, 20, 702–710. 10.1016/j.sbi.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Goryanova B.; Zhai X. Enzyme architecture: on the importance of being in a protein cage. Curr. Opin. Chem. Biol. 2014, 21, 1–10. 10.1016/j.cbpa.2014.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierenga R. K.; Kapetaniou E. G.; Venkatesan R. Triosephosphate isomerase: a highly evolved biocatalyst. Cell. Mol. Life Sci. 2010, 67, 3961–3982. 10.1007/s00018-010-0473-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He R.; Reyes A. C.; Amyes T. L.; Richard J. P. Enzyme Architecture: The Role of a Flexible Loop in Activation of Glycerol-3-phosphate Dehydrogenase for Catalysis of Hydride Transfer. Biochemistry 2018, 57, 3227–3236. 10.1021/acs.biochem.7b01282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett S. A.; Amyes T. L.; Wood B. M.; Gerlt J. A.; Richard J. P. Dissecting the Total Transition State Stabilization Provided by Amino Acid Side Chains at Orotidine 5′-Monophosphate Decarboxylase: A Two-Part Substrate Approach. Biochemistry 2008, 47, 7785–7787. 10.1021/bi800939k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Plache D. C.; Koudelka A. P.; Amyes T. L.; Gerlt J. A.; Richard J. P. Enzyme Architecture: Breaking Down the Catalytic Cage that Activates Orotidine 5′-Monophosphate Decarboxylase for Catalysis. J. Am. Chem. Soc. 2018, 140, 17580–17590. 10.1021/jacs.8b09609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschlag D. The role of induced fit and conformational changes of enzymes in specificity and catalysis. Bioorg. Chem. 1988, 16, 62–96. 10.1016/0045-2068(88)90038-7. [DOI] [Google Scholar]
- Wolfenden R. Enzyme catalysis. Conflicting requirements of substrate access and transition state affinity. Mol. Cell. Biochem. 1974, 3, 207–211. 10.1007/BF01686645. [DOI] [PubMed] [Google Scholar]
- Pillai B.; Cherney M. M.; Diaper C. M.; Sutherland A.; Blanchard J. S.; Vederas J. C.; James M. N. G. Structural insights into stereochemical inversion by diaminopimelate epimerase: an antibacterial drug target. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 8668–8673. 10.1073/pnas.0602537103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearne S. L. The interdigitating loop of the enolase superfamily as a specificity binding determinant or a flying buttress. Biochim. Biophys. Acta, Proteins Proteomics 2017, 1865, 619–630. 10.1016/j.bbapap.2017.02.006. [DOI] [PubMed] [Google Scholar]
- Gerlt J. A.; Babbitt P. C. Enzyme (re)design: lessons from natural evolution and computation. Curr. Opin. Chem. Biol. 2009, 13, 10–18. 10.1016/j.cbpa.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen K. N.; Dunaway-Mariano D. Markers of fitness in a successful enzyme superfamily. Curr. Opin. Struct. Biol. 2009, 19, 658–665. 10.1016/j.sbi.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S. D.; Zhang G.; Dai J.; Dunaway-Mariano D.; Allen K. N. Analysis of the Substrate Specificity Loop of the HAD Superfamily Cap Domain. Biochemistry 2004, 43, 2812–2820. 10.1021/bi0356810. [DOI] [PubMed] [Google Scholar]
- Whitty A.; Fierke C. A.; Jencks W. P. Role of binding energy with coenzyme A in catalysis by 3-oxoacid coenzyme A transferase. Biochemistry 1995, 34, 11678–11689. 10.1021/bi00037a005. [DOI] [PubMed] [Google Scholar]
- Fierke C. A.; Jencks W. P. Two functional domains of coenzyme A activate catalysis by coenzyme A transferase. Pantetheine and adenosine 3′-phosphate 5′-diphosphate. J. Biol. Chem. 1986, 261, 7603–7606. [PubMed] [Google Scholar]
- Plapp B. V.; Charlier H. A. Jr.; Ramaswamy S. Mechanistic implications from structures of yeast alcohol dehydrogenase complexed with coenzyme and an alcohol. Arch. Biochem. Biophys. 2016, 591, 35–42. 10.1016/j.abb.2015.12.009. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Zhai X.; Malabanan M. M. Reflections on the catalytic power of a TIM-barrel. Bioorg. Chem. 2014, 57, 206–212. 10.1016/j.bioorg.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pabis A.; Risso V. A.; Sanchez-Ruiz J. M.; Kamerlin S. C. Cooperativity and flexibility in enzyme evolution. Curr. Opin. Struct. Biol. 2018, 48, 83–92. 10.1016/j.sbi.2017.10.020. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Reyes A. C. Orotidine 5′-Monophosphate Decarboxylase: Probing the Limits of the Possible for Enzyme Catalysis. Acc. Chem. Res. 2018, 51, 960–969. 10.1021/acs.accounts.8b00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B. G.; Hassell A. M.; Wolfenden R.; Milburn M. V.; Short S. A. Anatomy of a proficient enzyme: the structure of orotidine 5′-monophosphate decarboxylase in the presence and absence of a potential transition state analog. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 2011–2016. 10.1073/pnas.030409797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman D. N.; Tu C.; Chen X.; Tanhauser S. M.; Kresge A. J.; Laipis P. J. Rate-equilibria relationships in intramolecular proton transfer in human carbonic anhydrase III. Biochemistry 1993, 32, 10757–10762. 10.1021/bi00091a029. [DOI] [PubMed] [Google Scholar]
- Paul S.; Taraphder S. Determination of the reaction coordinate for a key conformational fluctuation in human carbonic anhydrase II. J. Phys. Chem. B 2015, 119, 11403–11415. 10.1021/acs.jpcb.5b03655. [DOI] [PubMed] [Google Scholar]
- Pollack R. M. Enzymatic mechanisms for catalysis of enolization: ketosteroid isomerase. Bioorg. Chem. 2004, 32, 341–353. 10.1016/j.bioorg.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Schwans J. P.; Kraut D. A.; Herschlag D. Determining the catalytic role of remote substrate binding interactions in ketosteroid isomerase. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 14271–14275. 10.1073/pnas.0901032106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard J. P. Acid-base catalysis of the elimination and isomerization reactions of triose phosphates. J. Am. Chem. Soc. 1984, 106, 4926–4936. 10.1021/ja00329a050. [DOI] [Google Scholar]
- Ha N. C.; Kim M. S.; Lee W.; Choi K. Y.; Oh B. H. Detection of large pKa perturbations of an inhibitor and a catalytic group at an enzyme active site, a mechanistic basis for catalytic power of many enzymes. J. Biol. Chem. 2000, 275, 41100–41106. 10.1074/jbc.M007561200. [DOI] [PubMed] [Google Scholar]
- Kim S. W.; Cha S. S.; Cho H. S.; Kim J. S.; Ha N. C.; Cho M. J.; Joo S.; Kim K. K.; Choi K. Y.; Oh B. H. High-resolution crystal structures of Δ-5-steroid 3-ketosteroid isomerase with and without a reaction intermediate analogue. Biochemistry 1997, 36, 14030–14036. 10.1021/bi971546+. [DOI] [PubMed] [Google Scholar]
- Van der Kamp M. W.; Chaudret R.; Mulholland A. J. QM/MM modelling of ketosteroid isomerase reactivity indicates that active site closure is integral to catalysis. FEBS J. 2013, 280, 3120–3131. 10.1111/febs.12158. [DOI] [PubMed] [Google Scholar]
- Monod J.; Wyman J.; Changeux J. P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 1965, 12, 88–118. 10.1016/S0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Pauling L. The nature of forces between large molecules of biological interest. Nature 1948, 161, 707–709. 10.1038/161707a0. [DOI] [PubMed] [Google Scholar]
- Roston D.; Kohen A. A Critical Test of the “Tunneling and Coupled Motion” Concept in Enzymatic Alcohol Oxidation. J. Am. Chem. Soc. 2013, 135, 13624–13627. 10.1021/ja405917m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roston D.; Kohen A. Elusive transition state of alcohol dehydrogenase unveiled. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 9572–9577. 10.1073/pnas.1000931107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. P. Primary Deuterium Kinetic Isotope Effects: A Probe for the Origin of the Rate Acceleration for Hydride Transfer Catalyzed by Glycerol-3-Phosphate Dehydrogenase. Biochemistry 2018, 57, 4338–4348. 10.1021/acs.biochem.8b00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. P. Structure-Reactivity Effects on Intrinsic Primary Kinetic Isotope Effects for Hydride Transfer Catalyzed by Glycerol-3-Phosphate Dehydrogenase. J. Am. Chem. Soc. 2016, 138, 14526–14529. 10.1021/jacs.6b07028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhambra C.; Corchado J. C.; Sánchez M. L.; Gao J.; Truhlar D. G. Quantum Dynamics of Hydride Transfer in Enzyme Catalysis. J. Am. Chem. Soc. 2000, 122, 8197–8203. 10.1021/ja001476l. [DOI] [Google Scholar]
- Reyes A. C.; Amyes T. L.; Richard J. P. A reevaluation of the origin of the rate acceleration for enzyme-catalyzed hydride transfer. Org. Biomol. Chem. 2017, 15, 8856–8866. 10.1039/C7OB01652B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J.; Yu G.; Ator M. A.; Stubbe J. Pre-Steady-State and Steady-State Kinetic Analysis of E. coli Class I Ribonucleotide Reductase. Biochemistry 2003, 42, 10071–10083. 10.1021/bi034374r. [DOI] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Stubbe J. Site-Specific Replacement of Y356 with 3,4-Dihydroxyphenylalanine in the β2 Subunit of E.coli Ribonucleotide Reductase. J. Am. Chem. Soc. 2006, 128, 2522–2523. 10.1021/ja057776q. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman K. A.; Lei M.; Thai V.; Kerns S. J.; Karplus M.; Kern D. A hierarchy of timescales in protein dynamics is linked to enzyme catalysis. Nature 2007, 450, 913–916. 10.1038/nature06407. [DOI] [PubMed] [Google Scholar]
- Agarwal P. K. A Biophysical Perspective on Enzyme Catalysis. Biochemistry 2018, 10.1021/acs.biochem.8b01004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewar M. J. S. Multibond Reactions Cannot Normally be Synchronous. J. Am. Chem. Soc. 1984, 106, 209–219. 10.1021/ja00313a042. [DOI] [Google Scholar]
- Goryanova B.; Goldman L. M.; Ming S.; Amyes T. L.; Gerlt J. A.; Richard J. P. Rate and Equilibrium Constants for an Enzyme Conformational Change during Catalysis by Orotidine 5′-Monophosphate Decarboxylase. Biochemistry 2015, 54, 4555–4564. 10.1021/acs.biochem.5b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goryanova B.; Spong K.; Amyes T. L.; Richard J. P. Catalysis by Orotidine 5′-Monophosphate Decarboxylase: Effect of 5-Fluoro and 4′-Substituents on the Decarboxylation of Two-Part Substrates. Biochemistry 2013, 52, 537–546. 10.1021/bi301650d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Lorieau J.; McDermott A. E. Triosephosphate isomerase: 15N and 13C chemical shift assignments and conformational change upon ligand binding by magic-angle spinning solid-state NMR spectroscopy. J. Mol. Biol. 2010, 397, 233–248. 10.1016/j.jmb.2009.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozovsky S.; McDermott A. E. The time scale of the catalytic loop motion in triosephosphate isomerase. J. Mol. Biol. 2001, 310, 259–270. 10.1006/jmbi.2001.4672. [DOI] [PubMed] [Google Scholar]
- Massi F.; Wang C.; Palmer A. G. Solution NMR and computer simulation studies of active site loop motion in triosephosphate isomerase. Biochemistry 2006, 45, 10787–10794. 10.1021/bi060764c. [DOI] [PubMed] [Google Scholar]
- Porter D. J. T.; Short S. A. Yeast Orotidine-5′-Phosphate Decarboxylase: Steady-State and Pre-Steady-State Analysis of the Kinetic Mechanism of Substrate Decarboxylation. Biochemistry 2000, 39, 11788–11800. 10.1021/bi001199v. [DOI] [PubMed] [Google Scholar]
- Wood B. M.; Chan K. K.; Amyes T. L.; Richard J. P.; Gerlt J. A. Mechanism of the Orotidine 5′-Monophosphate Decarboxylase-Catalyzed Reaction: Effect of Solvent Viscosity on Kinetic Constants. Biochemistry 2009, 48, 5510–5517. 10.1021/bi9006226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacklow S. C.; Raines R. T.; Lim W. A.; Zamore P. D.; Knowles J. R. Triosephosphate isomerase catalysis is diffusion controlled. Biochemistry 1988, 27, 1158–1165. 10.1021/bi00404a013. [DOI] [PubMed] [Google Scholar]
- Brouwer A.; Kirsch J. Investigation of diffusion-limited rates of chymotrypsin reactions by viscosity variation. Biochemistry 1982, 21, 1302–1307. 10.1021/bi00535a030. [DOI] [PubMed] [Google Scholar]
- Deng N.; Forli S.; He P.; Perryman A.; Wickstrom L.; Vijayan R. S. K.; Tiefenbrunn T.; Stout D.; Gallicchio E.; Olson A. J.; Levy R. M. Distinguishing Binders from False Positives by Free Energy Calculations: Fragment Screening Against the Flap Site of HIV Protease. J. Phys. Chem. B 2015, 119, 976–988. 10.1021/jp506376z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genheden S.; Ryde U. Comparison of end-point continuum-solvation methods for the calculation of protein-ligand binding free energies. Proteins: Struct., Funct., Genet. 2012, 80, 1326–1342. 10.1002/prot.24029. [DOI] [PubMed] [Google Scholar]
- Pietrucci F.; Marinelli F.; Carloni P.; Laio A. Substrate Binding Mechanism of HIV-1 Protease from Explicit-Solvent Atomistic Simulations. J. Am. Chem. Soc. 2009, 131, 11811–11818. 10.1021/ja903045y. [DOI] [PubMed] [Google Scholar]
- Maganti L.; Open Source Drug Discovery Consortium; Ghoshal N. Probing the structure of Mycobacterium tuberculosisMbtA: model validation using molecular dynamics simulations and docking studies. J. Biomol. Struct. Dyn. 2014, 32, 273–288. 10.1080/07391102.2012.762752. [DOI] [PubMed] [Google Scholar]
- Schopf P.; Mills M. J. L.; Warshel A. The entropic contributions in vitamin B12 enzymes still reflect the electrostatic paradigm. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 4328–4333. 10.1073/pnas.1503828112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancia F.; Keep N. H.; Nakagawa A.; Leadlay P. F.; McSweeney S.; Rasmussen B.; secke P. B.; Diat O.; Evans P. R. How coenzyme B12 radicals are generated: the crystal structure of methylmalonyl-coenzyme A mutase at 2 Å resolution. Structure 1996, 4, 339–350. 10.1016/S0969-2126(96)00037-8. [DOI] [PubMed] [Google Scholar]
- Halpern J.; Kim S. H.; Leung T. W. Cobalt-carbon bond dissociation energy of coenzyme B12. J. Am. Chem. Soc. 1984, 106, 8317–8319. 10.1021/ja00338a065. [DOI] [Google Scholar]
- Gao J. Catalysis by enzyme conformational change as illustrated by orotidine 5′-monophosphate decarboxylase. Curr. Opin. Struct. Biol. 2003, 13, 184–192. 10.1016/S0959-440X(03)00041-1. [DOI] [PubMed] [Google Scholar]
- Gao J.; Byun K. L.; Kluger R. Catalysis by enzyme conformational change. Top. Curr. Chem. 2004, 238, 113–136. 10.1007/b94541. [DOI] [Google Scholar]
- Zhai X.; Amyes T. L.; Richard J. P. Role of Loop-Clamping Side Chains in Catalysis by Triosephosphate Isomerase. J. Am. Chem. Soc. 2015, 137, 15185–15197. 10.1021/jacs.5b09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai X.; Amyes T. L.; Richard J. P. Enzyme Architecture: Remarkably Similar Transition States for Triosephosphate Isomerase-Catalyzed Reactions of the Whole Substrate and the Substrate in Pieces. J. Am. Chem. Soc. 2014, 136, 4145–4148. 10.1021/ja501103b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kursula I.; Wierenga R. K. Crystal structure of triosephosphate isomerase complexed with 2-phosphoglycolate at 0.83-Å resolution. J. Biol. Chem. 2003, 278, 9544–9551. 10.1074/jbc.M211389200. [DOI] [PubMed] [Google Scholar]
- Richard J. P.; Amyes T. L.; Malabanan M. M.; Zhai X.; Kim K. J.; Reinhardt C. J.; Wierenga R. K.; Drake E. J.; Gulick A. M. Structure–Function Studies of Hydrophobic Residues That Clamp a Basic Glutamate Side Chain during Catalysis by Triosephosphate Isomerase. Biochemistry 2016, 55, 3036–3047. 10.1021/acs.biochem.6b00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Koudelka A. P.; Amyes T. L.; Richard J. P. Mechanism for Activation of Triosephosphate Isomerase by Phosphite Dianion: The Role of a Hydrophobic Clamp. J. Am. Chem. Soc. 2012, 134, 10286–10298. 10.1021/ja303695u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Amyes T. L.; Richard J. P. Mechanism for Activation of Triosephosphate Isomerase by Phosphite Dianion: The Role of a Ligand-Driven Conformational Change. J. Am. Chem. Soc. 2011, 133, 16428–16431. 10.1021/ja208019p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malabanan M. M.; Nitsch-Velasquez L.; Amyes T. L.; Richard J. P. Magnitude and origin of the enhanced basicity of the catalytic glutamate of triosephosphate isomerase. J. Am. Chem. Soc. 2013, 135, 5978–5981. 10.1021/ja401504w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodi P. J.; Knowles J. R. Neutral imidazole is the electrophile in the reaction catalyzed by triosephosphate isomerase: structural origins and catalytic implications. Biochemistry 1991, 30, 6948–6956. 10.1021/bi00242a020. [DOI] [PubMed] [Google Scholar]
- Zhai X.; Reinhardt C. J.; Malabanan M. M.; Amyes T. L.; Richard J. P. Enzyme Architecture: Amino Acid Side-Chains That Function To Optimize the Basicity of the Active Site Glutamate of Triosephosphate Isomerase. J. Am. Chem. Soc. 2018, 140, 8277–8286. 10.1021/jacs.8b04367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Althoff E. A.; Bolduc J.; Jiang L.; Moody J.; Lassila J. K.; Giger L.; Hilvert D.; Stoddard B.; Baker D. Structural Analyses of Covalent Enzyme-Substrate Analog Complexes Reveal Strengths and Limitations of De Novo Enzyme Design. J. Mol. Biol. 2012, 415, 615–625. 10.1016/j.jmb.2011.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter F.; Leaver-Fay A.; Khare S. D.; Bjelic S.; Baker D. De novo enzyme design using Rosetta3. PLoS One 2011, 6, e19230. 10.1371/journal.pone.0019230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolon D. N.; Voigt C. A.; Mayo S. L. De novo design of biocatalysts. Curr. Opin. Chem. Biol. 2002, 6, 125–129. 10.1016/S1367-5931(02)00303-4. [DOI] [PubMed] [Google Scholar]
- Zhang X.; DeChancie J.; Gunaydin H.; Chowdry A. B.; Clemente F. R.; Smith A. J. T.; Handel T. M.; Houk K. N. Quantum Mechanical Design of Enzyme Active Sites. J. Org. Chem. 2008, 73, 889–899. 10.1021/jo701974n. [DOI] [PubMed] [Google Scholar]