

Abstract
Testis weight is a genetically mediated trait associated with reproductive efficiency across numerous species. We sought to evaluate the genetically diverse, highly recombinant Diversity Outbred (DO) mouse population as a tool to identify and map quantitative trait loci (QTLs) associated with testis weight. Testis weights were recorded for 502 male DO mice and the mice were genotyped on the GIGAMuga array at ~143,000 SNPs. We performed a genome-wide association analysis and identified one significant and two suggestive QTLs associated with testis weight. Using bioinformatic approaches, we developed a list of candidate genes and identified those with known roles in testicular size and development. Candidates of particular interest include the RNA demethylase gene Alkbh5, the cyclin-dependent kinase inhibitor gene Cdkn2c, the dynein axonemal heavy chain gene Dnah11, the phospholipase D gene Pld6, the trans-acting transcription factor gene Sp4, and the spermatogenesis-associated gene Spata6, each of which has a human ortholog. Our results demonstrate the utility of DO mice in high--resolution genetic mapping of complex traits, enabling us to identify developmentally important genes in adult mice. Understanding how genetic variation in these genes influence testis weight could aid in the understanding of mechanisms of mammalian reproductive function.
Keywords: Diversity outbred mice, genome-wide association, testes, testis, QTL mapping, multiparental populations
Article Summary
Testicular size is a complex and physiologically important reproductive trait that is directly connected to male fertility. Phenotypic variation in complex traits such as testis weight is attributed to the combined effect of multiple genes, which may be identified via quantitative trait locus (QTL) mapping. Mapping precision can be greatly improved by the use of highly recombinant outbred populations, such as Diversity Outbred (DO) mice. Using DO mice, we identified three QTLs associated with testis weight, one of which replicates a previous finding and two of which have not been previously reported. Our findings demonstrate the advantages of DO mice in high-resolution genetic mapping of complex traits.
Introduction
Complex trait variation in natural and experimental populations is due to specific DNA sequence polymorphisms, environmental effects, and the interactions between these factors (Johannes et al. 2009). Testis weight is a complex trait that holds direct implications for reproductive success, as developmental abnormalities can lead to irregular sperm production and infertility in adulthood (Sharpe 2001).Variation in testis size has been linked to environmental factors such as social dominance, social organization and seasonal changes across numerous species. Specifically, laboratory and field studies show that testes weights are significantly higher in dominant males as compared to subordinates (Kruczek & Styrna 2009; Maruska & Fernald 2011; Setchell & Dixson 2001), are significantly higher in animals that engage in sperm competition as compared to monogamous species (Awata et al. 2006; Harcourt et al. 1981; Simmons and García-González 2008), and can be positively influenced by long photoperiods (Ortavant et al. 1988; Delgadillo et al. 2004).
There is also evidence suggesting the presence of a substantial genetic component (Chubb 1992; Soulsbury 2010; Zhang et al. 2017). For example, Le Roy et al. (2001) performed a quantitative genetic analysis across thirteen inbred strains of laboratory mice and found that heritability accounted for 82.3% of the total variance in testis weight. The heritability of testis size has also been identified in larger animals such as sheep and cattle, carrying clear ramifications for livestock fertility (Fossceco and Notter 1995; Coulter, Rounsaville, and Foote 1976). Several quantitative trait locus (QTL) studies have identified chromosomal regions associated with testis weight in adult mice (Storchova et al. 2004; Rocha et al. 2004; Suto 2008; Suto 2011). However, these studies used F2 intercrosses, backcrosses, consomic strains, and similar populations that typically lack the large amounts of recombination needed for gene identification (Parker et al. 2011; Flint 2011). This limitation can be resolved by using populations with more accumulated recombinations to improve mapping resolution. For example, Parker et al. (2016) utilized a commercially available outbred population of Carworth Farms White (CFW) mice to identify a narrow 3 Mb region associated with testis weight. This region contained the Inhba gene, which has been shown to affect testis morphogenesis and testis weight in mice (Mithraprabhu et al. 2010; Mendis et al. 2011; Zidek et al. 1998).
The mice used in the present study are of the Diversity Outbred (DO) stock, a genetically heterogeneous mouse population that displays a broad range of phenotypes (Churchill et al. 2012; French et al. 2015). While using CFW mice has shown to produce narrow QTLs, the DO population is even more genetically diverse and captures allelic variance not observed in CFW mice (Svenson et al. 2012). DO mice are derived from the same eight-way cross of common and wild-derived strains that produced the Collaborative Cross (CC) recombinant inbred panel (Churchill et al. 2012; Chesler et al. 2008). The high diversity in the CC population influenced many male reproductive traits, evidenced in many instances of male infertility during inbreeding where allelic incompatibilities become manifest (Shorter et al. 2017). In the outcross population we expect that these alleles are buffered and stabilized, rather than selected against, thereby enabling mapping of the genes and variants associated with these traits. Unlike the CC strains, the DO intercross strategy avoids mating of siblings or first cousins and distributes founder haplotypes throughout the population, thereby forming a highly recombinant outbred population (Chesler 2014). DO mice carry approximately 45 million segregating SNPs—around four times that of classical laboratory mouse strains—enabling the high-resolution genetic mapping that is essential in complex trait analysis (Svenson et al. 2012; Recla et al. 2014). Furthermore, the DO population is extendable: each individual in the population is genetically unique and additional cohorts can be added to studies as they proceed (Chesler 2014). This quality makes DO mice especially well-suited for genome-wide association studies (GWAS), which typically rely on extremely large samples. To date, DO mice have been successfully used to map QTLs associated with heart size (Shorter et al. 2017), chemotherapy-induced hematotoxicity (Gatti et al. 2017b), metabolic disease related traits (Tyler et al. 2017), benzene toxicity (Gatti et al. 2017a), atherosclerosis (Smallwood et al. 2014), acute pain sensitivity (Recla et al. 2014); anxiety-like behaviors and activity-related traits (Logan et al. 2013), and plasma cholesterol levels (Churchill et al. 2012; Svenson et al. 2012).
QTLs and candidate genes for testis weight have not yet been identified in studies using DO mice. In the current study, we demonstrate the utility of the DO population for high-resolution mapping of QTLs for paired testis weight and select candidate genes from within these loci for future functional validation. Our results hold implications for livestock fertility, sperm donation, and other fields in which reproductive success is of interest.
Materials and methods
Subjects
All procedures were approved by the Middlebury College Institutional Animal Care and Use Committee (IACUC) in accordance with NIH guidelines. We obtained male DO mice (n = 502; J:DO, JAX stock number 009376) from The Jackson Laboratory (Bar Harbor, ME). These DO mice were from generations 16–21 of outcrossing. Mice were singly housed in a temperature-controlled colony room (21 ± 1° C) with a 12:12 light/dark cycle and ad libitum access to food (Harlan Teklad Diet 2020X chow) and water. Virgin wood bedding was changed every other week and cages were ventilated with filtered air by an Enviro-Gard B environmental control system. All mice were involved in a separate behavioral study, after which they were euthanized for the collection of various tissues, including testes.
Phenotyping
Mice were euthanized when they were approximately five months old (M = 165 days, SD = 45 days) at the same time of day during the light phase of the day. First mice were weighed, then testes were removed and weighed in pairs. Weights were recorded to the nearest 0.0001 g using the Ohaus Adventurer Pro AV264 scale. The scale was professionally calibrated with a reference weight at least once per year. Testis weights and body weight were rankZ transformed prior to QTL analyses.
Tissue collection and genotyping
After euthanasia, tail tips were placed in 1.5 mL tubes containing 1 mL of saline buffer, immediately flash frozen in liquid nitrogen, then stored in a −80°C freezer. Samples were then loaded into 96-well plates and shipped on dry ice to Neogen (Neogen: GeneSeek Operations, Lincoln, NE, USA) for genotyping. SNPs were identified using the newly developed GigaMUGA, the third generation of the Mouse Universal Genotyping Array (MUGA) series. The GigaMUGA provides approximately 143,000 SNP markers built on the Illumina Infinium HD platform, with an average spacing of 22.5 Kb between probes (Morgan et al. 2015). The vast majority (98.5%) of probes target biallelic SNPs, and probe design is principally optimized for genetic mapping in the CC and DO populations (Chesler et al. 2016; Morgan et al. 2015).
QTL mapping
After obtaining the sample genotypes, DO genome reconstruction and QTL mapping were carried out using DOQTL software as described previously (Svenson et al. 2012; Church et al. 2015; Gatti et al. 2014; 2016). Briefly, the DOQTL software (Gatti et al. 2014) is an R package that is specialized for QTL mapping in DO mice and takes each of the eight founder strains into account.
Association mapping
For genome-wide association mapping, we imputed all single high quality SNPS from the Sanger Mouse Genome Project onto DO genomes and fit an additive genetic model at each SNP. We accounted for genetic relatedness between mice by using a kinship matrix based on the leave-one-chromosome-out (LOCO) method (Cheng and Palmer 2013). The LOCO method was chosen because traditional kinship calculations that include the causative marker are known to produce overly conservative mapping results (Yang et al. 2014; King and Long 2017). Generation was treated as a covariate to control for batch effects. The genome-wide significance thresholds for QTL were calculated using 1000 permutations to create a distribution for the null hypothesis. To determine statistical significance of the QTL peaks, a Bonferroni-corrected significance threshold was defined using a –log10(p-value). QTLs that exceeded p = 7.3 × 10−7 were considered to be significant at p < 0.05 and QTLs that reached p = 1.4 × 10−6 were considered to be suggestive at p < 0.1.
Linkage mapping
Next we performed linkage mapping by fitting an additive haplotype model with kinship correction at each locus to estimate founder effects for each QTL. This model provides estimates of the eight founder allele contributions at every marker. Generation was included as a covariate for linkage mapping. The genome-wide significance thresholds for QTL were calculated using 1000 permutations to create a distribution for the null hypothesis. Log of the odds ratio (LOD) was the reported mapping statistic for linkage mapping. Significant QTL were determined at a genome-wide p-value of < 0.05 and suggestive QTL were determined at a genome-wide p-value < 0.1. When a QTL peak was identified, a 95% Bayesian credible interval was used to determine the QTL region based on linkage results. If a given QTL had multiple peaks (as was the case for the QTL on chromosome 12), a 1.5-LOD support interval was used to determine the QTL region instead.
Bioinformatic analysis and gene prioritization
The Mouse Genome Informatics (MGI) Allele database (www.informatics.jax.org/allele; Blake et al. 2017) and the GeneNetwork2 mapping module (www.genenetwork.org; Wang et al. 2003; Chesler et al. 2004) were used to identify previously observed QTLs that shared overlapping confidence intervals with the loci reported in this study. We also used the MGI database to search for genes within the support intervals that were associated with relevant phenotypes, known to be expressed in testicular tissue, or linked to abnormal traits in knockout mice. We also used the NHGRI-EBI GWAS Catalog (MacArthur et al. 2017) to determine if genes within our support intervals had human homologs that were associated with testicular phenotypes such as testicular cancer, cryptorchidism, and testicular dysgenesis syndrome. The Mouse Phenome Database (MPD; http://phenome.jax.org) was accessed to identify genes within the QTLs that possessed coding polymorphisms. The MPD UNC-MMUGA2 dataset (Srivastava et al. 2017) was searched for nonsynonymous coding SNPs, stop-gain SNPs, stop-loss SNPs, SNPs resulting in frameshifts, and SNPs located in essential splice sites across the eight DO founder strains. Lastly, we downloaded dN/dS values from Ensembl to determine if any of the genes within the QTL regions also displayed dN/dS ratios > 1, providing evidence of positive selection. To prioritize among candidate genes within the QTL regions, we categorically scored any “hits” in each of these databases with a “1” versus “0” for those not found in these lists.
Mouse Genome Annotation
All gene locations are reported on mouse genome build GRCm38/mm10. Gene locations are provided on Ensembl 89. DO founder SNPs were extracted from the Sanger Mouse Genomes Project build REL-1505.
Results
Phenotypic analysis
Importantly, we observed considerable variation in mouse testis weight throughout the study sample. The Shapiro-Wilk test was used to assess the normality of the distribution and indicated that the assumption of normality was violated (p < 0.05). Figure 1 shows the distribution of testis weights in the DO population. In this histogram, scores appear to be reasonably normally distributed. Recorded values ranged from 0.07 g to 0.41 g, with an average of 0.22 g (SD = 0.04 g). Testis weight was only weakly correlated with body weight, (r = 0.199, n = 499, p < 0.001); and was not correlated with age (r = 0.018, n = 502, p = .688). There did not appear to be seasonal variation associated with testes weights; as a one-way ANOVA comparing testes weights across three seasons failed to reach significance (no mice were sacrificed in the fall).
Fig. 1.
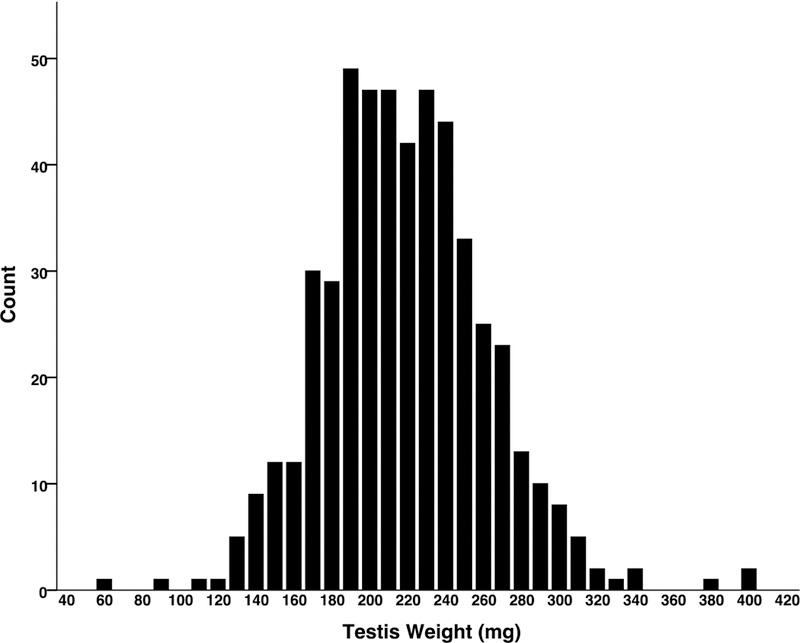
Testis weight distribution in DO mice (n = 502)
Graphics program: SPSS was used to create figure 1
QTL analysis
We identified one significant QTL on chromosome 4 that exceeded p = 7.3 × 10−7 (p < 0.05) and two suggestive QTLs on chromosomes 11 and 12 exceeding p = 1.4 × 10−6 (p < 0.1; Figure 2) that were associated with paired testis weight. Table 1 displays the position, peak LOD scores, number of protein coding genes in the interval, and percent variance explained for each QTL. The widths of the QTLs ranged between 3.44 and 7.55 Mb. The number of annotated genes within these intervals ranged from 10 to 129 genes. Figures 3, 4, and 5 display the LOD scores of SNPs within each of the identified QTLs. In addition to including Generation as a covariate, we also present mapping results with no covariates, Age + Generation, Age + Body Weight + Generation, Body Weight + Generation, and the ratio of testis weight to body weight + Generation (Supplementary Figures 1–5). These approaches have previously been used for QTL mapping of testis weight; however, they each produce different results in this population.
Fig. 2. Genome-wide association mapping of testis weight.
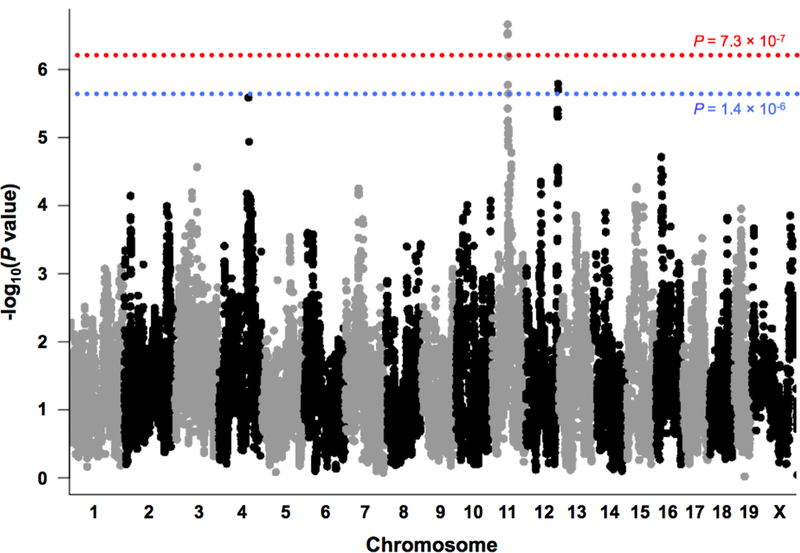
Each point plots the –log10(p- value) for the association between one SNP and testis weight. Values were plotted against their respective positions on the chromosomes. The dashed red and blue lines represent the genome-wide significant (p < 0.05) and genome-wide suggestive (p < 0.1) thresholds, respectively
Graphics programs: R and Microsoft PowerPoint were used to create figure 2
TABLE 1.
Testis weight QTLs
| QTL | Chr | CI (Mb) | Width (Mb) | Peak (Mb) | LOD | Genes in interval | % Var |
|---|---|---|---|---|---|---|---|
| TesWt4 | 4 | 106.81–114.36 | 7.55 | 110.154780 | 6.11 | 64 | 5.52 |
| TesWt11 | 11 | 57.91 – 63.06 | 5.15 | 61.072818 | 7.12 | 129 | 6.40 |
| TesWt12 | 12 | 116.58 −120.02 | 3.44 | 117.9868 | 5.80 | 10 | 5.25 |
The confidence interval (CI) and peak marker location are given in Mb position based on Build 38 of the mouse chromosome.
Fig. 3.
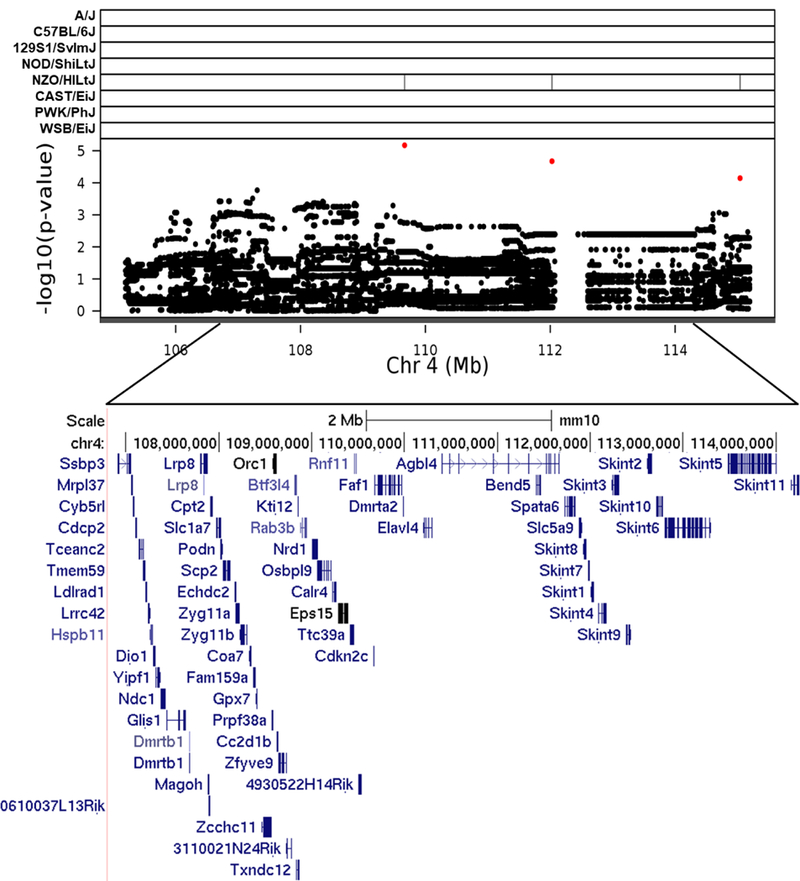
Association mapping of TestWt4 QTL. High resolution view of QTL support interval. Top panel shows SNP marker associations of the eight DO founders. The x-axis displays the distribution along the chromosome in physical distance. The y-axis displays the –log10(p-value) for the SNP mapping. Each data point shows the –log10(p-value) at one SNP; red data points indicate scores above the p < 0.1 threshold. Below each plot is an expanded view showing known genes within the QTL support interval. Gene data retrieved from the UCSC Genome Browser (https://genome.ucsc.edu)
Graphics programs: R and Microsoft PowerPoint were used to create figure 3
Fig. 4.
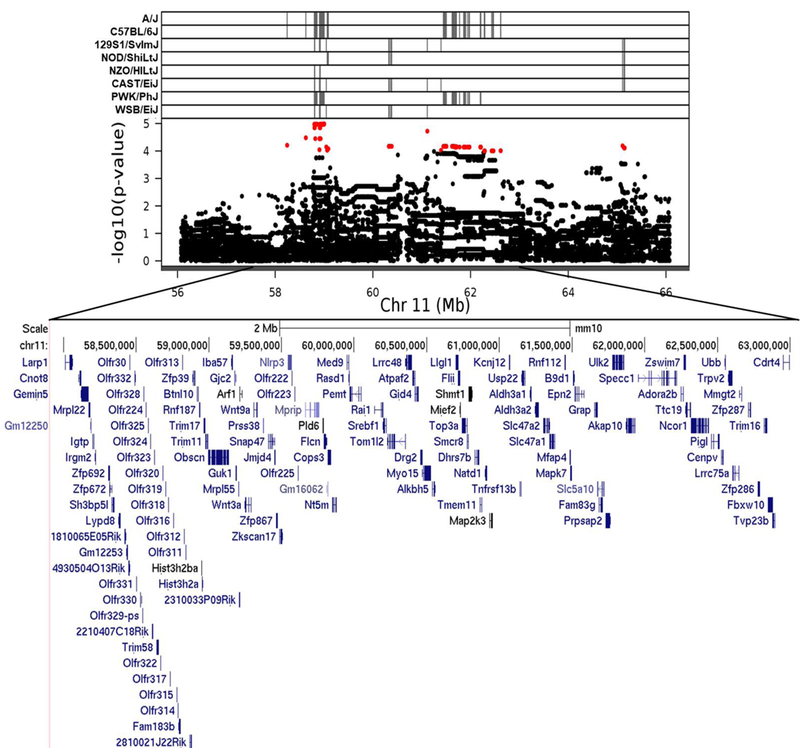
Association mapping of TestWt11 QTL. High resolution view of QTL support interval. Top panel shows SNP marker associations of the eight DO founders. The x-axis displays the distribution along the chromosome in physical distance. The y-axis displays the –log10(p-value) for the SNP mapping. Each data point shows the –log10(p-value) at one SNP; red data points indicate scores above the p < 0.1 threshold. Below each plot is an expanded view showing known genes within the QTL support interval. Gene data retrieved from the UCSC Genome Browser (https://genome.ucsc.edu)
Graphics programs: R and Microsoft PowerPoint were used to create figure 4
Fig. 5.
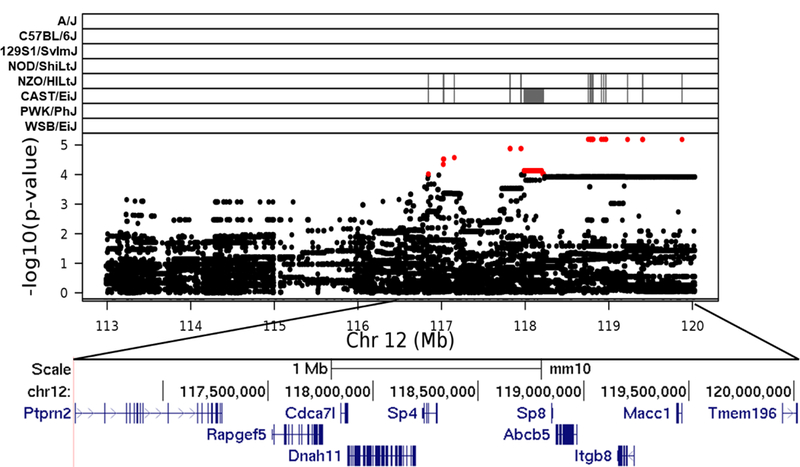
Association mapping of TestWt12 QTL. High resolution view of QTL support interval. Top panel shows SNP marker associations of the eight DO founders. The x-axis displays the distribution along the chromosome in physical distance. The y-axis displays the –log10(p-value) for the SNP mapping. Each data point shows the –log10(p-value) at one SNP; red data points indicate scores above the p < 0.1 threshold. Below each plot is an expanded view showing known genes within the QTL support interval. Gene data retrieved from the UCSC Genome Browser (https://genome.ucsc.edu)
Graphics programs: R and Microsoft PowerPoint were used to create figure 5
We estimated founder haplotype effects for each QTL. For the QTL on chromosome 4, NZO/HILtJ alleles were associated with increased testis weight (Figure 6). For the QTL on chromosome 11, we observed that PWK/PhJ, A/J, and C57BL/6J alleles contributed to increased testis weight, while CAST/EiJ and 129S1/SvlmJ alleles contributed to decreased testis weight (Figure 7). This pattern of allele effects suggests that there may be at least two distinct haplotypes underlying the chromosome 11 QTL. Founder coefficients from the linkage model on chromosome 12 show that the CAST/EiJ and NZO/HILtJ alleles were associated with increased testis weight at the QTL peak (~117 Mb; Figure 6).
Fig. 6.
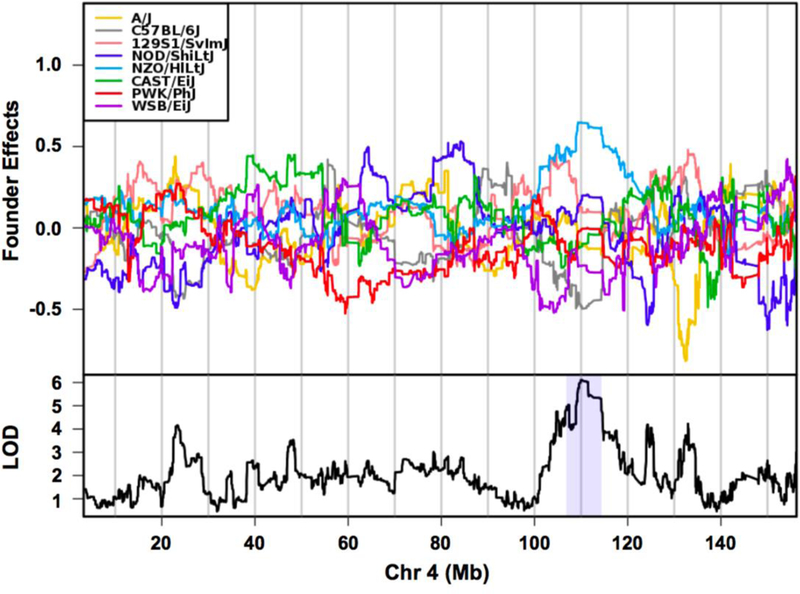
Haplotype effects on chromosome 4 QTL for testis weight. The top panel shows the eight DO founder allele effects (model coefficients) determined by linkage mapping. The x-axis is physical distance in Mb along the chromosome. The y-axis for the top panel is the effect coefficient, and the bottom panel is the LOD score. At ~110 Mb, the NZO/HILtJ alleles (light blue) are associated with larger testis weights. Shading in the bottom panel identifies the 95% credible interval.
Graphics programs: R and Microsoft PowerPoint were used to create figure 6
Fig. 7.

Haplotype effects on chromosome 11 QTL for testis weight. The top panel shows the eight DO founder allele effects (model coefficients) determined by linkage mapping. The x-axis is physical distance in Mb along the chromosome. The y-axis for the top panel is the effect coefficient, and the bottom panel is the LOD score. At ~61 Mb, PWK/PhJ (red), A/J (yellow), and C57BL/6J (grey) alleles contributed to increased testis weight, while CAST/EiJ (green) and 129S1/SvlmJ (pink) alleles contributed to decreased testis weight. Shading in the bottom panel identifies the 95% credible interval.
Graphics programs: R and Microsoft PowerPoint were used to create figure 7
Bioinformatic analysis and gene prioritization
Some of our QTLs overlapped with QTLs for related phenotypes that had previously been reported (Table 2) and we were able to use GeneNetwork2 to map suggestive QTLs that overlapped with those in this study. Within the three QTLs identified in this study, we found no mouse homologs of genes previously implicated in human GWAS for testicular phenotypes.
TABLE 2.
QTLs for related phenotypes sharing overlapping confidence intervals with regions identified in this study
| QTL Name/Locus | QTL Phenotype | Strain or Cross | Build 38 Location (Mb) | Reference |
|---|---|---|---|---|
| TesWt4 | ||||
| D4Mit199 | Testis weight | Smt x MOM F2 | 111.88 | Bolor et al. (2006) |
| Svw1 | Seminal vesicle weight | CC extinct lines | 90–119 | Shorter et al. (2017) |
| TesWt11 | ||||
| Epfq4 | Epididymal fat pad weight | M16i x L6 F2 | 12.32–68.61 | Rocha et al. (2004) |
| Spqq1 | Sperm quality | CBXE/EXCB RI strains | 54.17–63.43 | Golas et al. (2003) |
| N/A | Abnormal sperm head | IRCS 122D | 46.92–58.92 | L’Hôte et al. (2007) |
We interrogated multiple independent and complementary databases to prioritize the genes within our QTL intervals. Many of the genes within the QTL regions have been associated with peripheral traits of interest such as testis atrophy, male reproductive system physiology, Leydig cell hyperplasia, infertility, and abnormal spermatogenesis (Table S1, Table S2, and Table S3). Thirty-six genes have been directly implicated in testis weight variation, morphology, atrophy/degeneration, or testis growth/size. One gene (Obscn) was identified via MPD that contained nonsynonymous coding polymorphisms (Table S4, Table S5, and Table S6). Finally, we examined dN/dS ratios to compare the rate of substitution at silent sites (dS), which are presumed neutral, to the rate of substitutions at non-silent sites (dN), which possibly experience selection. Ratios above 1 suggest that natural selection is promoting changes in the protein sequence; whereas ratios below 1 indicate that changes in genetic sequence are actively selected against (Tables S7, S8, S9), In addition, testis-specific genes are reported to have the highest dN/dS values (Oliver et al. 2010). Thus, high dN/dS ratios may be consistent with testis-specific expression. We integrated these data to generate a list of the most promising candidate genes within each QTL (Table 3).
TABLE 3.
Candidate genes for testis weight
References are provided for genes that are linked to abnormal testis morphology, if a knockout mouse exists with an abnormal reproductive phenotype, or if implicated in male fertility
Discussion
We performed genome-wide mapping of QTLs underlying paired testis weight in the DO mouse population. We identified one significant QTL on chromosome 11 and two suggestive QTLs on chromosomes 4 and 12 that were associated with testis weight. Traditionally, mapping populations such as F2 intercrosses and consomic strains have been used to identify QTLs underlying phenotypic variation in animal models (Churchill et al. 2004; Flint 2011). These populations typically lack sufficient recombination and genetic variation to establish narrow chromosomal regions containing a small number of genes. Highly recombinant and genetically diverse populations such as heterogeneous stocks, some commercially available outbred mice, the hybrid mouse diversity panel, wild-derived “Montpellier strains”, the CC, and DO mice are complementary resources that can increase power and provide novel genetic variation for genetic mapping studies (Woods and Mott 2017; Parker et al. 2016; Lusis et al. 2016; Chang et al. 2017; Shorter et al. 2017; French et al. 2015). We used DO mice for genetic mapping in order to obtain a significant improvement in mapping resolution, as well as increased heterozygosity and more uniformly distributed genetic variation (Svenson et al. 2012; Smallwood et al. 2014). By using DO mice in conjunction with the recently developed DOQTL mapping software, we were able to map three QTLs for testis weight, two of which have not been previously reported.
Previous mapping studies in mice have found QTLs associated with testis weight, using both different populations and alternative statistical approaches. These approaches include mapping absolute testis weights (Zidek et al. 1998; Le Roy et al. 2001; Oka et al. 2004; White et al. 2012), transforming testis weights using a log adjustment (Rocha et al. 2004), conditionally including body weight and/or age as covariates (Shorter et al. 2017; Parker et al. 2016), or mapping testes weights relative to body weight (Bolor et al. 2006; Rocha et al. 2004; L’hote et al 2007; White et al. 2011; 2012). We applied a rankZ transformation in order to obtain a normal distribution and treated Generation as a covariate in order to avoid potential confounds across cohorts of mice. We also present QTL mapping results with no covariates, Age + Generation, Age + Body Weight + Generation, Body Weight + Generation, and the ratio of testis weight to body weight + Generation (Supplementary Figures 1–5). These five analyses produce different QTL mapping results, which may partly explain differences in QTLs identified for testis weight in past studies. Using either testis weight alone or Age + Generation for QTL mapping generally replicated the three QTLs we identified, although TesWt4 no longer reached the suggestive threshold. Mapping of either testis weight with Age + Body Weight + Generation or Body Weight + Generation identified a significant QTL on chromosome X. However, TesWt11 and TesWt12 no longer reached suggestive criteria with either approach (although Body Weight + Generation did continue to identify TesWt4 as suggestive). Mapping of the ratio of testis weight to body weight + Generation identified one suggestive QTL on chromosome 7, along with two significant QTLs on chromosomes 19 and X. Generally, the motivation to include covariates or otherwise adjust for correlated traits in GWAS is to discover loci associated with the primary phenotype independently of the correlated trait. However, adjusting for heritable covariates can introduce an unintended bias that can lead to false positives (Aschard et al 2015; Holmes & Smith 2017). Aschard and colleagues (2015) illustrate this point by examining results from a large meta-analysis of waist-to-hip ratio adjusted for body mass index (BMI). They observe that half of the reported associations for waist-to-hip ratio adjusted for BMI are likely influenced by a direct genetic association with BMI. Thus, the QTLs on chromosome 19 and X identified using body weight as a covariate (S3, S4) or by mapping testis weight relative to body weight (S5) should be interpreted with caution.
The widths of the QTLs identified in the present study are comparable to those reported in previous mapping studies that utilized DO mice (Logan et al. 2013; Recla et al. 2014; Smallwood et al. 2014; Gatti et al. 2017a; Gatti et al. 2017b; Shorter et al. 2017; Tyler et al. 2017). However, it should be noted that both the sample sizes and the statistical methods for determining support intervals varied across studies, which makes direct comparisons difficult. In addition, the number of significant QTLs reported for any given phenotype was also fairly consistent across studies. Broadly speaking, most DO QTL studies to date have reported 1–2 significant QTLs per trait.
Each QTL identified in the present study accounted for approximately 5–6% of the variance explained for testis weight. As predicted in power simulations performed by Gatti et al. (2014), 500 DO mice provide ~45% power to identify QTLs that explain 5% of the phenotypic variance. Thus, a larger sample size would significantly increase our ability to detect QTLs of small effect. Despite being underpowered, we still identified QTLs and obtained favorable mapping resolution. However, the effect size of individual QTL alleles is critical to assess power at a given sample size; and is almost never known in advance. Therefore, it is not possible to provide specific guidelines about the sample size needed for future DO mapping studies. Based on our current results, we predict that a sample size of 1,000 or more DO mice should be used for most traits.
Although our use of DO mice was intended to increase mapping precision, there is an inverse relationship between mapping resolution and statistical power (Parker and Palmer 2011). As we have previously reported, LD (r2) decays rapidly in DO mice as compared to other mapping populations such as heterogeneous stock (HS) mice, the hybrid mouse diversity panel (HMDP), 30 inbred strains, and a 34th generation advanced intercross line (AIL) derived from LG/J and SM/J strains (Parker et al. 2016). The mapping resolution of the DO population will only continue to improve with each additional outbreeding generation, but larger sample sizes may be required. Given the range of powerful genetic resources that exist, future replication, fine-mapping, and functional validation studies would benefit not only from an increased sample size of DO mice, but also from exploiting combinations of complementary crosses such as the wild-derived Montpellier strains or Collaborative Cross mice (Chang et al. 2017; Williams & Williams 2016).
The chromosome 4 QTL has previously been implicated in genetic mapping studies for testis weight and other related phenotypes, providing additional support for the involvement of this region in testicular growth and development. However, it is not clear if the same variants are segregating across different populations of mice. For example, Le Roy et al. (2001) reported a large QTL on chromosome 4 that was associated with absolute paired testis weight in C57BL/6By × NZB/BINJ F2 mice. Similarly, Bolor et al. (2006) reported a chromosome 4 QTL for relative testis weight in F2 mice derived from a cross between small testis mutant (Smt) females and males of the MOM strain derived from Japanese wild mice (Mus musculus molossinus) that overlaps with the TesWt4 QTL. To our knowledge, genotype data is unavailable for the founder strains that comprise either of these F2 crosses; so it is not possible to determine if the same alleles are influencing testis weight across populations. TesWt4 also closely overlaps with a chromosome 4 QTL for relative right testis weight in a reciprocal F2 cross derived from WSB/EiJ and PWD/PhJ mice (White et al. 2011) and in a reciprocal F2 cross derived from WSB/EiJ and CAST/EiJ mice (White et al. 2012). In both crosses, WSB/EiJ alleles contributed to increased testis weight, whereas NZO/HILtJ alleles were associated with increased testis weight in our DO mice (Figure 6). Using GeneNetwork2, we were able to identify a suggestive QTL that was associated with absolute testis weight in BXD mice that overlaps with TesWt4 (Zidek et al. 1998); the C57BL/6J alleles were associated with increased testis weight in the BXD panel. The identification of this QTL across multiple studies using different intra- and inter-subspecific crosses indicates that it may control normal variation in testis weight in mice. Lastly, TestWt4 shares overlapping regions with a QTL for seminal vesicle weight recently reported by Shorter et al. (2017) in Collaborative Cross (CC) mice. In the CC mice, PWK/PhJ alleles were associated with decreased seminal vesicle weight at the chromosome 4 QTL, while NZO/HILtJ alleles were associated with increased seminal vesicle weight. We observed similar allele effects of NZO/HILtJ, but not PWK/PhJ alleles for testis weight in our DO mice.
To our knowledge, neither the chromosome 11 or chromosome 12 QTLs have previously been implicated in testis weight. This is not particularly surprising, given that most genetic mapping populations will have different genetic variants segregating compared to DO mice. Thus, even if a gene strongly affects testis weight, it is likely that different variants will be segregating in different mapping populations, and thus QTL including those of commonly used laboratory strains such as A/J and C57BL/6J, both of which cause an increase in testis weight. Although we did not observe any overlap with other testis weight QTLs, the region on chromosome 11 has been associated with related phenotypes: it overlaps with a QTL for epididymal fat pad weight in an F2 intercross derived from lines of mice selected for high and low growth (Rocha et al. 2004), with a sperm quality QTL identified in CBXE/EXCB recombinant inbred strains (Golas et al. 2008), and with a QTL for abnormal sperm head morphology in congenic mice (L’Hôte et al. 2007). Such co-mapping may indicate the presence of a gene with pleiotropic effects, or may demonstrate that testis weight is influenced by alternative reproductive factors.
Interestingly, Shorter et al. (2017) identified a QTL for testis weight in extinct lines of CC mice on chromosome 1 that was not replicated in the present study. Also of note, the mean testis weight for the DO mice (0.22 g) was considerably larger than the mean testis weight observed for CC extinct fertile lines (0.15 g), CC extinct infertile lines (0.13 g), and each of the eight founder strains (Shorter et al. 2017), consistent with previous studies of testes weight observed among inbred strains reported in the Mouse Phenome Database (Handel M. Male reproductive parameters in 14 inbred strains of mice. MPD:Handel1. Mouse Phenome Database web resource (RRID:SCR_003212), The Jackson Laboratory, Bar Harbor, Maine USA. https://phenome.jax.org [Cited (10/30/2017)]). CAST/EiJ mice had the lightest testis (0.11 g) of all founders, which is inconsistent with our finding that the CAST/EiJ allele is associated with increased testis weight on chromosome 12. Some of these differences in testes weights may be due to the age at which mice were tested. For example, the DO mice in the present study were between 85–225 days old, the CC extinct lines ranged in age from 179–862 days, and the inbred strains reported in MPD were approximately 56–70 days old at the time of testing. However, it is also possible that the observed discrepancies reflect critical genetic differences between the CC and DO populations, which could have arisen over the multiple generations of outbreeding in the DO stock (Churchill et al. 2012; Gatti et al. 2014).
Each of the three QTLs encompassed a diverse set of protein-coding genes, some of which have been previously associated with pertinent reproductive traits (Table S1, S2, S3). Within TesWt4, we identified the spermatogenesis-associated gene Spata6 as a promising candidate gene. Spata6 encodes a 49.7 kDa protein (SPATA6) that has been suggested to play an important role in testicular germ cell tumor formation (Huo et al. 2015). Both Spata6 and its human ortholog SPATA6 are known to be expressed exclusively in the testis in adults (Oh et al. 2003; Huo et al. 2015). In addition, it has been reported that SPATA6 is required for assembly of the sperm connecting piece and tight head-tail conjunction, and that inactivation of Spata6 causes acephalic spermatozoa and sterility in male mice (Yuan et al. 2015). While Spata6 has not been previously identified as a candidate gene for testis weight variation, it is worth noting that Spata6 lies just outside the proximal boundary of a QTL for testis weight reported by Bolor et al. (2006). Therefore, the potential of a role for Spata6 in testicular development must be considered. Another TestWt4 candidate gene of particular interest was the cyclin-dependent kinase (CDK) inhibitor gene Cdkn2c, which encodes the CDK inhibitor CDKN2C. The function of CDKN2C in somatic cell division regulation is well-studied (Solomon et al. 2008; Broxmeyer et al. 2011; Rowell et al. 2014), but there is also evidence of a role in spermatogenesis (Forand et al. 2009; Xu et al. 2013). In addition, Zindy et al. (2001) found that Cdkn2c-null mice develop Leydig cell hyperplasia, leading to decreased levels of testosterone production. Other studies using Cdkn2c knockout mice reported abnormal reproductive phenotypes as well, including testicular neoplasia and Leydig cell tumorigenesis (Franklin et al. 1998; Latres et al. 2000).
Within TesWt11, we identified the alkB homolog 5, RNA demethylase gene Alkbh5 as a top candidate gene, and it was the only candidate gene to be directly associated with testis weight variation. Zheng et al. (2013) reported reduced testis weight in Alkbh5 mouse knockouts (of an unspecified strain) that were generated via Cre-Lox recombination (Hardy et al. 1997). These Alkbh5-deficient males also showed impaired fertility caused by apoptotic metaphase-stage spermatocytes (Zheng et al. 2013). However, no data was reported for Alkbh5 expression in testis in the MGI Gene Expression Database. One candidate gene within TesWt11 that is known to be expressed in testis is the phospholipase D family, member 6 gene Pld6 (also known as Mitopld). Mitopld-null mice fail to produce progeny, display abnormal spermatocytes at P16, exhibit meiotic arrest during spermatogenesis, and have smaller testes than heterozygous and wild-type littermates (Watanabe et al. 2011; Huang et al. 2011).
The prospective influence of candidate genes within TesWt12 is less clear. One candidate gene is the trans-acting transcription factor 4 gene, Sp4. Sp4 is a zinc finger transcription factor that is known to be expressed in testis. While Sp4 knockout mice show failures of copulation, they do have histologically intact testes and exhibit complete spermatogenesis (Supp et al. 1996; Göllner et al. 2001). The trans-acting transcription factor 8 gene, Sp8 is also located within TesWt12, although it is not known whether or not it is expressed in testis. It is believed to mediate the canonical WNT signaling pathway to promote outgrowth in limbs and external genitalia (Lin et al. 2013). Another TesWt12 candidate gene is the dynein, axonemal, heavy chain 11 gene, Dnah11. It is known to be expressed in adult testis and mutations in DNAH11 were associated with sterility and asthenozoospermia in men (Zuccarello et al. 2008; Pennarun et al. 1999; Guichard et al. 2001). Asthenozoospermia is a common cause of male infertility characterized by reduced or absent sperm motility possibly due to absent or shortened arms of dyneins. Other genes within the TestWt12 interval could be promising candidates based on their known function but have not been previously associated with reproductive traits: these included genes that coded for transmembrane proteins, integrins, or were associated with the cell division cycle.
Although we were able to identify some compelling and functionally relevant candidates via bioinformatic analysis, there may be other genes within the QTL regions that influence testis weight but whose function is unknown or whose link to testis is not intuitive. Ultimately, our goal is to discern which variants are responsible for trait variation, and how they act. To do so beyond the SNP associations performed herein, technologies such as genome editing can be used to evaluate the specific functional effects of particular variants, perhaps enabling us to establish precise molecular sites of variant action. Expression regulatory variants can also be detected through eQTL analysis but knowledge of the time and tissue in which the variant exhibits effects is required. Transcriptome analysis of gene expression is an exceptionally useful tool for hypothesis-free parsing among candidate genes identified in QTL mapping and has been successful in other outbred mouse populations (Mulligan et al. 2012; Parker et al. 2016). This strategy may be utilized in future RNA sequencing studies of testis weight variation and could be especially useful for identifying novel candidate genes with no previously known relationship to testis weight. Given that none of the genes within our QTL regions with dN/dS ratios > 1 (consistent with testis-specific expression) have known relationships to testis relevant traits, we believe this approach may be especially valuable in looking beyond the “usual suspects”. However, it is possible that some genes influencing testis weight are only expressed during development, and not in adult mice. Therefore, eQTL mapping using tissue from the adult males in the present study may still not provide a complete understanding of the relationships between DNA sequence, gene expression values and testis weights.
In the present study, performed in the context of an unrelated behavioral analysis, we considered only testis weight. From this readily obtainable measure, reproductive biologists may further assess the impact of variants we have identified on phenotypes such as sperm morphology, Sertoli cell development, and testicular inflammation. Integrating these traits directly into QTL mapping will provide a more comprehensive dataset, which will allow us to gain a mechanistic understanding of the role of the underlying genetic pathways in place. Further, mapping of additional phenotypes may have allowed us to identify additional loci for which traits co-mapped, implicating genetic variants that are linked to several reproductive phenotypes. Finally, we did not assess the fertility status of the animals, so we cannot confirm if the variation in testis weight impacted reproductive success in DO mice. While a positive correlation between testis weight and male fertility has been observed in CC mice (Shorter et al. 2017), such a relationship can only be speculated for our sample.
In summary, we have demonstrated the use of DO mice for high-resolution mapping of QTLs underlying testis weight variation. Using a series of bioinformatic approaches, we developed a list of prioritized candidate genes and identified those with potential roles in testicular size and development. The use of the highly recombinant DO population greatly improved our ability to refine the size of QTLs from broad chromosomal regions to narrow intervals spanning between 3.44 and 7.55 Mb. These findings can be used to guide further identification of genetic determinants of testis weight and demonstrate the advantages of the DO population in such studies.
Supplementary Material
Fig. 8.
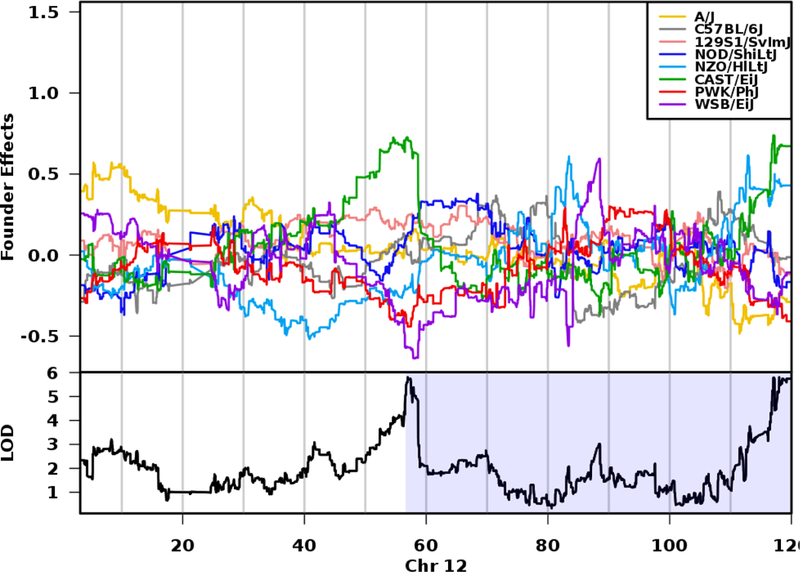
Haplotype effects on chromosome 12 QTL for testis weight. The top panel shows the eight DO founder allele effects (model coefficients) determined by linkage mapping. The x-axis is physical distance in Mb along the chromosome. The y-axis for the top panel is the effect coefficient, and the bottom panel is the LOD score. At ~117 Mb, the CAST/EiJ (green) and NZO/HILtJ alleles (light blue) are associated with larger testis weights. Shading in the bottom panel identifies the 95% credible interval.
Graphics programs: R and Microsoft PowerPoint were used to create figure 8
Footnotes
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Aschard H, Vilhjálmsson BJ, Joshi AD, Price AL, & Kraft P (2015). Adjusting for heritable covariates can bias effect estimates in genome-wide association studies. American Journal of Human Genetics, 96(2), 329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awata S, Heg D, Munehara H, & Kohda M (2006). Testis size depends on social status and the presence of male helpers in the cooperatively breeding cichlid Julidochromis ornatus. Behavioral Ecology, 17(3), 372–379. [Google Scholar]
- Beffert U, Farsian FN, Masiulis I, Hammer RE, Yoon SO, Giehl KM, & Herz J (2006). ApoE receptor 2 controls neuronal survival in the adult brain. Current biology, 16(24), 2446–2452. [DOI] [PubMed] [Google Scholar]
- Blake JA, Eppig JT, Kadin JA, Richardson JE, Smith CL, & Bult CJ (2017). Mouse Genome Database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic acids research, 45(D1), D723–D729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolor H, Wakasugi N, Zhao WD, & Ishikawa A (2006). Detection of quantitative trait loci causing abnormal spermatogenesis and reduced testis weight in the small testis (Smt) mutant mouse. Experimental animals, 55(2), 97–108. [DOI] [PubMed] [Google Scholar]
- Broxmeyer HE, Franklin DS, Cooper S, Hangoc G, & Mantel C (2011). Cyclin dependent kinase inhibitors differentially modulate synergistic cytokine responsiveness of hematopoietic progenitor cells. Stem cells and development, 21(10), 1597–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PL, Kopania E, Keeble S, Sarver BAJ, Larson E, Orth A, Belkhir K, Boursot P, Bonhomme F, Good JM, Dean MD (2017). Whole exome sequencing of wild-derived inbred strains of mice improves power to link phenotype and genotype. Mammalian Genome, 28(9–10), 416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng R, & Palmer AA (2013). A simulation study of permutation, bootstrap, and gene dropping for assessing statistical significance in the case of unequal relatedness. Genetics, 193(3), 1015–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler EJ, Miller DR, Branstetter LR, Galloway LD, Jackson BL, Philip VM, Voy BH, Culiat CT, Threadgill DW, Williams RW, Churchill GA, Johnson DK, & Manly KF (2008). The Collaborative Cross at Oak Ridge National Laboratory: developing a powerful resource for systems genetics. Mammalian Genome, 19(6), 382–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler EJ (2014). Out of the bottleneck: the Diversity Outcross and Collaborative Cross mouse populations in behavioral genetics research. Mammalian Genome, 25(1–2), 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler EJ, Gatti DM, Morgan AP, Strobel M, Trepanier L, Oberbeck D, McWeeny S, Hitzemann R, Ferris M, Clayshultle A, Bell TA, Manuel de Villena FP, & Churchill GA (2016). Diversity Outbred Mice at 21: Maintaining Allelic Variation in the Face of Selection. G3: Genes| Genomes| Genetics, 6(12), 3893–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler EJ, Lu L, Wang J, Williams RW, & Manly KF (2004). WebQTL: rapid exploratory analysis of gene expression and genetic networks for brain and behavior. Nature neuroscience, 7(5), 485–486. [DOI] [PubMed] [Google Scholar]
- Chubb C (1992). Genes regulating testes size. Biol Reprod, 47(1): 29–36. [DOI] [PubMed] [Google Scholar]
- Church RJ, Gatti DM, Urban TJ, Long N, Yang X, Shi Q, … & Navarro V (2015). Sensitivity to hepatotoxicity due to epigallocatechin gallate is affected by genetic background in diversity outbred mice. Food and Chemical Toxicology, 76, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill GA, Airey DC, Allayee H, Angel JM, Attie AD, Beatty J, … & Bleich A (2004). The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nature genetics, 36(11), 1133–1137. [DOI] [PubMed] [Google Scholar]
- Churchill GA, Gatti DM, Munger SC, & Svenson KL (2012). The diversity outbred mouse population. Mammalian genome, 23(9–10), 713–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter GH, Rounsaville TR, & Foote RH (1976). Heritability of testicular size and consistency in Holstein bulls. Journal of animal science, 43(1), 9–12. [DOI] [PubMed] [Google Scholar]
- Delgadillo JA, Cortez ME, Duarte G, Chemineau P, & Malpaux B (2004). Evidence that the photoperiod controls the annual changes in testosterone secretion, testicular and body weight in subtropical male goats. Reproduction, Nutrition, Development, 44(3), 183–193. [DOI] [PubMed] [Google Scholar]
- Flint J (2011). Mapping quantitative traits and strategies to find quantitative trait genes. Methods, 53(2), 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forand A, Fouchet P, Lahaye JB, Chicheportiche A, Habert R, & Bernardino-Sgherri J (2009). Similarities and differences in the in vivo response of mouse neonatal gonocytes and spermatogonia to genotoxic stress. Biology of reproduction, 80(5), 860–873. [DOI] [PubMed] [Google Scholar]
- Fossceco SL, & Notter DR (1995). Heritabilities and genetic correlations of body weight, testis growth and ewe lamb reproductive traits in crossbred sheep. Animal Science, 60(02), 185–195. [Google Scholar]
- Franklin DS, Godfrey VL, Lee H, Kovalev GI, Schoonhoven R, Chen-Kiang S, … & Xiong Y (1998). CDK inhibitors p18INK4c and p27Kip1 mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes & development, 12(18), 2899–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French JE, Gatti DM, Morgan DL, Kissling GE, Shockley KR, Knudsen GA, Shepard KG, Price HC, King D, Witt KL, & Pedersen LC (2015). Diversity outbred mice identify population-based exposure thresholds and genetic factors that influence benzene-induced genotoxicity. Environmental Health Perspectives (Online), 123(3), 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti DM, Svenson KL, Shabalin A, Wu LY, Valdar W, Simecek P, … & Chesler EJ (2014). Quantitative trait locus mapping methods for diversity outbred mice. G3: Genes| Genomes| Genetics, 4(9), 1623–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatti DM, French JE, & Schughart K (2016). QTL Mapping and Identification of Candidate Genes in DO Mice: A Use Case Model Derived from a Benzene Toxicity Experiment. Methods in molecular biology (Clifton, NJ), 1488, 265–281. [DOI] [PubMed] [Google Scholar]
- Gatti D, French JE, & Schughart K (2017a). QTL Mapping and Identification of Candidate Genes in DO Mice: A Use Case Model Derived from a Benzene Toxicity Experiment. In Systems Genetics (pp. 265–281). Humana Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- Gatti DM, Weber SN, Goodwin NC, Lammert F, & Churchill GA (2017b). Genetic background influences susceptibility to chemotherapy-induced hematotoxicity. The pharmacogenomics journal [DOI] [PMC free article] [PubMed]
- Golas A, Dzieza A, Kuzniarz K, & Styrna J (2008). Gene mapping of sperm quality parameters in recombinant inbred strains of mice. International Journal of Developmental Biology, 52(2–3), 287–293. [DOI] [PubMed] [Google Scholar]
- Göllner H, Bouwman P, Mangold M, Karis A, Braun H, Rohner I, … & Cutforth T (2001). Complex phenotype of mice homozygous for a null mutation in the Sp4 transcription factor gene. Genes to cells, 6(8), 689–697. [DOI] [PubMed] [Google Scholar]
- Guichard C, Harricane MC, Lafitte JJ, Godard P, Zaegel M, Tack V, … & Bouvagnet P (2001). Axonemal dynein intermediate-chain gene (DNAI1) mutations result in situs inversus and primary ciliary dyskinesia (Kartagener syndrome). The American Journal of Human Genetics, 68(4), 1030–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handel MA, Lane PW, Schroeder AC, & Davisson MT (1988). New mutation causing sterility in the mouse. Molecular Reproduction and Development, 21(4), 409–423. [DOI] [PubMed] [Google Scholar]
- Harcourt AH, Harvey PH, Larson SG, & Short RV (1981). Testis weight, body weight and breeding system in primates. Nature, 293(5827), 55–57. [DOI] [PubMed] [Google Scholar]
- Hardy S, Kitamura M, Harris-Stansil T, Dai Y, & Phipps ML (1997). Construction of adenovirus vectors through Cre-lox recombination. Journal of virology, 71(3), 1842–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes MV, & Smith GD (2018). Problems in interpreting and using GWAS of conditional phenotypes illustrated by ‘alcohol GWAS’. Molecular Psychiatry (epub ahead of print). [DOI] [PMC free article] [PubMed]
- Huang H, Gao Q, Peng X, Choi SY, Sarma K, Ren H, … & Frohman MA (2011). piRNA-associated germline nuage formation and spermatogenesis require MitoPLD profusogenic mitochondrial-surface lipid signaling. Developmental cell, 20(3), 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo S, Du W, Peng Shi YS, & Zhao S (2015). The role of spermatogenesis-associated protein 6 in testicular germ cell tumors. International journal of clinical and experimental pathology, 8(8), 9119. [PMC free article] [PubMed] [Google Scholar]
- Johannes F, Porcher E, Teixeira FK, Saliba-Colombani V, Simon M, Agier N, … & Bouchez D (2009). Assessing the impact of transgenerational epigenetic variation on complex traits. PLoS Genet, 5(6), e1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch S, Acebron SP, Herbst J, Hatiboglu G, & Niehrs C (2015). Post-transcriptional Wnt signaling governs epididymal sperm maturation. Cell, 163(5), 1225–1236. [DOI] [PubMed] [Google Scholar]
- Latres E, Malumbres M, Sotillo R, Martín J, Ortega S, Martín‐Caballero J, … & Barbacid M (2000). Limited overlapping roles of P15INK4b and P18INK4c cell cycle inhibitors in proliferation and tumorigenesis. The EMBO journal, 19(13), 3496–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Roy I, Tordjman S, Migliore-Samour D, Degrelle H, & Roubertoux PL (2001). Genetic architecture of testis and seminal vesicle weights in mice. Genetics, 158(1), 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L’Hôte D, Serres C, Laissue P, Oulmouden A, Rogel-Gaillard C, Montagutelli X, & Vaiman D (2007). Centimorgan-range one-step mapping of fertility traits using interspecific recombinant congenic mice. Genetics, 176(3), 1907–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Yin Y, Bell SM, Veith GM, Chen H, Huh SH, … & Ma L (2013). Delineating a conserved genetic cassette promoting outgrowth of body appendages. PLoS genetics, 9(1), e1003231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan RW, Robledo RF, Recla JM, Philip VM, Bubier JA, Jay JJ, … & Churchill GA (2013). High‐precision genetic mapping of behavioral traits in the diversity outbred mouse population. Genes, Brain and Behavior, 12(4), 424–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusis AJ, Seldine MM, Allayee H, Bennett BJ, Civelek M, Davis RC, Eskin E, Farber CR, Hui S, Mehrabian M, Norheim F, Pan C, Parks B, Rau CD, Smith DJ, Vallim T, Wang Y, & Wang J (2016). The Hybrid Mouse Diversity Panel: a resource for systems genetics analyses of metabolic and cardiovascular traits. Journal of Lipid Research, 57(6), 925–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King EG, & Long AD (2017). The Beavis effect in next-generation mapping panels in Drosophila melanogaster. G3: Genes Genomes Genet 7(6): 1643–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruczek M, & Styrna J (2009). Semen quantity and quality correlate with bank vole males’ social status. Behavioural Processes, 82(3), 279–285. [DOI] [PubMed] [Google Scholar]
- MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, … & Pendlington ZM (2017). The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic acids research, 45(D1), D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruska KP, & Fernald RD (2011). Plasticity of the reproductive axis caused by social status change in an african cichlid fish: II. testicular gene expression and spermatogenesis. Endocrinology, 152(1), 291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Somlo S, Makova S, Tian X, & Brueckner M (2003). Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell, 114(1), 61–73. [DOI] [PubMed] [Google Scholar]
- Mendis SH, Meachem SJ, Sarraj MA, & Loveland KL (2011). Activin A balances Sertoli and germ cell proliferation in the fetal mouse testis. Biology of reproduction, 84(2), 379–391. [DOI] [PubMed] [Google Scholar]
- Meng G, Zhang F, Fuss I, Kitani A, & Strober W (2009). A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity, 30(6), 860–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithraprabhu S, Mendis S, Meachem SJ, Tubino L, Matzuk MM, Brown CW, & Loveland KL (2010). Activin Bioactivity Affects Germ Cell Differentiation in the Postnatal Mouse Testis In Vivo 1. Biology of reproduction, 82(5), 980–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan AP, Fu CP, Kao CY, Welsh CE, Didion JP, Yadgary L, … & Giusti-Rodriguez P (2015). The mouse universal genotyping array: from substrains to subspecies. G3: Genes| Genomes| Genetics, 6(2), 263–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Wang X, Adler AL, Mozhui K, Lu L, & Williams RW (2012). Complex control of GABA (A) receptor subunit mRNA expression: variation, covariation, and genetic regulation. PLoS One, 7(4), e34586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh C, Aho H, Shamsadin R, Nayernia K, Müller C, Sancken U, … & Adham IM (2003). Characterization, expression pattern and chromosomal localization of the spermatogenesis associated 6 gene (Spata6). Molecular human reproduction, 9(6), 321–330. [DOI] [PubMed] [Google Scholar]
- Oka A, Mita A, Sakurai-Yamatani N, Yamamoto H, Takagi N, Takano-Shimizu T, …..& Shiroishi T (2004). Hybrid breakdown caused by substitution of the X chromosome between two mouse subspecies. Genetics, 166(2), 913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver TA, Garfield DA, Manier MK, Haygood R, Wray GA, Palumbi SR (2010). Whole-genome positive selection and habitat-driven evolution in a shallow and deep-sea urchin. Genome Biology and Evolution, 2, 800–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortavant R, Bocquier F, Pelletier J, Ravault JP, Thimonier J, & Volland-Nail P (1988). Seasonality of reproduction in sheep and its control by photoperiod. Australian journal of biological sciences, 41(1), 69–86. [PubMed] [Google Scholar]
- Parker CC, & Palmer AA (2011). Dark matter: are mice the solution to missing heritability? Frontiers in Genetics, 2(32). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CC, Cheng R, Sokoloff G, Lim JE, Skol AD, Abney M, & Palmer AA (2011). Fine-mapping alleles for body weight in LG/J× SM/J F2 and F34 advanced intercross lines. Mammalian genome, 22(9–10), 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CC, Gopalakrishnan S, Carbonetto P, Gonzales NM, Leung E, Park YJ, … & Lionikas A (2016). Genome-wide association study of behavioral, physiological and gene expression traits in outbred CFW mice. Nature genetics, 48(8), 919–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennarun G, Escudier E, Chapelin C, Bridoux AM, Cacheux V, Roger G, … & Duriez B (1999). Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. The American Journal of Human Genetics, 65(6), 1508–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recla JM, Robledo RF, Gatti DM, Bult CJ, Churchill GA, & Chesler EJ (2014). Precise genetic mapping and integrative bioinformatics in Diversity Outbred mice reveals Hydin as a novel pain gene. Mammalian Genome, 25(5–6), 211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha JL, Eisen EJ, Van Vleck LD, & Pomp D (2004). A large-sample QTL study in mice: II. Body composition. Mammalian Genome, 15(2), 100–113. [DOI] [PubMed] [Google Scholar]
- Rowell EA, Wang L, Chunder N, Hancock WW, & Wells AD (2014). Regulation of T Cell Differentiation and Alloimmunity by the Cyclin-Dependent Kinase Inhibitor p18ink4c. PloS one, 9(3), e91587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KY, Sinnar SA, Reinholdt LG, Vaccari S, Hall S, Garcia MA, … & Conti M (2008). The mouse polyubiquitin gene Ubb is essential for meiotic progression. Molecular and cellular biology, 28(3), 1136–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpe RM (2001). Hormones and testis development and the possible adverse effects of environmental chemicals. Toxicology letters, 120(1), 221–232. [DOI] [PubMed] [Google Scholar]
- Setchell JM, & Dixson AF (2001). Changes in the secondary sexual adornments of male mandrills (Mandrillus sphinx) are associated with gain and loss of alpha status. Hormones and Behavior 39(3), 177–184. [DOI] [PubMed] [Google Scholar]
- Shorter JR, Odet F, Aylor DL, Pan W, Kao CY, Fu CP, … & Feathers RW (2017). Male Infertility Is Responsible for Nearly Half of the Extinction Observed in the Mouse Collaborative Cross. Genetics, 206(2), 557–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorter JR, Huang W, Beak JY, Hua K, Gatti DM, de Villena FPM, … & Jensen BC (2017). Quantitative trait mapping in Diversity Outbred mice identifies two genomic regions associated with heart size. Mammalian Genome, 1–10. [DOI] [PMC free article] [PubMed]
- Simmons LW, & García‐González F (2008). Evolutionary reduction in testes size and competitive fertilization success in response to the experimental removal of sexual selection in dung beetles. Evolution, 62(10), 2580–2591. [DOI] [PubMed] [Google Scholar]
- Smallwood TL, Gatti DM, Quizon P, Weinstock GM, Jung KC, Zhao L, … & Bennett BJ (2014). High-resolution genetic mapping in the diversity outbred mouse population identifies Apobec1 as a candidate gene for atherosclerosis. G3: Genes, Genomes, Genetics, 4(12), 2353–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DA, Kim JS, Jenkins S, Ressom H, Huang M, Coppa N, … & Waldman T (2008). Identification of p18INK4c as a tumor suppressor gene in glioblastoma multiforme. Cancer research, 68(8), 2564–2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulsbury CD (2010). Genetic patterns of paternity and testes size in mammals. PLos One 5(3): e9581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Morgan AP, Najarian ML, Sarsani VK, Sigmon JS, Shorter JR, … & Ferris MT (2017). Genomes of the mouse collaborative cross. Genetics, 206(2), 537–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storchova R, Gregorová S, Buckiova D, Kyselova V, Divina P, & Forejt J (2004). Genetic analysis of X-linked hybrid sterility in the house mouse. Mammalian Genome, 15(7), 515–524 [DOI] [PubMed] [Google Scholar]
- Supp DM, Witte DP, Branford WW, Smith EP, & Potter SS (1996). Sp4, a member of the Sp1-family of zinc finger transcription factors, is required for normal murine growth, viability, and male fertility. Developmental biology, 176(2), 284–299. [DOI] [PubMed] [Google Scholar]
- Suto JI (2008). Genetic dissection of testis weight in a mouse strain having an extremely large testis: major testis weight determinants are autosomal rather than Y-linked on the basis of comprehensive analyses in Y-chromosome consomic strains. Proceedings of the Japan Academy, Series B, 84(9), 393–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suto JI (2011). Genetic dissection of testis weight in mice: quantitative trait locus analysis using F2 intercrosses between strains with extreme testis weight, and association study using Y-consomic strains. Mammalian genome, 22(11–12), 648–660. [DOI] [PubMed] [Google Scholar]
- Svenson KL, Gatti DM, Valdar W, Welsh CE, Cheng R, Chesler EJ, … & Churchill GA (2012). High-resolution genetic mapping using the Mouse Diversity outbred population. Genetics, 190(2), 437–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, … & Herz J (1999). Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell, 97(6), 689–701. [DOI] [PubMed] [Google Scholar]
- Tyler AL, Ji B, Gatti DM, Munger SC, Churchill GA, Svenson KL, & Carter GW (2017). Epistatic networks jointly influence phenotypes related to metabolic disease and gene expression in diversity outbred mice. Genetics, 206(2), 621–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Williams RW, Manly KF (2003) WebQTL: web-based complex trait analysis. Neuroinformatics 1:299–308 [DOI] [PubMed] [Google Scholar]
- Watanabe T, Chuma S, Yamamoto Y, Kuramochi-Miyagawa S, Totoki Y, Toyoda A, … & Noce T (2011). MITOPLD is a mitochondrial protein essential for nuage formation and piRNA biogenesis in the mouse germline. Developmental cell, 20(3), 364–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Sanger Institute. (2009). Alleles produced for the KOMP project by the Wellcome Trust Sanger Institute. MGI Direct Data Submission
- White MA, Steffy B, Wiltshire T, Payseur BA (2011). Genetic dissection of a key reproductive barrier between nascent species of house mice. Genetics, 189(1), 289–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Stubbings M, Dumont BL, & Payseur BA (2012). Genetics and evolution of hybrid male sterility in house mice. Genetics, 191(3) 917–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RW, & Williams EG (2016). Resources for systems genetics. In Systems Genetics, Methods in Molecular Biology, 1488: 3–29. [DOI] [PubMed] [Google Scholar]
- Woods LC, & Mott R (2017). Heterogeneous Stock Populations for Analysis of Complex Traits. Methods in Molecular Biology, 1488, 31–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Xia W, Zohar Y, & Gui JF (2013). Zebrafish dmrta2 regulates the expression of cdkn2c in spermatogenesis in the adult testis. Biology of reproduction, 88(1). [DOI] [PubMed] [Google Scholar]
- Yang J, Zaitlen NA, Goddard ME, Visscher PM, & Price AL (2014). Advantages and pitfalls in the application of mixed-model association methods. Nature Genetics 46(2), 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S, Stratton CJ, Bao J, Zheng H, Bhetwal BP, Yanagimachi R, & Yan W (2015). Spata6 is required for normal assembly of the sperm connecting piece and tight head–tail conjunction. Proceedings of the National Academy of Sciences, 112(5), E430–E439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Na W, Zhang HL, Wang N, Du ZQ, Wang SZ, Wang ZP, Zhang Z, & Li H (2017). TCF21 is related to testis growth and development in broiler chickens. Genet Sel Evol, 49(1):25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Murphy MW, Gearhart MD, Bardwell VJ, & Zarkower D (2014). The mammalian Doublesex homolog DMRT6 coordinates the transition between mitotic and meiotic developmental programs during spermatogenesis. Development, 141(19), 3662–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, … & Lu Z (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Molecular cell, 49(1), 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidek V, Musilova A, Pintir J, Simakova M, Pravenec M (1998). Genetic dissection of testicular weight in the mouse with BXD recombinant inbred strains. Mammalian Genome, 9(7), 503–505. [DOI] [PubMed] [Google Scholar]
- Zindy F, den Besten W, Chen B, Rehg JE, Latres E, Barbacid M, … & Roussel MF (2001). Control of spermatogenesis in mice by the cyclin D-dependent kinase inhibitors p18Ink4c and p19Ink4d. Molecular and cellular biology, 21(9), 3244–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccarello D, Ferlin A, Cazzadore C, Pepe A, Garolla A, Moretti A, Cordeschi G, Francavilla S, Foresta C (2008). Mutations in dynein genes in patients affected by isolated non-syndromic asthenozoospermia. Human Reproduction, 23(8), 1957–1962. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.