

Summary
Background:
Tissue factor (TF) is the essential cell-surface initiator of coagulation and mediates cell signaling through protease activated receptor (PAR) 2. Having a diverse cellular distribution, TF is involved in many biological pathways and pathologies. Our earlier work identified host-cell derived TF on the envelope covering several viruses and showed its involvement toward enhanced cell infection in vitro.
Objective:
In the current study, we evaluated the in vivo role of virus surface TF on infection and on the related modulator of infection PAR2.
Methods:
Using herpes simplex virus type 1 (HSV1) as a model enveloped virus, purified HSV1 was generated with or without envelope TF through propagation in a TF-inducible cell line. Infection was studied after IV inoculation of BALB/c, C57BL/6J or C57BL/6J PAR2 knock out (KO) mice with 5 × 105 plaque forming units of HSV1 mimicking viremia. Three days post-inoculation, organs were processed, and virus was quantified by plaque forming assays and quantitative real-time polymerase chain reaction.
Results:
Infection of brain, lung, heart, spinal cord, and liver by HSV1 required viral TF. Demonstrating promise as a therapeutic target, infection was inhibited by virus-specific anti-TF monoclonal antibodies or small molecule inhibitors of coagulation. PAR2 modulates HSV1 in vivo as demonstrated using PAR2 knock-out mice and PAR2 agonist peptide.
Conclusion:
TF is a constituent of many permissive host cells types. Therefore, the results presented here may explain why many viruses are correlated to hemostatic abnormalities and denote TF as a novel pan-specific envelope antiviral target.
Keywords: anticoagulant, coagulation, protease activated receptor, tissue factor, virus infection
Introduction
Tissue factor (TF) is a multi-functional transmembrane receptor essential for mammalian life [1–3]. It is critically positioned as a cofactor that enhances the protease factor (F) VIIa at the intersection of blood coagulation and the vascular inflammatory response. The activation of FX to FXa is triggered by TF/FVIIa, which initiates the procoagulant response to vascular damage. As a constituent of the nascent TF/FVIIa/FXa complex, TF furthermore coordinates protease activated receptor (PAR) 2-mediated cell signaling with diverse effects on health and disease, including angiogenesis, tissue repair, diabetes, pain, cell proliferation and innate immunity [4–9]. To localize these protease-activating and cell-signaling functions, TF activity is restricted to sites where cells are stimulated, such as by vascular damage, and on the surface of related microvesicles [10–12]. Adding to the functional repertoire of TF, our earlier work identified its constitutive activity and antigen on the host cell-originated envelope structure of three herpesvirus family members, herpes simplex virus type 1 (HSV1), HSV2 and cytomegalovirus, each purified from several cultured cell types [13]. These viruses thus bypass the physiological localization of coagulation protease activation by TF, which helps to explain their reported links to vascular disease [14–17].
To investigate the effects of virus envelope TF on cell infection, we use HSV1 as a model envelope virus, which establishes latency with frequent reactivation and a coincident inflammatory response [18]. Although commonly recognized as the “cold sore virus”, HSV1 has numerous effects on health. In addition to its correlation with vascular disease [14–17], HSV1 is the leading cause of infectious blindness [19], sporadic encephalitis [20] and genital herpes [21], and is associated with intestinal dysregulation [22]. As TF is multifunctional, we previously explored the contribution of envelope TF in infection using purified HSV1 that either bears host cell-derived TF (HSV1/TF+) or is deficient in TF (HSV/TF−) [23]. These in vitro studies showed that infection of endothelial cell monolayers under serum-free culture conditions was enhanced by the availability of virus envelope TF and the mechanism involved protease activated receptor (PAR) 2. Therefore, we hypothesized that in vivo infection may be impacted by viral TF facilitating coagulation enzyme activation. In the current study we investigated the effects of TF on the surface of blood-borne HSV1 in mice. Against the conventional paradigm that virus-encoded gene products primarily control virus surface-host communication and thus infectivity, we report that the availability of host-encoded TF as a constituent of the HSV1 envelope induces permissiveness to infection in mice.
Materials and methods
Virus Production
HSV1/TF+ and HSV/TF− were propagated in TF-inducible human melanoma cells [24] and purified as previously described [23]. Comparable in vitro plaque forming units (PFU) per virus particle (VP) number was ensured [25]. Thus, each virus preparation had similar numbers of VP/mL and PFU/mL. This was achieved by introducing UV-inactivated virus into higher PFU/VP preparations. Viruses were purified by sucrose gradient ultracentrifugation, quality assured for lack of cellular debris and quantified to derive VP/mL by negative staining electron microscopy [25]. All cells were shown to be mycoplasma free. To derive the number of infectious viruses, plaque assays on African green monkey kidneys cells (Vero, CCL-81; ATCC, Manassas, VA) were conducted, as previously described [23].
Animals
Animal experiments were approved by the Animal Care Committee at the University of British Columbia. Two mouse strains were compared; BALB/c mice, which are relatively vulnerable to infection and C57BL/6J mice, which are relatively resistant [26,27]. Eight-week-old female BALB/c or wild type C57BL/6J mice were obtained from Jackson Laboratories (Bar Harbor, ME). Additional experiments were conducted with wild type C57BL/6J and age and sex-matched PAR2 knockout (PAR2-) mice backcrossed for 9 generations with C57BL/6J [28]. The number of mice in each experiment was chosen to provide 95% statistical power with a 5% error level. Animals were randomly allocated to each treatment group and excluded from the study if injection of the virus was unsuccessful.
HSV1 Inoculation of Mouse Strains
BALB/c mice were inoculated with a sublethal dose (5 × 105 PFU) of either HSV1/TF+ or HSV/TF− by intravenous tail vein injection. Identical experiments were conducted where 0.33 mg each of three anti-TF antibodies were injected intraperitoneally 4 hours before inoculation with HSV1/TF+. These included monoclonal anti-TF 5G9 [29], 9C3, and 6B4 [30–32], as previously prepared and characterized elsewhere. Irrelevant anti-T antigen (TIB115, ATCC) provided an IgG1 isotype control (1 mg/animal). Additionally, BALB/c mice were simultaneously injected with HSV1/TF+ and either apixaban, nematode anticoagulant protein c2 [33] (NAPc2; Corvas, San Diego, CA), or hirudin (Hypen Biomed, Burlington, ON), all at 1mg/kg, or PAR2 activating peptide (PAR2ap) (3 µmol/kg; SLIGRL-NH2; Product Number 1468, Tocris, Oakville, ON). The doses used for NAPc2 (1 mg/kg) [34] and hirudin (1 mg/kg) [35] are well established in the literature. However, mouse studies involving apixaban are uncommon. The dose selected for apixaban (1 mg/kg) is based on studies conducted in mice that reported anticoagulation with no evidence of gastrointestinal [36] or cerebral bleeding [37] at 1.2 or 2 mg/kg, respectively.
Previous reports have shown that new HSV1 production is detectable three days post-inoculation of BALB/c mice [38,39]. Since this timing also enables clearance of the initial virus inoculum [38,39], mice were sacrificed in the current study three days post-inoculation. The lungs, heart, liver, spinal cord and brain were surgically removed, frozen on dry ice, and stored at −80oC. Prior to analysis individual organs were weighed into 1 ml of cell media and processed through a 40 µm cell strainer to separate the larger cell debris. The lysate was used to infect Vero cell monolayers and quantified by standard plaque assay to determine the amount of infectious virus (PFU) per gram of tissue.
RT-PCR Analysis
The cell lysate was also used in some cases to determine the amount of viral genome per mg of tissue by quantitative real time polymerase chain reaction (qRT-PCR). Viral DNA was extracted using QIAamp cador Pathogen Mini Kit (Qiagen, Montreal, QC) according to the manufacture’s protocol. Standard curve samples were prepared using purified HSV1 DNA and mouse lung genomic DNA as a source of beta actin gene (Actb). The standard curve samples were run in triplicate with the wells having 101 - 109 copies of DNA. The limit of quantification for the qRT-PCR assay was 10 copies of HSV-1 DNA. For the determination of viral titer, mouse organ samples were run in triplicate along with standardized samples. Those that did not yield a positive signal in duplicate wells following 40 cycles of amplification were considered negative. The HSV1 DNA copy number per gram of tissue sample was normalized and expressed as log10 DNA copies per 105 Actb genes. The primers for HSV-1 amplified from mouse organs were as follows: Forward, 5’-CTCCTGGAGGTGATGGACAGTCTC-3’; Reverse, 5’-GCAGGTGTAGAAGTACCTCTCGGG-3’; Probe, 5’-ACTGCAAGGTTCGCAAGGCGACCAA-3’. For the Actb housekeeping gene, primers and FAM probe reporter dye were obtained from TaqMan Assay Mm00607939_s1 (Applied Biosystems, Foster City, CA). Reactions were performed on 2 µL of DNA in 20 µL reaction mixtures using TaqMan Fast Advanced gene expression master mix and qRT-PCR was performed on an ABI 7900HT Sequence Detection System by selecting the appropriate detector (Applied Biosystems). The cycle program was set at 50°C for 2 minutes, 95°C for 20 seconds, followed by 40 cycles of 95°C for 1 second and 60°C for 20 seconds. Data were analyzed using SDS 2.3 software.
Statistical Analysis
The data are presented as the mean ± SEM of at least 4 separate animals measured in triplicate and experiments were conducted on at least 2 different litters. Data are presented as mean ± SEM, and Student’s t test (two-tailed distribution) was used to calculate p-values.
Results
Infection Th2-immuno-dominant mice is enhanced by TF on HSV1.
The potential effect of virus surface TF on infection was evaluated in vivo by comparing HSV1/TF+ to HSV1/TF− infection of BALB/c mice. This strain of mouse is permissive to infection because their immune response is largely driven by Th2 cell pathways and is characterized by high antibody titers [26,27,40]. Three days post-inoculation, five organs were recovered and evaluated for virus infiltration; heart, lung, spinal cord, brain and liver. HSV1 was quantified by plaque-forming assays and each organ was shown to accumulate infectious virus. Infectivity was found to be highly dependent on the presence of envelope TF (Fig. 1A, Virus). These functional data were supported by measurement of the virus genome copy number by qRT-PCR (Figure 1A, Genome), which showed that production of infectious virus was inhibited when envelope TF was absent. Therefore, envelope TF enhances the infection cycle at or prior to DNA replication.
Fig. 1.
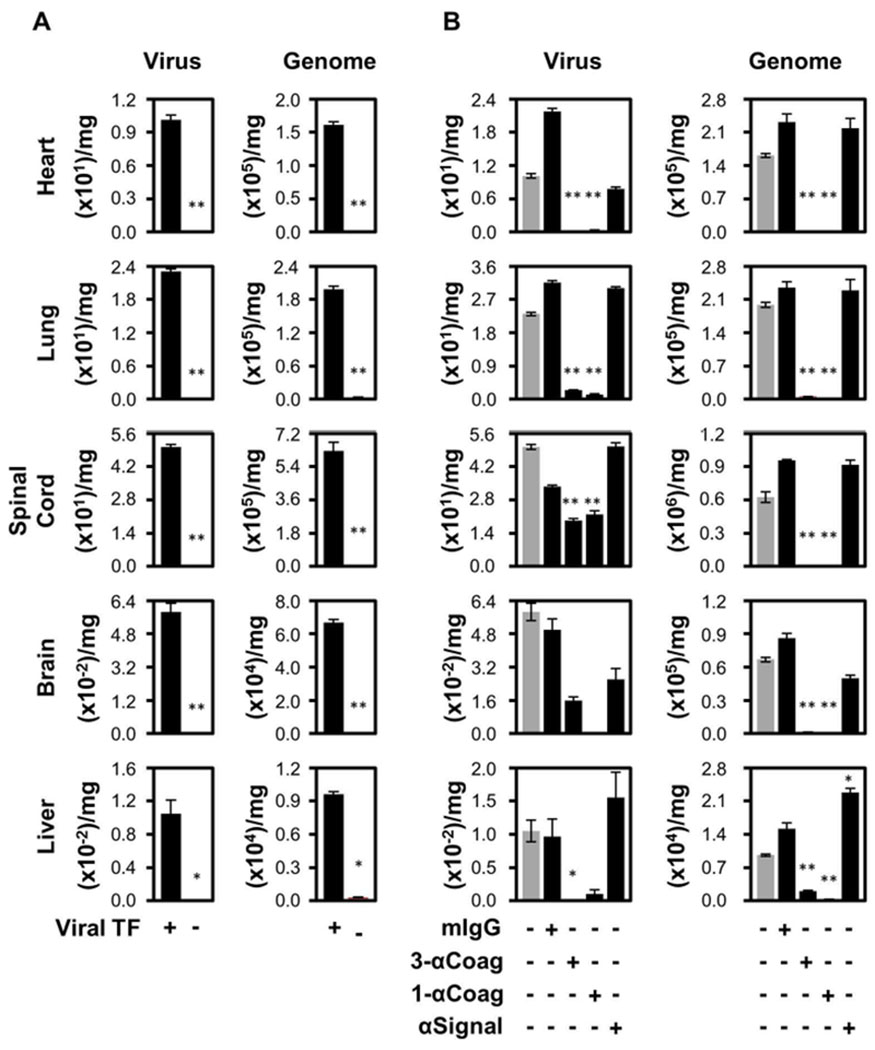
Infection of Th2 immuno-dominant mice (BALB/c) is enhanced by TF on HSV1. (A) Eight-week-old female BALB/c mice were inoculated intravenously with 5 × 105 PFU of either TF competent (TF+; n=24) or deficient (TF−; n=13) HSV1 via the tail vein. Three days post-infection, the mice were processed and the amount of infectious virus (Virus; PFU/mg) and HSV1 genetic material (Genome; genome copies/mg) were determined. (B) Additional experiments were conducted after pre-immunization of mice with a mixture of three anti-TF IgG1 monoclonal antibodies, 5G9, 9C3 and 6B4 (3-αCoag; at 0.33 mg each/mouse; n=10), 5G9 alone (1-αCoag; 1 mg/mouse; n=6) or 10H10 alone (αSignal; 1 mg/mouse; n=6), 4 hours prior to injection of the virus. Purified anti-Simian virus 40 large T antigen (mIgG; 1 mg/mouse; n=8) was used as an irrelevant monoclonal IgG1 subclass-matched negative control. Grey bars represent data from HSV1/TF+ as depicted in Panel A. In all panels data are expressed as mean ± SEM. As determined by Student’s t test, **P ≤ 0.05 and *P ≤ 0.10 when compared to the TF+ virus.
Further addressing the role of viral TF, BALB/c mice received a combination of three well-characterized anticoagulant monoclonal antibodies [30,32] that recognize different epitopes on only the human TF incorporated into the purified HSV1 envelope (3-αTF). This treatment reduced the infectious HSV1 (Figure 1B, Virus) and total virus genome copy number (Fig. 1B, Genome) detectable in each organ. Comparable results were observed using a single TF-specific antibody, 5G9 (Fig. 1B, 1-αTF), excluding the possibility that multivalent antibody presentation may facilitate complement-mediated virus inactivation. Since there was no difference between the triple or single anti-TF treatments suggests that only one of the functional regions of TF requires inhibition for an antiviral effect, in this case FX-binding. A human anti-TF monoclonal antibody that has no effect on the procoagulant cofactor function of TF but inhibits PAR2 signalling was evaluated (Fig. 1B, αSignal). At three days post-infection this antibody had minimal effect on the amount of infectious virus measured in each organ. These plaque data were corroborated by qRT-PCR (Fig. 1B, Genome). Furthermore, an isotype control injected at the same concentration of specific antibody had no effect (Fig. 1B, mIgG).
Infection of BALB/c mice is inhibited by anticoagulants.
The involvement of TF in FXa production and the consequent downstream generation of thrombin and effects on HSV1 infection of BALB/c mice were investigated using specific coagulation protease inhibitors. Since infection of BALB/c by HSV1/TF− (Fig. 1A, Virus, TF−) had mostly undetectable levels of infection, only HSV1/TF+ mice were evaluated. Fig. 2 shows that all three anticoagulants studied blocked HSV1/TF+ infection. These were: hirudin, a thrombin-specific inhibitor; NAPc2, which inhibits FXa and the TF/FVIIa/FXa complex; and apixaban, a small molecule FXa-directed oral anticoagulant therapeutic. All mice appeared healthy with no evidence of spontaneous bleeding up to the experimental end-point (3 days).
Fig. 2.

Infection of BALB/c mice is inhibited by anticoagulation. Eight-week-old female BALB/c mice were inoculated intravenously with 5 × 105 PFU of TF competent (n=24) HSV1 alone (grey bar as depicted in Figure 1, Panel A) or simultaneously with hirudin (1 mg/kg, n=9), NAPc2 (1 mg/kg, n=18), or apixaban (1 mg/kg, n=18) via the tail vein. In each case, three days post infection, the mice were processed and the amount of infectious virus (PFU/mg) was determined in each organ. In all panels data are expressed as mean ± SEM. As determined by Student’s t test, **P ≤ 0.05 and *P ≤ 0.10 when compared to the TF+ virus.
Infection of Th1 immuno-dominant mice (C57BL/6J) is enhanced by TF on HSV1.
Mouse strains differ in their susceptibility to viral infection [41,42]. Contrasting BALB/c mice, C57BL/6J mice are resistant to virus infection because their immune response is predominantly Th1-driven, triggering a rapid innate immune response that involves phagocytic cells [26,27]. Predictably, we found that infection of C57BL/6J by HSV1/TF+ was reduced by approximately one log in most organs studied relative to Th2 immuno-dominant BALB/c mice (Fig. 3, HSV/TF+ compared to Fig. 1A, HSV/TF+). Nevertheless, detection of infectious HSV1 by plaque assays three days after inoculation of C57BL/6J was enhanced in each organ studied when TF was available on the virus surface (Fig. 3, HSV1/TF+ compared to HSV1/TF−). The exception was brain, which was not permissive to infection by HSV/TF+ or HSV1/TF−, presumably because of the added protective effects of the blood-brain barrier. Interestingly, unlike the PFU measurement of virus infection, the genome copy number for infection of C57BL/6J mice was not appreciably dependent on the viral TF phenotype (Fig. 3, Genome). Thus, the presence of envelope TF increases the proportion of infectious to non-infectious virus that is produced in C57BL/6J mice, suggesting inhibition due to the lack of envelope TF is at or post viral DNA replication.
Fig. 3.

Infection of Th1 immuno-dominant mice (C57BL/6J) is enhanced by TF on HSV1. Eight week old female C57BL/6J mice were inoculated intravenously with 5 × 105 PFU of either TF competent (TF+, n=7) or deficient (TF−; n=8) HSV1 via the tail vein. Three days post-infection, the mice were processed and the amount of infectious virus (Virus; PFU/mg) and HSV1 genetic material (Genome; genome copies/mg) were determined. In all panels data are expressed as mean ± SEM. As determined by Student’s t test, **P ≤ 0.05 and *P ≤ 0.10 when compared to the TF+ virus.
Infection of C57BL/6J mice is enhanced by PAR2 deficiency.
Given the known TF role in PAR-mediated signaling, we infected PAR2 KO mice with purified HSV1/TF+. Deleting PAR2 expression in C57BL/6J mice caused an increase of HSV1 infection in the heart, lung, brain and liver (Fig. 4, HSV1/TF+, PAR2+ compared to HSV1/TF+, PAR2-). These data were corroborated by measuring genome copy number (Fig. 4, Genome) except for spinal cord, which may reflect the accumulation of non-infectious virus due to specific mechanisms involved in neurotropism. These combined data indicate that PAR2-competence in wild type mice is protective against infectious virus production.
Fig. 4.
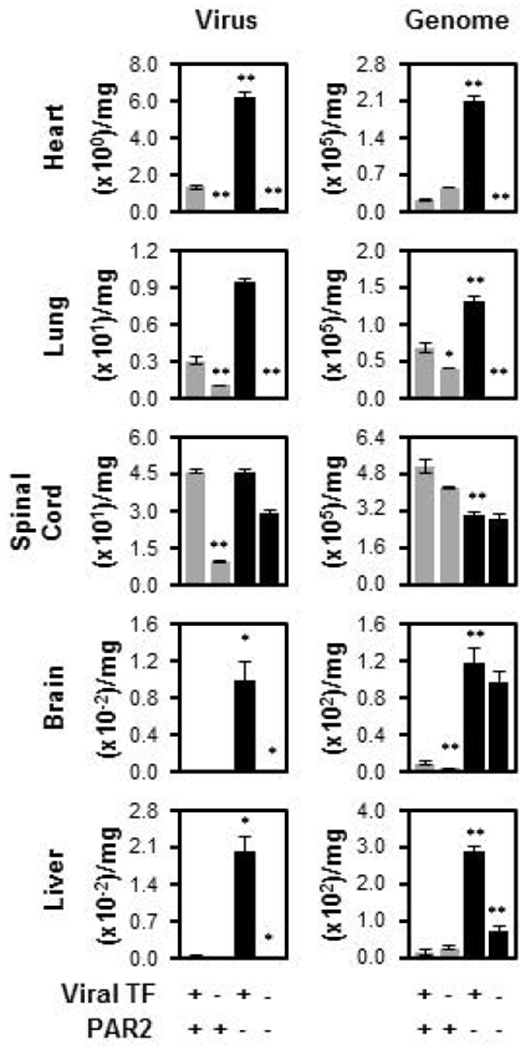
Infection of C57BL/6J is enhanced by PAR2 deficiency. Wild type C57BL/6J mice inoculated with either TF competent (TF+, n=7, grey bar, as in Fig. 3, TF+) or deficient (TF−; n=8, grey bar, as in Fig. 3, TF−) HSV1 via the tail vein were compared to PAR2 KO (PAR2−; n=6) mice and the combined effect of PAR2 knock out and viral TF-deficiency (n=5). Three days post-inoculation, the mice were processed and the amount of infectious virus (Virus; PFU/mg) or genome copies (Genome; genome copies/mg) was determined. In all panels data are expressed as mean ± SEM. As determined by Student’s t test, **P ≤ 0.05 and *P ≤ 0.10 and when compared to the TF+ virus except for the TF−/PAR2− which is compared to the TF− virus.
Although C57BL/6J mice are relatively resistant to infection and more so in the absence of viral TF, we studied the effect of PAR2 deficiency in mice combined with the lack of TF on the virus surface (Fig. 4). When TF was excluded from the virus surface and infection in PAR2 KO mice was followed, diminished infection was anticipated since envelope TF was important for HSV1 infection of C57BL/6J wild type mice (Fig. 3). Consistently, heart, lung, spinal cord, brain and liver trended toward reduced infection by HSV1/TF− compared to HSV1/TF+ when PAR2 was genetically deleted and measured by virus plaque assays or genome.
Infection of BALB/c mice is inhibited by PAR2 activating peptide.
Synthetic PAR2 activating peptide (PAR2ap) that triggers G protein-coupled cell signaling was added to the HSV1/TF+ inoculum to study the outcome of a systemic gain-of-function on PAR2 stimulation. Since HSV1 infection is weak in C57BL/6J mice and the genetically deficient mice suggested that PAR2 activation in wild type will lead to inhibition of infection, the Th2 immuno-dominant strain was used. Plaque formation assays revealed that PAR2ap attenuated infection in BALB/c mice by 80-90% when measured three days post-inoculation (Fig. 5).
Fig. 5.

Infection of BALB/c mice is inhibited by PAR2 activating peptide. Experiments were conducted as in Fig. 1 using BALB/c mice to investigate the simultaneous injection of TF competent HSV1 (grey bar, from Fig. 1A, Virus, TF+) with PAR2ap (3 umol/kg, n=7). In all panels data are expressed as mean ± SEM. As determined by Student’s t test, **P ≤ 0.05 and *P ≤ 0.10 compared to no addition of PAR2ap.
Discussion
Relatively little is known about the function of virus envelope constituents obtained from the host cell [43,44]. In the current work we investigated the effects of host cell membrane-derived HSV1 envelope TF on hemostasis and infection in vivo. Using a model of viremia where a sublethal dose of purified HSV1/TF+ or HSV1/TF− was injected into the tail vein, two mouse strains demonstrated that viral TF contributes greatly to infection regardless of whether a Th1 or Th 2 immune response is prevalent. The extreme was in the higher susceptibility Th2 immuno-dominant strain, BALB/c, where envelope TF was obligate for detection of infectious HSV1 in all organs tested, heart, lung, spinal cord, brain and liver. Significant enhancement due to viral TF was also observed in C57BL/6J mice, which favor a Th1 immune response and are consequently less vulnerable to HSV1 infection than BALB/c mice. Therefore, the physiological initiator of coagulation, TF, associated with the HSV1 envelope is a previously unrecognized host cell-derived constituent that serves an important function during the in vivo virus infection cycle.
The role of TF on the virus was further dissected using species-specific monoclonal antibodies that recognize the human TF on HSV1/TF+ but not the TF intrinsic to the mouse [45]. These well-characterized anticoagulant antibodies [30] reduced the infectious virus in heart, lung, liver, spinal cord, brain and liver, identifying envelope TF function as an antiviral target. This possibility was supported using NAPc2, which forms a quaternary inhibition-complex with TF, FXa and FVIIa [33,46], and thus inhibits all extrinsic pathway coagulation factors simultaneously. More effective than the TF-specific anticoagulant antibodies, NAPc2 reduced the infectious HSV1 in all tissues evaluated to a nearly undetectable level. However, unlike the antibodies used here, NAPc2 not only affects viral TF but also the TF-complexes intrinsic to the mouse, which may be involved in the virus infection process.
TF is found throughout the body due to its prevalent constitutive expression by fibroblasts, pericytes, smooth muscle cells, epithelial cells, astrocytes and cardiomyocytes, and inducibility on other cells like endothelial and monocyte lineage cells [3]. Thus, TF-bearing cells are permissive to infection by many clinically important enveloped viruses from different virus families, including Ebola [47], influenza [48], human immunodeficiency virus [49], dengue [50], Zika [51], hepatitis C [52] and others. As a pervasive host-cell encoded transmembrane protein, TF is conceivably a common in vivo envelope constituent. As an example, NAPc2 has been used to successfully treat infection of rhesus monkeys by the filovirus, Ebola [53,54], although TF being on the virus envelope was not considered by these authors. Thus, in combination with our current in vivo and prior in vitro data [13], TF may participate across numerous virus families because of its wide cell distribution, thereby serving as a broad-spectrum anti-envelope virus target.
Thrombin is the ultimate coagulation protease and is produced once TF mediates FXa generation. Facilitating PAR1, 3 and 4 activation, feedback regulation of hemostasis and clot formation [55,56], thrombin is a major hemostasis regulator. To investigate the possibility that thrombin has an additional function, HSV1 infection was measured in the presence of the highly specific thrombin inhibitor, hirudin. Indeed, hirudin attenuated in vivo infection, indicating the virus exploits thrombin activity for infection. However, inhibition of infection was also facilitated by NAPc2, suggesting that upstream FXa may also be involved. Therefore, the effect of a FXa-directed anticoagulant was investigated. At the approximate therapeutic dose used in humans (1.0 versus ~0.4 mg/kg), apixaban, was as effective as NAPc2 at reducing HSV1 infection, suggesting that inhibition of FXa may be the dominant antiviral effector. While avoiding the induction of bleeding may be a challenge, an unforeseen application of apixaban, or other routinely used anticoagulant therapeutic, may be to control virus infection.
We previously reported that the infection of cultured human endothelial cell monolayers by HSV1 was enhanced by a brief pretreatment with PAR2ap and inhibited by a TF-specific antibody (10H10) that disrupts TF-mediated PAR2 signaling [23]. The experimental conditions excluded the participation of plasma proteins (e.g. activated coagulation proteases), and immune and other cells that would normally be present during infection in vivo. To investigate the biological role of PAR2 in HSV1 infection, the current study showed that C57BL/6J PAR2 KO mice were more vulnerable to infection three days post-inoculation. Therefore, PAR2 competence protected the animal against HSV1 infection. PAR2ap predictably inhibited infection in mice. This involvement of PAR2 is likely due to the recruitment of innate immune-responsive cells but is opposite to the enhancement observed in vitro.
Although TF stimulates PAR2 [8], the results reported here suggest that the increased infection in mice caused by viral TF is not explained by stimulation of PAR2 intrinsic to the mouse. A disconnect between viral TF and PAR2 in vivo is further supported by the finding that 10H10, which is specific to the human TF [45] on the virus surface, had no effect on infection by the third day after inoculation. Thus, when combined with our earlier in vitro studies where the involvement of viral TF was studied one hour after inoculation [23] a compartmentalized model is emerging. First, early in the infection cycle when the virus initially docks with the target cell, envelope TF enhances infection through PAR2 that requires FXa as shown by in vitro studies [23]. Thus, when TF−, FXa- or thrombin-specific anticoagulants were present at the time of mouse inoculation, this first compartment was affected, inhibiting infection. A second compartment is proposed to evolve later in the infection cycle (by day three) as functional cellular TF induced by infection [57] and virus envelope TF promote FXa and thrombin production, which not only affect the target cell but additional cells in circulation. The role of PAR2 may consequently switch from pro-infection to antiviral. The involvement of PAR2 in influenza A virus (IAV) infection has been studied and shown to enhance [58,59] or reduce infection [60]. To explain the inconsistency, cellular protein expression is dynamic during virus infection [44], suggesting sampling time or virus load may alter the respective roles of PARs during infection. Like HSV1, IAV is an envelope virus. Therefore, differing envelope factors derived from chicken eggs [61] or canine epithelial cells [62], such as TF, may also affect the roles of PAR2 during envelope virus infection.
The data presented here strongly suggest that host-derived TF on the virus surface and the related production of FXa and thrombin play important roles in HSV1 infection in vivo that involve PAR stimulation. Since TF is derived from the host cell where virus replicates, a virus may undergo various envelope surface transformations depending on the host cell from which it egresses. Acquisition of envelope TF or other yet unidentified host proteins may incline the infection of previously protected tissue and affect tropism. An example is brain, which was not infectible in mice unless TF was a constituent of the HSV1 surface, providing an explanation for occurrences of sporadic encephalitis.
Essentials.
The coagulation initiator, tissue factor (TF), is on the herpes simplex virus 1 (HSV1) surface.
HSV1 surface TF was examined in mice as an antiviral target since it enhances infection in vitro.
HSV1 surface TF facilitated infection of all organs evaluated and anticoagulants were antiviral.
Protease activated receptor 2 inhibited infection in vivo and its pre-activation was antiviral.
Acknowledgements
This work was supported by the Canadian Institutes of Health Research grant 273985 (E. L. G. Pryzdial) and National Institutes of Health grant HL60742 (W. Ruf). A. Y. Simon was supported by a Canadian Blood Services Postdoctoral Fellowship.
Footnotes
Disclosure of Conflicts of Interests
The authors state that they have no conflict of interest.
References
- (1).Rosen ED, Chan JC, Idusogie E, Clotman F, Vlasuk G, Luther T, Jalbert LR, Albrecht S, Zhong L, Lissens A, Schoonjans L, Moons L, Collen D, Castellino FJ, Carmeliet P. Mice lacking factor VII develop normally but suffer fatal perinatal bleeding. Nature 1997;390:290–294. [DOI] [PubMed] [Google Scholar]
- (2).Carmeliet P, Mackman N, Moons L, Luther T, Gressens P, Van V I, Demunck H, Kasper M, Breier G, Evrard P, Muller M, Risau W, Edgington T, Collen D Role of tissue factor in embryonic blood vessel development. Nature 1996;383:73–75. [DOI] [PubMed] [Google Scholar]
- (3).Butenas S Tissue factor structure and function. Scientifica (Cairo) 2012;2012:964862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Xu Z, Xu H, Ploplis VA, Castellino FJ. Factor VII deficiency impairs cutaneous wound healing in mice. Mol Med 2010;16:167–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Eisenreich A, Zakrzewicz A, Huber K, Thierbach H, Pepke W, Goldin-Lang P, Schultheiss HP, Pries A, Rauch U. Regulation of pro-angiogenic tissue factor expression in hypoxia-induced human lung cancer cells. Oncol Rep 2013;30:462–470. [DOI] [PubMed] [Google Scholar]
- (6).Badeanlou L, Furlan-Freguia C, Yang G, Ruf W, Samad F. Tissue factor-protease-activated receptor 2 signaling promotes diet-induced obesity and adipose inflammation. Nat Med 2011;17:1490–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lam DK, Schmidt BL. Serine proteases and protease-activated receptor 2-dependent allodynia: a novel cancer pain pathway. Pain 2010;149:263–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Witkowski M, Landmesser U, Rauch U. Tissue factor as a link between inflammation and coagulation. Trends Cardiovasc Med 2016;26:297–303. [DOI] [PubMed] [Google Scholar]
- (9).Schaffner F, Ruf W. Tissue factor and PAR2 signaling in the tumor microenvironment. Arterioscler Thromb Vasc Biol 2009;29:1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Grover SP, Mackman N. Tissue factor: An essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol 2018;38:709–725. [DOI] [PubMed] [Google Scholar]
- (11).Hisada Y, Alexander W, Kasthuri R, Voorhees P, Mobarrez F, Taylor A, McNamara C, Wallen H, Witkowski M, Key NS, Rauch U, Mackman N. Measurement of microparticle tissue factor activity in clinical samples: A summary of two tissue factor-dependent FXa generation assays. Thromb Res 2016;139:90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Davizon P, Lopez JA. Microparticles and thrombotic disease. Curr Opin Hematol 2009;16:334–341. [DOI] [PubMed] [Google Scholar]
- (13).Pryzdial EL, Sutherland MR, Ruf W. The procoagulant envelope virus surface: contribution to enhanced infection. Thromb Res 2014;133 Suppl 1:S15–S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Espinola-Klein C, Rupprecht HJ, Blankenberg S, Bickel C, Kopp H, Victor A, Hafner G, Prellwitz W, Schlumberger W, Meyer J. Impact of infectious burden on progression of carotid atherosclerosis. Stroke 2002;33:2581–2586. [DOI] [PubMed] [Google Scholar]
- (15).Epstein SE, Zhu J, Burnett MS, Zhou YF, Vecellotti G, Hajjar D. Infection and atherosclerosis: Potential roles of pathogen burden and molecular mimicry. Arterioscler Thromb Vasc Biol 2000;20:1417–1420. [DOI] [PubMed] [Google Scholar]
- (16).Zhu J, Nieto FJ, Horne BD, Anderson JL, Muhlestein JB, Epstein SE. Prospective study of pathogen burden and risk of myocardial infarction or death. Circulation 2001;103:45–51. [DOI] [PubMed] [Google Scholar]
- (17).Siscovick DS, Schwartz SM, Corey L, Grayston JT, Ashley R, Wang SP, Patsy BM, Tracy RP, Kuller LH, Kronmal RA. Chlamydia pneumoniae, herpes simplex virus type 1, and cytomegalovirus and incident myocardial infarction and coronary heart disease death in older adults - The Cardiovascular Health Study. Circulation 2000;102:2335–2340. [DOI] [PubMed] [Google Scholar]
- (18).Zuckerman RA. The clinical spectrum of herpes simplex viremia. Clin Infect Dis 2009;49:1302–1304. [DOI] [PubMed] [Google Scholar]
- (19).Farooq AV, Shukla D. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Surv Ophthalmol 2012;57:448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Stahl JP, Mailles A, Dacheux L, Morand P. Epidemiology of viral encephalitis in 2011. Med Mal Infect 2011;41:453–464. [DOI] [PubMed] [Google Scholar]
- (21).Malkin JE. Epidemiology of genital herpes simplex virus infection in developed countries. Herpes 2004;11 Suppl 1:2A–23A. [PubMed] [Google Scholar]
- (22).Khoury-Hanold W, Yordy B, Kong P, Kong Y, Ge W, Szigeti-Buck K, Ralevski A, Horvath TL, Iwasaki A. Viral spread to enteric neurons links genital HSV-1 infection to toxic megacolon and lethality. Cell Host Microbe 2016;19:788–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sutherland MR, Ruf W, Pryzdial ELG. Tissue factor and glycoprotein C on herpes simplex virus type 1 are protease-activated receptor 2 cofactors that enhance infection. Blood 2012;119:3638–3645. [DOI] [PubMed] [Google Scholar]
- (24).Dorfleutner A, Hinterman E, Tarui T, Takada Y, Ruf W. Cross-talk of integrin alpha3beta1 and tissue factor in cell migration. Molec Biol Cell 2004;15:4416–4425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sutherland MR, Raynor CM, Leenknegt H, Wright JF, Pryzdial ELG. Coagulation initiated on herpesviruses. Proc Natl Acad Sci USA 1997;94:13510–13514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Sellers RS, Clifford CB, Treuting PM, Brayton C. Immunological variation between inbred laboratory mouse strains: Points to consider in phenotyping genetically immunomodified mice. Vet Pathol 2012;49:32–43. [DOI] [PubMed] [Google Scholar]
- (27).Chen X, Oppenheim JJ, Howard OMZ. BALB/c mice have more CD4+CD25+ T regulatory cells and show greater susceptibility to suppression of their CD4+CD25− responder T cells than C57BL/6 mice. J Leukoc Biol 2005;78:114–121. [DOI] [PubMed] [Google Scholar]
- (28).Damiano BP, Cheung WM, Santulli RJ, Fung-Leung WP, Ngo K, Ye RD, Darrow AL, Derian CK, De Garavilla L, Andrade-Gordon P. Cardiovascular responses mediated by protease-activated receptor-2 (PAR-2) and thrombin receptor (PAR-1) are distinguished in mice deficient in PAR-2 or PAR-1. J Pharmacol Exp Ther 1999;288:671–678. [PubMed] [Google Scholar]
- (29).Huang MD, Syed R, Stura EA, Stone MJ, Stefanko RS, Ruf W, Edgington TS, Wilson IA. The mechanism of an inhibitory antibody on TF-initiated blood coagulation revealed by the crystal structures of human tissue factor, Fab5G9 and TF.5G9 complex. J Mol Biol 1998;275:873–894. [DOI] [PubMed] [Google Scholar]
- (30).Morrissey JH, Fair DS, Edgington TS. Monoclonal antibody analysis of purified and cell-associated tissue factor. Thromb Res 1988;52:247–261. [DOI] [PubMed] [Google Scholar]
- (31).Ahamed J, Versteeg HH, Kerver M, Chen VM, Mueller BM, Hogg PJ, Ruf W. Disulfide isomerization switches tissue factor from coagulation to cell signaling. Proc Natl Acad Sci USA 2006;103:13932–13937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ruf W, Edgington TS. An anti-tissue factor monoclonal antibody which inhibits the TF-VIIa complex is a potent anticoagulant in plasma. Thromb Haemost 1991;66:529–533. [PubMed] [Google Scholar]
- (33).Bergum PW, Cruikshank A, Maki SL, Kelly CR, Ruf W, Vlasuk GP. Role of zymogen and activated factor X as scaffolds for the inhibition of the blood coagulation factor VIIa-tissue factor complex by recombinant nematode anticoagulant protein c2. J Biol Chem 2001;276:10063–10071. [DOI] [PubMed] [Google Scholar]
- (34).Uusitalo-Jarvinen H, Kurokawa T, Mueller BM, Andrade-Gordon P, Friedlander M, Ruf W. Role of protease activated receptor 1 and 2 signaling in hypoxia-induced angiogenesis. Arterioscler Thromb Vasc Biol 2007;27:1456–1462. [DOI] [PubMed] [Google Scholar]
- (35).Pawlinski R, Pedersen B, Schabbauer G, Tencati M, Holscher T, Boisvert W, Andrade-Gordon P, Frank RD, Mackman N. Role of tissue factor and protease-activated receptors in a mouse model of endotoxemia. Blood 2004;103:1342–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bushi D, Chapman J, Wohl A, Stein ES, Feingold E, Tanne D. Apixaban decreases brain thrombin activity in a male mouse model of acute ischemic stroke. J Neurosci Res 2018;96:1406–1411. [DOI] [PubMed] [Google Scholar]
- (37).Wei H, Shang J, Keohane C, Wang M, Li Q, Ni W, O’Neill K, Chintala M. A novel approach to assess the spontaneous gastrointestinal bleeding risk of antithrombotic agents using Apc(min/+) mice. Thromb Haemost 2014;111:1121–1132. [DOI] [PubMed] [Google Scholar]
- (38).Lubinski J, Wang L, Mastellos D, Sahu A, Lambris JD, Friedman HM. In vivo role of complement-interacting domains of herpes simplex virus type 1 glycoprotein C. J Exp Med 1999;190:1637–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lubinski JM, Jiang M, Hook L, Chang Y, Sarver C, Mastellos D, Lambris JD, Cohen GH, Eisenberg RJ, Friedman HM. Herpes simplex virus type 1 evades the effects of antibody and complement in vivo. J Virol 2002;76:9232–9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Becker KJ. Strain-Related Differences in the Immune Response: Relevance to human stroke. Transl Stroke Res 2016;7:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Engler H, Zawatzky R, Kirchner H, Armerding D. Experimental-infection of inbred mice with herpes-simplex virus: Comparison of interferon-production and natural-killer cell-activity in susceptible and resistant adult mice. Arch Virol 1982;74:239–247. [DOI] [PubMed] [Google Scholar]
- (42).Yao HW, Ling P, Chen SH, Tung YY, Chen SH. Factors affecting herpes simplex virus reactivation from the explanted mouse brain. Virology 2012;433:116–123. [DOI] [PubMed] [Google Scholar]
- (43).Lippe R Deciphering novel host-herpesvirus interactions by virion proteomics. Front Microbiol 2012;3:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Lum KK, Cristea IM. Proteomic approaches to uncovering virus-host protein interactions during the progression of viral infection. Expert Rev Proteomics 2016;13:325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Versteeg HH, Schaffner F, Kerver M, Petersen HH, Ahamed J, Felding-Habermann B, Takada Y, Mueller BM, Ruf W. Inhibition of tissue factor signaling suppresses tumor growth. Blood 2008;111:190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Buddai SK, Toulokhonova L, Bergum PW, Vlasuk GP, Krishnaswamy S. Nematode anticoagulant protein c2 reveals a site on factor Xa that is important for macromolecular substrate binding to human prothrombinase. J Biol Chem 2002;277:26689–26698. [DOI] [PubMed] [Google Scholar]
- (47).Martines RB, Ng DL, Greer PW, Rollin PE, Zaki SR. Tissue and cellular tropism, pathology and pathogenesis of Ebola and Marburg viruses. J Pathol 2015;235:153–174. [DOI] [PubMed] [Google Scholar]
- (48).La Gruta NL, Kedzierska K, Stambas J, Doherty PC. A question of self-preservation: immunopathology in influenza virus infection. Immunol Cell Biol 2007;85:85–92. [DOI] [PubMed] [Google Scholar]
- (49).Clayton KL, Garcia JV, Clements JE, Walker BD. HIV infection of macrophages: Implications for pathogenesis and cure. Pathog Immun 2017;2:179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Dalrymple NA, Mackow ER. Virus interactions with endothelial cell receptors: implications for viral pathogenesis. Curr Opin Virol 2014;7:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Mladinich MC, Schwedes J, Mackow ER. Zika virus persistently infects and Is basolaterally released from primary human brain microvascular endothelial cells. MBio 2017;8:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Revie D, Salahuddin SZ. Role of macrophages and monocytes in hepatitis C virus infections. World J Gastroenterol 2014;20:2777–2784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Geisbert TW, Hensley LE, Jahrling PB, Larsen T, Geisbert JB, Paragas J, Young HA, Fredeking TM, Rote WE, Vlasuk GP. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet 2003;362:1953–1958. [DOI] [PubMed] [Google Scholar]
- (54).Ruf W Emerging roles of tissue factor in viral hemorrhagic fever. Trends Immunol 2004;25:461–464. [DOI] [PubMed] [Google Scholar]
- (55).Zhao P, Metcalf M, Bunnett NW. Biased signaling of protease-activated receptors. Front Endocrinol (Lausanne) 2014;5:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Pryzdial ELG, Lee FMH, Lin BH, Carter RLR, Tegegn TZ, Belletrutti MJ. Blood coagulation dissected. Transfus Apher Sci 2018;DOI: 10.1016/j.transci.2018.07.003. [DOI] [PubMed] [Google Scholar]
- (57).Key NS, Bach RR, Vercellotti GM, Moldow CF. Herpes simplex virus type 1 does not require productive infection to induce tissue factor in human umbilical vein endothelial cells. Lab Invest 1993;68:645–651. [PubMed] [Google Scholar]
- (58).Nhu QM, Shirey K, Teijaro JR, Farber DL, Netzel-Arnett S, Antalis TM, Fasano A, Vogel SN. Novel signaling interactions between proteinase-activated receptor 2 and Toll-like receptors in vitro and in vivo. Mucosal Immunol 2010;3:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Khoufache K, LeBouder F, Morello E, Laurent F, Riffault S, Andrade-Gordon P, Boullier S, Rousset P, Vergnolle N, Riteau B. Protective role for protease-activated receptor-2 against influenza virus pathogenesis via an IFN-gamma-dependent pathway. J Immunol 2009;182:7795–7802. [DOI] [PubMed] [Google Scholar]
- (60).Feld M, Shpacovitch V, Ehrhardt C, Fastrich M, Goerge T, Ludwig S, Steinhoff M. Proteinase-activated receptor-2 agonist activates anti-influenza mechanisms and modulates IFNgamma-induced antiviral pathways in human neutrophils. Biomed Res Int 2013;2013:879080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Antoniak S, Owens AP, Baunacke M, Williams JC, Lee RD, Weithauser A, Sheridan PA, Malz R, Luyendyk JP, Esserman DA, Trejo J, Kirchhofer D, Blaxall BC, Pawlinski R, Beck MA, Rauch U, Mackman N. PAR-1 contributes to the inate immune response during viral infection. J Clin Inv 2013;123:1310–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Khoufache K, Berri F, Nacken W, Vogel AB, Delenne M, Camerer E, Coughlin SR, Carmeliet P, Lina B, Rimmelzwaan GF, Planz O, Ludwig S, Riteau B. PAR1 contributes to influenza A virus pathogenicity in mice. J Clin Invest 2013;123:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]