

Abstract
Endurance exercise has been shown to be a positive regulator of skeletal muscle metabolic function. Changes in mitochondrial dynamics (fusion and fission) have been shown to influence mitochondrial oxidative capacity. We therefore tested whether genetic disruption of mitofusins (Mfns) affected exercise performance in adult skeletal muscle. We generated adult-inducible skeletal muscle-specific Mfn1 (iMS-Mfn1KO), Mfn2 (iMS-Mfn2KO), and Mfn1/2 (iMS-MfnDKO) knockout mice. We assessed exercise capacity by performing a treadmill time to exhaustion stress test before deletion and up to 8 wk after deletion. Analysis of either the iMS-Mfn1KO or the iMS-Mfn2KO did not reveal an effect on exercise capacity. However, analysis of iMS-MfnDKO animals revealed a progressive reduction in exercise performance. We measured individual electron transport chain (ETC) complex activity and observed a reduction in ETC activity in both the subsarcolemmal and intermyofibrillar mitochondrial fractions specifically for NADH dehydrogenase (complex I) and cytochrome-c oxidase (complex IV), which was associated with a decrease in ETC subunit expression for these complexes. We also tested whether voluntary exercise training would prevent the decrease in exercise capacity observed in iMS-MfnDKO animals (n = 10/group). However, after 8 wk of training we did not observe any improvement in exercise capacity or ETC subunit parameters in iMS-MfnDKO animals. These data suggest that the decrease in exercise capacity observed in the iMS-MfnDKO animals is in part the result of impaired ETC subunit expression and ETC complex activity. Taken together, these results provide strong evidence that mitochondrial fusion in adult skeletal muscle is important for exercise performance.
NEW & NOTEWORTHY This study is the first to utilize an adult-inducible skeletal muscle-specific knockout model for Mitofusin (Mfn)1 and Mfn2 to assess exercise capacity. Our findings reveal a progressive decrease in exercise performance with Mfn1 and Mfn2 deletion. The decrease in exercise capacity was accompanied by impaired oxidative phosphorylation specifically for complex I and complex IV. Furthermore, voluntary exercise training was unable to rescue the impairment, suggesting that normal fusion is essential for exercise-induced mitochondrial adaptations.
Keywords: electron transport chain, exercise, mitochondria, mitofusin, skeletal muscle
INTRODUCTION
Skeletal muscle mitochondria are extremely plastic organelles capable of responding to both physiological and pathological stimuli (29, 62). These stimuli influence the content (biogenesis), interconnectivity (fusion/fission), and turnover (mitophagy) of mitochondria, ultimately affecting their metabolic function and oxidative capacity (29). Mitochondrial dysfunction in skeletal muscle has been suggested as being a major contributor to the development of metabolic diseases, such as type 2 diabetes and obesity (1, 29, 48, 62). In contrast, endurance exercise has been shown to increase mitochondrial content and/or functionality by increasing mitochondrial biogenesis, improving mitochondrial efficiency (phosphate-to-oxygen ratio), modulating fusion/fission, and mitophagy (6, 18, 61). Furthermore, endurance exercise has been proven to be the best treatment and prevention of metabolic disease (25, 26). However, the individual contribution of these mitochondrial changes to improving exercise performance and oxidative capacity is not fully delineated. Therefore, understanding the molecular changes that contribute to the multiple facets of mitochondrial remodeling in skeletal muscle is important to fully understanding the benefit of exercise.
Mitochondrial dynamics (fusion/fission) has emerged in recent years as an important cellular process involved in maintaining mitochondrial quality control and oxidative capacity (17, 27, 39). Mitochondria undergo fusion and fission to exchange their mitochondrial DNA and oxidative enzymes, as well as remove dysfunctional mitochondria from the network (58). The process of mitochondrial fusion utilizes GTPase transmembrane proteins mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) located on the outer mitochondrial membrane to fuse the outer membrane of the mitochondria. The optic protein atrophy 1 (Opa1) protein located in the inner mitochondrial membrane is responsible for fusion of the inner membrane of the mitochondria (9). On the other hand, mitochondrial fission utilizes dynamin-like protein 1 (Drp1), as well as membrane-bound adaptor proteins including fission 1 (Fis1), mitochondrial fission factor (Mff), and mitochondrial division (MiD) proteins, to cleave the outer mitochondrial membrane (9, 31).
Recent studies have begun to highlight the important contribution of mitochondrial dynamics in skeletal muscle and the role they play in the exercise response. In both rodents and humans, endurance exercise has been shown to increase the expression of genes involved in fusion and fission (8, 20, 28, 33). Furthermore, in vivo imaging of skeletal muscle mitochondria after bouts of exercise reveals that they exist in a more fused state (40). Skeletal muscle overexpression of the fission regulator Drp1 resulted in a decrease in exercise performance (57). Constitutive deletion of Mfn2 in skeletal muscle did not affect exercise performance at 6 mo of age but exhibited a decrease in time to exhaustion after Mfn2 had been deleted for 22 mo (53). Congenital deletion of both Mfn1 and Mfn2 in skeletal muscle has been previously reported (12). However, these animals are severely developmentally runted, and the limited assessment of exercise performance suggested signs of exercise intolerance. Moreover, given that both Mfns were deleted early during development, interpreting the true consequence of such an early deletion for mature skeletal muscle becomes difficult. Furthermore, transgenic mice overexpressing Opa1 exhibit a mild increase in exercise performance, suggesting that increasing mitochondrial fusion is beneficial to exercise performance (13). In contrast and somewhat contradictory, mice heterozygous for Opa1 deletion or muscle-specific deletion of Opa1 exhibited increases in exercise performance, as these models have a negative effect on mitochondrial fusion (7, 13, 43). These apparent contradictory observations reveal a gap in our understanding of the factors that contribute to mitochondrial fusion in skeletal muscle. However, the consequence of impaired mitochondrial fusion for exercise performance in adult skeletal muscle is unknown.
Here we sought to investigate the contribution of Mfn1 and Mfn2 in adult skeletal muscle exercise performance. To address this question, we generated adult-inducible skeletal muscle-specific Mfn1 and/or Mfn2 knockout animals. Our data suggest that removal of both Mfn1 and Mfn2 in skeletal muscle resulted in a significant reduction in exercise performance. The decrease in exercise performance was due to reduction in mitochondrial electron transport chain (ETC) activity in NADH dehydrogenase (complex I) and cytochrome-c oxidase (complex IV). Moreover, prophylactic exercise training was unable to prevent the reduction in exercise performance with Mfn1 and Mfn2 deletion. Taken together, these data highlight the importance of Mfn1 and Mfn2 in skeletal muscle and the important role they play in exercise performance and normal mitochondrial function.
METHODS
Animal studies.
All animal experiments were performed according to procedures approved by the University of Alabama at Birmingham (UAB) Institutional Animal Care and Use Committee. To obtain inducible skeletal muscle-specific Mfn1 and/or Mfn2 double-knockout animals, Mfn1 floxed and/or Mfn2 double-floxed animals (11) were intercrossed with the tamoxifen-inducible Cre recombinase under the control of the human skeletal actin promoter (HSA-MerCreMer) (36), hereinafter referred to as iMS-Mfn1KO, iMS-Mfn2KO, and iMS-MfnDKO. Deletion of the mitofusins was obtained by administering tamoxifen (Sigma catalog no. T5648) by intraperitoneal injection (2 mg/day for 5 consecutive days) as previously described (4, 36). Unless otherwise stated, 20- to 22 wk-old female mice on a C57BL/6 background were used for all experiments.
Real-time qPCR analysis.
Total mRNA was isolated from skeletal muscle beds with TRIzol (Invitrogen) followed by RNA Cleanup (Qiagen). Samples were subjected to reverse transcription (Thermo Fisher) followed by real-time qPCR with SYBRgreen (Bio-Rad). Relative expression levels were determined by the comparative threshold method, normalizing to the following housekeeping genes: hypoxanthine phosphoribosyltransferase (HPRT), TATA-box binding protein (TBP), and ribosomal protein large P0 (36B4).
Western blotting analysis.
Total protein was isolated from indicated skeletal muscle beds with modified radioimmunoprecipitation assay buffer supplemented with protease and phosphatase inhibitors (Thermo Fisher). Protein extracts (~30 µg) were subjected to electrophoresis on 4–20% TGX gels (Bio-Rad) and transferred to nitrocellulose membranes (Bio-Rad) for Western blot analysis. The following antibodies were used for the present study: mitofusin 1 (Abcam catalog no. 126575), mitofusin 2 (Abcam catalog no. ab56889), Total OXPHOS Rodent Antibody Cocktail (Abcam catalog no. ab110413), and α-tubulin (Developmental Studies Hybridoma Bank). Bands were detected by chemiluminescence and imaged on a digital imager. Densitometry analysis was performed with ImageJ software by normalizing the band of interest to tubulin or Ponceau S staining.
Transmission electron micrograph analysis.
Skeletal muscle from the soleus was dissected, trimmed (1 mm × 2 mm), and placed into fixative. Samples were processed for transmission electron micrographs as previously described (4). Images were acquired from random fields with a digital camera connected to a FEI-Tecnai T12 Spirit 20-120kv. Individual mitochondrial areas of subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondria were quantified computationally with ImageJ software. All quantifications were performed in a blinded manner.
Mitochondrial DNA copy and mitochondrial DNA damage analysis.
Nuclear (nucDNA) and mitochondrial (mtDNA) genomic DNA were isolated with DNeasy (Qiagen) according to the manufacturer’s instructions. mtDNA-to-nucDNA ratio was determined by real-time qPCR using SYBRgreen with primers specific to the mitochondrial genome and to the nuclear genome (45). mtDNA damage was determined by performing PCR for a long (16.2 kb) and a short (0.22 kb) amplicon against the mtDNA genome as previously described (3). mtDNA lesion frequency was calculated as previously described (3, 47).
Electron transport chain complex activity.
Skeletal muscle was isolated and snap frozen in liquid nitrogen until ready for measurement of complex activity. Mitochondrial fractions from either the SS or IMF fraction were isolated as previously described (32, 46). Individual ETC complex activity was measured in the SS and IMF mitochondrial fractions by absorbance spectrophotometry as previously described (4). Briefly, complex I activity was measured with 2,6-dichloroindophenol (DCIP) as the terminal electron acceptor with the oxidation of NADH-reducing artificial substrate Coenzyme Q10. Complex II activity was measured by reduction rates of DCIP with succinate. Complex III activity was measured by reduction rates of cytochrome-c oxidase. Complex IV activity was measured by the oxidation rates of cytochrome-c oxidase. Complex V activity was measured by monitoring the oxidation of NADH.
Treadmill and voluntary wheel exercise.
Treadmill exhaustive stress tests were performed as previously described (4). Briefly, exercise stress tests were performed at 10 m/min with a 10% grade for 5 min and then subsequently increased by 2 m/min every 2 min until mice reached exhaustion or a maximum speed of 36 m/min. Time to exhaustion was recorded, and total distance run and work performed were calculated as previously described (24). The experimenter was blinded to the genotype of the animals during the course of the study. For voluntary wheel exercise, animals were housed individually with unfettered access to running wheels connected to an electronic monitoring system with total distance recorded. Wheel cages were locked 12–14 h before tissue collection to prevent acute exercise effects. All mice were housed individually and had access to food and water ad libitum.
Kinematic gait analysis.
Gait analysis was performed with a CatWalk system (Noldus Information Technology) as previously described (23, 35). Paw prints were collected on a computer running CatWalk acquisition software (version 9.1). Trials on the catwalk in which animals stopped or turned around were excluded from analysis. Data are presented as the average of three uninterrupted trials down the catwalk.
Statistical analysis.
Data in bar graphs are represented as means ± standard error (SE) where indicated. Data in whisker plots are represented as median, minimum value, maximum value, lower quartile, and upper quartile. P values were determined by two-tailed Student’s t-test. P values of <0.05 were considered statistically significant. For multiple pairwise comparisons, two-way ANOVAs were performed, followed by a Tukey adjustment.
RESULTS
Exercise performance in adult-inducible muscle-specific Mfn knockouts.
Here we sought to determine the role of Mfn1 and Mfn2 in adult skeletal muscle on exercise performance. We first generated an adult skeletal muscle-specific Mfn1-knockout animal model (iMS-Mfn1KO). We first confirmed the effect on Mfn1 expression 8 wk after deletion. We observed a 75–85% reduction in the mRNA expression of Mfn1, with no effect on Mfn2 expression (Fig. 1A). We also observed similar reductions (90–95%) at the protein level (Fig. 1, B and C). We next sought to determine whether loss of Mfn1 in adult skeletal muscle affected exercise performance. Twelve- to fourteen-week-old iMS-Mfn1KO animals were subjected to a progressive endurance exercise stress test. We measured exercise capacity before deletion of Mfn1 and then again 8 wk after deletion. The iMS-Mfn1KO animals did not exhibit any differences in time to exhaustion before or 8 wk after deletion (Fig. 1D). We observed similar levels of calculated total distance run and work performed (Fig. 1, E and F). These data suggest that removal of Mfn1 in skeletal muscle was not sufficient to negatively influence exercise performance.
Fig. 1.

Effect of adult deletion of either Mitofusin (Mfn)1 or Mfn2 on exercise performance. A–F: Mfn1 and Mfn2 mRNA expression in different skeletal muscle beds (A); Mfn1 and Mfn2 protein expression (B); densitometry quantification of Mfn1 and Mfn2 protein expression normalized to tubulin and relative to control in gastrocnemius (GAS) muscle (C; n = 3 per group), baseline time to exhaustion (D); total distance run (E); and calculated total work (F; n = 7 per group) in adult-inducible skeletal muscle-specific Mfn1 knockout mice (iMS-Mfn1KO) and control littermates. G–L: Mfn1 and Mfn2 mRNA expression (G); Mfn1 and Mfn2 protein expression (H); densitometry quantification of Mfn1 and Mfn2 protein expression normalized to tubulin (Tuba) and relative to control in GAS muscle (I); baseline time to exhaustion (J); total distance run (K); and calculated total work (K) in adult-inducible skeletal muscle-specific Mfn2 knockout mice (iMS-Mfn2KO) and control littermates. Data are means ± SE. *P < 0.05 compared with control as determined by unpaired t-test.
Mfn1 and Mfn2 are both expressed in skeletal muscle; however, Mfn2 is the predominantly expressed mitofusin (52). We therefore determined the consequence of deleting Mfn2 in adult skeletal muscle (iMS-Mfn2KO) in our system. Mfn2 deletion was obtained as described above for Mfn1. We observed a significant reduction of Mfn2 mRNA expression (80–90%) (Fig. 1G). The decrease in Mfn2 mRNA expression was associated with a concomitant decrease (90–95%) in protein expression (Fig. 1, H and I). We did not observe any change in expression at either the mRNA or protein level for Mfn1 in the iMS-Mfn2 KO animals (Fig. 1, G–I). We next determined whether deletion of Mfn2 would have a negative effect on exercise performance. The iMS-Mfn2KO animals were subjected to an exercise stress test before and 8 wk after deletion. We did not observe any differences in time to exhaustion, total distance run, and work performed (Fig. 1, J–L). These observations were similar to what was previously reported by another group studying Mfn2 in adult skeletal muscle (53). Taken together, these observations suggest that single deletion of either Mfn1 or Mfn2 in skeletal muscle was not sufficient to affect exercise performance.
Given the significant homology between the two mitofusins and possible functional redundancy, single deletions of either family member might not be sufficient (10, 51). We therefore generated an adult-induced skeletal muscle deleted for both mitofusins (iMS-MfnDKO), as described above. We observed an 80–90% reduction in the mRNA of Mfn1 and Mfn2 in all skeletal muscle beds analyzed (Fig. 2A). We also observed a similar reduction at the protein level in the iMS-MfnDKO animals (Fig. 2, B and C). We next sought to determine whether adult skeletal muscle deletion of Mfn1 and Mfn2 had any effect on exercise capacity. We performed weekly exhaustive exercise stress test before deletion, on the last day of tamoxifen administration (postdeletion), and once a week afterward for 8 wk. We observed a progressive decrease in exercise performance in the iMS-MfnDKO animals beginning at ∼2–3 wk after deletion (Fig. 2, D and E). The decrease in exercise performance plateaued at ~70% reduction by 7–8 wk (Fig. 2, D and E). The decrease in time to exhaustion was reflected in a decrease in calculated total distance run and the amount of work performed (Fig. 2, F and G).
Fig. 2.

Effect of adult deletion of both Mitofusin (Mfn)1 and Mfn2 on exercise performance: Mfn1 and Mfn2 mRNA expression in various skeletal muscle beds: soleus (SOL), extensor digitorum longus (EDL), tibialis anterior (TA), triceps (TRI), quadriceps (QUAD), gastrocnemius (GAS), and diaphragm (DIA) (A); Mfn1 and Mfn2 protein expression (B); densitometry quantification of Mfn1 and Mfn2 protein expression normalized to tubulin and relative to control in GAS muscle (C; n = 3 per group); longitudinal exercise stress test (D); time to exhaustion (E); total distance run (F); and calculated total work in adult-inducible skeletal muscle-specific Mfn1 and Mfn2 double knockout mice (iMS-MfnDKO) and control littermates (G; n = 7 per group) before deletion and 8 wk after deletion. Data are means ± SE. *P < 0.05; **P < 0.01 compared with control as determined by unpaired t-test.
Mutations in Mfn2 are associated with the development of the type 2A form of the neurodegenerative disease Charcot-Marie-Tooth disease (CMT), which is known to affect skeletal muscle function (63). We next determined whether this decrease in exercise performance was due to movement impairment in the iMS-MfnDKO animals. We performed gait kinematic analysis on iMS-MfnDKO after 8 wk of deletion of Mfn1 and Mfn2 (Fig. 3). We observed only a mild impairment in hindlimb stride length (Fig. 3F); notably, a similar defect was observed in mice with hemizygous Mfn2 CMT2A-causing mutations (5). Taken together, these observations suggest that both Mfn1 and Mfn2 in skeletal muscle are required for normal exercise performance.
Fig. 3.

Mild impairment to gait kinematics with muscle deletion of Mitofusin (Mfn)1 and Mfn2: paw print intensity (A); stand time (B); swing time (C); step cycle (D); swing speed (E); and stride length (F) in adult-inducible skeletal muscle-specific Mfn1 and Mfn2 double knockout mice (iMS-MfnDKO; n = 5) and control littermates (n = 5) 8 wk after deletion. Data are means ± SE. **P < 0.01 compared with control as determined by unpaired t-test.
Disruption of mitochondrial network and integrity.
We next sought to determine what was responsible for the decrease in exercise performance in the iMS-MfnDKO animals. Skeletal muscle mitochondria exist as an interconnected network (15) but can be segregated into two different compartments based on their subcellular location, SS and IMF, with distinct biochemical properties (14). We first determined whether deletion of both mitofusins disrupted the mitochondrial network between the SS and IMF mitochondria. Given the large concentration of mitochondria found in the highly oxidative soleus muscle, we generated transmission electron micrographs from this muscle bed. We observed a significant decrease in the size of the individual mitochondria in the SS fraction (Fig. 4, A and B) and a significant increase in the IMF fraction (Fig. 4, C and D). We next determined whether disruption of the mitochondrial network affected mtDNA integrity. We performed a mtDNA damage assay and calculated the number of lesions per mitochondrial genome (47). We observed a significant increase in mtDNA lesions in the iMS-MfnDKO animals (Fig. 4E). We also assessed mtDNA-to-nucDNA ratio as a surrogate of mitochondrial mass and content. We observed an ~50% reduction in mtDNA-to-nucDNA ratio (Fig. 4F), suggesting a decrease in mitochondrial content in the iMS-MfnDKO animals. These observations are similar to what was previously reported in congenital skeletal muscle deletion for Mfn1 and Mfn2 (12), albeit to a lesser extent. We also measured the mRNA expression of mitochondrial-encoded subunits of the ETC in skeletal muscle deleted for both Mfn1 and Mfn2. We observed a significant reduction (75–90%) in expression of all of the measured mitochondrial-encoded subunits (Fig. 4G). We did not observe any significant difference in mtDNA damage lesion formation or mtDNA-to-nucDNA ratio in either of the single Mfn1 or Mfn2 knockouts (Fig. 4, H–K). Taken together, these observations confirm a disruption of the mitochondrial network with deletion of mitofusins in adult skeletal muscle. Furthermore, these observations confirm a dysregulation of mtDNA stability, supporting the observations made with congenital deletion of both Mfn1 and Mfn2 (12).
Fig. 4.

Mitochondrial morphology with deletion of mitofusins (Mfns) in skeletal muscle. A–F: representative transmission electron micrograph (TEM) of subsarcolemmal (SS) mitochondria (A); quantification of individual mitochondrial area in the SS fraction (B); representative TEM of intermyofibrillar (IMF) mitochondria (C); quantification of individual mitochondrial area in the IMF fraction (D); mitochondrial DNA (mtDNA) damage represented as lesion frequency (E); mtDNA-to-nuclear DNA (nucDNA) ratio (F); and mRNA expression of mitochondrial-encoded electron transport chain subunits NADH dehydrogenase 5 (ND5), cytochrome b (CYTb), and ATP synthase subunit a (ATP6) in soleus (G) of adult-inducible skeletal muscle-specific Mfn1 and Mfn2 double knockout mice (iMS-MfnDKO) and control littermates. H and I: mitochondrial DNA damage represented as lesion frequency (H) and mtDNA-to-nucDNA ratio (I) of adult-inducible skeletal muscle-specific Mfn1 knockout mice (iMS-Mfn1KO) and control littermates. J and K: mtDNA damage represented as lesion frequency (J) and mtDNA-to-nucDNA ratio (I) of adult-inducible skeletal muscle-specific Mfn2 knockout mice (iMS-Mfn2KO) and control littermates. Data in B and D are presented as a whisker plot with the minimum and maximum value, median, lower and upper quartile based on n = 4; N = 500–1,000 mitochondria. Data in E and F are means ± SE; n = 3–7 per group; *P < 0.05; **P < 0.01 compared with control as determined by unpaired t-test.
We next sought to determine what effect this would have on ETC activity within the two mitochondrial pools of the iMS-MfnDKO animals. We observed a significant reduction (~60–75%) in complex I and complex IV activity in both the IMF and SS fractions of the iMS-MfnDKO animals compared with control animals (Fig. 5, A and D). However, the IMF fraction was significantly affected for complex II and complex V (~60–70% reduction) in the iMS-MfnDKO animals (Fig. 5, B and E). Notably, complex III and citrate synthase activity was not affected in either the SS or the IMF fraction in the iMS-MfnDKO animals (Fig. 5, C and F). These data suggest that disruption of the interconnectivity between SS and IMF mitochondrial pools has differential effects on ETC enzymatic activity with disruption of Mfn1 and Mfn2 in skeletal muscle.
Fig. 5.

Electron transport chain complex activity with skeletal muscle deletion of Mitofusin (Mfn): NADH dehydrogenase (complex I) activity (A); succinate dehydrogenase (complex II) activity (B); cytochrome c reductase (complex III) activity (C); cytochrome-c oxidase (complex IV) activity (D); ATP synthase (complex V) activity (E); and citrate synthase (CS) activity (F) in the subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondrial fractions from gastrocnemius muscle of adult-inducible skeletal muscle-specific Mfn1 and Mfn2 double knockout mice (iMS-MfnDKO; n = 5) and control littermates (n = 5). Data are means ± SE. *P < 0.05; **P < 0.01 compared with control as determined by unpaired t-test.
Impaired mitochondrial function with Mfn1 and 2 disruption.
We next sought to determine what molecular changes were contributing to the decrease in exercise performance observed in the iMS-MfnDKO animals. Given the observed reduction in mtDNA-to-nucDNA ratio, which is used as an indicator of mitochondrial biogenesis, we asked whether this process was disrupted. We first measured the expression of key transcriptional regulators involved in mitochondrial biogenesis and oxidative phosphorylation (OXPHOS). We did not observe any significant changes in mRNA expression of the key transcriptional coactivators of the peroxisome proliferator-activated receptor γ coactivator 1 family members [PGC-1α, PGC-1β, and PGC-1-related coactivator (PRC)] or their downstream transcription factors estrogen-related receptor α (Errα), nuclear respiratory factor 1 (Nrf-1), and mitochondrial transcription factor A (Tfam) (Fig. 6A). We also measured the expression of some downstream components of these transcription factors that are involved in the ETC, where we observed a 40% reduction in cytochrome c, somatic (Cycs). However this decrease in Cycs expression may be due to its role in other cellular processes such as apoptosis (30).
Fig. 6.

Loss of complex I and complex IV subunit expression with Mitofusin (Mfn) deletion in skeletal muscle. A–F: gastrocnemius (GAS) muscle of adult-inducible skeletal muscle-specific Mfn1 and Mfn2 double knockout mice (iMS-MfnDKO; n = 5) and control (n = 5) littermates. A: mRNA expression of transcriptional regulators peroxisome proliferator-activated receptor γ coactivator 1 [PGC-1α, PGC-1β, and PGC-1-related coactivator (PRC)], estrogen-related receptor alpha (Errα), nuclear respiratory factor 1 (Nrf1), and mitochondrial transcription factor A (Tfam). B: mRNA expression of nuclear-encoded electron transport chain (ETC) subunits NADH dehydrogenase 1 beta subunit 5 (ndufb5), cytochrome c, somatic (cycs), cytochrome c oxidase subunit 5B (cox5b), and ATP synthase subunit O (atp5o). C: protein expression of ETC subunits ATP synthase F1 subunit alpha (Atp5a), Ubiquinol-cytochrome c reductase protein 2 (Uqcrc2), mitochondrial-encoded cytochrome-c oxidase 1 (MtCO1), Succinate dehydrogenase complex iron sulfur subunit B (Sdhb), and NADH:Ubiquinone oxidoreductase subunit B8 (Ndufb8). D: densitometry quantification of ETC subunit protein expression normalized to tubulin (Fig. 2B) and relative to control. E: mRNA expression of ETC subunits represented in C. F: mRNA expression of mitochondrial-encoded ETC subunits NADH dehydrogenase 2 and 5 (ND2 and ND5), cytochrome b (CYTb), cytochrome oxidase 2 (COX2), and ATP synthase subunit membrane subunit 6 (ATP6). G–J: GAS muscle of adult-inducible skeletal muscle-specific Mfn2 knockout mice (iMS-Mfn2KO; n = 3) and control (n = 3) littermates. G: protein expression of ETC subunits. H: densitometry quantification of ETC subunit protein expression normalized to tubulin (Fig. 1B) and relative to control. I: protein expression of ETC subunits. J: densitometry quantification of ETC subunit protein expression normalized to tubulin (Fig. 1H) and relative to control. Data are presented as means ± SE. *P < 0.05 compared with control as determined by unpaired t-test.
We next measured the protein levels of some of the ETC subunits involved in OXPHOS. We observed an 80–90% reduction in the expression of NADH:Ubiquinone Oxidoreductase Subunit B8 (Ndufb8-complex I) and the mitochondrial-encoded Cytochrome-c oxidase 1 (MtCO1-complex IV) (Fig. 6, C and D). We next measured the transcript level of Ndufb8 and MtCO1 to determine whether the decrease in expression was at either the transcriptional or translational level. Surprisingly, the mRNA expression of the nuclear-encoded Ndufb8 was not affected whereas mtCO1 was significantly reduced (80%) in the iMS-MfnDKO animals (Fig. 6E). Analysis of other mitochondrial-encoded OXPHOS subunits also revealed 75–90% reduction in mRNA expression (Fig. 6F). We did not observe any change in protein expression of any of the measured ETC subunits in either the iMS-Mfn1 KO or iMS-Mfn2 KO animals (Fig. 6, G–J). These data suggest that the decrease in Ndufb8 protein expression was not due to changes in mRNA expression but at the translational level. Furthermore, the decrease in mtCO1 protein was at least in part due to a significant reduction in mRNA expression in the iMS-MfnDKO animals.
Exercise performance in skeletal muscle Mfn double knockouts.
Exercise has been proven to be a powerful intervention in the treatment and reversal of mitochondrial dysfunction (18, 25, 32, 49). We next determined whether exercise training could prevent the mitochondrial dysfunction observed in iMS-MfnDKO animals. We subjected the iMS-MfnDKO animals and control littermates to an exercise exhaustive stress test before induction of deletion; animals were then randomly placed in cages with free running wheels on the last day of the tamoxifen injections. The animals were subjected to exhaustive exercise stress every 2 wk up to 8 wk after deletion. In response to voluntary training the control animals exhibited a time-dependent increase, plateauing at approximately twofold increase in time to exhaustion (Fig. 7, A and B). In contrast, the iMS-MfnDKO animals showed a mild benefit of voluntary training around 2 wk of training (Fig. 7A). However, this training benefit diminished thereafter, with a precipitous decline in time to exhaustion, achieving levels similar to sedentary iMS-MfnDKO animals (Fig. 7, A and B). The iMS-MfnDKO animals averaged ~10 rpm on the voluntary wheels compared with 20 rpm in the control animals. This suggests that the iMS-MfnDKO animals were able to run on the voluntary wheels, albeit to a lesser extent than the control animals, despite their poor aerobic capacity (Fig. 2), D and E.
Fig. 7.

Decreased exercise performance with training in Mitofusin (Mfn) skeletal muscle-deficient animals. A–D: 8-wk longitudinal exercise stress test (A), time to exhaustion (B), total distance run (C), and calculated total work performed (D) in adult-inducible skeletal muscle-specific Mfn1 and Mfn2 double knockout mice (iMS-MfnDKO) and control littermates. Data are presented as means ± SE; n = 10 per group; *P < 0.05 compared with sedentary control, #P < 0.05 compared with exercised control as determined by 2-way ANOVA with pairwise comparisons (Tukey adjustment).
Endurance training has been shown to induce metabolic adaptations in skeletal muscle including increasing the expression of OXPHOS genes (18, 49). Endurance exercise has also been reported to alter the metabolic profiles of the SS and IMF mitochondrial fractions (14). We therefore determined the effect of 8 wk of voluntary training on mitochondrial parameters of OXPHOS expression and activity. We observed an approximately two- to fourfold increase in mRNA expression of ETC subunits in control animals in response to exercise; this increase was blunted in the exercised iMS-MfnDKO animals (Fig. 8A). We next measured the enzymatic activity of complex I and complex IV in both the SS and IMF fractions in control and iMS-MfnDKO animals. In response to exercise, the IMF fraction exhibited an approximately twofold increase in both complex I and complex IV activity in the control animals; this increase was absent in the iMS-MfnDKO animals (Fig. 8, B and C). These data suggest that voluntary exercise training is unable to circumvent skeletal muscle Mfn1 and 2 deficiency and they are both required for normal exercise performance. Taken together, these observations highlight the importance of mitochondrial fusion in skeletal muscle and exercise performance.
Fig. 8.
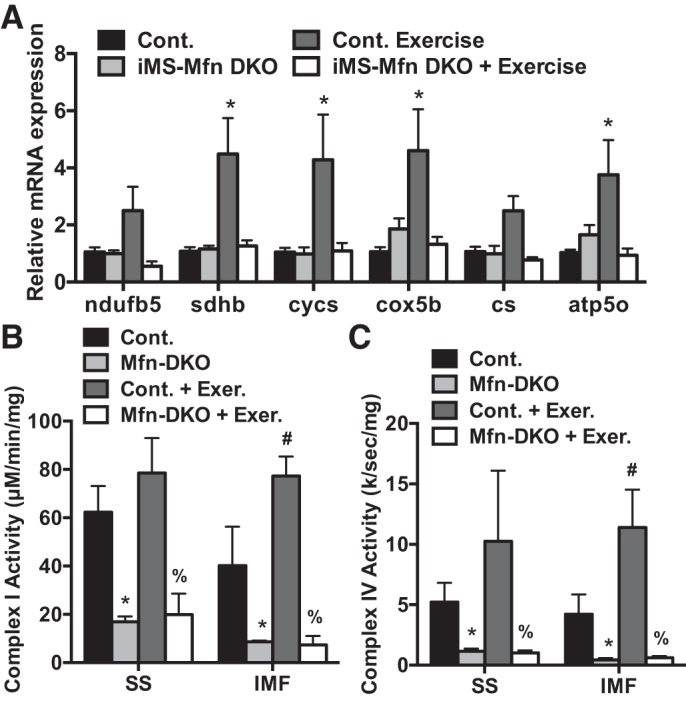
Impaired mitochondrial oxidative phosphorylation response to exercise training with Mitofusin (Mfn) deletion. A–C: mRNA expression of nuclear-encoded electron transport chain (ETC) subunits NADH dehydrogenase 1 beta subunit 5 (ndufb5), succinate dehydrogenase complex iron sulfur subunit B (sdhb), cytochrome c, somatic (cycs), cytochrome-c oxidase subunit 5B (cox5b), citrate synthase (cs), and ATP synthase subunit O (atp5o) (A) and NADH dehydrogenase (complex I) (B) and cytochrome-c oxidase (complex IV) (C) activity in the subsarcolemmal (SS) and intermyofibrillar (IMF) mitochondrial fractions from gastrocnemius muscle of adult-inducible skeletal muscle-specific Mfn1 and Mfn2 double knockout mice (iMS-MfnDKO; n = 5) and control littermates (n = 4). Data are presented as means ± SE. *P < 0.05 compared with sedentary control; #P < 0.05 compared with sedentary iMS-MfnDKO; %P < 0.05 compared with exercised control. P values were determined by 2-way ANOVA with pairwise comparisons (Tukey adjustment).
DISCUSSION
Endurance exercise has been shown to induce a variety of adaptations in skeletal muscle to keep up with the increase in energy demands (19). The remodeling that occurs in skeletal muscle includes, but is not limited to, changes in fiber type composition, vascular density, and mitochondrial adaptations (19, 49). Much emphasis has been placed on changes in mitochondrial parameters that increase oxidative capacity and exercise performance. These changes in skeletal muscle mitochondria include increased mitochondrial biogenesis, efficiency, dynamics, and mitophagy (6, 18, 61). Mitochondrial biogenesis, the process of increasing mitochondrial mass through de novo synthesis, has received the most attention, as this increases the total amount of mitochondria available. However, recent studies have questioned the extent to which mitochondrial biogenesis is absolutely required for increasing exercise performance.
The transcriptional coactivator PGC-1α has been shown to be a potent inducer of mitochondrial biogenesis in skeletal muscle (59). In addition, PGC-1α has been shown to be induced in response to endurance-based exercise in both humans and rodents (2, 44, 50). However, we have recently reported that simultaneous deletion of both PGC-1α and -1β in skeletal muscle did not affect the ability to respond positively to exercise training (4). Furthermore, the ability to respond positively to training occurred despite the lack of increases in mitochondrial biogenesis with voluntary training. Other studies have shown in humans that other things such as increased oxygen uptake with training enhance oxidative capacity independent of mitochondrial biogenesis (60). These observations suggest that other mitochondrial adaptations are more important for exercise performance and training than solely increases in mitochondrial biogenesis.
In this study, we sought to determine the contribution of mitochondrial dynamics, specifically fusion in mitochondria in skeletal muscle in response to exercise. To this end, we generated adult-inducible skeletal muscle-specific Mfn1 and/or Mfn2 knockouts and determined their contribution to exercise performance in skeletal muscle. We have concluded that 1) single deletion of either Mfn1 or Mfn2 in skeletal muscle is not sufficient to impair exercise performance; 2) simultaneous deletion of both Mfn1 and Mfn2 results in decreased exercise capacity; 3) the decrease in exercise performance is due in part to impaired mitochondrial function, specifically complex I and complex IV; and 4) voluntary exercise training is not sufficient to improve the deficit in exercise performance with mitofusin deficiency, suggesting that mitochondrial fusion is essential to exercise-induced training adaptation in skeletal muscle.
Our data reveal for the first time that induced adult deletion of either Mfn1 or Mfn2 in skeletal muscle was not sufficient to affect exercise performance (Fig. 1). Mfn1 and Mfn2 are both expressed in skeletal muscle, and therefore removal of either family member is not sufficient to reveal a phenotype (52). Furthermore, studies in cardiac muscle have arrived at similar conclusions regarding the need to remove both mitofusins to assess the role of mitochondrial fusion (42, 54, 55). The observation that removal of both Mfn1 and Mfn2 in adult skeletal muscle resulted in a profound exercise performance phenotype in as little as 8 wk reveals the importance of mitochondrial fusion in adult skeletal muscle. Others have reported skeletal muscle Mfn double knockouts that also showed a decrease in exercise performance (12, 40). However, as these were either congenital deletions or deletions induced in satellite cells, the effects on exercise performance could be confounded by molecular compensations that occurred in development.
In the present study, we also provide some insight into the functional half-life of the mitochondrial network in skeletal muscle exercise performance. In our iMS-MfnDKO animals we did not observe any significant decrease in exercise performance until 2 wk after deletion (Fig. 2D). This observation suggests that the functional half-life of the mitochondrial network in skeletal muscle is ∼2 wk, which is in line with what has been proposed in other tissues including cardiac muscle (37). In our system, by impairing outer mitochondrial fusion the balance would be shifted toward increased fission and presumably increased mitophagy (41). However, although we did not explicitly test this in the present study, further studies will be needed to fully elucidate the contribution.
Two recent studies have assessed the consequence of disruption of mitochondrial fusion in adult skeletal muscle by deleting the inner mitochondrial fusogen Opa1 (43, 56); however, only one study measured exercise performance. Pereira et al. reported that Opa1 deletion for 16 wk did not negatively affect exercise performance and after 36 wk exhibited an increase in exercise performance (43). Notably, this increase in exercise performance was similar to what was reported in mice hemizygous for deletion of Opa1 (7). These observations are in contrast to what we observed in the iMS-MfnDKO animals in both the effect on exercise performance and the time to phenotype development. Furthermore, the extent of mitochondrial function was different between these two models and the iMS-MfnDKO animals reported here. The work by Tezze et al. revealed a mild decrease in the expression of ETC complex I subunits and enzymatic activity; none of the remaining complexes appeared to be affected (56). In contrast, Pereira et. al. did not report any decreases in ETC subunit expression but an overall decrease in ATP production, with no effect on exercise performance (43). It is also worth mentioning that transgenic overexpression of Opa1, which would be pro-mitochondrial fusion, also exhibits a mild increase in exercise performance (13). These observations suggest that disruption of outer mitochondrial fusion has a more profound effect on exercise performance than simply disrupting inner mitochondrial fusion.
The observation that disruption of both Mfn1 and Mfn2 results in a decrease in complex I and complex IV activity linked to exercise performance is interesting, as impairments in both complex I and complex IV are connected to exercise intolerance and decreased exercise performance (16). Furthermore, complex I and complex IV, although separate enzymatic complexes, actually exist as a supercomplex with complex III in intact mitochondria (38). Disruption of both Mfn1 and Mfn2 did not affect the mRNA expression of the nuclear-encoded subunit for Ndufb8, but its expression at the protein level was significantly decreased (Fig. 6). These observations suggest that changes at the posttranslational level are contributing to the decrease in expression of Ndufb8. However, we cannot exclude the possibility of the destabilized mtDNA integrity and the overall decrease in the ETC subunits expressed from the mitochondrial genome as being the major contributor. Furthermore, the observation that voluntary exercise training in the iMS-MfnDKO animals failed to prevent the negative effect of adult loss of mitofusins (Fig. 7) suggests that normal mitochondrial fusion is a critical component of exercise-induced mitochondrial adaptations. Notably, we did not observe any changes in ETC mRNA expression or complex I and complex IV activity in response to exercise in the iMS-MfnDKO animals (Fig. 8). One possible explanation for this blunted response to voluntary training is that the animals were unable to pass the threshold needed for increasing oxidative capacity (21).
The contribution of mitochondrial dynamics in skeletal muscle to exercise training is largely unknown. However, the present study sought to shed light on the role of mitochondrial fusion in skeletal muscle. Studies in both humans and rodents have revealed that exercise training increased the expression of mitochondrial fusion genes in skeletal muscle (8, 33). Furthermore, skeletal muscle inactivity has been shown to have a negative effect on mitochondrial fusion. Chronic disuse of skeletal muscle results in an increase in fragmented mitochondria and a decrease in mitochondrial fusion genes (28). Overall, fused mitochondria are more efficient in producing energy, whereas fragmented mitochondria are less energy efficient (34). These studies would suggest that exercise training improves skeletal muscle oxidative capacity by altering mitochondrial dynamics. However, we cannot exclude the possibility that mitochondrial dynamics play a role in other adaptations important for exercise performance such as reactive oxygen species production and uncoupled proton leak (22, 61).
In summary, we believe that we have shown that adult skeletal muscle disruption of mitochondrial fusion leads to increased mitochondrial fragmentation. This in turn results in an increase in mtDNA instability, ultimately leading to a decrease in expression of mtDNA-encoded subunits. The effect on mtDNA levels and subunit expression alters the stoichiometry of the ETC complex assembly and activity. The decrease in ETC activity explains in part the decrease in exercise performance and impairs the ability to respond to training (Fig. 9).
Fig. 9.
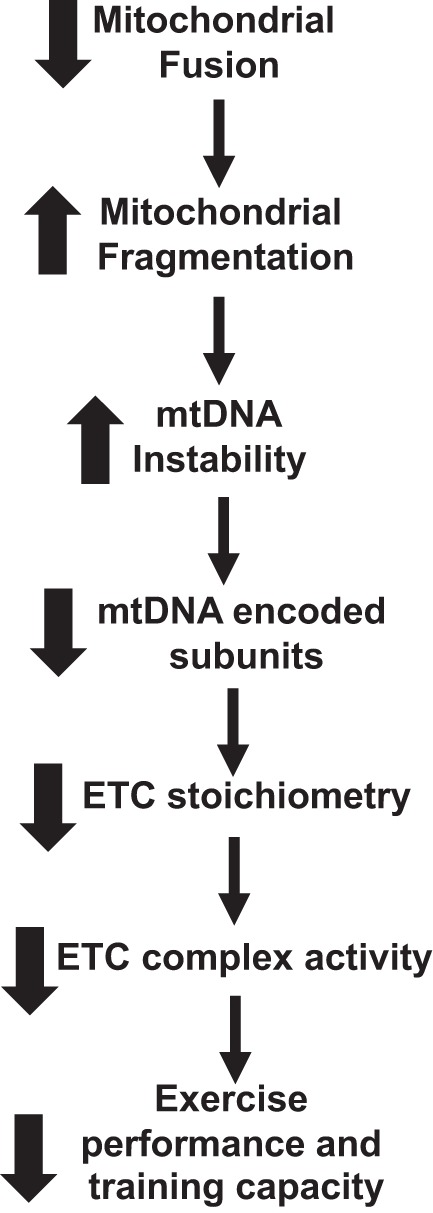
Flow chart of the effect of disrupting mitochondrial fusion on exercise capacity. Deleting both Mitofusin (Mfn)1 and Mfn2 in skeletal muscle impairs exercise performance and training capacity. ETC, electron transport chain; mtDNA, mitochondrial DNA.
Finally, the present study highlights the importance of intact mitochondrial fusion in adult skeletal muscle on exercise performance. Endurance exercise still remains a powerful tool in the treatment of metabolic diseases and reversing mitochondrial dysfunction. Therefore, identifying factors that can increase mitochondrial fusion could provide new mechanistic targets in increasing exercise performance and help treat metabolic diseases.
GRANTS
This work was supported in part by NIH Grant AR-0062128 to G. C. Rowe and NIH T32 Fellowship HD-071866 to M. B. Bell.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
M.B.B., Z.B., and G.R.M. performed experiments; M.B.B., Z.B., G.R.M., and G.C.R. analyzed data; M.B.B., G.R.M., and G.C.R. interpreted results of experiments; G.C.R. prepared figures; M.B.B. and G.C.R. drafted manuscript; M.B.B., Z.B., G.R.M., and G.C.R. edited and revised manuscript; M.B.B., Z.B., G.R.M., and G.C.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We are grateful to the UAB Diabetes Research Center Bioanalytical REDOX Biology Core, supported by National Institutes of Health (NIH) Grant P30 DK-079626, for help with electron transport complex activity assays as well as the UAB Neuroscience Behavioral Assessment Core supported by NIH Grant P30 NS-47466.
Present address of G. R. McGinnis: Dept. of Kinesiology and Nutrition Sciences, University of Nevada, Las Vegas, NV 89154.
REFERENCES
- 1.Abel ED. Mitochondrial dynamics and metabolic regulation in cardiac and skeletal muscle. Trans Am Clin Climatol Assoc 129: 266–278, 2018. [PMC free article] [PubMed] [Google Scholar]
- 2.Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP, Holloszy JO. Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC-1. FASEB J 16: 1879–1886, 2002. doi: 10.1096/fj.02-0367com. [DOI] [PubMed] [Google Scholar]
- 3.Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, McIntyre K, Runge MS. Mitochondrial integrity and function in atherogenesis. Circulation 106: 544–549, 2002. doi: 10.1161/01.CIR.0000023921.93743.89. [DOI] [PubMed] [Google Scholar]
- 4.Ballmann C, Tang Y, Bush Z, Rowe GC. Adult expression of PGC-1α and -1β in skeletal muscle is not required for endurance exercise-induced enhancement of exercise capacity. Am J Physiol Endocrinol Metab 311: E928–E938, 2016. doi: 10.1152/ajpendo.00209.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bannerman P, Burns T, Xu J, Miers L, Pleasure D. Mice hemizygous for a pathogenic mitofusin-2 allele exhibit hind limb/foot gait deficits and phenotypic perturbations in nerve and muscle. PLoS One 11: e0167573, 2016. [Erratum in PLoS One 13: e0204536, 2018.] doi: 10.1371/journal.pone.0167573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Booth FW, Ruegsegger GN, Toedebusch RG, Yan Z. Endurance exercise and the regulation of skeletal muscle metabolism. Prog Mol Biol Transl Sci 135: 129–151, 2015. doi: 10.1016/bs.pmbts.2015.07.016. [DOI] [PubMed] [Google Scholar]
- 7.Caffin F, Prola A, Piquereau J, Novotova M, David DJ, Garnier A, Fortin D, Alavi MV, Veksler V, Ventura-Clapier R, Joubert F. Altered skeletal muscle mitochondrial biogenesis but improved endurance capacity in trained OPA1-deficient mice. J Physiol 591: 6017–6037, 2013. doi: 10.1113/jphysiol.2013.263079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cartoni R, Léger B, Hock MB, Praz M, Crettenand A, Pich S, Ziltener JL, Luthi F, Dériaz O, Zorzano A, Gobelet C, Kralli A, Russell AP. Mitofusins 1/2 and ERRalpha expression are increased in human skeletal muscle after physical exercise. J Physiol 567: 349–358, 2005. doi: 10.1113/jphysiol.2005.092031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol 22: 79–99, 2006. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 10.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160: 189–200, 2003. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen H, McCaffery JM, Chan DC. Mitochondrial fusion protects against neurodegeneration in the cerebellum. Cell 130: 548–562, 2007. doi: 10.1016/j.cell.2007.06.026. [DOI] [PubMed] [Google Scholar]
- 12.Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 141: 280–289, 2010. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Civiletto G, Varanita T, Cerutti R, Gorletta T, Barbaro S, Marchet S, Lamperti C, Viscomi C, Scorrano L, Zeviani M. Opa1 overexpression ameliorates the phenotype of two mitochondrial disease mouse models. Cell Metab 21: 845–854, 2015. doi: 10.1016/j.cmet.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cogswell AM, Stevens RJ, Hood DA. Properties of skeletal muscle mitochondria isolated from subsarcolemmal and intermyofibrillar regions. Am J Physiol Cell Physiol 264: C383–C389, 1993. doi: 10.1152/ajpcell.1993.264.2.C383. [DOI] [PubMed] [Google Scholar]
- 15.Dahl R, Larsen S, Dohlmann TL, Qvortrup K, Helge JW, Dela F, Prats C. Three-dimensional reconstruction of the human skeletal muscle mitochondrial network as a tool to assess mitochondrial content and structural organization. Acta Physiol (Oxf) 213: 145–155, 2015. doi: 10.1111/apha.12289. [DOI] [PubMed] [Google Scholar]
- 16.Dimauro S, Andreu AL. Mutations in mitochondrial DNA as a cause of exercise intolerance. Ann Med 33: 472–476, 2001. doi: 10.3109/07853890109002096. [DOI] [PubMed] [Google Scholar]
- 17.Dorn GW 2nd, Kitsis RN. The mitochondrial dynamism-mitophagy-cell death interactome: multiple roles performed by members of a mitochondrial molecular ensemble. Circ Res 116: 167–182, 2015. doi: 10.1161/CIRCRESAHA.116.303554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drake JC, Wilson RJ, Yan Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J 30: 13–22, 2016. doi: 10.1096/fj.15-276337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17: 162–184, 2013. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 20.Fealy CE, Mulya A, Lai N, Kirwan JP. Exercise training decreases activation of the mitochondrial fission protein dynamin-related protein-1 in insulin-resistant human skeletal muscle. J Appl Physiol (1985) 117: 239–245, 2014. doi: 10.1152/japplphysiol.01064.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Granata C, Oliveira RS, Little JP, Renner K, Bishop DJ. Mitochondrial adaptations to high-volume exercise training are rapidly reversed after a reduction in training volume in human skeletal muscle. FASEB J 30: 3413–3423, 2016. doi: 10.1096/fj.201500100R. [DOI] [PubMed] [Google Scholar]
- 22.Grassi B, Rossiter HB, Zoladz JA. Skeletal muscle fatigue and decreased efficiency: two sides of the same coin? Exerc Sport Sci Rev 43: 75–83, 2015. doi: 10.1249/JES.0000000000000043. [DOI] [PubMed] [Google Scholar]
- 23.Hamers FP, Koopmans GC, Joosten EA. CatWalk-assisted gait analysis in the assessment of spinal cord injury. J Neurotrauma 23: 537–548, 2006. doi: 10.1089/neu.2006.23.537. [DOI] [PubMed] [Google Scholar]
- 24.Handschin C, Chin S, Li P, Liu F, Maratos-Flier E, Lebrasseur NK, Yan Z, Spiegelman BM. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1alpha muscle-specific knock-out animals. J Biol Chem 282: 30014–30021, 2007. doi: 10.1074/jbc.M704817200. [DOI] [PubMed] [Google Scholar]
- 25.Heo JW, No MH, Park DH, Kang JH, Seo DY, Han J, Neufer PD, Kwak HB. Effects of exercise on obesity-induced mitochondrial dysfunction in skeletal muscle. Korean J Physiol Pharmacol 21: 567–577, 2017. doi: 10.4196/kjpp.2017.21.6.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hesselink MK, Schrauwen-Hinderling V, Schrauwen P. Skeletal muscle mitochondria as a target to prevent or treat type 2 diabetes mellitus. Nat Rev Endocrinol 12: 633–645, 2016. doi: 10.1038/nrendo.2016.104. [DOI] [PubMed] [Google Scholar]
- 27.Hyde BB, Twig G, Shirihai OS. Organellar vs cellular control of mitochondrial dynamics. Semin Cell Dev Biol 21: 575–581, 2010. doi: 10.1016/j.semcdb.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iqbal S, Ostojic O, Singh K, Joseph AM, Hood DA. Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle Nerve 48: 963–970, 2013. doi: 10.1002/mus.23838. [DOI] [PubMed] [Google Scholar]
- 29.Jelenik T, Roden M. Mitochondrial plasticity in obesity and diabetes mellitus. Antioxid Redox Signal 19: 258–268, 2013. doi: 10.1089/ars.2012.4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang X, Wang X. Cytochrome C-mediated apoptosis. Annu Rev Biochem 73: 87–106, 2004. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 31.Kasahara A, Scorrano L. Mitochondria: from cell death executioners to regulators of cell differentiation. Trends Cell Biol 24: 761–770, 2014. doi: 10.1016/j.tcb.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 32.Kelly NA, Ford MP, Standaert DG, Watts RL, Bickel CS, Moellering DR, Tuggle SC, Williams JY, Lieb L, Windham ST, Bamman MM. Novel, high-intensity exercise prescription improves muscle mass, mitochondrial function, and physical capacity in individuals with Parkinson’s disease. J Appl Physiol (1985) 116: 582–592, 2014. doi: 10.1152/japplphysiol.01277.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kryściak K, Majerczak J, Kryściak J, Łochyński D, Kaczmarek D, Drzymała-Celichowska H, Krutki P, Gawedzka A, Guzik M, Korostynski M, Szkutnik Z, Pyza E, Jarmuszkiewicz W, Zoladz JA, Celichowski J. Adaptation of motor unit contractile properties in rat medial gastrocnemius to treadmill endurance training: relationship to muscle mitochondrial biogenesis. PLoS One 13: e0195704, 2018. doi: 10.1371/journal.pone.0195704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liesa M, Shirihai OS. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab 17: 491–506, 2013. doi: 10.1016/j.cmet.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lucas EK, Reid CS, McMeekin LJ, Dougherty SE, Floyd CL, Cowell RM. Cerebellar transcriptional alterations with Purkinje cell dysfunction and loss in mice lacking PGC-1α. Front Cell Neurosci 8: 441, 2015. doi: 10.3389/fncel.2014.00441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McCarthy JJ, Srikuea R, Kirby TJ, Peterson CA, Esser KA. Inducible Cre transgenic mouse strain for skeletal muscle-specific gene targeting. Skelet Muscle 2: 8, 2012. doi: 10.1186/2044-5040-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menzies RA, Gold PH. The turnover of mitochondria in a variety of tissues of young adult and aged rats. J Biol Chem 246: 2425–2429, 1971. [PubMed] [Google Scholar]
- 38.Milenkovic D, Blaza JN, Larsson NG, Hirst J. The enigma of the respiratory chain supercomplex. Cell Metab 25: 765–776, 2017. doi: 10.1016/j.cmet.2017.03.009. [DOI] [PubMed] [Google Scholar]
- 39.Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol 212: 379–387, 2016. doi: 10.1083/jcb.201511036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mishra P, Varuzhanyan G, Pham AH, Chan DC. Mitochondrial dynamics is a distinguishing feature of skeletal muscle fiber types and regulates organellar compartmentalization. Cell Metab 22: 1033–1044, 2015. doi: 10.1016/j.cmet.2015.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ni HM, Williams JA, Ding WX. Mitochondrial dynamics and mitochondrial quality control. Redox Biol 4: 6–13, 2015. doi: 10.1016/j.redox.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papanicolaou KN, Kikuchi R, Ngoh GA, Coughlan KA, Dominguez I, Stanley WC, Walsh K. Mitofusins 1 and 2 are essential for postnatal metabolic remodeling in heart. Circ Res 111: 1012–1026, 2012. doi: 10.1161/CIRCRESAHA.112.274142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pereira RO, Tadinada SM, Zasadny FM, Oliveira KJ, Pires KM, Olvera A, Jeffers J, Souvenir R, Mcglauflin R, Seei A, Funari T, Sesaki H, Potthoff MJ, Adams CM, Anderson EJ, Abel ED. OPA1 deficiency promotes secretion of FGF21 from muscle that prevents obesity and insulin resistance. EMBO J 36: 2126–2145, 2017. doi: 10.15252/embj.201696179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pilegaard H, Saltin B, Neufer PD. Exercise induces transient transcriptional activation of the PGC-1alpha gene in human skeletal muscle. J Physiol 546: 851–858, 2003. doi: 10.1113/jphysiol.2002.034850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Quiros PM, Goyal A, Jha P, Auwerx J. Analysis of mtDNA/nDNA ratio in mice. Curr Protoc Mouse Biol 7: 47–54, 2017. doi: 10.1002/cpmo.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rasmussen HN, Andersen AJ, Rasmussen UF. Optimization of preparation of mitochondria from 25-100 mg skeletal muscle. Anal Biochem 252: 153–159, 1997. doi: 10.1006/abio.1997.2304. [DOI] [PubMed] [Google Scholar]
- 47.Redmann M, Benavides GA, Wani WY, Berryhill TF, Ouyang X, Johnson MS, Ravi S, Mitra K, Barnes S, Darley-Usmar VM, Zhang J. Methods for assessing mitochondrial quality control mechanisms and cellular consequences in cell culture. Redox Biol 17: 59–69, 2018. doi: 10.1016/j.redox.2018.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rovira-Llopis S, Bañuls C, Diaz-Morales N, Hernandez-Mijares A, Rocha M, Victor VM. Mitochondrial dynamics in type 2 diabetes: pathophysiological implications. Redox Biol 11: 637–645, 2017. doi: 10.1016/j.redox.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rowe GC, Safdar A, Arany Z. Running forward: new frontiers in endurance exercise biology. Circulation 129: 798–810, 2014. doi: 10.1161/CIRCULATIONAHA.113.001590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Russell AP, Feilchenfeldt J, Schreiber S, Praz M, Crettenand A, Gobelet C, Meier CA, Bell DR, Kralli A, Giacobino JP, Dériaz O. Endurance training in humans leads to fiber type-specific increases in levels of peroxisome proliferator-activated receptor-gamma coactivator-1 and peroxisome proliferator-activated receptor-alpha in skeletal muscle. Diabetes 52: 2874–2881, 2003. doi: 10.2337/diabetes.52.12.2874. [DOI] [PubMed] [Google Scholar]
- 51.Santel A. Get the balance right: mitofusins roles in health and disease. Biochim Biophys Acta 1763: 490–499, 2006. doi: 10.1016/j.bbamcr.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 52.Santel A, Frank S, Gaume B, Herrler M, Youle RJ, Fuller MT. Mitofusin-1 protein is a generally expressed mediator of mitochondrial fusion in mammalian cells. J Cell Sci 116: 2763–2774, 2003. doi: 10.1242/jcs.00479. [DOI] [PubMed] [Google Scholar]
- 53.Sebastián D, Sorianello E, Segalés J, Irazoki A, Ruiz-Bonilla V, Sala D, Planet E, Berenguer-Llergo A, Muñoz JP, Sánchez-Feutrie M, Plana N, Hernández-Álvarez MI, Serrano AL, Palacín M, Zorzano A. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J 35: 1677–1693, 2016. doi: 10.15252/embj.201593084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Song M, Franco A, Fleischer JA, Zhang L, Dorn GW 2nd. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metab 26: 872–883.e5, 2017. doi: 10.1016/j.cmet.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song M, Mihara K, Chen Y, Scorrano L, Dorn GW 2nd. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21: 273–286, 2015. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tezze C, Romanello V, Desbats MA, Fadini GP, Albiero M, Favaro G, Ciciliot S, Soriano ME, Morbidoni V, Cerqua C, Loefler S, Kern H, Franceschi C, Salvioli S, Conte M, Blaauw B, Zampieri S, Salviati L, Scorrano L, Sandri M. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab 25: 1374–1389.e6, 2017. doi: 10.1016/j.cmet.2017.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Touvier T, De Palma C, Rigamonti E, Scagliola A, Incerti E, Mazelin L, Thomas JL, D’Antonio M, Politi L, Schaeffer L, Clementi E, Brunelli S. Muscle-specific Drp1 overexpression impairs skeletal muscle growth via translational attenuation. Cell Death Dis 6: e1663, 2015. doi: 10.1038/cddis.2014.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb Perspect Biol 5: a011072, 2013. doi: 10.1101/cshperspect.a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J 282: 647–672, 2015. doi: 10.1111/febs.13175. [DOI] [PubMed] [Google Scholar]
- 60.Zoladz JA, Grassi B, Majerczak J, Szkutnik Z, Korostyński M, Karasiński J, Kilarski W, Korzeniewski B. Training-induced acceleration of O2 uptake on-kinetics precedes muscle mitochondrial biogenesis in humans. Exp Physiol 98: 883–898, 2013. doi: 10.1113/expphysiol.2012.069443. [DOI] [PubMed] [Google Scholar]
- 61.Zoladz JA, Koziel A, Woyda-Ploszczyca A, Celichowski J, Jarmuszkiewicz W. Endurance training increases the efficiency of rat skeletal muscle mitochondria. Pflugers Arch 468: 1709–1724, 2016. doi: 10.1007/s00424-016-1867-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zorzano A, Liesa M, Palacín M. Mitochondrial dynamics as a bridge between mitochondrial dysfunction and insulin resistance. Arch Physiol Biochem 115: 1–12, 2009. doi: 10.1080/13813450802676335. [DOI] [PubMed] [Google Scholar]
- 63.Züchner S, Mersiyanova IV, Muglia M, Bissar-Tadmouri N, Rochelle J, Dadali EL, Zappia M, Nelis E, Patitucci A, Senderek J, Parman Y, Evgrafov O, Jonghe PD, Takahashi Y, Tsuji S, Pericak-Vance MA, Quattrone A, Battologlu E, Polyakov AV, Timmerman V, Schröder JM, Vance JM. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot-Marie-Tooth neuropathy type 2A. Nat Genet 36: 449–451, 2004. [Erratum in Nat Genet 36: 660, 2004.] doi: 10.1038/ng1341. [DOI] [PubMed] [Google Scholar]