

Abstract
Cognitive dysfunction is increasingly recognized as an important comorbidity of diabetes mellitus. Different stages of diabetes-associated cognitive dysfunction can be discerned, with different cognitive features, affected age groups, prognosis, and likely also different underlying mechanisms. Relatively subtle, slowly progressive cognitive decrements occur in all age groups. More severe stages, particularly mild cognitive impairment and dementia, with progressive deficits, occur primarily in older individuals. The latter are clearly most relevant for patient management and are the focus of this review.
Evolving insights from studies on risk factors, brain imaging, and neuropathology provide important clues on mechanisms. In the majority of patients multiple etiologies likely determine the cognitive phenotype. Although both the risk of -clinically diagnosed- Alzheimer’s disease and that of vascular dementia is increased in association with diabetes, the cerebral burden of the prototypical Alzheimer’s pathologies is not. A major challenge is therefore to pinpoint from the spectrum of diabetes-related disease processes those that affect the brain and contribute to development of dementia beyond Alzheimer’s pathologies. Observations from experimental models can help to meet that challenge, but this requires further improving the synergy between experimental and clinical scientists. Development of targeted treatment and preventive strategies depends on these translational efforts.
Worldwide the prevalence of diabetes is increasing, both in absolute and relative numbers1. Particularly for type 2 diabetes (T2DM), this is attributed to changing lifestyle factors, such as diet, overweight and physical inactivity2. Another key factor that adds to the prevalence of T2DM is increased longevity and aging of populations around the world. The latter is particularly evident in low and middle income countries1. These trends are expected to continue over the years to come. For dementia, we see very similar population trends3. As a consequence, there is an increased cooccurrence of diabetes and dementia. It has become evident, however, that diabetes and dementia concur more frequently than would be expected by chance alone. Epidemiological studies have established an increased risk of dementia among individuals with diabetes4. Diabetes is also linked to less severe forms of cognitive dysfunction5. This has important implications for patient management, particularly in older individuals where dementia and pre-dementia stages of cognitive impairment most commonly occur.
This review addresses the different manifestations of diabetes-associated cognitive dysfunction. Emphasis will be on dementia and pre-dementia stages of cognitive impairment in T2DM. Throughout the manuscript we will use the term diabetes if we refer to diabetes in general and T1DM or T2DM if we refer to these specific subtypes. We will show that studies on risk factors and on neuroimaging and neuropathology correlates of cognitive dysfunction provide important clues on underlying mechanisms, although many questions still remain. Experimental models may help to further unravel the etiology and identify treatment targets. A key strength of experimental models is clearly that they can single out individual causative pathways, in ways and at a level of detail that is impossible in humans. Technical progress provided tools that can boost studies of these pathways, from the level of specific molecular interactions to systems biology. It is of fundamental importance, however, that potential mechanisms identified in models are also evaluated in the context of other morbidities with which they may cooccur in patients. We will make the point that further improving synergy between clinical and experimental scientists can foster innovation in designing animal models that accurately replicate the complexities of the interaction between diabetes and dementia in humans.
While awaiting these further research developments, cognitive dysfunction in diabetes does already affect daily clinical care today. The final section of this review addresses clinical implications of the latest data on diabetic brain injury and future perspectives.
Cognitive dysfunction and diabetes: scope of the problem
There is strong epidemiological evidence for links between diabetes and cognitive dysfunction 5–7. Importantly, cognitive dysfunction in relation to diabetes should not be regarded as a unitary construct. Manifestations and prognosis of diabetes-associated cognitive dysfunction vary depending on diabetes type and age8.
In children with T1DM, for example, there may be subtle changes in cognitive development, particularly in those with an onset of diabetes before the age of 79. Adults with T1DM also present subtle decrements in cognitive performance relative to age-matched controls, particularly affecting the domains intelligence (effect size Cohen’s d= 0.7), psychomotor efficiency (d =0.6), and cognitive flexibility (d = 0.5)10. These decrements generally remain quite stable over time 11, although there may be subgroups of patients, particularly those with advanced microvascular complications, in whom the severity of cognitive impairment may worsen substantially over time9,12.
In adults with T2DM, deficits in cognitive functioning can roughly be divided in three different stages, according to severity: diabetes-associated cognitive decrements, mild cognitive impairment (MCI), and dementia5. The term diabetes-associated decrements refers to subtle changes in cognitive function, that might give rise to cognitive complaints (usually expressed only by the patient), but should not affect activities of daily life or diabetes self-management5. The subtle cognitive changes may concern one or several domains, including processing speed, executive function, and memory, typically with a Cohen’s effect size of 0.2–0.5 relative to people without diabetes at the group level (systematic reviews 13,14). These decrements are likely to have an onset in pre-diabetic stages15 and evolve only very slowly over the course of many years, at a rate that is up to 50% faster than that of normal cognitive ageing 13,15–18.
Mild cognitive impairment and dementia
Diagnostic constructs for mild cognitive impairment and dementia and their etiologies in people with diabetes are the same as in people without diabetes (text box 1)
TEXT Box 1: Diagnostic constructs for mild cognitive impairment and dementia.
Mild cognitive impairment (MCI) refers to acquired objective cognitive impairment (mostly defined as a performance below 1 to 1.5 SD units of normative values) affecting one or more cognitive domains with largely preserved activities of daily life19,20. This construct captures a stage between normal cognition and dementia that identifies individuals at high risk of transition to dementia. Dementia, in turn, is defined as acquired objective cognitive impairment affecting multiple cognitive domains, severe enough to affect activities of daily life21.
Of note, the diagnostic labels MCI and dementia do not refer to a particular etiology. In clinical practice as well as in most epidemiological studies, assumptions on the likely etiology are primarily based on the nature of the symptoms (e.g. acquired episodic memory deficit is suggestive of Alzheimer’s disease) , while excluding other causes (e.g. a brain tumor). It is clear, however, that this approach has insufficient specificity and sensitivity in determining the actual etiology22. Therefore, particularly in research settings, the etiology of MCI and dementia is increasingly based on biomarkers (e.g. amyloid in the cerebrospinal fluid (CSF) or on PET-scans to demonstrate Alzheimer pathology)23.
Two prospective population-based studies, have reported quite comparable findings on the risk of MCI in patients with diabetes. One observed a hazard ratio (HR) of 1.5 (95% confidence interval (CI) 1.0–2.2) for amnesic MCI and of 1.2 (0.9–1.8) for nonamnesic MCI24. The other reported a HR of 1.6 ( 1.2–2.2) form amnesic and 1.4 (0.8–2.2) for nonamnesic MCI25. Prognosis of MCI is worse in patients with diabetes. Two meta-analyses, each containing seven - not completely overlapping - studies, reported a relative risk (RR) of conversion to dementia of 1.7 (1.1–2.4)26 and 1.7 (1.1–2.6)27 for patients with MCI and diabetes, compared to patients with MCI without diabetes.
There have been many studies on the risk of dementia in relation to diabetes. Systematic reviews and meta-analyses4,6,7,28, including over 25 original studies with well over 2 million participants, estimate the RR for all type dementia at 1.73 (1.65–1.82)6, for Alzheimer’s disease (AD) at 1.53, (1.42–1.63)7, and for vascular dementia at 2.27 (1.94–2.66)6 for people with diabetes compared to those without. A large recent cohort study from Canada indicates that dementia risk is already increased in those with newly diagnosed diabetes (HR 1.16 (1.15–1.18))29. Moreover, elevated glucose levels in individuals without diabetes have also been linked to increased dementia risk30.
When stratified by ethnicity, the RR of AD in Western populations and Eastern populations were 1.36 (1.18–1.53) and 1.62 (1.49–1.75), respectively7. When stratified by sex, the RR of all type dementia in women with diabetes was 1.62 (1.45–1.80) and in men 1.58 (1.38–1.81)28. For vascular dementia the RR was 2.34 (1.86–2.94) in women and 1.73 (1.61–1.85) in men, translating into a 19% greater risk for the development of vascular dementia in women with diabetes than men28. Of note, few previous studies have addressed such potential modifying effects of sex and ethnicity (e.g. 7,28,31), and these topics needs further exploration.
Trajectories of cognitive dysfunction
Based on current evidence, we would argue that the different stages of cognitive dysfunction in patients with diabetes should not be regarded as a continuum15. Diabetes- associated decrements, the mildest stage, can occur in all age groups, from young adults and even adolescents with T2DM32,33 to the oldest old34. Further cognitive decline over time is generally slow over the course of many years15. These decrements affect the patients with diabetes as a group. In other words, there is a subtle shift in average cognitive performance across individuals and the difference compared to people without diabetes is not due to poor performance of a small subgroup of patients pulling the average performance of the patients down. At an individual level, due to the subtle nature of the deficits, it is difficult to establish if a patient is affected based on formal cognitive testing. Dementia, on the other hand, is characterized by poor cognitive performance where an individual stands apart from what is considered normal cognitive performance. In other words, it affects individuals. It is primarily a condition of old age3 and mostly involves relentless, year-by-year, cognitive decline. Although diabetes may also increase the risk of young-onset dementia (i.e. before the age of 65)35,36, the vast majority of individuals with diabetes who develop dementia are well over the age of 65, just like people without diabetes3. Hence, considering the marked differences in affected age-groups clinical manifestation, and trajectories of cognitive decline, diabetes-associated cognitive decrements and dementia should be regarded as different entities, likely with different underlying mechanisms.
Mechanisms of cognitive dysfunction – observations in humans
In light of the increasing prevalence of diabetes, population trends in ageing, and the impact of cognitive dysfunction on affected individuals and society as a whole, preventive treatment is warranted. However, insight in potential therapeutic targets and underlying mechanisms is still incomplete, although there is an evolving scientific literature that does provide important clues.
An important trend is that recent studies not only provide data on risk factors and brain imaging correlates of diabetes-associated cognitive decrements, but increasingly also on dementia. While the latter is clearly most clinically relevant, it is also much more challenging to address with epidemiological studies. It requires huge cohorts to acquire a sufficient number of cases of patients with T2DM and incident dementia31. Fortunately, large collaborative autopsy studies37 and novel biomarkers of dementia etiology have also stimulated progress in this field. This section will summarize this literature, focusing on T2DM.
Risk factors
The main picture that emerges from the many studies on the risk factors for cognitive dysfunction in diabetes is that there are many different factors involved, with (very) small effects each15,38.
Glycaemic control has received a lot of attention. Converging evidence shows that higher A1C levels are associated with diabetes-associated cognitive decrements, but the strength of the relation is weak39. A1C levels (or repeated glucose measurements) have also been linked to dementia risk in people without diabetes30. Whether A1C levels are also related to dementia risk among people with diabetes is less clear. There are few available studies 39 and there are indications of non-linearity, where both low and high levels related to dementia risk30. There also is an emerging literature indicating that apart from chronically elevated glucose levels fluctuations or peaks in glucose levels may be linked to cognitive decrements as well as dementia risk39,40.
Observational studies have reported potential benefit for cognition of some glucose lowering compound over others41, which suggests the need to assess outcomes other than blood glucose and A1C levels to understand the effect of anti-hyperglycemia drugs on cognition A recent large registry study in veterans with T2DM <75 years of age42, for example, found metformin use to be associated with a lower risk of subsequent dementia than sulfonylurea use, while adjusting for known confounders. Yet, randomized controlled intervention studies thus far do not support that intensive glycaemic control or any particular glucose lowering agent is associated with better cognitive functioning39,43, or dementia44. Occurrence of hypoglycemic episodes, on the other hand, is clearly linked to cognitive decline and increased dementia risk29,31,38,39.
Vascular risk factors, in particular hypertension and dyslipidemia, may be associated with cognitive decrements among people with T2DM, although the evidence is inconsistent despite a substantial number of studies38. Few studies have addressed how vascular risk factors affect dementia risk among patients with T2DM29,38. Yet, because many studies in the general population have demonstrated the importance of (midlife) vascular risk factors for dementia risk 45,46, and because T2DM is associated with an adverse vascular risk factor profile, also in prediabetic stages1, it is reasonable assume that these factors contribute to dementia risk among patients with T2DM and are thus a lead for prevention. It is also clear that patients with manifestations of microvascular (e.g. diabetic retinopathy) or macrovascular disease (e.g. myocardial infarction, stroke) are more likely to have worse cognitive performance15,38 and are at increased dementia risk29,31. Other studies have identified insulin resistance, inflammation, and depression as potential risk factors for cognitive dysfunction in people with diabetes38,39.
All in all, it is clear that multiple risk factors are involved38. It is also clear that there are still substantial knowledge gaps on how these interconnect, translate to potentially modifiable mechanisms, and also on which genetic factors are involved.
Patterns of brain injury
There is an expanding literature on brain imaging studies in patients with diabetes47,48, although it should be noted that few studies included patients who were affected by MCI or dementia. As a framework for the interpretation of the findings, it is important to distinguish between imaging markers that primarily reflect brain injury, markers that reflect specific etiological processes, and markers that reflect both. Markers of injury include, for example, measures of atrophy and microstructural white matter integrity. Although patterns of injury may be suggestive of a particular etiology, they are by no means etiologically specific (e.g. medial temporal lobe atrophy cannot be taken as proof for AD as a primary etiology). Markers of etiological processes include for example amyloid PET and measures of cerebral blood flow, although the latter may also change as a consequence of brain injury.
It is clear from the literature that T2DM is associated with brain atrophy (figure 1), although the regional pattern of brain volume changes varies between studies47,48. The magnitude of the volume reduction is modest, with effect sizes of 0.2–0.6 SD units, comparable with 3–5 years of normal aging47. Another emerging marker of brain injury in T2DM is diffusion tensor imaging (DTI). This technique allows to explore microstructural integrity of the white matter and related changes in brain networks. DTI studies show widespread changes in white matter microstructure and connectivity in relation to T2DM, which are clearly related to cognitive dysfunction47,48.
FIG 1.
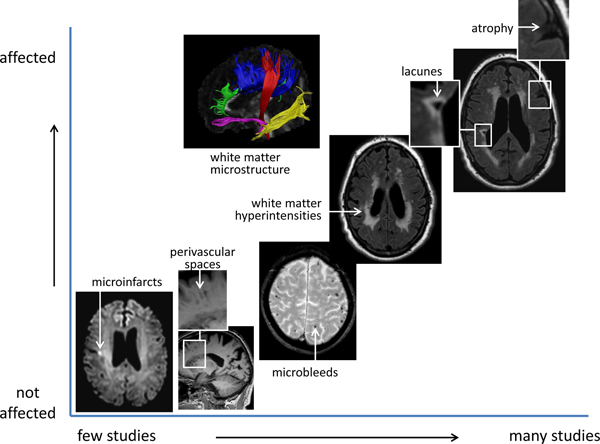
Brain imaging findings in patients with T2DM
The figure summarizes findings form brain imaging studies in T2DM (for details and literature references see text). The position of each imaging marker on the X-axis reflects how intensively it has been studied in relation to T2DM. The position on the Y-axis reflects to which extent a marker is affected in individuals with T2DM relative to controls, based on the evidence from available studies. Image of white matter microstructure courtesy of Y Reijmer, UMC Utrecht.
Given the links between diabetes and vascular disease, manifestations of so-called cerebral small vessel disease on MRI are clearly of interest. These include lacunes, white matter hyperintensities, visible perivascular spaces, microbleeds and microinfarcts49. Although widely accepted as markers of vascular injury, these MRI visible lesions have limited specificity for particular underlying etiological processes49. White matter hyperintensities, for example, can develop as a consequence of different underlying vascular pathologies, but also as a consequence of non-vascular processes such as inflammation50. Based on the current literature, T2DM is associated with an increased occurrence of lacunes and a modest increase in the volume of white matter hyperintensities (Figure 1)47. There may be an increased occurrence of microbleeds in patients with T2DM, but evidence is not consistent47,51 52. There have been very few imaging studies on the relation between T2DM and perivascular spaces53,54 and microinfarcts55 thus far.
Etiological markers and neuropathology
As also indicated in the preceding section, diabetes is clearly associated with vascular brain injury on MRI. Indeed, neuropathological studies also report an increased burden of cerebrovascular lesions in people with diabetes. This particularly concerns lacunes37,56. In contrast, neuropathological studies do not report a clear increase in the burden of large artery infarcts or microinfarcts in patients with diabetes37,56. The increased occurrence of lacunes might be attributable to abnormalities in the small cerebral perforating arterioles, such as arteriolosclerosis, lipohyalinosis, or fibrinoid necrosis57. Indeed, there are studies in human autopsy material that show arteriolar abnormalities in patients with diabetes58,59, but it should be noted that to date the impact of diabetes on the different blood vessel types of the brain has not been assessed systematically. Also with regard to cerebrovascular dysfunction in T2DM there are still uncertainties. There are reports of reduced cerebral perfusion and impaired cerebrovascular reactivity, but results of different studies have been conflicting, likely due to differences in study populations, imaging techniques and variation in dealing with confounding factors like cerebral atrophy60.
Evidently, AD is another key etiology to consider. Converging evidence from brain autopsy studies from the past decade shows that the core neuropathological features of AD are not more common in subjects with than in those without T2DM61. Several recent studies of large autopsy cohorts report that the occurrence of neuritic amyloid plaques (Odds ratio (OR) 0.96 (95% CI 0.68–1.36)37; 1.08 (0.84–1.38)56; 0.97 (0.68–1.38)62) and tau tangles (0.82 (0.61–1.11)37; 0.85 (0.66–1.11)56 1.12 (0.81–1.54)62), p = 0.48) is not increased in T2DM. Studies on in vivo biomarkers of AD pathology are completely in line with these observations. T2DM is neither associated with CSF or PET biomarkers of cerebral amyloid nor tau pathology63–65. Yet, despite these findings, T2DM is associated with higher levels of MRI and PET biomarkers of neurodegeneration65, suggesting that T2DM accelerates neurodegeneration via non-AD mechanisms.
Another emerging concept in mechanistic studies is the potential role of cerebral insulin resistance66,67. Insulin signaling in the brain has important roles in brain physiology and cognition66,67. Moreover, disturbances in insulin signaling have been noted in brain tissue of people with AD, irrespective of T2DM (review67). This gives rise to the possibility that a core feature of T2DM, disturbed insulin signaling, causing insulin resistance, not only affects systemic metabolism, but also directly impacts the brain, by disturbing cerebral insulin pathways. Other etiological leads from studies in humans that warrant further investigation are accumulation of advanced glycation end products (AGEs)68 – for which skin autofluorescence is a non-invasive proxy – and increased blood-brain barrier (BBB) permeability69, pointing to possible roles of inflammation and endothelial dysfunction.
Diverging observations, converging pathways?
The preceding sections on studies in humans clearly do not point to a single mechanism underlying diabetes-associated cognitive dysfunction. The different stages of cognitive dysfunction in T2DM differ in severity, prognosis and likely have different underlying etiologies. Moreover, while diabetes is associated with several different imaging-manifestations of cerebral injury, one patient may show one manifestation and the next patient another. Furthermore, how can we understand that diabetes increases the risk of a clinical diagnosis of AD, while biomarker and neuropathological studies clearly indicate that the burden of AD pathologies is not increased? The likely explanation is that in the majority of individuals with diabetes the clinical phenotype of cognitive dysfunction or dementia is due to multiple pathologies (figure 2). Hence, although AD pathologies are not increased in T2DM, they are still considered the commonest cause of dementia, also in people with T2DM: over 40% of individuals with T2DM have intermediate to severe AD pathology in their brain at the time of death37,62. Yet, the elevated dementia risk in T2DM should be attributed to pathologies other than AD, which may often evolve on a background of AD pathology. The major challenge for etiological studies is thus to pinpoint from the diverse spectrum of diabetes-related disease processes those specific mechanisms affecting the brain beyond AD pathology. This clearly includes vascular disease, but likely also non-AD mechanism of neurodegeneration. The next section will summarize how animal models may contribute in meeting that challenge.
FIG 2.
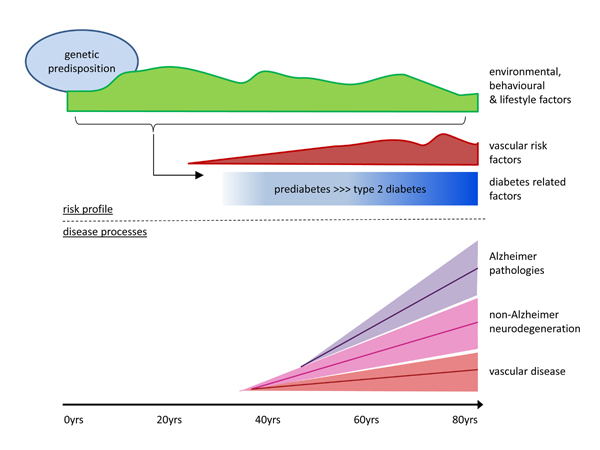
risk factors and underlying pathologies for dementia in T2DM
The figure provides a life course perspective on risk factors and disease processes contributing to the development of T2DM and dementia. T2DM most commonly develops in mid- or late life, in the context of environmental, behavioural and lifestyle factors – that vary over the course of life - on a background of genetic risk. People with T2DM frequently have an adverse vascular risk factor profile, including obesity, hypertension and dyslipidemia, often already in prediabetic stages. Many of the factors that predispose and co-occur with T2DM, as well as factors that are related to having T2DM (e.g. elevated glucose, glucose lowering treatment) may affect the brain. Evidently, with so many factors involved there are marked interindividual differences in risk factor profiles and exposures.
Brain pathologies contributing to the development of dementia accumulate over the course of decades in the context of an individual’s risk profile. In the majority of cases multiple pathologies cooccur, with variable proportions between individuals. Vascular pathologies are more common in individuals with T2DM than in those without and may thus contribute to the elevated dementia risk in T2DM. Of note, while Alzheimer pathologies are a key contributor to dementia in people with T2DM, the burden of these pathologies is not increased compared to people with T2DM. The excess dementia risk in people with T2DM is thus likely to be also attributable to additional non-Alzheimer neurodegenerative pathologies, which are yet to be identified (for details and references see main text).
Mechanisms of cognitive dysfunction: experimental models
The diverse spectrum of findings identified in patients with diabetes with or without dementia is mechanistically explored using experimental models, including cell lines, organoids and animal models ranging from rodents to non-human primates. Rodents are commonly used in both diabetes and dementia research owing to their genetic similarities to humans, including genome size, number of genes (99%) and synteny. Neither diabetes nor AD-like pathology develops spontaneously in rodents, unless specific gene manipulations or pharmacological interventions are used. Depending on the intervention, conditions associated with diabetes, dementia, or both can be induced in rodents (see Table 1). For the most part, insights from these interventions have been restricted to cerebral effects of inducing diabetes in normal rodents70–81 (non-AD rodent models; Table 1) and in rodents genetically modified to accumulate β-amyloid (Aβ) in the brain82–86 (AD rodent models; Table 1).
TABLE 1.
Cerebral effects of inducing diabetes or insulin resistance in normal rodents (i.e., non-AD rodent models) and in rodents genetically modified to accumulate Aβ in the brain (i.e., AD rodent models). Common interventions to induce diabetic conditions in rodents included recessive mutations in the obesity gene (ob, also known as Lep), defects in the leptin receptor (Ob-R), diet, and administration of streptozotocin. Rodents with pancreatic overexpression of human amylin spontaneously develop both type-2 diabetes and dementia-like pathology.
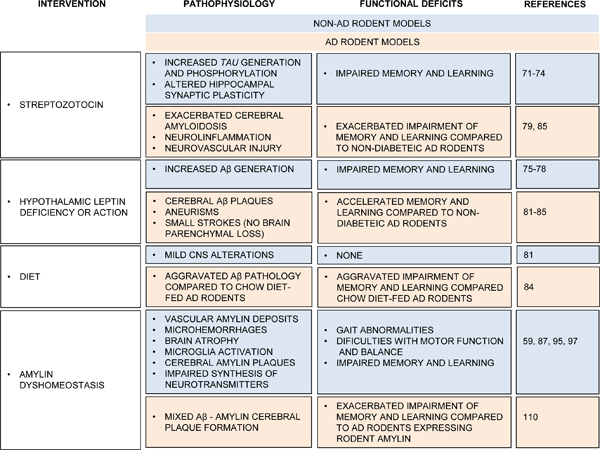
|
Here, we reviewed these rodent models with the objective of identifying pathophysiological processes that may contribute to an AD phenotype without entailing AD pathology. We also suggested characteristics that need to be captured in novel animal models in order to optimize our chances to uncover mechanisms underlying the dementia risk in diabetes.
Crosstalk between diabetes and AD-related processes
In mice without pre-existing AD pathology (Table 1; non-AD rodent models), induction of diabetes, genetically, pharmacologically (i.e., streptozotocin injection) or by diet, is associated with increased Aβ generation75–78 and hyperphosphorylation of tau protein71–74. Similarly, diabetic AD mice showed accelerated cerebral Aβ formation84,85 and cerebrovascular pathologies82,83, including aneurisms and small strokes (Table 1; AD rodent models). In contrast to humans however, brains of diabetic AD mice had no brain atrophy 83. In brains from diabetic AD mice generated by crossing AD mice with db/db mice, an increased vascularization was observed83, which probably compensated for the leptin deficiency-mediated vascular disruption. Thus, inducing diabetic states lowers the threshold for neurodegeneration in AD mice via mechanisms that involve cerebrovascular pathologies82–86.
Cerebral insulin resistance in rodent models of type 2 diabetes and AD
As mentioned earlier in this review, data from experimental models of demonstrate commonalities in abnormalities in signaling pathways between cerebral an in systemic insulin resistance, providing potential pathways that can link metabolic and cerebral changes in T2DM67. Moreover, experimental models show that brain insulin resistance may contribute to AD by promoting Aβ generation and hyperphosphorylation of tau protein 71–78,84,85. Increased brain levels of Aβ correlated with altered insulin signal transduction and autophagy and increased beta-site amyloid precursor protein cleaving enzyme (BACE)1/β-secretase and γ-secretase activities77,78. The results suggest a role of insulin resistance and subsequent hyperinsulinemia in impairing Aβ clearance. Streptozotocin injection in mice, which results in insulin deficiency, characteristic of an advanced diabetic state, appears linked to abnormal brain levels of hyperphosphorylated tau protein71,72,74,79.
Stimulating hippocampal insulin receptors by direct administration of insulin into the hippocampus improved learning ability in normal rodents87,88. A similar treatment had less effect in diabetic rodents88. Brain levels of Aβ and tau hyperphosphorylation were reduced in AD mice by treatments that improved insulin availability or/and sensitivity (experimental work reviewed in66). Thus, brain insulin resistance plays a complex role in promoting AD pathology and is a promising therapeutic target to slow the progression of cognitive decline in humans.
Non-AD processes contributing to an AD phenotype in diabetes
Induction of diabetic states in non-AD rodents can cause memory and learning impairments59,70,79–81,89. Possible non-AD processes contributing to cognitive dysfunction in rodent models of diabetes are discussed below (see Table 2).
TABLE 2.
Non-AD processes contributing to an AD phenotype in diabetes, as suggested from experimental studies in rodents. Color code: brown – AD rodent models; blue gray – Non-AD rodent models.
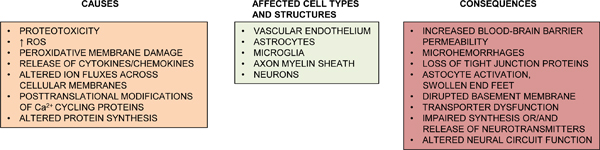
|
Vascular endothelial dysfunction
In diabetes, endothelial dysfunction is linked to vascular accumulation of toxic lipids90, AGEs products91 or/and aggregated proteins59. Proteinaceous deposition on blood vessel walls damages endothelial cells59,91, increases the production of reactive oxygen species (ROS)92,93 and impairs production of vasodilatory substances92, which reduces the cerebral blood flow. Stalled blood flow can lead to neurovascular uncoupling and hypoxic neuronal injury92–94. Elevated ROS production can further damage cellular structures and activate matrix metalloproteinases inducing cytoskeletal reorganization and vascular remodeling93. Cytoskeletal reorganization affects the stability of tight junction proteins resulting in increased capillary permeability, depletion of energy resources and altered neural viability92,93.
Inflammation and blood-brain barrier injury
Vascular endothelial dysfunction upregulates inflammatory mediators which can disrupt the BBB59,89,93,94. BBB disruption exposes the brain parenchyma to potentially neurotoxic blood proteins, thrombin, fibrin, plasmin and hemoglobin, as well as the iron from lysed red blood cells. A leaky BBB induces abnormal neuronal activity93.
White matter disease of vascular origin
White matter disease has been clinically associated with vascular contributions to cognitive impairment and dementia93,94. It is also known as small blood vessel disease and may be the result of long term endothelial dysfunction, capillary loss and subsequent ischemia93,94. Indeed, recent results59 from a rat model of T2DM demonstrated the association of periventricular white matter injury with chronic vascular endothelial dysfunction, microhemorrhages and reduced brain perfusion.
Demyelination and axonal loss
Compared to normal rats, brains of diabetic rats have myelin loss, abundant white matter vacuoles and smaller volumes59. Demyelination and loss of axons can alter synthesis or/and release of neurotransmitters in the brain, which can further accentuate white matter disease and brain atrophy. Brain phenylalanine and tyrosine (precursors of catecholamine) were reduced by >50% in diabetic rats compared to normal rats95. Thus, experimental diabetes can cause impairments of protein synthesis in the brain.
Peroxidative membrane injury, mitochondrial dysfunction and neurodegeneration
Exposure of unsaturated fatty acids to cytosolic ROS generates reactive aldehydes, such as 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA)96. Elevated reactive aldehyde levels cause peroxidative membrane injury and have been used as biomarkers for neuronal oxidative damage96. Indeed, brain tissues from diabetic rats and from patients with diabetes and AD showed intraneural accumulation of 4-HNE-based adducts97, indicating the peroxidative cell damage as a contributor to neurodegeneration in diabetes.
Accumulating evidence from experimental models of insulin resistance and T2DM indicates systemic mitochondrial dysfunction as a pathological mechanism contributing to health deterioration and cognitive decline. Specific mechanisms linking mitochondrial and metabolic dysfunction with neurodegeneration and AD were discussed in a recent review98.
Neuronal Ca2+ mishandling, posttranslational modification of Ca2+-dependent proteinkinases
In diabetic rats, altered Ca2+ signaling contributes to neuron dysfunction via multiple mechanisms99. A recent study in brains (and hearts) of humans with diabetes and AD and in brains of diabetic rats identified posttranslational modification of Ca2+/calmodulin-dependent proteinkinase II (CaMKII)100. It was shown that diabetes induces O-GlcNAcylation (covalent binding of O-linked Nacetylglucosamine) of CaMKII, which activates the kinase100. Overactivity of CaMKII can cause neuronal excitotoxicity and dysfunction of ion channels involved in gene transcription and viability.
Amylin dyshomeostasis: a bridge between AD and non-AD processes?
Amylin is a pancreatic β-cell hormone co-secreted with insulin and plays a role in normal glucose homeostasis101. Amylin from humans (but not rodents101) is amyloidogenic and aggregates quickly when overexpressed. The majority of individuals with T2DM have large deposits of aggregated amylin in the pancreatic islets101, kidneys102 and heart103. Aggregated amylin induces cell dysfunction and apoptosis101. Accumulating data from several laboratories have confirmed that the brains of patients with T2DM and AD contain an abnormally increased level of aggregated amylin and mixed amylin-Aβ plaques104–107 (see figure 3). A recent epidemiological study indicated also a genetic risk for developing mixed Aβ-amylin plaques in the brain108. The results indicate amylin dyshomeostasis as a possible new link between T2DM and increased risk of AD104–108.
FIG 3.
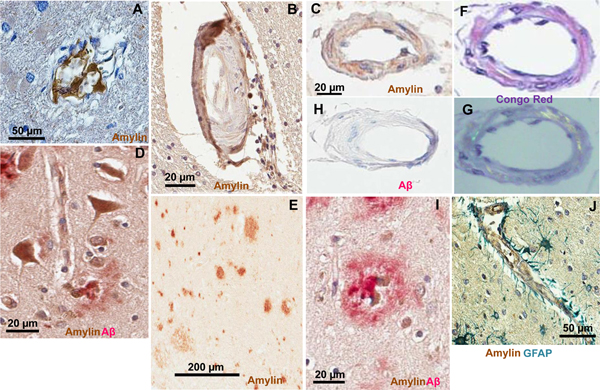
Pancreatic amylin forms amyloid and interacts with Aβ in the brains of patients with T2DM Sections through the brains of patients with T2DM and AD showing amylin-positive vascular wall contours in capillaries (A), small arterioles (B and C), neurons (D) and plaques (E). Vascular amylin deposits in (C) shows apple-green birefringence in the Congo red stain (F and G). Same blood vessel shows no Aβ immunoreactivity (H). Amylin interacts with Aβ forming cerebral mixed amylin-Aβ deposits (D and I). In J, vascular deposition of amylin (brown) and astroglial reaction (green stain for glial fibrillary acidic protein; GFAP) are shown. A and J are from Ref.59, B,C, F, G, H and I are from Ref. 104, D and E are from Ref. 97.
Amylin is oversecreted in individuals with prediabetic insulin resistance (i.e., hyperinsulinemia coincides always with hyperamylinemia) 101. Overexpression (3-fold) of human amylin in the pancreatic β-cells of rodents (HIP rats and mice) results in pancreatic amyloid, β-cell apoptosis and overt hyperglycemia109. In addition to the development of late-onset T2DM, HIP rats showed vestibulomotor dysfunction, altered balance, and impaired memory and learning ability59,89. Brain dysfunction in HIP rats correlated with amylin deposition in brain blood vessels59,89 and brain parenchyma59. In contrast to diabetic AD mice generated by crossing AD mice with db/db mice83, which showed brain microhemorrhages without parenchymal loss, HIP rats have brain microhemorrhages associated with white matter rarefaction and brain atrophy59 (see Table 1). A recent study110 demonstrated that HIP mice expressing a mutant form of the APP in neurons develop cross-seeding of amylin-Aβ pathology leading to accelerated brain dysfunction compared to transgenic mice expressing only amylin or the Aβ protein. These results suggest that systemic amylin dyshomeostasis is a trigger of mixed vascular-amylin-Aβ pathologies. Interaction of amylin with Aβ pathology was also documented in brains of patients with diabetes and AD104,105. These results suggest that not increased Aβ burden per se, but perhaps amyloid with a different composition may develop in brains of patients with diabetes and AD.
Next steps, challenges and further improvements of experimental models
Each animal model (Table 1) has certain limitations and no experimental model exists that accurately phenocopies the human brain condition in diabetes and AD. For example, transgenic mouse models of AD overexpressing the amyloid precursor protein (APP) do not only show exacerbated Aβ, but also elevated full length of APP and other fragments of the Aβ processing111,112. This might explain why the amyloid burden is increased in diabetic AD mice but not in patients with AD and T2DM.
The pathophysiology of T2DM encompasses a complex interplay of multiple deficiencies involving insulin resistance, relative insulin deficiency and pancreatic β‐cell dysfunction that ultimately result in multiorgan impairments. Although AD is primarily a neurodegenerative process, it often occurs in the context of vascular risk factors, systemic cardiovascular disease and cerebral vascular pathology in humans 45,46,113.. Mice that accumulate Aβ in the brain do not demonstrate these vascular co-morbidities. Thus, to achieve progress in investigating and validating causative mechanisms of increased risk of cognitive decline in patients with T2DM, we think a vital tool will be animal models carrying significant heterogeneity of diabetes pathology along with a broad spectrum of phenotypes seen in patients with dementia. Novel lines of transgenic mice that are engineered to achieve inducible and reversible expression of human proteins involved with diabetic brain injury could be an important step to identify a cerebral pathologic substrate of diabetes.
Current implications for patient management
Manifestations of cognitive dysfunction in diabetes, as reviewed herein, are increasingly recognized. Recent clinical diabetes guidelines start to provide suggestions how cognitive impairment should be detected and how this should affect diabetes management114–116.
Clearly, detection and management of cognitive dysfunction in T2DM is not “ One size fits all”. The different stages of cognitive dysfunction, due to their different features and impact, require different approaches. Diabetes-associated cognitive decrements are by definition subtle, without clearly affecting social or occupational functioning or diabetes self management5. It therefore suffices to act on cognitive complaints, rather than to strive for active detection strategies like screening. Approaches to diagnose and manage diabetes-associated cognitive decrements and to differentiate these subtle cognitive deficits from more severe stages of cognitive dysfunction, in particular MCI and dementia, have been proposed before5. First of all, age of the patient provides important context, as cognitive decrements occur in all age groups, whereas MCI and dementia rarely occur under the age of 60 to 6520. Secondly, the nature of the complaints should be compatible with decrements, in that the patient may express worries about his or her cognitive abilities, often focusing on memory, but that there are no obvious mishaps. Finally, the complaints should have developed insidiously, with limited progression over time, and there should be no alternative explanations. In such cases, it may often be sufficient to explain the patient that the complaints may be due to diabetes-associated cognitive decrements, that the complaints can be annoying, but that further marked decline is not expected to occur, particularly in patients under the age of 60 to 65. Yet, it should be acknowledged that a diagnostic label of “diabetes-associated cognitive decrements” always remains a probable diagnosis, purely based on the symptoms, as there are no definite signs on which it can be based5. Hence, reevaluation of the patient after 6 to 12 months is generally warranted, to verify if the course is indeed compatible with the diagnosis.
MCI and dementia clearly warrant another approach. These stages of cognitive dysfunction are associated with worse diabetes self-management and glycaemic control, with an increased frequency of hospital admissions and occurrence of severe hypoglycemic episodes, and with an increased occurrence of major cardiovascular events and death in patients with diabetes44,117,118. In order to try to avoid these adverse outcomes, screening for cognitive impairment in older adults with diabetes is being advocated116. Nevertheless, it should be acknowledged that there are still open questions regarding the actual target group and frequency for screening, the appropriate screening instrument, and – importantly – if early identification of cognitive impairment can indeed avert these adverse outcomes. With regard to the diagnostic approach of patients who are actually suspected of cognitive impairment the picture is much clearer: that should be no different from that in patients without diabetes5. As there are no diabetes specific features to MCI and dementia the same diagnostic tests are indicated as in patients without diabetes. Yet, the evaluation should consider how the cognitive deficits impact diabetes management. Particularly in patients with MCI, there should be serial assessments over time to monitor for changes in cognitive status20, as some patients may progress to dementia, whereas others may remain stable or even improve.
At present there are no treatments that can halt the processes that underlie cognitive impairment, except from adequate cardiovascular risk factor management. Importantly, such prevention strategies apply to patients of all age groups. While we have argued that there may be little benefit of actively screening for cognitive deficits in young adults or in midlife, vascular risk factor management and lifestyle modifications likely have the highest impact if started early and maintained throughout life. Of note, in patients without earlier cardiovascular events guidelines for primary prevention apply. Yet, if an MRI is performed and manifestations of small vessel disease detected, cardiovascular risk factor treatment may be modified according to recent recommendations119. Finally, it has been proposed that the less favorable risk benefit ratio of intensive glycaemic control in older individuals with diabetes with cognitive impairment is an argument to set glycaemic targets higher (for details see recent guidelines115,120).
Future perspectives
Cognitive dysfunction is now accepted as an important and common comorbidity -or even complication- of diabetes mellitus. Research over the past decades has delineated the clinical features and brain imaging correlates of diabetes-associated cognitive dysfunction in different age groups across the lifespan5,8. Insights derived from clinical research are increasingly being translated to daily clinical care for individual patients with diabetes, but there are still knowledge gaps. Current challenges include improving the delineation of the diagnostic construct of diabetes-associated cognitive decrements and development of effective strategies to detect undiagnosed frank cognitive impairment in vulnerable subjects.
Course-modifying treatment and prevention strategies for diabetes-associated cognitive dysfunction, in particular MCI and dementia, remain the highest unmet needs. Therapies should target diabetes-specific mechanisms of cognitive dysfunction. However, other contributing disease processes, not unique to diabetes, also need to be elucidated further, because disease processes that are not specific to diabetes and may not even be accelerated by diabetes, like AD, are still likely to be important contributors to cognitive dysfunction in people with diabetes, just like they are in people without diabetes. Thus, developments in the etiological treatment of AD and other dementia etiologies outside the field of diabetes are also highly relevant (e.g.121). From a prevention perspective, individuals with T2DM and a particularly elevated dementia risk can be identified with established risk scores (e.g.31). They may constitute a particularly relevant target group in dementia prevention trials. In the meantime, it is important that randomized controlled trials on prevention of diabetic complications consider cognitive outcomes, if not as a primary outcome, than at least as secondary.
TEXT Box 2. Translational potential – enhancing cross-talk between clinical and experimental studies.
Key features of cognitive dysfunction and dementia in humans with T2DM to be addressed:
Cognitive dysfunction and dementia in T2DM are due to mixed etiologies, which mostly occur in the context of brain ageing
Molecular or cellular processes involved in multiple etiologies (i.e. converging pathways) would be key targets for therapy
T2DM does not accelerate the occurrence of AD pathologies. Yet, because AD pathologies are common, also in patients with T2DM, other etiologies will often occur on a background of AD pathology
In addition to vascular pathologies, non-AD mechanisms of neurodegeneration should be a key focus of etiological research.
Insights from experimental studies to be considered:
The intervention used to induce diabetes (Table 1) can impact the cerebral phenotype in rodent models, also apart from diabetes. Animal models that adequately capture the heterogeneity of diabetes seen in humans are essential to uncover a pathological substrate for cognitive dysfunction/dementia in T2DM.
A number of non-AD processes (Table 2) appear to induce an AD-like phenotype in diabetic rodent models. For example, vascular lesions and mixed amylin-Aβ plaque formation occur in both rodent models of amylin dyshomeostasis and humans with dementia and T2DM. Understanding how these various pathways translate to cognitive dysfunction in humans with T2DM needs further investigation.
Key points.
Cognitive dysfunction in diabetes can manifest itself as diabetes-associated cognitive decrements, mild cognitive impairment (MCI), and dementia
Because of marked differences in affected age-groups and trajectories of cognitive decline, diabetes-associated cognitive decrements and dementia should be regarded as different entities, likely with different underlying mechanisms
Mechanisms of MCI and dementia in diabetes have mainly been studied in patients with T2DM and involve mixed vascular and neurodegenerative pathologies, often on a background of Alzheimer pathology, although T2DM does not increase the burden of the latter
Key causative pathways in diabetes-associated cognitive dysfunction need to be identified in order to develop course modifying therapies
Experimental models can single out individual causative pathways, in ways and at a level of detail that is impossible in humans
It is of fundamental importance that potential mechanisms of brain dysfunction identified in experimental models of diabetes are also evaluated in the complex setting of other morbidities with which they may cooccur in patients
Acknowledgements/funding
The research of GJB is supported by Vici Grant 918.16.616 from the Netherlands Organisation for Scientific Research (NWO). FD acknowledges funding from NIH (R01AG053999 and R01AG057290) Alzheimer’s Association (VMF-15-363458) and American Stroke Association (16GRNT310200).
GJB consults for and receives research support from Boehringer Ingelheim. All financial compensation for these services is transferred to his employer, the UMCU Utrecht. FD has no potential conflict of interest to disclose.
Footnotes
Literature selection
Due to the wide scope of this review, the references cited represent a selection of the available literature. Where possible, we referred to published (systematic) reviews, that provide a complete overview of available original studies. When original studies on a particular topic were quoted, and multiple studies were available, we quoted the first landmark studies and/or the most recent comprehensive studies that – in our view – represent a major advance to the field. For further background on specific topics the reader is encouraged to read the quoted papers and also explore the additional references provided in those papers.
References
- 1.International Diabetes Federation, IDF Diabetes Atlas, Eighth edition, 2017, <http://www.diabetesatlas.org/resources/2017-atlas.html> (2017). [PubMed]
- 2.Kahn SE, Cooper ME & Del Prato S Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet 383, 1068–1083 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prince M et al. The global prevalence of dementia: a systematic review and metaanalysis. Alzheimers Dement 9, 63–75 e62, doi: 10.1016/j.jalz.2012.11.007 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Biessels GJ, Staekenborg S, Brunner E, Brayne C & Scheltens P Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol 5, 64–74, doi: 10.1016/S1474-4422(05)70284-2 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Koekkoek PS, Kappelle LJ, van den Berg E, Rutten GE & Biessels GJ Cognitive function in patients with diabetes mellitus: guidance for daily care. Lancet Neurol 14, 329–340, doi: 10.1016/S1474-4422(14)70249-2 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Gudala K, Bansal D, Schifano F & Bhansali A Diabetes mellitus and risk of dementia: A meta-analysis of prospective observational studies. J Diabetes Investig 4, 640–650, doi: 10.1111/jdi.12087 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang J et al. An updated meta-analysis of cohort studies: Diabetes and risk of Alzheimer’s disease. Diabetes Res Clin Pract 124, 41–47, doi: 10.1016/j.diabres.2016.10.024 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Biessels GJ, Deary IJ & Ryan CM Cognition and diabetes: a lifespan perspective. Lancet Neurol 7, 184–190, doi: 10.1016/S1474-4422(08)70021-8 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Ryan CM, van Duinkerken E & Rosano C Neurocognitive consequences of diabetes. Am Psychol 71, 563–576, doi: 10.1037/a0040455 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Brands AM, Biessels GJ, de Haan EH, Kappelle LJ & Kessels RP The effects of type 1 diabetes on cognitive performance: a meta-analysis. Diabetes Care 28, 726–735 (2005). [DOI] [PubMed] [Google Scholar]
- 11.The Diabetes C, Complications Trial/Epidemiology of, D. & Complications Study Research, G. Long-term effect of diabetes and its treatment on cognitive function. N.Engl.J.Med. 356, 1842–1852 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nunley KA et al. Clinically Relevant Cognitive Impairment in Middle-Aged Adults With Childhood-Onset Type 1 Diabetes. Diabetes Care 38, 1768–1776, doi: 10.2337/dc15-0041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Monette MC, Baird A & Jackson DL A meta-analysis of cognitive functioning in nondemented adults with type 2 diabetes mellitus. Can J Diabetes 38, 401–408, doi: 10.1016/j.jcjd.2014.01.014 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Palta P, Schneider AL, Biessels GJ, Touradji P & Hill-Briggs F Magnitude of cognitive dysfunction in adults with type 2 diabetes: a meta-analysis of six cognitive domains and the most frequently reported neuropsychological tests within domains. J.Int.Neuropsychol.Soc. 20, 278–291 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Biessels GJ, Strachan MW, Visseren FL, Kappelle LJ & Whitmer RA Dementia and cognitive decline in type 2 diabetes and prediabetic stages: towards targeted interventions. Lancet Diabetes Endocrinol 2, 246–255, doi: 10.1016/S2213-8587(13)70088-3 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Bangen KJ et al. Relationship Between Type 2 Diabetes Mellitus and Cognitive Change in a Multiethnic Elderly Cohort. J Am Geriatr Soc 63, 1075–1083, doi: 10.1111/jgs.13441 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pappas C, Andel R, Infurna FJ & Seetharaman S Glycated haemoglobin (HbA1c), diabetes and trajectories of change in episodic memory performance. J Epidemiol Community Health 71, 115–120, doi: 10.1136/jech-2016-207588 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Yaffe K et al. Diabetes, glucose control, and 9-year cognitive decline among older adults without dementia. Arch.Neurol. 69, 1170–1175 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petersen RC Mild cognitive impairment as a diagnostic entity. J.Intern.Med. 256, 183–194 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Petersen RC et al. Practice guideline update summary: Mild cognitive impairment: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology 90, 126–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.American Psychiatric A Diagnostic and Statistical Manual of Mental Disorders, Fourth edition (DSM-IV). (1994). [Google Scholar]
- 22.Scheltens P et al. Alzheimer’s disease. Lancet (2016). [Google Scholar]
- 23.Dubois B et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol 9, 1118–1127, doi: 10.1016/S1474-4422(10)70223-4 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Luchsinger JA et al. Relation of diabetes to mild cognitive impairment. Arch Neurol 64, 570–575, doi: 10.1001/archneur.64.4.570 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Roberts RO et al. Association of diabetes with amnestic and nonamnestic mild cognitive impairment. Alzheimers Dement 10, 18–26, doi: 10.1016/j.jalz.2013.01.001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cooper C, Sommerlad A, Lyketsos CG & Livingston G Modifiable predictors of dementia in mild cognitive impairment: a systematic review and meta-analysis. Am J Psychiatry 172, 323–334, doi: 10.1176/appi.ajp.2014.14070878 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Li JQ et al. Risk factors for predicting progression from mild cognitive impairment to Alzheimer’s disease: a systematic review and meta-analysis of cohort studies. J Neurol Neurosurg Psychiatry 87, 476–484, doi: 10.1136/jnnp-2014-310095 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Chatterjee S et al. Type 2 Diabetes as a Risk Factor for Dementia in Women Compared With Men: A Pooled Analysis of 2.3 Million People Comprising More Than 100,000 Cases of Dementia. Diabetes Care 39, 300–307, doi: 10.2337/dc15-1588 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haroon NN et al. Risk of dementia in seniors with newly diagnosed diabetes: a population-based study. Diabetes Care 38, 1868–1875, doi: 10.2337/dc15-0491 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Crane PK et al. Glucose levels and risk of dementia. N.Engl.J.Med. 369, 540–548 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Exalto LG et al. Risk score for prediction of 10 year dementia risk in individuals with type 2 diabetes: a cohort study. Lancet Diabetes Endocrinol 1, 183–190, doi: 10.1016/S2213-8587(13)70048-2 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brady CC et al. Obese adolescents with type 2 diabetes perform worse than controls on cognitive and behavioral assessments. Pediatr Diabetes 18, 297–303, doi: 10.1111/pedi.12383 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Yates KF, Sweat V, Yau PL, Turchiano MM & Convit A Impact of metabolic syndrome on cognition and brain: a selected review of the literature. Arterioscler Thromb Vasc Biol 32, 2060–2067, doi: 10.1161/ATVBAHA.112.252759 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van den Berg E, de Craen AJ, Biessels GJ, Gussekloo J & Westendorp RG The impact of diabetes mellitus on cognitive decline in the oldest of the old: a prospective population-based study. Diabetologia 49, 2015–2023, doi: 10.1007/s00125-006-0333-1 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Kadohara K, Sato I & Kawakami K Diabetes mellitus and risk of early-onset Alzheimer’s disease: a population-based case-control study. Eur J Neurol 24, 944–949, doi: 10.1111/ene.13312 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Heath CA, Mercer SW & Guthrie B Vascular comorbidities in younger people with dementia: a cross-sectional population-based study of 616 245 middle-aged people in Scotland. J Neurol Neurosurg Psychiatry 86, 959–964, doi: 10.1136/jnnp-2014-309033 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Abner EL et al. Diabetes is associated with cerebrovascular but not Alzheimer’s disease neuropathology. Alzheimers Dement 12, 882–889, doi: 10.1016/j.jalz.2015.12.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feinkohl I, Price JF, Strachan MW & Frier BM The impact of diabetes on cognitive decline: potential vascular, metabolic, and psychosocial risk factors. Alzheimers Res Ther 7, 46, doi: 10.1186/s13195-015-0130-5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geijselaers SLC, Sep SJS, Stehouwer CDA & Biessels GJ Glucose regulation, cognition, and brain MRI in type 2 diabetes: a systematic review. Lancet Diabetes Endocrinol 3, 75–89, doi: 10.1016/S2213-8587(14)70148-2 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Rawlings AM et al. Glucose Peaks and the Risk of Dementia and 20-Year Cognitive Decline. Diabetes Care 40, 879–886, doi: 10.2337/dc16-2203 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patrone C, Eriksson O & Lindholm D Diabetes drugs and neurological disorders: new views and therapeutic possibilities. Lancet Diabetes Endocrinol. 2, 256–262 (2014). [DOI] [PubMed] [Google Scholar]
- 42.Orkaby AR, Cho K, Cormack J, Gagnon DR & Driver JA Metformin vs sulfonylurea use and risk of dementia in US veterans aged >/=65 years with diabetes. Neurology (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Areosa Sastre A, Vernooij RW, Gonzalez-Colaco Harmand M & Martinez G Effect of the treatment of Type 2 diabetes mellitus on the development of cognitive impairment and dementia. Cochrane Database Syst Rev 6, CD003804 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.de Galan BE et al. Cognitive function and risks of cardiovascular disease and hypoglycaemia in patients with type 2 diabetes: the Action in Diabetes and Vascular Disease: Preterax and Diamicron Modified Release Controlled Evaluation (ADVANCE) trial. Diabetologia 52, 2328–2336 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Norton S, Matthews FE, Barnes DE, Yaffe K & Brayne C Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 13, 788–794 (2014). [DOI] [PubMed] [Google Scholar]
- 46.de Bruijn RF et al. The potential for prevention of dementia across two decades: the prospective, population-based Rotterdam Study. BMC.Med. 13, 132 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Biessels GJ & Reijmer YD Brain changes underlying cognitive dysfunction in diabetes: what can we learn from MRI? Diabetes 63, 2244–2252, doi: 10.2337/db14-0348 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Moran C et al. Neuroimaging and its Relevance to Understanding Pathways Linking Diabetes and Cognitive Dysfunction. J Alzheimers Dis 59, 405–419, doi: 10.3233/JAD-161166 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Wardlaw JM et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 12, 822–838 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wardlaw JM, Valdes Hernandez MC & Munoz-Maniega S What are white matter hyperintensities made of? Relevance to vascular cognitive impairment. J Am Heart Assoc 4, 001140, doi: 10.1161/JAHA.114.001140 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qiu C et al. Diabetes, markers of brain pathology and cognitive function: the Age, Gene/Environment Susceptibility-Reykjavik Study. Ann Neurol 75, 138–146, doi: 10.1002/ana.24063 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Caunca MR et al. Cerebral Microbleeds, Vascular Risk Factors, and Magnetic Resonance Imaging Markers: The Northern Manhattan Study. J Am Heart Assoc 5, doi: 10.1161/JAHA.116.003477 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gutierrez J, Rundek T, Ekind MS, Sacco RL & Wright CB Perivascular spaces are associated with atherosclerosis: an insight from the Northern Manhattan Study. AJNR Am J Neuroradiol 34, 1711–1716, doi: 10.3174/ajnr.A3498 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bouvy WH et al. Perivascular spaces on 7 Tesla brain MRI are related to markers of small vessel disease but not to age or cardiovascular risk factors. J Cereb Blood Flow Metab 36, 1708–1717, doi: 10.1177/0271678X16648970 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Veluw SJ et al. Detection, risk factors, and functional consequences of cerebral microinfarcts. Lancet Neurol 16, 730–740, doi: 10.1016/S1474-4422(17)30196-5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pruzin JJ et al. Diabetes, Hemoglobin A1C, and Regional Alzheimer Disease and Infarct Pathology. Alzheimer Dis Assoc Disord 31, 41–47, doi: 10.1097/WAD.0000000000000172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wardlaw JM, Smith C & Dichgans M Mechanisms of sporadic cerebral small vessel disease: insights from neuroimaging. Lancet Neurol. 12, 483–497 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nelson PT et al. Human cerebral neuropathology of Type 2 diabetes mellitus. Biochim.Biophys.Acta 1792, 454–469 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ly H et al. Brain microvascular injury and white matter disease provoked by diabetes-associated hyperamylinemia. Ann Neurol 82, 208–222, doi: 10.1002/ana.24992 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Brundel M, Kappelle LJ & Biessels GJ Brain imaging in type 2 diabetes. Eur Neuropsychopharmacol 24, 1967–1981, doi: 10.1016/j.euroneuro.2014.01.023 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Arvanitakis Z et al. Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology 67, 1960–1965 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Dos Santos Matioli MNP et al. Diabetes is Not Associated with Alzheimer’s Disease Neuropathology. J Alzheimers Dis 60, 1035–1043, doi: 10.3233/JAD-170179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gottesman RF et al. Association Between Midlife Vascular Risk Factors and Estimated Brain Amyloid Deposition. JAMA 317, 1443–1450, doi: 10.1001/jama.2017.3090 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moran C et al. Type 2 diabetes mellitus and biomarkers of neurodegeneration. Neurology 85, 1123–1130, doi: 10.1212/WNL.0000000000001982 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vemuri P et al. Age, vascular health, and Alzheimer disease biomarkers in an elderly sample. Ann Neurol 82, 706–718, doi: 10.1002/ana.25071 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Biessels GJ & Reagan LP Hippocampal insulin resistance and cognitive dysfunction. Nat Rev Neurosci 16, 660–671, doi: 10.1038/nrn4019 (2015). [DOI] [PubMed] [Google Scholar]
- 67.Arnold SE et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol 14, 168–181, doi: 10.1038/nrneurol.2017.185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Moran C et al. Type 2 diabetes, skin autofluorescence, and brain atrophy. Diabetes 64, 279–283 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Janelidze S et al. Increased blood-brain barrier permeability is associated with dementia and diabetes but not amyloid pathology or APOE genotype. Neurobiol Aging 51, 104–112, doi: 10.1016/j.neurobiolaging.2016.11.017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Biessels GJ & Gispen WH The impact of diabetes on cognition: What can be learned from rodent models? Neurobiol.Aging 26 Suppl 1, 36–41 (2005). [DOI] [PubMed] [Google Scholar]
- 71.Clodfelder-Miller BJ, Zmijewska AA, Johnson GV & Jope RS Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes 55, 3320–3325 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de la Monte SM, Tong M, Lester-Coll N, Plater M Jr. & Wands JR Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer’s disease. J Alzheimers Dis 10, 89–109 (2006). [DOI] [PubMed] [Google Scholar]
- 73.Planel E et al. Insulin dysfunction induces in vivo tau hyperphosphorylation through distinct mechanisms. The Journal of Neuroscience 27, 13635–13648 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim B, Backus C, Oh S, Hayes JM & Feldman EL Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology 150, 5294–5301, doi: 10.1210/en.2009-0695 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li ZG, Zhang W & Sima AA Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes 56, 1817–1824, doi: 10.2337/db07-0171 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Puig KL, Floden AM, Adhikari R, Golovko MY & Combs CK Amyloid precursor protein and proinflammatory changes are regulated in brain and adipose tissue in a murine model of high fat diet-induced obesity. PLoS One 7, e30378, doi: 10.1371/journal.pone.0030378 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu Y et al. Impaired amyloid beta-degrading enzymes in brain of streptozotocin-induced diabetic rats. J Endocrinol Invest 34, 26–31, doi: 10.3275/6995 (2011). [DOI] [PubMed] [Google Scholar]
- 78.Son SM et al. Accumulation of autophagosomes contributes to enhanced amyloidogenic APP processing under insulin-resistant conditions. Autophagy 8, 1842–1844, doi: 10.4161/auto.21861 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang JQ et al. Brain aging and AD-like pathology in streptozotocin-induced diabetic rats. J Diabetes Res 2014, 796840, doi: 10.1155/2014/796840 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heyward FD et al. Adult mice maintained on a high-fat diet exhibit object location memory deficits and reduced hippocampal SIRT1 gene expression. Neurobiol Learn Mem 98, 25–32, doi: 10.1016/j.nlm.2012.04.005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ramos-Rodriguez JJ et al. Differential central pathology and cognitive impairment in pre-diabetic and diabetic mice. Psychoneuroendocrinology 38, 2462–2475, doi: 10.1016/j.psyneuen.2013.05.010 (2013). [DOI] [PubMed] [Google Scholar]
- 82.Takeda S et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A 107, 7036–7041, doi: 10.1073/pnas.1000645107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Niedowicz DM et al. Obesity and diabetes cause cognitive dysfunction in the absence of accelerated beta-amyloid deposition in a novel murine model of mixed or vascular dementia. Acta Neuropathol Commun 2, 64, doi: 10.1186/2051-5960-2-64 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ramos-Rodriguez JJ et al. Prediabetes-induced vascular alterations exacerbate central pathology in APPswe/PS1dE9 mice. Psychoneuroendocrinology 48, 123–135, doi: 10.1016/j.psyneuen.2014.06.005 (2014). [DOI] [PubMed] [Google Scholar]
- 85.Devi L, Alldred MJ, Ginsberg SD & Ohno M Mechanisms underlying insulin deficiency-induced acceleration of beta-amyloidosis in a mouse model of Alzheimer’s disease. PLoS One 7, e32792, doi: 10.1371/journal.pone.0032792 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gao C, Liu Y, Li L & Holscher C New animal models of Alzheimer’s disease that display insulin desensitization in the brain. Rev Neurosci 24, 607–615, doi: 10.1515/revneuro-2013-0034 (2013). [DOI] [PubMed] [Google Scholar]
- 87.Bell GA & Fadool DA Awake, long-term intranasal insulin treatment does not affect object memory, odor discrimination, or reversal learning in mice. Physiol Behav 174, 104–113, doi: 10.1016/j.physbeh.2017.02.044 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marks DR, Tucker K, Cavallin MA, Mast TG & Fadool DA Awake intranasal insulin delivery modifies protein complexes and alters memory, anxiety, and olfactory behaviors. J Neurosci 29, 6734–6751, doi: 10.1523/JNEUROSCI.1350-09.2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Srodulski S et al. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol Neurodegener 9, 30, doi: 10.1186/1750-1326-9-30 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Beckman JA & Creager MA Vascular Complications of Diabetes. Circ Res 118, 1771–1785, doi: 10.1161/CIRCRESAHA.115.306884 (2016). [DOI] [PubMed] [Google Scholar]
- 91.Basta G, Schmidt AM & De Caterina R Advanced glycation end products and vascular inflammation: implications for accelerated atherosclerosis in diabetes. Cardiovasc Res 63, 582–592, doi: 10.1016/j.cardiores.2004.05.001 (2004). [DOI] [PubMed] [Google Scholar]
- 92.Quaegebeur A, Lange C & Carmeliet P The neurovascular link in health and disease: molecular mechanisms and therapeutic implications. Neuron 71, 406–424, doi: 10.1016/j.neuron.2011.07.013 (2011). [DOI] [PubMed] [Google Scholar]
- 93.Iadecola C The pathobiology of vascular dementia. Neuron 80, 844–866, doi: 10.1016/j.neuron.2013.10.008 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Viswanathan A, Rocca WA & Tzourio C Vascular risk factors and dementia: how to move forward? Neurology 72, 368–374 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ilaiwy A et al. Human amylin proteotoxicity impairs protein biosynthesis, and alters major cellular signaling pathways in the heart, brain and liver of humanized diabetic rat model in vivo. Metabolomics 12, doi: 10.1007/s11306-016-1022-9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mattson MP & Rydel RE Alzheimer’s disease. Amyloid ox-tox transducers. Nature 382, 674–675, doi: 10.1038/382674a0 (1996). [DOI] [PubMed] [Google Scholar]
- 97.Verma N et al. Intraneuronal Amylin Deposition, Peroxidative Membrane Injury and Increased IL-1beta Synthesis in Brains of Alzheimer’s Disease Patients with Type-2 Diabetes and in Diabetic HIP Rats. J Alzheimers Dis 53, 259–272, doi: 10.3233/JAD-160047 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Neth BJ & Craft S Insulin Resistance and Alzheimer’s Disease: Bioenergetic Linkages. Front Aging Neurosci 9, 345, doi: 10.3389/fnagi.2017.00345 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Biessels GJ, ter Laak MP, Hamers FP & Gispen WH Neuronal Ca (2+) disregulation in diabetes mellitus. Eur.J.Pharmacol. 447, 201–209 (2002). [DOI] [PubMed] [Google Scholar]
- 100.Erickson JR et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 502, 372–376, doi: 10.1038/nature12537 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Westermark P, Andersson A & Westermark GT Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev 91, 795–826, doi: 10.1152/physrev.00042.2009 (2011). [DOI] [PubMed] [Google Scholar]
- 102.Gong W et al. Amylin deposition in the kidney of patients with diabetic nephropathy. Kidney Int 72, 213–218, doi: 10.1038/sj.ki.5002305 (2007). [DOI] [PubMed] [Google Scholar]
- 103.Despa S et al. Hyperamylinemia contributes to cardiac dysfunction in obesity and diabetes: a study in humans and rats. Circ Res 110, 598–608, doi: 10.1161/CIRCRESAHA.111.258285 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jackson K et al. Amylin deposition in the brain: A second amyloid in Alzheimer’s disease? Ann.Neurol. (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Oskarsson ME et al. In vivo seeding and cross-seeding of localized amyloidosis: a molecular link between type 2 diabetes and Alzheimer disease. Am J Pathol 185, 834–846, doi: 10.1016/j.ajpath.2014.11.016 (2015). [DOI] [PubMed] [Google Scholar]
- 106.Schultz N, Byman E, Fex M & Wennstrom M Amylin alters human brain pericyte viability and NG2 expression. J Cereb Blood Flow Metab 37, 1470–1482, doi: 10.1177/0271678X16657093 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fawver JN et al. Islet amyloid polypeptide (IAPP): a second amyloid in Alzheimer’s disease. Curr Alzheimer Res 11, 928–940 (2014). [DOI] [PubMed] [Google Scholar]
- 108.Roostaei T et al. Genome-wide interaction study of brain beta-amyloid burden and cognitive impairment in Alzheimer’s disease. Mol Psychiatry 22, 287–295, doi: 10.1038/mp.2016.35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Matveyenko AV & Butler PC Islet amyloid polypeptide (IAPP) transgenic rodents as models for type 2 diabetes. ILAR J 47, 225–233 (2006). [DOI] [PubMed] [Google Scholar]
- 110.Moreno-Gonzalez I et al. Molecular interaction between type 2 diabetes and Alzheimer’s disease through cross-seeding of protein misfolding. Mol Psychiatry 22, 1327–1334, doi: 10.1038/mp.2016.230 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang Z et al. Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J Neurosci 29, 10788–10801, doi: 10.1523/JNEUROSCI.2132-09.2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Young-Pearse TL, Chen AC, Chang R, Marquez C & Selkoe DJ Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev 3, 15, doi: 10.1186/1749-8104-3-15 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA & Schneider JA Relation of cerebral vessel disease to Alzheimer’s disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol 15, 934–943, doi: 10.1016/S1474-4422(16)30029-1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.International Diabetes Federation, Global Guideline for Managing Older People with Type 2 Diabetes, <https://www.idf.org/e-library/guidelines/78-global-guideline-for-managing-older-people-with-type-2-diabetes.html> (2013).
- 115.Committee Report: Glycemic targets for elderly patients with diabetes: Japan Diabetes Society (JDS)/Japan Geriatrics Society (JGS) Joint Committee on Improving Care for Elderly Patients with Diabetes. J Diabetes Investig 8, 126–128, doi: 10.1111/jdi.12599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.America Diabetes Association, Standards of Medical Care in Diabetes-2017. Diabetes Care 40, S1–S142, doi: 10.2337/dc17-S003 (2017). [DOI] [PubMed] [Google Scholar]
- 117.Munshi MN Cognitive Dysfunction in Older Adults With Diabetes: What a Clinician Needs to Know. Diabetes Care 40, 461–467, doi: 10.2337/dc16-1229 (2017). [DOI] [PubMed] [Google Scholar]
- 118.Feil DG et al. Risk of hypoglycemia in older veterans with dementia and cognitive impairment: implications for practice and policy. J.Am.Geriatr.Soc. 59, 2263–2272 (2011). [DOI] [PubMed] [Google Scholar]
- 119.Smith EE et al. Prevention of Stroke in Patients With Silent Cerebrovascular Disease: A Scientific Statement for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 48, e44–e71, doi: 10.1161/STR.0000000000000116 (2017). [DOI] [PubMed] [Google Scholar]
- 120.American Diabete Association, Standards of Medical Care in Diabetes-2017: Section 11. Older Adults. Diabetes Care 40, S99–S104, doi: 10.2337/dc17-S003 (2017). [DOI] [PubMed] [Google Scholar]
- 121.Sevigny J et al. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537, 50–56, doi: 10.1038/nature19323 (2016). [DOI] [PubMed] [Google Scholar]