

Abstract
Crohn’s disease and ulcerative colitis, collectively known as inflammatory bowel disease (IBD), are chronic inflammatory disorders of the gastrointestinal tract. Although the etiopathogenesis of IBD is largely unknown, it is widely thought that diet has a crucial role in the development and progression of IBD. Epidemiological and genetic association studies have identified a few promising dietary and genetic risk factors. These preliminary studies have promted significant interest in investigating the complex interaction between diet, host genetics, the gut microbiota and immune function in the pathogenesis of IBD. In this Review, we discuss the recent epidemiological, gene–environment interaction, microbiome, and animal studies that have explored the relationship between diet and the risk of IBD. Additionally, we will highlight the limitations of these prior studies, in part explaining their contradictory findings, and review future directions.
Table of Contents Blurb
Emerging data implicate diet as a crucial environmental factor in the development and progression of inflammatory bowel disease (IBD). Here, the authors discuss findings from epidemiological, animal, gene–environment interaction and gut microbiome studies that have investigated the role of diet in the etiopathogenesis of IBD.
Introduction
Crohn’s disease and ulcerative colitis, collectively known as inflammatory bowel disease (IBD), are chronic inflammatory disorders of the gastrointestinal tract that have an increasing incidence12 and affect over one million individuals in the US and 2.5 million in Europe 13, 14, resulting in substantial morbidity15, health care expenses and loss of productivity 16, 17. Although the pathophysiology of IBD is largely unknown, it is thought to be related to an inappropriate immune response to commensal bacteria in genetically susceptible hosts (FIG. 1). A role for genetic risk factors in the development of IBD has been highlighted by the identification of >200 susceptibility loci in genome-wide association studies (GWAS)18, 19. In addition to the genetic contribution to IBD pathophysiology, environmental factors also seem to substantially contribute to both the development and progression of IBD. For instance, twin studies have demonstrated a <50% concordance in the development of Crohn’s disease and ulcerative colitis20, 21. In addition, epidemiological studies have confirmed the rapid increase in the incidence of IBD in the US and developing countries that have witnessed a dramatic ‘westernization’ of lifestyle with the worldwide prevalence of the disease surpassing 0.3% at the turn of 21st century. 12, 22. Moreover, studies have shown that the incidence of IBD in immigrants to developed countries exceeds that of individuals from their country of origin23–26.
Figure 1 |. The pathophysiology of inflammatory bowel disease.
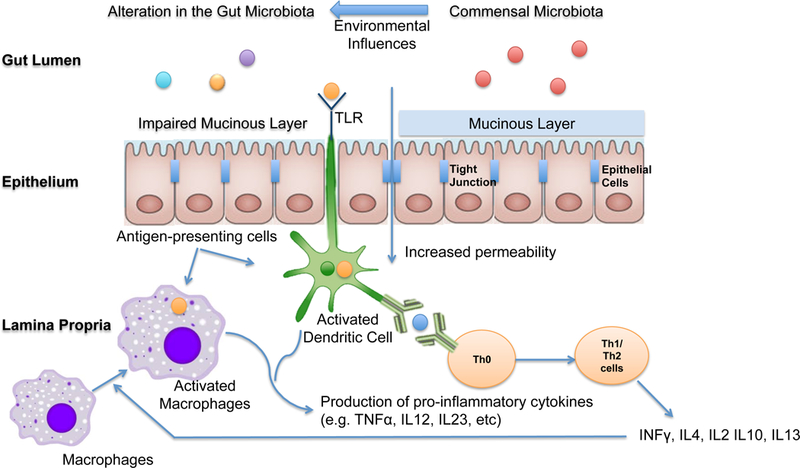
The pathophysiology of inflammatory bowel disease (IBD) is related to an inappropriate host immune response to commensal bacteria in genetically susceptible individuals. Environmental influences alter the composition of the gut microbiota and change mucosal barrier function. Dendritic cells and macrophages are antigen-presenting cells involved in activation of T cells and production of pro-inflammatory cytokines. Dendritic cells are activated through the the recognition of luminal antigens by Toll-like receptors (TLRs) and are in turn necessary for activation of naïve T cells. Macrophages could also serve as antigen-presenting cells once stimulated by INFγ secreted by T cells. Activated macrophages and dendritic cells also produce pro-inflammatory cytokines including TNF-alpha, IL212, and IL23. The result of this pro-inflamamtory cytokine cascade is loss of immune tolerance to commensal bacteria and the production of pro-inflammatory cytokines by activated T cells. INF, Inteferon-gamma;TNF, tumour necrosis factor; IL-312, interleukin-12; IL-23, interleukin-23;
Among environmental factors, diet is widely thought to have a pivotal role in the development of IBD. Although the exact pathophysiological mechanisms remain unknown, a number of plausible explanations have been proposed (FIG. 2) Firstly, diet has a key role in defining the composition of the human gut microbiota and, consequently, that of microbial metabolites 27. Secondly, food and nutrients associated with a Western diet, characterized by high intakes of red meat, sugary desserts, high-fat foods, and refined grains have been linked to increased mucosal inflammation as measured by stool calprotectin in human subjects 28. Lastly, animal studies have demonstrated that dietary composition regulates mucosal barrier function, a crucial factor in the pathogenesis of IBD29, 30. Despite these data, human observational studies investigating the role of diet in IBD have yielded contradictory and inconclusive results.
Figure 2 |. Potential mechanisms underpinning the relationship between diet and inflammatory bowel disease.
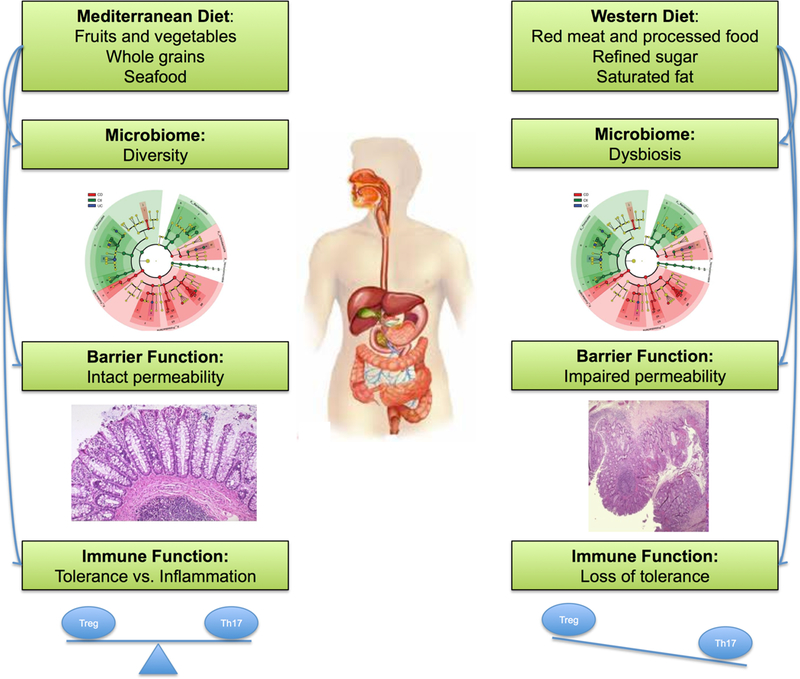
As compared to Mediterranean diet which is characterized by high intake of fruits and vegetables, whole grains, and sea good, western diet characterized by high intake of red and processed meat, saturated fat, and refined sugar is widely thought to increase risk of IBD. Although the exact mechanism underpinning the association between diet and risk of IBD is unknown, a number of plausible mechanisms have been proposed. Specifically, western diet has been linked to changes in the gut microbiome and epithelial barrier function and appears to have a direct impact on the immune function triggering a pro-inflammatory environment characterized by an imbalance in the TH17/Treg axis.
In this Review, we will discuss the available data on the role of diet in the pathogenesis of IBD, with a focus on epidemiological,, gene–environment interaction, intervention, gut microbiome, and animal studies. We will also highlight limitation of prior studies, particularly epidemiological and gene-environment studies and review future directions that build on these preliminary studies.
Intervention studies
Recently, there has been a resurgence of interest in the potential of dietary intervention for the treatment of IBD. Although a comprehensive discussion of prior dietary intervention studies is beyond the scope of this Review and has been previously published31, the early success of these studies further supports a role for diet in the pathogenesis of IBD. For example, several studies have demonstrated that exclusive enteral nutrition (EEN), which involves the administration of a liquid diet formula for a defined period of time, improves both clinical symptoms and intestinal inflammation in patients with Crohn’s disease; one randomized controlled trial demonstrated that a short-term EEN was superior to corticosteroids in promoting mucosal healing in paediatric patients with Crohn’s disease32, 33. Nevertheless, similar studies in adults, where compliance to such a restrictive diet might be challenging, have not been as promising33. Lastly, a number of uncontrolled human intervention studies have also reported promising results for other diets in the treatment of IBD including specific carbohydrate diet (SCD), low-fermentable oligosaccharide, disaccharide, monosaccharide, and polyol (FODMAP) diet and Paleolithic diet 34. Although large, well-designed, randomized studies in humans are needed to fully examine the therapeutic effects of these diets in IBD. These early results suggest a role for diet in IBD pathogenesis and support the need for further investigation in this area.
Epidemiological studies
A limited number of prior studies have examined the association between specific dietary factors and risk of IBD35–37. Unfortunately, many of these studies have a number of drawbacks. Specifically, most prior studies were retrospective case–control analyses, and are therefore subject to numerous limitations including recall and selection biases. Recall bias of diet before the onset of IBD symptoms is particularly problematic. For example, if individuals are recruited after an extended period following diagnosis, they may have significantly changed their diet or preferentially recall consumption of specific foods that exacerbate their symptoms. Similarly, recall of diet within a very brief interval post-diagnosis is subject to concerns about reverse causation, whereby symptoms that immediately precede the formal diagnosis might alter dietary intake. In addition, the majority of previous studies collected dietary data at a single time point and were therefore unable to account for variations in dietary intake over time. There have also been very few studies that have rigorously validated dietary assessments within their study populations. Finally, because of the relatively low absolute incidence of IBD in the general population, most prior cohort studies have been limited by sample size. Taken together, these limitations might contribute to the inconsistent associations between dietary factors and the risk of IBD that is generally seen in the scientific literature.
To address these limitations, over the past 10 years, a number of large prospective cohort studies have attempted to better characterize the link between diet and risk of IBD by leveraging validated and updated dietary data collected during long-term follow-up (TABLE 1). The majority of these studies used the semiquantitative food frequency questionnaire (SFFQ) to estimate long-term dietary intake. The SFFQ is a method of assessing food and nutrient intake over a specified period of time by obtaining data on the frequency — and in some cases portion sizes — of food and beverage consumption38. The SFFQ has several advantages over other forms of dietary assessment (such as 24 hour recall or dietary records) in that it focuses on patterns of dietary intake over longer periods of time and therefore avoids biases that could be introduced through the episodic or occasional consumption of specific foods and beverages. In addition, SFFQs are relatively easy to complete by participants, therefore providing an ideal method to conduct large-scale epidemiological studies of diet in chronic diseases. Lastly, the SFFQ has been extensively validated against diet records (Box 1), allowing for the use of statistical methods to account for measurement errors. By contrast, the SFFQ is limited by its use in specific populations with well-characterized patterns of dietary consumption (i.e. Western societies) and is subject to over-reporting of consumption of healthy foods as a consequence of social desirability bias.
Table 1|.
Prospective studies of diet and inflammatory bowel disease in the EPIC, NHS and NHSII cohorts
| Study | Dietary exposure(s) | Outcome( s) |
Population and design |
Key findings* | Ref s |
|---|---|---|---|---|---|
| Hart et al. 2008 | Intake of carbohydrates; fatty acids; protein; fiber; alcohol; and micronutrients, including vitamin C, vitamin D, vitamin E, calcium and iron. | UC | • Nested case– control study using prospectively-collected dietary data from the EPIC cohort. • 139 cases of incident UC matched 1:4 to 556 controls. |
• No statistically significant associations between dietary factor intakes and UC. • A nonsignificant trend towards higher risk of UC associated with PUFA as percentage of total energy intake (OR 1.56, 95% CI 0.87–2.77 for highest versus lowest quartile; Ptrend=0.07 ). |
55 |
| Hart et al. 2009 | Intake of linoleic acid; α-linoleic acid; oleic acid; DHA; and EPA. | UC | • Nested case– control study using prospectively- collected dietary data from the EPIC cohort. • 126 cases of incident UC over matched 1:4 to 504 controls. |
• Higher linoleic acid intake was associated with increased risk of UC (OR 2.49, 95% CI 1.23–5.07 for highest versus. lowest quartile; Ptrend=0.02 ). • Higher DHA intake was associated with lower risk of UC (OR 0.23, 95% CI 0.06–0.97 for highest versus lowest quartile; Ptrend=0.03) |
56 |
| Ananthakrishn an et al. 2013 | Energy-adjusted, cumulative average intake of total dietary fiber; and fiber intake from different dietary sources. | CD and UC | • Prospective analysis of 170,776 w omen enrolled in the NHS and NHS II. • 269 incident cases of CD and 338 incident cases of UC during follow-up |
• Total fiber intake was associated with lower risk of CD (HR 0.59, 95% CI 0.39–0.90 for highest versus lowest quintile; Ptrend=0.08 ). • The association was strongest for fiber from fruit sources (HR 0.57, 95% CI 0.38–0.85; Ptrend=0.02). • No statistically significant associations were observed between fiber intake and risk of UC. |
40 |
| Ananthakrishn an et al. 2014 | Energy-adjusted, cumulative average intake of total fat; saturated fats; unsaturated fats; and n-6 and n-3 PUFAs. | CD and UC | • Prospective analysis of 170,805 women enrolled in the NHS and NHS II. • 269 incident cases of CD and 338 incident cases of UC during follow-up. |
• No associations were observed between intakes of total fat, saturated fat, monounsatur ated fat or total PUFAs and risk of CD or UC. • Higher n-3:n-6 PUFA ratio was associated with a lower risk of UC (HR 0.69, 95% CI 0.49–0.98 for highest versus lowest quintile; Ptrend=0.03). |
41 |
| Chan et al. 2014 | Intake of total fat; linoleic acid; α-linoleic acid; oleic acid; DHA; and EPA. | CD | • Nested case– control study using prospectively- collected dietary data from the EPIC cohort. • 73 cases of incident CD matched 1:4 to 292 controls. |
• Increasing intake of DHA was associated with a lower risk of CD (OR 0.07, 95% CI 0.02–0.81 for highest versus lowest quintile; Ptrend =0.04). • No statistically significant associations were observed for other fatty acids or total fat intake. |
59 |
| Chan et al. 2014 | Intake of total carbohydrates, simple sugars; and starch. | CD and UC | • Nested case– control study using prospectively- collected dietary data from the EPIC cohort. • 110 cases of incident CD and 244 cases of incident UC matched 1:4 to 440 and 976 controls, respective ly. |
• No statistically significant associations were observed between total dietary carbohydrate, sugar or starch intakes and the risk of CD or UC. | 60 |
| Ananthakrishn an et al. 2015 | Total zinc intake (from both dietary and supplemental sources). | CD and UC | • Prospective analysis of 170, 809 women enrolled in the NHS and NHS II. • 269 incident cases of CD and 338 incident cases of UC during follow-up. |
• Higher intake of zinc was associated with lower risk of CD (HR 0.57, 95% CI 0.38–0.86 for highest versus lowest quintile; Ptrend=0.003). • Reduction in risk of CD was observed with a daily zinc intake of up to 15mg. |
52 |
| Ananthakrishn an et al. 2015 | ‘Prudent’ and ‘Western’ high-school dietary patterns (at age 13–17 years). | CD and UC | • Prospective analysis of 39,511 women enrolled in NHS II. • 70 cases of CD and 103 cases of UC during follow-up. |
• Higher prudent diet score during adolescence was associated with lower risk of CD (HR 0.47, 95% CI 0.23–0.98 for highest versus lowest score quartiles; Ptrend =0.04) but not UC. | 117 |
| Khalili et at. 2016 | Dietary potassium and sodium intake. | CD and UC | • Prospective analysis of 194,711 women enrolled in the NHS and NHS II. • 273 incident cases of CD and 335 incident cases of UC during follow-up. |
• Increasing dietary potassium intake was associated with lower risk of CD (HR 0.62, 95% CI 0.40–0.95 for highest versus lowest quintile; Ptrend=0.005). • There was a trend toward an inverse association between potassium intake and risk of UC (Ptrend=0.08). |
51 |
| Racine et al. 2016 | aMED score and three a posteriori derived principal component dietary patterns. | CD and UC | • Nested case– control study using prospectively- collected dietary data from the EPIC cohort. • 117 cases of incident CD and 256 cases of incident UC matched 1:4 to 468 and 1022 controls, respective ly. |
• No association was detected between the aMED score and risk of CD or UC. • There was an association between the ‘sugar and soft drinks’ dietary pattern and risk of UC (IRR 1.31, 95% CI 0.85– 2.02 for the highest versus the lowest quintile; Ptrend =0.05). |
61 |
| Opstelten et al. 2016 | Intake of milk; cheese; yoghurt; and total dairy products. Dietary calcium intake (without supplements). | CD and UC | • Nested case – control study using prospectively-collected dietary data from the EPIC cohort. • 110 cases of incident CD and 244 cases of incident UC matched 1:4 to 440 controls, respectively. |
• No statistically significant associations with risk of CD or UC were observed across quartiles of total or individual dairy product intakes or dietary calcium intake. • Compared to those who did not drink milk, any milk consumption was associated with a lower risk of CD (OR 0.30, 95% CI 0.13–0.65) but not UC |
62 |
| Khalili et al. 2017 | Dietary total iron and heme iron intakes. Red and processed meat intakes. | CD and UC | • Prospective analysis of 150,387 women enrolled in the NHS and NHS II. 260 incident cases of CD and 319 incident cases of UC during follow-up. | • There was a trend toward higher risk of UC associated with dietary heme iron intake (HR 1.26, 95%CI 0.86–1.83 for highest versus lowest quintile; Ptrend=0.10). • In a lagged analysis, red meat intake was associated with higher risk of UC (HR 1.15, 95% CI 1.01–1.30 for each additional weekly serving of red meat). |
42 |
| Bergman et al. 2017 | Habitual alcohol consumption during the 12 months prior to recruitment and at ages 20, 30, 40 and 50 years. | CD and UC | • Nested case– control study using prospectively-collected dietary data from the EPIC cohort. • 84 cases of incident CD and 198 cases of incident UC matched 1:4 to 336 and 792 controls, respectively. |
• No statistically significant associations between alcohol consumption at recruitment, lifetime average alcohol use or pattern of lifetime alcohol use and risk CD or UC. | 118 |
Multivariable estimates are presented with the P value for linear trend across quantiles of the exposure of interest where available. aMED, adapted Mediterranean Diet Score; EPA, eicosapentaenoic acid; EPIC, European Prospective Investigation into Cancer; DHA, docosahexaenoic acid; HR, hazard ratio; IRR, incidence rate ratio; NHS, Nurses’ Health Study; OR, odds ratio; PUFA, polyunsaturated fatty acid.
Box 1 : SFFQ validation studies.
This SFFQ has been extensively validated in several studies, including observations that : nutrient intake assessed by the SFFQ strongly correlated with detailed dietary records (r=0.65)1–3; specific items between 2 questionnaires administered 1 year apart were strongly correlated (r=0.67)1–4; nutrients calculated from the SFFQ correlated with biochemical indicators of dietary intake, including plasma levels of beta-carotene (r=0.30–0.42) and vitamin E (r=0.41–0.53), as well as red blood cell levels of folate (r=0.55)5–8; estimates of fatty acid intake correlated with adipose tissue levels of linoleic acid (r=0.35–0.37), trans-fatty acid (r=0.51) and n-3 fatty acids (r=0.48–0.49)9,10. Repeated validation studies of the FFQ in the NHS have yielded similar and stable correlations with diet records over time. In a validation study published in 2017, which was conducted in 632 women from the NHS cohort between 2010 and 2012, the FFQ was correlated with two 7-day dietary records that were administered one year apart (r=0.62) and automated self-administered 24-hour recalls (r=0.63) over a one year period, suggesting that the SFFQ provides reasonably valid estimates of intakes of a wide variety of dietary variables11. These correlations probably underestimate the validity of the SFFQ because they also incorporate the errors related to use of the 6-day dietary record for estimating long-term intake
Dietary studies in the Nurses’ Health Study.
The Nurses’ Health Study (NHS) is a prospective cohort study that began in 1976 when 121,701 female registered nurses aged 30–55 years completed a mailed health questionnaire39. Similar baseline questionnaires were returned in 1989 from a parallel cohort of 116,686 female nurses aged 25–42 years that were enrolled in NHSII. Questionnaires have been mailed to members of both cohorts every two years, starting in 1976 in NHS and 1989 in NHSII, to obtain updated information on diet, lifestyle and medical diagnoses, with a follow-up rate of >90% until 2012 for the NHS and until 2013 for the NHSII. Dietary data is collected every four years in these cohorts through a 161-item SFFQ. This SFFQ has been extensively validated in several studies, including observations that : nutrient intake assessed by the SFFQ strongly correlated with detailed dietary records (r=0.65)1–3; specific items between 2 questionnaires administered 1 year apart were strongly correlated (r=0.67)1–4; nutrients calculated from the SFFQ correlated with biochemical indicators of dietary intake, including plasma levels of beta-carotene (r=0.30–0.42) and vitamin E (r=0.41–0.53), as well as red blood cell levels of folate (r=0.55)5–8; estimates of fatty acid intake correlated with adipose tissue levels of linoleic acid (r=0.35–0.37), trans-fatty acid (r=0.51) and n-3 fatty acids (r=0.48–0.49)9, 10. Repeated validation studies of the FFQ in the NHS have yielded similar and stable correlations with diet records over time. In a validation study published in 2017, which was conducted in 632 won from the NHS cohort between 2010 and 2012, the FFQ was correlated with two 7-day dietary records that were administered one year apart (r=0.62) and automated self-administered 24-hour recalls (r=0.63) over a one year period, suggesting that the SFFQ provides reasonably valid estimates of intakes of a wide variety of dietary variables11. These correlations probably underestimate the validity of the SFFQ because they also incorporate the errors related to use of the 6-day dietary record for estimating long-term intake.
Early work from the NHS and NHSII showed a low risk of Crohn’s disease among individuals who consume high amounts of fiber, particularly fiber from vegetables and fruits (Table 1)40. By contrast, a high n-3:n-6 fatty acids ratio was associated with a low risk of ulcerative colitis, whereas a high intake of red meat was associated with an increased risk of ulcerative colitis (Table 1)41, 42. Owing to the increasingly recognized role of specific nutrients and electrolytes such as heme, potassium and zinc in regulating the intestinal barrier and immune function43–50, over the past 5 years studies have focused on examining the relationship between quantity of nutrient consumption and risk of IBD. Preliminary data from these cohorts have shown an inverse association between dietary zinc and potassium intake and risk of Crohn’s disease, whereas dietary sodium, heme and iron intake were not associated with IBD risk (Table 1)42, 51, 52.
Dietary studies in the European Prospective Investigation in Cancer study.
The European Prospective Investigation in Cancer (EPIC) study, which investigated the link between dietary intake and the risk of common malignant diseases in the general population, is comprised of 519,978 men and women recruited from 23 centers in 10 European countries53. The EPIC–IBD study is a subcohort of the EPIC study that involved a total of 401,326 initially healthy men and women aged 20–80 years who did not have Crohn’s disease or ulcerative colitis and who were recruited from 12 centers in 8 European countries between 1991 and 1998. EPIC–IBD participants provided information on age, gender and lifestyle factors (such as smoking, physical activity levels and dietary information) through the self-completion of questionnaires at baseline. The follow-up rate until 2010 in EPIC–IBD was >98%. Dietary assessment in EPIC–IBD was conducted at baseline when participants reported dietary intake using country-specific validated questionnaires53. In most centers, a self-administered FFQ was used to assess dietary intake (with 88–266 food-related questions ) over the previous 12 months. Interviewer- administered diet questionnaires were used in Greece, Spain and in Ragusa, Italy. In the EPIC study, almost 10% of the cohort (~37,000 individuals) were assessed using a single highly-standardized, 24-hour recall of actual food consumption to account for any between-cohort differences that might exist in systematic exposure measurement error as well as for within-cohort attenuation biases in relative risk estimates. The energy-adjusted intakes were used to estimate correlations between FFQ and 24-hour recall and the mean intake levels estimated from 24-hour recall were used to account for within-cohort and between-cohort variations, respectively54.
Prior to the establishment of EPIC-IBD cohort, one study from EPIC that consisted of 260,686 participants from 10 centers across 5 countries, investigated the associations between intake of a total of 18 nutrients, vitamins and minerals and the risk of ulcerative colitis55. No associations were detected, with the exception of a weak positive association between increased total polyunsaturated fatty acid (PUFA) intake as a percentage of total energy intake and increased risk of developing ulcerative colitis. A similar broad analysis has yet to be published for Crohn’s disease in this particular cohort. However, following these initial results, a subsequent study from the EPIC–IBD study further investigated the associations between intake of PUFAs and ulcerative colitis56. On the basis of findings by other groups that n-6 PUFAs are metabolized to pro-inflammatory lipid mediators57, 58, this study hypothesized that individuals with the highest dietary intakes of n-6 PUFAs would have an increased risk of developing ulcerative colitis. Indeed, the highest quintile for dietary intakes of the n-6 PUFA linoleic acid, as measured by the FFQ, was associated with an increased risk of developing ulcerative colitis (OR 2.49, 95% CI 1.23–5.07) 56 (Table 1). Conversely, the same study found that participants with the highest quintile for dietary intakes of docosahexaenoic acid (DHA), an n-3 PUFA that is metabolized to anti-inflammatory lipid mediators, had a decreased risk of developing ulcerative colitis (OR 0.32, 95% CI 0.06–0.97). Although similar findings for DHA intake and risk of Crohn’s disease have been reported in the EPIC–IBD study 59, no associations between n-6 PUFA intake and Crohn’s disease have been found in EPIC-IBD to date 59 (Table 1).
In contrast, investigations into the role of monosaccharide, disaccharide and starch intake and the risk of developing ulcerative colitis or Crohn’s disease in development of Crohn’s disease and ulcerative colitis using EPIC-IBD have not demonstrated any consistent associations60 (Table 1). Interestingly, however, EPIC-IBD study participants in the highest quintile for diets rich in sugar and soft drinks had an increased risk of developing ulcerative colitis (IRR 1.68, 95% CI 1.00–2.82) 61 (Table 1). This finding was mainly attributed to those individuals with a dietary pattern of high intake of sugar and soft drinks who had intakes of vegetables, legumes and fruit that were below that of the median population (OR 2.80, 95 % CI 1.54–5.09) 61. High consumption of milk has been shown to be associated with decreased risk of developing Crohn’s disease but not ulcerative colitis, independent of dietary calcium intake62. However, the association did not show a dose-response relationship and there was no association between total intake of dairy products, yogurt or cheese with either ulcerative colitis or Crohn’s disease. Finally, investigations into alcohol intake and the risk of developing ulcerative colitis or Crohn’s disease in EPIC-IBD have not reported any statistically significant associations63, despite speculation that isoflavones in alcoholic beverages accumulated during fermentation process64 might have antioxidant and immune modulatory effects based on preclinical studies65.
Limitations of prospective cohort studies and future directions.
Despite offering unique opportunities to study dietary factors in relation to IBD incidence, both the EPIC and NHS studies are limited by the under-representation of individuals with a early onset of IBD (age < 40 years). This drawback limits the generalizability of the findings of these studies, particularly regarding individuals who were diagnosed with IBD before the age of 40 years. Although dietary data derived from these cohorts has been extensively validated, the correlation between FFQ and dietary records, considered to be the gold standard, is modest for many foods and nutrients, which in turn can be a source of considerable measurement errors. In addition, despite being the largest cohort studies to date that have examined the link between diet and risk of IBD, the number of incident cases of IBD in these studies is limited, precluding the identification of more modest associations.
Thus, there continues to be an unmet need for a comprehensive, prospective examination of association between diet and risk of IBD. Such an effort could perhaps be achieved through the establishment of a large international consortium and could be used to achieve a number of important aims. Firstly, to improve our understanding of the associations between dietary patterns and risk of IBD across cohort studies comprised of different populations with a wide distribution of participant characteristics. Secondly, to examine the presence of dose-response relationships across a wide exposure range. Thirdly, to generate summary estimates that have greater precision than the individual studies due to the larger sample size, therefore allowing examination of the population attributable risk of dietary factors in IBD. Finally, such a collaborative effort could allow for examinations of whether associations differ within specific population subgroups. These analyses are particularly important as they might identify subgroups of the population that might benefit the most from specific lifestyle interventions. In addition, given that IBD is a heterogeneous disease, the evaluation of whether associations vary by disease subtype might lead to a better understanding of the aetiology of IBD.
Animal studies
Dietary factors are known to induce insulin resistance, influence the balance of fatty acids that elicit or reduce inflammation of the gut epithelium, modify intestinal permeability and contribute to sulfur production which in turn alter the gut microbiota by promoting the growth of sulfate-reducing bacteria 66, 67. Animal studies have shown that a Western diet, increases the susceptibility of mice to dextran sodium sulfate (DSS)-induced colitis and increases the infiltration of macrophages compared with a Mediterranean diet, characterized by a high intake of fruits, vegetables, whole-grain foods, poultry and fish68. In addition, a Western diet in mice alters host intestinal barrier function by increasing the colonization of adherent-invasive Escherichia coli29. Similarly in mice, a high-fat diet was shown to disrupt the gut barrier function by modifying the composition of luminal bile salts 30.
Although dietary fiber has been shown to exert its anti-inflammatory effect by modulating the composition of the gut microbiota, increasing colonic fermentation and promoting the production of short-chain fatty acids, studies in mice models of colitis have also shown that fiber might have a direct immunomodulatory effect through inhibition of pro-inflammatory cytokine production in peripheral blood mononuclear cells 69. Additionally, in a model of Citrobacter rodentium-induced acute colitis, a fiber-rich diet reduced inflammation by modifying the host microbiota to increase production of short-chain fatty acids, and by altering host immune mechanisms of bacterial recognition and response to promote host health. In contrast to fiber, dietary red meat exacerbates DSS-induced colitis in mice 70. Although the exact mechanism underlying this observation is unclear, one potential explanation is that animal proteins increase luminal levels of sphingosine-1-phosphate, a sphingolipid involved in cell signaling, which has been shown to increase inflammation in mice models of colitis 71–73.
Over the past 5 years, there has been a growing interest on the role of dietary sodium in regulating the immune response. Several animal studies have shown a role for dietary sodium intake in the development and progression of autoimmune disorders through IL-23–IL-23R-mediated activation of pathogenic pro-inflammatory type 17 T helper (TH17) cells, which have a critical role in development of IBD45–47. In addition to enhancing the induction of pro-inflammatory TH17 cells, sodium also appears to suppress immune tolerance through inhibiting the immunosuppressvie functions of forkhead box protein P3 positive (FOXP3)+ T regulatory (Treg) cells 74. Sodium might also lead to activation of pro-inflammatory macrophages 75. In contrast to sodium, a study published in 2016 demonstrated the profound suppressive effect of potassium, released into the extracellular fluid as a result of necrosis in human tumors, on T cell effector function44. In turn, the study also demonstrated that augmentation of potassium efflux in tumor-specific T cells enhances their effector function and promotes tumor clearance. Together, these findings demonstrate the critical role of sodium and potassium in regulating the TH17/Treg balance.
Gene–environment studies
Studies investigating the interaction between environmental components and genetic variants in functionally-annotated genes have been increasingly used to help infer causal associations76 and provide insight into potential biological pathways through which an environmental factor such as diet might contribute to the etiopathogenesis of chronic diseases, including IBD. Genetic loci associated with IBD risk can be broadly categorized into those involving abnormalities in the innate and/or adaptive immune response and mucosal barrier function77. Experimental data also suggest that many of these pathways are probably influenced by dietary factors78. Thus, it is biologically plausible that specific dietary components have differential effects on the incidence of IBD according to the genetic background.
Data on the influence of gene–environment interaction on IBD risk is sparse. This paucity is largely due to the limited availability of well-characterized cohorts with detailed, updated and validated data on dietary and lifestyle factors with simultaneous annotation of genetic variants. In addition, due to the low incidence of IBD in the general population, many cohort studies have a limited number of participants, precluding the possibility of assessing gene–environment interactions. Nevertheless, the investigation of gene–environment interaction is an area of great unmet need as results from such studies will not only shed light on the complex interaction between diet, genetic susceptibility and immune function but could also help to inform dietary recommendations for individuals at a high risk of IBD or those with established disease.
Preliminary analyses of gene–environment interaction in studies incorporating dietary factors in IBD have been promising51, 79–81 (TABLE 2). An analysis from the NHS and NHSII, which included 149 individuals with Crohn’s disease and 172 individuals with ulcerative colitis matched to 650 control participants, examined the interaction between total dietary intake of iron and heme iron and genetic variants associated with risk of IBD42. This study demonstrated an interaction between iron and heme iron intake and the ulcerative colitis susceptibility locus rs1801274, a coding variant in the FCGR2A(encoding low affinity immunoglobulin γ Fc region receptor II-a) gene. Specifically, among women with the GG genotype, increasing heme iron intake was associated with a substantially lower risk of UC (OR 0.11, 95% CI 0.03–0.37 for each 1 g increase in heme iron intake) (TABLE 2). In contrast, among women with the TT genotype, increasing heme iron intake was associated with an almost 3-fold higher risk of UC (OR 2.76, 95% CI 1.02–7.48). Due to the crucial role of FCGR2A in regulating humoral response to infection82 and the known importance of the rs1801274 variant in altering the binding capacity of the encoded protein product to C-reactive protein (CRP) and immunoglobulin G2 (IgG2)83–85, this data provides evidence for an intriguing interaction between dietary heme intake and immune function in the development of ulcerative colitis.
Table 2|.
Gene-environment studies of diet in inflammatory bowel disease
| Study | Dietary exposure(s ) |
Genetic variant(s) |
Outcome( s) |
Populatio n and Design | Key findings* | Ref s |
|---|---|---|---|---|---|---|
| Costea et al. 2014 | Dietary n-6:n-3 fatty acid intake ratio | Fifteen SNPs in 3 PUFA metabolism genes (FADS1, FADS2 and CYP4F3) | CD | • Case–control study • 182 newly diagno sed pediatr ic patient s with CD, match ed to 250 control s. |
• Statistically significant interactions involving 6 of the candidate SNPs and the n-6:n-3 fatty acid ratio (Pinteraction ≤0.04 for all comparisons ). • Higher n-6:n-3 ratio was associated with an increased risk of CD among children with the GG genotype at rs1290617 in CYP4F3 (OR 2.91, 95%CI 0.90–9.39), but not among those with the TT genotype (OR 0.92, 95% CI 0.44–1.89; Pinteraction=0. 04). |
81 |
| Khalili et at. 2016 | Dietary potassium and sodium intake | Six SNPs in Th17 pathway genes (JAK2, STAT3, CCR6, IL21, IL10 and IL23R) | CD and UC | • Nested case–control study among women enrolled in the NHS and NHS II. • 169 individ uals with CD match ed to 740 control particip ants, and 202 individuals with UC match ed to 740 control particip ants. Yes |
• The association between dietary potassium intake and risk of CD was modified by the rs7657746 variant in IL21 (Pinteraction=0 .004). • Among women with the GG genotype, increasing potassium intake was associated with a higher risk of CD (OR 1.58, 95% CI 1.15– 2.16 per 200 mg increase in potassium intake). • By contrast, increasing potassium intake was associated with a lower risk of CD among those with the AA genotype (OR 0.90, 95% CI 0.82–0.98). |
51 |
| Khalili et al. 2017 | Total dietary intakes of iron, heme iron and red meat. | 180 GWAS-identified CD and UC risk variants | CD and UC | • Nested Case– control study among women enrolle d in the NHS and NHS II. • 169 individ uals with CD match ed to 740 control particip ants, and 202 individ uals with UC match ed to 740 control particip ants. |
• Statistically significant interaction between heme iron intake and the UC risk variant rs1801212 74 in FCGR2A (Pinteraction=7 x10−5). • Among women with the GG genotype, increasing heme iron intake was associated with a substantiall y lower risk of UC (OR 0.11, 95% CI 0.03–0.37 for each 1 g increase in heme iron intake). • Among women with the TT genotype, increasing heme iron intake was associated with an almost 3-fold higher risk of UC (OR 2.76, 95% CI 1.02–7.48). |
42 |
| Ananthakrishn an et al. 2017 | Dietary n-3:n-6 fatty acid ratio | Eight SNPs in 3 PUFA metabolism genes (FADS1, FADS2 and CYP4F3) | CD and UC | • Nested case–control study among women enrolle d in the NHS and NHS II. • 101 individ uals with CD and139 individuals with UC match ed to 495 control particip ants. |
• The association between n-3:n-6 PUFA intake ratio and UC was modified by the rs4646904 SNP in CYP4F3 (Pinteraction=0 .049). • A high (≥ median) n-3:n-6 intake ratio was associated with a lower risk of UC among women with the GG or AG genotypes (OR 0.57, 95% CI 0.32–0.99), but not among those with the AA genotype (OR 0.95, 95% CI 0.47–1.93). |
88 |
Estimates are from multivariable-adjusted models. P for interaction is for multiplicative interaction between the dietary exposure and the genetic variant of interest. CD, Crohn’s disease; NHS, Nurses’ Health Study; OR, odds ratio; PUFA, polyunsaturated fatty acid; SNP, single nucleotide polymorphism; UC ulcerative colitis.
Similarly, in a nested case–control study of 202 individuals with ulcerative colitis and 169 individuals with Crohn’s disease in the NHS and NHSII cohorts matched to 740 control participants based on age, menopausal status, month of blood collection and fasting status, an interaction between dietary potassium intake and genetic variants in the IL-23 pathway that have been previously associated with risk of IBD in GWAS was identified77. Specifically, the rs7657746 variant of IL21 (encoding IL-21) seemed to modify the association between potassium intake and risk of IBD (Table 2). Each 200 mg increase in dietary potassium intake was inversely associated with risk of ulcerative colitis (OR 0.90, 95 CI 0.83–0.98) among participants with the CC genotype, but not among those with the AG or GG genotypes. Similar findings were observed in patients with Crohn’s disease in this study. As IL-21 has a key role in the development of TH17 cells through signal transducer and activator of transcription 3 (STAT3), a transcription factor required for the differentiation of TH17 cells in vivo, the findings from gene–environment interaction studies suggest a potential mechanism for the observed association86. IL-21 and IL-23 induce expression of the orphan nuclear receptor RORγ, which, in synergy with STAT3, promotes IL-17 expression in CD4+ T cells, leading to the activation of TH17 cells 86. In addition, IL-21 inhibits the TGF-β-dependent generation of FOXP3+ Treg cells, induces Th17 cell activation87. Interestingly, in this study, the gene–environment interaction finding was further supported by in vitro studies demonstrating that potassium induces FOXP3 expression in naïve and memory T cells and pro-inflammatory TH17 cells. This effect was present even in the presence of pro-inflammatory cytokine suggesting that potassium may suppress inflammation in a pro-inflammatory milieu.
Lastly, data from the NHS and NHSII have also demonstrated that two variants in CYP4F3, which encodes a cytochrome P450 enzyme (CYP4F3) involved in PUFA metabolism, might modify the association between n-3 and n-6 PUFA intake and risk of ulcerative colitis88 (Table 2). Specifically, the association between n-3:n-6 PUFA intake ratio and UC was modified by the rs4646904 SNP in CYP4F3 (Pinteraction=0.049). A high (≥ median) n-3:n-6 intake ratio was associated with a lower risk of UC among women with the GG or AG genotypes (OR 0.57, 95% CI 0.32–0.99), but not among those with the AA genotype (OR 0.95, 95% CI 0.47–1.93). Indeed, similar findings were previously reported in a case–control study of children with newly diagnosed Crohn’s disease 81, suggesting that the interaction appears to be robust.
Limitations of prior gene–environment studies and future directions.
Despite the early promising results from gene–environment studies, it is important to acknowledge the limitations of these data. First, the majority of these studies had a limited sample size and were therefore not adequately powered to broadly assess (that is, on a genome-wide level ) the presence of gene–environment interactions. Secondly, despite the presence of stringent P value thresholds, in these preliminary studies, as demonstrated in previous validation studies, even robust preliminary findings may still represent spurious associations further emphasizing the need for validation studies in gene-environment studies89, 90. Thirdly, due to the limited sample size and the adjustment of most of these studies for multiple variables, these preliminary studies might have been prone to type II error. Lastly, although gene–environment interaction analyses might elucidate potential biological mechanisms underpinning the link between diet and IBD, statistical interaction does not imply biological modification; thus, further mechanistic studies are needed to fully characterize the results of these studies. Despite these limitations, preliminary findings highlight the need for larger scale analyses of the interaction between diet and genetics on risk of IBD, reinforcing the need for large international consortia composed of well-characterized cohort studies with simultaneous assessment of genetic and dietary data.
Gut microbiota studies
Diet is widely linked to the composition of the human gut microbiome and microbial metabolites91. Although much of the microbial diversity seen in the adult gut might be attained by the age of 4 years, the adult microbiome remains susceptible to the influence of diet92. Indeed, dietary patterns have been proposed to explain greater than half of the variation in the adult intestinal microbiome92. Accordingly, the gut microbiome has a crucial role in the pathogenesis of IBD, as evidenced by observations that infusion of intestinal luminal contents into excluded ileum triggers post-operative recurrence in patients with Crohn’s disease93. Furthermore, in mice models of colitis, microbial colonization is required for the development of active inflammation94, 95. Thus, diet, through its modulatory effect on the gut microbiota, might modify the risk of developing IBD (Figure 2)
Patients with established IBD have dysbiosis of the gut microbiome, characterized by reduced bacterial diversity, enrichment of bacteria of the family Enterobacteriaceae and reduced abundance of bacteria of the phylum Firmicutes and the genus Bacteroides (Figure 2)92, 96–98. Interestingly, similar patterns of bacterial enrichment have been reported in observational studies of healthy volunteers fed a high-fat, low-fiber diet, suggesting that prior epidemiological findings demonstrating an inverse association between fiber intake and risk of Crohn’s disease might in part be explained by diet-mediated alteration of the gut microbiota in genetically susceptible individuals99, 100.
The majority of prior studies investigating the interaction between diet, the microbiome and intestinal inflammation have been conducted in animal models of colitis. In mice, dietary heme iron was shown to directly injure the colonic surface epithelium through the generation of cytotoxic and oxidative stress101, 102. Moreover, increased dietary heme iron intake has also been associated with marked changes in the composition of the gut microbiota, with an increased ratio of Gram-negative:Gram-positive bacteria in mice103. This effect was primarily driven by the increased abundance of Gram-negative species, including those belonging to the genus Bacteroides and Akkermansia, leading to a marked increase in lipopolysaccharide production103. In a 2017 study, Akkermansia muciniphila was shown in mice to be a pathobiont, promoting colitis in genetically susceptible hosts104. Interestingly, alteration in the function of the gut mucosal barrier related to dietary intake of heme iron seems to be dependent on the presence of sulfide-producing and mucin-degrading bacteria (for example, Akkermansia)101. By contrast, in a mouse model of spontaneous ileitis, depletion of luminal iron altered gut microbial composition to promote inflammation105. Finally, a high-fat diet has been linked to changes in phospholipid profile and bacteria taxa in the gut, highlighting the complex interactions between the host and the gut microbiome in response to high fat intake106, 107. More specifically, high intake of saturated fat altered the gut microbiome, characterized by an increase in the sulfite-reducing pathobiont Bilophila wadsworthia, and induced colonic inflammation in Il10-knockout mice 108. This data provided a potential mechanism by which diets high in saturated fat might increase the risk of IBD in a susceptible host.
Human observational studies examining the interaction between diet, the microbiome and IBD are scarce. Among these studies is the ongoing multinational Genetics, Environment and Microbiome study that aims to identify factors that increase the risk of IBD109, 110. This study is examining several environmental exposures, such as being breastfed and the composition of the microbiome, in a cohort of patients at high risk of IBD. Interestingly, preliminary data from this study has not shown a correlation between intestinal permeability110 — a key pathogenic pathway in IBD that is known to be influenced by diet78 — and the gut microbiota. Specific patterns of association have been reported in a large Dutch cohort, in which total carbohydrate intake and features of a Western diet, including high caloric intake and consumption of sugar-sweetened beverages, were negatively associated with gut microbiome diversity 111. By contrast, features of a Mediterranean diet, such as consumption of fruits, vegetables and red wine, were associated with increased diversity of the gut microbiome. Red wine consumption was also associated with the increased abundance of Faecalibacterium prausnitzii, which has been proposed to have anti-inflammatory properties in patients with IBD112 .
Several studies have examined the correlation between host genetics and the gut microbiome in healthy individuals and patients with IBD113–115. Generally, these studies have established a potential association between several genetic variants, including IBD-specific risk variants, and the gut microbiome. However, perhaps the most intriguing finding has been the observation that gene–diet interaction could in part regulate the composition of human gut microbiome. Using metagenomic data from >1500 healthy individuals, it was found that the abundance of bacteria of the genus Bifidobacterium was regulated by a functional variant within the LCT gene, which encodes lactase, the enzyme responsible for the breakdown of lactose116. Interestingly, this association was further modified by dairy intake. These studies further illustrate the interaction between diet, host genetics and the gut microbiome in human health and diseases of the gastrointestinal tract and highlights the complex relationship between these factors in studying IBD pathogenesis (FIG. 3).
Figure 3 |. The complex causal relationship between diet and inflammatory bowel disease-.
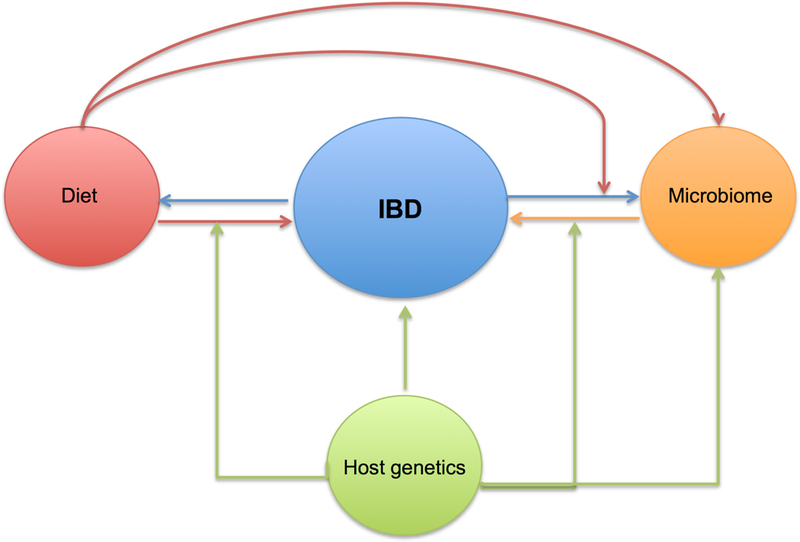
The pathophysiology of IBD is thought to be related to inappropriate immune response to the gut microbiome in the genetically susceptible host. The composition of the gut microbiome is primarily dictated by environmental determinants particularly diet and the host genetics. In turn dietary factors may have differential influence on the immune function and risk of IBD according to the host genetics. Studies examining the role of diet, gut microbiome, and host genetics on risk of IBD are particularly complicated by the reciprocal effect of active disease on the gut microbiome and individuals’ dietary choices.
Conclusions
There is growing body of evidence suggesting a role for diet in the development of IBD, particularly among genetically susceptible individuals. In addition, data from animal studies suggest that dietary modification might alter the risk of IBD. Nevertheless, there continues to be a substantial need for a comprehensive evaluation of the role of diet in the etiopathogenesis of IBD. Such studies will be challenging because of the complex nature of the relationship between diet, genetics, the microbiome and disease activity. A number of large international collaborations are currently in the planning stages and hope to provide an in-depth prospective examination of the role of specific diets in IBD and overcome many of the shortcomings of prior studies. The successful execution of these studies will represent a critical first step in advancing our understanding of the role of diet in the pathogenesis of IBD.
Key points.
1) Epidemiological studies have identified a few potential dietary risk factors for Crohn’s disease and ulcerative colitis.
2) Early results from dietary intervention studies in treatment of Crohn’s disease and ulcerative colitis have been promising particularly in pediatrics but high quality randomized trials are needed to assess their efficacy
3) Preliminary gene-environment and microbiome studies have demonstrated an interaction between diet, host genetics, and the gut microbiota in the etiopathogenesis of Crohn’s disease and ulcerative colitis.
4) Future large-scale studies to prospectively examine the role of diet in Crohn’s disease and ulcerative colitis in the context of host genetics and gut microbiota are needed.
Acknowledgements
HK is funded by the National Institute of Diabetes and Digestive and Kidney Diseases (K23 DK099681 and R03 ), ATC is funded by NIH K24 DK098311, a Stuart and Suzanne Steele MGH Research Scholar Award, and a Senior Investigator Award from the Crohn’s and Colitis Foundation.
Financial Disclosures: H. K. has received consulting fees from Abbvie and Samsung Bioepis. H. K. also receives funding from Takeda. S. S. M. C. has received consulting fees from Abbvie and Ferring Pharmaceuticals. P. L. is supported by a career development grant by CCF. A. N. A. is a member of the scientific advisory board for Exact Sciences, Abbvie, and Cubist pharmaceuticals. A. T. C. has served as a consultant for Bayer Healthcare, Pfizer Inc., and Takeda.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
ONLINE ONLY-
Subject Categories
Health sciences / Gastroenterology / Gastrointestinal diseases / Inflammatory bowel disease Health sciences / Gastroenterology / Gastrointestinal system / Microbiota Health sciences / Health care / Public health / Epidemiology
Display items
REFERENCES
- 1.Feskanich D et al. Reproducibility and validity of food intake measurements from a semiquantitative food frequency questionnaire. J Am Diet Assoc 93, 790–6 (1993). [DOI] [PubMed] [Google Scholar]
- 2.Rimm E et al. Reproducibility and validity of an expanded self-administered semiquantitative food questionnaire among health professionals. Am J Epidemiol 135, 1114–1126 (1992). [DOI] [PubMed] [Google Scholar]
- 3.Rockett HR et al. Validation of a youth/adolescent food frequency questionnaire. Prev Med 26, 808–16 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Rockett HR, Wolf AM & Colditz GA Development and reproducibility of a food frequency questionnaire to assess diets of older children and adolescents. J Am Diet Assoc 95, 336–40 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Ascherio A et al. Correlations of vitamin A and E intakes with the plasma concentrations of carotenoids and tocopherols among American men and women. J Nutr 122, 1792–801 (1992). [DOI] [PubMed] [Google Scholar]
- 6.Romieu I et al. Food predictors of plasma beta-carotene and alpha-tocopherol: validation of a food frequency questionnaire. Am J Epidemiol 131, 864–76 (1990). [DOI] [PubMed] [Google Scholar]
- 7.Willett WC et al. Validation of dietary questionnaire with plasma carotenoid and alpha-tocopherol level. Am J Clin Nutr 38, 631–639 (1983). [DOI] [PubMed] [Google Scholar]
- 8.Giovannucci E et al. Folate, methionine, and alcohol intake and risk of colorectal adenoma. J Natl Cancer Inst 85, 875–84. (1993). [DOI] [PubMed] [Google Scholar]
- 9.Hunter DJ et al. Comparison of measures of fatty acid intake by subcutaneous fat aspirate, food frequency questionnaire, and diet records in a free-living population of US men. Am J Epidemiol 135, 418–27 (1992). [DOI] [PubMed] [Google Scholar]
- 10.London SJ et al. Fatty acid composition of subcutaneous adipose tissue and diet in postmenopausal US women. Am J Clin Nutr 54, 340–5 (1991). [DOI] [PubMed] [Google Scholar]
- 11.Yuan C et al. Validity of a Dietary Questionnaire Assessed by Comparison With Multiple Weighed Dietary Records or 24-Hour Recalls. Am J Epidemiol 185, 570–584 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molodecky NA et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology 142, 46–54 e42; quiz e30 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Burisch J, Jess T, Martinato M, Lakatos PL & EpiCom E The burden of inflammatory bowel disease in Europe. J Crohns Colitis 7, 322–37 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Kappelman MD et al. The prevalence and geographic distribution of Crohn’s disease and ulcerative colitis in the United States. Clin Gastroenterol Hepatol 5, 1424–9 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Ferguson A, Sedgwick DM & Drummond J Morbidity of juvenile onset inflammatory bowel disease: effects on education and employment in early adult life. Gut 35, 665–8 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kappelman MD et al. Direct health care costs of Crohn’s disease and ulcerative colitis in US children and adults. Gastroenterology 135, 1907–13 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longobardi T, Jacobs P & Bernstein CN Work losses related to inflammatory bowel disease in the United States: results from the National Health Interview Survey. The American journal of gastroenterology 98, 1064–72 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Franke A et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nature genetics 42, 1118–25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kenny EE et al. A genome-wide scan of Ashkenazi Jewish Crohn’s disease suggests novel susceptibility loci. PLoSgenetics 8, e1002559 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kugathasan S & Amre D Inflammatory bowel disease-environmental modification and genetic determinants. Pediatric clinics of North America 53, 727–49 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Tysk C, Lindberg E, Jarnerot G & Floderus-Myrhed B Ulcerative colitis and Crohn’s disease in an unselected population of monozygotic and dizygotic twins. A study of heritability and the influence of smoking. Gut 29, 990–6 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ng SC et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet 390, 2769–2778 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Williams CN Does the incidence of IBD increase when persons move from a low- to a high-risk area? Inflammatory bowel diseases 14 Suppl 2, S41–2 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Probert CS, Jayanthi V, Pinder D, Wicks AC & Mayberry JF Epidemiological study of ulcerative proctocolitis in Indian migrants and the indigenous population of Leicestershire. Gut 33, 687–93 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benchimol EI et al. Inflammatory bowel disease in immigrants to Canada and their children: a population-based cohort study. Am J Gastroenterol 110, 553–63 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Li X, Sundquist J, Hemminki K & Sundquist K Risk of inflammatory bowel disease in first- and second-generation immigrants in Sweden: a nationwide follow-up study. Inflamm Bowel Dis 17, 1784–91 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Albenberg LG & Wu GD Diet and the intestinal microbiome: associations, functions, and implications for health and disease. Gastroenterology 146, 1564–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poullis A, Foster R, Shetty A, Fagerhol MK & Mendall MA Bowel inflammation as measured by fecal calprotectin: a link between lifestyle factors and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev 13, 279–84 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Martinez-Medina M et al. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut 63, 116–24 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Stenman LK, Holma R, Eggert A & Korpela R A novel mechanism for gut barrier dysfunction by dietary fat: epithelial disruption by hydrophobic bile acids. Am J Physiol Gastrointest Liver Physiol 304, G227–34 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Halmos EP & Gibson PR Dietary management of IBD--insights and advice. Nat Rev Gastroenterol Hepatol 12, 133–46 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Borrelli O et al. Polymeric diet alone versus corticosteroids in the treatment of active pediatric Crohn’s disease: a randomized controlled open-label trial. Clin Gastroenterol Hepatol 4, 744–53 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Zachos M, Tondeur M & Griffiths AM Enteral nutritional therapy for induction of remission in Crohn’s disease. Cochrane Database Syst Rev, CD000542 (2007). [DOI] [PubMed] [Google Scholar]
- 34.Charlebois A, Rosenfeld G & Bressler B The Impact of Dietary Interventions on the Symptoms of Inflammatory Bowel Disease: A Systematic Review. Crit Rev Food Sci Nutr 56, 1370–8 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Persson PG, Ahlbom A & Hellers G Diet and inflammatory bowel disease: a case-control study. Epidemiology 3, 47–52 (1992). [DOI] [PubMed] [Google Scholar]
- 36.Chapman-Kiddell CA, Davies PS, Gillen L & Radford-Smith GL Role of diet in the development of inflammatory bowel disease. Inflamm Bowel Dis 16, 137–51 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Tragnone A et al. Dietary habits as risk factors for inflammatory bowel disease. Eur J Gastroenterol Hepatol 7, 47–51 (1995). [PubMed] [Google Scholar]
- 38.Willett WC, Reynolds RD, Cottrell-Hoehner S, Sampson L & Browne ML Validation of a semi-quantitative food frequency questionnaire: comparison with a 1-year diet record. J Am Diet Assoc 87, 43–7 (1987). [PubMed] [Google Scholar]
- 39.Colditz GA, Manson JE & Hankinson SE The Nurses’ Health Study: 20-year contribution to the understanding of health among women. J Womens Health 6, 49–62 (1997). [DOI] [PubMed] [Google Scholar]
- 40.Ananthakrishnan AN et al. A prospective study of long-term intake of dietary fiber and risk of Crohn’s disease and ulcerative colitis. Gastroenterology 145, 970–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ananthakrishnan AN et al. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn’s disease. Gut 63, 776–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khalili H et al. Dietary Iron and Heme Iron Consumption, Genetic Susceptibility, and Risk of Crohn’s Disease and Ulcerative Colitis. Inflamm Bowel Dis 23, 1088–1095 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bettelli E et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441, 235–8 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Eil R et al. Ionic immune suppression within the tumour microenvironment limits T cell effector function. Nature 537, 539–543 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kleinewietfeld M et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496, 518–22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu C et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496, 513–7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yosef N et al. Dynamic regulatory network controlling TH17 cell differentiation. Nature 496, 461–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bao S et al. Zinc modulates the innate immune response in vivo to polymicrobial sepsis through regulation of NF-kappaB. Am J Physiol Lung Cell Mol Physiol 298, L744–54 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Finamore A, Massimi M, Conti Devirgiliis L & Mengheri E Zinc deficiency induces membrane barrier damage and increases neutrophil transmigration in Caco-2 cells. J Nutr 138, 1664–70 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Prasad AS Zinc: role in immunity, oxidative stress and chronic inflammation. Curr Opin Clin Nutr Metab Care 12, 646–52 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Khalili H et al. Identification and Characterization of a Novel Association between Dietary Potassium and Risk of Crohn’s Disease and Ulcerative Colitis. Front Immunol 7, 554 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ananthakrishnan AN et al. Zinc intake and risk of Crohn’s disease and ulcerative colitis: a prospective cohort study. Int J Epidemiol 44, 1995–2005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Riboli E et al. European Prospective Investigation into Cancer and Nutrition (EPIC): study populations and data collection. Public Health Nutr 5, 1113–24 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Ferrari P et al. Within- and between-cohort variation in measured macronutrient intakes, taking account of measurement errors, in the European Prospective Investigation into Cancer and Nutrition study. Am J Epidemiol 160, 814–22 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Hart AR et al. Diet in the aetiology of ulcerative colitis: a European prospective cohort study. Digestion 77, 57–64 (2008). [DOI] [PubMed] [Google Scholar]
- 56.Investigators., I.i.E.S. et al. Linoleic acid, a dietary n-6 polyunsaturated fatty acid, and the aetiology of ulcerative colitis: a nested case-control study within a European prospective cohort study. Gut 58, 1606–11 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Calder PC Fatty acids and inflammation: the cutting edge between food and pharma. EurJ Pharmacol 668 Suppl 1, S50–8 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Simopoulos AP Evolutionary aspects of diet, the omega-6/omega-3 ratio and genetic variation: nutritional implications for chronic diseases. Biomed Pharmacother 60, 502–7 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Chan SS et al. Association between high dietary intake of the n-3 polyunsaturated fatty acid docosahexaenoic acid and reduced risk of Crohn’s disease. Aliment Pharmacol Ther 39, 834–42 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan SS et al. Carbohydrate intake in the etiology of Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis 20, 2013–21 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Racine A et al. Dietary Patterns and Risk of Inflammatory Bowel Disease in Europe: Results from the EPIC Study. Inflamm Bowel Dis 22, 345–54 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Opstelten JL et al. Dairy Products, Dietary Calcium, and Risk of Inflammatory Bowel Disease: Results From a European Prospective Cohort Investigation. Inflamm Bowel Dis 22, 1403–11 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Bergmann MM et al. No association of alcohol use and the risk of ulcerative colitis or Crohn’s disease: data from a European Prospective cohort study (EPIC). Eur J Clin Nutr 71, 512–518 (2017). [DOI] [PubMed] [Google Scholar]
- 64.Gavaler JS, Rosenblum ER, Deal SR & Bowie BT The phytoestrogen congeners of alcoholic beverages: current status. Proc Soc Exp Biol Med 208, 98–102 (1995). [DOI] [PubMed] [Google Scholar]
- 65.Seifried HE, Anderson DE, Fisher EI & Milner JA A review of the interaction among dietary antioxidants and reactive oxygen species. J Nutr Biochem 18, 567–79 (2007). [DOI] [PubMed] [Google Scholar]
- 66.Yamamoto T, Nakahigashi M & Saniabadi AR Review article: diet and inflammatory bowel disease--epidemiology and treatment. Aliment Pharmacol Ther 30, 99–112 (2009). [DOI] [PubMed] [Google Scholar]
- 67.Barton LL, Ritz NL, Fauque GD & Lin HC Sulfur Cycling and the Intestinal Microbiome. Dig Dis Sci 62, 2241–2257 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Kim IW et al. Western-style diets induce macrophage infiltration and contribute to colitis-associated carcinogenesis. J Gastroenterol Hepatol 25, 1785–94. [DOI] [PubMed] [Google Scholar]
- 69.Breton J et al. Intrinsic immunomodulatory effects of low-digestible carbohydrates selectively extend their anti-inflammatory prebiotic potentials. Biomed Res Int 2015, 162398 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Le Leu RK, Young GP, Hu Y, Winter J & Conlon MA Dietary red meat aggravates dextran sulfate sodium-induced colitis in mice whereas resistant starch attenuates inflammation. Dig Dis Sci 58, 3475–82 (2013). [DOI] [PubMed] [Google Scholar]
- 71.Degagne E et al. Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J Clin Invest 124, 5368–84 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jantchou P, Morois S, Clavel-Chapelon F, Boutron-Ruault MC & Carbonnel F Animal protein intake and risk of inflammatory bowel disease: The E3N prospective study. Am J Gastroenterol 105, 2195–201 (2010). [DOI] [PubMed] [Google Scholar]
- 73.Liang J et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 23, 107–20 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hernandez AL et al. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J Clin Invest 125, 4212–22 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang WC et al. High salt primes a specific activation state of macrophages, M(Na). Cell Res 25, 893–910 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hunter DJ Gene-environment interactions in human diseases. Nat Rev Genet 6, 287–98 (2005). [DOI] [PubMed] [Google Scholar]
- 77.Khor B, Gardet A & Xavier RJ Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–17 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leone VA, Cham CM & Chang EB Diet, gut microbes, and genetics in immune function: can we leverage our current knowledge to achieve better outcomes in inflammatory bowel diseases? Curr Opin Immunol 31, 16–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Van Der Sloot KW et al. Visceral Adiposity, Genetic Susceptibility, and Risk of Complications Among Individuals with Crohn’s Disease. Inflamm Bowel Dis 23, 82–88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ananthakrishnan AN et al. Genetic polymorphisms in fatty acid metabolism modify the association between dietary n3:n6 intake and risk of ulcerative colitis. Inflamm Bowel Dis Accepted (May 2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Costea I et al. Interactions between the dietary polyunsaturated fatty acid ratio and genetic factors determine susceptibility to pediatric Crohn’s disease. Gastroenterology 146, 929–31 (2014). [DOI] [PubMed] [Google Scholar]
- 82.Hargreaves CE et al. Fcgamma receptors: genetic variation, function, and disease. Immunol Rev 268, 6–24 (2015). [DOI] [PubMed] [Google Scholar]
- 83.Stein MP et al. C-reactive protein binding to FcgammaRIIa on human monocytes and neutrophils is allele-specific. J Clin Invest 105, 369–76 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Stein MP, Mold C & Du Clos TW C-reactive protein binding to murine leukocytes requires Fc gamma receptors. J Immunol 164, 1514–20 (2000). [DOI] [PubMed] [Google Scholar]
- 85.Bharadwaj D, Stein MP, Volzer M, Mold C & Du Clos TW The major receptor for C-reactive protein on leukocytes is fcgamma receptor II. J Exp Med 190, 585–90 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhou L et al. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nature immunology 8, 967–74 (2007). [DOI] [PubMed] [Google Scholar]
- 87.Fantini MC et al. IL-21 regulates experimental colitis by modulating the balance between Treg and Th17 cells. Eur J Immunol 37, 3155–63 (2007). [DOI] [PubMed] [Google Scholar]
- 88.Ananthakrishnan AN et al. Genetic Polymorphisms in Fatty Acid Metabolism Modify the Association Between Dietary n3: n6 Intake and Risk of Ulcerative Colitis: A Prospective Cohort Study. Inflamm Bowel Dis 23, 1898–1904 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Caspi A et al. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science 301, 386–9 (2003). [DOI] [PubMed] [Google Scholar]
- 90.Risch N et al. Interaction between the serotonin transporter gene (5-HTTLPR), stressful life events, and risk of depression: a meta-analysis. JAMA 301, 2462–71 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kau AL, Ahern PP, Griffin NW, Goodman AL & Gordon JI Human nutrition, the gut microbiome and the immune system. Nature 474, 327–36 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Brown K, DeCoffe D, Molcan E & Gibson DL Diet-induced dysbiosis of the intestinal microbiota and the effects on immunity and disease. Nutrients 4, 1095–119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.D’Haens GR et al. Early lesions of recurrent Crohn’s disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology 114, 262–7 (1998). [DOI] [PubMed] [Google Scholar]
- 94.Sellon RK et al. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infect Immun 66, 5224–31 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hudcovic T, Stepankova R, Cebra J & Tlaskalova-Hogenova H The role of microflora in the development of intestinal inflammation: acute and chronic colitis induced by dextran sulfate in germ-free and conventionally reared immunocompetent and immunodeficient mice. Folia Microbiol (Praha) 46, 565–72 (2001). [DOI] [PubMed] [Google Scholar]
- 96.Morgan XC et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13, R79 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nagalingam NA & Lynch SV Role of the microbiota in inflammatory bowel diseases. Inflamm Bowel Dis 18, 968–84 (2012). [DOI] [PubMed] [Google Scholar]
- 98.Spor A, Koren O & Ley R Unravelling the effects of the environment and host genotype on the gut microbiome. Nat Rev Microbiol 9, 279–90 (2011). [DOI] [PubMed] [Google Scholar]
- 99.Wu GD et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.De Filippo C et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 107, 14691–6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ijssennagger N et al. Gut microbiota facilitates dietary heme-induced epithelial hyperproliferation by opening the mucus barrier in colon. Proc Natl Acad Sci U S A 112, 10038–43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ijssennagger N, de Wit N, Muller M & van der Meer R Dietary heme-mediated PPARalpha activation does not affect the heme-induced epithelial hyperproliferation and hyperplasia in mouse colon. PLoS One 7, e43260 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.N IJ et al. Dietary heme alters microbiota and mucosa of mouse colon without functional changes in host-microbe cross-talk. PLoS One 7, e49868 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Seregin SS et al. NLRP6 Protects Il10(−/−) Mice from Colitis by Limiting Colonization of Akkermansia muciniphila. Cell Rep 19, 733–745 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Werner T et al. Depletion of luminal iron alters the gut microbiota and prevents Crohn’s disease-like ileitis. Gut 60, 325–33 (2011). [DOI] [PubMed] [Google Scholar]
- 106.Turnbaugh PJ, Backhed F, Fulton L & Gordon JI Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3, 213–23 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.David LA et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Devkota S et al. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 487, 104–8 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kevans D et al. IBD Genetic Risk Profile in Healthy First-Degree Relatives of Crohn’s Disease Patients. J Crohns Colitis 10, 209–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kevans D et al. Determinants of intestinal permeability in healthy first-degree relatives of individuals with Crohn’s disease. Inflamm Bowel Dis 21, 879–87 (2015). [DOI] [PubMed] [Google Scholar]
- 111.Zhernakova A et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352, 565–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sokol H et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci US A 105, 16731–6 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Imhann F et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Knights D et al. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med 6, 107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Turpin W et al. Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet 48, 1413–1417 (2016). [DOI] [PubMed] [Google Scholar]
- 116.Bonder MJ et al. The effect of host genetics on the gut microbiome. Nat Genet 48, 1407–1412 (2016). [DOI] [PubMed] [Google Scholar]
- 117.Ananthakrishnan AN et al. High School Diet and Risk of Crohn’s Disease and Ulcerative Colitis. Inflamm Bowel Dis 21, 2311–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bergmann MM et al. No association of alcohol use and the risk of ulcerative colitis or Crohn’s disease: data from a European Prospective cohort study (EPIC). Eur J Clin Nutr 71, 566 (2017). [DOI] [PubMed] [Google Scholar]