

Abstract
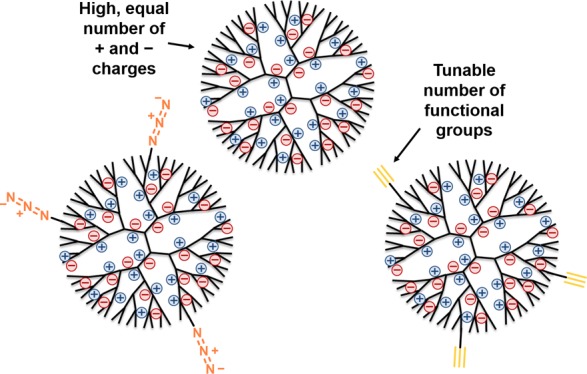
Dendrimers are interesting candidates for various applications because of the high level of control over their architecture, the presence of internal cavities, and the possibility for multivalent interactions. More specifically, zwitterionic dendrimers modified with an equal number of oppositely charged groups have found use in in vivo biomedical applications. However, the design and control over the synthesis of these dendrimers remains challenging, in particular with respect to achieving full modification of the dendrimer. In this work, we show the design and subsequent synthesis of dendrimers that are highly charged while having zero net charge, that is zwitterionic dendrimers that are potential candidates for biomedical applications. First, we designed and fully optimized the synthesis of charge-neutral carboxybetaine and sulfobetaine zwitterionic dendrimers. Following their synthesis, the various zwitterionic dendrimers were extensively characterized. In this study, we also report for the first time the use of X-ray photoelectron spectroscopy as an easy-to-use and quantitative tool for the compositional analysis of this type of macromolecules that can complement techniques such as nuclear magnetic resonance and gel permeation chromatography. Finally, we designed and synthesized zwitterionic dendrimers that contain a variable number of alkyne and azide groups that allow straightforward (bio)functionalization via click chemistry.
1. Introduction
Dendrimers are multivalent (macro)molecules in which the repeat units are not connected in a linear fashion, but form a well-defined, branched three-dimensional architecture of nanometer size.1−4 As a result, they possess a distinct molecular architecture that consists of a central core, branches, and terminal functional groups present at the outer surface of the macromolecule. Because of their unique, highly defined 3D structure, dendrimers have found use in a wide variety of applications ranging from dyes5 to catalysts,6 and from magnetic resonance imaging (MRI) contrast agents7 to sensors for small molecules.8 Furthermore, the high level of control over dendritic architectures, the presence of internal cavities and the possibility for multivalent binding have made dendrimers ideal candidates for carriers in biomedical applications.3,4,9 Therefore, over the past years, the use of dendrimers for gene and drug delivery has been extensively reported.9−12 However, most commonly used dendrimers, which include PPI (poly(propylene imine)) and PAMAM (poly(amido amine)),13 by themselves are not biocompatible and can induce cytotoxic effects.10,14 As a result, for in vivo use the exterior of dendrimers needs to be modified with, for instance, polyethylene glycol15−17 or charged groups18−23 to reduce their toxicity.
While such dendrimers modified with charged groups have found use in biomedical applications such as drug delivery, sensing, and MRI contrast agents,21−30 the design and control over the synthesis of these dendrimers remains challenging and is still subject to further optimization. So far, only partially zwitterionic dendrimers (ZIDs) (modification of either only the interior or of the exterior of the dendrimer), or dendrimers with nonpermanent, pH-sensitive charged groups (i.e. protonated amines) have been reported.21−30
Given these limitations, it is therefore still highly relevant to design and characterize dendrimers that are highly charged, yet could intrinsically have a zero net charge, that is ZID. We expect that the complete charge neutrality of such ZIDs can further diminish undesired interactions within a living system, thus making them more suitable for in vitro and in vivo applications, just like their larger, but less well-defined polymer counterparts, zwitterionic polymers.21,31,32 Building blocks for such a new type of zwitterionic materials should allow for a high density of oppositely charged moieties, creating a strong zwitterionic character, while remaining overall neutral. In addition, their inherently multivalent nature should, in principle, allow that ZIDs are functionalized in a controlled manner with multiple functional groups, including for example biorecognition elements. Within this project, specifically PPI dendrimers were found to be interesting candidates because of their high density of quaternizable amine groups and commercial availability.1,13 Furthermore, they have been found to more stable than PAMAM dendrimers, which can undergo retro-Michael reactions (β-eliminations) at high temperature or pH, which may be detrimental during the synthesis of the ZID.33,34
Here, we report the first synthesis of a series of ZIDs as well as their characterization. We prepare second and third generation ZIDs with a near-equal number of positively charged (quaternary) nitrogen atoms and negatively charged carboxylates, so as to make an electrically neutral oligocarboxybetaine. Of course, such carboxylate moieties could be functionalized—for example via standard NHS/EDC chemistry—but only with concomitant loss of charge neutrality, which for such relatively small molecules would easily increase the interaction with biomolecules. To thus allow for a precisely controlled functionalization of intrinsically charge-neutral ZIDs, we subsequently developed a synthetic strategy that allows incorporation of a specific number of clickable azido or alkyne functional groups. In this work, we also present for the first time the use of X-ray photoelectron spectroscopy (XPS) for the quantitative compositional analysis of such three-dimensional oligomers and show the power of this technique for the analysis of the chemical composition of large organic molecules. Because of their highly branched nature, dendrimeric building blocks have a large number of functional groups that all need to be converted during the synthesis. However, from an analytical perspective, it is often far from trivial to accurately determine this degree of conversion. To complement limitations with conventionally used analytical methods such as nuclear magnetic resonance (NMR), mass spectrometry (MS), gel permeation chromatography (GPC), and infrared (IR), we thus invoked XPS. This technique, traditionally used for surface analysis, allowed us to precisely determine the nature and electronic environment of specific elements in the relevant functional group (nitrogen atoms in our case), from which we could accurately determine the overall conversion.
2. Experimental Section
2.1. Materials
Milli-Q water was purified by a Barnsted water purification system, with a resistivity of <18.3 MΩ·cm. The reported plasma cleaner was a Diener Femto plasma system. Molecular sieves (10 Å) were oven-dried (120 °C, overnight) prior to use. Sonication steps were performed in an Elmasonic P 30 H ultrasonic unit at 80 kHz. Float-a-lyzer G2 dialysis membranes (VWR) with a 500–1000 D CE (default) or 1000–5000 D CE (when specifically mentioned) were used for the final purification step. Commercially available reagents were used without purification, unless mentioned otherwise: PPI dendrimers G2, G3, and G4 (PPI, SyMOChem); ethanol (EtOH, absolute, dried over molecular sieves, Merck); hydrochloric acid (HCl, 37% in water, Acros Organics); dichloromethane (DCM, GPR Rectapur, Fisher Scientific); n-hexane (≥99%, Sigma-Aldrich); acetone (Semiconductor grade, Sigma-Aldrich); deuterium oxide (D2O, 99.9 atom % D, Sigma-Aldrich); 3-(trimethylsilyl)-1-propanesulfonic acid-d6 sodium salt (TMS salt, 98 atom % D, Sigma-Aldrich); formaldehyde (37 wt % in water, 10–15% methanol, Fisher Scientific); formic acid (99%, VWR); tert-butyl bromoacetate (98%, Sigma-Aldrich); tert-butyl 2-iodoacetate (Sigma-Aldrich); trifluoroacetic acid (Biosolve B.V.); methyl bromoacetate (96%, Sigma-Aldrich); methyl 2-iodoacetate (95%, Sigma-Aldrich); methyl iodide (99%, stabilized, Fisher Scientific); sodium bromoacetate (98%, Sigma-Aldrich); sodium iodoacetate (≥98%, Sigma-Aldrich); sodium 2-bromoethylsulfonate (98%, Sigma-Aldrich); sodium 3-bromopropanesulfonate (≥97%, Sigma-Aldrich); propargyl-N-hydroxysuccinimidyl ester (NHS-alkyne, Sigma-Aldrich); azido-PEG8-NHS ester (NHS-PEG-azide, Sigma-Aldrich); sodium hydroxide (NaOH, 98.5% pellets, Fisher Scientific); sodium sulfate (Na2SO4, anhydrous, Fisher Scientific); triethylamine (99%, distilled, on KOH, Fisher Scientific); azide-PEG3-biotin (Sigma-Aldrich); sodium l-ascorbate (sodium ascorbate, ≥98%, Sigma-Aldrich); and copper sulfate pentahydrate (≥98%, Sigma-Aldrich).
3. Methods
3.1. Nuclear Magnetic Resonance
1H NMR measurements were recorded on a Bruker AVANCE III NMR at 400 MHz, 13C NMR spectra were recorded at 100 MHz. For the 1H–15N HMBC (heteronuclear multiple bond correlation) measurements settings of 600 and 60 MHz were used, respectively, on a 600 MHz Bruker AVANCE III Ultrashield Plus equipped with a cryoprobe. Chemical shifts are reported in parts per million (ppm) and are referred to the methyl signal of the sodium salt of 3-(trimethylsilyl)-1-propanesulfonic acid-d6 (δ = 0).
3.2. Infrared
IR analyses were performed on a Bruker Tensor 27 spectrometer with platinum attenuated total reflection accessory.
3.3. X-ray Photoelectron Spectroscopy
Samples for dendrimer analysis were prepared by concentrating the dendrimers (in Milli-Q water) and dropcasting 3 μL of this suspension onto a piece of Si(111) (Siltronix, N-type, phosphorus doped), which was cleaned by rinsing and sonicating for 5 min in semiconductor grade acetone followed by oxygen plasma treatment (Diener electronic, Femto A) for 1 min at 100% power. The dropcast samples were subsequently dried in vacuum overnight before XPS measurements were started. XPS spectra were obtained using a JPS-9200 photoelectron spectrometer (JEOL, Japan) with monochromatic Al Kα X-ray radiation at 12 kV and 20 mA. The obtained spectra were analyzed using CASA XPS software (version 2.3.16 PR 1.6). In C 1s and N 1s narrow-range spectra, the positions are set to 285 and 400 eV for the C–C and N–C signals, respectively.
3.4. Gel Permeation Chromatography
The polymer molecular weight and polydispersity index (PDI) were determined using GPC (Agilent G5654A quaternary pump, G7162A refractive index detector), where a PSS SUPREMA Combination medium (P/N 206–0002) 1000 Å single porosity column was employed (0.05% NaN3 in Milli-Q water as eluent, 1 mL/min). Dendrimer in Milli-Q solutions were freshly prepared. Twenty microliters was used for each analysis. An Agilent PL2080-0101 PEO calibration kit was used for calibration purposes.
3.5. Mass Spectrometry
MS data were recorded on an exactive high-resolution MS instrument (Thermo Scientific) equipped with an electrospray ionization (ESI) probe. The MS was calibrated daily using ProteoMass LTQ/FT-hybrid ESI Pos. Mode Cal Mix and Pierce ESI Neg. Ion Cal. solutions. Thermo Xcalibur Browser software (version 4.0.27.19) was used for instrument control, data acquisition, and data processing.
3.6. Synthesis of 2 PPI-Me
A solution of 0.500 g of 1 PPI G3 dendrimer (0.296 mmol) in 10 mL Milli-Q water was prepared. A 100 mL 3-neck round bottom flask with a cooler and a stirring bar was flushed with argon by applying 3 vacuum–argon cycles, ending with a final argon refill. Under argon overpressure, 20 mL Milli-Q water, 6.35 mL formaldehyde (37% aqueous solution; 75 mmol, 15 equiv per PPI primary amine), and 6.12 mL formic acid (150 mmol, 30 equiv per PPI primary amine) were added. The mixture was cooled on ice before the 1 PPI G3 in 10 mL Milli-Q water was added dropwise. The reaction mixture was allowed to warm up to room temperature, after which the setup was closed under argon and refluxed using an oil bath for 5 continuous days to assure full conversion.
Afterwards, the mixture was cooled on ice and the pH was increased to 11 by the slow addition of a saturated NaOH solution. The solution became cloudy because the methylated dendrimers were less water soluble after deprotonation at this concentration. The aqueous solution was extracted with DCM for three times. The combined organic layers were washed with water and dried over Na2SO4. After evaporation of the solvent, 0.474 g (0.22 mmol) of a yellow oil was obtained with a yield of 80%.
For the synthesis of 2 PPI-Me G2 and G4, the amounts were adjusted in order to retain 15 equiv of formaldehyde and 30 equiv formic acid per PPI primary amine.
3.7. Synthesis of 3 PPI-CB1
2 PPI-Me (0.200 g; 0.094 mmol) was dissolved in 3 mL aqueous NaOH solution at pH 10 by stirring in a 10 mL round bottom flask. Sodium iodoacetate (2.08 g; 10 mmol) was added and the solution was stirred at room temperature in the dark for 3 days. Afterward, the pH was adjusted to ∼7 using an HCl solution to assure compatibility with the dialysis membrane and the volume of the mixture was increased to 10 mL by addition of Milli-Q water. The mixture was dialyzed against 500 mL Milli-Q water for 3 days with three medium exchanges. After evaporation of the solvent and lyophilization, 0.238 g of a fluffy white powder was obtained with a yield of 68%.
For the synthesis of 3 PPI-CB1 G2 and G4 with sodium iodoacetate, the amounts were adjusted in order to retain 4 equiv of sodium iodoacetate per 2 PPI-Me tertiary amine.
3.8. Optimization Menschutkin Alkylation of Methylated Dendrimers Using Different Alkyl Halides
To optimize the conversion of the alkylation reaction, we tested different dendrimer sizes and alkylation agents and studied the results by XPS N 1s high-resolution scans (see Figures S2 and S3). For the sodium salts sodium bromoacetate, sodium bromoethane sulfonate, and sodium bromopropane sulfonate, a similar procedure as described for sodium iodoacetate was used, keeping the same equivalents: 0.200 g of 2 PPI (0.094 mmol) was dissolved in 3 mL aqueous NaOH solution at pH 10 by stirring in a 10 mL round bottom flask. The alkyl halide sodium salt (10 mmol) was added and the solution was stirred at room temperature in the dark for 3 days. Afterward, the pH was adjusted to ∼7 using an aqueous HCl solution to assure compatibility with the dialysis membrane and the volume of the mixture was increased to 10 mL by addition of Milli-Q water. The mixture was dialyzed against 500 mL Milli-Q water for 3 days with three medium exchanges. After evaporation of the solvent and lyophilization, the products were obtained with various yields and conversions (see Figure S3).
For the reaction with protected acids or methyl iodide instead of free acetates (tert-butyl iodoacetate, tert-butyl bromoacetate, methyl iodoacetate, methyl bromoacetate), an adjusted protocol was followed, using the same equivalents. For solubility reasons, acetonitrile was used as a solvent instead of aqueous NaOH solution. Afterward, the solvent and other volatiles were evaporated in vacuo and a deprotection step was performed before purification. To this end, tert-butyl-protected CB1 dendrimers were stirred in 50 mmol trifluoroacetic acid (TFA) in 15 mL acetonitrile (3.3 M) at room temperature for 3 days, followed by evaporation of solvent, TFA, and tert-butanol. After dissolving the product in Milli-Q water, the pH was adjusted to ∼7 using a NaOH solution before purification by dialysis. Deprotection of methyl-protected CB1 dendrimers was achieved by refluxing in a pH 10 NaOH solution for 3 days, and the pH was adjusted to ∼7 using an HCl solution before purification by dialysis. The dendrimer solutions were dialyzed against 500 mL Milli-Q water for 3 days with three medium exchanges. After evaporation of the solvent and lyophilization, the products were obtained with various conversions (see Table 1 and Figure S3).
Table 1. XPS Results for the Optimization of the Alkylation/Charge–Neutralization Reaction on 2, via Either the Sodium Salt (a–d) or the Protected Acid (e–h) of the Indicated Alkyl halides (See Figure S3).
| reactant | conversiona (%) | |
|---|---|---|
| a | sodium iodoacetate | 87 |
| b | sodium bromoacetate | 54 |
| c | sodium bromoethane sulfonate | 74 |
| d | sodium bromopropane sulfonate | 70 |
| e | tert-butyl iodoacetate + deprotection | 85 |
| f | tert-butyl bromoacetate + deprotection | 93 |
| g | methyl iodoacetate + deprotection | 79 |
| h | methyl bromoacetate + deprotection | 77 |
Based on conversion to quaternary amine as measured by XPS N 1s narrow scans.
3.9. Synthesis of Alkyne-Functionalized PPI Dendrimer 4a
1 PPI G3 (50 mg, 0.029 mmol) was dissolved in 5 mL dry acetonitrile by stirring in an argon-flushed 25 mL round bottom flask. Then, 9 μL (0.062 mmol, n = 2), 13 μL (0.093 mmol, n = 3), or 26 μL (0.186 mmol, n = 6) of triethylamine was added and the solution was cooled on ice. A solution of 13.5 mg (0.060 mmol, n = 2), 20.3 mg (0.090 mmol, n = 3), or 40.5 mg (0.180 mmol, n = 6) of alkyne-NHS in 5 mL dry acetonitrile was slowly added to the 1 PPI G3 solution while stirring vigorously to assure an even distribution of the functional click handles over the dendrimers. The mixture was allowed to warm up to room temperature and stirring was continued overnight under argon. The solvent and triethylamine were removed by rotavap and oil pump vacuum until a viscous colorless oil was left. About 10% of the crude was purified using dialysis as described before for MS analysis purposes. From this fraction, an average yield of ∼90% could be calculated. The rest of the crude product was used for the next reaction step without further purification.
3.10. Synthesis of Alkyne-Functionalized PPI-Me 5a
The functionalized dendrimer 4a was methylated as described before for compound 2, using the same equivalents, conditions, and purification by extraction (note: after extraction there were still some minor impurities present in NMR, which were fully removed after extensive dialysis in the next modification step). This led to methylated, functionalized dendrimers with typical yields of ∼70%.
3.11. Synthesis of Alkyne-Functionalized PPI-CB1 6a
The methylated, functionalized dendrimers were alkylated using sodium iodoacetate as described previously for compound 3. This yielded alkyne-modified ZID with typical yields of 30% (not taking into account that the conversion in the last step is not quantitative).
3.12. Synthesis of Azide-Functionalized PPI Dendrimer 4b
1 PPI G3 (50 mg; 0.029 mmol) was dissolved in 5 mL dry acetonitrile by stirring in an argon-flushed 25 mL round bottom flask. Then, 13 μL (0.093 mmol, n = 3) or 26 μL (0.186 mmol, n = 6) of triethylamine was added and the solution was cooled on ice. A solution of 50.8 mg (0.090 mmol, n = 3) or 101.6 mg (0.180 mmol, n = 6) of azide-PEG-NHS in 5 mL dry acetonitrile was slowly added to the 1 PPI G3 solution while stirring vigorously to assure an even distribution of the functional handles over the dendrimers. The mixture was allowed to warm up to room temperature, and stirring was continued overnight under argon. The solvent and triethylamine were removed by rotavap and oil pump vacuum until a viscous colorless oil was left. The crude product was used for the next reaction step without further purification.
3.13. Synthesis of Azide-Functionalized PPI-Me 5b
The functionalized dendrimer 4b was methylated as described before for compound 2, using the same equivalents, conditions, and purification by extraction (note: after extraction there were still some minor impurities present in NMR, which were fully removed after extensive dialysis in the next modification step). This led to methylated, functionalized dendrimers with an average yield of ∼89%.
3.14. Synthesis of Azide-Functionalized PPI-CB1 6b
The methylated, functionalized dendrimers were alkylated using sodium iodoacetate as described previously for compound 3. This yielded alkyne-modified ZID with an average yield of ∼68%.
3.15. Synthesis of Biotin-Functionalized PPI Dendrimer 7
A solution of 21.6 mg (0.0055 mmol) alkyne-functionalized ZID 6a (n = 3) in 1.6 mL Milli-Q water was prepared. Next, 0.2 mL of a 1 mg/mL solution (0.001 mmol) of copper(II) sulfate pentahydrate in Milli-Q water was mixed with 0.2 mL of a 200 mg/mL solution (0.2 mmol) of sodium ascorbate in Milli-Q water. This mixture was added to the dendrimer 6a solution after which 10.8 mg (0.024 mmol) azide-PEG3-biotin was added. The solution was stirred overnight at room temperature. The mixture was dialyzed (MWCO 1000–5000 Da) against 500 mL Milli-Q water for 3 days with three medium exchanges. After evaporation of the solvent and lyophilization, 20.6 mg of a fluffy light yellow powder was obtained.
4. Results and Discussion
Previously, PAMAM22,23 or PPI dendrimers22 and polyethyleneimine polymers35 have been modified to become exteriorly zwitterionic by reacting only the outer primary amines with zwitterionic monomers in order to reduce their cytotoxicity. Alternatively, Hu et al. investigated the interior zwitterionic modification of both PPI and PAMAM dendrimers for drug delivery purposes by first acetylating the outer amines and subsequently reacting the inner tertiary amines with 1,3-propane sultone in DMF.19 However, the conversion of their last step was only moderate (below 50%), probably because their solvent of choice was not compatible with the formed highly zwitterionic product, leading to premature precipitation before the fully zwitterionic product could be formed. A drawback of partial zwitterionic modification is the relatively low density of charges and—more importantly—the presence of unreacted amines that can be protonated, leading to an overall positively charged dendrimer.
To introduce zwitterionic character to the PPI dendrimer, several synthetic routes can be followed. However, not all will necessarily lead to an uncharged final product. For example, Carr et al. have reported the synthesis of zwitterionic crosslinkers for hydrogels by performing a Michael addition on a secondary amine-containing monomer with a protected acid (which was deprotected in the end), followed by N-alkylation using methyl iodide.20 While this approach could also be applied to PAMAM or PPI dendrimers, it would lead to an overall charge of −2 (meaning two noncompensated carboxylate negative charges), regardless of the size of the initial dendrimer (see Supporting Information, Figure S1). Apart from being a conceptual concession, this would likely be detrimental for their performance in biological systems,36 as it could attract positively charged biomaterials. As a result, we rather aim for the design of fully zwitterionic, charge-neutral dendrimers, and for this it is crucial that the installation of the negatively charged group occurs simultaneously with the creation of the positivity charged group (in case of PPI dendrimers, this is the ammonium group). This means that before introduction of the negatively charged group by alkylation, all amine groups in the PPI dendrimer need to be tertiary in nature (thus becoming quaternary upon alkylation; see Figure S1). To assure this, we choose the first step in the modification of the PPI dendrimer to be the double methylation of the outer, primary amines using the Eschweiler–Clarke reaction, as this can be run to full conversion to yield dendrimers with tertiary amines only (Scheme 1).37,38 In contrast, direct alkylation of both the primary and tertiary amines of the PPI dendrimer with an appropriate alkyl halide (that also provides a negative charge) would make the dendrimers over-alkylated and thus bearing an overall negatively charged, as the alkylation agent would alkylate the primary amines up to three times. Via the Eschweiler–Clarke reaction this is prevented, and the dendrimers can subsequently be made truly zwitterionic by reacting all the tertiary amines with an alkyl halide with a protected or free anionic group, possibly followed by a deprotection step (Scheme 1).39 For the synthesis of ZIDs, we chose to work with PPI generation 3 (G3) dendrimers (in line with the definition by Meijer and co-workers, a PPI dendrimer with 16 end groups is called a third-generation dendrimer40) because of their size and the trade-off between the stability under relevant conditions as mentioned before and low number of defects (as the latter increases with generation growth).41
Scheme 1. Synthesis of Zwitterionic PPI-CB1 Dendrimers (Colors for the Different Compounds Are Also Used in the Results & Discussion Section To Label Experimental Data to the Appropriate Compound).

4.1. Synthesis of Zwitterionic Dendrimer 3 PPI-CB1
As explained above, the first step in the synthesis of the zwitterionic PPI-based dendrimers is the methylation of the primary amine groups under Eschweiler–Clarke conditions, leading to intermediate 2 PPI-Me (Scheme 1).
This first intermediate, 2, was subsequently characterized using NMR, IR, and MS. The 1H NMR spectrum of intermediate 2 showed the appearance of a new, sharp singlet at 2.48 ppm with corresponding integrals that could be assigned to the installed methyl groups, while the existing methylene peaks of the dendrimer showed the expected downfield shift (peak a in Figure 1b). Furthermore, IR spectroscopy also showed the presence of methyl groups as indicated by C–H bending vibrations at 1456 cm–1 for intermediate 2. Indirectly, the intensity decrease of the N–H stretch (3360 and 3280 cm–1) and N–H bend (1592 cm–1) bands also indicated the disappearance of the primary amines of starting material 1 (Figure 1a).
Figure 1.

IR (a) and 1H NMR results (b) with corresponding peak assignment (c) of 1 (PPI—red), 2 (PPI–Me—green) and 3 (PPI-CB1—blue); and DOSY (d) results of product 3 PPI-CB1. Full assignment of NMR and IR spectra is provided in the Supporting Information.
Dendrimer 2 can be converted into a ZID, by reacting it with an appropriate alkylating agent that also features a negatively charged group (Scheme 1). Initially, we selected sodium iodoacetate as the alkylation agent to introduce both the positive and negative charges in a single step. After the reaction, all dendrimer backbone peaks showed a downfield shift in the 1H NMR spectrum (Figure 1b). In addition, a new peak was observed at 3.97 ppm, corresponding to the CH2 group of the carboxybetaines (peak e in Figure 1b,c). The presence of the carboxylate group was also confirmed by the C=O stretch peak at 1600 cm–1 in the IR spectrum (Figure 1a). Diffusion-ordered spectroscopy (DOSY) also indicated that all the assigned signals belonged to one macromolecular structure by showing the same diffusion coefficient (Figure 1d). Applying the Stokes–Einstein equation to the data obtained by DOSY, we estimate the hydrodynamic diameter for 3 to be 3.2 nm (Table S1), which is in agreement with values reported in the literature for related dendrimers of similar mass.42,43
GPC data also confirmed the estimated mass and desired monodispersity of product 3. The observed molecular weight (Mw, 4.1 × 103 g/mol) was in reasonable agreement with the proposed molecular weight at full conversion (3877), keeping in mind that the Mw of the zwitterionic, dendritic structure 3 was calculated using a linear, nonzwitterionic calibration system. The measured polydispersity (PDI) of dendrimer 3 was low (1.102), especially given the multiple consecutive modification steps that led to 3.
Determining the exact conversion of this second reaction step was found to be challenging because IR is not a quantitative technique and 1H NMR signals became broad, preventing accurate integration. Therefore, we initially used 1H–15N HMBC to indirectly measure the 15N NMR spectrum of compound 3 PPI-CB1 (Figure S10). The indicative methyl 1H peak at 3.28 ppm (also visible in Figure 1b) was found to correlate with multiple 15N NMR peaks. This implied that after modification still different N atoms were present, which suggests the presence of unreacted tertiary amines and incomplete conversion.
To be able to quantify this conversion, we made use of the unique information that XPS can provide about the conversion from tertiary to quaternary amines, as XPS can reveal the electronic configuration of the nitrogen atoms. XPS provides both quantitative and qualitative information on the elemental composition and chemical environment of elements on a surface. The binding energy of the core electrons of an element is measured, and from the obtained spectra, the amount of the element present can be calculated.44,45 XPS is generally used to investigate the chemical composition of ca. the top-10 nm of surfaces and is not employed as a standard characterization method for organic molecules. However, following earlier research in our labs that focused on the extensive characterization of polymers that are covalently bound to surfaces46−48 or that are strongly adsorbed onto surfaces,49 we hypothesized that this technique might also provide unique information in the characterization of large (meaning: nonvolatile) but well-defined compounds. We therefore used the technique to characterize organic macromolecules by dropcasting and drying them from solution on a silicon surface.
In Figure 2 the XPS C 1s and N 1s narrow-range scans are displayed. By scanning the element peaks with high resolution, valuable quantitative information about the chemical state of the element involved can be obtained, which can be related to the electronic effects of its substituents and thereby its chemical structure. After methylation, the C 1s scan of intermediate 2 showed solely C–C (285.0 eV) and C–N (286.0 eV) carbon species, in accordance with the structure of 2. In contrast, after alkylation with sodium iodoacetate, an additional peak in the C 1s narrow scan is visible at 287.7 eV, which points to the presence carboxylate carbons (−C(=O)–O. Furthermore, the C–N peak also broadened because of the increase in the variation of this type of carbon. More importantly, the XPS N 1s scans provided us with unique information about the fraction of quaternary nitrogen atoms. Before alkylation, structure 2 shows one sharp peak for tertiary amines (400.0 eV). After alkylation, compound 3 showed an additional major peak at 403.0 eV, corresponding to quaternary amines. The ratio of these two peaks shows that a conversion of approximately 80% was achieved when reacting 2 with sodium iodoacetate.
Figure 2.

Narrow scan XPS C 1s (left) and N 1s (right) data of 1 (top), 2 (middle), and 3 (bottom).
Unfortunately, the quantitative information about the chemical state of N as obtained by XPS was unique with respect to the accuracy it offered in determining the degree of conversion. Such accuracy could not be achieved—and hence not compared—with results obtained by other, well known, techniques (e.g., NMR or MS).
As the conversion of the last step was not 100%, there will be tertiary amines left that can be protonated under physiological conditions, which will lead to dendrimers that are not permanently charge-neutral as the carboxylate group is needed to compensate for the positively charged amine. Furthermore, because it is known that the performance of zwitterionic materials is enhanced with an increase in hydration of the brush that is linked to its charge density,50 we set out to systematically explore a range of reaction conditions to optimize this last reaction step.
XPS provided a very powerful and sensitive method to systematically screen conditions under which the last reaction step could be improved. In particular, we used XPS (Table 1, Figures S2, S3, and S4) to study the influence of the following parameters:
4.1.1. Steric Hindrance within the Dendrimer
Generations 2, 3, and 4 of the PPI dendrimer were reacted with sodium iodoacetate. Steric hindrance within the dendrimer was found not to play a significant role because the conversion did not increase significantly with decreased dendrimer size (Figure S2).
4.1.2. Solvent Effect
The use of both methyl- and tert-butyl-protected carboxylates allowed us to use an aprotic polar solvent because both the reactant and the intermediate product (before deprotection) are soluble in such a solvent, enhancing the stability of the alkyl halide precursors. We indeed observed an increase in conversion up to roughly 90% when using protected acids (e,f), possibly due to the stability of the halide precursors in an aprotic solvent. However, when using this approach, an extra deprotection is necessary, which then needs to be quantitative to yield a charge-neutral ZID (e,h).
4.1.3. Reactivity
We found a clear difference in conversion when comparing sodium iodo- and bromoacetate (87% vs 54%) in water, which is in line with expectation as iodide is a better leaving group in this reaction (a,b). However, the corresponding reactions with protected acids in organic solvents showed no significant difference when comparing halide leaving group (e–h). We postulate that due to the lack of (water-induced) competitive elimination reactions and the long reaction time (3 days), in this case both groups eventually reach the same high conversion.
4.1.4. Nature and Size of the Anionic Group
We evaluated both the nature of the anionic head group, which was either a carboxylate or a sulfonate group, as well as the carbon spacer length between the opposite charges of the zwitterion pair. We chose to synthesize CB1 (carboxybetaine, one-carbon spacer) ZID because it does not allow for Hofmann elimination (resulting in loss of C2H3COO–), in contrast to CB2 (carboxybetaine, two-carbon spacer) species.51 On top of that, we compared SB2 and SB3 (sulfobetaine, with two- and three-carbon spacers, respectively) to study steric effects caused by the precursor. We indeed found a lower conversion when using the slightly bigger SB3 compound (c,d). Unfortunately, the precursor for SB1 was not available.
To further evaluate the effect of sterics, we also reacted an ∼80% converted ZID with methyl iodide as a small, strong alkylation agent, resulting in near 100% conversion toward quaternary amines (Figure S4). This suggests that indeed for steric reasons it is practically hard to reach full conversion (for 2, a 93% conversion of the 30 N atoms effectively means that 28 N atoms will be quaternized). While the final product after reaction with methyl iodide is fully quaternized, it is no longer charge-neutral, likely thus on average +2, and therefore we did not use it as such for further studies.
Overall, from this comparative study, we concluded that the sodium iodoacetate reaction on 1 was most efficient. For this reaction, the highest conversion was obtained without the need for an additional deprotection step. A reaction efficiency of 87% implies for an N30-molecule like 3 that typically 26 tertiary N atoms have been converted to quaternary N atoms. As a result, under physiological conditions likely one or two N atoms will be protonated. The determination of the precise charge and charge distribution under these conditions are topics of future investigations in our labs.
4.2. Synthetic Design of Functional, Zwitterionic Dendrimers
In order to use the ZIDs for biological or biomedical applications, functional groups at the dendrimer’s periphery that allow easy modification are needed. In order to do this, fast, easy, orthogonal and water-compatible chemistry is required. Given the intrinsic multivalent nature of the PPI dendrimers, introduction of a functional group can be achieved through reaction with a predefined number of amine groups of the dendrimer, provided that both the installed bond and the functional group are compatible with the conditions of the subsequent reaction steps that alkylate and add the zwitterionic moieties (Scheme 1). On the basis of these provisions, we relied on the reaction of PPI with activated esters to form amide bonds to introduce a new functional group of choice. We chose to functionalize the dendrimers with alkyne and azido groups as they allow for a range of click chemistries (Scheme 2).52
Scheme 2. Synthesis of Alkyne-Functionalized Zwitterionic PPI-CB1 Dendrimers.

Note: n = 3 is shown as example, but other degrees of functionalization are possible; this synthesis was also performed with n = 2 and n = 6; see Supporting Information for details.
To install an alkyne click handle, PPI dendrimer 1 was first reacted with a predetermined number of equivalents of an NHS-activated ester featuring an alkyne group connected with a short linker (forming intermediate product 4a). The formed product was directly reacted toward the fully methylated intermediate 5a, using the Eschweiler–Clarke conditions previously established for the nonfunctional dendrimer 3 (Scheme 1). Finally, the functionalized, ZIDs with an alkyne (6a) handle were obtained through reaction with sodium iodoacetate (again following procedures previously employed in the synthesis of 3).
In the 1H NMR spectrum (Figure S11) of the reaction product 6a, we observed characteristic signals for the alkyne groups (3.33 ppm), as well as for the formed amide bonds (CH2 next to the amide, 3.36 ppm). On the basis of NMR integration, an average degree of incorporation of 2.9 for 6a (n = 3) could be determined. To confirm with another technique and to obtain information about the distribution of the number of functional groups per dendrimer, we used MS. Unfortunately, the ESI–MS of methylated intermediate 5a led to fragmentation due to rearrangements within the dendrimer, most probably due to ionization53 or methylation.54 This prompted us to isolate intermediates 4a (both n = 2 and n = 3) for MS analysis to study the distribution of functional groups over the ZID. MS data nicely showed a fairly small range of distribution of n = 2 and n = 3 with the desired number of n as center of the distribution (Figures S20 and S21). The final product 6a itself could not be measured, which we attribute to ionization difficulties of the highly ZIDs; unfortunately, also with matrix-assisted laser desorption ionization–time of flight we were unable to get signal.
GPC confirmed the estimated mass and desired PDI of products 3 and 6a (both for n = 2 and n = 3). The observed molecular weight was in good agreement with the molecular weight at full conversion. As also observed for 3, the PDIs of all three products; 1.102 (3), 1.193 (6an = 2), and 1.138 (6an = 6) were rather low, which is even more remarkable as these are macromolecules that underwent several consecutive modification steps (Figures S22, S23 and S24).
To quantify the incorporation of the functional groups, XPS again proved to be a valuable technique. Because the carbonyl–carbon that forms upon forming the amide bond gives a signal that is found at a sufficiently high binding energy (∼288 eV) to be discerned as a separate peak in C 1s XPS spectra, it is possible to determine the relative number of C=O carbons present in the dendrimer.
In Figure 3 the XPS C 1s data confirm an average incorporation of 2 and 3 alkyne groups by showing a similar amount of carbonyl–carbon signals (n = 2 carbonyls out of 128 carbons in total = 1.6% theoretically, 1.9% is observed) (n = 3 carbonyls out of 132 carbons in total = 2.2% theoretically, 3.6% is observed) at 288.2 and 288.1 eV in the upper graphs for intermediate structure 5a. Furthermore, both C 1s scans of compound 6an = 2 and n = 3 show a similar transformation upon alkylation as described before for compound 3 (Figure 2).
Figure 3.

XPS narrow scan C 1s data of 5a (top) and 6a (bottom) of n = 2 (left) and n = 3 (right) alkyne-modified dendrimer. For reference, a characteristic part of the modified dendrimer is shown.
To extend the versatility of click reaction-based dendrimer modification, we also synthesized ZIDs with azide groups. By reacting PPI dendrimer 1 with an NHS-activated ester that was linked to an azide group via a short oligo(ethylene oxide) chain (NHS-EO8-azide) group, azide-functionalized intermediate 4b was formed. This intermediate 4b was further reacted following procedures previously employed in the synthesis of both 3 and 6a to eventually yield product 6b (see Figure 4 and Scheme S3 in Supporting Information).
Figure 4.

Proposed schematic structure of azide-functionalized zwitterionic PPI-CB1 dendrimer 6b (n = 3). Please note that this dendrimer was also synthesized with n = 2 and n = 6.
In the 1H NMR spectrum (Figure S13) of reaction product 6b (n = 3), we observed characteristic signals for the ethylene glycol spacers (3.69 ppm), as well as for the formed amide bonds (3.37 ppm). DOSY indicated that all the assigned signals, including the functional groups, belonged to one macromolecular structure by showing the same diffusion coefficient (Figure 5), confirming that the low-molecular weight functional groups were successfully linked to the high-molecular weight dendrimer. We also observed a trend in increased diffusion constant with increasing size and expected molecular weight of the ZID (Figure 5): applying the Stokes–Einstein equation, the hydrodynamic diameters for 3, 6a, and 6b were estimated to be 3.2, 4.1, and 5.1 nm, respectively (Table S1). Unfortunately, dynamic light scattering proved to be impossible to use as a complementary technique to determine the dimensions of our dendrimers, which is most likely the result of the relatively small size of the dendrimers. As a result, we also could not determine the zeta potential to get more insight into the charge distribution within the dendrimer structures.
Figure 5.

DOSY spectra of unmodified PPI-CB1 dendrimer 3, alkyne-modified 6a PPI-CB1, and azide-modified 6b PPI-CB1 (both n = 3). In the functionalized dendrimers, the most indicative 1H signal is labeled by a star (*). In all three spectra, the signals in the highlighted band correspond to the ZID, and the signals at 4.8 and 0.0 ppm represent water and TMS, respectively. Furthermore, theoretical molecular weights (at full conversion) are given.
Unfortunately, though not fully unexpectedly, in the IR spectrum, the alkyne and azide signals could hardly be discerned for 6a and 6b (both n = 2 or n = 3) because the abundance of these groups in the large dendrimer is relatively low (namely only 2 or 3 alkyne or azide groups per 4000–5000 g/mol structure). Fortunately, because of the modularity of the synthetic route (Scheme 2), the synthesis of functional dendrimers with a higher number of functional groups (reaction with 6 equiv instead of 2 or 3 equiv) was straightforward. For these n = 6 dendrimers, it was possible to observe by IR spectroscopy the alkyne and azide groups at 2115 and 2106 cm–1, respectively (Figure S19). Hence, these IR measurements provided further experimental evidence for the successful incorporation of the functional alkyne and azide moieties.
As a proof-of-principle experiment to demonstrate that our installed click handles can indeed be used for (bio)functionalization, we introduced a biotin moiety onto the alkyne-containing ZID 6a (n = 3) by reacting the latter with an azide-functionalized PEG-biotin derivative as shown in Scheme S4. NMR data nicely showed the corresponding signal for the proton in the formed triazole at 8.13 ppm in the 1H NMR spectrum after extensive dialysis, providing evidence that the biotin was successfully coupled to the ZID (see Figures S15 and S16).
5. Conclusions
In this work, we showed the design and optimized synthesis of (nearly) charge-neutral, multivalently clickable ZID. The effect of different parameters on the conversion toward a fully zwitterionic system was established, leading to an optimized synthesis protocol. In this optimization study, XPS, which so far was mainly employed for surface characterization, proved to be a very valuable technique to analyze macromolecules by providing information about the degree of conversion in these highly functional dendrimer structures. Furthermore, we developed a modular synthetic approach to incorporate a variable number of alkyne or azide functional groups, which allow for covalent (bio)functionalization. Proof-of-principle coupling of an azide-biotin conjugate by click chemistry showed that the ZID indeed can be further functionalized. The fact that multiple, or ultimately even different, functional groups can be incorporated will further enhance the (bio)applicability of this kind of ZIDs.
Acknowledgments
The authors thank Dr. Pieter de Waard and Prof. Aldrik Velders for assistance with the 15N and 2D-NMR measurements, and Margaux Tellez and Maaike van Slagmaat for their contributions to this work. This project was supported by NWO (LIFT program, grant 731.015.042) with Surfix BV as a partner.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b03521.
More detailed information about materials and methods; additional reaction schemes; and IR, NMR, MS, and GPC data and calculations (PDF)
The authors declare the following competing financial interest(s): Dr Scheres is CEO and co-owner of Surfix BV - this company funded part of this research.
Supplementary Material
References
- Boas U.; Christensen J. B.; Heegaard P. M. H. Dendrimers: Design, Synthesis and Chemical Properties. J. Mater. Chem. 2006, 16, 3785–3798. 10.1039/b611813p. [DOI] [Google Scholar]
- Sato K.; Anzai J.-I. Dendrimers in Layer-by-Layer Assemblies: Synthesis and Applications. Molecules 2013, 18, 8440–8460. 10.3390/molecules18078440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbasi E.; Aval S.; Akbarzadeh A.; Milani M.; Nasrabadi H.; Joo S.; Hanifehpour Y.; Nejati-Koshki K.; Pashaei-Asl R. Dendrimers: Synthesis, Applications, and Properties. Nanoscale Res. Lett. 2014, 9, 247. 10.1186/1556-276x-9-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. C.; MacKay J. A.; Fréchet J. M. J.; Szoka F. C. Designing Dendrimers for Biological Applications. Nat. Biotechnol. 2005, 23, 1517–1526. 10.1038/nbt1171. [DOI] [PubMed] [Google Scholar]
- Froehling P. E. Dendrimers and dyes—a review. Dyes Pigm. 2001, 48, 187–195. 10.1016/s0143-7208(00)00099-1. [DOI] [Google Scholar]
- Knapen J. W. J.; van der Made A. W.; de Wilde J. C.; van Leeuwen P. W. N. M.; Wijkens P.; Grove D. M.; van Koten G. Homogeneous catalysts based on silane dendrimers functionalized with arylnickel(II) complexes. Nature 1994, 372, 659–663. 10.1038/372659a0. [DOI] [Google Scholar]
- Kobayashi H.; Brechbiel M. Nano-Sized MRI Contrast Agents with Dendrimer Cores. Adv. Drug Delivery Rev. 2005, 57, 2271–2286. 10.1016/j.addr.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Fernandes E. G. R.; Vieira N. C. S.; de Queiroz A. A. A.; Guimarães F. E. G.; Zucolotto V. Immobilization of Poly(propylene imine) Dendrimer/Nickel Phtalocyanine as Nanostructured Multilayer Films To Be Used as Gate Membranes for SEGFET pH Sensors. J. Phys. Chem. C 2010, 114, 6478–6483. 10.1021/jp9106052. [DOI] [Google Scholar]
- Nimesh S.Gene Therapy: Potential Applications of Nanotechnology; Woodhead Publishing, 2013. [Google Scholar]
- Tsai H.-C.; Imae T. Fabrication of Dendrimers Toward Biological Application. Prog. Mol. Biol. Transl. Sci. 2011, 104, 101–140. 10.1016/b978-0-12-416020-0.00003-6. [DOI] [PubMed] [Google Scholar]
- Sharma A. K.; Gothwal A.; Kesharwani P.; Alsaab H.; Iyer A. K.; Gupta U. Dendrimer Nanoarchitectures for Cancer Diagnosis and Anticancer Drug Delivery. Drug Discovery Today 2017, 22, 314–326. 10.1016/j.drudis.2016.09.013. [DOI] [PubMed] [Google Scholar]
- Wang T.; Zhang Y.; Wei L.; Teng Y. G.; Honda T.; Ojima I. Design, Synthesis, and Biological Evaluations of Asymmetric Bow-Tie PAMAM Dendrimer-Based Conjugates for Tumor-Targeted Drug Delivery. ACS Omega 2018, 3, 3717–3736. 10.1021/acsomega.8b00409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newkome G. R.; Shreiner C. D. Poly(amidoamine), polypropylenimine, and related dendrimers and dendrons possessing different 1→2 branching motifs: An overview of the divergent procedures. Polymer 2008, 49, 1–173. 10.1016/j.polymer.2007.10.021. [DOI] [Google Scholar]
- Otto D. P.; de Villiers M. M. Poly(Amidoamine) Dendrimers as a Pharmaceutical Excipient. Are We There Yet?. J. Pharm. Sci. 2018, 107, 75–83. 10.1016/j.xphs.2017.10.011. [DOI] [PubMed] [Google Scholar]
- Stasko N. A.; Johnson C. B.; Schoenfisch M. H.; Johnson T. A.; Holmuhamedov E. L. Cytotoxicity of Polypropylenimine Dendrimer Conjugates on Cultured Endothelial Cells. Biomacromolecules 2007, 8, 3853–3859. 10.1021/bm7008203. [DOI] [PubMed] [Google Scholar]
- Jevprasesphant R.; Penny J.; Jalal R.; Attwood D.; McKeown N. B.; D’Emanuele A. The Influence of Surface Modification on the Cytotoxicity of PAMAM Dendrimers. Int. J. Pharm. 2003, 252, 263–266. 10.1016/s0378-5173(02)00623-3. [DOI] [PubMed] [Google Scholar]
- Geiger B. C.; Wang S.; Padera R. F.; Grodzinsky A. J.; Hammond P. T. Cartilage-Penetrating Nanocarriers Improve Delivery and Efficacy of Growth Factor Treatment of Osteoarthritis. Sci. Transl. Med. 2018, 10, eaat8800 10.1126/scitranslmed.aat8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodewein L.; Schmelter F.; Di Fiore S.; Hollert H.; Fischer R.; Fenske M. Differences in Toxicity of Anionic and Cationic PAMAM and PPI Dendrimers in Zebrafish Embryos and Cancer Cell Lines. Toxicol. Appl. Pharmacol. 2016, 305, 83–92. 10.1016/j.taap.2016.06.008. [DOI] [PubMed] [Google Scholar]
- Hu J.; Su Y.; Zhang H.; Xu T.; Cheng Y. Design of Interior-Functionalized Fully Acetylated Dendrimers for Anticancer Drug Delivery. Biomaterials 2011, 32, 9950–9959. 10.1016/j.biomaterials.2011.09.016. [DOI] [PubMed] [Google Scholar]
- Carr L. R.; Zhou Y.; Krause J. E.; Xue H.; Jiang S. Uniform Zwitterionic Polymer Hydrogels with a Nonfouling and Functionalizable Crosslinker Using Photopolymerization. Biomaterials 2011, 32, 6893–6899. 10.1016/j.biomaterials.2011.06.006. [DOI] [PubMed] [Google Scholar]
- Xiong Z.; Wang Y.; Zhu J.; Li X.; He Y.; Qu J.; Shen M.; Xia J.; Shi X. Dendrimers Meet Zwitterions: Development of a Unique Antifouling Nanoplatform for Enhanced Blood Pool, Lymph Node and Tumor CT Imaging. Nanoscale 2017, 9, 12295–12301. 10.1039/c7nr03940a. [DOI] [PubMed] [Google Scholar]
- Svenningsen S. W.; Janaszewska A.; Ficker M.; Petersen J. F.; Klajnert-Maculewicz B.; Christensen J. B. Two for the Price of One: PAMAM-Dendrimers with Mixed Phosphoryl Choline and Oligomeric Poly(Caprolactone) Surfaces. Bioconjugate Chem. 2016, 27, 1547–1557. 10.1021/acs.bioconjchem.6b00213. [DOI] [PubMed] [Google Scholar]
- Wang L.; Wang Z.; Ma G.; Lin W.; Chen S. Reducing the Cytotoxity of Poly(Amidoamine) Dendrimers by Modification of a Single Layer of Carboxybetaine. Langmuir 2013, 29, 8914–8921. 10.1021/la400623s. [DOI] [PubMed] [Google Scholar]
- Ramireddy R. R.; Subrahmanyam A. V.; Thayumanavan S. Zwitterionic Moieties from the Huisgen Reaction: A Case Study with Amphiphilic Dendritic Assemblies. Chem.—Eur. J. 2013, 19, 16374–16381. 10.1002/chem.201302442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L.; Wang Y.; Ji F.; Wen Y.; Li J.; Yang B.; Yao F. Synthesis and Characterization of Dendritic Star-Shaped Zwitterionic Polymers as Novel Anticancer Drug Delivery Carriers. J. Biomater. Sci., Polym. Ed. 2014, 25, 1641–1657. 10.1080/09205063.2014.936994. [DOI] [PubMed] [Google Scholar]
- Wang L.; Zhang J.; Lin W.; Wang Z.; Chen S. Development of a Protein Mimic with Peptide Ligands to Enhance Specific Sensing and Targeting by the Zwitterionic Surface Engineering of Poly(Amido Amine) Dendrimers. Adv. Mater. Interfaces 2013, 1, 1300059. 10.1002/admi.201300059. [DOI] [Google Scholar]
- Wang Y.; Li L.; Li J.; Yang B.; Wang C.; Fang W.; Ji F.; Wen Y.; Yao F. Stable and PH-Responsive Polyamidoamine Based Unimolecular Micelles Capped with a Zwitterionic Polymer Shell for Anticancer Drug Delivery. RSC Adv. 2016, 6, 17728–17739. 10.1039/c5ra25505h. [DOI] [Google Scholar]
- Huang D.; Yang F.; Wang X.; Shen H.; You Y.; Wu D. Facile Synthesis and Self-Assembly Behaviour of pH-Responsive Degradable Polyacetal Dendrimers. Polym. Chem. 2016, 7, 6154–6158. 10.1039/c6py01511e. [DOI] [Google Scholar]
- Cao W.; Huang J.; Jiang B.; Gao X.; Yang P. Highly Selective Enrichment of Glycopeptides Based on Zwitterionically Functionalized Soluble Nanopolymers. Sci. Rep. 2016, 6, 29776. 10.1038/srep29776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y.; Qian Y.; Zhou X.; Hu H.; Liu X.; Zhou Z.; Tang J.; Shen Y. Facile synthesis of zwitterionic polyglycerol dendrimers with a β-cyclodextrin core as MRI contrast agent carriers. Polym. Chem. 2016, 7, 6354–6362. 10.1039/c6py01404f. [DOI] [Google Scholar]
- Zhang P.; Sun F.; Tsao C.; Liu S.; Jain P.; Sinclair A.; Hung H.-C.; Bai T.; Wu K.; Jiang S. Zwitterionic Gel Encapsulation Promotes Protein Stability, Enhances Pharmacokinetics, and Reduces Immunogenicity. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 12046–12051. 10.1073/pnas.1512465112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramasamy T.; Ruttala H. B.; Gupta B.; Poudel B. K.; Choi H.-G.; Yong C. S.; Kim J. O. Smart Chemistry-Based Nanosized Drug Delivery Systems for Systemic Applications: A Comprehensive Review. J. Controlled Release 2017, 258, 226–253. 10.1016/j.jconrel.2017.04.043. [DOI] [PubMed] [Google Scholar]
- Astruc D.; Boisselier E.; Ornelas C. Dendrimers Designed for Functions: From Physical, Photophysical, and Supramolecular Properties to Applications in Sensing, Catalysis, Molecular Electronics, Photonics, and Nanomedicine. Chem. Rev. 2010, 110, 1857–1959. 10.1021/cr900327d. [DOI] [PubMed] [Google Scholar]
- Patil M. L.; Zhang M.; Taratula O.; Garbuzenko O. B.; He H.; Minko T. Internally Cationic Polyamidoamine PAMAM-OH Dendrimers for SiRNA Delivery: Effect of the Degree of Quaternization and Cancer Targeting. Biomacromolecules 2009, 10, 258–266. 10.1021/bm8009973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J.; Zeng F.; Jian H.; Wu S. Conjugation with Betaine: A Facile and Effective Approach to Significant Improvement of Gene Delivery Properties of PEI. Biomacromolecules 2013, 14, 728–736. 10.1021/bm301826m. [DOI] [PubMed] [Google Scholar]
- Magin C. M.; Cooper S. P.; Brennan A. B. Non-Toxic Antifouling Strategies. Mater. Today 2010, 13, 36–44. 10.1016/s1369-7021(10)70058-4. [DOI] [Google Scholar]
- Pine S. H.; Sanchez B. L. Formic Acid-Formaldehyde Methylation of Amines. J. Org. Chem. 1971, 36, 829–832. 10.1021/jo00805a022. [DOI] [Google Scholar]
- Zhou X.; Chen Y.; Han J.; Wu X.; Wang G.; Jiang D. Betaine Ester-Shell Functionalized Hyperbranched Polymers for Potential Antimicrobial Usage: Guest Loading Capability, PH Controlled Release and Adjustable Compatibility. Polymer 2014, 55, 6261–6270. 10.1016/j.polymer.2014.10.020. [DOI] [Google Scholar]
- Wu L.; Jasinski J.; Krishnan S. Carboxybetaine, Sulfobetaine, and Cationic Block Copolymer Coatings: A Comparison of the Surface Properties and Antibiofouling Behavior. J. Appl. Polym. Sci. 2011, 124, 2154–2170. 10.1002/app.35233. [DOI] [Google Scholar]
- Bosman A. W.; Janssen H. M.; Meijer E. W. About Dendrimers: Structure, Physical Properties, and Applications. Chem. Rev. 1999, 99, 1665–1688. 10.1021/cr970069y. [DOI] [PubMed] [Google Scholar]
- de Brabander-van den Berg E. M. M.; Meijer E. W. Poly(Propylene Imine) Dendrimers: Large-Scale Synthesis by Hetereogeneously Catalyzed Hydrogenations. Angew. Chem., Int. Ed. Engl. 1993, 32, 1308–1311. 10.1002/anie.199313081. [DOI] [Google Scholar]
- Wrobel D.; Appelhans D.; Signorelli M.; Wiesner B.; Fessas D.; Scheler U.; Voit B.; Maly J. Interaction Study between Maltose-Modified PPI Dendrimers and Lipidic Model Membranes. Biochim. Biophys. Acta, Biomembr. 2015, 1848, 1490–1501. 10.1016/j.bbamem.2015.03.033. [DOI] [PubMed] [Google Scholar]
- Müller R.; Laschober C.; Szymanski W. W.; Allmaier G. Determination of Molecular Weight, Particle Size, and Density of High Number Generation PAMAM Dendrimers Using MALDI–TOF–MS and nES–GEMMA. Macromolecules 2007, 40, 5599–5605. 10.1021/ma062599e. [DOI] [Google Scholar]
- Briggs D.; Grant J. T.. Perspectives and Basic Principles. Surface Analysis by Auger and X-ray Photoelectron Spectroscopy; IM Publications and SurfaceSpectra Limited, 2003; pp 1–88. [Google Scholar]
- Giesbers M.; Marcelis A. T. M.; Zuilhof H. Simulation of XPS C1s Spectra of Organic Monolayers by Quantum Chemical Methods. Langmuir 2013, 29, 4782–4788. 10.1021/la400445s. [DOI] [PubMed] [Google Scholar]
- Joshi S.; Pellacani P.; van Beek T. A.; Zuilhof H.; Nielen M. W. F. Surface Characterization and Antifouling Properties of Nanostructured Gold Chips for Imaging Surface Plasmon Resonance Biosensing. Sens. Actuators, B 2015, 209, 505–514. 10.1016/j.snb.2014.11.133. [DOI] [Google Scholar]
- van Andel E.; de Bus I.; Tijhaar E. J.; Smulders M. M. J.; Savelkoul H. F. J.; Zuilhof H. Highly Specific Binding on Antifouling Zwitterionic Polymer-Coated Microbeads as Measured by Flow Cytometry. ACS Appl. Mater. Interfaces 2017, 9, 38211–38221. 10.1021/acsami.7b09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A. T.; Baggerman J.; Paulusse J. M. J.; Zuilhof H.; van Rijn C. J. M. Bioconjugation of Protein-Repellent Zwitterionic Polymer Brushes Grafted from Silicon Nitride. Langmuir 2011, 28, 604–610. 10.1021/la2031363. [DOI] [PubMed] [Google Scholar]
- Slagman S.; Jonkers W. A.; Zuilhof H.; Franssen M. C. R. Elucidating the Mechanism behind the Laccase-Mediated Modification of Poly(Ethersulfone). RSC Adv. 2018, 8, 27101–27110. 10.1039/c8ra04402c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Q.; Mi L.; Han X.; Bai T.; Liu S.; Li Y.; Jiang S. Differences in Cationic and Anionic Charge Densities Dictate Zwitterionic Associations and Stimuli Responses. J. Phys. Chem. B 2014, 118, 6956–6962. 10.1021/jp503473u. [DOI] [PubMed] [Google Scholar]
- Cao B.; Li L.; Tang Q.; Cheng G. The Impact of Structure on Elasticity, Switchability, Stability and Functionality of an All-in-One Carboxybetaine Elastomer. Biomaterials 2013, 34, 7592–7600. 10.1016/j.biomaterials.2013.06.063. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]
- Weener J.-W.; van Dongen J. L. J.; Meijer E. W. Electrospray Mass Spectrometry Studies of Poly(Propylene Imine) Dendrimers: Probing Reactivity in the Gas Phase. J. Am. Chem. Soc. 1999, 121, 10346–10355. 10.1021/ja984432+. [DOI] [Google Scholar]
- Alder R. W.; Colclough D.; Mowlam R. W. Fragmentation During the Formic Acid/Formaldehyde (Eschweiler-Clarke) Methylation of Polyamines. Tetrahedron Lett. 1991, 32, 7755–7758. 10.1016/0040-4039(91)80583-r. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.