

Abstract
Signal peptide peptidase (SPP) and the four homologous SPP‐like (SPPL) proteases constitute a family of intramembrane aspartyl proteases with selectivity for type II‐oriented transmembrane segments. Here, we analyse the physiological function of the orphan protease SPPL2c, previously considered to represent a non‐expressed pseudogene. We demonstrate proteolytic activity of SPPL2c towards selected tail‐anchored proteins. Despite shared ER localisation, SPPL2c and SPP exhibit distinct, though partially overlapping substrate spectra and inhibitory profiles, and are organised in different high molecular weight complexes. Interestingly, SPPL2c is specifically expressed in murine and human testis where it is primarily localised in spermatids. In mice, SPPL2c deficiency leads to a partial loss of elongated spermatids and reduced motility of mature spermatozoa, but preserved fertility. However, matings of male and female SPPL2c −/− mice exhibit reduced litter sizes. Using proteomics we identify the sarco/endoplasmic reticulum Ca2+‐ATPase (SERCA2)‐regulating protein phospholamban (PLN) as a physiological SPPL2c substrate. Accumulation of PLN correlates with a decrease in intracellular Ca2+ levels in elongated spermatids that likely contribute to the compromised male germ cell differentiation and function of SPPL2c −/− mice.
Keywords: intramembrane proteolysis, phospholamban, signal peptide peptidase‐like proteases, spermatogenesis, tail‐anchored proteins
Subject Categories: Development & Differentiation; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Intramembrane proteases cleave substrate proteins within the hydrophobic environment of the phospholipid bilayer 1. Thereby, cleavage fragments are released into the cytoplasm, which can act as transcriptional regulators after nuclear translocation as exemplified by the paradigmatic SREBP and Notch pathways critically regulating cholesterol homeostasis and cellular differentiation, respectively 2. In addition, intramembrane proteolysis controls the homeostasis of membrane‐bound substrate proteins 3.
Signal peptide peptidase (SPP) and the SPP‐like proteases SPPL2a‐c and SPPL3 are aspartyl intramembrane proteases with homology to presenilin 4, 5, the catalytic subunit of the γ‐secretase complex. However, SPP/SPPL proteases show specificity for type II‐oriented substrate proteins, i.e. single‐pass transmembrane proteins with their N‐terminus intracellularly as opposed to extracellularly (type I) 6, 7. SPP was initially discovered based on its ability to cleave signal peptides in the endoplasmic reticulum (ER) 4. Since then, multiple functions of this protease have been unravelled 6 including the processing of viral proteins like the hepatitis C virus (HCV) core protein 8 and a role in ER‐associated protein degradation (ERAD) 9, 10, 11. Recently, selected tail‐anchored (TA) proteins like heme oxygenase 1 (HO‐1) were identified as proteolytic substrates of SPP 11, 12. With their type II‐oriented transmembrane domain and short luminal C‐terminus, TA proteins can be cleaved independent of any preceding ectodomain processing, which is necessary for SPP‐catalysed turnover of signal peptides 13, or recruitment of the ERAD factor Derlin‐1 as has been shown for the type II membrane protein XBP1u 9.
However, so far it has not been reported if TA proteins can also be substrates of any of the four SPPL proteases. In contrast to the ER‐resident SPP, SPPL2a, SPPL2b and SPPL3 reside in lysosomes/late endosomes, the plasma membrane and the Golgi apparatus, respectively. SPPL3 acts as a major regulator of protein glycosylation and glycosaminoglycan biosynthesis by shedding various glycosyltransferases 14, 15. Its ability to cleave substrates with large ectodomains is a unique property within the SPPL family and distinguishes it from SPPL2a and SPPL2b. In cell‐based overexpression set‐ups, substrate spectra of SPPL2a and SPPL2b overlap significantly suggesting similar catalytic properties of both proteases. Whereas SPPL2a is of critical importance for the development of B lymphocytes and dendritic cells 16, 17, 18, the in vivo function of SPPL2b is currently less clear 19 and the identification of physiological substrates of SPPL2b is still pending.
In contrast to the other SPPL2 family members, very little is known so far about SPPL2c. Based on its intronless gene structure, it was hypothesised to represent a non‐expressed pseudogene 20, 21. Upon heterologous expression of the SPPL2c open reading frame, the resulting protein was observed in the ER 21. However, endogenous expression of SPPL2c has not been demonstrated so far. SPPL2c exhibits the catalytic YD/FD and GxGD signature motifs, conserved in all intramembrane aspartyl proteases 4, 5. Nevertheless, proteolytic activity of SPPL2c has not been revealed yet. Conspicuously, the proposed ER localisation of SPPL2c suggests that its intracellular distribution overlaps with that of SPP. This leads to the question why two SPP/SPPL proteases in the same cellular compartment have evolved and to what extent their functions overlap.
Here, we have systematically analysed expression and function of the orphan intramembrane protease SPPL2c. We demonstrate that SPPL2c is an ER‐resident protein, which is specifically expressed in murine and human testis under endogenous conditions. There, it is present in developing germ cells with the highest abundance in spermatids. Consequently, differentiation and function of male germ cells are compromised in SPPL2c‐deficient mice. We demonstrate for the first time that SPPL2c exhibits proteolytic activity. Similar to SPP, SPPL2c cleaves selected TA proteins, however with a distinct, only partially overlapping substrate spectrum. Using proteomics, we have identified the SERCA regulating protein phospholamban (PLN) as physiological substrate of SPPL2c, presumably leading to a dysregulation of intracellular Ca2+ handling in SPPL2c −/− male germ cells.
Results
SPPL2c is a testis‐specific ER‐resident intramembrane protease
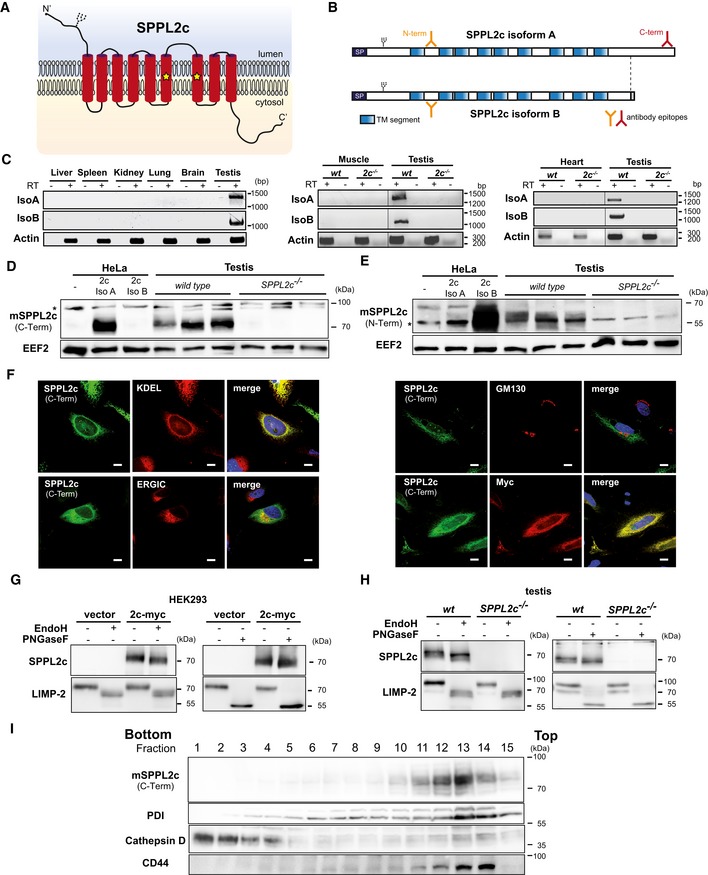
As predicted from the amino acid sequence, the murine SPPL2c protein exhibits a nine‐transmembrane domain topology (Fig 1A and B) with the active site consensus FD and GFGD motifs present in transmembrane segments 6 and 7 21. The coding sequence of the canonical SPPL2c isoform A protein is present as a contiguous block in the genomic locus (Fig EV1A). In addition, databases list a second SPPL2c transcript, isoform B, with a distinct 3′‐end, which codes for a protein with a shorter C‐terminus. We screened for endogenous expression of SPPL2c in a variety of murine tissues (Fig 1C) by RT–PCR using primer pairs specific for the two isoforms. We detected mRNA of both SPPL2c isoforms in testis, but not in heart, skeletal muscle, brain, spleen, kidney, liver and lung. Polyclonal antisera against two different epitopes (N‐Term, C‐Term) of the SPPL2c protein were generated as indicated in Fig 1B and were validated using HEK293 cells overexpressing the two isoforms (Fig EV1B). As expected from the position of the epitopes, the C‐terminal antibody specifically recognised the longer isoform A due to the lack of this epitope in the C‐terminally truncated isoform B, whereas the N‐terminal antiserum was capable of detecting both. With these antibodies, we detected the endogenous SPPL2c protein in testis lysates from wild‐type mice by Western blotting (Fig 1D and E) finally excluding SPPL2c being a non‐expressed pseudogene. To control for specificity, we analysed samples from SPPL2c −/− mice where the entire coding region had been deleted (Fig EV1A). Thus, we provide evidence for endogenous expression of both SPPL2c isoforms at protein level in murine testis. We also assessed several other tissues from wild‐type and SPPL2c −/− mice (Fig EV1C). In support of the transcriptional data (Fig 1C), we did not detect SPPL2c protein in any additional tissue under basal conditions. In agreement with previous reports about human SPPL2c 21, ectopically expressed murine SPPL2c was detected in the ER (Fig 1F), but not in the Golgi. In addition, partial co‐localisation with a marker protein of the ER‐Golgi intermediate compartment (ERGIC) could be observed.
Figure 1. SPPL2c is a testis‐specific ER‐resident intramembrane protease.
-
APredicted topology of murine SPPL2c. Positions of the predicted N‐glycosylation site and critical aspartate residues of the active centre (asterisks) are indicated.
-
BScheme of the two murine SPPL2c isoforms A and B. Epitopes (N‐term/C‐term) used for generation of antisera are marked. SP, predicted signal peptide.
-
CRT–PCR analysis of SPPL2c expression in different murine tissues. Total RNA from the indicated tissues was either transcribed into cDNA prior to PCR amplification (+RT) or, as negative control, used directly as template (−RT). As indicated in Fig EV1A, a common forward primer in combination with an isoform‐specific reverse primer was employed. A fragment of the actin ORF was amplified as control.
-
D, EWestern Blot analysis of total lysates from testis isolated from wild type or SPPL2c −/− using antibodies against C‐ (D) or N‐terminal (E) epitopes of SPPL2c. Lysates from HeLa cells transiently expressing the SPPL2c isoforms were analysed in parallel. Full Western blots are shown in Fig EV1B. *, non‐specific band.
-
FSPPL2c was visualised by indirect immunofluorescence in HeLa cells transiently expressing C‐terminally Myc‐tagged murine SPPL2c isoform A. For detection of SPPL2c, either the anti‐Myc or the SPPL2c antiserum (C‐terminal epitope) was employed as indicated together with anti‐KDEL, anti‐GM130 or anti‐ERGIC53 as indicated in order to label the ER, the cis‐Golgi apparatus or the ER‐Golgi intermediate compartment (ERGIC), respectively. Scale bars, 10 μm.
-
GLysates from HEK293 cells transiently transfected with murine SPPL2c‐myc (isoform A) were treated with the glycosidases PNGase F or Endo H as indicated prior to Western blot analysis with anti‐SPPL2c (C‐term). Deglycosylation of endogenously expressed LIMP2 was analysed as control.
-
HN‐glycosylation of endogenous SPPL2c is similar to the overexpressed protein. Total lysates from testis of wild‐type or SPPL2c −/− mice were treated and analysed as in (G).
-
ISubcellular fractionation of post‐nuclear supernatant from murine testis using a self‐forming Percoll density gradient. Fractions were collected from bottom to top and subjected to Western blot analysis for SPPL2c. Protein disulphide isomerase (PDI), cathepsin D and CD44 were detected as organelle marker proteins.
Source data are available online for this figure.
Figure EV1. Generation and validation of SPPL2c −/− mice and SPPL2c antisera.
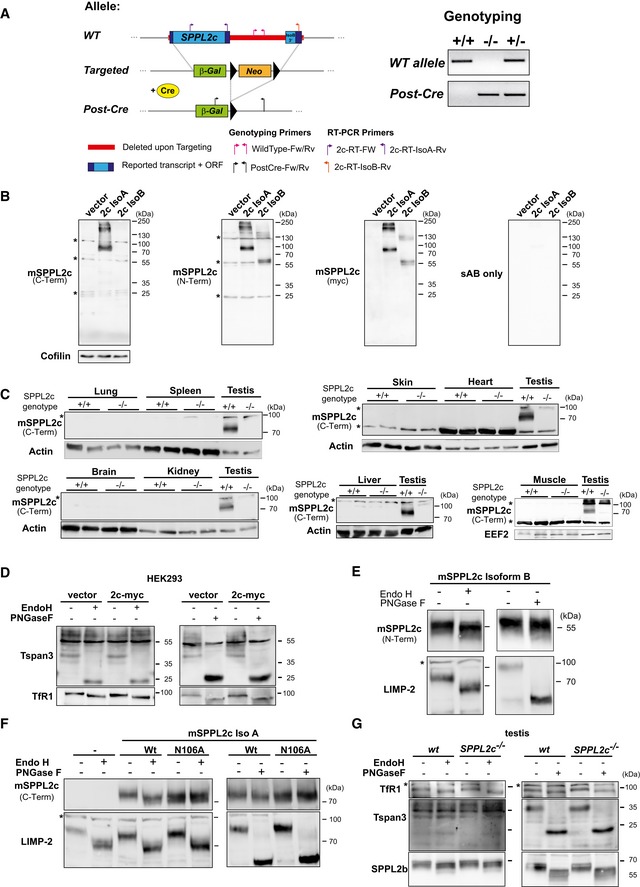
- Scheme of the murine SPPL2c genomic locus and the employed targeting strategy of the allele designed by Velocigene. Upon deletion of the entire SPPL2c coding region, a β‐galactosidase reporter gene (β‐Gal) and a floxed neomycin resistance cassette (Neo) were inserted. The latter was excised by breeding with Cre‐Deleter mice. Prior to analysis of SPPL2c −/− mice, the Cre transgene was removed again by breeding. Positions of primers used for genotyping or RT–PCR are indicated. For genotyping, two PCRs amplifying specific fragments from the wild type (WT) or the post‐Cre Knockout allele were performed as depicted from a representative set of mice.
- HEK293 cells were transiently transfected with SPPL2c isoform A or B fused to a C‐terminal Myc epitope or empty vector. Western blot detection was performed with the newly generated, affinity‐purified antisera against an N‐ or C‐terminal epitope of SPPL2c. In parallel, anti‐Myc was employed as a control and cofilin was detected to confirm equal protein loading. *non‐specific band.
- SPPL2c protein was not detected in the major murine tissues. Total tissue lysates from wild‐type (+/+) or SPPL2c −/− mice as indicated were analysed by Western blotting using the SPPL2c antiserum generated against the C‐terminus of the protein. Testis lysates were included as positive control, and Actin or EEF2 was visualised to confirm equal protein loading. *non‐specific band.
- Additional deglycosylation controls for Fig 1E. To control for proper deglycosylation of lysates of SPPL2c‐transfected HEK cells, shifts in the bands for Tetraspanin‐3 (Tspan3) and Transferrin receptor 1 (TfR1) were detected by Western blotting using specific antibodies.
- N‐glycosylation of SPPL2c isoform B is similar to that of isoform A. Lysates of HEK293 cells transiently expressing murine SPPL2c isoform B were treated with endoglycosidase H (Endo H) or peptidyl N‐glycosidase F (PNGase F) prior to Western blot analysis with the SPPL2c antiserum against an N‐terminal epitope, which also detects isoform B. As control for successful deglycosylation, we also visualised the lysosomal integral membrane protein LIMP‐2. *, non‐specific band.
- Murine SPPL2c is N‐glycosylated at N106. HEK293 cells were transiently transfected with wild‐type SPPL2c isoform A (Iso A) or a N106A mutant inactivating the putative N‐glycosylation consensus site. To reveal N‐glycosylation of the expressed proteins, lysates were treated with Endo H or PNGase F prior to Western blotting. SPPL2c was detected with the antiserum against the C‐terminal epitope validate in (B) and LIMP‐2 served as control for the deglycosylation. *, non‐specific band.
- Additional deglycosylation controls for Fig 1F. To control for proper deglycosylation of murine testis lysates, shifts in the bands for Tspan3, TfR1 and SPPL2b were detected by Western blotting using specific antibodies. *, non‐specific band.
Source data are available online for this figure.
Based on the presence of an N‐glycosylation consensus site within the N‐terminal luminal domain of the protein (Fig 1B), we confirmed glycosylation of overexpressed SPPL2c by treatment with N‐glycosidase F (PNGase F) and endoglycosidase H (Endo H) (Figs 1G and EV1D and E). The observed sensitivity of SPPL2c to EndoH treatment indicates that its glycans are not of the complex type in accordance with the detected ER localisation of the protein (Figs 1G and EV1D and E). Mutagenesis of N106 completely abolished N‐glycosylation of the SPPL2c protein and thereby confirmed glycosylation of this specific site (Fig EV1F). Importantly, we could show that also SPPL2c from murine testis was modified with EndoH‐sensitive N‐glycans providing a first indication that also endogenous SPPL2c resides in the ER (Figs 1H and EV1G). This was further supported by co‐sedimentation of SPPL2c with the ER marker protein disulphide isomerase (PDI) in a subcellular fractionation of murine testis (Fig 1I).
SPPL2c cleaves distinct tail‐anchored proteins with a substrate spectrum partially overlapping with that of SPP
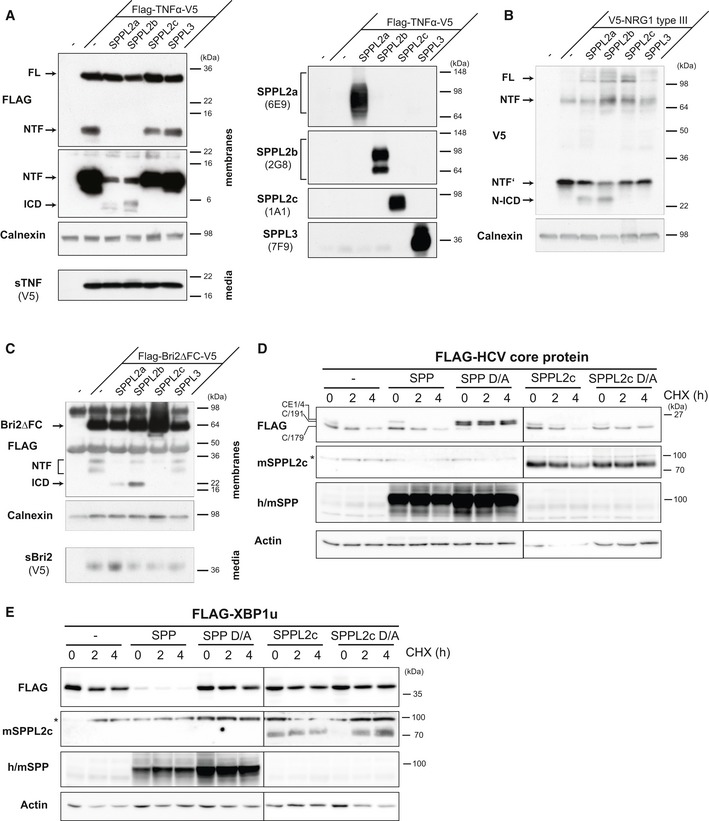
We aimed to unravel proteolytic activity of SPPL2c and therefore assessed a potential cleavage of known substrates of SPPL2a/b, which are the most closely related proteases within the SPP/SPPL family. Since we have so far not identified any cell line with endogenous SPPL2c expression, we used co‐expression assays to assess a potential processing of tumour necrosis factor (TNF‐α) (Fig EV2A) 22, 23, neuregulin 1 type III (Fig EV2B) 24 and Bri2 (Fig EV2C) 25 by SPPL2c. In contrast to SPPL2a and SPPL2b, no cleavage indicated by a major NTF depletion and/or release of any intracellular domain (ICD) of the proteins tested was detected in cells co‐expressing catalytically active SPPL2c. Since processing of these proteins takes place in the late secretory pathway or at the plasma membrane, where SPPL2c is not present, we also assessed known substrates of the ER‐resident homologue SPP. The HCV core protein was very efficiently processed by endogenous SPP 8, which was blocked by expression of catalytically inactive (D/A) SPP (Fig EV2D) that has been shown previously to act as a dominant negative construct 26. In contrast, expression of catalytically inactive SPPL2c (SPPL2c D/A) did not abolish processing of the HCV core protein (Fig EV2D), indicating that SPPL2c does not functionally interact with the HCV core protein. Similarly, also the SPP substrate XBP1u 9 was not cleaved by SPPL2c (Fig EV2E).
Figure EV2. SPPL2c does not cleave known substrates of SPPL2a, SPPL2b and SPP.
-
A–CThe SPPL2a/b substrates TNF‐α (A), neuregulin 1 (NRG1) type III (B) and Bri2 (C) are not processed by co‐expressed SPPL2c. T‐Rex™‐293 cells stably expressing catalytically active human SPPL2a, SPPL2b, SPPL2c or SPPL3 under a doxycycline‐inducible promoter were transiently transfected with the indicated epitope‐tagged substrates. Expression of the proteases upon doxycycline induction was confirmed by Western blotting using the indicated protein‐specific antibodies. Full‐length (FL) and processed forms (NTF, ICD) of substrates were detected by Western blotting of total membrane preparations. To detect Bri2ΔFC and N‐terminal fragments (NTFs), an immunoprecipitation with anti‐FLAG was performed prior to Western blot analysis. In addition, cell culture media of TNF‐α‐ and Bri2ΔFC‐expressing cells (A, C) were analysed to confirm proteolytic release of the ectodomain (sTNF‐α, sBri2), which is a prerequisite for the cleavage by SPPL2a/b. Western blot detection with anti‐FLAG (TNF‐α, Bri2) or anti‐V5 (NRG1 type III; sTNF‐α and sBri2) directed against the respective tag‐epitopes was conducted. By this means, full‐length forms of the substrates as well as the N‐terminal fragments (NTF) derived from processing of the ectodomain (sTNF‐α, sBri2) or the hairpin structure of NRG1 type III were revealed. Upon expression of SPPL2a or SPPL2b, an intracellular domain (ICD) was released from all three substrates, which was not observed upon SPPL2c expression.
-
D, EHEK293 cells were transiently transfected with N‐terminally FLAG‐tagged HCV core‐E1/4 protein (D) or XBP1u (E) either alone or in combination with active or inactive (D/A) murine SPP or SPPL2c. To compare the turnover and stability of the respective substrates in the absence or presence of the protease, cycloheximide (CHX) chase experiments were performed. Therefore, transfected cells were treated for 0, 2 or 4 h with 100 μg/ml CHX to block protein synthesis prior to cell harvest. The overexpressed murine SPPL2c was detected with anti‐SPPL2c (C‐terminal epitope). For detection of overexpressed murine (m) SPP, a polyclonal antiserum cross‐reacting with endogenous human SPP was employed. In (D), the different processed forms (C/191, C/179)) of the HCV core‐E1/4 protein (CE1/4) were labelled as described previously 13. In these experiments, we predominantly detected a dimeric form of SPP. *, non‐specific band.
Source data are available online for this figure.
In contrast, SPPL2c efficiently processed the TA protein HO‐1 that has recently been identified as endogenous SPP substrate 11. Like SPP, SPPL2c co‐expression efficiently reduced overall levels of murine HO‐1 (Fig 2A). In support, HO‐1 was also identified in a proteomic substrate screen in SPPL2c‐overexpressing HEK cells (accompanying manuscript by Papadopoulou et al 27). Upon co‐expression of human HO‐1 with the murine proteases, even a potential cleavage product with a slightly lower molecular weight was detected (Fig EV3A). Without any relevant luminal domain, such a minor shift in size would be consistent with an intramembrane cleavage releasing HO‐1 from the ER membrane. In agreement, we detected HO‐1 in the cytosol and in part also in the nucleus of HeLa cells overexpressing both HO‐1 and active, but not inactive SPPL2c (Fig 2B). Having identified HO‐1 as SPPL2c substrate, we assessed additional TA proteins as performed previously for SPP 11. The ubiquitin‐conjugating enzyme E2 J1 (Ube2J1) was processed by neither SPP nor SPPL2c indicating that cleavage of TA proteins is selective and that the TA topology on its own is not sufficient for cleavage (Fig 2C). Cytochrome b5 (Cyb5a) and the stress‐associated ER‐associated proteins 1 + 2 RAMP4 and RAMP4‐2 were all proteolysed by SPP (Fig 2D) 11. All three substrate candidates were localised to the ER overlapping with the distribution of co‐expressed SPPL2c (Fig EV3B). However, among these three potential substrates, we found only RAMP4‐2 to be cleaved efficiently by SPPL2c under these conditions leading to its near‐complete depletion whereas the other two proteins exhibited no or just a very minor reduction (Fig 2D). We also evaluated cleavage of all tested TA proteins by the shorter SPPL2c isoform B (Fig EV3C). With regard to these proteins, we did not obtain any evidence for a relevant difference in cleavage specificity between the two SPPL2c isoforms.
Figure 2. SPPL2c cleaves selected tail‐anchored (TA) proteins with a substrate spectrum distinct from SPP.
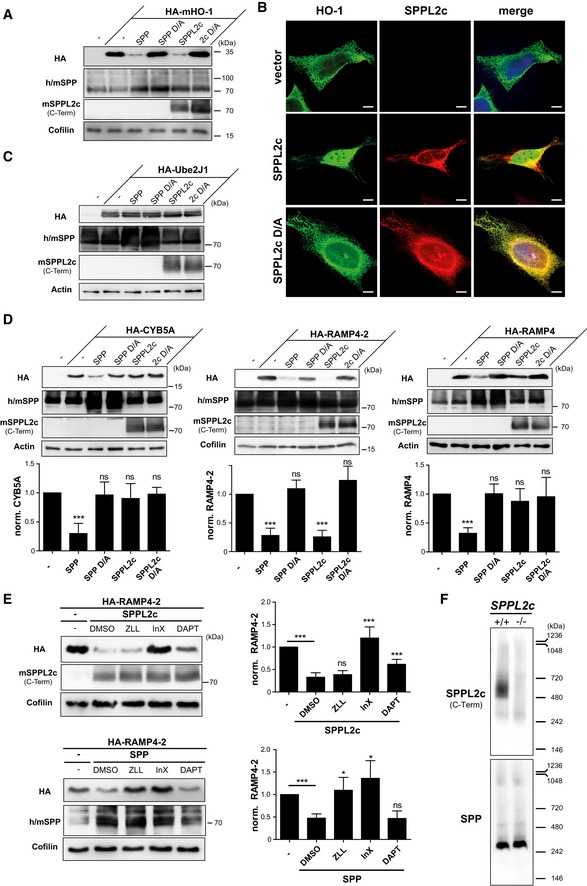
- HEK293 cells were transiently transfected with murine N‐terminally HA‐tagged heme oxygenase 1 (HA‐mHO‐1) alone or in combination with active or inactive (D/A) murine SPP or SPPL2c. Substrate levels were determined by Western blot analysis with anti‐HA. For detection of SPP, an antiserum recognising both endogenous human (h) and overexpressed murine (m) SPP was employed. Murine SPPL2c was visualised using the antiserum generated against the C‐terminus of the protein. Actin was detected to control for equal protein loading.
- Immunofluorescence analysis of HeLa cells transiently expressing murine HO‐1 (HA‐mHO‐1) alone or together with active or inactive (D/A) SPPL2c‐myc. Substrate and proteases were visualised using the HA and Myc epitopes, respectively. Scale bars, 10 μm.
- The TA protein Ube2J1 is not proteolysed by co‐expressed SPP or SPPL2c in HEK293 cells. Western blot analysis was performed as in (A).
- Differential cleavage of CYB5a, RAMP4‐2 and RAMP4 by SPPL2c and SPP. Substrates and proteases were co‐expressed in HEK293 cells followed by Western blot analysis as described above. Densitometric quantification from at least n = 3 independent experiments was performed. Substrate levels were normalised and compared to cells without protease overexpression. Mean ± SD; unpaired Student's t‐test; ***P ≤ 0.001; ns, non‐significant.
- HEK293 cells expressing HA‐RAMP4‐2 alone or in combination with SPPL2c or SPP were treated with 100 μM (Z‐LL)2‐ketone (ZLL), 5 μM inhibitor X (InX), 5 μM DAPT or DMSO as control. RAMP4‐2 band intensity was quantified from blots of three independent experiments and normalised to cells just overexpressing the substrate. Mean ± SD, unpaired Student's t‐test; ***P ≤ 0.001, *P ≤ 0.05; ns, non‐significant.
- Microsomes isolated from testis of wild‐type and SPPL2c −/− mice were solubilised in 1% digitonin, and proteins were separated by blue‐native PAGE. After transfer to PVDF membrane, SPPL2c and SPP were detected using the polyclonal antisera introduced above.
Source data are available online for this figure.
Figure EV3. SPPL2c cleaves selected tail‐anchored (TA) proteins.
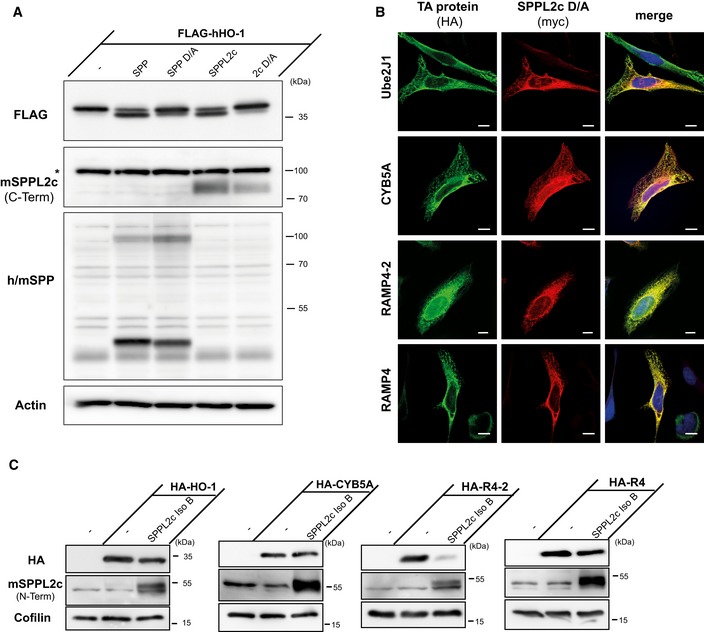
- Ectopically expressed murine SPPL2c also cleaves human heme oxygenase 1 (HO‐1). HEK293 cells were transiently transfected with human N‐terminally FLAG‐tagged HO‐1 (FLAG‐hHO‐1) alone or in combination with active or inactive (D/A) murine SPP or SPPL2c. Western blot analysis was performed with anti‐FLAG. SPP was detected with a polyclonal serum detecting the overexpressed murine and the endogenous human (h/m) SPP. SPPL2c was visualised with the antiserum against the C‐terminus of the protein. To control for equal protein loading, actin was visualised. *, non‐specific band.
- All TA proteins analysed in Fig 2 co‐localise with SPPL2c. HeLa cells were transiently transfected with N‐terminally HA‐tagged Ube2J1, CYB5A, RAMP4‐2 or RAMP4 together with inactive (D/A) murine SPPL2c fused to a Myc epitope at its C‐terminus. Substrates and the inactive proteases were visualised with anti‐HA and anti‐Myc, respectively, in conjunction with fluorophore‐conjugated secondary antibodies. Scale bars, 10 μm.
- Substrate specificity of SPPL2c Isoform B is similar to isoform A. The indicated proteins were transiently expressed in HEK293 cells either alone or together with murine SPPL2c isoform B (IsoB). The expressed substrate candidate proteins were detected with anti‐HA. Expression of SPPL2c was confirmed with anti‐SPPL2c directed against an N‐terminal epitope. Cofilin was used to confirm equal protein loading.
Source data are available online for this figure.
Having demonstrated proteolytic activity of SPPL2c, we aimed to determine its susceptibility to known inhibitors of SPP/SPPL and/or other GxGD proteases which we applied to HEK293 cells co‐expressing RAMP4‐2 as a model substrate and SPPL2c or SPP as control (Fig 2E). Surprisingly, the SPP inhibitor (Z‐LL)2‐ketone that also blocks SPPL2a/b but shows no effect on SPPL3 and γ‐secretase 4, 25, 28 did not inhibit the SPPL2c‐mediated cleavage of RAMP4‐2, though it significantly blocked cleavage by SPP. In contrast, the γ‐secretase inhibitor X reduced the activity of SPPL2c efficiently and in a very comparable way to that of SPP. DAPT, a non‐active site inhibitor of γ‐secretase, has so far not been found to target any SPP/SPPL protease 25, 28, 29 and did also not inhibit SPP‐triggered degradation of RAMP4‐2 (Fig 2E). However, application of DAPT partially blocked cleavage by SPPL2c (Fig 2E). Thus, in comparison with SPP and the other SPPL2 proteases, SPPL2c exhibits a very unique inhibitory profile.
SPP has been described to be part of high molecular weight complexes in the ER 9, 30. Upon blue‐native PAGE separation of murine testis lysates, we predominantly observed a digitonin‐resistant SPP complex with an apparent molecular weight of ~250 kDa (Fig 2F). Very minor amounts of SPP were present in assemblies of ~500 and ~900 kDa, respectively. In contrast, the bulk of SPPL2c was part of a digitonin‐resistant complex with a size of ~500 kDa. This indicates that SPPL2c like SPP has the intrinsic ability to form high molecular weight complexes in the membrane of the ER. However, the complex organisation we observed for both proteases in murine testis was distinct. Though it remains possible that antibody epitopes in these native complexes have been masked preventing protease detection, our findings strongly suggest that SPP and SPPL2c are not part of the same high molecular weight assemblies. Altogether, we conclude that the biochemical and functional properties of both proteases differ despite the overlapping subcellular localisation and the shared ability to cleave selected TA proteins. Based on this, unique in vivo functions of SPPL2c may be postulated, which do not overlap with that of SPP or any other of the SPPL proteases.
SPPL2c has a critical function in spermatids
In support of a specific physiological function of SPPL2c, there was no compensatory upregulation of SPP, SPPL2a or SPPL2b at the transcriptional level in testis of SPPL2c‐deficient mice (Fig EV4A). For SPP, we confirmed this at the protein level (Fig EV4B). To define the physiological function of SPPL2c in testis, we aimed to determine in which cell type SPPL2c is expressed in this organ. Therefore, we utilised an E. coli β‐galactosidase reporter, which replaced the SPPL2c coding sequence in the SPPL2c knockout allele and is thus under control of the endogenous SPPL2c promotor (Figs 3A and EV1A and EV4C). This approach revealed expression of SPPL2c within the seminiferous tubules, where spermatogenesis takes place. To refine this and to determine in which stage(s) of differentiating germ cells SPPL2c is present, we FACS‐sorted testis suspensions based on their DNA content using Hoechst 33342 staining (Fig EV4D) thereby discriminating 1C (spermatids, spermatozoa), 2C (spermatogonia, secondary spermatocytes, Sertoli cells, other somatic cells) and 4C (primary spermatocytes, G2 spermatogonia) populations. By this means, in particular different meiotic stages of germ cells can be separated. Western Blotting revealed highest expression of SPPL2c in the haploid (1C) cell population (Fig 3B). However, also in the 2C and 4C populations, SPPL2c was detected indicating that SPPL2c expression starts early in developing germ cells before reaching a maximum in spermatids. Furthermore, we visualised endogenous SPPL2c with our antiserum in testis sections from wild‐type mice (Fig 3C). This confirmed the presence of SPPL2c in spermatids with most intense labelling being observed in cells directly surrounding the lumen of the tubules and thus representing elongated spermatids.
Figure EV4. Analysis of SPPL2c −/− testis.
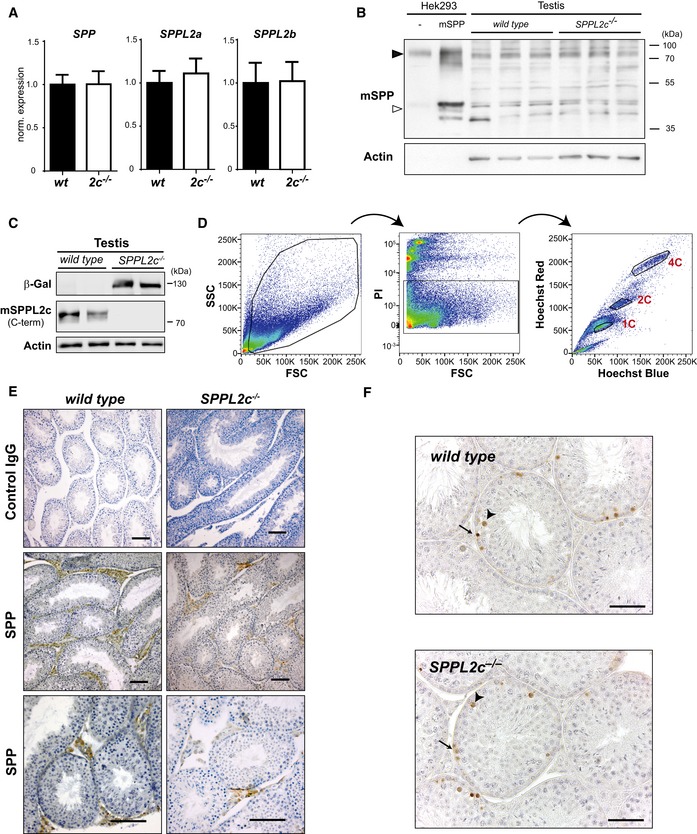
- No change in SPP, SPPL2a and SPPL2b expression in testis of SPPL2c‐deficient mice. Total RNA was isolated from testis of wild‐type and SPPL2c −/− mice (n = 3). In the resulting cDNA, SPP, SPPL2a and SPPL2b transcript abundance was quantified by qRT–PCR and normalised to that of Tuba1a. Bars depict mean values normalised to those of wild‐type samples ± SD.
- Western Blot analysis of SPP protein in total lysates from wild‐type and SPPL2c −/− testis. To facilitate identification of murine endogenous SPP, HEK293 cells transiently expressing murine SPP carrying a C‐terminal Myc epitope or just transfected with empty vector (−) were analysed. Due to the strong expression in these cells, only 1/8 of the protein amount was loaded from these samples as compared to the testis lysates. In addition to the SPP monomer (open arrowhead), we predominantly detected dimeric SPP (closed arrowhead), which exhibits a high stability under these experimental conditions. Actin was detected as control for protein loading.
- β‐galactosidase reporter expression in SPPL2c −/− testis. Total lysates from wild‐type and SPPL2c −/− testis were analysed by Western blotting for E. coli β‐galactosidase, SPPL2c and Actin.
- Gating strategy for sorting of individual germ cell populations based on their DNA content as determined by Hoechst 33342 staining. Cells were first roughly gated based on their forward (FSC) and sideward scatter (SSC) prior to exclusion of PI‐positive dead cells. Finally, 1C (haploid cells including spermatids), 2C (spermatogonia, secondary spermatocytes, Sertoli cells, other somatic cells) and 4C cells (primary spermatocytes, G2 spermatogonia) were gated based on their individual Hoechst staining as depicted.
- Immunohistochemical visualisation of SPP in paraffin sections from Bouin‐fixed wild‐type and SPPL2c −/− testis. Prior to immunostaining, sections were subjected to epitope retrieval in citrate buffer. A polyclonal SPP antiserum generated against an internal epitope of murine SPP was used as primary antibody. As a control for antibody specificity, stainings with normal rabbit polyclonal IgG were performed in parallel. Scale bars, 100 μm.
- The reduction of SPPL2c‐deficient spermatids is not caused by apoptosis. TUNEL staining was performed on paraffin sections from wild‐type and SPPL2c −/− testis (n = 3 per genotype), as shown here from a representative example. Labelled apoptotic cells were observed among the spermatogonia (arrow) and primary spermatocytes (arrowhead), however at a similar frequency in both genotypes. No relevant labelling was seen in spermatids neither in wild‐type nor in SPPL2c −/− sections. Scale bars, 100 μm.
Source data are available online for this figure.
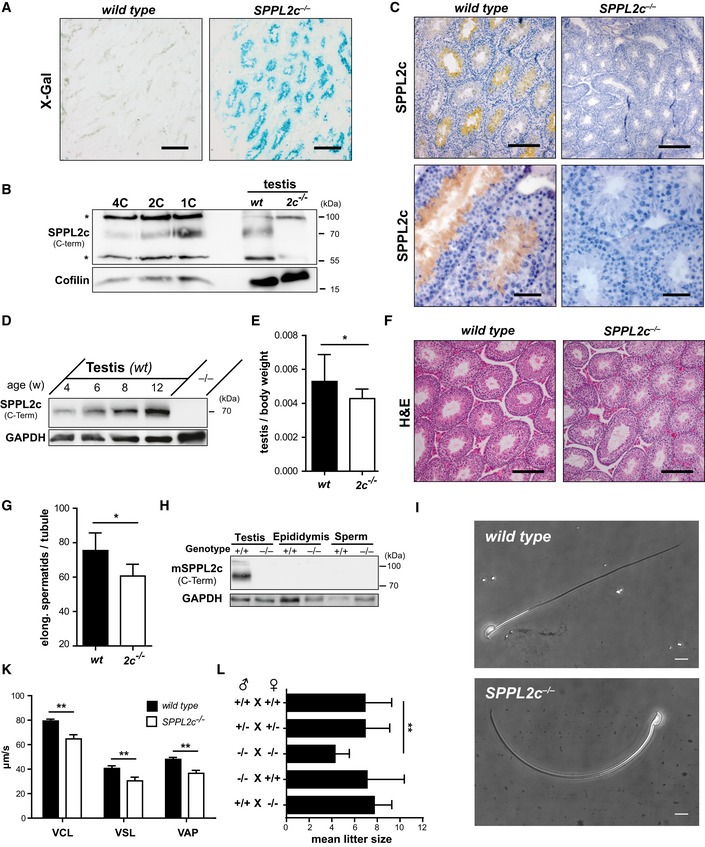
Figure 3. SPPL2c has a critical function in elongated spermatids.
-
AHistochemical visualisation of β‐galactosidase (β‐gal) reporter activity revealed SPPL2c expression within seminiferous tubules. Cryosections from PFA‐fixed wild‐type and SPPL2c −/− testis were stained with X‐Gal as β‐gal substrate. Scale bar, 500 μm.
-
BSPPL2c protein levels were analysed in germ cell populations from testis of wild‐type mice sorted for their DNA content which was determined by Hoechst 33342 staining as depicted in Fig EV4D. While for 4C, 2C and 1C fractions, 10 μg of protein was loaded, for the testis control samples (= total lysates), 20 μg of protein was subjected to electrophoresis. *non‐specific band.
-
CImmunohistochemical detection of SPPL2c was performed on cryosections of Bouin‐fixed wild‐type and SPPL2c −/− testis. The SPPL2c antiserum directed against a C‐terminal epitope of the protease and diaminobenzidine as peroxidase substrate was employed. Nuclei were stained with haematoxylin. Scale bar, 100 μm (upper panel) or 50 μm (lower panel).
-
DSPPL2c levels were compared in total lysates prepared from testis of wild‐type mice sacrificed at the age of 4, 6, 8 or 12 weeks by Western blotting. A testis lysate from an adult SPPL2c −/− mouse was used as negative control.
-
EThe weight of both testicles of wild‐type and SPPL2c −/− mice (n = 12 per genotype) was determined and normalised to the body weight. Mean ± SD; unpaired Student's t‐test; *P ≤ 0.05.
-
FParaffin sections from Bouin‐fixed wild‐type and SPPL2c −/− testis were stained with haematoxylin and eosin (H&E). Scale bars, 100 μm.
-
GIn H&E stained paraffin sections from Bouin‐fixed wild‐type and SPPL2c −/− testis, the mean number of elongated spermatids per seminiferous tubule was determined. Cells were counted in 20 tubuli in sections from n = 6 mice per genotype. Mean ± SD; unpaired Student's t‐test; *P ≤ 0.05.
-
HSPPL2c is not present in mature spermatozoa. Western blot analysis of total lysates of wild‐type (+/+) and SPPL2c −/− testis, epididymis and spermatozoa recovered from the cauda epididymidis/vas deferens for SPPL2c.
-
IMature sperm were isolated from the epididymis of wild‐type or SPPL2c‐deficient mice and analysed by phase‐contrast microscopy. Representative pictures of morphologically normal spermatozoa are depicted. Scale bars, 10 μm.
-
KReduced motility of SPPL2c −/− sperm cells. Spermatozoa were recovered from the cauda epididymidis of wild‐type and SPPL2c −/− mice (n = 3) and subjected to computer‐assisted sperm analysis. Average path velocity (VAP), straight line velocity (VSL) and curvilinear velocity (VCL) were determined in micrometre per second. Mean ± SD; unpaired Student's t‐test; **P ≤ 0.01.
-
LMean litter sizes for different breeding schemes. The data depicted are based on n = 12 (+/+ × +/+), n = 57 (+/− × +/−), n = 11 (−/− × −/−), n = 10 (−/− × +/+) and n = 11 (+/+ × −/−) different breedings, respectively. Mean ± SD; one‐way ANOVA with Newman–Keuls post hoc testing; **P ≤ 0.01.
Source data are available online for this figure.
In agreement with the observed expression in germ cells, we observed that protein levels of SPPL2c increased during the onset of sexual maturity (Fig 3D). We also analysed the distribution of SPP in murine testis by immunohistochemistry (Fig EV4E). The vast majority of labelling was associated with interstitial cells between the seminiferous tubules, presumably including Leydig cells, and no significant expression was detected within the germ cell compartment. Though limitations with regard to sensitivity or specificity of the available antibodies may not be excluded, this suggests that the distribution of SPPL2c and SPP within murine testis differs significantly. Importantly, the observed SPP expression pattern was comparable in testis from wild‐type and SPPL2c −/− mice.
Based on the described SPPL2c distribution, we analysed if SPPL2c deficiency affects spermatogenesis. As a first indication, we observed that the normalised testis weight of SPPL2c −/− mice was significantly reduced compared to controls (Fig 3E). However, upon histological examination of SPPL2c −/− testis, no gross abnormalities of germ cell differentiation were detected (Fig 3F). With regard to the restricted expression pattern of SPPL2c, we specifically quantified those cells where this protease exhibits its highest abundance in wild‐type testis. When comparing the number of elongated spermatids per seminiferous tubule between wild‐type and SPPL2c‐deficient mice, we observed a significant reduction in the latter (Fig 3G). Using TUNEL stainings, we could exclude increased apoptosis within this cell type as underlying mechanism (Fig EV4F). Based on the lower number of elongated spermatids, it seems likely that the overall production of mature spermatozoa is reduced in SPPL2c −/− mice; however, this parameter cannot be reliably quantified in mice.
We assessed whether the SPPL2c protein persists in spermatozoa after their release from the testis. Therefore, we performed Western blot analysis of epididymis as well as of mature sperm recovered from the cauda epididymidis (Fig 3H). We failed to detect the SPPL2c protein in these samples indicating that SPPL2c is lost during further sperm maturation. Therefore, we conclude that the latest step of expression and therefore also proteolytic function of SPPL2c in spermatogenesis is presented by the stage of elongated spermatids. Nevertheless, we assessed sperm morphology and motility experimentally. No obvious morphological differences between wild‐type and SPPL2c −/− sperm as well as in the abundance of dysmorphic sperm, which were observed in both genotypes to a comparable degree, could be observed by phase‐contrast microscopy (Fig 3I). Representative pictures of morphologically normal spermatozoa are depicted in Fig 3I. However, analysis of epididymal sperm motility by computer‐assisted sperm analysis (CASA) revealed that sperm from SPPL2c‐deficient mice moved significantly slower (Fig 3K). Thus, the loss of SPPL2c in sperm progenitor cells is associated with a detectable functional impairment of mature sperm cells where SPPL2c is not present also under wild‐type conditions.
To define the pathophysiological consequences, we analysed litter sizes from different mating schemes. The mean litter size of homozygous breedings of SPPL2c −/− mice (−/− × −/−) was significantly reduced versus that of heterozygous (+/− × +/−) or wild‐type mice (+/+ × +/+) (Fig 3L), indicating a physiologically relevant subfertility of SPPL2c Ko × Ko matings. However, when mating male SPPL2c −/− mice with wild‐type females, the resulting litter sizes did not differ significantly from that of wild type. The same was observed when crossing SPPL2c‐deficient females with wild‐type males. This may indicate that in addition to the described impact of SPPL2c deficiency on spermatogenesis and sperm function, which on its own does not impair fertility, other not yet discovered mechanisms, possibly in the female reproductive tract, may contribute to the subfertility of the −/− × −/− matings.
SPPL2c regulates Ca2+ homeostasis by cleaving phospholamban
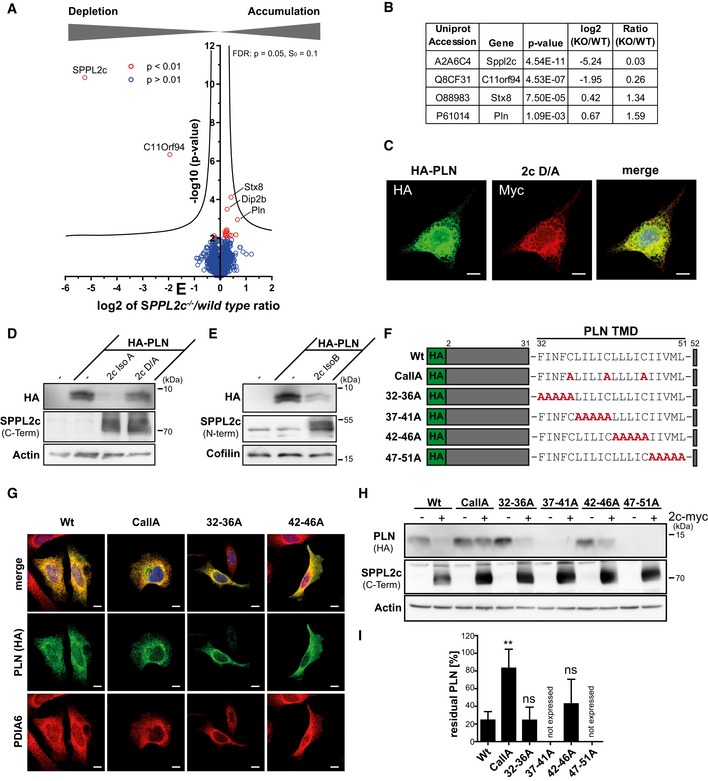
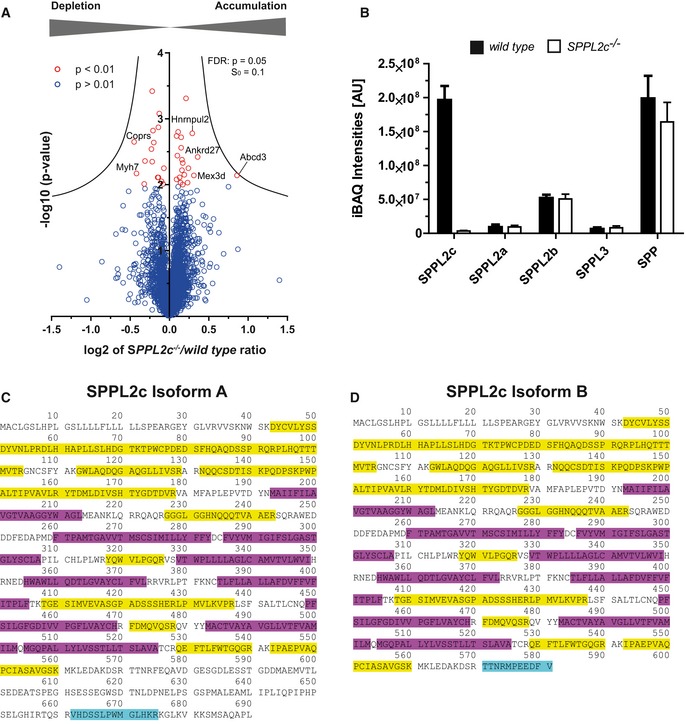
Having confirmed proteolytic activity of SPPL2c in cell‐based systems, we aimed to identify physiological substrates of this protease in testis, which would help to explain the described phenotype in the knockout mice. Therefore, we performed an unbiased label‐free quantitative proteomic analysis of carbonate‐washed total membrane and cytosolic fractions isolated from wild‐type and SPPL2c −/− testis (Figs 4A and EV5A). On average, 4,915 and 4,400 proteins were identified by at least two peptides in the membrane and cytoplasmic fraction, respectively (Dataset EV1). Finally, 4,191 (membrane fraction) and 3,741 proteins (cytosolic fraction) could be relatively quantified. No major changes were detected in the cytosolic fraction (Fig EV5A). In the membrane fraction, two proteins, SPPL2c and C11orf94, were found to be significantly reduced in samples from SPPL2c −/− mice (Fig 4A and B). This further validates SPPL2c deficiency of our knockout mice. The abundance ratio of 3% compared to the wild‐type levels in SPPL2c −/− samples might be explained by little carry‐over of peptides between the measurements. C11orf94 is an uncharacterised protein which appeared to be about fourfold reduced in the SPPL2c −/− testis. Why this is the case will require further investigations.
Figure 4. Identification of phospholamban (PLN) as novel substrate of SPPL2c.
-
ACarbonate‐washed total membrane fractions from wild‐type and SPPL2c −/− testis (n = 5) were subjected to a label‐free quantitative proteome analysis. For each quantified protein, a log2 intensity ratio between SPPL2c‐deficient and wild‐type samples was calculated. Negative values indicate a depletion and positive values an enrichment in SPPL2c −/− samples. In the depicted volcano plot, the negative log10 of the P value (y axis) is plotted versus the log2 intensity ratio of SPPL2c −/− versus wild‐type samples (x axis). All proteins above the significance level of P < 0.01 (unpaired Student's t‐test) are coloured in red. The hyperbolic curve indicates the threshold for a permutation‐based FDR correction for multiple hypotheses with P = 0.05 and s0 = 0.1.
-
BQuantification of SPPL2c, C11orf94, Syntaxin 8 (Stx8) and Phospholamban (PLN) in the proteomic dataset. A two‐sided Student's t‐test was performed.
-
CHeLa cells were transiently transfected with N‐terminally HA‐tagged PLN (HA‐PLN) and inactive (D/A) SPPL2c‐myc. The expressed proteins were visualised by indirect immunofluorescence via their HA and Myc epitopes. Scale bars, 10 μm.
-
D, EHEK293 cells were transiently transfected with HA‐PLN alone or in combination with active or inactive (D/A) murine SPPL2c isoform A (D) or isoform B (E). Substrate processing was analysed by Western blotting with anti‐HA. SPPL2c expression was confirmed with the antiserum against the C‐terminal epitope. Actin was detected to confirm equal protein loading.
-
FScheme of mutants generated for analysis of determinants within the PLN transmembrane domain (TMD) required for SPPL2c‐mediated intramembrane proteolysis.
-
GSubcellular localisation of the PLN mutants was analysed by indirect immunofluorescence using anti‐HA in combination with anti‐protein disulphide isomerase A6 (PDIA6) to stain the ER. The 37–41A and 47–51 PLN mutants were not expressed in relevant amounts. Scale bars, 10 μm.
-
HHEK293 cells were transiently transfected with wild type or mutated HA‐PLN alone or together with SPPL2c isoform A.
-
IQuantification of n = 5 experiments as shown in (H). For each PLN variant, the residual PLN in SPPL2c co‐expressing cells is depicted as % of PLN in the absence of the protease. Mean ± SD; one‐way ANOVA with Dunnett's post hoc test. **P ≤ 0.01, ns = not significant.
Source data are available online for this figure.
Figure EV5. Identification of endogenous SPPL2c substrates by proteomics.
-
AA label‐free quantitative proteome analysis of cytosolic fractions from wild‐type and SPPL2c −/− testis (n = 5) was performed. For each quantified protein, a log2 intensity ratio between wild‐type and SPPL2c‐deficient samples was calculated. In the depicted volcano plot for each protein, the negative log10 of the P‐value (y‐axis) is plotted versus the log2 ratio of intensity values of SPPL2c −/− versus wild‐type samples (x‐axis). All proteins above the significance level of P < 0.01 (Student's t‐test) are coloured in red. The hyperbolic curve indicates the threshold for a permutation‐based FDR correction for multiple hypotheses with P = 0.05 and s0 = 0.1.
-
BComparison of the abundance of different SPPL proteases and SPP in membrane preparations from wild‐type and SPPL2c −/− testis. Protein levels were estimated on the basis of the MS data using intensity‐based absolute quantification (iBAQ). Mean ± SD, n = 5 per genotype.
-
C, DMapping of peptides identified by mass spectrometry in testis membrane preparations from wild‐type mice to isoform 1 (C, UniProt A2A6C4‐1) and isoform 2 (D, UniProt A2A6C4‐2) of murine SPPL2c. Identified peptide sequences are indicated in yellow. Isoform‐specific peptides for the respective isoforms of SPPL2c are marked in turquoise. Transmembrane domains as annotated in UniProt are indicated in purple.
By applying intensity‐based absolute quantification (iBAQ) 31, the proteomic data set allowed an estimation of the protein levels of different SPP/SPPL proteases in murine testis (Fig EV5B). Despite the restricted expression pattern in this organ, the determined SPPL2c abundance was in the same range as that of SPP, but higher than that of the other SPPL family members. This indicates that SPPL2c is strongly expressed in murine testis. Based on the detection of isoform‐specific peptides (Fig EV5C and D), we could confirm the presence of both isoform A and isoform B of SPPL2c as revealed already by Western blotting (Fig 1D and E). For potential substrates, we expected an accumulation in the protease‐deficient samples. The three proteins phospholamban (PLN), syntaxin 8 (STX8) and Disco‐interacting protein 2 homologue B (Dip2b) were significantly enriched in SPPL2c −/− testis with enrichment factors of 1.59, 1.34 and 1.21, respectively (Fig 4B, Dataset EV1). Since the sequence of Dip2b does not contain any predicted transmembrane segments and presumably represents a soluble protein, we excluded it as a SPPL2c substrate and assume that its accumulation is a secondary effect of SPPL2c depletion. In contrast, STX8 and PLN are TA proteins and thus fulfil the basic requirements of representing candidate substrates. STX8 was also identified in an overexpression‐based SPPL2c substrate screen and could be further validated as SPPL2c substrate 27.
PLN is a small protein of 52 amino acids with a transmembrane segment between residue 32 and the C‐terminus in agreement with a TA topology 32. PLN interacts with and modulates the activity of the SERCA2 Ca2+‐ATPase and is therefore primarily known as a regulator of cardiac Ca2+ handling, contractility and function 32. To assess a potential cleavage of PLN, we analysed co‐localisation with SPPL2c as prerequisite for processing by indirect immunofluorescence in transiently transfected HeLa cells. Under these conditions, both PLN and SPPL2c localised in the ER (Fig 4C). Next, we expressed HA‐PLN in HEK293 cells alone or together with active or inactive (D/A) SPPL2c (Fig 4D). Active SPPL2c significantly depleted PLN as described above for RAMP4‐2 and HO‐1, which was not observed upon expression of SPPL2c D/A (Fig 4D). The same effect was obtained with SPPL2c isoform B (Fig 4E).
To gain further insights into SPPL2c‐mediated intramembrane proteolysis of PLN, we analysed which part of the PLN transmembrane segment would be critical for this process. Since it has been demonstrated that helix‐destabilising residues promote cleavage by SPPL2 proteases 33, 34, we aimed to modulate the stability of the PLN transmembrane segment by introducing blocks of five amino acids (32–36A, 37–41A, 42–46A and 47–51A). Additionally, we generated an alanine mutant of three cysteine residues evenly distributed within this part of PLN (CallA, Fig 4F). Comparable to the wild‐type protein, all of these mutants except of the non‐expressed 37–41A and 47–51A versions showed a clear co‐localisation with the ER marker PDIA6 excluding an interference of the introduced mutations with the subcellular sorting of the PLN variants (Fig 4G). Based on these observations, we conducted cleavage assays by overexpressing the different PLN mutants or the wild‐type protein in the presence or absence of co‐expressed SPPL2c in HEK293 cells. While the 32–36A and 42–46A mutants presented no major differences in their ability to be processed by SPPL2c, simultaneous alanine‐substitution of the three conspicuous cysteine residues within the PLN TMD reduced cleavage efficiency to about 20% of the wild‐type protein thereby clearly arguing for a substantial role of these residues for SPPL2c‐mediated PLN intramembrane proteolysis (Fig 4H and I).
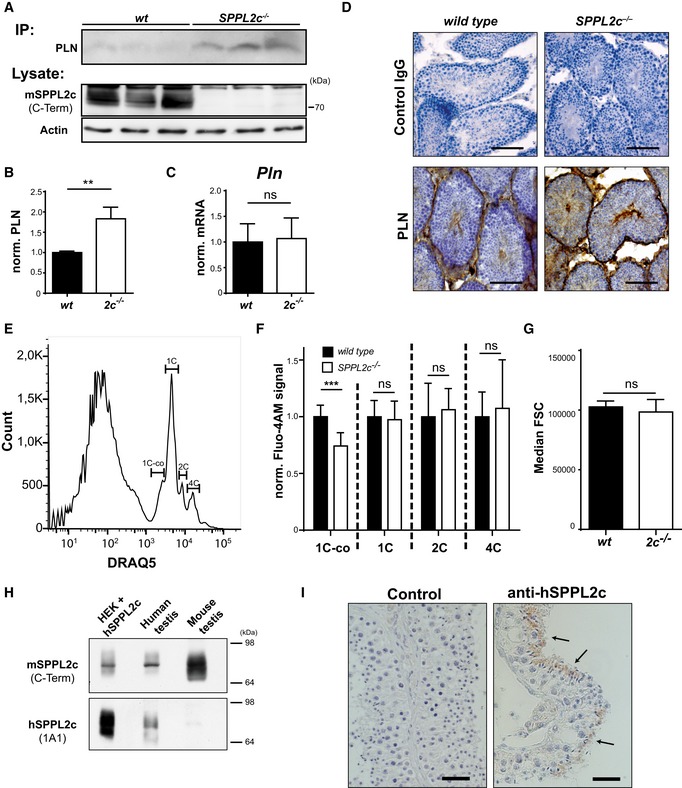
We sought to provide further evidence that PLN cleavage by SPPL2c takes place under endogenous conditions in vivo. Therefore, we confirmed PLN accumulation in SPPL2c −/− testis, which had been observed in the proteomic data set, by immunoprecipitation in combination with Western blotting (Fig 5A and B). In addition, we excluded differences in PLN mRNA expression in testis of both genotypes by qRT–PCR (Fig 5C). In order to represent a physiological substrate of SPPL2c, expression of PLN in germ cells and in particular elongated spermatids would be a prerequisite. When analysing the distribution of PLN by immunohistochemistry in wild‐type and SPPL2c −/− testis (Fig 5D), we observed prominent labelling of spermatids in addition to some minor signals in germ cells localised towards the periphery of the seminiferous tubules. Furthermore, considerable PLN expression was detected in peritubular and interstitial cells. The additional presence of PLN in non SPPL2c‐expressing cell types presumably is the reason why the overall PLN enrichment in total membrane preparations (Fig 4A and B) or lysates (Fig 5A and B) from SPPL2c‐deficient testis was limited to 1.5‐fold. However, the overlapping distributions in spermatids strongly suggest that PLN represents a physiological substrate of SPPL2c in this specific cell type.
Figure 5. SPPL2c regulates Ca2+ homeostasis in spermatids by cleaving phospholamban.
-
AAccumulation of endogenous PLN in the testis of SPPL2c −/− mice. PLN was immunoprecipitated (IP) from equal protein amounts of total lysates of wild‐type or SPPL2c −/− testis with the mouse monoclonal antibody. Western blot analysis of the IPs was performed with the rabbit monoclonal PLN antibody. SPPL2c and actin were detected from total lysates directly.
-
BQuantification of normalised PLN/Actin ratios from n = 3 biological replicates per genotype. Mean ± SD, unpaired Student's t‐test; **P ≤ 0.01.
-
CExpression of PLN in murine testis from wild‐type (wt) SPPL2c −/− (2c −/−) was quantified by qPCR. n = 6. Unpaired Student's t‐test. ns = not significant.
-
DImmunohistochemical detection of PLN in paraformaldehyde (PFA)‐fixed testis from wild‐type and SPPL2c −/− mice. Cryosections were stained using a rabbit monoclonal PLN antibody or normal rabbit IgG as negative control and diaminobenzidine as peroxidase substrate. Scale bars, 100 μm.
-
ETestis suspensions were stained with 2.5 μM DRAQ5 to determine cellular DNA amount. 1C‐co = elongated spermatids, 1C = other haploid cells including round spermatids, 2C = spermatogonia, secondary spermatocytes, Sertoli cells, other somatic cells, 4C = primary spermatocytes, G2 spermatogonia.
-
FIntracellular Ca2+ concentrations were analysed in testis suspensions from either wild‐type or SPPL2c‐deficient mice using the Ca2+‐sensitive probe Fluo4‐AM. Individual germ cell populations were identified as shown in (E). Bars indicate Median Fluo4‐AM fluorescence ± SD of 8 individual samples from three mice per genotype normalised to those of wild‐type samples. Unpaired Student's t‐test. ***P ≤ 0.001, ns = not significant.
-
GMedian forward scatter (FSC) was controlled in the 1C‐co population from the data sets shown in (F). Median FSC ± SD of 8 individual samples from three mice per genotype. Unpaired Student's t‐test. ns = not significant.
-
H, IExpression of SPPL2c in human testis. (H) Western blot analysis of lysates from HEK293 cells expressing human (h) SPPL2c, human and murine testis using antisera against a C‐terminal epitope of murine SPPL2c or human SPPL2c as indicated. (I) Immunohistochemical detection of SPPL2c with the monoclonal 1A1 antibody in paraffin sections from Bouin's‐fixed human testis. In addition, sections were incubated with control immunoglobulins. Arrows indicate elongated spermatids. Scale bars, 30 μm.
Source data are available online for this figure.
With regard to the phenotypes described above, we asked what the pathophysiological consequences of increased PLN levels in SPPL2c‐deficient spermatids could be. In cardiomyocytes, PLN controls Ca2+ loading of intracellular Ca2+ stores 35. To evaluate a potential impact of SPPL2c deficiency on intracellular Ca2+ handling, we analysed basal Ca2+ homeostasis in wild‐type and SPPL2c −/− murine testicular cells by labelling dissociated cells with the Ca2+‐sensitive probe Fluo4‐AM. In this set up, individual male germ cell populations were identified by their DNA content as described previously 36, 37 (Fig 5E). As shown in Fig 5F, basal Ca2+ levels were reduced by about 25% specifically in the 1C‐co population of SPPL2c‐deficient animals primarily containing haploid cells with condensed nuclei representing elongated spermatids where SPPL2c is strongly expressed in wild‐type mice. In contrast to this, no alterations of basal Ca2+ levels could be observed in the 1C (other haploid cells including round spermatids), 2C (spermatogonia, secondary spermatocytes, Sertoli cells, other somatic cells) and 4C (primary spermatocytes, G2 spermatogonia) populations where in wild‐type mice, much lower levels of SPPL2c could be detected. Importantly, we could exclude that the observed differences in basal Fluo4‐AM labelling were caused by a size difference in elongated spermatids between the two genotypes as indicated by comparable median forward scatters (FSC) of the 1C‐co populations (Fig 5G). These data strongly argue for a novel role of SPPL2c in fine‐tuning intracellular calcium homeostasis of elongated spermatids by controlling abundance of the SERCA‐regulator PLN, and this confirms PLN as a physiologically relevant substrate of this protease.
Finally, we investigated whether our findings describing a novel role of SPPL2c in male germ cell development in mice may also have implications in humans. Using the antiserum against murine SPPL2c, which cross‐reacts with the human protein, and a novel human‐specific SPPL2c monoclonal antibody 27, we demonstrate endogenous expression of SPPL2c in human testis lysates (Fig 5H). Furthermore, also human SPPL2c was detected in elongated spermatids (Fig 5I). We consider this identical expression pattern as a strong indication that the functional impact of SPPL2c in spermatogenesis, which we have described here, is also preserved in humans.
Discussion
This study has provided the first proof of proteolytic activity of the orphan intramembrane protease SPPL2c and identified PLN as a physiologically relevant substrate. Loss of SPPL2c in mice impairs differentiation and motility of male germ cells. In comparison with SPPL2a/b 19, SPPL2c exhibits a highly restricted expression pattern in mice being exclusively present in developing male germ cells with highest levels in elongated spermatids under basal conditions. Apparently, we cannot exclude that we missed expression in certain low‐abundant cell types in other tissues due to limitations in detection sensitivity or in tissues not tested. Furthermore, it is conceivable that certain cell types upregulate SPPL2c expression following certain stimuli or under distinct pathophysiological conditions. In a previous microarray‐based expression analysis, human SPPL2c expression was detected in several additional tissues including brain, heart and skin 21. This may indicate species differences and a broadened SPPL2c expression pattern in humans. However, those findings could also be false positives resulting from traces of genomic DNA in the analysed RNA samples because the SPPL2c gene has no introns. Similar to our results, an extensive study of the human proteome identified SPPL2c only in testis 38. Based on the published phenotypes of SPPL2a/b‐ and SPPL3‐deficient mice, these proteases play a significant role in the immune system 7. In this regard, the newly described role of SPPL2c extends the in vivo relevance of SPP/SPPL to the reproductive system. A possible impact of the other family members on germ cell differentiation and fertility remains to be investigated. Beyond SPPL2c, also other intramembrane proteases have been found to be expressed in testis and/or influence spermatogenesis in mice or other model organisms. This includes the γ‐secretase complex 39, 40, 41, 42, the rhomboid protease RHBDD1 also known as RHBDL4 43, 44 and also the SPPL protease SPPL3 15, 45.
From a cell biological perspective, it is interesting why with SPP and SPPL2c two different ER‐resident SPP/SPPL intramembrane proteases have evolved. Our results strongly suggest that they fulfil independent physiological functions despite a certain functional overlap regarding their substrate specificity profiles. This view is supported by the highly divergent expression pattern of SPP and SPPL2c within the testis. However, due to limitations of the available antibodies, weak expression of SPP in germ cells and even elongated spermatids may not be excluded at this stage. Since constitutive SPP knockout mice are not viable 46, conditional genetic approaches will be required to assess whether loss of SPP affects spermatogenesis and/or potentially aggravates the phenotype of SPPL2c‐deficient mice. Despite this, the observed impairment of spermatogenesis and sperm motility in SPPL2c −/− mice that cannot be compensated by SPP or other pathways demonstrates a non‐redundant function of this protease. Furthermore, also the suggested different complex organisation of SPP and SPPL2c in testis points to distinct functions. In addition to a 200 kDa homo‐tetramer, SPP forms complexes with the pseudoprotease Derlin‐1 and the E3 ubiquitin ligase TRC8 9, 46, which is critical for its role in the ERAD process. If these and/or any other proteins are part of the SPPL2c complex remains to be determined. The inhibitory profile of SPPL2c indicates two remarkable differences with regard to other SPP/SPPL proteases. In the performed RAMP4‐2 cleavage assay, SPPL2c was not inhibited by the active site inhibitor (Z‐LL)2‐ketone, which acts on all SPP/SPPL proteases with the exception of SPPL3 4, 7, 25, 28. In contrast, the γ‐secretase inhibitor DAPT partially blocked cleavage by SPPL2c. DAPT binds to γ‐secretase at a site different from the catalytic centre 47 and does not inhibit any other SPP/SPPL family member 7, 29. Since so far we do not have any cell line with endogenous SPPL2c expression at hand, inhibitor testing has to rely on SPPL2c overexpression for the time being. Validating our results from the RAMP4‐2 assay with additional substrates will be important. Nevertheless, these findings strongly suggest that SPPL2c exhibits relevant differences to the other SPP/SPPL family members with regard to structure and/or cleavage mechanism.
To our knowledge, expression of PLN in testis has not been reported before and spermatogenesis in PLN knockout mice has not been analysed. Since these mice are fertile 48 (www.mmrrc.org, RRID:MMRRC_000027‐MU), gross alterations seem unlikely. PLN can act as an inhibitor of SERCA2 activity, the calcium pump involved in filling the ER calcium stores 32, 35. By this means, PLN is a major regulator of cardiomyocyte function and contractility. The affinity of PLN to SERCA2 is significantly influenced by phosphorylation of PLN. Furthermore, initial studies have revealed that triggered degradation of PLN by autophagy or the ubiquitin–proteasome system may play an additional regulatory role 49, 50, 51. However, an involvement of intramembrane proteases in the control of PLN as we report it here has not been reported before. Whether SPPL2c primarily controls levels of PLN or whether the cytosolic cleavage product, which would comprise about 30 amino acids, fulfils a biological function is subject to further analysis.
Transgenic overexpression of PLN in cardiomyocytes has been found to alter intracellular calcium handling, filling of the ER and cellular contractility, which also affects cardiac function in vivo 52, 53. It can be speculated that an altered PLN homeostasis as consequence of impaired degradation has similar effects. However, very little is known about the role of calcium during specific stages of spermatogenesis 54. Expression of the SERCA2 calcium pump, which is regulated by PLN in the heart, starts in spermatocytes and becomes more pronounced in spermatids 55. This corresponds to the pattern we have observed for PLN in mouse testis (Fig 5D). In addition, late spermatids specifically presented with reduced intracellular Ca2+ levels, which might be directly connected to the observed accumulation of PLN in these cells (Fig 5F). Despite the still enigmatic function of SERCA2 and PLN in spermatids, there is growing evidence that Ca2+ is critically involved in regulation of germ cell differentiation. For example, the transmembrane protein 203 (TMEM203) plays a role in the regulation of ER calcium stores, although its precise molecular function is not known yet 36. Mice deficient for this protein fail to produce mature spermatozoa. In testis, they exhibit a subtle, relative reduction in numbers of late stage post‐meiotic spermatids, which display degenerative intracytoplasmic vacuolar changes and fail to be released from the germinal epithelium 36. Calcium handling in TMEM203 −/− testicular cells was different from that of wild‐type cells in several respects. As further supported by the analysis of CIB1 −/− mice 56, these observations strongly indicate that defects in calcium handling can impair male germ cell differentiation. Although the phenotype of SPPL2c −/− mice is much milder, it seems possible that the observed loss of elongated spermatids is caused by the impaired Ca2+ handling based on the PLN accumulation.
Another interesting question is whether proteolysis of PLN in spermatids has implications for the functionality of mature spermatozoa. We failed to detect PLN in mature epididymal sperm irrespective of the genotype. This indicates that PLN also in wild‐type cells similar to other ER proteins is ultimately extruded from the cells and that its specific function like that of SPPL2c is limited to the stage of spermatids. It is conceivable that timed proteolytic PLN turnover is required to facilitate efficient filling of intracellular calcium stores and that a disturbance of this process may account for the motility defect of spermatozoa from SPPL2c −/− mice. Though the requirement of calcium for sperm movement is undisputed 54, 57, the specific contribution of intracellular stores in this context is less clear. Therefore, to define the precise impact of PLN and SPPL2c on this process will require further investigations.
However, the role of SPPL2c in germ cells is not limited to cleaving PLN 27. As identified in a cell‐based substrate screen utilising SPPL2c overexpression, SPPL2c is capable of processing selected SNARE proteins of the early secretory pathway. Overlapping with the substrates revealed by Papadopoulou et al 27, we observed an accumulation of STX8 in SPPL2c‐deficient testis in our proteomic analysis. It seems likely that other relevant substrates have escaped our proteomic screening since the effect of SPPL2c deficiency in the analysis of total testis membrane preparations has been diluted by non‐ or low‐SPPL2c‐expressing cells. Targeted Western blot analysis of selected candidate substrates from Papadopoulou et al 27 in sorted spermatids may therefore help to resolve this. SPPL2c‐mediated cleavage of SNAREs affects the ER exit of selected cargo and leads to a mistrafficking of selected glycosyltransferases. Consequently, the Golgi morphology within spermatids and the glycosylation pattern of mature spermatozoa are altered in SPPL2c‐deficient mice 27. Overall, this strongly indicates that the in vivo function of SPPL2c is not limited to a single substrate, but rather impacts on different processes in parallel.
We showed that the expression pattern of SPPL2c is conserved in humans. It may be worth mentioning that SPPL2c is one of the six genes affected in the Koolen‐de Vries 17q21 microdeletion syndrome 58, 59. While the highly testis‐specific expression of SPPL2c makes a direct contribution of this protease to the mental retardation phenotype of Koolen‐de Vries‐patients rather unlikely, deletion of SPPL2c might contribute to disease‐associated infertility that was recently reported for a single male patient 60. This finding in combination with SPPL2c expression in elongated spermatids of humans, as demonstrated in this study, points towards a conserved function of SPPL2c in human spermatogenesis. Therefore, considering this gene as a candidate locus in cases of male subfertility is certainly justified. The SPPL2C gene is located on chromosome 17q21 in a chromosomal region, for which a 900 kb inversion polymorphism has been identified 61, 62. This leads to two major haplotypes H2 and H1, of which the H2 haplotype has probably undergone positive selection in Europe and is found in about 20% of the individuals. As analysed by Stefansson et al 61 in an Icelandic population, females carrying the H2 haplotype had a higher number of children for which the reason, however, is currently incompletely understood. For other genes encompassed by this translocation, differential expression levels in individuals with the H1 or H2 haplotype have been shown 63. Based on this, it is tempting to speculate that these H1 and H2 alleles also influence expression of SPPL2c and thereby modulate fertility of the individuals. Homozygous breedings of male and female SPPL2c −/− mice were clearly subfertile with reduced litter sizes. However, the observed breeding statistics strongly suggests that the role of SPPL2c in male germ cell differentiation is only part of the explanation and that its function in the reproductive process is more extensive than currently appreciated. Initial attempts to localise SPPL2c expression in the female reproductive tract have been unsuccessful. However, considering the stage‐specific SPPL2c expression during spermatogenesis, its detection in maturing oocytes may be challenging. Additional work will be needed to fully unravel the impact of SPPL2c on the reproductive capacity of mice and humans.
Materials and Methods
Plasmids
Expression plasmids for murine SPPL2c and putative substrate proteins were amplified from suitable cDNAs and subcloned into pcDNA3.1/Hygro+ vector (Invitrogen). Constructs of human SPPL2c and other human SPPL2 proteases were in pcDNA3.1/Zeo‐ (Invitrogen) as described before 15 and in accompanying manuscript by Papadopoulou et al 27. As indicated, murine SPPL2c and SPP were expressed with a C‐terminal Myc epitope, whereas the substrate candidates were expressed with N‐terminal HA, FLAG or V5 epitopes as indicated. Murine SPPL2c open reading frames (ORFs) utilised in this study for heterologous overexpression corresponded to the two isoforms A (NM_199019.2) and B (NM_001082535.1). If not specifically stated otherwise in the figure legends, isoform A was used in SPPL2c overexpression experiments. A construct encoding the SPPL2a/b substrate Bri2 devoid of the furin‐cleaved pro‐peptide (Bri2‐ΔFC) was employed as described 25. The ORF encoding the N‐terminal 195 residues of the HCV polyprotein (Glasgow strain genotype 1a) comprising the core protein and 4 amino acids of the envelope protein E1 was amplified from pSV‐Sport1/CE1/4 13 and subcloned with an N‐terminal triple FLAG‐tag into pcDNA3.1/Neo+ (Invitrogen). For ectopic expression of human XBP1u, a pcDNA3.1/Neo+ expression constructs with an N‐terminal triple FLAG‐tag harbouring a silent mutation in the 3′‐IRE1‐splice‐site was used that had been described previously 9. Human HO‐1 (full‐ORF Gateway clone 187935150) was cloned with an N‐terminal triple FLAG‐tag into pcDNA3.1/Neo+.
Cell culture and transfection
HEK293 and HeLa cells (both from DSMZ) were maintained in DMEM (GIBCO) supplemented with 10% foetal calf serum (FCS) (Biochrom) as well as 100 U/ml penicillin (Sigma) and 100 μg/ml streptomycin (Sigma). T‐Rex™‐293 cell lines inducibly expressing human SPPL2a and SPPL2b have been described before 25. Generation of cells expressing SPPL2c is described in Ref. 27. Cells were transfected with Turbofect (Thermo Scientific) or Lipofectamin 2000 according to manufacturer's recommendations. Alternatively, cells were transfected using 25 kDa linear polyethylenimine as has been described before 9.
Generation of SPPL2c−/− mice
Chimeric mice (Strain ID: 4933407P14Riktm1(KOMP)Vlcg) were obtained from the UC Davis KOMP Repository. Mice were produced from targeted ES cells generated by Velocigene Regeneron Pharmaceuticals (Project ID: VG12958) based on the VGB6 Parent Line (C57BL/6N Tac background). The obtained chimeras were bred with C57BL6/N Crl wild‐type mice. The floxed neomycin resistance was excised by breeding with mice carrying a Cre‐Deleter allele 64, which was removed prior to analysis of the SPPL2c −/− mouse strain. All SPPL2c‐deficient mice used in this study carried the post‐Cre allele as indicated in Fig EV1A. Genotyping was performed with PCRs specifically detecting the wild type (wt‐Fw, 5′‐AAAACTGAGTCCTCTAGAAGCGGGG‐3′; wt‐Rv, 5′‐GCAATCTGAGCAGGGATAGCATTGG‐3′) and knockout allele (ko‐Fw, 5′‐ACTTGCTTTAAAAAACCTCCCACA‐3′; ko‐Rv, 5′‐AAATTCTGGTCTCTGTCCTCCCACC‐3′) amplifying fragments of 270 bp (wt) or 823 bp (ko), respectively.
RT–PCR
Total RNA was extracted from tissues using the NucleospinRNA Kit (Macherey Nagel) or in case of muscle and heart TRIzol (SIGMA) according to the manufacturer's instructions. In all cases, cDNA was generated from 1 μg total RNA with random hexamer primers applying the RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific). To demonstrate expression of the different SPPL2c isoforms, a common forward primer (2c‐RT‐Fw, 5′‐GGATTTCACACCAGCCATGAC‐3′) and isoform‐specific reverse primers in the 3′‐untranslated sequence of the SPPL2c Isoform A (2c‐RT‐IsoA‐Rv, 5′‐AGCATCCCCGTGCAGTGTTCTG‐3′) or B (2c‐RT‐IsoB‐Rv, 5′‐GTGGGAAGG AAGAGGCATCCTC‐3′) were used for amplification. As control, a fragment of the actin ORF was amplified using the following primers: fw, 5′‐GTTACAACTGGGACGAC ATGG‐3′; rv, 5′‐GATGGCTACGTACATGGCTG‐3′.
qRT–PCR
To quantitatively detect mRNA levels of SPP/SPPL2 family members as well as PLN in murine testis, real‐time PCR was performed using 0.5 μl of cDNA generated as described above using the Universal Probe Library System (Roche) with a LightCycler 480 Instrument II (Roche). The following primers and probes were applied as follows: SPP (fw: 5′‐GTCCTGGTGGCACTAGCC‐3′, rv: 5′‐AGGATTACGGCCGAGGACT‐3′, probe 7); SPPL2a (fw: 5′‐GAGAGCATCATGGTGGAACTT‐3′, rv: TGGCACCCTGATAACTACTGG‐3′, probe 107); SPPL2b (fw: 5′‐TCTTCCGAAATGAGGACCAG‐3′, rv: 5′‐CGGATGGTCTTCAGCATGTA‐3′, probe 29); PLN (fw: 5′‐TGAGCTTTCCTGCGTAACAG‐3′, rv: 5′‐TGGTCAAGAGAAAGATAAAAAGTTGA‐3′, probe 27) and Tuba1a (fw: 5′‐CTGGAACCCACGGTCATC‐3′, rv: 5′‐GTGGCCACGAGCATAGTTATT‐3′, probe 88).
Protein extraction and Western blotting
Cell and tissue lysates were prepared as described before 19, 65. Protein concentration was determined with a bicinchoninic acid (BCA) protein assay (Thermo Scientific). SDS PAGE was performed using tris‐glycine 66 or tris‐tricine 67 buffer systems as indicated. Protein transfer to nitrocellulose or PVDF membranes and immunodetection using peroxidase‐conjugate secondary antibodies and a LAS4000 imaging system for visualisation of chemiluminescence (GE Healthcare) has been described before 68. For deglycosylation, aliquots of lysates were treated with endoglycosidase H (Endo H, Roche) or peptidyl N‐glycosidase F (PNGase F, Roche) according to manufacturer's instructions prior to Western blot analysis. For detection of murine SPPL2c, polyclonal rabbit antisera against the following synthetic peptides were generated and subsequently affinity‐purified: GHNQQQTVAAERSQRAWEDD (N‐Term, res. 232–251) and LHKRKGLKVKKSMSAQAPL (C‐Term, res. 672–690). Murine SPP was detected with a cross‐reacting antiserum generated against a C‐terminal epitope of human SPP 69. The monoclonal antibody for detection of human SPPL2a (6E9), SPPL2b (3F9) and SPPL3 (7F9) has been described earlier 25, 28, and the monoclonal antibody 1A1 utilised for detection of human SPPL2c is described in detail in Ref. 27. The polyclonal antibody directed against tetraspanin‐3 has been described previously by Ref. 70. Monoclonal antibodies against the HA epitope (3F10) and PLN (D9W8M) were obtained from Roche and Cell Signaling, respectively. Antibodies against protein disulphide isomerase (ab11432) and CD44 (IM7) were purchased from Abcam and BioLegend, respectively. Anti‐transferrin receptor 1 (ab84036) was obtained from Abcam. Polyclonal antisera against murine cathepsin D (SII.10) 71 and the lysosomal integral membrane protein LIMP‐2 72 were described before. Anti‐E. coli β‐galactosidase (Z3781) was obtained from Promega. To confirm equal protein loading, eukaryotic elongation factor 2 (EEF2, ab33523 Abcam), β‐actin (A2066, Sigma), GAPDH (FL‐355, Santa Cruz) or cofilin (D3F9, Cell Signaling) were used as control.
Immunoprecipitation
Testis lysates were prepared as described above, and aliquots (2 mg protein) were diluted in 50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1% Triton X‐100, 4 mM EDTA, supplemented with complete protease inhibitor cocktail (Roche), 4 mM Pefabloc (Roth) and 0.5 μg/ml pepstatin A (Sigma). PLN was immunoprecipitated with a mouse monoclonal antibody (2D12) and protein G agarose (both from Thermo). For Western blot analysis of the immunoprecipitates, a rabbit monoclonal PLN antibody (Cell Signalling) was employed.
Immunofluorescence
Cells adherent to glass coverslips were fixed with 4% paraformaldehyde in PBS and subjected to immunocytochemical stainings according to 65. The Myc epitope was detected with the 9B11 monoclonal antibody from Cell Signaling. The ER was visualised with anti‐KDEL (Enzo) and the ER‐Golgi intermediate compartment (ERGIC) with anti‐ERGIC‐53 (clone OTI1A8, Enzo). HA‐tagged TA proteins were detected using the 3F10 antibody described above. Alexa 488‐ and 594‐conjugated secondary antibodies were obtained from Molecular Probes. Nuclei were visualised with DAPI (4‐,6‐diamidino‐2‐phenylindole from Sigma‐Aldrich). An Olympus FV1000 confocal laser scanning microscope was used for acquisition of images which were further processed with Olympus Fluoview and Adobe Photoshop software.
Microsome isolation from mouse testis and blue‐native (BN) PAGE
Testes from two mice were homogenised in cold 1.5 ml buffer A (250 mM sucrose, 50 mM HEPES/KOH, pH 7.6, 50 mM potassium acetate, 6 mM magnesium acetate, 1 mM EDTA, 1 mM DTT) containing 10 μg/ml PMSF and EDTA‐free complete protease inhibitor cocktail (Roche) using a glass–Teflon potter at full speed. The homogenate was centrifuged at 1,000 × g for 10 min at 4°C. The supernatant was collected and kept on ice, and the pellet was resuspended in 1.5 ml buffer A followed by homogenisation and centrifugation as described above. The collected supernatants were combined and centrifuged at 10,000 × g for 10 min to remove mitochondria followed by centrifugation at 142,000 × g for 1 h. The membrane pellet from the high‐speed centrifugation was resuspended in buffer B (8 mM Tris/HCl, pH 7.4, 0.6 mM MgCl2, 0.4 mM EGTA, 20 mM NaCl, 4% glycerol) and solubilised for 20 min on ice by adding 1% digitonin (Calbiochem) from a 10× stock solution in buffer B. Non‐solubilised proteins were removed by centrifugation for 20 min at 100,000 g. 1/40 BN‐sample buffer (500 mM 6‐aminohexanoic acid, 100 mM Tris/HCl, pH 7, 5% Coomassie G250) was added prior to separation on a 3–12% BN‐acrylamide gel (Invitrogen). For size determination, Native‐Mark Unstained Protein Standard (Invitrogen) was used. The gel was blotted on PVDF membrane, which was destained with methanol, immersed in 5% (w/v) milk in TBS‐T and analysed by immunodetection.
Subcellular fractionation of murine testis
After removal of the tunica albuginea, testicles from two wild‐type mice were minced with an ultraturrax in 250 mM sucrose, 10 mM HEPES‐NaOH, pH 7.4 and 1 mM EDTA (HB, homogenisation buffer) and then further homogenised by eight strokes of a Potter homogeniser. For sedimentation of nuclei, the tissue homogenate was centrifuged at 750 × g for 10 min. The resulting post‐nuclear supernatant was adjusted to a final concentration of 30% (w/v) Percoll in a final volume of 9.5 ml and underlayered with 0.5 ml 65% (w/v) sucrose in 10 mM HEPES‐NaOH using a glass capillary fitted to a peristaltic pump. Centrifugation was performed in a 70.1 Ti fixed angle rotor (Beckman Coulter) at 40,500 × g max for 40 min at 4°C. Fifteen fractions were collected from bottom (fraction 1) to top (fraction 15) and analysed by Western blotting.
Histological analysis
Murine testis was fixed by immersion in Bouin solution (Sigma) or 4% (w/v) PFA in 0.1 M phosphate buffer, pH 7.4, as indicated. Prior to cryosectioning, tissue samples were equilibrated in 30% sucrose in 0.1 M phosphate buffer, pH 7.4. Alternatively, paraffin embedding and haematoxylin–eosin (H&E) staining of sections were performed according to standard procedures. Activity of the β‐galactosidase reporter was visualised with X‐Gal in cryosections of PFA‐fixed tissue as described before 19, 68. Immunohistochemical stainings of cryo‐ or paraffin sections were performed utilising the ABC System (Vector Laboratories) and diaminobenzidine as peroxidase substrate. In some cases, deparaffinised and rehydrated sections were subjected to epitope retrieval with 10 mM sodium citrate, pH 6, for 5 min in a microwave oven. Employed antibodies include anti‐mSPPL2c (C‐Term), which was described above, and anti‐PLN (Rabbit monoclonal D9W8M, Cell Signalling). SPP was visualised with the pre‐described 69 polyclonal antiserum. To control for specificity, rabbit polyclonal control IgG (BioLegend) was employed in parallel. After completion of immunostaining, nuclei were visualised with haematoxylin.
TUNEL staining
For detection of apoptotic cells, deparaffinised and rehydrated sections from wild‐type and SPPL2c −/− testis were stained by using the ApopTag Peroxidase In Situ Apoptosis Detection Kit (Merck, Darmstadt, Germany). In brief, after rinsing with TBS (1 × 5 min), sections were treated with citrate buffer (pH 6) for 15 min in microwave oven followed by washing with TBS. After deactivating endogenous peroxidase with H2O2 (3%), the slides were washed with TBS (15 min) followed by equilibration buffer (10 min). Sections were stained by TdT (terminal deoxynucleotidyl‐transferase) enzyme in a humidified chamber at 37° for 60 min. After stop/wash buffer (10 min) and washing in TBS (3× 1 min), sections were incubated with anti‐digoxigenin‐conjugate (30‐min room temperature), washed in TBS (4× 2 min) and incubated with DAB (5 min). Sections were washed in aqua bidest (3× 5 min), counterstained with haematoxylin, dehydrated and embedded using Eukitt. Controls were performed by omitting TdT.
Analysis of SPPL2c in human testis
The samples and the methods employed were previously described 73. For immunohistochemical staining, sections from three fixed and paraffin‐embedded human testicular biopsies of men with normal spermatogenesis were used. They were stained with the monoclonal 1A1 antibody (1:50). Negative controls included incubation with IgG and omission of the primary antibody. Samples were counterstained with haematoxylin. Small testicular samples were derived from two additional men and were homogenised and protein used for Western blotting. For the scientific use of the samples, all patients had granted written informed consent; the ethical committee (Ethikkommission, Technische Universität München, Fakultät für Medizin, München, project number 5158/11) has approved the study. The experiments were performed in accordance with relevant guidelines and regulations.
Sperm preparation and computer‐assisted sperm analysis (CASA)
Sperm preparation was performed as previously described 74, 75. In brief, mice were euthanised by asphyxiation with CO2. After preparation of the caudae epididymides and vasa deferentia, the organs were rinsed with medium HS (135 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 30 mM HEPES, 10 mM glucose) adjusted to pH 7.4 with NaOH. Semen was allowed to exude from caudae epididymides and vasa deferentia for 15 min by 5% CO2 after transfer to 1 ml of medium HS containing 5 mg/ml bovine serum albumin and 15 mM NaHCO3. Cells were washed twice by dilution with 4 ml HS and sedimentation (400 × g; 5 min). Sperm were resuspended in HS to a final volume of 500 μl and stored at room temperature. CASA was performed as previously described 74. In brief, the CASA system (Medical Technology, Hamburg, Germany) analysed the sperm samples, mixed 1:1 with HS/5% BSA to retrieve average path velocity (VAP), straight line velocity (VSL) and curvilinear velocity (VCL) in micrometre per second. For analysis of sperm morphology, mature sperm isolated from the epididymis were diluted 1:4 in HS buffer and subsequently air‐dried on microscope slides for 1 h. Cells were then fixed for 15 min in methanol and again air‐dried for 1 h. Sperm morphology was analysed by phase‐contrast microscopy using a Leica DMi8 microscope equipped with a Leica CFC7000 T camera.
Proteomic analysis of testis membrane preparations
Post‐nuclear supernatants were prepared from testis of wild‐type and SPPL2c −/− mice (n = 5 per genotype) as described above. Total membrane fractions were obtained by ultracentrifugation at 146,944 × g max for 1 h at 4°C. The supernatant was recovered as the cytosolic fraction for further analysis. The sedimented membranes were resuspended in 0.1 M Na2CO3, pH 11.5, incubated on ice for 1 h and centrifuged again as described above. Membranes were washed once in 10 mM HEPES‐NaOH, pH 7.4, and resuspended in the same buffer after a final sedimentation. Aliquots of both the cytosolic and the carbonate‐washed membrane fractions were subjected to proteomic analysis. A protein amount of 15 μg per sample was subjected to proteolytic digestion with 0.3 μg LysC (Promega) and 0.15 μg trypsin (Promega) using the filter‐assisted sample preparation (FASP) protocol 76 with 30 kDa Vivacon spin filters (Sartorius). Proteolytic peptides were desalted by stop and go extraction with C18 tips 77. The purified peptides were dried by vacuum centrifugation. Samples were dissolved in 20 μl 0.1% formic acid. Peptides were analysed on an Easy nLC 1000 nanoHPLC (Thermo Scientific) which was coupled online via a Nanospray Flex Ion Source (Thermo Scientific) equipped with a PRSO‐V1 column oven (Sonation) to a Q‐Exactive HF mass spectrometer (Thermo Scientific). An amount of 1.3 μg of peptides was separated on an in‐house packed C18 column (30 cm × 75 μm ID, ReproSil‐Pur 120 C18‐AQ, 1.9 μm, Dr. Maisch GmbH) using a binary gradient of water (A) and acetonitrile (B) supplemented with 0.1% formic acid (0 min, 2% B; 3:30 min, 5% B; 137:30 min, 25% B; 168:30 min, 35% B; 182:30 min, 60% B) at 50°C column temperature. A data‐dependent acquisition method was used. Full MS scans were acquired at a resolution of 120,000 (m/z range: 300–1,400, AGC target: 3E+6). The 15 most intense peptide ions per full MS scan were selected for peptide fragmentation (resolution: 15,000, isolation width: 1.6 m/z, AGC target: 1E+5, NCE: 26%). A dynamic exclusion of 120 s was used for peptide fragmentation.
The data were analysed with the software MaxQuant (maxquant.org, Max‐Planck Institute Munich) version 1.5.5.1 78. The MS data were searched against a reviewed FASTA database of Mus musculus from UniProt including isoforms (download: 11 April 2017, 24992 entries). Trypsin was defined as protease. Two missed cleavages were allowed for the database search. The option first search was used to recalibrate the peptide masses within a window of 20 ppm. For the main search, peptide and peptide fragment mass tolerances were set to 4.5 and 20 ppm, respectively. Carbamidomethylation of cysteine was defined as static modification. Acetylation of the protein N‐term as well as oxidation of methionine was set as variable modifications. The false discovery rate for both peptides and proteins was adjusted to < 1%. Label‐free quantification (LFQ) of proteins required at least two ratio counts of razor peptides. Only razor and unique peptides were used for quantification. LFQ normalisation was done separately for the membrane and cytosolic fraction. The software Perseus (version 1.5.8.5) was used for further data analysis. The protein LFQ intensities were log2 transformed, and a two‐sided Student's t‐test was applied to evaluate the significance of proteins with changed abundance. Additionally, a permutation‐based false discovery rate estimation was used 79. Protein levels of the different SPPL proteases and SPP (HM13) were estimated on the basis of the MS data using intensity‐based absolute quantification (iBAQ) which is proportional to protein copy numbers and allows direct comparisons of protein abundances between different proteins 31.
Sorting of germ cell populations from murine testis
Testes were taken from C57BL/6 wild‐type mice, and the tunica albuginea was removed to isolate seminiferous tubules. Tubules were then digested for 20 min at 32°C in 1 ml of digestion buffer (HBSS supplemented with 20 mM HEPES, pH 7.2, 1.2 mM MgSO4, 1.3 mM CaCl2, 4 μg/ml DNase, 1 μg/ml collagenase type I, 0.05% lactate). Tubules were rinsed once with PBS and again digested for 20 min at 32°C in 1 ml of digestion buffer. The cell suspension was then filtered through a 40‐μm mesh, and cells were pelleted by centrifugation. Cells were resuspended in FACS incubation buffer (HBSS supplemented with 20 mM HEPES, pH 7.2, 1.2 mM MgSO4, 1.3 mM CaCl2, 6.6 mM sodium pyruvate, 0.05% lactate, glutamine and 1% foetal calf serum) and incubated with 5 μg/ml of Hoechst 33342 at 32°C for 1 h. Propidium iodide was added at a concentration of 2 μg/ml to exclude dead cells from further analysis. Sorting of meiocytes was performed with the 85‐μm nozzle in an ARIA II (BD Biosciences). Cells with Hoechst and PI staining were excited by 355 and 488 nm lasers, respectively. PI signal was detected by using 685 LP and 710/40 BP filter. Hoechst blue signal was detected using 600 LP and 620/10 BP filters in front of first detector, and Hoechst red was detected with 440/40 BP filter in front of the second detector. Cells were collected in PBS and frozen at −80°C after centrifugation for subsequent analysis. FlowJo (Tree Star Inc.) and FACSDiva software were used for cell sorting and data analysis. Cells were finally resuspended in 60 μl lysis buffer and processed further for Western Blotting.
Calcium measurement in testis suspensions
To estimate the intracellular calcium concentrations in different murine germ cell populations, testes were dissected, decapsulated and the tubuli subsequently suspended in 1 ml suspension buffer (HBSS (GIBCO) + 1% FCS + 1.2 mM CaCl2 + 0.9 mM MgCl2) by gently pipetting up and down. The cell suspension was passed through a 100‐μm Cell Strainer (BD Biosciences) and centrifuged for 5 min at 800 g. The resulting pellet was resuspended in 800 μl suspension buffer, and cell concentrations were adjusted so that in the final samples, an optimised detection rate of approx. 3,000 events/s on a FacsCanto II (BD Biosciences) flow cytometer was achieved. 50 μl of the obtained cell suspensions was loaded with the Ca2+‐sensitive probe Fluo‐4AM (2 μM, Biomol) in a total volume of 200 μl for 30 min at 37°C and then sedimented for 5 min at 800 g. Next, cellular DNA was stained using DRAQ5 (BioLegend) at a final concentration of 2.5 μM in suspension buffer without calcium and magnesium for 15 min in a total volume of 500 μl. Finally, sample volume was adjusted to 1 ml and Fluo4‐AM‐ and DRAQ5‐dependent fluorescence was measured in the presence of 1 mM EGTA for 1 min with a FACS Canto II applying the FACSDiva software. Further analysis was performed using the FlowJo software (FlowJo LLC). Individual germ cell populations were gated based on their DNA content according to Ref. 36, 37 as depicted in Fig 5E. Median Fluo4‐AM signals of each population were normalised to those observed for the respective wild‐type control. In addition, median forward scatter of the 1C‐co population was compared between wild‐type controls or SPPL2c‐deficient animals to ensure that differences in Fluo4‐AM staining were not caused by differences in cell size.
Statistical analysis
If not stated elsewise, all bar diagrams show mean values ± SD. Tests applied for assessment of statistical significance are depicted in the respective figure legends.
Author contributions
JN, TM, RH, SAM, RM, AP, VD, MH‐K and VA performed the experiments and analysed data. RL‐R, AM and MB conducted immunohistochemistry and histological analysis of testis. GW performed CASA analysis of sperm motility. UB and RJ contributed expertise and experimental set‐ups for flow cytometric sorting of testicular cell suspensions. SAM and SFL contributed the proteomic‐based substrate screen. PS provided general support. MKL and RF gave conceptual advice and contributed to the design of the study. BS designed, conceptualised and supervised the research and analysed data. BS and TM wrote the manuscript. All authors contributed to the editing of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgements
We thank Andreas Meinhardt, Institute of Anatomy, Justus‐Liebig University Giessen for help with the histological analysis of testis and quantification of spermatids. Furthermore, we thank Sebastian Held, Kai Wilkens and Anna Berghofer for excellent technical assistance and Michael Schwake for providing the LIMP‐2 antiserum. We are grateful to J. Ullrich Schwarzer and Frank‐Michael Köhn for providing human testis samples and Astrid Tiefenbacher for technical assistance. We thank the Z3 Imaging unit of the SFB 877 for support. This work was supported by the Deutsche Forschungsgemeinschaft as part of the SFB877/B7 (project B7, to B.S.), the research group FOR2290 (R.F; S.L.; M.K.L.) as well as grants SCHR 1284/1‐1, SCHR 1284/2‐1, FL 635/2‐2 and MA 1080/23‐1 and MA 1080/23‐2. In addition, this project received support from the Breuer Foundation Research Award (to S.L.), the Helmholtz Israel Program and the Centers of Excellence in Neurodegeneration (S.L.).
EMBO Reports (2019) 20: e46449
See also: AA Papadopoulou et al (March 2019) and HS Young & MJ Lemieux (March 2019)
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 80 partner repository with the data set identifier PXD011922.
References
- 1. Urban S (2016) SnapShot: cartography of intramembrane proteolysis. Cell 167: 1898–1898 e1891 [DOI] [PubMed] [Google Scholar]
- 2. Urban S, Freeman M (2002) Intramembrane proteolysis controls diverse signalling pathways throughout evolution. Curr Opin Genet Dev 12: 512–518 [DOI] [PubMed] [Google Scholar]
- 3. Avci D, Lemberg MK (2015) Clipping or extracting: two ways to membrane protein degradation. Trends Cell Biol 25: 611–622 [DOI] [PubMed] [Google Scholar]
- 4. Weihofen A, Binns K, Lemberg MK, Ashman K, Martoglio B (2002) Identification of signal peptide peptidase, a presenilin‐type aspartic protease. Science 296: 2215–2218 [DOI] [PubMed] [Google Scholar]
- 5. Ponting CP, Hutton M, Nyborg A, Baker M, Jansen K, Golde TE (2002) Identification of a novel family of presenilin homologues. Hum Mol Genet 11: 1037–1044 [DOI] [PubMed] [Google Scholar]
- 6. Mentrup T, Fluhrer R, Schroder B (2017) Latest emerging functions of SPP/SPPL intramembrane proteases. Eur J Cell Biol 96: 372–382 [DOI] [PubMed] [Google Scholar]
- 7. Mentrup T, Loock AC, Fluhrer R, Schroder B (2017) Signal peptide peptidase and SPP‐like proteases ‐ Possible therapeutic targets? Biochim Biophys Acta 1864: 2169–2182 [DOI] [PubMed] [Google Scholar]
- 8. McLauchlan J, Lemberg MK, Hope G, Martoglio B (2002) Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J 21: 3980–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen CY, Malchus NS, Hehn B, Stelzer W, Avci D, Langosch D, Lemberg MK (2014) Signal peptide peptidase functions in ERAD to cleave the unfolded protein response regulator XBP1u. EMBO J 33: 2492–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Avci D, Fuchs S, Schrul B, Fukumori A, Breker M, Frumkin I, Chen CY, Biniossek ML, Kremmer E, Schilling O et al (2014) The yeast ER‐intramembrane protease Ypf1 refines nutrient sensing by regulating transporter abundance. Mol Cell 56: 630–640 [DOI] [PubMed] [Google Scholar]
- 11. Boname JM, Bloor S, Wandel MP, Nathan JA, Antrobus R, Dingwell KS, Thurston TL, Smith DL, Smith JC, Randow F et al (2014) Cleavage by signal peptide peptidase is required for the degradation of selected tail‐anchored proteins. J Cell Biol 205: 847–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hsu FF, Yeh CT, Sun YJ, Chiang MT, Lan WM, Li FA, Lee WH, Chau LY (2015) Signal peptide peptidase‐mediated nuclear localization of heme oxygenase‐1 promotes cancer cell proliferation and invasion independent of its enzymatic activity. Oncogene 34: 2360–2370 [DOI] [PubMed] [Google Scholar]
- 13. Lemberg MK, Martoglio B (2002) Requirements for signal peptide peptidase‐catalyzed intramembrane proteolysis. Mol Cell 10: 735–744 [DOI] [PubMed] [Google Scholar]
- 14. Kuhn PH, Voss M, Haug‐Kroper M, Schroder B, Schepers U, Brase S, Haass C, Lichtenthaler SF, Fluhrer R (2015) Secretome analysis identifies novel signal Peptide peptidase‐like 3 (Sppl3) substrates and reveals a role of Sppl3 in multiple Golgi glycosylation pathways. Mol Cell Proteomics 14: 1584–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Voss M, Kunzel U, Higel F, Kuhn PH, Colombo A, Fukumori A, Haug‐Kroper M, Klier B, Grammer G, Seidl A et al (2014) Shedding of glycan‐modifying enzymes by signal peptide peptidase‐like 3 (SPPL3) regulates cellular N‐glycosylation. EMBO J 33: 2890–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schneppenheim J, Dressel R, Hüttl S, Lüllmann‐Rauch R, Engelke M, Dittmann K, Wienands J, Eskelinen EL, Hermans‐Borgmeyer I, Fluhrer R et al (2013) The intramembrane protease SPPL2a promotes B cell development and controls endosomal traffic by cleavage of the invariant chain. J Exp Med 210: 41–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beisner DR, Langerak P, Parker AE, Dahlberg C, Otero FJ, Sutton SE, Poirot L, Barnes W, Young MA, Niessen S et al (2013) The intramembrane protease Sppl2a is required for B cell and DC development and survival via cleavage of the invariant chain. J Exp Med 210: 23–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bergmann H, Yabas M, Short A, Miosge L, Barthel N, Teh CE, Roots CM, Bull KR, Jeelall Y, Horikawa K et al (2013) B cell survival, surface BCR and BAFFR expression, CD74 metabolism, and CD8‐dendritic cells require the intramembrane endopeptidase SPPL2A. J Exp Med 210: 31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schneppenheim J, Hüttl S, Mentrup T, Lüllmann‐Rauch R, Rothaug M, Engelke M, Dittmann K, Dressel R, Araki M, Araki K et al (2014) The intramembrane proteases signal Peptide peptidase‐like 2a and 2b have distinct functions in vivo . Mol Cell Biol 34: 1398–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Golde TE, Wolfe MS, Greenbaum DC (2009) Signal peptide peptidases: a family of intramembrane‐cleaving proteases that cleave type 2 transmembrane proteins. Semin Cell Dev Biol 20: 225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Friedmann E, Lemberg MK, Weihofen A, Dev KK, Dengler U, Rovelli G, Martoglio B (2004) Consensus analysis of signal peptide peptidase and homologous human aspartic proteases reveals opposite topology of catalytic domains compared with presenilins. J Biol Chem 279: 50790–50798 [DOI] [PubMed] [Google Scholar]
- 22. Friedmann E, Hauben E, Maylandt K, Schleeger S, Vreugde S, Lichtenthaler SF, Kuhn PH, Stauffer D, Rovelli G, Martoglio B (2006) SPPL2a and SPPL2b promote intramembrane proteolysis of TNFalpha in activated dendritic cells to trigger IL‐12 production. Nat Cell Biol 8: 843–848 [DOI] [PubMed] [Google Scholar]
- 23. Fluhrer R, Grammer G, Israel L, Condron MM, Haffner C, Friedmann E, Bohland C, Imhof A, Martoglio B, Teplow DB et al (2006) A gamma‐secretase‐like intramembrane cleavage of TNFalpha by the GxGD aspartyl protease SPPL2b. Nat Cell Biol 8: 894–896 [DOI] [PubMed] [Google Scholar]
- 24. Fleck D, Voss M, Brankatschk B, Giudici C, Hampel H, Schwenk B, Edbauer D, Fukumori A, Steiner H, Kremmer E et al (2016) Proteolytic processing of neuregulin 1 type III by three intramembrane‐cleaving proteases. J Biol Chem 291: 318–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Martin L, Fluhrer R, Reiss K, Kremmer E, Saftig P, Haass C (2008) Regulated intramembrane proteolysis of Bri2 (Itm2b) by ADAM10 and SPPL2a/SPPL2b. J Biol Chem 283: 1644–1652 [DOI] [PubMed] [Google Scholar]
- 26. Dev KK, Chatterjee S, Osinde M, Stauffer D, Morgan H, Kobialko M, Dengler U, Rueeger H, Martoglio B, Rovelli G (2006) Signal peptide peptidase dependent cleavage of type II transmembrane substrates releases intracellular and extracellular signals. Eur J Pharmacol 540: 10–17 [DOI] [PubMed] [Google Scholar]
- 27. Papadopoulou AA, Müller SA, Mentrup T, Shmueli MD, Niemeyer J, Haug‐Kröper M, von Blume J, Mayerhofer A, Feederle R, Schröder B, Lichtenthaler SF (2019) Signal Peptide Peptidase‐Like 2c (SPPL2c) impairs vesicular transport and cleavage of SNARE proteins. EMBO Rep 20: e46451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Voss M, Fukumori A, Kuhn PH, Kunzel U, Klier B, Grammer G, Haug‐Kroper M, Kremmer E, Lichtenthaler SF, Steiner H et al (2012) Foamy virus envelope protein is a substrate for signal peptide peptidase‐like 3 (SPPL3). J Biol Chem 287: 43401–43409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Weihofen A, Lemberg MK, Friedmann E, Rueeger H, Schmitz A, Paganetti P, Rovelli G, Martoglio B (2003) Targeting presenilin‐type aspartic protease signal peptide peptidase with gamma‐secretase inhibitors. J Biol Chem 278: 16528–16533 [DOI] [PubMed] [Google Scholar]
- 30. Schrul B, Kapp K, Sinning I, Dobberstein B (2010) Signal peptide peptidase (SPP) assembles with substrates and misfolded membrane proteins into distinct oligomeric complexes. Biochem J 427: 523–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (2011) Global quantification of mammalian gene expression control. Nature 473: 337–342 [DOI] [PubMed] [Google Scholar]
- 32. Cerra MC, Imbrogno S (2012) Phospholamban and cardiac function: a comparative perspective in vertebrates. Acta Physiol 205: 9–25 [DOI] [PubMed] [Google Scholar]
- 33. Fluhrer R, Martin L, Klier B, Haug‐Kroper M, Grammer G, Nuscher B, Haass C (2012) The alpha‐helical content of the transmembrane domain of the British dementia protein‐2 (Bri2) determines its processing by signal peptide peptidase‐like 2b (SPPL2b). J Biol Chem 287: 5156–5163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hüttl S, Helfrich F, Mentrup T, Held S, Fukumori A, Steiner H, Saftig P, Fluhrer R, Schröder B (2016) Substrate determinants of signal peptide peptidase‐like 2a (SPPL2a)‐mediated intramembrane proteolysis of the invariant chain CD74. Biochem J 473: 1405–1422 [DOI] [PubMed] [Google Scholar]
- 35. MacLennan DH, Kranias EG (2003) Phospholamban: a crucial regulator of cardiac contractility. Nat Rev Mol Cell Biol 4: 566–577 [DOI] [PubMed] [Google Scholar]
- 36. Shambharkar PB, Bittinger M, Latario B, Xiong Z, Bandyopadhyay S, Davis V, Lin V, Yang Y, Valdez R, Labow MA (2015) TMEM203 is a novel regulator of intracellular calcium homeostasis and is required for spermatogenesis. PLoS ONE 10: e0127480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rotgers E, Cisneros‐Montalvo S, Jahnukainen K, Sandholm J, Toppari J, Nurmio M (2015) A detailed protocol for a rapid analysis of testicular cell populations using flow cytometry. Andrology 3: 947–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H et al (2014) Mass‐spectrometry‐based draft of the human proteome. Nature 509: 582–587 [DOI] [PubMed] [Google Scholar]
- 39. Dirami G, Ravindranath N, Achi MV, Dym M (2001) Expression of Notch pathway components in spermatogonia and Sertoli cells of neonatal mice. J Androl 22: 944–952 [DOI] [PubMed] [Google Scholar]
- 40. Gosney R, Liau WS, Lamunyon CW (2008) A novel function for the presenilin family member spe‐4: inhibition of spermatid activation in Caenorhabditis elegans . BMC Dev Biol 8: 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamaguchi F, Yamaguchi K, Tokuda M (2000) Presenilin‐1 protein specifically expressed in Leydig cells with its expression level increased during rat testis development. Int J Biochem Cell Biol 32: 81–87 [DOI] [PubMed] [Google Scholar]
- 42. Murta D, Batista M, Trindade A, Silva E, Henrique D, Duarte A, Lopes‐da‐Costa L (2014) In vivo notch signaling blockade induces abnormal spermatogenesis in the mouse. PLoS ONE 9: e113365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Y, Guan X, Fok KL, Li S, Zhang X, Miao S, Zong S, Koide SS, Chan HC, Wang L (2008) A novel member of the Rhomboid family, RHBDD1, regulates BIK‐mediated apoptosis. Cell Mol Life Sci 65: 3822–3829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang Y, Song W, Li S, Guan X, Miao S, Zong S, Koide SS, Wang L (2009) GC‐1 mRHBDD1 knockdown spermatogonia cells lose their spermatogenic capacity in mouse seminiferous tubules. BMC Cell Biol 10: 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tang T, Li L, Tang J, Li Y, Lin WY, Martin F, Grant D, Solloway M, Parker L, Ye W et al (2010) A mouse knockout library for secreted and transmembrane proteins. Nat Biotechnol 28: 749–755 [DOI] [PubMed] [Google Scholar]
- 46. Aizawa S, Okamoto T, Sugiyama Y, Kouwaki T, Ito A, Suzuki T, Ono C, Fukuhara T, Yamamoto M, Okochi M et al (2016) TRC8‐dependent degradation of hepatitis C virus immature core protein regulates viral propagation and pathogenesis. Nat Commun 7: 11379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Morohashi Y, Kan T, Tominari Y, Fuwa H, Okamura Y, Watanabe N, Sato C, Natsugari H, Fukuyama T, Iwatsubo T et al (2006) C‐terminal fragment of presenilin is the molecular target of a dipeptidic gamma‐secretase‐specific inhibitor DAPT (N‐[N‐(3,5‐difluorophenacetyl)‐L‐alanyl]‐S‐phenylglycine t‐butyl ester). J Biol Chem 281: 14670–14676 [DOI] [PubMed] [Google Scholar]
- 48. Luo W, Grupp IL, Harrer J, Ponniah S, Grupp G, Duffy JJ, Doetschman T, Kranias EG (1994) Targeted ablation of the phospholamban gene is associated with markedly enhanced myocardial contractility and loss of beta‐agonist stimulation. Circ Res 75: 401–409 [DOI] [PubMed] [Google Scholar]
- 49. Nakagawa T, Yokoe S, Asahi M (2016) Phospholamban degradation is induced by phosphorylation‐mediated ubiquitination and inhibited by interaction with cardiac type Sarco(endo)plasmic reticulum Ca(2+)‐ATPase. Biochem Biophys Res Commun 472: 523–530 [DOI] [PubMed] [Google Scholar]
- 50. Teng AC, Miyake T, Yokoe S, Zhang L, Rezende LM Jr, Sharma P, MacLennan DH, Liu PP, Gramolini AO (2015) Metformin increases degradation of phospholamban via autophagy in cardiomyocytes. Proc Natl Acad Sci USA 112: 7165–7170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yokoe S, Asahi M (2017) Phospholamban is downregulated by pVHL‐mediated degradation through oxidative stress in failing heart. Int J Mol Sci 18: E2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kadambi VJ, Ponniah S, Harrer JM, Hoit BD, Dorn GW II, Walsh RA, Kranias EG (1996) Cardiac‐specific overexpression of phospholamban alters calcium kinetics and resultant cardiomyocyte mechanics in transgenic mice. J Clin Invest 97: 533–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Neumann J, Boknik P, DePaoli‐Roach AA, Field LJ, Rockman HA, Kobayashi YM, Kelley JS, Jones LR (1998) Targeted overexpression of phospholamban to mouse atrium depresses Ca2+ transport and contractility. J Mol Cell Cardiol 30: 1991–2002 [DOI] [PubMed] [Google Scholar]
- 54. Darszon A, Nishigaki T, Beltran C, Trevino CL (2011) Calcium channels in the development, maturation, and function of spermatozoa. Physiol Rev 91: 1305–1355 [DOI] [PubMed] [Google Scholar]
- 55. Lawson C, Dorval V, Goupil S, Leclerc P (2007) Identification and localisation of SERCA 2 isoforms in mammalian sperm. Mol Hum Reprod 13: 307–316 [DOI] [PubMed] [Google Scholar]
- 56. Yuan W, Leisner TM, McFadden AW, Clark S, Hiller S, Maeda N, O'Brien DA, Parise LV (2006) CIB1 is essential for mouse spermatogenesis. Mol Cell Biol 26: 8507–8514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rahman MS, Kwon WS, Pang MG (2014) Calcium influx and male fertility in the context of the sperm proteome: an update. Biomed Res Int 2014: 841615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Koolen DA, Vissers LE, Pfundt R, de Leeuw N, Knight SJ, Regan R, Kooy RF, Reyniers E, Romano C, Fichera M et al (2006) A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet 38: 999–1001 [DOI] [PubMed] [Google Scholar]
- 59. Shaw‐Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S, Curley R, Cumming S, Dunn C, Kalaitzopoulos D et al (2006) Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet 38: 1032–1037 [DOI] [PubMed] [Google Scholar]
- 60. Moradalibeigi A, Asgarzade N, Rezaei H (2018) A case of Koolen de vries syndrome or 17q21.31 microdeletion syndrome associated with infertility: a case report. SM J Case Rep 4: 1080 [Google Scholar]
- 61. Stefansson H, Helgason A, Thorleifsson G, Steinthorsdottir V, Masson G, Barnard J, Baker A, Jonasdottir A, Ingason A, Gudnadottir VG et al (2005) A common inversion under selection in Europeans. Nat Genet 37: 129–137 [DOI] [PubMed] [Google Scholar]
- 62. Steinberg KM, Antonacci F, Sudmant PH, Kidd JM, Campbell CD, Vives L, Malig M, Scheinfeldt L, Beggs W, Ibrahim M et al (2012) Structural diversity and African origin of the 17q21.31 inversion polymorphism. Nat Genet 44: 872–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. de Jong S, Chepelev I, Janson E, Strengman E, van den Berg LH, Veldink JH, Ophoff RA (2012) Common inversion polymorphism at 17q21.31 affects expression of multiple genes in tissue‐specific manner. BMC Genom 13: 458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schwenk F, Baron U, Rajewsky K (1995) A cre‐transgenic mouse strain for the ubiquitous deletion of loxP‐flanked gene segments including deletion in germ cells. Nucleic Acids Res 23: 5080–5081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schröder B, Wrocklage C, Hasilik A, Saftig P (2010) Molecular characterisation of “transmembrane protein 192” (TMEM192), a novel protein of the lysosomal membrane. Biol Chem 391: 695–704 [DOI] [PubMed] [Google Scholar]
- 66. Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685 [DOI] [PubMed] [Google Scholar]
- 67. Schagger H, Von JG (1987) Tricine‐sodium dodecyl sulfate‐polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem 166: 368–379 [DOI] [PubMed] [Google Scholar]
- 68. Nguyen TL, Schneppenheim J, Rudnik S, Lullmann‐Rauch R, Bernreuther C, Hermans‐Borgmeyer I, Glatzel M, Saftig P, Schröder B (2017) Functional characterization of the lysosomal membrane protein TMEM192 in mice. Oncotarget 8: 43635–43652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Urny J, Hermans‐Borgmeyer I, Gercken G, Schaller HC (2003) Expression of the presenilin‐like signal peptide peptidase (SPP) in mouse adult brain and during development. Gene Expr Patterns 3: 685–691 [DOI] [PubMed] [Google Scholar]
- 70. Seipold L, Damme M, Prox J, Rabe B, Kasparek P, Sedlacek R, Altmeppen H, Willem M, Boland B, Glatzel M et al (2017) Tetraspanin 3: a central endocytic membrane component regulating the expression of ADAM10, presenilin and the amyloid precursor protein. Biochim Biophys Acta Mol Cell Res 1864: 217–230 [DOI] [PubMed] [Google Scholar]
- 71. Claussen M, Kubler B, Wendland M, Neifer K, Schmidt B, Zapf J, Braulke T (1997) Proteolysis of insulin‐like growth factors (IGF) and IGF binding proteins by cathepsin D. Endocrinology 138: 3797–3803 [DOI] [PubMed] [Google Scholar]
- 72. Zachos C, Blanz J, Saftig P, Schwake M (2012) A critical histidine residue within LIMP‐2 mediates pH sensitive binding to its ligand beta‐glucocerebrosidase. Traffic 13: 1113–1123 [DOI] [PubMed] [Google Scholar]
- 73. Mayer C, Adam M, Glashauser L, Dietrich K, Schwarzer JU, Kohn FM, Strauss L, Welter H, Poutanen M, Mayerhofer A (2016) Sterile inflammation as a factor in human male infertility: involvement of Toll like receptor 2, biglycan and peritubular cells. Sci Rep 6: 37128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Wandernoth PM, Mannowetz N, Szczyrba J, Grannemann L, Wolf A, Becker HM, Sly WS, Wennemuth G (2015) Normal fertility requires the expression of carbonic anhydrases II and IV in sperm. J Biol Chem 290: 29202–29216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wennemuth G, Westenbroek RE, Xu T, Hille B, Babcock DF (2000) CaV2.2 and CaV2.3 (N‐ and R‐type) Ca2+ channels in depolarization‐evoked entry of Ca2+ into mouse sperm. J Biol Chem 275: 21210–21217 [DOI] [PubMed] [Google Scholar]
- 76. Wisniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362 [DOI] [PubMed] [Google Scholar]
- 77. Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix‐assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75: 663–670 [DOI] [PubMed] [Google Scholar]
- 78. Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Vizcaino JA, Csordas A, del‐Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez‐Riverol Y, Reisinger F, Ternent T et al (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44: D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 80 partner repository with the data set identifier PXD011922.