

Abstract
Extracellular vesicles (EVs) have been shown to carry microbial components and function in the host defense against infections. In this study, we demonstrate that Mycobacterium tuberculosis (M.tb) RNA is delivered into macrophage‐derived EVs through an M.tb SecA2‐dependent pathway and that EVs released from M.tb‐infected macrophages stimulate a host RIG‐I/MAVS/TBK1/IRF3 RNA sensing pathway, leading to type I interferon production in recipient cells. These EVs also promote, in a RIG‐I/MAVS‐dependent manner, the maturation of M.tb‐containing phagosomes through a noncanonical LC3 pathway, leading to increased bacterial killing. Moreover, treatment of M.tb‐infected macrophages or mice with a combination of moxifloxacin and EVs, isolated from M.tb‐infected macrophages, significantly lowered bacterial burden relative to either treatment alone. We hypothesize that EVs, which are preferentially removed by macrophages in vivo, can be combined with effective antibiotics as a novel approach to treat drug‐resistant TB.
Keywords: extracellular vesicles, immunotherapy, LC3‐associated phagosome, mycobacterial RNA, Mycobacterium tuberculosis
Subject Categories: Immunology; Membrane & Intracellular Transport; Microbiology, Virology & Host Pathogen Interaction
Introduction
Mycobacterium tuberculosis (M.tb), the causative agent of tuberculosis (TB), has been a major source of human suffering since antiquity. Presently, over 2 billion people are infected by M.tb worldwide, leading to an estimated 10.4 million active TB cases and 1.7 million deaths in 2016 1. As an airborne pathogen, M.tb primarily infects alveolar macrophages which are exposed to various virulence factors and pathogen‐associated molecular patterns (PAMPs). The M.tb PAMPs are detected by host germline‐encoded pattern‐recognition receptors (PRRs) leading to the production of proinflammatory cytokines such as TNF‐α and IL‐1β, which are essential for an effective immune response 2. During an M.tb infection, PRR activation also initiates non‐transcriptional responses such as the induction of phagocytosis and autophagy in infected macrophages 3. However, there is limited knowledge on the intercellular trafficking of M.tb PAMPs and corresponding activation of the host PRR‐dependent pathways in uninfected cells.
Extracellular vesicles (EVs) are membrane‐bound vesicles released by both eukaryotic and prokaryotic cells. These vesicles play an important role in intercellular communication regulating various cellular functions of recipient cells. Based on their origin and size, EVs released by eukaryotic cells are divided into three main categories: exosomes, microvesicles, and apoptotic bodies 4. Previous studies found that M.tb‐infected macrophages release exosomes and microvesicles carrying M.tb PAMPs including mycobacterial proteins, lipids, and nucleic acids. These EV‐carrying M.tb PAMPs may be detected by PRRs on recipient cells to activate or attenuate cellular responses 5, 6, 7, 8, 9, 10. More recently, EVs released by M.tb‐infected human neutrophils were also found to regulate proinflammatory response in recipient cells 11. EVs from M.tb‐infected macrophages trigger the TNF‐α production in THP‐1 human macrophages and naïve mouse bone marrow‐derived macrophages (BMMs) 5, 12. A similar result was detected in Raw 264.7 cells treated with EVs from M. bovis BCG‐infected macrophages 7. In contrast, these vesicles also suppress the expression of major histocompatibility complex (MHC) class II molecules through a TLR2‐dependent pathway in mouse BMMs 13. In the context of the adaptive immune response, M.tb antigens carried in host cell‐derived EVs may be delivered to the antigen processing and presentation pathway in recipient cells. EVs from Mycobacterium bovis BCG‐infected or M.tb culture filtrate protein (CFP)‐pulsed macrophages activate an M.tb Ag‐specific CD4+ and CD8+ T cell response in naïve mice or mice previously vaccinated with M. bovis BCG. The EV‐vaccinated mice were also protected from a low‐dose aerosol M.tb infection 14, 15. The recent identification of mycobacterial RNA within EVs 8 suggests that host RNA sensors may also be activated in EV‐recipient cells.
In the present study, we found that the transport of M.tb RNA to EVs is dependent on the expression of the mycobacterial SecA2 secretion system and that EVs carrying M.tb RNA stimulate IFN‐β production in recipient BMMs. Moreover, EVs also promote LC3‐associated M.tb phagosome maturation in a RIG‐I/MAVS‐dependent pathway. Finally, we found EVs from M.tb‐infected BMMs work synergistically with antibiotics to decrease bacterial load within infected macrophages and following an in vivo mouse infection and do so in a MAVS‐dependent manner.
Results
EVs released by M.tb‐infected macrophages stimulate RIG‐I/MAVS‐dependent type I interferon production in macrophages
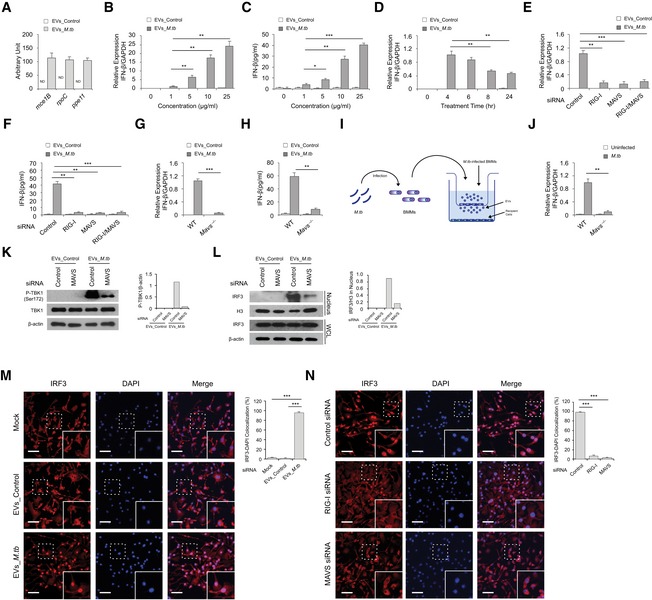
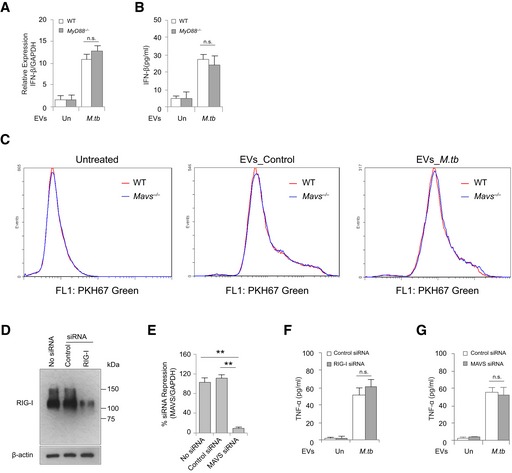
Our previous study identified M.tb RNA in EVs isolated from M.tb‐infected Raw 264.7 cells in vitro 8. As shown in Fig 1A, mycobacterial RNA was also detected in EVs released by mouse BMMs infected with M.tb. However, the functional consequence of this EV‐associated mycobacterial RNA in the context of an M.tb infection was not defined in this earlier study. During a viral infection, viral RNA is an important PAMP in driving type I IFN production in host cells 16. Therefore, we hypothesized that EVs containing M.tb RNA may stimulate the host nucleic acid sensing pathways, triggering type I IFN production in recipient cells. As shown in Fig 1B and C, EVs secreted from M.tb‐infected BMMs induced in naive BMMs a dose‐dependent production of IFN‐β. Maximum IFN‐β mRNA transcription was observed 4 h post‐treatment when using a concentration of 10 μg/ml EVs isolated from M.tb‐infected macrophages (Fig 1D). In contrast, no IFN‐β mRNA induction was detected in cells treated with EVs from uninfected cells (Fig 1D). MAVS and MyD88 are two critical adaptor proteins in host RNA sensing pathways that perceive cytosolic and endosomal foreigner RNA, respectively, and drive type I IFN production 16. To test their roles in EV‐induced type I IFN production, IFN‐β expression was measured in MAVS‐knockdown and MyD88‐deficient BMMs following EV treatment. As shown in Fig 1E and F, when using MAVS‐knockdown BMMs as the recipient cells, EVs isolated from M.tb‐infected macrophages failed to induce IFN‐β production. However, no significant difference in IFN‐β production was seen between MyD88‐deficient and WT BMMs (Fig EV1A and B), suggesting a role for the host cytosolic RNA sensing pathway in EV‐induced type I IFN production. Similarly, a loss of EV‐induced IFN‐β production was also detected in Mavs −/− BMMs (Fig 1G and H). To test whether the decreased IFN‐β production was caused by an attenuated EV uptake in Mavs −/− BMMs, we analyzed the uptake rate of EVs by WT and Mavs −/− cells. As shown in Fig EV1C, Mavs −/− BMMs showed a comparable EV uptake rate as WT BMMs. MAVS is activated following interaction with either of the two cytosolic RNA sensors RIG‐I and MDA5 that recognize foreign RNA 16. The importance of RIG‐I in type I IFN production during a bacterial infection has been assessed in Listeria monocytogenes and M.tb 17, 18. To test whether RIG‐I is also involved in EV‐induced type I IFN production, we measured the IFN‐β production in RIG‐I‐knockdown BMMs treated with EVs from M.tb‐infected macrophages. Similar to the Mavs −/−, knockdown of RIG‐I in BMMs significantly diminished IFN‐β production following treatment with EVs from M.tb‐infected macrophages compared to control siRNA‐treated cells (Figs 1E and F, and EV1D). In contrast, neither RIG‐I nor MAVS knockdown (Fig EV1E) has significant effect on the TNF‐α production in BMMs treated with EVs secreted from M.tb‐infected macrophages (Fig EV1F and G). To better mimic how uninfected macrophages may be exposed to EVs released from infected macrophages in vivo, WT M.tb‐infected BMMs were co‐cultured with uninfected WT and Mavs −/− BMMs using a transwell system (Fig 1I). IFN‐β mRNA abundance in cells of the bottom chamber was measured. In Mavs −/− BMMs, the IFN‐β expression was detectible but significantly lower compared to IFN‐β produced by WT BMMs (Fig 1J).
Figure 1. EVs released by M.tb‐infected macrophages stimulate RIG‐I/MAVS‐dependent type I interferon expression in host cells.
-
AQuantitative RT–PCR analysis of M.tb RNA in EVs from uninfected (EVs_Control) or M.tb‐infected (EVs_M.tb) BMMs. ND, not detected.
-
B, CQuantitative RT–PCR analysis for IFN‐β mRNA (B) or IFN‐β protein (C) levels in wild‐type BMMs 5 and 24 h after treatment with EVs, respectively (1 μg/ml equals approximately 3 × 108 vesicles/ml).
-
D–F(D) IFN‐β mRNA level was quantified in wild‐type BMMs treated with EVs at a concentration of 10 μg/ml for the times indicated. Quantitative RT–PCR analysis for IFN‐β mRNA (E) or IFN‐β protein (F) levels in either WT or MAVS‐ or RIG‐I‐knockdown BMMs treated for 4 h (E) or 24 h (F) with 10 μg/ml EVs from uninfected or M.tb‐infected macrophages. Control: negative control siRNA.
-
G, HSimilar to the above except using WT and Mavs −/− BMMs.
-
ISchematic of transwell assay for measuring IFN‐β induction. BMMs were uninfected or infected with WT M.tb at an MOI of 5. Twenty‐four hours post‐infection, cells were transferred into the top chamber and co‐cultured with naïve BMMs (bottom chamber). The IFN‐β mRNA levels in BMMs (bottom) were quantified by qRT–PCR after 24 h.
-
JTop chamber: WT BMMs; bottom chamber: WT or Mavs −/− BMMs.
-
KWestern blot analysis for phospho‐TBK1 (Ser172) in WT and MAVS‐knockdown BMMs treated for 4 h with EVs from uninfected or M.tb‐infected macrophages. β‐Actin served as a loading control.
-
LWestern blot analysis of IRF3 under the conditions described above for TBK1. Histone H3 (H3) and β‐actin were used as loading controls for nuclear fraction and whole‐cell lysate (WCL), respectively. Densitometry of the Western blots for (K) and (L) is shown.
-
MFluorescence microscopy analysis for nuclear translocation of IRF3 in wild‐type BMMs untreated (Mock) or treated with EVs from uninfected or M.tb‐infected macrophages. Scale bar, 20 μm.
-
NIRF3 nuclear translocation was analyzed in RIG‐I‐ or MAVS‐knockdown BMMs. Scale bar, 20 μm.
Figure EV1. Evaluating the role of MyD88, MAVS and Rig‐1 in EV‐induced cytokine production and EV uptake.
-
AQuantitative real‐time PCR for IFN‐β expression in WT and MyD88 −/− BMMs at 5 h after treatment with EVs isolated from uninfected (Un) or M.tb‐infected (M.tb) macrophages.
-
BSimilar to (A), but IFN‐β protein in BMM culture supernatant was measured using ELISA at 24 h.
-
CEV uptake assay. WT and Mavs −/− BMMs were treated with PKH67 green‐labeled EVs for 4 h, and then, EV uptake rate was determined by flow cytometry.
-
D, EWestern blot (D) and quantitative real‐time PCR (E) to determine siRNA knockdown efficiency for RIG‐I (D) and MAVS (E), respectively, in BMMs.
-
F, GELISA analysis for TNF‐α in the culture supernatant of BMMs pre‐treated with control, RIG‐I (F), or MAVS (G) siRNA, followed by EV treatment for 24 h.
EVs released by M.tb‐infected BMMs activate TBK1 and IRF3 in macrophages
Protein kinase TBK1 and transcriptional regulator IRF3 are two critical factors downstream of the MAVS‐dependent RNA signaling pathway during a viral infection. Following stimulation, IRF3 is phosphorylated by TBK1 and subsequently transported into the nucleus to initiate transcription of type I IFNs 16. The EVs released by M.tb‐infected macrophages induced TBK1 phosphorylation at Ser172 as well as IRF3 nuclear translocation (Fig 1K and L). TBK1 phosphorylation and IRF3 nuclear translocation decreased by 95 and 85%, respectively, in MAVS‐knockdown BMMs when compared to WT BMMs after treatment with EVs from M.tb‐infected macrophages. A minimal response was also observed in BMMs treated with EVs from uninfected macrophages. Immunofluorescence microscopy analysis also showed a high level of IRF3 nuclear translocation in WT BMMs following exposure with EVs from M.tb‐infected macrophages but only limited translocation in RIG‐I‐ or MAVS‐knockdown BMMs (Fig 1M and N).
EV‐induced type I production in BMMs requires the M.tb SecA2 secretion system
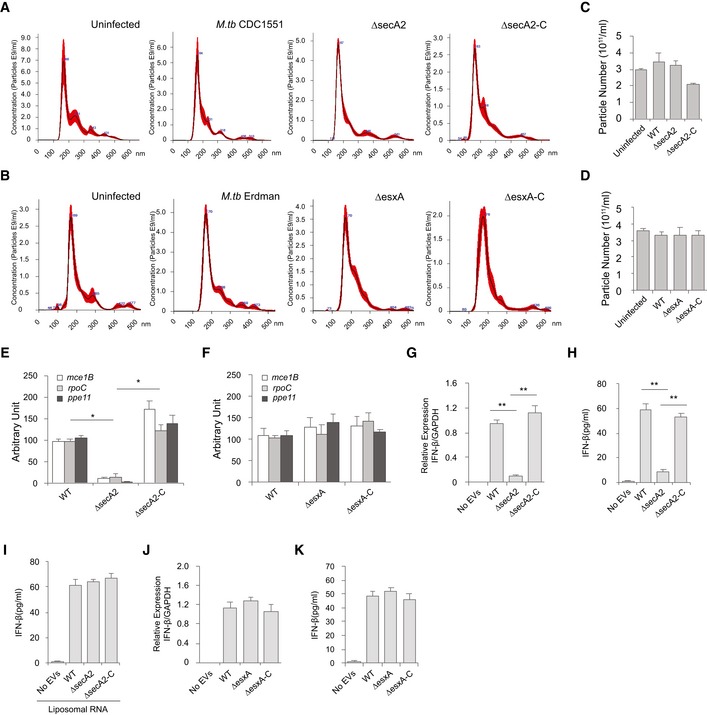
The SecA2 and Esx‐1 protein secretion systems are important for mycobacterial virulence 19, 20. Recently, SecA2 was shown to be required for the secretion of Listeria monocytogenes nucleic acids, while Esx‐1 was required for mycobacterial DNA release into the cytosol of infected cells 17, 21. Both Esx‐1 and SecA2 were required for release of M.tb RNA into the cytosol of infected macrophages 18. A decrease in mycobacterial RNA release was also observed in the supernatant of cultured SecA2‐deficient compared to WT M.tb (Fig EV2A). To determine whether these secretion systems were also required for M.tb RNA trafficking into EVs within infected macrophages, we analyzed EVs released by macrophages infected with a ΔsecA2 or ΔesxA M.tb. As shown in Fig 2A–D, the deficiency of either the secA2 or esxA had no significant effect on the EV biogenesis by infected macrophages. EVs isolated from macrophages infected with the M.tb strains maintain a similar size profile as those from uninfected macrophages (Fig 2A and B). Additionally, a similar EV yield was achieved across all samples (Fig 2C and D). Quantitative RT–PCR was performed to define the amount of M.tb RNA in the isolated EVs. A significant decrease in M.tb RNA was observed in EVs released from macrophages infected with the ΔsecA2 M.tb when compared to vesicles released from cells infected with WT or secA2‐complemented M.tb strains (Fig 2E). In contrast, RNA bioanalyzer analysis indicated that EVs from all three M.tb strain‐infected macrophages carry a similar quantity of total RNA (i.e., both host and mycobacterial RNA species) (Fig EV2B and C), suggesting that the amount of M.tb RNA in EVs is only a small percentage of the total RNA. In additional experiments, it was found that EVs released from macrophages infected with any of the three M.tb strains had similar amount of total proteins (Fig EV2D) and SecA2 deficiency had no effect on the delivery of M.tb lipoarabinomannan and 19‐kDa lipoprotein (LpqH) to EVs during M.tb infection (Fig EV2E). While there is a slight decrease in ΔsecA2 M.tb survival in macrophages relative to WT and secA2‐complemented stain when EVs were harvested (24 h post‐infection) (Fig EV2F), it is clear that this limited decrease in bacterial numbers was not responsible for the sharp decline of mycobacterial RNA in host cell‐released EVs. No significant difference in M.tb RNA abundance was detected among EVs purified from macrophages infected with WT, ∆esxA, or esxA‐complemented M.tb strains (Fig 2F). Furthermore, EVs released from macrophages infected with the ∆secA2 M.tb failed to induce IFN‐β production in recipient BMMs (Fig 2G and H). This lack of IFN‐β production by EV‐treated BMMs was rescued by adding liposome‐encapsulated RNA using RNA that was isolated from M.tb cells, mycobacterial culture supernatant, or EVs released by WT M.tb‐infected macrophages. It was not rescued using liposomes containing RNA isolated from EVs from uninfected or ∆secA2 M.tb‐infected cells (Figs 2I and EV3A–C). No significant difference in IFN‐β production was detected in BMMs treated with EVs from macrophages infected with WT, ∆esxA, or esxA‐complemented M.tb strains (Fig 2J and K).
Figure EV2. Analyzing the affect of SecA2‐deletion on mycobacterial and host RNA/protein/glycolipid transport to EVs.
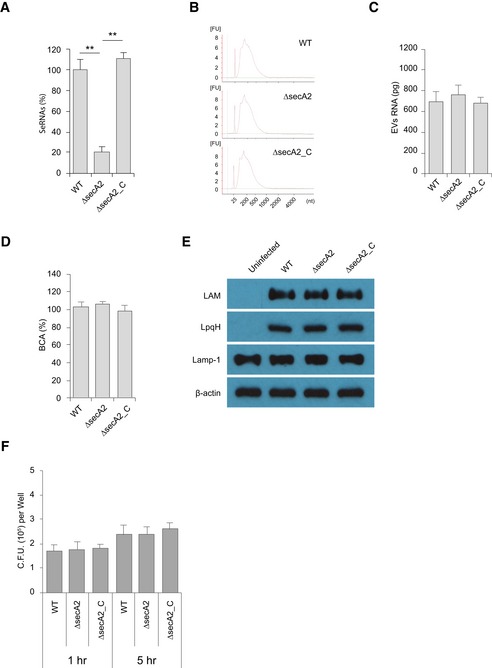
- Released M.tb RNA in the culture supernatant from the same number of mycobacteria.
- RNA bioanalyzer analysis for total RNA isolated from EVs released by WT, ΔsecA2, or secA2‐complemented (ΔsecA2_C) M.tb strains.
- Quantification of EV total RNA analyzed in (B).
- Quantification of total proteins of EVs from various M.tb strain‐infected BMMs.
- Western blot analysis for host and M.tb proteins in EVs.
- M.tb survival assay in wild‐type mouse BMMs.
Figure 2. EV‐induced type I production in macrophages requires the M.tb SecA2 secretion system.
-
ANanoSight analysis for EVs isolated from BMMs infected with WT, ∆secA2, or secA2‐complemented (∆secA2‐C) M.tb CDC 1551 strains. Red highlight indicates the observed variation between 5 independent Nanosight analysis of a given sample.
-
BSimilar to (A), but EVs from cells infected with WT, ∆esxA, or esxA‐complemented (∆esxA‐C) M.tb Erdman strains.
-
CYield of purified EVs from BMMs infected with various M.tb CDC 1551 strains based on NanoSight analysis.
-
DSimilar to (C), but EVs from cells infected with WT, ∆esxA, or esxA‐complemented (∆esxA‐C) M.tb Erdman strains.
-
E, FqRT–PCR analysis for M.tb RNA in EVs from BMMs infected with various M.tb CDC 1551 strains (E) or Erdman strains (F). ND, not detected.
-
G, HqRT–PCR analysis for IFN‐β mRNA (G) or IFN‐β protein (H) levels in WT BMMs treated with EVs for 4 h and 24 h, respectively. The EVs were isolated from BMMs that were infected with the different M.tb CDC 1551 strains.
-
IELISA analysis for IFN‐β secreted by BMMs treated with EVs plus liposomes containing M.tb RNA.
-
J, KqRT–PCR analysis for IFN‐β mRNA (J) or protein (K) levels in WT BMMs treated with EVs for 4 and 24 h, respectively. The EVs were isolated from BMMs that were infected with the different M.tb Erdman strains.
Figure EV3. EV‐induced IFN‐β production by BMMs requires the mycobacterial RNA to be lipid encapsulated.
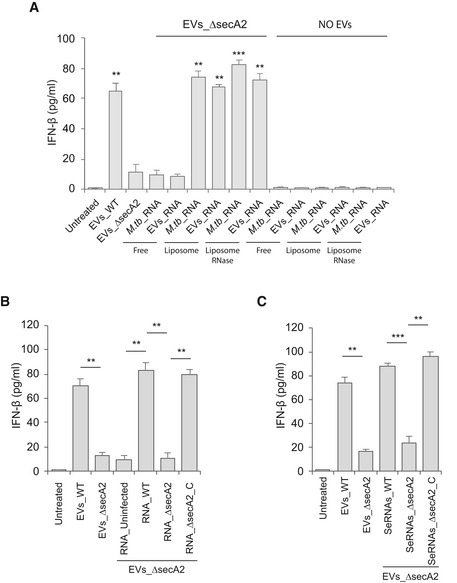
- ELISA analysis for IFN‐β secreted by BMMs treated with EVs from ΔsecA2‐infected macrophages in combination with free or liposomal (with or without RNase treatment) M.tb RNA or RNA from EVs released by WT M.tb‐infected macrophages. EVs_WT and EVs_ΔsecA2, EVs from WT or ΔsecA2 M.tb‐infected macrophages, respectively; M.tb_RNA, RNA from M.tb cells; EVs_RNA, RNA from EVs released by WT M.tb‐infected macrophages. **P < 0.01 and ***P < 0.001, compared to EVs_ΔsecA2, by two‐tailed Student's t‐test.
- Similar to (A), but BMMs were co‐treated with EVs_ΔsecA2 and liposomal RNA from EVs released by uninfected (RNA_Uninfected), WT (RNA_WT), ΔsecA2 (RNA_ΔsecA2), or secA2‐complemented (RNA_ΔsecA2_C) M.tb strains.
- Similar to (B), but BMMs were co‐treated with EVs_ΔsecA2 and liposomal RNA released (SeRNAs) by WT (SeRNAs_WT), ΔsecA2 (SeRNAs_ΔsecA2), or secA2‐complemented (SeRNAs_ΔsecA2_C) M.tb strains.
EVs released by M.tb‐infected BMMs restrict M.tb replication in host cells by activating the M.tb RNA/RIG‐I/MAVS signaling pathway
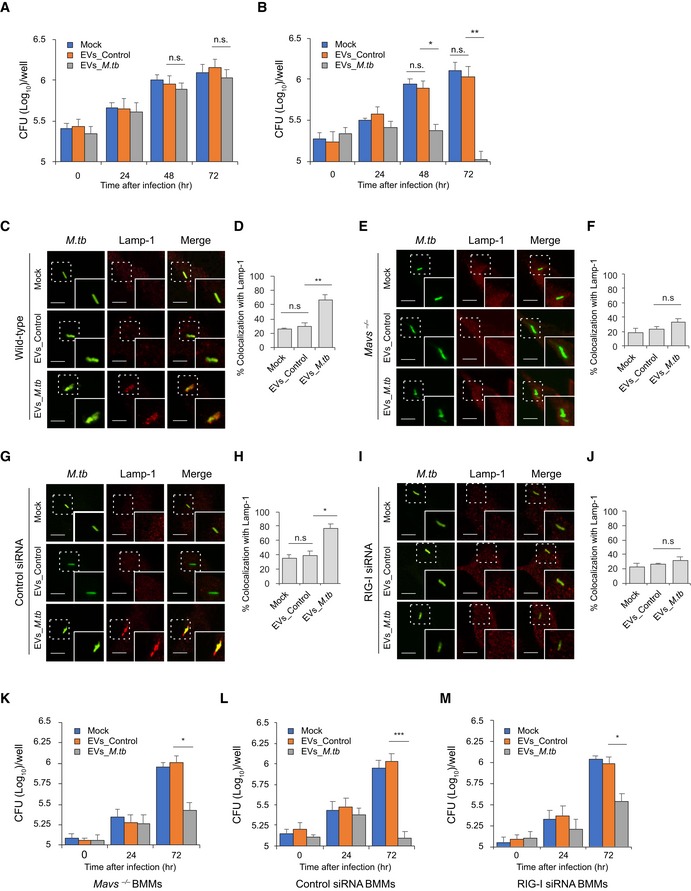
To test the contribution of EVs in the control of M.tb infection, we measured M.tb CFU in BMMs pre‐treated with EVs from uninfected or M.tb‐infected macrophages. EVs from M.tb‐infected macrophages had no significant effect on M.tb replication in BMMs in the absence of IFN‐γ (Fig 3A). In contrast, M.tb numbers were significantly lower in BMMs pre‐treated with EVs released by M.tb‐infected macrophages in combination with IFN‐γ, a key cytokine in controlling an M.tb infection (Fig 3B). A key survival strategy for M.tb is its capacity to inhibit phagosome maturation within infected macrophages 2. To begin evaluating the M.tb compartment post‐EV treatment, we stained for the late endosome/lysosome marker Lamp‐1. We found elevated colocalization of M.tb with Lamp‐1 when infected macrophages were pre‐treated with IFN‐γ plus EVs from M.tb‐infected macrophages relative to IFN‐γ plus EVs from uninfected macrophages (Fig 3C and D). The rate of Lamp‐1 colocalization was comparable in M.tb‐infected BMMs that were untreated or pre‐treated with EVs from uninfected macrophages. Our previous study showed that EVs from both M.tb‐infected or uninfected macrophages carry host Lamp‐1 protein 8. To determine whether Lamp‐1 that colocalized with M.tb (Fig 3C) was derived from EVs, we analyzed M.tb‐Lamp‐1 colocalization in BMMs expressing Lamp‐1‐mCherry. As shown in Appendix Fig S1A and B, a similar percentage of M.tb‐Lamp‐1‐mCherry colocalization was detected in cells pre‐treated with IFN‐γ and EVs from M.tb‐infected macrophages, indicating that the Lamp‐1 observed in Fig 3C originated from host cells and not EVs. To examine whether the host cytosolic RNA sensing pathway plays a role in the EV‐induced phagosome maturation, M.tb trafficking was assessed in Mavs −/− BMMs pre‐treated with EVs. As shown in Fig 3E and F, the elevated M.tb colocalization with Lamp‐1 following IFN‐γ and EV treatment was absent when using Mavs −/− BMMs. Diminished Lamp‐1 colocalization was also seen in RIG‐I‐knockdown M.tb‐infected BMMs (Fig 3G–J). Consistent with the Lamp‐1 colocalization, M.tb burden in Mavs −/− BMMs and RIG‐I siRNA‐treated BMMs was higher relative to control BMMs when cells were pre‐treated with IFN‐γ and EVs from M.tb‐infected macrophages (Fig 3B, K–M). However, even in the absence of the RNA signaling pathway, EVs from M.tb‐infected BMMs reduce M.tb burden within infected cells relative to untreated BMMs, indicating that EVs have additional effects on host macrophages which impact bacterial survival and/or replication.
Figure 3. EVs released by M.tb‐infected macrophages restrict M.tb replication in host cells by activating the M.tb RNA/RIG‐I/MAVS signaling pathway.
-
A, BM.tb CFU in WT mouse BMMs pre‐treated with EVs minus (A) or plus (B) co‐treatment with IFN‐γ. BMMs were treated with EVs from uninfected (EVs_Control) or M.tb‐infected (EVs_M.tb) cells for 0, 24, 48, and 72 h following a 1‐h M.tb infection. Mock, no EV treatment.
-
C–FImmunofluorescence microscopy analysis for colocalization of M.tb (GFP) with lysosome marker Lamp‐1 in WT (C and D) or Mavs −/− (E and F) mouse BMMs. Cells were pre‐treated for 5 h with EVs from uninfected or M.tb‐infected macrophages plus IFN‐γ and then infected with GFP‐expressing M.tb for 24 h prior to immunostaining. (D and F) Quantitative analysis of M.tb colocalization with Lamp‐1 in WT and Mavs −/− mouse BMMs, respectively.
-
G–JImmunofluorescence microscopy as described above except using control siRNA‐treated (G and H) or RIG‐I siRNA‐treated (I and J) BMMs. (H and J) Quantitative analysis of M.tb colocalization with Lamp‐1 in control siRNA‐treated or RIG‐I siRNA‐treated mouse BMMs, respectively.
-
K–MM.tb CFU in infected Mavs −/− (K), control siRNA‐treated (L), or RIG‐I siRNA‐treated (M) mouse BMMs pre‐treated with IFN‐γ plus EVs from uninfected (EVs_Control) or M.tb‐infected (EVs_M.tb) BMMs. CFU was determined immediately after the 1‐h infection or 24 and 72 h post‐infection.
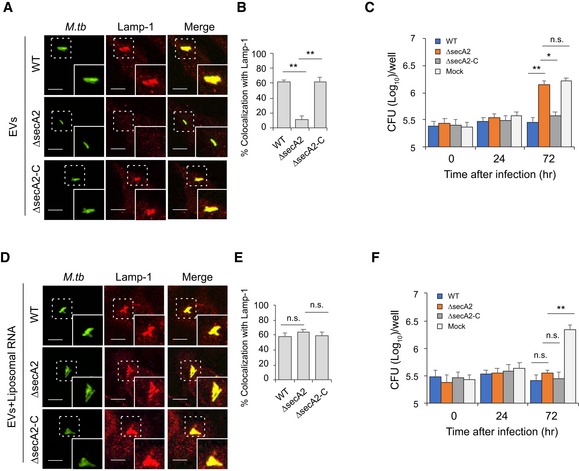
To investigate whether the M.tb RNA in EVs is required for the RIG‐I/MAVS‐dependent M.tb‐killing pathway, WT BMMs were pre‐treated with EVs isolated from macrophages that were infected with WT, ∆secA2, or secA2‐complemented strains. In contrast to EVs from WT or secA2‐complemented strain‐infected macrophages, EVs from ∆secA2 M.tb‐infected macrophages failed to promote phagosome maturation and suppress M.tb replication in BMMs (Fig 4A–C). This deficiency of EVs from ∆secA2 M.tb‐infected macrophages was rescued by adding liposome‐encapsulated RNA, with the RNA isolated from either EVs released from M.tb‐infected BMMs (Fig 4D–F) or using total M.tb RNA (Appendix Fig S2A and B). Adding this RNA resulted in increased phagosome maturation and decreased M.tb survival, indicating that EV‐associated M.tb RNA was driving the anti‐mycobacterial response in recipient cells.
Figure 4. EV‐stimulated M.tb phagolysosome maturation in BMMs when using EVs released from macrophages infected with M.tb‐expressing SecA2.
-
AImmunofluorescence microscopy analysis for colocalization of M.tb with Lamp‐1 in WT BMMs pre‐treated with EVs from macrophages infected with WT, ∆secA2, or secA2‐complemented (∆secA2‐C) M.tb CDC 1551 strains. The cells were pre‐treated with EVs supplemented with recombinant mouse IFN‐γ for 5 h and subsequently infected for 24 h with GFP‐expressing M.tb.
-
BQuantitative analysis for the colocalization of M.tb with Lamp‐1.
-
CM.tb CFU in WT BMMs pre‐treated with recombinant mouse IFN‐γ and EVs from macrophages infected with WT, ∆secA2, or secA2‐complemented (∆secA2‐C) M.tb CDC 1551 strains.
-
D, EAs described in (A) and (B), but liposomes containing RNA isolated from EVs released from BMMs infected with WT M.tb were also added to cells 5 h prior to infection.
-
FSimilar to (C), but liposome‐encapsulated RNA isolated from EVs released from BMMs infected with WT M.tb was included during EV and IFN‐γ pretreatment.
EVs released by M.tb‐infected BMMs activate LC3‐associated phagocytosis pathway in BMMs during M.tb infection
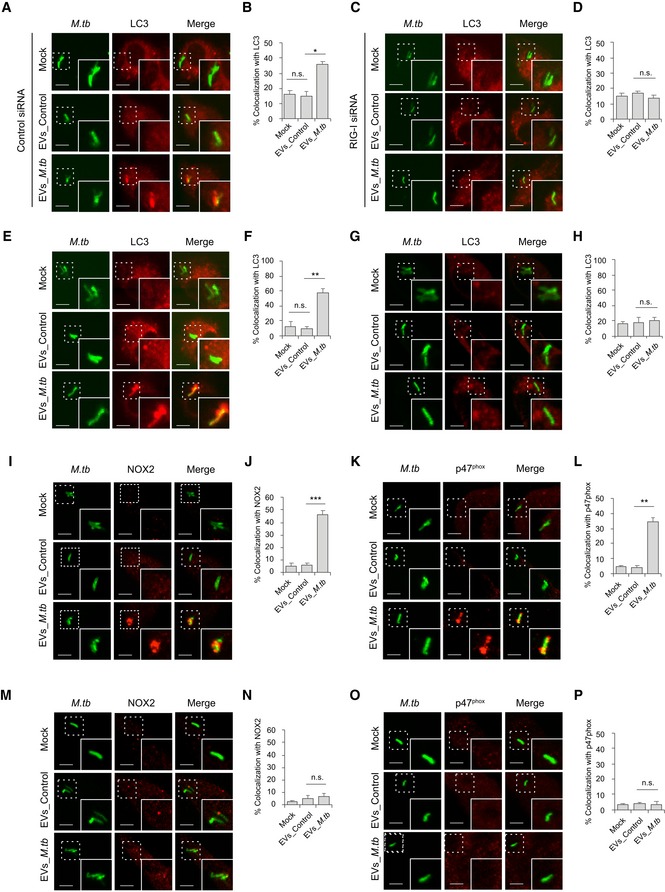
Autophagy plays a key role in the clearance of intracellular pathogens. Recently, it was found that ubiquitin (Ub)‐mediated autophagy contributes to the control of Mycobacterium bovis BCG and M.tb infection in host cells through a TBK1‐regulated pathway 3, 22. The MAVS‐dependent activation of TBK1 by EVs from M.tb‐infected macrophages suggests that these EVs may regulate this autophagic pathway. To test this hypothesis, the colocalization of M.tb with the autophagosome biomarker LC3 and Ub was investigated by immunofluorescence microscopy. As shown in Fig 5, pretreatment of control siRNA‐treated (Fig 5A and B) BMMs with EVs from M.tb‐infected cells plus IFN‐γ significantly increased colocalization of M.tb with LC3 compared to the untreated BMMs or BMMs treated with EVs from uninfected macrophages. This increased phagosome maturation was not seen in either RIG‐I‐knockdown (Fig 5C and D). A similar result was detected between WT (Fig 5E and F) and Mavs −/− BMMs (Fig 5G and H). In contrast, neither EVs from uninfected BMMs nor those from M.tb‐infected BMMs promoted the trafficking of M.tb into Ub‐positive vesicles in BMMs (Appendix Fig S3A). Furthermore, a knockdown of TBK1 had no significant effect on the colocalization of M.tb with LC3‐positive vesicles in WT BMMs treated with IFN‐γ plus EVs (Appendix Fig S3B and C), suggesting an alternative LC3‐dependent autophagic pathway.
Figure 5. EVs released by M.tb‐infected macrophages activate LC3‐associated phagosome maturation in BMMs via a RIG‐I/MAVS‐dependent pathway.
-
A, BImmunofluorescence microscopy and quantitative analysis for colocalization of M.tb with autophagosome marker LC3 in control siRNA‐treated BMMs that were untreated or pre‐treated for 5 h with recombinant mouse IFN‐γ and EVs from uninfected (EVs_Control) or M.tb‐infected (EVs_M.tb) macrophages followed by a 24‐h infection with GFP‐expressing M.tb. Mock, untreated.
-
C, DSimilar to (A), but in RIG‐I siRNA‐treated BMMs.
-
E, FImmunofluorescence microscopy and quantitative analysis for colocalization of M.tb with marker LC3 in WT BMMs that were left untreated or pre‐treated for 5 h with recombinant mouse IFN‐γ and EVs from uninfected (EVs_Control) or M.tb‐infected (EVs_M.tb) macrophages, followed by a 24‐h infection with GFP‐expressing M.tb. Mock, untreated.
-
G, HSimilar to (E and F), but in Mavs −/− BMMs.
-
I–LImmunofluorescence microscopy quantitative analysis for colocalization of M.tb with NOX2 (I and J) and p47phox (K and L) as described above in WT BMMs.
-
M–PSimilar to (I–L), but using Mavs −/− BMMs. NOX2 (M and N) and p47phox (O and P).
LC3‐associated phagocytosis (LAP), a Ub‐independent process, was recently defined as a host defense against bacterial infection 23. After engagement/activation of the PRR by the bacterial PAMPs, NOX2 NADPH oxidase complex was recruited to the phagosomal membrane to stimulate the production of reactive oxygen species (ROS). The generation of ROS promoted recruitment of LC3 to the phagosome, facilitating phagosome–lysosome fusion 24. The NOX2 NADPH oxidase constitutes a membrane‐bound subunit (NOX2/gp91phox, and p22phox) and three cytosolic components (p67phox, p47phox, and p40phox). To test whether LAP is involved in EV‐triggered phagosome–lysosome fusion in M.tb‐infected BMMs, we analyzed the colocalization of NOX2 and p47phox with M.tb. Similar to LC3, pretreatment of BMMs with IFN‐γ and EVs from M.tb‐infected macrophages significantly increased colocalization of M.tb with NOX2 (Fig 5I and J) and p47phox (Fig 5K and L). This effect of EVs relies on the host RNA sensing pathway as this increased colocalization of M.tb with NOX2 (Fig 5M and N) and p47phox (Fig 5O and P) was not observed in Mavs −/− BMMs.
EVs released by M.tb‐infected BMMs synergistically attenuate M.tb survival in BMMs when combined with moxifloxacin
The ability of EVs from M.tb‐infected macrophages to inhibit M.tb survival in host cells suggests these vesicles may have potential in anti‐TB therapy. To test this hypothesis, WT mouse BMMs were first infected with wild‐type M.tb, and 24 h post‐infection, cells were treated with a combined regimen consisting of IFN‐γ, EVs from M.tb‐infected macrophages, and moxifloxacin, a key antibiotic against MDR‐TB. As shown in Fig 6A, an EV–moxifloxacin combination significantly increased M.tb trafficking to a Lamp‐1‐positive compartment compared to EVs or moxifloxacin alone. In contrast, in Mavs −/− BMMs, EVs from M.tb‐infected macrophages failed to enhance the effect of moxifloxacin as a similar number of M.tb colocalized with Lamp‐1 in cells treated with moxifloxacin alone compared to the EV–moxifloxacin combination (Fig 6B). Moreover, moxifloxacin combined with EVs from M.tb‐infected macrophages also resulted in increased trafficking of M.tb to LC3 (Fig 6C)‐, NOX2 (Fig 6D)‐, and p47phox (Fig 6E)‐positive compartments when compared to moxifloxacin or EV treatment alone. Additional studies indicated that the effect of these EVs on M.tb trafficking to LC3 (Fig 6F)‐, NOX2 (Fig 6G)‐, and p47phox (Fig 6H)‐positive compartments was MAVS dependent. An effect on M.tb survival was also observed within infected BMMs as EVs from M.tb‐infected macrophages significantly enhanced the efficacy of moxifloxacin (Fig 6I). Consistent with the Lamp‐1 colocalization results, no difference in bacterial load was detected in Mavs −/− BMMs between moxifloxacin treatment alone and drug plus EVs (Fig 6J). Similar to the EV pretreatment studies, there was a similar level of colocalization between Ub and M.tb in untreated BMMs compared to post‐exposure treatments with EVs (Appendix Fig S4A), and no TBK1 involvement was apparent in the delivery of M.tb to a LC3‐positive compartment (Appendix Fig S4B).
Figure 6. EVs released by M.tb‐infected BMMs attenuate M.tb survival in macrophages when combined with moxifloxacin.
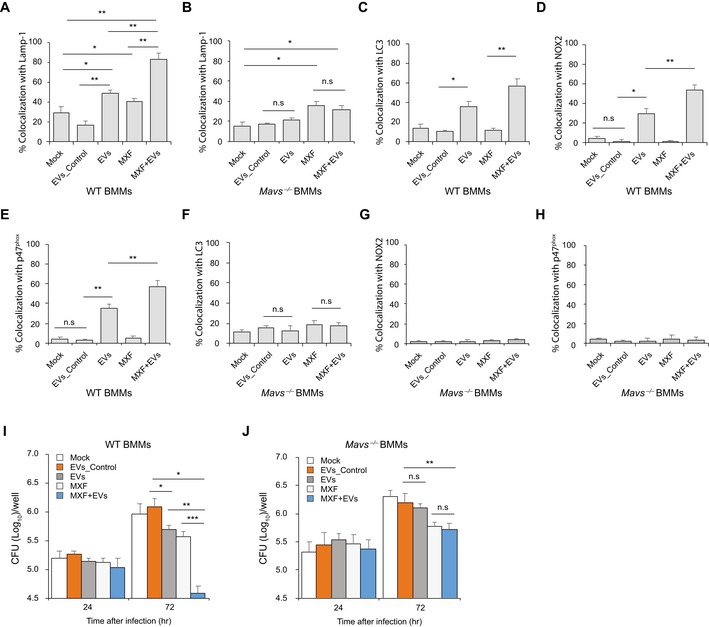
-
A, BQuantitative analysis of immunofluorescence microscopy images for colocalization of M.tb with Lamp‐1 in WT (A) and Mavs −/− (B) BMMs infected with M.tb for 24 h and then treated for an additional 24 h with moxifloxacin (MXF) and/or EVs from M.tb‐infected BMMs (EVs+MXF) in the presence of recombinant mouse IFN‐γ. Mock, no EV or moxifloxacin treatment; EVs_Control, EVs from uninfected BMMs.
-
C–ESimilar to (A), but LC3 (C), NOX2 (D), and p47phox (E) colocalization with M.tb was analyzed.
-
F–HAs described above except Mavs −/− BMMs were used and colocalization of M.tb with LC3 (F), NOX2 (G), and p47phox (H) was quantified.
-
I, JM.tb CFU analysis in WT (I) and Mavs −/− (J) BMMs infected with M.tb for 24 h followed by treatment with EVs, moxifloxacin, or a combination in the presence of IFN‐γ for 24 and 72 h.
EVs released by M.tb‐infected BMMs significantly decreased M.tb survival in mice when combined with moxifloxacin
The decreased bacterial numbers observed in WT BMMs after treatment with moxifloxacin plus EVs suggest that host cell‐derived EVs might be an effective immunotherapy in combination with anti‐TB drugs. To test this hypothesis, WT C57BL/6 mice were low‐dose aerosol‐infected with M.tb, which was followed 3 weeks later with a 2‐week treatment with moxifloxacin and a single‐dose EV treatment given 4 weeks post‐infection (Fig 7A). As shown in Fig 7B, mice treated with moxifloxacin or EVs from M.tb‐infected macrophages or with a combination therapy had smaller granuloma‐like lesions in the lung when compared to untreated mice or those receiving EVs from uninfected macrophages. Consistent with histopathological results, these groups of mice had significantly lower mycobacterial burden in the lung and spleen (Fig 7C). Interestingly, moxifloxacin–EVs combined treatment was more effective than moxifloxacin or EVs alone (Fig 7B and C). To determine whether EV‐based immunotherapy is dependent on MAVS, we performed the combination treatment using M.tb‐infected Mavs −/− mice. Consistent with the in vitro results, EVs from M.tb‐infected macrophages failed to boost moxifloxacin‐based chemotherapy in M.tb‐infected Mavs −/− mice. No significant histopathological difference was seen between the lungs of the different M.tb‐infected groups in Mavs −/− mice (Fig 7D), and a similar M.tb count was seen in the lung and spleen of Mavs −/− mice receiving only moxifloxacin compared to mice treated with the combination of moxifloxacin and EVs (Fig 7E). Cytokine levels were also affected by EVs as higher levels of IFN‐β were found in the serum of M.tb‐infected mice following treatment with EVs from M.tb‐infected macrophages (Fig 7F). This EV‐stimulated IFN‐β production in mice was dependent on MAVS (Fig 7G). Finally, EVs from M.tb‐infected macrophages also induced increased levels of TNF‐α and IL‐1β production in M.tb‐infected mice via a MAVS‐independent pathway but had no effect on IFN‐γ production during the infection period (Fig 7F and G).
Figure 7. EVs released by M.tb‐infected macrophages significantly decrease M.tb survival in mice when combined with moxifloxacin.
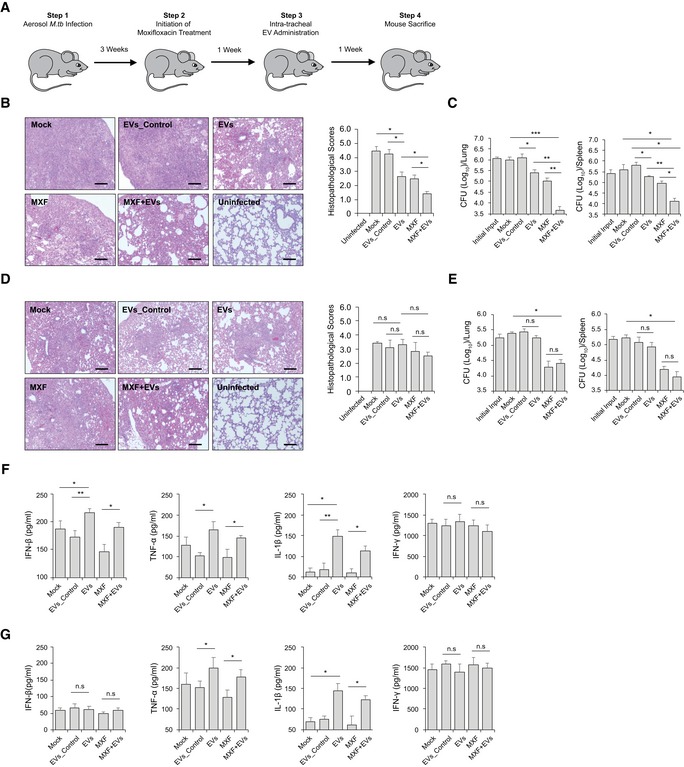
-
ASchematic for EV‐based adjunctive immunotherapy and moxifloxacin‐based chemotherapy in M.tb‐infected mice.
-
B–ERepresentative histopathological analysis for lung sections of WT (B) and Mavs −/− (D) mice that were infected with M.tb and subsequently left untreated (Mock) or treated with EVs from uninfected BMMs (EVs_Control), EVs from M.tb‐infected BMMs (EVs), moxifloxacin (MXF), or a combination of EVs and MXF (EVs+MXF). M.tb CFU in the lung and spleen of WT (C) or Mavs −/− (E) mice treated with EVs, MXF, or a combination of both.
-
F, GELISA analysis for IFN‐β, TNF‐α, IL‐1β, and IFN‐γ protein level in serum isolate from M.tb‐infected WT (F) or Mavs −/− (G) mice treated with EVs, MXF, or a combination of both. Data shown are representative of two independent experiments.
Discussion
Cell‐to‐cell communication plays a critical role in host defense against microbial infections. For intracellular pathogens, communication between infected host cells and cells of the immune system is mediated through cell–cell contact or release of soluble factors by the infected cell including cytokines, chemokines, and various inflammatory mediators. Recently, EVs are recognized as key players in intercellular communication and may transfer pathogen‐derived nucleic acids and proteins to bystander cells. However, there remains limited information on how these EVs modulate the host response to infection 4. Previous studies from our laboratory and others have begun to characterize the role of EVs in intercellular communication during an M.tb infection using both infected macrophages and mouse infection models. For example, EVs from M.tb‐infected macrophages induce the production of multiple cytokines including TNF‐α in recipient cells through a MyD88‐dependent pathway 5, 7, 12. In the present study, we found that EVs containing M.tb RNA may deliver bacterial nucleic acids into uninfected cells, leading to the activation of the RIG‐I/MAVS‐dependent RNA sensing pathway. Together these data suggest that various M.tb PAMPs or host signal molecules are carried in EVs from M.tb‐infected macrophages, and these molecules dictate the effect of EVs on recipient cells. However, the lack of a suitable animal model that is impaired or deficient in EV biogenesis has hampered the in vivo studies to address the positive or negative effect of EVs on infection.
Our study indicates that M.tb RNA released during a macrophage infection requires expression of the mycobacterial SecA2 protein. Unlike SecA1, SecA2 is dispensable for growth and exports only a limited number of proteins. These SecA2‐dependent secreted proteins are involved in bacterial pathogenesis and cellular responses to environmental stress 19. The SecA2 protein has been identified in all mycobacterial strains and some Gram‐positive bacteria including Bacillus, Clostridium, Corynebacterium, Listeria, Staphylococcus, and Streptococcus species 25. In L. monocytogenes, the deficiency of the SecA2 protein significantly decreases bacterial RNA release during bacterial culture 17. In M.tb, we also found that the SecA2 protein is critical for mycobacterial RNA release into the cytosol of infected macrophages and during growth in culture media 18. These results suggest that the secretion of bacterial RNA into the extracellular environment might exist as a ubiquitous pathway for bacteria expressing a SecA2 secretion system. Moreover, although we only evaluated the transfer of bacterial RNA to EVs during the course of an M.tb infection, it is possible that the intercellular transfer of bacterial RNA via host cell‐derived EVs may also be observed for other pathogens that express a SecA2 expression system.
EVs from M.tb‐infected macrophages promoted phagosome maturation in M.tb‐infected macrophages when used as pretreatment agent or after an M.tb infection, leading to reduced mycobacterial replication. Our results also found that EVs from M.tb‐infected macrophages trigger M.tb‐containing phagosome maturation through a LC3‐associated pathway 26. LAP represents an alternative autophagy‐dependent antimicrobial pathway in host cells, where LC3‐modified vesicles fuse with lysosomes, promoting microbial degradation 26. Unlike classical autophagy, LAP is a Ub‐independent process and only utilizes a subset of autophagy machinery components for the modification of microbe‐containing vesicles by the LC3‐conjugation system 27, 28. As an established intracellular bacterial pathogen, M.tb has evolved an inhibitory mechanism for evading LAP through release of CpsA, a LytR‐CpsA‐Psr (LCP) domain‐containing protein that may interfere with the recruitment of NOX2 NADPH oxidase to M.tb‐containing phagosomes 29. Nevertheless, our data indicate that this M.tb‐mediated inhibition can be overcome by treating infected macrophages with EVs isolated from M.tb‐infected macrophages and IFN‐γ. Both signals are needed as EVs or IFN‐γ alone failed to induce phagosome maturation through a LC3‐associated pathway. Recently, EVs from M.tb‐infected human neutrophils were found to promote M.tb‐associated autophagy in human macrophages by triggering superoxide anion production and TLR2/6 signaling pathway 11. It is presently unclear how the EVs from M.tb‐infected neutrophils stimulate reactive oxygen species and whether classic or alternative autophagic pathway is activated by EVs. Nevertheless, these studies, likes ours, indicate the involvement of host cell‐derived EVs in the activation of autophagic pathway. Interestingly, EV‐mediated LC3 conjugation of M.tb‐containing phagosomes requires the host RIG‐I/MAVS cytosolic RNA sensing pathway. Our study highlights a previously undefined role for the host RNA sensing pathways in noncanonical LC3‐associated phagosome maturation in host cells during the course of an M.tb infection.
Drug‐resistant TB is becoming a major threat in the global TB control 1. Globally in 2016, MDR/RR‐TB was diagnosed in an estimated 4.1% of new cases and about 19% of previously treated cases. Among these, approximately 6.2% of cases were XDR‐TB. An estimated treatment success rate for MDR/RR‐TB and XDR‐TB was 54 and 30%, respectively. Treatment for MDR/RR‐TB and XDR‐TB requires a longer therapeutic duration with less effective, more expensive, and more toxic drugs, leading to a higher rate of treatment failure and mortality. To stop the global spread of MDR/RR‐TB and XDR‐TB, new anti‐TB drugs or combined regimens are urgently needed. Recently, a combined therapeutic strategy consisting of an adjunct immunotherapy and anti‐mycobacterial drugs has been proposed and investigated 30. The agents most commonly used in TB immunotherapy include various immune mediators such as all‐trans retinoic acid, which is known to deplete myeloid‐derived suppressor cells as well as increase expression of CD1d on antigen‐presenting cells. When all‐trans retinoic acid in combination with the CD1b ligand alpha‐galactosylceramide was administered to mice along with antibiotics, there was significant improvement in bacterial clearance and lower relapse rates than when treated with isoniazid, rifampicin, and pyrazinamide alone 31. Other immune‐based therapies have targeted various pathways that are responsible for driving a host inflammatory response following a mycobacterial infection with the goal of promoting the right balance between too little and too much inflammation 32. Such host targets include inhibiting PGE2 production which is associated with increased IL‐10 production. Recent work has also focused on ways to stimulate angiogenesis promoting increased blood supply which will allow for more efficient drug penetration and increase the access of host immune cells to the granuloma 33.
In the present study, we investigated an alternative approach that consists of an EV‐based immunotherapy combined with a mycobacterial antibiotic. Unlike the agents investigated previously, the EVs derived from M.tb‐infected host cells will have a more limited target cell population as prior studies indicate a predisposition for EV uptake by macrophage and DCs 5. Our present data support the uptake of EVs by macrophages and DCs but not T cells or neutrophils when administered by intratracheal injection (Fig EV4). Targeting mycobacterial PAMPs and antigens into M.tb‐infected macrophages or uninfected host cells may trigger anti‐mycobacterial pathways such as LAP and provide M.tb antigens for activation of an acquired immune response 5, 29. We found that EVs containing M.tb PAMPs such as mycobacterial RNA are able to elicit an effective anti‐mycobacterial response in macrophages. This suggested that EVs may promote clearance in vivo and provide additional benefit to antibiotic treatment. Indeed, we observed both decreased bacterial load and limited lung pathology in M.tb‐infected mice treated with EVs and moxifloxacin compared to either alone. Previous studies have supported this concept that EVs, which when targeted to specific cell populations, can have significantly higher therapeutic activity. For example, exosomes isolated from HEK293 cells were pre‐loaded with synthetic let‐7a miRNA, a tumor suppressor. When these exosomes expressed the transmembrane domain of platelet‐derived growth factor receptor fused to the GE11 peptide, they were specifically targeted to xenograft breast cancer cells via a GE11–EGFR interaction resulting in reduced tumor growth in RAG2 −/− mice 34. Our study extends the potential application of EVs as immunotherapeutic agents, especially as an adjunctive therapy for currently drug‐resistant infections caused by intracellular pathogens such as M.tb.
Figure EV4. EV uptake assay.
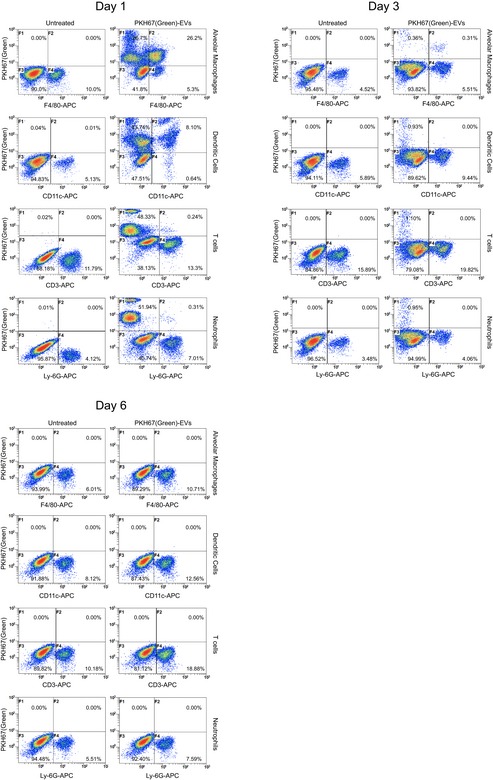
Wild‐type C57BL/6 mice (n = 3 per group) were aerosol‐infected with WT M.tb at a dose of 100–150 CFU/lung. Three weeks post‐infection, PKH67 green‐labeled EVs isolated from WT M.tb‐infected macrophages were intratracheally injected into mice and the distribution of labeled EVs in different cell types of bronchoalveolar lavage (BAL) was determined by flow cytometry. Alveolar macrophages, APC‐F4/80; DCs, APC‐CD11c; T cells, APC‐CD3; neutrophils, APC‐Ly‐6G. Data shown are representative of two independent experiments.
In summary, we found that the presence of M.tb RNA in EVs released from infected macrophages is dependent on the bacteria's SecA2 secretion system. Further, these EVs carrying M.tb RNA can activate the host RIG‐I/MAVS/TBK1/IRF3 RNA sensing signaling pathway in recipient macrophages, leading to the production of type I IFNs. Additionally, a RIG‐I/MAVS‐dependent phagosome maturation is induced by EVs from M.tb‐infected macrophages, resulting in an increased trafficking of M.tb into LC3‐ and Lamp‐1‐positive vesicles and increased bacterial killing. Finally, we found that EVs can synergize with TB antibiotics to promote bacterial clearance and limit lung pathology suggesting a novel immunotherapeutic approach to treat drug‐resistant M.tb.
Materials and Methods
Mice
Wild‐type C57BL/6 and MyD88 −/− mice have been described previously 5. Mavs −/− mice on a C57BL/6 background were obtained as a generous gift from Dr. Stanley Perlmanb (University of Iowa, USA) 35. All mice were housed and bread at the institutional animal facility under specific pathogen‐free conditions. Mice were housed with standard housing in cages containing 3–5 mice per cage. The University of Notre Dame is accredited through the Animal Welfare Assurance (#A3093‐01). All animal experiments were approved by the Institutional Animal Care and Use Committees (IACUCs) of University of Notre Dame.
Bacterial strains
All M.tb strains were grown in Middlebrook 7H9 broth (Cat. No. 271310; BD) supplemented with 10% (v/v) Middlebrook oleic acid–albumin–dextrose–catalase (OADC) (Cat. No. 211886; BD) and 0.2% glycerol until mid‐exponential phase, and washed with complete medium for macrophages or ddH2O plus 0.05% Tween‐80 when required.
Cell culture
Bone marrow‐derived macrophages (BMMs) were isolated from wild‐type C57BL/6, Mavs −/−, or MyD88 −/− mice (female, 6–8 weeks) as described previously 36, and cells were grown in DMEM supplemented with 10% (v/v) heat‐inactivated FBS, 20% L929 cell‐conditioned medium as a source of macrophage colony‐stimulating factor, and 100 U/ml penicillin and 100 U/ml streptomycin (SV30010; HyClone) at 37°C and 5% CO2.
siRNA transfection
Mouse BMMs (3 × 105 cells/well) were transfected with AllStars Negative Control siRNA (Cat. No. 1027280; Qiagen), RIG‐I (5′‐GAAGCGUCUUCUAAUAAUU‐3′), MAVS (5′‐GAUCAAGUGACUCGAGUUU‐3′ and 5′‐GGACCAAAUAGCAGUAUCA‐3′), and TBK1 (SMARTpool: ON‐TARGETplus Tbk1 siRNA; Dharmacon) siRNA oligos (25 pmol/3 × 105 cells) in 24‐well plates using Lipofectamine 2000 (Cat. No. 11668‐027; Invitrogen) following the manufacturer's protocol. The transfected cells were cultured in BMM complete medium for 48 h before use.
Macrophage‐derived EV isolation
BMMs were infected with various M.tb strains at an MOI of 5 for 4 h and washed with pre‐warm PBS (1×) three times to remove the remaining M.tb. Infected cells were incubated in DMEM supplemented with 10% EV‐free FBS for an additional 72 h, and exosome‐enriched EVs were isolated as described previously 15. Isolated EVs were quantified using BCA protein assay and the NanoSight LM10 (Malvern Panalytical, UK). EVs were pre‐treated with RNase A and DNase I before use.
Survival assay of M.tb strains in BMMs
For EV pretreatment experiments, BMMs were treated with EVs at 10 μg/ml for 5 h and subsequently infected with M.tb strains at an MOI of 5 for 1 h at 37°C and 5% CO2. The cells were then washed with complete BMM medium three times and further incubated for another 24, 48, and 72 h at 37°C and 5% CO2. Finally, cells were washed with pre‐cold PBS 3× and lysed in 0.05% SDS. A series of dilution of cell lysates in PBS (1×) were added onto 7H11 agar plates (Cat. No. 7244A; Acumedia) supplemented with 10% (v/v) OADC and 0.2% glycerol. Plates were incubated at 37°C for 3–4 weeks until counting. For EV and moxifloxacin treatment post‐M.tb infection, BMMs were first infected with M.tb for 1 h at 37°C and 5% CO2 and then washed with complete BMM medium three times. The cells were incubated for 24 h at 37°C and 5% CO2 before treatment with EVs at 5 μg/ml and moxifloxacin at 1.0 μg/ml for 24 and 72 h at 37°C and 5% CO2 in the presence of recombinant mouse IFN‐γ (200 units, Cat. No. 14‐8311‐63; Invitrogen).
Transwell assay
Wild‐type BMMs were infected with WT M.tb strain at an MOI of 5 for 1 h at 37°C and 5% CO2 and then washed with complete BMM medium three times. Cells were incubated for another 24 h at 37°C and 5% CO2 before transferring into transwell inserts (pore size, 0.4 μm; Cat. No. 3413; Corning) that were subsequently co‐incubated with WT or Mavs −/− BMMs pre‐seeded in the lower compartment. The IFN‐β mRNA level within BMMs in the lower chamber was analyzed by qRT–PCR.
RNA purification
To determine mycobacterial RNA in macrophage‐derived EVs, total RNA in EVs was isolated using mirVana™ miRNA Isolation Kit (AM1560; Invitrogen). For IFN‐β mRNA measurement, BMMs were treated with isolated EVs at 37°C and 5% CO2 for various times as required, and then, total cellular RNA was purified using Qiagen RNeasy Plus Mini Kit (Cat. No. 74136; Qiagen).
qRT–PCR
RNAs were initially treated with DNase I (Cat. No. 18068015; Invitrogen) following the manufacturer's instructions. For mycobacterial RNA in EVs, cDNA was synthesized with AMV Reverse Transcriptase (Cat. No. M0277; NEB) and a mixture of M.tb reverse primers: mce1B: Forward, 5′‐GAGATCGGCAAGGTCAAAGC‐3′, Reverse: 5′‐GCGGTCGTGGACTGATACAA‐3′; rpoC: Forward, 5′‐ATGGTGACCGGGCTGTACTA‐3′, Reverse: 5′‐CGCTTCGGCCGGCGAAGA‐3′; and ppe11: Forward, 5′‐CGGCACCGCAAGCAACGAG‐3′, Reverse: 5′‐GCGGTCCCAAGTTCCCAAGT‐3′. For IFN‐β analysis, Forward, 5′‐TCCGAGCAGAGATCTTCAGGAA‐3′; Reverse: 5′‐TGCAACCACCACTCATTCTGAG‐3′, cDNA was synthesized using AMV Reverse Transcriptase and Oligo(dT)20 primer. Quantitative PCR was performed using PerfeCTa SYBR® Green SuperMix (Cat. No. 95054; Quantabio) and specific primers on StepOnePlus Real‐Time PCR System (Applied Biosystems). GAPDH (Forward, 5′‐TCGTCCCGTAGACA AAATGG‐3′; Reverse: 5′‐TTGAGGTCAATGAAGGGGTC‐3′) was used as an input control.
Liposome RNA treatment
EV RNA from WT M.tb‐infected BMMs was prepared as described above using mirVana™ miRNA Isolation Kit (AM1560, Invitrogen), and purified RNA was packed using Lipofectamine 2000 (Cat. No. 11668‐027; Invitrogen) following the manufacturer's protocol. WT BMMs were treated with EVs (10 μg/ml) from macrophages infected with WT, ∆secA2, or secA2‐complemented strains in the presence of liposome RNA at a final concentration of 10 pg/ml when required.
Whole‐cell lysates and nuclear fraction preparation
BMMs were treated with EVs from uninfected or M.tb‐infected macrophages at a dose of 10 μg/ml for 4 h at 37°C and 5% CO2 and then washed with pre‐cold PBS three times. Whole‐cell lysates (WCL) were prepared by adding WCL lysis buffer (50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1.0% Triton X‐100) containing 1× protease inhibitor cocktail (P8340; Sigma‐Aldrich) and incubated on ice for 30 min. Nuclear fraction was prepared as described previously 37. Briefly, cells were washed with pre‐cold PBS three times and lysis buffer (10 mM HEPES (pH 7.8), 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA) once. Cell pellets were then resuspended in 500 μl of lysis buffer containing 1× protease inhibitor cocktail and incubated in ice for 15 min before adding 25 μl of 10% NP‐40 and mixed thoroughly. Cell lysates were centrifuged at 1,200 × g for 10 min at 4°C. The pellets (nuclear fraction) were washed with lysis buffer three times and resuspended in WCL lysis buffer and incubated on ice for 30 min before adding SDS‐loading buffer (5×).
Immunoblotting
WCL and nuclear fraction were denatured at 95°C for 10 min and separated by 12.0% SDS–PAGE gel. Proteins were transferred onto nitrocellulose membranes and probed with rabbit anti‐IRF3 (Cat. No. A303‐384A; Bethyl Laboratories Inc.), anti‐TBK1 (Cat. No. 3504; Cell Signaling Technology), anti‐phospho‐TBK1 (Ser172) (Cat. No. 5483; Cell Signaling Technology), anti‐β‐actin (Cat. No. 4970; Cell Signaling Technology), and anti‐Histone H3 (Cat. No. 9717; Cell Signaling Technology) antibodies, followed by goat anti‐rabbit IgG‐HRP (Cat. No. 31460; Thermo Scientific). For M.tb lipid and protein, mouse anti‐LAM mAb (Cat. No. NR‐13811; BEI Resources) and mouse anti‐LpqH mAb (Cat. No. NR‐13792; BEI Resources) were used.
Immunofluorescent microscopy analysis
For EV pretreatment assay, mouse BMMs (1 × 105 cells/well) were seeded onto glass coverslips overnight and then pre‐treated with 10 μg/ml EVs for 5 h before infected by M.tb strains at an MOI of 5 at 37°C and 5% CO2 for 1 h, followed by three washes with complete BMM medium. The infected cells were incubated at 37°C and 5% CO2 for another 24 h and fixed in 4% paraformaldehyde (PFA) at RT for 2 h. The fixed cells were permeabilized in PBS containing 0.2% Triton X‐100 and then blocked in PBS plus 2% FBS for 30 min at RT and incubated in primary rabbit anti‐Lamp‐1 (Cat. No. sc‐5570; Santa Cruz Biotechnology, Inc.), anti‐LC3 (Cat. No. 12741; Cell Signaling Technology), anti‐NOX2 (Cat. No. 611414; BD), anti‐p47phox (Cat. No. SC‐17844; Santa Cruz Biotechnology), or mono‐ and polyubiquitinylated conjugates monoclonal antibody (FK2) (Cat. No. BML‐PW8810‐0100; Enzo Life Sciences) for 1 h at RT. The cells were then washed with PBS three times and incubated with Cy3‐conjugated donkey anti‐rabbit (Cat. No. 711‐165‐152; Jackson ImmunoResearch) or Texas Red‐conjugated goat anti‐mouse (Cat. No. T‐6390; Invitrogen) IgG secondary antibody for 1 h at RT. The coverslips were washed in PBS three times and mounted onto the glass slides. Alternatively, wild‐type BMMs were transfected with LAMP1‐mCherry‐expressing plasmid (kindly provided by Dr. Haoxing Xu, University of Michigan) using Lipofectamine 2000. Twenty‐four hours post‐transfection, cells were infected with GFP‐expressing M.tb and fixed with PFA as described above. For post‐exposure treatment, BMMs were infected with M.tb at an MOI of 5 at 37°C and 5% CO2 for 1 h before being washed with complete BMM medium three times. After 24 h, M.tb‐infected cells were treated with EVs (5 μg/ml), moxifloxacin (1.0 μg/ml), or a combination for another 24 h before immunostaining. To determine IRF3 localization in the nucleus, BMM cells were treated with EVs at 37°C and 5% CO2 for 4 h and then probed using rabbit anti‐IRF3 antibody and Cy3‐conjugated goat anti‐rabbit IgG secondary antibody as described above. The nuclei of cells were stained with DAPI. The slides were analyzed using Nikon C2+ confocal laser scanning microscope at Optical Microscopy Core, University of Notre Dame. For quantitative analysis, at least 100 cells per condition were counted in three independent areas of slides.
Combination treatment with EVs and moxifloxacin in M.tb‐infected mice
Mavs −/− and wild‐type C57BL/6 mice (8–10 weeks old, female) were infected with wild‐type M.tb H37Rv via a low‐dose aerosol infection, 100–150 CFUs in the lung, using a Glas‐Col Inhalation Exposure System (Glas‐Col, Terre haute, IN) as described previously 38. Mice were housed in disposable cages (1–3 mice/cage) using standard bedding with no restriction on access to sterile food or water. M.tb input in the lung of mice was determined at day 1. Three weeks post‐infection, infected mice were treated by oral gavage with moxifloxacin at a dose of 50 mg/kg daily 6 days per week for 2 weeks, and one dose of EVs (5 μg/mouse, in 50 μl PBS) was administered intratracheally at 4 weeks post‐M.tb infection as described previously 39. After the treatment, mouse serum was harvested via cardiac puncture and prepared using BD Microtainer Serum Separator Tube (BD), and cytokine production was measured by ELISA as described below. In parallel, mouse lungs and spleens were harvested, homogenized, and plated onto Middlebrook 7H11 agar plates, and mycobacterial colonies were counted after 3–4 weeks of incubation at 37°C and expressed as log10 CFUs per organ. For pathological analysis, mouse lung sections were prepared and stained with hematoxylin and eosin (H&E) at the Histology Core Facility of University of Notre Dame and scored as described previously 38. M.tb infections were carried out in the biosafety level 3 laboratory.
EV uptake assay
EVs were labeled using PKH67 Fluorescent Cell Linker Kits (Cat. No. MINI67; Sigma‐Aldrich) following the manufacturer's instructions. For the assay in BMM culture, WT or Mavs −/− BMMs were treated with PKH67‐labeled EVs for 4 h at 37°C and 5% CO2 as described above. For the assay in mice, wild‐type mice were infected with WT M.tb as described above. Three weeks post‐infection, mice were intratracheally injected with PKH67‐labeled EVs (5 μg/mouse, in 30 μl PBS) from M.tb‐infected macrophages. At days 1, 3, and 6, mouse bronchoalveolar lavage (BAL) was harvested and isolated single cells were labeled using cell type‐specific surface biomarkers and analyzed by the Cytomics FC 500 Flow Cytometer (Beckman Coulter): alveolar macrophages, APC anti‐mouse F4/80 Ab (Cat. No. 123115; Biolegend); DCs, APC anti‐mouse CD11c Ab (Cat. No. 117309; Biolegend); T cells, APC anti‐mouse CD3 Ab (Cat. No. 100236; Biolegend); and neutrophils, APC anti‐mouse Ly‐6G Ab (Cat. No. 127613; Biolegend).
ELISA
IFN‐β and TNF‐α levels were measured in BMM culture supernatants 24 h after EV treatments. ELISA was performed using avidin‐HRP (Cat. No. 18‐4100‐94; ebioscience) and TMB (Cat. No. 00‐4201‐56; ebioscience) according to the manufacturer's instructions. For TNF‐α measurement, capture antibody (Cat. No. 14‐7341‐85; ebioscience), detection antibody (Cat. No. 13‐7341‐85; ebioscience), and TNF‐α Standard (Cat. No. 39‐8321‐60; ebioscience) were used. For IFN‐β measurement, capture antibody (Purified anti‐mouse IFN‐β Antibody, Cat. No. 519202; Biolegend), detection antibody (Biotin anti‐mouse IFN‐β Antibody, Cat. No. 508105; Biolegend), and IFN‐β standard (Cat. No. 581309; Biolegend) were used. Mouse IFN‐γ and IL‐1β were measured using IFN‐gamma (Cat. No. 88‐7314‐22; ebioscience) and IL‐1 beta (Cat. No. 88‐7013‐22; ebioscience) mouse ELISA kit, respectively.
Statistical analysis
Statistical analysis was performed to determine differences between groups by two‐tailed Student's t‐tests using GraphPad Prism software (version 5.04; GraphPad Software). P < 0.05 was considered significant.
Author contributions
YC was responsible for the experiments performed in this study and assisted in the writing of the manuscript. JSS aided in the design of the experiments and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1
Acknowledgements
Funds for this work were provided by Grant AI052439 from the National Institute of Allergy and Infectious Diseases. We are deeply grateful to Dr. Stanley Perlman from University of Iowa for providing the MAVS−/− mice. We are also very appreciative to the TARGET Program at the John Hopkins University School of Medicine for providing us the SecA2 CDC1551 mutant.
EMBO Reports (2019) 20: e46613
See also: M Biton et al (March 2019)
References
- 1. WHO WHO | Global tuberculosis report 2017.
- 2. Philips JA, Ernst JD (2012) Tuberculosis pathogenesis and immunity. Annu Rev Pathol 7: 353–384 [DOI] [PubMed] [Google Scholar]
- 3. Watson RO, Manzanillo PS, Cox JS (2012) Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA‐sensing pathway. Cell 150: 803–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schorey JS, Cheng Y, Singh PP, Smith VL (2015) Exosomes and other extracellular vesicles in host–pathogen interactions. EMBO Rep 16: 24–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bhatnagar S, Shinagawa K, Castellino FJ, Schorey JS (2007) Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo . Blood 110: 3234–3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Giri PK, Kruh NA, Dobos KM, Schorey JS (2010) Proteomic analysis identifies highly antigenic proteins in exosomes from M. tuberculosis‐infected and culture filtrate protein‐treated macrophages. Proteomics 10: 3190–3202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walters SB, Kieckbusch J, Nagalingam G, Swain A, Latham SL, Grau GER, Britton WJ, Combes V, Saunders BM (2013) Microparticles from mycobacteria‐infected macrophages promote inflammation and cellular migration. J Immunol 190: 669–677 [DOI] [PubMed] [Google Scholar]
- 8. Singh PP, Li L, Schorey JS (2015) Exosomal RNA from Mycobacterium tuberculosis‐infected cells is functional in recipient macrophages. Traffic 16: 555–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hare NJ, Chan B, Chan E, Kaufman KL, Britton WJ, Saunders BM (2015) Microparticles released from Mycobacterium tuberculosis‐infected human macrophages contain increased levels of the type I interferon inducible proteins including ISG15. Proteomics 15: 3020–3029 [DOI] [PubMed] [Google Scholar]
- 10. Li L, Cheng Y, Emrich S, Schorey J (2018) Activation of endothelial cells by extracellular vesicles derived from Mycobacterium tuberculosis infected macrophages or mice. PLoS ONE 13: e0198337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alvarez‐Jiménez VD, Leyva‐Paredes K, García‐Martínez M, Vázquez‐Flores L, García‐Paredes VG, Campillo‐Navarro M, Romo‐Cruz I, Rosales‐García VH, Castañeda‐Casimiro J, González‐Pozos S et al (2018) Extracellular vesicles released from Mycobacterium tuberculosis‐infected neutrophils promote macrophage autophagy and decrease intracellular mycobacterial survival. Front Immunol 9: 272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Singh PP, Smith VL, Karakousis PC, Schorey JS (2012) Exosomes isolated from mycobacteria‐infected mice or cultured macrophages can recruit and activate immune cells in vitro and in vivo . J Immunol 189: 777–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Singh PP, LeMaire C, Tan JC, Zeng E, Schorey JS (2011) Exosomes released from M. tuberculosis infected cells can suppress IFN‐γ mediated activation of naïve macrophages. PLoS ONE 6: e18564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Giri PK, Schorey JS (2008) Exosomes derived from M. Bovis BCG infected macrophages activate antigen‐specific CD4+ and CD8+ T cells in vitro and in vivo . PLoS ONE 3: e2461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cheng Y, Schorey JS (2013) Exosomes carrying mycobacterial antigens can protect mice against Mycobacterium tuberculosis infection. Eur J Immunol 43: 3279–3290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu J, Chen ZJ (2014) Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol 32: 461–488 [DOI] [PubMed] [Google Scholar]
- 17. Abdullah Z, Schlee M, Roth S, Mraheil MA, Barchet W, Böttcher J, Hain T, Geiger S, Hayakawa Y, Fritz JH et al (2012) RIG‐I detects infection with live Listeria by sensing secreted bacterial nucleic acids. EMBO J 31: 4153–4164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cheng Y, Schorey JS (2018) Mycobacterium tuberculosis‐induced IFN‐β production requires cytosolic DNA and RNA sensing pathways. J Exp Med 215: 2919–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Feltcher ME, Braunstein M (2012) Emerging themes in SecA2 protein export. Nat Rev Microbiol 10: 779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gröschel MI, Sayes F, Simeone R, Majlessi L, Brosch R (2016) ESX secretion systems: mycobacterial evolution to counter host immunity. Nat Rev Microbiol 14: 677–691 [DOI] [PubMed] [Google Scholar]
- 21. Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS (2012) Mycobacterium tuberculosis activates the DNA‐dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11: 469–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pilli M, Arko‐Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB et al (2012) TBK‐1 promotes autophagy‐mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37: 223–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Martinez J, Malireddi RS, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan J‐L, Tan H, Peng J et al (2015) Molecular characterization of LC3‐associated phagocytosis (LAP) reveals distinct roles for Rubicon, NOX2, and autophagy proteins. Nat Cell Biol 17: 893–906 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24. Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MAO, Glogauer M, Grinstein S, Brumell JH (2009) Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci USA 106: 6226–6231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Green ER, Mecsas J (2016) Bacterial secretion systems – an overview. Microbiol Spectr 4: 0012‐2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mitchell G, Isberg RR (2017) Innate immunity to intracellular pathogens: balancing microbial elimination and inflammation. Cell Host Microbe 22: 166–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lam GY, Cemma M, Muise AM, Higgins DE, Brumell JH (2013) Host and bacterial factors that regulate LC3 recruitment to Listeria monocytogenes during the early stages of macrophage infection. Autophagy 9: 985–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hubber A, Kubori T, Coban C, Matsuzawa T, Ogawa M, Kawabata T, Yoshimori T, Nagai H (2017) Bacterial secretion system skews the fate of Legionella‐containing vacuoles towards LC3‐associated phagocytosis. Sci Rep 7: 44795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Köster S, Upadhyay S, Chandra P, Papavinasasundaram K, Yang G, Hassan A, Grigsby SJ, Mittal E, Park HS, Jones V et al (2017) Mycobacterium tuberculosis is protected from NADPH oxidase and LC3‐associated phagocytosis by the LCP protein CpsA. Proc Natl Acad Sci USA 114: E8711–E8720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Uhlin M, Andersson J, Zumla A, Maeurer M (2012) Adjunct immunotherapies for tuberculosis. J Infect Dis 205: S325–S334 [DOI] [PubMed] [Google Scholar]
- 31. Mourik BC, Leenen PJM, de Knegt GJ, Huizinga R, van der Eerden BCJ, Wang J, Krois CR, Napoli JL, Bakker‐Woudenberg IAJM, de Steenwinkel JEM (2016) Immunotherapy added to antibiotic treatment reduces relapse of disease in a mouse model of tuberculosis. Am J Respir Cell Mol Biol 56: 233–241 [DOI] [PubMed] [Google Scholar]
- 32. Kiran D, Podell BK, Chambers M, Basaraba RJ (2016) Host‐directed therapy targeting the Mycobacterium tuberculosis granuloma: a review. Semin Immunopathol 38: 167–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dartois V (2014) The path of anti‐tuberculosis drugs: from blood to lesions to mycobacterial cells. Nat Rev Microbiol 12: 159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ohno S, Takanashi M, Sudo K, Ueda S, Ishikawa A, Matsuyama N, Fujita K, Mizutani T, Ohgi T, Ochiya T et al (2013) Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Mol Ther 21: 185–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Suthar MS, Ramos HJ, Brassil MM, Netland J, Chappell CP, Blahnik G, McMillan A, Diamond MS, Clark EA, Bevan MJ et al (2012) The RIG‐I‐like receptor LGP2 controls CD8(+) T cell survival and fitness. Immunity 37: 235–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roach SK, Schorey JS (2002) Differential regulation of the mitogen‐activated protein kinases by pathogenic and nonpathogenic mycobacteria. Infect Immun 70: 3040–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Flaherty DM, Monick MM, Carter AB, Peterson MW, Hunninghake GW (2002) Oxidant‐mediated increases in redox factor‐1 nuclear protein and activator protein‐1 DNA binding in asbestos‐treated macrophages. J Immunol 168: 5675–5681 [DOI] [PubMed] [Google Scholar]
- 38. Cheng Y, Moraski GC, Cramer J, Miller MJ, Schorey JS (2014) Bactericidal activity of an imidazo[1, 2‐a]pyridine using a mouse M. tuberculosis infection model. PLoS ONE 9: e87483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cheng Y, Liepert LD, Waller J, Leevy WM, Schorey JS (2017) A novel method for intratracheal injection of infectious agents into mice. Lab Anim 51: 530–533 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
Review Process File
Source Data for Figure 1