

Abstract
Genome‐wide studies in tumor cells have indicated that chromatin‐modifying proteins are commonly mutated in human cancers. The lysine‐specific methyltransferase 2C (KMT2C/MLL3) is a putative tumor suppressor in several epithelia and in myeloid cells. Here, we show that downregulation of KMT2C in bladder cancer cells leads to extensive changes in the epigenetic status and the expression of DNA damage response and DNA repair genes. More specifically, cells with low KMT2C activity are deficient in homologous recombination‐mediated double‐strand break DNA repair. Consequently, these cells suffer from substantially higher endogenous DNA damage and genomic instability. Finally, these cells seem to rely heavily on PARP1/2 for DNA repair, and treatment with the PARP1/2 inhibitor olaparib leads to synthetic lethality, suggesting that cancer cells with low KMT2C expression are attractive targets for therapies with PARP1/2 inhibitors.
Keywords: DNA repair, epigenetic regulation, KMT2C, PARPi sensitivity
Subject Categories: Cancer; Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination
Introduction
It is well established that epigenetic dysregulation is an integral component of cancer etiology and progression 1. Therefore, it is not surprising that numerous epigenetic modifiers, such as DNMT3A, EZH2, and the MLL proteins, are frequently found genetically altered in cancer 2, 3. Lysine (K)‐specific methyltransferase 2C (KMT2C, also known as MLL3) belongs to the mixed‐lineage leukemia (MLL) family of histone methyltransferases which methylate the histone 3 tail at lysine 4 (H3K4) 4 as part of the complex proteins associated with Set 1 (COMPASS) complex 5. Although originally identified as oncogenic fusions in leukemia 6, recent genome‐wide mutation studies have revealed frequent, presumably loss‐of‐function, mutations in various members of the MLL family, including MLL2/KMT2B, MLL3/KMT2C, and MLL4/KMT2D in a variety of malignancies, particularly solid tumors 7, 8, 9, 10, 11. Mouse studies have also uncovered a tumor suppressor role for KMT2C in acute myeloid leukemia (AML) 12 and urothelial tumorigenesis 13. Mechanistic studies of KMT2C in normal cells have focused primarily on its role in enhancer regulation 14, 15 by deposition of H3K4me1 marks. Interestingly, recent reports also indicate roles for KMT2C in transcription regulation, which are independent of its H3K4 monomethylation activity on enhancers 16, 17. However, its role in tumorigenesis remains largely undefined.
Bladder cancer is the fifth most common human malignancy and the second most frequently diagnosed genitourinary tumor after prostate cancer 18. The majority of malignant tumors arising in the urinary bladder are urothelial carcinomas. Superficial carcinoma accounts for approximately 75% of the newly diagnosed cases while the remaining 25% represents muscle‐invasive bladder cancer 19. The latter, often originating from superficial carcinoma, is a life‐threatening disease with high metastatic potential. Recent genome‐wide studies on superficial and muscle‐invasive urothelial carcinoma have indicated that epigenetic regulators, including KMT2C, are commonly mutated in both types 11, 20. Here, we show that KMT2C is downregulated in neoplastic tissue in several epithelial cancers including urothelial carcinoma. As expected, KMT2C knockdown leads to epigenetic and expression changes. Of interest, genes involved in DNA damage response (DDR) and DNA repair, particularly homologous recombination (HR)‐mediated DNA repair, are downregulated. This leads to increased DNA damage and chromosomal instability, highlighted by generation of micronuclei and numerical/regional chromosome losses. In our experiments, cells with reduced KMT2C expression are highly dependent on the alternative end‐joining (alt‐EJ) pathway for repair of double‐strand breaks (DSBs), while inhibition of PARP1/2 causes synthetic lethality.
Results
KMT2C is downregulated in human epithelial cancers
Mutational data from published studies show that the majority of KMT2C mutations cluster within the plant homeodomain (PHD) fingers 1–3 located in the N‐terminus of the protein (Catalogue of Somatic Mutations in Cancer—COSMIC). KMT2C PHD fingers 1–3 act as “readers” of the histone methylation status, recognizing monomethylated H3K4 (H3K4me1), while the catalytic Su(var)3‐9, Enhancer of zeste, Trithorax (SET) domain, located in the C‐terminus, is the “writer” that adds methyl‐ groups to complete the methylation process 21. KMT2C is commonly mutated in high‐grade muscle‐invasive urothelial carcinoma 7, in which mutations were recently found equally distributed within the two major subtypes, luminal papillary and basal squamous 11. Little is known, however, about low‐grade/early‐stage tumors, including superficial papillomas. To address this issue, we sequenced the N‐ and C‐terminus of the KMT2C transcript in tumors and matching normal tissues from a cohort of 72 patients diagnosed with superficial or muscle‐invasive urothelial cancer of variable grade 22. We identified mutations primarily within PHD fingers 1–3 (Fig 1A), which showed no statistical preference with respect to grade and stage (mutations were found in 12/43 high grade vs. 4/29 low grade, and 9/32 invasive vs. 7/40 superficial tumors). Interestingly, a recent study on non‐invasive bladder cancer also identified a high frequency (15%) of KMT2C likely loss‐of‐function mutations in non‐invasive bladder cancer 20, indicating that KMT2C inactivation might occur early in carcinogenesis. In our mutation detection study, both frameshift and missense mutations were identified, the vast majority of which are predicted to be damaging (Fig 1A and Table EV1). Recently identified missense mutations within the PHD fingers 1–3 have been shown to disrupt the interaction between KMT2C and BAP1 leading to reduced recruitment of KMT2C to gene enhancers 1. Our KMT2C expression analysis in 104 matched normal/cancer tissue pairs from an expanded bladder cancer patients cohort (n = 138; Appendix Tables S1 and S2) revealed that, in comparison with normal tissues, KMT2C expression is downregulated in the majority of tumors at both the RNA and protein levels (71/104, P < 0.001; Fig 1B and C).
Figure 1. KMT2C downregulation in cancer tissue.
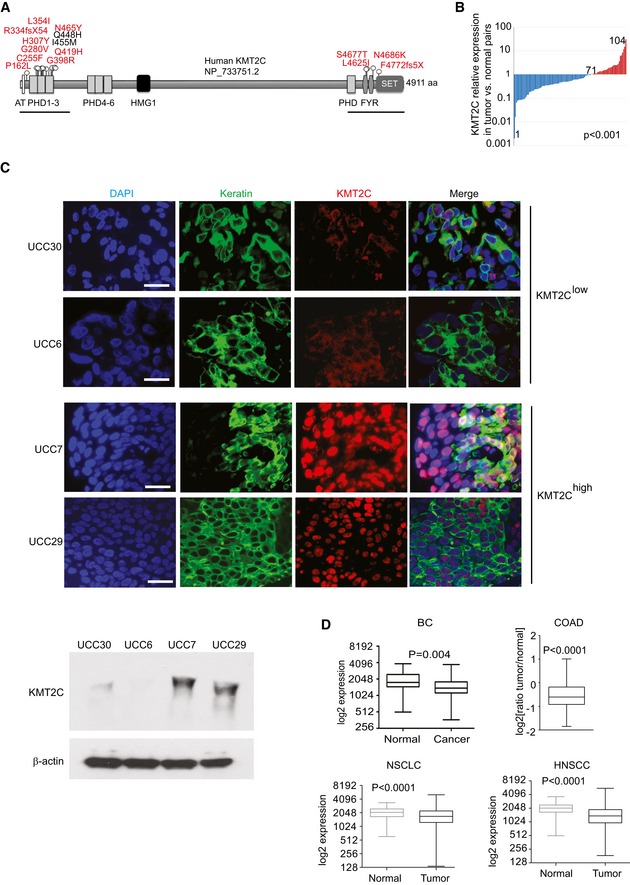
- Comparison of KMT2C expression in cancer/healthy matched tissue pairs (n = 104) of the study cohort. Expression is presented as log(ratio tumor/healthy) in the y‐axis. Data obtained from qRT–PCR analysis. P value calculated by Wilcoxon signed‐rank test.
- Immunofluorescence (top) and Western blot analysis (bottom) against KMT2C on representative human bladder cancers with variable KMT2C transcript levels: 11th, 4th, 93rd, and 79th percentiles for UCC30, 6, 7, and 29, respectively (Appendix Table S2), from the differential expression analysis of the study cohort. Antibodies against KRT5 or KRT20 were used to stain urothelial cells and DAPI as nuclear counterstain. β‐Actin is used as loading control in Western blots. Scale bars indicate 50 μm.
- Comparison of KMT2C expression in human healthy and cancer tissues from bladder cancer (BC, n = 136), colorectal adenocarcinoma (COAD, n = 128), non‐small‐cell lung cancer (NSCLC, n = 341), and head and neck squamous cell carcinoma (HNSCC, n = 174) patients. For NSCLC analysis, separate cohorts from adenocarcinoma and squamous cell carcinoma were combined. Separate analysis of the two NSCLC subtypes (adenocarcinoma and squamous cell carcinoma) yielded the same results. For COAD, the y‐axis is the log2(ratio tumor/normal) of KMT2C expression as assessed with Affymetrix microarray. All expression data were obtained from TCGA through cbioportal.org. P values calculated by Mann–Whitney U‐test. The middle lines inside the boxes indicate the median (50th percentile). The lower and the upper box boundaries represent the 25th percentile and the 75th percentile, respectively. The lower and upper whiskers extend to the lowest and highest values, respectively, within the 1.5× interquartile range (box height) from the box boundaries.
Source data are available online for this figure.
KMT2C is mutated in several epithelial cancers 8, implying a general role as a tumor suppressor. To investigate this hypothesis, we performed a meta‐analysis on publicly available RNA‐seq data from The Cancer Genome Atlas (TCGA) Consortium 23, 24, 25, 26. We found that similarly to bladder cancer (BC), KMT2C is downregulated in comparison with normal tissue in colorectal adenocarcinoma (COAD), non‐small‐cell lung cancer (NSCLC), and head and neck squamous cell carcinoma (HNSCC; Fig 1D). These data indicate that KMT2C downregulation is a rather common event in tumorigenesis in several human epithelial tissues. On the other hand, a recent report 27 and our own meta‐analysis of non‐epithelial cancers with the use of the GEPIA web server 28 indicated that, in comparison with respective healthy tissue, KMT2C is expressed at higher levels in glioblastoma multiforme (GBM), brain lower grade glioma (LGG), diffuse large B‐cell lymphomas (DLBL), acute myeloid leukemia (AML), and sarcomas (SARC; Appendix Fig S1). This is in agreement with the fact that KMT2C truncating mutations account for only 0.6% in these cancer types (2/397, 2/512, 0/41, 3/200, and 2/254 cases, respectively; not shown).
Our meta‐analysis of publicly available DNA methylation data 7 obtained from the MethHC database 29 indicates that two Illumina methylation detection probes (cg17322443 and cg19258062) located within a CpG island (chr7:152435133–152437025, assembly GRCh38/hg38, ENCODE) spanning the KMT2C proximal promoter are subject to DNA methylation in bladder tumor samples, while remaining methylation‐free in normal tissue (Fig EV1A and B), confirming a previously published report 30. More importantly, the same CpG island within the KMT2C proximal promoter is also hypermethylated in tumor samples from COAD, NSCLC, and HNSCC (Fig EV1C). Collectively, these data indicate that both mutational inactivation and transcriptional downregulation via promoter methylation of KMT2C might contribute to reduced activity facilitating tumor development in several epithelial cancers.
Figure EV1. KMT2C promoter methylation in human cancers.
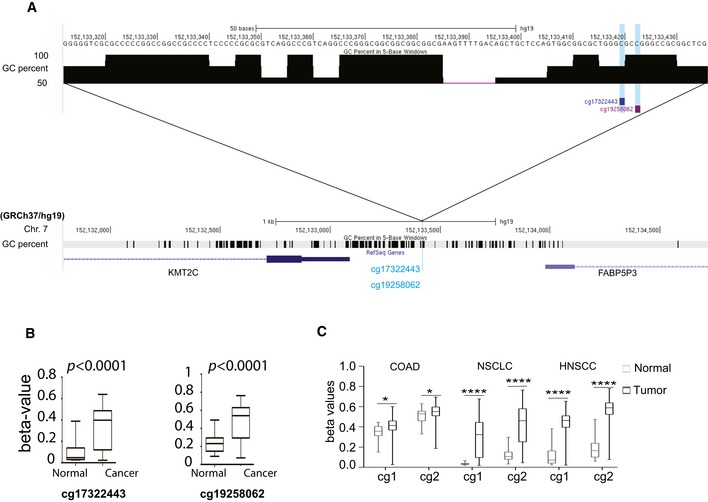
- Schematic of the upstream promoter region of the KMT2C locus indicating the position and sequence of methylation detection probes within the CpG island (located at chr7:152435133–152437025, assembly GRCh38/hg38) that encompasses the KMT2C promoter region.
- Comparison of the methylation levels of the above probes in tumor samples and normal bladder tissue. Methylation data were obtained from TCGA through the MethHC database for n = 21 healthy/tumor pairs. Wilcoxon matched‐pairs signed‐rank test was used.
- Tumor vs. normal paired comparison of the methylation levels in the KMT2C promoter in various cancer types; cg1: cg17322443; cg2: cg19258062. Methylation data were obtained from the MethHC database (Huang et al 29). BC: n = 21, COAD: n = 21, NSCLC: n = 70, HNSCC: n = 50. For NSCLC analysis, separate cohorts from adenocarcinoma and squamous cell carcinoma were combined. Separate analysis of the two NSCLC subtypes yielded the same results. Wilcoxon matched‐pairs signed‐rank test was used. * designates P‐value < 0.05 and ****P‐value < 0.0001.
KMT2C loss affects enhancer activity and gene expression in a subset of genes
To investigate its role in urothelial carcinoma cells, we used two independent shRNAs (KD1/KD2) to knock down KMT2C levels in human BC cell lines (Fig 2A). While the loss of KMT2C activity did not affect cell proliferation or apoptosis (Appendix Fig S2), RNA‐seq experiments in HTB9 cells showed that, directly or indirectly, 3,324 genes were transcriptionally affected upon KMT2C silencing (1.4‐fold and higher change in expression levels). Of those, 1,846 were downregulated while 1,478 were upregulated. Gene ontology (GO) analysis indicated that many of the affected genes are involved in DDR, DNA repair, DNA replication, cell cycle control, and apoptosis, all of which are considered hallmarks of cancer, and are associated with tumor aggressiveness 31 (Fig 2B). In order to study directly the role of KMT2C and to circumvent the lack of chromatin immunoprecipitation (ChIP)‐grade anti‐KMT2C antibodies, we exogenously expressed a Flag‐tagged KMT2C protein (fKMT2C) in HTB9/KD1 cells (Fig 2C).
Figure 2. KMT2C loss leads to extensive epigenetic changes in human bladder cancer cells.
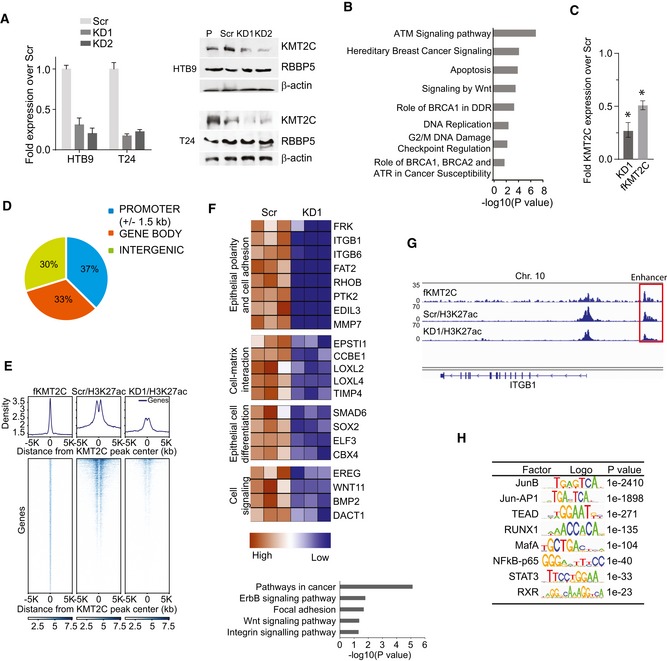
- KMT2C transcript (left) and protein (right) levels in human bladder cancer cell lines stably transduced with lentiviral vectors expressing shRNAs against KMT2C (KD1 and KD2) in comparison with Scr control cells expressing scrambled shRNAs (Scr). RBBP5, another COMPASS complex protein used as internal control and b‐actin as loading control. Transcript levels were assessed by qRT–PCR in triplicates, and values shown represent mean ± SEM.
- Bar graph showing selected biological processes and signaling pathways obtained from Gene Ontology (GO) enrichment analysis for the 3,324 differentially expressed genes between Scr control and KMT2C/KD1 HTB9 cells. Expression values were obtained from RNA‐seq data.
- Quantitative RT–PCR for KMT2C in HTB9/KD1 cells, and HTB9/KD1 cells stably transfected with a plasmid expressing a Flag‐tagged full‐length KMT2C protein (fKMT2C). Expression levels are shown in the y‐axis as respective ratios over KMT2C expression in Scr control cells (Scr expression corresponds to 1). Experiments were performed in triplicates and analyzed with Mann–Whitney U‐test. Values shown represent mean ± SEM. * designates P‐value < 0.05.
- Genome distribution of KMT2C peaks in HTB9/KD1 cells complemented with fKMT2C. Data obtained from ChIP‐seq experiments.
- Density plot indicating KMT2C binding and H3K27ac levels on active enhancers in Scr control and KD1 HTB9 cells.
- Bar graph showing selected biological processes and signaling pathways obtained from Gene Ontology (GO) enrichment analysis for 253 genes in proximity to active enhancers affected by KMT2C knockdown and heatmap of their expression (> 1.5‐fold H3K27ac and mRNA downregulation). Data obtained from ChIP‐seq and RNA‐seq experiments.
- Bedgraph indicating KMT2C binding and H3K27ac at a putative enhancer of the ITGB1 locus before and after KMT2C knockdown in HTB9 cells.
- Transcription factor binding motif analysis on active enhancers affected by KMT2C knockdown. Data obtained from ChIP‐seq experiments.
Source data are available online for this figure.
To gain further insight into the function of KMT2C in gene transcription regulation, we used fKMT2C‐complemented HTB9/KD1 cells to map KMT2C binding genome‐wide through ChIP‐sequencing (ChIP‐seq). In addition, to measure the epigenetic effects of KMT2C loss we performed ChIP‐seq experiments for histone 3 lysine 27 acetylation (H3K27ac), histone 3 lysine 4 trimethylation (H3K4me3), and histone 3 lysine 9 acetylation (H3K9ac) histone modifications on HTB9 KMT2C/KD1 and control Scr cells. ChIP‐seq experiments performed with anti‐Flag antibodies indicated that KMT2C binding sites are equally dispersed among promoter, gene body, and intergenic regions (12,417, 10,882, and 9,885 peaks, respectively; Fig 2D).
In agreement with its role in enhancer regulation, KMT2C colocalizes with the active enhancer mark H3K27ac on intergenic sites likely representing active enhancers 32 (Fig 2E). Our ChIP‐Seq analysis identified 2,808 genes proximally located to enhancers that are characterized by KMT2C binding and significant H3K27ac loss upon KMT2C silencing. GO analysis on genes of this group that are also downregulated upon KMT2C silencing (1.5‐fold or higher reduction) revealed an enrichment in processes such as focal adhesion and integrin‐mediated adhesion as well as ErbB and Wnt signaling pathways (Fig 2F). More specifically, we identified genes that encode proteins which are critical for cell adherence to the epithelial basement membrane: ITGB1, ITGB6, RHOB, a putative tumor suppressor also commonly mutated in BC 7, 20, MMP7; (Fig 2G); the extracellular matrix organization LOXL2, LOXL4 and TIMP4, an epigenetically silenced putative tumor suppressor in bladder carcinoma 33, and epithelial development and differentiation (SMAD6, SOX2, EREG, WNT11, BMP2). Interestingly, KMT2D/MLL4 was recently reported to regulate the enhancers of genes involved in cell–cell and cell–matrix adhesion as well as in differentiation of keratinocytes affecting the expression of ITGB2, ITGB4, LOXL1, LOXL2, SOX7, WNT10A genes by a similar way 34. An analysis of transcription factor binding motifs in KMT2C peaks, located at enhancers that are characterized by significant H3K27ac loss upon KMT2C silencing, identified JUNB, TEAD, RUNX1, and MAFA as the most enriched transcription factors (Fig 2H).
KMT2C localizes at promoters and controls the expression of DNA damage response and repair genes
Interestingly, our ChIP‐seq experiments also revealed 12417 fKMT2C binding sites enriched at transcription start site proximal regions (TSS ± 1,500 bp) that contain large domains of H3K4me3 H3K9ac and H3K27ac marks (Fig 3A and B). KMT2C silencing was associated with transcriptional suppression of 1,368 genes, which are characterized by promoter‐only KMT2C binding. This finding indicates that besides enhancer regulation, KMT2C is also involved in promoter activation in cancer cells. Transcription factor binding motif analysis of fKMT2C‐bound regions yielded a totally different set of transcription factors from those identified in enhancers. The most prominent of these is ELK1 (Fig 3C), a prominent RAS/MAPK target controlling components of the basal transcriptional machinery, the spliceosome and the ribosome 35.
Figure 3. KMT2C controls the expression of DDR and DNA repair genes in BC cells.
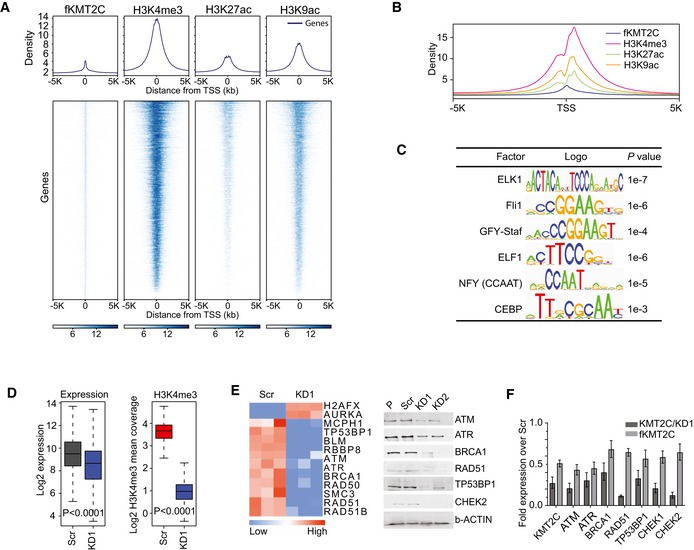
- Density plot indicating KMT2C binding and H3K4me3, H3K27ac, and H3K9ac levels on transcription start sites (TSS) in HTB9 cells.
- Histogram indicating distribution of histone modifications around transcription start sites (TSS ± 5,000 bp). Data obtained from ChIP‐seq with antibodies against the indicated histone modifications.
- Transcription factor binding motif analysis on TSS of genes transcriptionally affected by KMT2C knockdown. Data obtained from ChIP‐seq experiments.
- Boxplot indicating expression (left) and H3K4me3 levels (right) of genes with KMT2C presence on their promoters in Scr control and KMT2C/KD1 cells. Median comparison of Expr/K4 m3 values was performed with two‐tailed paired Wilcoxon rank sum test with continuity correction. The middle lines inside the boxes indicate the median (50th percentile). The lower and the upper box boundaries represent the 25th percentile and the 75th percentile, respectively. The lower and upper whiskers extend to the lowest and highest values, respectively, within the 1.5× interquartile range (box height) from the box boundaries.
- Heatmap comparison of the expression levels of genes implicated in DDR between control (Scr) and KMT2C‐knockdown (KD1) HTB9 cells (left). Expression data were obtained from RNA‐seq experiments. Western blot analysis of selected proteins in control (Scr) and KMT2C‐knockdown (KD1 and KD2) HTB9 cells (right).
- Expression level restoration of selected genes in KMT2C/KD1 HTB9 cells complemented with exogenously expressed Flag‐tagged KMT2C (fKMT2C). Data obtained by qRT–PCR. Experiments were performed in triplicates, and values shown represent mean ± SEM.
Source data are available online for this figure.
Our ChIP‐seq and RNA‐seq data indicated that upon KMT2C silencing, the subgroup of genes showing reduced expression levels also show reduced H3K4me3 levels at the respective TSSs (Fig 3D). GO analysis on this group of the 1,368 downregulated genes revealed several processes such as DDR and DSB repair by HR, which interestingly presented the highest score (see also Fig 2B). More specifically, KMT2C silencing was associated with decreased expression of key components of DDR (ATM, ATR) and the HR DNA repair pathway (BRCA1, BRCA2, RAD50, RAD51; Fig 3E). Interestingly, restoration of KMT2C activity by means of exogenous expression of fKMT2C also restored the expression levels of these genes (Fig 3F).
Our own ChIP‐seq data as well as ENCODE data indicate that KMT2C and the COMPASS complex component RBBP5 colocalize together with ELK1 upon the TSS of ATM, ATR, BRCA1, and BRCA2 genes (Fig 4A). Moreover, KMT2C levels modulate positively the H3K4me3 enrichment on TSS of these genes, indicating an important role for this histone methyltransferase on their transcriptional activation. More specifically, upon KMT2C silencing, H3K4me3 levels were significantly reduced, whereas restoration of KMT2C activity also restored H3K4me3 levels. Promoter region immunoprecipitation either as direct binding or through long‐range enhancer interactions has previously been reported for both KMT2C and KMT2D 36, 37. KMT2C binding upon the promoter region of the ATM, ATR, BRCA1, and BRCA2 genes is independently corroborated in a recently published analysis 38 (Appendix Fig S3). Interestingly in the same study, a 32% of KMT2C is located within promoter regions, indicating roles for KMT2C besides enhancer H3K4 monomethylation.
Figure 4. KMT2C controls the expression of DDR and DNA repair genes in various cancers.
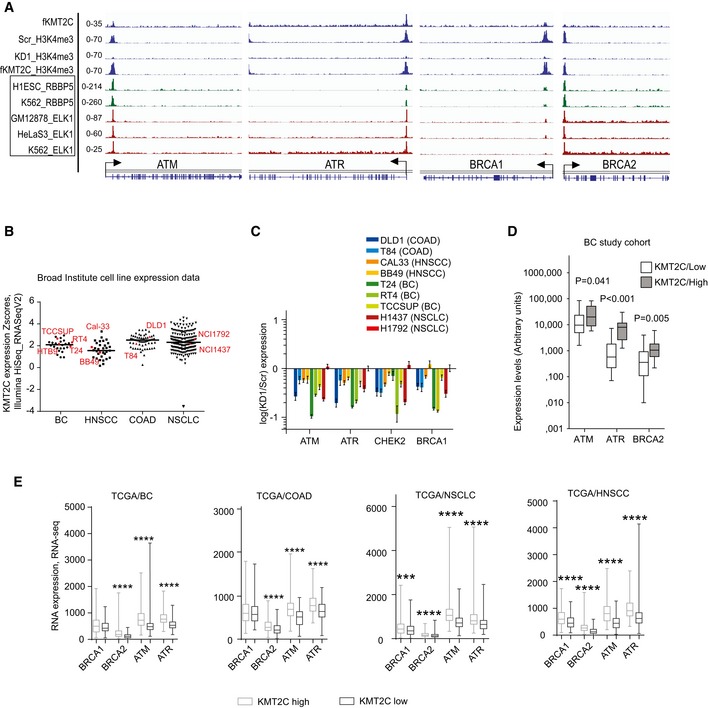
- Bedgraphs indicating KMT2C and H3K4me3 binding at the TSS of indicated loci in HTB9 cells; also, from published studies available at the ENCODE, the binding of the COMPASS complex member RBBP5 and the transcription factor ELK1 is indicated in the same loci.
- KMT2C expression (y‐axis) in various human cell lines (x‐axis). Cell lines under study are indicated as red geometrical schemes. Data were obtained directly from the Broad Institute CCLE server.
- Expression levels of indicated genes in indicated cell lines upon KMT2C knockdown. Expression is shown as log(KD1/Scr) in the y‐axis. Remaining KMT2C transcript levels for all knockdown experiments can be found in Table EV2. Note that H1792 which shows poor KD1 (˜25%) also shows no change in ATM, ATR, BRCA1, and BRCA2 expression (light red bar appearing last in each set). Experiments were performed in triplicates. In plots, bars represent mean ± SEM.
- Correlation in expression levels between KMT2C and indicated genes in our study cohort of superficial and muscle‐invasive BC. Data obtained from qRT–PCR. Experiments were performed in duplicates. P values were calculated by Mann–Whitney U‐test.
- Correlation in expression levels between KMT2C and indicated genes in BC, COAD, NSCLC, and HNSCC tumors. RNA‐seq data were obtained from the TCGA through cbioportal.org. Mann–Whitney U‐test was used. *** designates P‐value < 0.001 and ****P‐value < 0.0001.
This observation prompted us to knock down KMT2C expression in a wide panel of BC, COAD, HNSCC, and NSCLC cell lines which according to publicly available data showed variable KMT2C expression levels (Fig 4B). Quantitative RT–PCR experiments revealed an invariable downregulation of DDR and HR components (Fig 4C). Finally, expression analysis of our cohort of bladder cancer tumors (Fig 4D), as well as meta‐analysis of publicly available TCGA expression data from BC, COAD, NSCLC, and HNSCC, indicated that KMT2C levels strongly correlate with the expression of the same genes (Fig 4E). Interestingly, a positive correlation between ATM, ATR, BRCA1, BRCA2, and KMT2C expression is also derived from TCGA data GBM, LGG, AML, DLBL, SARC, and breast invasive carcinoma (BRIC) RNA‐seq data (Appendix Fig S4). Altogether, these data indicate that KMT2C controls the epigenetic status of genes involved in DDR and DNA repair and directly or indirectly their expression levels, even in tissues in which a tumor suppressor role of KMT2C has yet to be established. Deficiencies in DNA repair due to germline or somatic mutations is a common event in cancer 39, while reduced expression of DNA repair components due to epigenetic control, primarily DNA methylation, is also observed 40, 41. In this case, however, loss of KMT2C seems to affect en bloc the expression of multiple key components of the DDR and DNA repair pathways.
Bladder cancer cells lacking KMT2C are HR‐deficient and present high levels of genomic instability
The observation that KMT2C loss affects genes involved in DDR and DNA repair prompted an in‐depth cytogenetic analysis which revealed that both HTB9 and T24 cells lacking KMT2C show increased DNA damage as indicated by higher frequency of nuclear foci staining for the DNA damage marker γH2AX. DNA damage levels are comparable to those measured in BRCA1‐knockdown cells (Figs 5A and EV2A). Cisplatin is known to cause DSBs which, in cells beyond the G1 phase, are repaired by the HR machinery. To assess the contribution of HR in DSB repair, we treated HTB9 and T24 cells with cisplatin and immunostained against γH2AX and the HR repair protein RAD51. While both Scr control and KD cells showed the same frequency of γH2AX foci, RAD51‐positive nuclei were significantly fewer in the latter (Figs 5B and EV2B). Moreover, sister chromatid exchange (SCE) assays upon cisplatin treatment clearly demonstrated that, while Scr control cells are HR competent, their KD counterparts show low levels of HR DSB repair (Figs 5C and EV2C). It is known that HR factors stabilize stalled forks by protecting them from nucleolytic degradation, help restarting DNA synthesis from stalled forks, and repair DSBs generated by collapsed forks 42, 43, 44, 45, 46. To investigate the ability of KMT2C/KD cells to resolve stalled forks in S phase, we used DNA fiber assays. In the presence of the DNA replication inhibitor hydroxyurea (HU), KMT2C/KD cells show a behavior similar to that of BRCA1‐deficient cells, i.e., inability to resolve stalled forks (Figs 5D and EV2D). These functional data indicate that loss of KMT2C leads to HR deficiency due to downregulation of multiple HR components, as well as compromisation of DNA replication under genotoxic stress.
Figure 5. Cells lacking KMT2C are HR‐deficient.
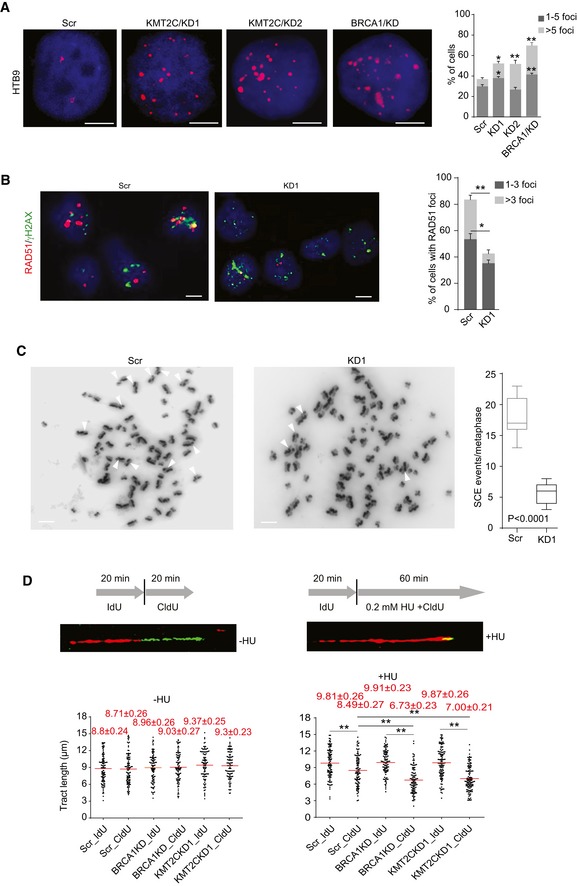
- Immunofluorescence of γH2AX foci (left) and quantitation (right) in control (Scr) and KMT2C‐knockdown (KMT2C/KD1 and KD2) HTB9 cells. BRCA1 knockdown (BRCA1/KD) is used as control. The y‐axis indicates added percentage of cells with 1–5 and > 5 foci for each cell type. Scale bars indicate 5 μm. All comparisons have been performed against Scr control cells. Values in the bargraph represent mean ± SEM from three experiments. Student's t‐test was used. * designates P‐value < 0.05, and ** designates P‐value < 0.01.
- Frequency of RAD51 foci in cisplatin‐treated HTB9 control (Scr) and KMT2C‐knockdown (KD1) cells. The y‐axis indicates added percentage of cells with 1–3 and 3 foci. Scale bars indicate 10 μm. Values in the bargraph represent mean ± SEM from three experiments. Student's t‐test was used. * designates P‐value < 0.05, and ** designates P‐value < 0.01.
- Sister chromatid exchange (SCE) assay with cisplatin‐treated HTB9 control (Scr) and KMT2C‐knockdown (KD1) cells. Red arrowheads indicate sister chromatid exchange events. Results were obtained from 15 metaphases per group. Mann–Whitney U‐test was used.
- DNA fiber assay on control (Scr) and KMT2C‐knockdown (KD1) HTB9 cells. BRCA1‐knockdown cells are used as controls. Experiments performed with or without hydroxyurea (HU) treatment under the conditions indicated in the schematic. Examples of DNA fibers from HTB9/KD1 cells are shown. The length of minimum 100 fibers from each condition was measured. Values in the plot are means ± SEM. Mann–Whitney U‐test was used. ** designates P‐value < 0.01.
Figure EV2. Cells lacking KMT2C are HR‐deficient.
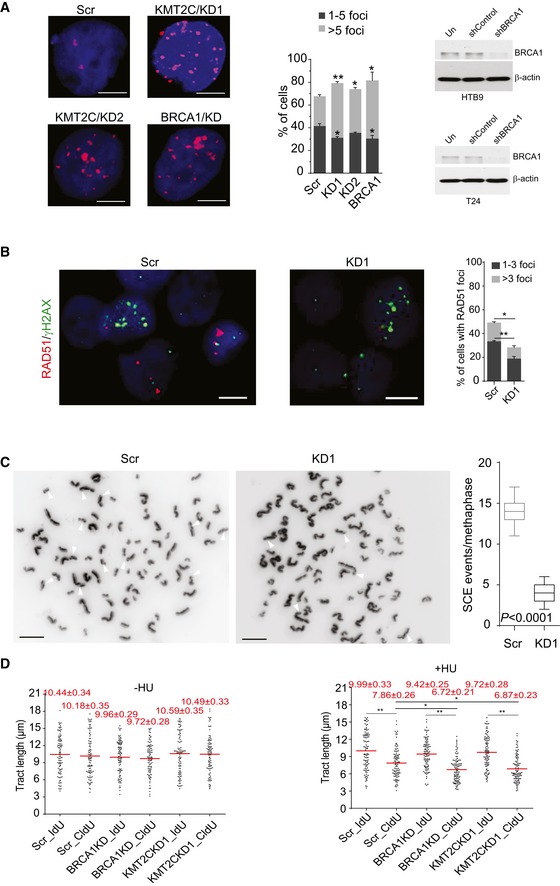
- Immunofluorescence of γH2AX foci and quantitation in control (Scr) and KMT2C‐knockdown (KMT2C/KD1 and KD2) T24 cells. BRCA1 knockdown (BRCA1/KD) is used as control. Scale bars indicate 10 μm. Values in the plot correspond to mean ± SEM. Data from three experiments were analyzed with Student's t‐test. * designates P‐value < 0.05, and ** designates P‐value < 0.01. Remaining protein levels of BRCA1 are also shown for both HTB9 (referring to Fig 5A) and T24 are also shown.
- Frequency of RAD51 foci in cisplatin‐treated T24 control (Scr) and KMT2C‐knockdown (KD1) cells. Scale bars indicate 10 μm. Values in the plot correspond to mean ± SEM. Data from three experiments were analyzed with Student's t‐test. * designates P‐value < 0.05, and ** designates P‐value < 0.01.
- Sister chromatid exchange assay with cisplatin‐treated T24 control (Scr) and KMT2C‐knockdown (KD1) cells. Results were obtained from 15 metaphases per group. White arrowheads indicate sister chromatid exchange events.
- DNA fiber assay on control (Scr) and KMT2C‐knockdown (KD1) T24 cells. BRCA1‐knockdown cells are used as controls. Experiments performed with or without hydroxyurea (HU) treatment under the conditions indicated in Fig 5D. The length of minimum 100 fibers from each condition was measured. Red horizontal lines indicate the median tract length in each group.
Source data are available online for this figure.
Interestingly, stalled or collapsed replication forks are a major source of DSBs and endogenous genomic instability in dividing cells 47. In cancer, oncogene‐induced replication stress contributes critically to DNA damage while cancer cells with HR deficiency are characterized by extensive genomic instability 48, 49. Chromosomal instability, as a type of genomic instability, has been also linked to HR deficiency and mitotic defects 50, 51. In KMT2C/KD cells, the increased frequency of micronuclei, chromosome bridges, lagging chromosomes, and chromosome congression (Fig 6A and B) implies gross defects in mitotic fidelity and genome integrity safeguarding. To assess the chromosomal status of KMT2C/KD cells, we used gross karyotypic analysis which revealed that both HTB9 and T24 KMT2C/KD cells had lower chromosomal count in total and per individual chromosome (Fig 6C and D). Moreover, chromosomal microarray analysis (CMA) on HTB9 cells indicated that chromosomal losses were more frequent and more extensive than respective gains (Fig 6E and F).
Figure 6. KMT2C loss leads to genomic instability.
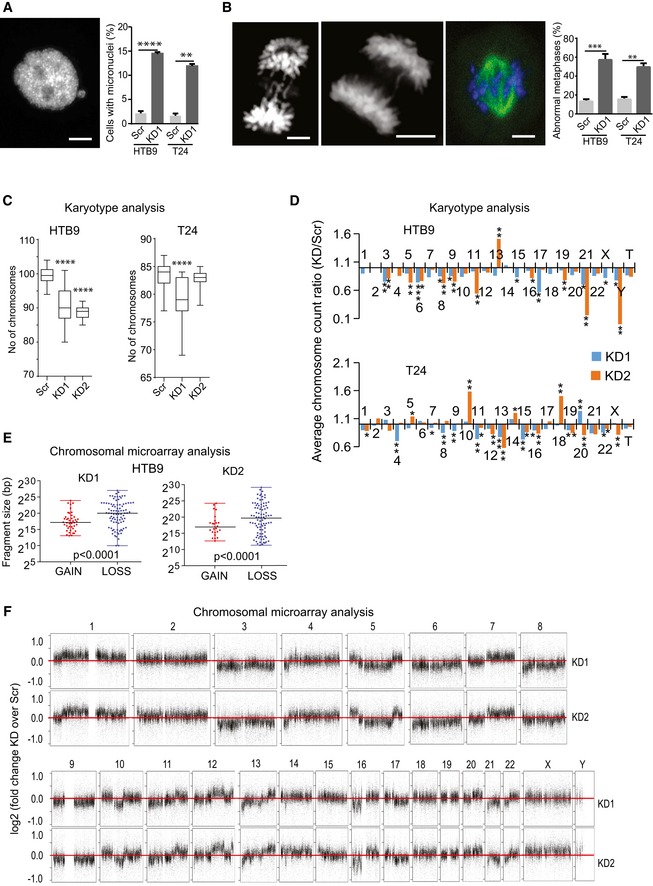
- Representative image (from HTB9/KD1 cells) and frequency of both HTB9 and T24 control (Scr) and KMT2C/KD1 cells with micronuclei. Scale bar indicates 5 μm. Values represent mean ± SEM from three experiments. Student's t‐test was used. ** designates P‐value < 0.01 and ****P‐value < 0.0001.
- Representative images (from HTB9 cells) and frequency of abnormal metaphases presenting lagging chromosomes, chromosome bridges, and chromosome congression in HTB9 and T24 control (Scr) and KMT2C/KD1 cells. Scale bar indicates 5 μm. Values represent mean ± SEM from three experiments. Student's t‐test was used. **designates P‐value < 0.01 and *** P‐value < 0.001.
- Karyotypic analysis and chromosomal count in control (Scr) and KMT2C/KD1 and KD2 cells. All comparisons performed against Scr control cells. Metaphases studied: HTB9 Scr, n = 20; HTB9 KD1, n = 11; HTB9 KD2, n = 12; T24 Scr, n = 20; T24 KD1, n = 15; T24 KD2, n = 12. Mann–Whitney U‐test was used. **** designates P‐value < 0.0001.
- Average chromosome count ration KD/Scr obtained from Giemsa‐stained metaphase spreads of HTB9 and T24 KD1 and KD2 cells. Number of metaphases studied: HTB9 Scr, n = 20; HTB9 KD1, n = 11; HTB9 KD2, n = 12; T24 Scr, n = 20; T24 KD1, n = 15; T24 KD2, n = 12. Mann–Whitney U‐test was used. * designates P‐value < 0.05, **P‐value < 0.01, and ***P‐value < 0.001.
- Fragment size of gains and losses obtained from CMA on HTB9/KD1 and KD2 cells. Mann–Whitney U‐test was used. One sample from each cell type was used in CMA.
- Copy number gains and losses of HTB9 KD1 and KD2 cells in comparison with HTB9/Scr controls. Data obtained from CMA. Values as presented as log(KD1/Scr) in the y‐axis. The horizontal red line indicates log value 0, which corresponds to no change. Note that for the majority of chromosomes there are losses in the KD1 cells.
KMT2C loss leads to PARP1/2 dependence for DNA repair
Our expression and cytogenetic data clearly indicate that the HR repair machinery is compromised in KMT2C/KD cells. HR deficiency is known to skew the balance toward canonical nonhomologous end joining (c‐NHEJ) with the participation of the TP53BP1 protein 52. However, the number of TP53BP1 foci in cisplatin‐treated KMT2C/KD cells is comparable with Scr control cells (Figs 7A and EV3A), possibly implying that the activity of c‐NHEJ is not elevated. To this direction, we compared the activity of NHEJ pathway between KMT2C/KD1 and Scr control cells by counting chromosomal fusion events in a dicentric assay. Repair of DSBs induced by ionizing radiation (IR) in this assay generated chromosomal fusions with equal frequency between KMT2C/KD1 and Scr control cells (Figs 7B and EV3B), implying that both employ non‐HR mechanisms for DNA repair equally. It is widely accepted that ligase IV participates in the final stages of the c‐NHEJ 53, while poly(adenosine diphosphate [ADP]–ribose) polymerase 1 (PARP1) is an integral component of the alt‐EJ pathway 54, 55. To assess the individual contribution of c‐NHEJ and alt‐EJ in DNA repair, we induced DSBs via IR in both Scr control and KMT2C/KD1 cells and measured the frequency of chromosomal fusions in the presence of the ligase IV inhibitor SCR7 56 or the PARP1/2 inhibitor olaparib 57. Inhibition of PARP1/2 in KMT2C/KD cells led to a significant reduction (P < 0.01) in the number of chromosome fusions, while ligase IV inhibition had a lesser effect (Figs 7C and EV3C). This implies that KMT2C/KD cells rely heavily on alt‐EJ for DSB repair. PARP1/2 inhibition in BRCA‐deficient cells is known to lead to accumulation of chromosome fragments and radial structures, a phenotype associated with c‐NHEJ 52, 58, 59. As Fig 7D indicates, blocking the alt‐EJ pathway with olaparib leads to significantly (P = 0.021) more radial chromosomes in comparison with Scr controls, while simultaneous treatment with olaparib and SCR7 ameliorated this phenotype. Comparable results were obtained when the c‐NHEJ and alt‐EJ pathways were genetically inhibited through shRNA knockdown of ligase IV and ligase III, respectively (Fig EV4A and B). This further supports the hypothesis that, in KMT2C/KD cells, the alt‐EJ pathway plays a more important role in DSB repair.
Figure 7. KMT2C loss leads to PARP1/2 dependence for DNA repair.
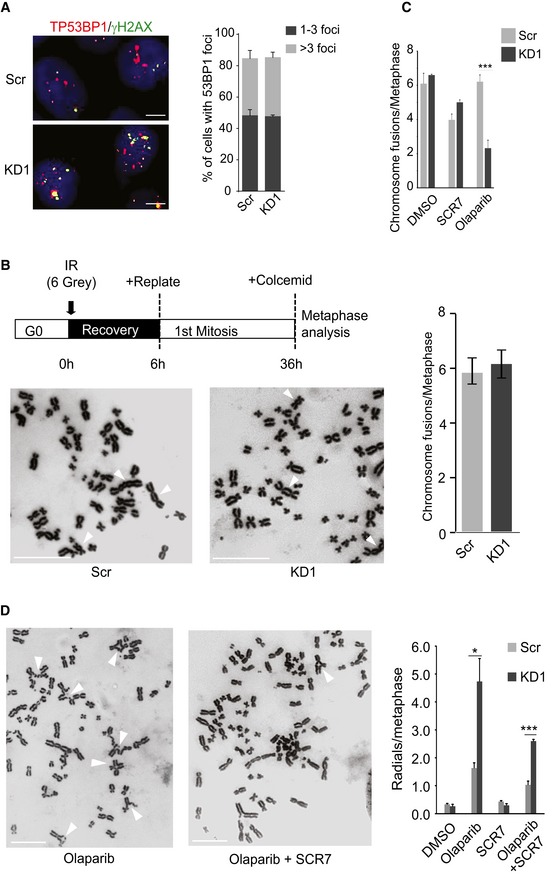
- Frequency of TP53BP1 foci in cisplatin‐treated HTB9 control (Scr) and KMT2C‐knockdown (KD1) cells. Size bars in microscopy panels correspond to 5 μm. In the plot, bars represent mean ± SEM from n = 3 experiments.
- Frequency of chromosome fusions obtained from IR‐treated (schematic) HTB9 control (Scr) and KMT2C‐knockdown (KD1) cells. Representative karyotypes are shown. Size bars in karyotype panels correspond to 10 μm. White arrows indicate chromosome fusion events. In the plot, bars represent mean ± SEM from n = 3 experiments.
- Frequency of chromosome fusions in IR‐treated HTB9 control (Scr) and KMT2C‐knockdown (KD1) cells upon treatment with SCR7 (30 μM) and olaparib (15 μM). Bars represent mean ± SEM from n = 3 experiments. *** designates P‐value < 0.001.
- Frequency of radial chromosomes in IR‐treated HTB9 control (Scr) and KMT2C‐knockdown (KD1) cells upon treatment with SCR7 (30 μM), olaparib (15 μM), or both. Representative karyotypes are shown. Size bars in karyotype panels correspond to 10 μm. White arrows indicate radial structures. In the plot, bars represent mean ± SEM from n = 3 experiments. * designates P‐value < 0.05 and ***P‐value < 0.001.
Figure EV3. KMT2C loss leads to PARP1/2 dependence for DNA repair.
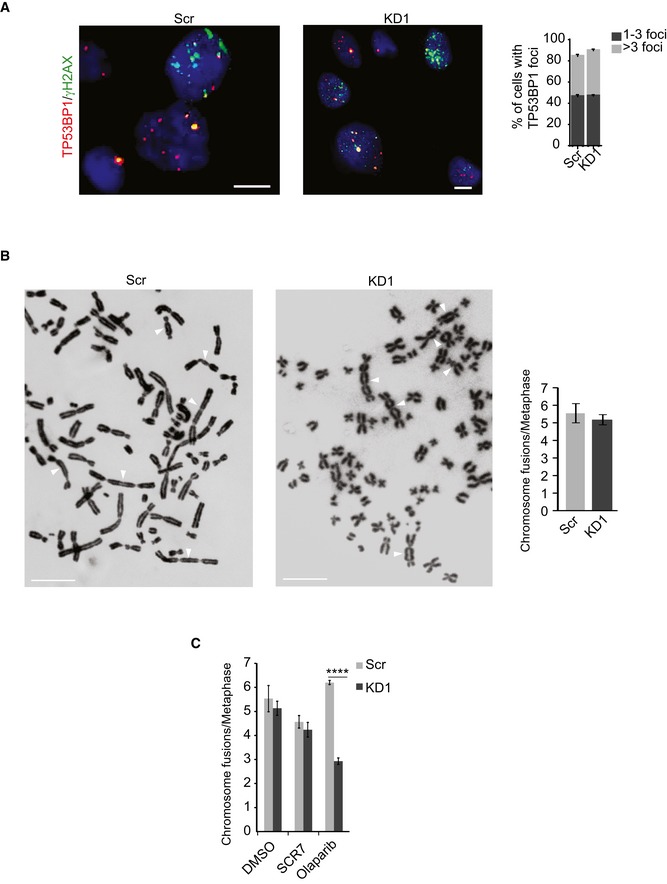
- Frequency of TP53BP1 foci in cisplatin‐treated T24 control (Scr) and KMT2C‐knockdown (KD1) cells. Size bars in microscopy panels correspond to 10 μm. In the plot, bars represent mean ± SEM from n = 3 experiments.
- Frequency of chromosome fusions obtained from IR‐treated (schematic) T24 control (Scr) and KMT2C‐knockdown (KD1) cells. Representative karyotypes are shown. Size bars in karyotype panels correspond to 5 μm. White arrows indicate chromosome fusions. In the plot, bars represent mean ± SEM from n = 3 experiments.
- Frequency of chromosome fusions in IR‐treated T24 control (Scr) and KMT2C‐knockdown (KD1) cells upon treatment with SCR7 and olaparib. Bars represent mean ± SEM from n = 3 experiments. Throughout the figure, Mann–Whitney U‐test was used.
Figure EV4. KMT2C loss leads to PARP1/2 dependence for DNA repair.
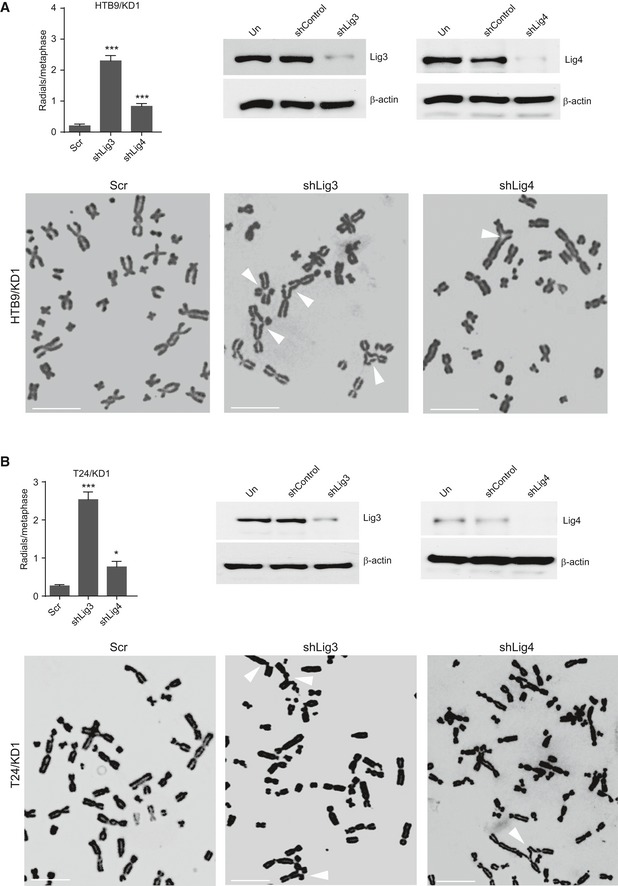
-
A, BFrequency of radial structures in IR‐treated HTB9/KD1 (A) and T24/KD1 (B) cells upon ligase III and ligase IV knockdown (top left), and Western blot analysis indicating respective leftover protein levels (top right). Representative karyotypes are shown. Values in the plot indicate mean ± SEM. Analysis of three experiments was performed using Student's t‐test. * designates P‐value < 0.05 and ***P‐value < 0.001.
Source data are available online for this figure.
Tumors with KMT2C loss are sensitive to PARP1/2 inhibition
Previous reports have shown that HR‐deficient cells are sensitive to PARP1/2 inhibitors 58, 60. In fact, recently published results from a Phase II clinical trial indicated that patients with castration‐resistant prostate cancer that carry mutations in DNA repair genes, such as BRCA1/2, ATM, Fanconi anemia components, and CHEK2, show positive response to olaparib, indicating a dependence on PARP1/2 for survival upon DNA damage 61. In agreement with this, KMT2C/KD cells show increased sensitivity to olaparib (Fig 8A). This observation was also confirmed in long‐term treatments in clonogenic assays with three different concentrations of olaparib (Fig EV5). Moreover, generation of DSBs through IR is detrimental for olaparib‐treated KMT2C/KD cells, underscoring the dependence of these on PARP1/2 for DNA repair (Fig 8B). These findings are independently corroborated from publicly available data from the cancerrxgene.org database 62, which show that cell lines from BC, NSCLC, HNSCC, and COAD with reduced expression of KMT2C are more sensitive to PARPi (Fig 8C).
Figure 8. KMT2C loss leads to PARP1/2 dependence in vitro and in vivo .
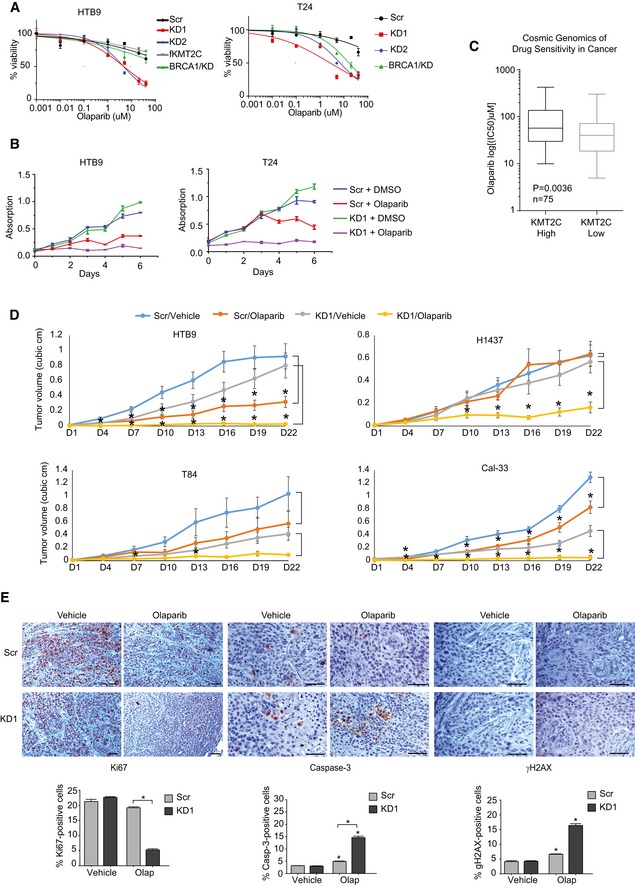
- MTT assays on untreated control (Scr) and KMT2C/KD cells. BRCA1/KD cells are used as controls. Values represent mean ± SEM from three experiments.
- MTT assays with IR‐treated control (Scr) and KMT2C/KD1 cells treated with 15 μM olaparib. Values represent mean ± SEM from three experiments.
- Boxplot indicating olaparib IC50 of BLCA, HNSCC, COAD, and NSCLC cell lines from publicly available data (https://www.cancerrxgene.org/). KMT2Chigh and KMT2Clow indicate that KMT2C expression of the cell line is at the top and bottom 50% of the cohort, respectively (data obtained from cbioportal.org; Cancer Cell Line Encyclopedia). Mann–Whitney U‐test was used.
- Tumor volume obtained from xenografts of control and KMT2C/KD1 cells treated with vehicle or olaparib. The number of mice analyzed for each cohort and raw measurements are provided in Appendix Table S3. The following tumor weight averages (in grams) were obtained ±SEM for vehicle and olaparib, respectively: HTB9/Scr, 0.791 ± 0.155 and 0.468 ± 0.097; HTB9/KD1, 0.862 ± 0.156 and 0.072 ± 0.023; T84/Scr, 1.032 ± 0.217 and 0.413 ± 0.097; T84/KD1, 0.562 ± 0.159 and 0.105 ± 0.032; H1437/Scr, 0.661 ± 0.133 and 0.780 ± 0.133; H1437/KD1, 0.723 ± 0.099 and 0.363 ± 0.108; Cal‐33/Scr, 0.439 ± 0.051 and 0.301 ± 0.029; Cal‐33/KD1, 0.584 ± 0.224 and 0.05 ± 0.016. Statistically significant pairwise comparison with respective vehicle for each day is indicated with star. Mann–Whitney U‐test was used. * designates P‐value < 0.05. All statistical values including those between Scr and KD1 cells are provided in Appendix Table S4.
- Immunohistochemistry with the indicated antibodies on tumor sections from control (Scr) and KMT2C/KD1 HTB9 cells grown subcutaneously in NOD/SCID mice which were treated with vehicle or olaparib. Statistically significant pairwise comparison with respective vehicle is indicated with stars on top of each column. All other statistically significant comparisons are indicated with squared brackets connecting pairs under comparison. In microscopy images, scale bars indicate 50 μm. In bargraphs, values correspond to mean ± SEM from n = 3 experiments. Student's t‐test was used for the analysis. * designates P‐value < 0.05.
Figure EV5. In vitro clonogenic assays indicating PARPi sensitivity of KMT2C/KD cells.
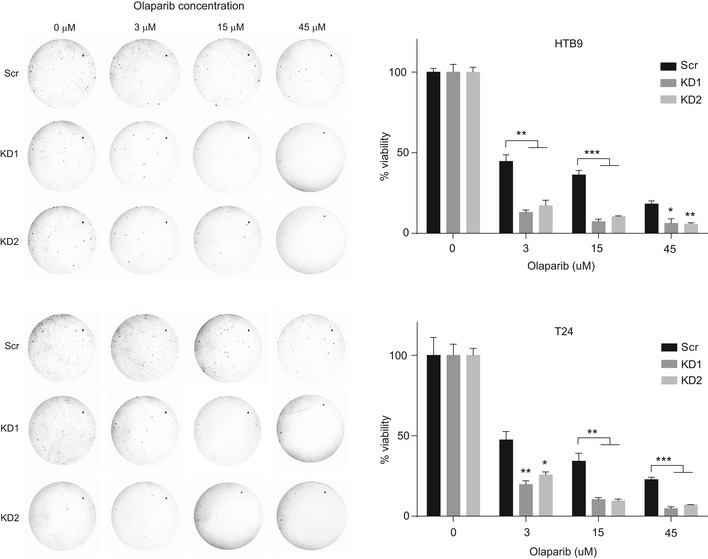
Representative photographs (left) and number of colonies (y‐axis) generated by HTB9 and T24 (Scr and KMT2C/KD1) cells treated with increasing concentrations of olaparib (x‐axis). Cells were seeded and grown for 20 days at which point the experiment was concluded and dishes were photographed. Values in the plot indicate mean number of colonies ± SEM. Analysis of three experiments was performed using Student's t‐test. * designates P‐value < 0.05, ** designates P‐value < 0.01, and *** designates P‐value < 0.001.
To corroborate our finding in vivo, we used one cell line from each tumor type under study in xenograft experiments (HTB9/BC, H1437/NSCLC, T84/COAD, and Cal‐33/HNSCC). Although T84 and Cal‐33 KMT2C/KD cells grew somewhat slower in vivo in comparison with the respective Scr controls, KD1 cells are more sensitive to olaparib which totally suppressed tumor grown in mice (Fig 8D). This was associated with reduced proliferation, high DNA damage, and severe apoptosis in KD1 cells in all cell lines tested (Fig 8E). Altogether, our in vitro and in vivo experiments indicate that KMT2C/KD cells rely heavily upon alt‐EJ for DSB repair. Although we cannot exclude alt‐EJ‐independent PARP1/2 functions in DNA repair, we hypothesize that upon inhibition of alt‐EJ, KMT2C/KD cells rely exclusively upon c‐NHEJ for DSB repair. This, however, is either insufficient or too error prone to deal with elevated DNA damage, eventually leading to cell death.
Discussion
Histone‐modifying enzymes have emerged as critical players in tumor biology in recent years. H3K4 methyltransferases have been implicated in tumorigenesis as both oncogenes and tumor suppressors in a variety of neoplasias. Bladder cancer presents some of the highest reported mutation rates in KMT2C and KMT2D, and to a lesser extent in KMT2B 7, 11, 63. A high percentage of reported mutations lead to truncated protein products with presumably impaired functionality. Recent reports, however, indicate that loss of the catalytic activity of KMT2C and KMT2D has a less severe effect on transcription regulation than the respective complete gene knockout 16, 17, implying that these proteins may have additional roles in transcriptional regulation beyond H3K4 monomethylation.
In support of this, loss of the catalytic activity of the Drosophila homolog Trithorax (Trr) has negligible effect on fly development, while its complete loss leads to embryonic lethality 64, 65. Therefore, somatic mutations, even those truncating the protein from its catalytic activity, might not be the only MLL‐related genetic events associated with cancer. These observations prompted us to focus our studies on the expression levels of KMT2C and its role in already transformed cells. We report for the first time that the epigenetic regulator KMT2C is significantly downregulated in many different types of cancer. We thus speculate that loss‐of‐function mutations in combination with progressively reduced gene expression due to promoter methylation limit KMT2C activity in cancer cells. Thus, in tumor evolution, promoter methylation of KMT2C may provide a selective advantage to emerging KMT2C‐mutated cells by reducing wild‐type protein levels. In support of this model, KMT2C mutations were recently identified as late events in subclones of lung adenocarcinomas during tumor evolution 66 and in metastatic breast cancer subclones 67.
Functionally, KMT2C and the related KMT2D protein direct H3K4 monomethylation, which poises enhancers for activation and transcription factor binding, thus regulating the transcription of neighbor genes 68, 69. In addition to its catalytic role in H3K4me1 deposition, KMT2C interacts with the histone acetyltransferase complex CBP/p300 and the H3K27 demethylase UTX to promote H3K27 acetylation and enhancer activation 68. The precise control of transcriptional networks through enhancers is important for the tissue‐specific expression pattern of developmental genes and plays a crucial role in establishing and maintaining cell fate and identity 70. Recent studies, however, have shown that alterations in the enhancer epigenetic landscape also correlate with tumorigenesis 71, 72, 73. The reduced expression of KMT2C in bladder epithelial tumor cells leads to a substantial loss of H3K27 acetylation in a subset of active enhancers that control expression of genes involved in focal adhesion, adherens junctions, migration, epithelial cell development, and differentiation. In a recent study by Lin‐Shiao et al 34, loss of KMT2D activity by shRNA silencing in primary neonatal human epidermal keratinocytes (NHEKs) and spontaneously immortalized human epidermal keratinocytes (HaCaTs) revealed an important regulatory role of KMT2D in enhancer activity of genes involved in the same processes. This may imply that KMT2C and KMT2D proteins exert coordinated and synergistic functions in enhancer elements and their loss during carcinogenesis deregulates cell adhesion and signaling with profound effects to tumor progression and invasion. Previous reports have shown that concomitant loss of KMT2C and the tumor suppressor protein TP53 expedites tumor formation in mice 13, implying a preferential collaboration between the two. Our own meta‐analysis of the publicly available TCGA RNA‐seq and mutation data failed to substantiate any consistent correlation between TP53 mutation status and KMT2C expression levels or mutation status (Appendix Fig S5A and B). Moreover, a similar meta‐analysis on cancer cell lines also failed to identify any correlation between TP53 mutation status and KMT2C expression (Appendix Fig S5C). Whether the reported connection between KMT2C and TP53 is species‐specific or tissue‐specific and whether a more universal connection between the proteins exists are questions that would require further experiments to be addressed.
Upon KMT2C reduction, profound gene expression changes are observed. Several genes involved in DDR and DNA repair are downregulated, seemingly due to loss of KMT2C binding on their proximal promoters. Expression downregulation in these cases is associated with reduction in H3K4me3 levels. Although other H3K4 methyltransferases have been found to regulate promoter activity 74, 75, this is the first time that KMT2C is found upon promoter regions and implicated in transcription activation of genes, including those encoding DDR and DNA repair proteins. Whether transcription factors mediating oncogenic programs in cancer cells, such as ELK1 downstream of the RAS/MAPK cascade, are responsible for KMT2C recruitment onto promoter regions is a hypothesis that warrants further investigation. It is relevant in this respect that the implication of KMT2C in transcriptional regulation of these genes is confirmed in published TCGA datasets.
Cells with reduced KMT2C levels behave as HR‐deficient despite the fact that BRCA1/2 proteins and other HR components are not mutated. HR deficiency as a result of epigenetic regulation of BRCA1/2 expression levels has also been described (reviewed by Konstantinopoulos et al 76). On the other hand, the alt‐EJ pathway assumes a critical role, potentially due to HR deficiency. This explains the increased sensitivity of KMT2C/KD cells to PARP1/2 inhibition and offers a promising treatment alternative for KMT2Clow cases. Because PARP1/2 participates in the repair of single‐strand breaks (SSB; also induced during DNA replication), we cannot exclude the possibility that the increased sensitivity of KMT2C/KD cells to PARPi is also due to unrepaired SSBs which contribute to excess DNA damage. Interestingly, synthetic lethality between PARP1/2 inhibition and BRCA1/2 loss has been solidly established and already exploited in the clinic 77 while a similar link has been established in preclinical models lacking ATM 78. Our results will hopefully trigger further studies aiming to investigate the relationship between the epigenetic landscape and DNA damage response.
Materials and Methods
Human specimens
Bladder tissue specimens were obtained from 138 patients diagnosed with primary urothelial carcinoma of the bladder. Bladder cancer patients underwent transurethral resection of bladder tumors (TURBT) for non‐muscle‐invasive bladder cancer (Ta, T1) or radical cystectomy (RC) for muscle‐invasive bladder cancer (T2–T4). All patients were treated at “Laiko” General Hospital, Athens, Greece (Appendix Tables S1 and S2). Whenever feasible, normal adjacent tissue specimens from 104 of the same patients were included in the study as reference samples, following pathologist's evaluation for absence of CIS and dysplasia. Tissue samples (tumors and normal adjacent specimens) were sectioned into two mirror‐image specimens, one of which was submitted to pathologist's evaluation, while the other one was immediately frozen in liquid nitrogen and stored at −80°C until further processing. All patients were diagnosed with urothelial carcinoma on the basis of histopathological criteria, and none of the patients had received any kind of neoadjuvant therapy prior to surgery. No inclusion or exclusion criteria were used other than tissue quality after thawing. Our study was performed according to the ethical standards of the 1975 Declaration of Helsinki, as revised in 2008, and was approved by the ethics committee of “Laiko” General Hospital. Informed consent was obtained from all the participating patients.
Sanger sequencing
Total RNA from bladder tissue from a previously described cohort 22 was reverse transcribed with random primers and used in PCRs to obtain overlapping amplicons 600–800 bp that cover the portion of KMT2C coding region of interest. The oligonucleotide sequences used are provided in Appendix Table S5. PCR fragments were sequenced in both strands with standard Sanger sequencing procedures.
Cell culture
All cell lines were originally purchased from ATCC. Cells were cultured in Dulbecco's Modified Eagle's Medium (Sigma‐Aldrich, cat. D6429) supplemented with 10% heat‐inactivated fetal bovine serum (Biosera, cat. FB‐1001/500) and penicillin (100 units/ml)/streptomycin (100 μg/ml; Thermo Fischer Scientific, cat. 15140122) at 37°C with 5% CO2, with the exception of H1437 and H1792 which were cultured in RPMI‐1640 medium (Sigma‐Aldrich, cat. R8758).
Lentivirus production, infection, and shRNA knockdown
Scramble, anti‐KMT2C, and anti‐BRCA1 short hairpin RNA‐producing DNA sequences were cloned in PLKO.1‐puro‐IRES‐gfp plasmids. To produce replication‐incompetent lentivirus, 293T cells were co‐transfected with either Lenti‐Scr‐GFP or Lenti‐shKMT2C‐GFP constructs in combination with the pMD2.G and psPAX2 plasmids (Addgene, cat. #12259 and #12260) using the CaCl2 precipitation method. Twelve hours later, growth medium was replenished. Viral supernatants were harvested 36 and 70 h post‐transfection. Cell lines were infected overnight with filtered viral supernatants. Three days post‐infection, cells were selected with 5–10 μg/ml puromycin (Sigma‐Aldrich, cat. P8833) over a period of 7 days. TRCN0000008744 and TRCN000000743 (Sigma‐Aldrich) shRNA clones against KMT2C were used for generation of KMT2C/KD1 and KMT2C/KD2 cells, respectively. TRCN0000039834 (Sigma‐Aldrich) shRNA clone against BRCA1 was used for generation of BRCA1/KD cells. Scramble, anti‐DNA ligase III (TRCN0000048502), and anti‐ligase IV (TRCN0000009847) short hairpin RNA‐producing DNA sequences (Sigma‐Aldrich) were cloned in PLKO.1‐blast plasmid (Addgene: #26655). Lentiviral supernatants generated using these plasmids were used to infect HTB9 and T24 KMT2C/KD1 cell lines. Three days post‐infection, cells were selected with 10 μg/ml blasticidin over a period of 12 days for the generation of KMT2C/KD1 cell lines with DNA ligase III or IV knockdown.
ChIP‐seq preparation and analysis
Chromatin was prepared from Scr control and KMT2C/KD HTB9 cells with the SimpleChIP® Enzymatic Chromatin IP Kit (Cell Signaling, cat. 9003) according to the manufacturer's instructions. Libraries were prepared in Greek Genome Center (GGC) Biomedical Research Foundation of Academy of Athens (BRFAA) as previously described 79 and sequenced on the Illumina platform. Single‐end 85‐bp reads for H3, H3K27ac, H3K4me3, and H3K9ac were generated with the NextSeq500 in the GGC. All ChIP‐seq data were aligned to human genome version GCCh37/hg19 with the use of bowtie2 (version 2.1.0) 80 and «–very‐sensitive» parameter. Samtools (version 0.1.19) 81 was used for data filtering and file format conversion. MACS (version 1.4.2) algorithm 82 was used for peak calling with H3 ChIP as control. Gene annotation and genomic distribution of the peaks identified by MACs were performed with BEDTools 83, and graph representation (heatmaps) of the tag read density around TSS was performed with seqMiner (version 1.3.3) software 84. ChIP‐seq data have been deposited in the Short Read Archive (SRA) under the BioProject ID: PRJNA508740.
RNA‐seq preparation and analysis
Library preparation for RNA‐seq was carried out in the GGC of BRFAA. RNA was isolated from Scr control and KMT2C/KD cells, and RNA‐seq libraries were prepared using the TruSeq RNA kit using 1 μg of total RNA. The libraries were constructed according to Illumina's protocols, and equal amounts were mixed and run in the Illumina NextSeq500 in the GGC. Single‐end 85‐bp reads for three Scr control and three KMT2C/KD samples were generated. RNA‐seq raw sequencing data were aligned to human genome version GCCh37/hg19 with the use of TopHat (version 2.0.9) 85 with the use of «–b2‐very‐sensitive» parameter. Samtools (version 0.1.19) 81 was used for data filtering and file format conversion, while HT‐seq count (version 0.6.1p1) algorithm 86 was performed for assigning aligned reads into exons using the following command line «htseq‐count –s no –m intersection ‐nonempty». Finally, differentially expressed genes were identified with the use of the DESeq R package 87 and genes with fold change cutoff 1.5 and P‐adj ≤ 0.05 were considered to be differentially expressed genes (DEGs). Gene ontology and pathway analysis was performed in the DEGs with the DAVID knowledge base 88 and Ingenuity Pathway Analysis software (IPA). Only pathways and biological processes with P‐value ≤ 0.05 were considered significantly enriched. RNA‐seq data have been deposited in the Short Read Archive (SRA) under the BioProject ID: PRJNA508526.
CMA
The chromosomal microarray analysis (CMA) was performed with the high‐resolution 2 × 400 K G3 CGH+SNP microarray platform (G4842A, Design ID 028081, Agilent Technologies, Santa Clara, CA, USA). The specific platform features a total of 292,097 oligonucleotide CGH probes covering the whole genome, with a median CGH probe spacing of 7 kb, as well as 118,955 single nucleotide polymorphism (SNP) probes for the detection of copy neutral loss of heterozygosity (CN‐LOH), resulting in a resolution of 5–10 Mb for CN‐LOH. The wet‐lab protocol was according to the manufacturer's instructions (Agilent Oligonucleotide Array‐Based CGH for Genomic DNA Analysis) and consisted of enzymatic digestion of genomic DNA in parallel with a sex‐matched reference DNA (Agilent Technologies) with restriction enzymes AluI and RsaI, followed by differential labeling with Cy3 and Cy5 fluorescent dyes for sample and reference, respectively. Following purification, the combined labeled DNA samples were applied to the microarray (hybridization for 40 h at 67°C), washed, and scanned at 3 micron resolution on the Agilent High‐Resolution Microarray Scanner (G2505C, Agilent Technologies). The images were extracted and analyzed using the Agilent Feature Extraction software and the CytoGenomics v.3.0 software suite. The ADM‐1 aberration detection algorithm was utilized, and the minimum number of probes required for a call was set to 4.
Real‐time qPCR
For human tissue samples, total RNA was isolated, following the pulverization of 40–100 mg of bladder tissue specimens, with the use of TRI reagent (Molecular Research Center, Inc., Cincinnati, OH, USA) and reverse transcribed with MMLV reverse transcriptase (Invitrogen, Carlsbad, CA, USA) using oligo‐dT primers. For cell lines, total RNA was isolated with the use of TRI reagent and reverse transcribed with PrimeScript™ RT reagent Kit (Takara, RR037A) using oligo‐dT and random primers. Quantitative PCR was performed in the 7500 Real‐Time PCR System using the sequence detection software (Applied Biosystems, Carlsbad, CA, USA). The 10 μl reaction mixture consists of Kapa SYBR Fast Universal 2× qPCR Master Mix (Kapa Biosystems, Inc., Woburn, MA, USA). Melting curve analysis was performed following the amplification in order to distinguish specific reaction products from non‐specific ones or primer dimers. Gene expression analysis was carried out using the relative quantification method 89. Duplicate or triplicate reactions were performed for each tested sample, and the average C T was calculated for the quantification analysis. HPRT1 was used as an endogenous reference control. Oligonucleotide sequences are provided in Appendix Table S6.
Immunofluorescence experiments
Cells were plated on poly‐L‐lysine (Sigma, cat. P1274)‐coated coverslips. Cells were fixed by 10‐min incubation in 4% paraformaldehyde (Alfa Aesar, 30525‐89‐4) at room temperature, permeabilized for 4 min in 1× PBS/0.5% Triton X‐100, washed with PBS, and blocked in 1% bovine serum albumin (Applichem, cat. A1391,0100) and 10% fetal bovine serum in PBS. Cells were incubated with primary antibody (Appendix Table S7) overnight at 4°C, followed by incubation with a fluorescent secondary antibody for 1 h at room temperature as previously described 90. Antibody solutions were made in PBS with 1% bovine serum albumin. Coverslips were mounted on glass slides using VECTASHIELD Antifade Mounting Medium with 49,6‐diamidino‐2‐phenylindole (DAPI) for DNA staining (Vektor, cat. H‐1200). For DNA repair experiments, cells were treated with 2 μΜ cisplatin for 6 h. For tissue stainings, tumors were fixed in 4% formaldehyde at 4°C, thoroughly washed in PBS, placed in 30% sucrose overnight, and frozen in optimal cutting temperature (OCT) compound (Tissue Tek, Sakura). Frozen 10 μm sections were obtained using a Leica (CM1950) cryostat. Sectioned tissues were washed three times in PBS, blocked for 1 h, and incubated with primary and secondary antibodies as described. Image processing and foci counts were performed using ImageJ.
Cytogenetics and FISH
Standard procedures were used for chromosome preparation and staining 91. Briefly, cells were treated with 10 μg/ml Colcemid™ (Thermo Fisher Scientific, catalog No. 15210040) for 1 h, harvested, treated with 75 mM KCl for 20 min, fixed in methanol/glacial acetic acid (3:1, v/v), and processed for cytogenetic analysis. Imaging and karyotyping were performed via microscopy and computer imaging techniques. At least 30 metaphases per cell line were karyotyped. Karyotypes were analyzed according to the International System for Human Cytogenetic Nomenclature (ISCN) 2013.
MTT cell viability assays
For cell viability assays, cells were plated at 6–10 × 103 per well in 48‐well plates and incubated with complete Dulbecco's Modified Eagle's Medium (DMEM) or RPMI‐1640 medium containing different concentrations of olaparib as indicated in the respective figure legend. Assays were performed using the standard MTT colorimetric assay (Sigma, cat. M5655) according to the manufacturer's instructions. Measurements were analyzed using GraphPad Prism v6.
Soft‐agar clonogenic assays
Basal anchorage‐independent growth inhibition of olaparib was assessed by a double‐layer soft‐agar assay. Cells (5 × 104) were suspended in complete medium containing 0.35% agar and increasing olaparib concentrations, and seeded in triplicate in 24‐well plates onto a base layer of complete medium containing 1% agar. Medium was replenished every 3–5 days for 15–20 days, before colony counting. Image processing and colony counts were performed using ImageJ.
Mice
Male NOD/SCID mice were purchased from the Jackson repository and bred in individually ventilated cages at the Animal House Facility of the Foundation for Biomedical Research Foundation of the Academy of Athens (Athens, Greece) under veterinarian supervision. All procedures for care and treatment of animals were approved by the Institutional Committee on Ethics of Animal Experiments and the Greek Ministry of Agriculture. Cells were injected when mice reached the age of 4–6 weeks. For in vivo treatments, cells were injected in the flanks, and when tumors reached a palpable size (2–3 mm in diameter or dimension), mice were randomly assigned to groups. No exclusion criterion was applied. The olaparib therapy group was intraperitoneally administered with either vehicle or olaparib injection (AZD2281, MedChem express, at a dose of 50 mg/kg in PBS solution containing 12.5% DMSO and 12.5% kolliphor) following the cycling dosing scheme OROOR (O: olaparib or Vehicle, R: Rest) for 21 days. Tumors were dissected and weighed 24 h after the last treatment. Tumor volume measurements were performed every 3 days using a caliper.
Ionizing radiation experiments
Irradiation was carried out in a GammaCell 220 irradiator (Atomic Energy of Canada Ltd., Ottawa, Canada) at room temperature. For chromosomal fusion events analysis, cells synchronized at the G0/G1 phase by growing the cell cultures to confluency followed by serum deprivation for 48 h (0.1–0.25% serum) 92. Cells were exposed to ionizing radiation (6 Gy) and incubated at 37°C for 6 h to recover. At 6 h post‐irradiation, cells were trypsinized and cultured with fresh medium for 30 h. Subsequently, cells were treated with colcemid for 1 h to arrest dividing cells at metaphase and processed for chromosome preparation and staining as previously described. For drug treatments, cells were exposed to 15 μΜ olaparib and/or 30 μΜ SCR7 (MedChem Express, cat. HY‐12742) 24 h prior to irradiation until metaphase harvesting. For each experiment, 30 metaphases were scored. Experiments were repeated thrice. Light microscopy was coupled to an image analysis system (MetaSystems, Altlussheim, Germany) to facilitate scoring.
Protein extraction and Western blot analysis
Cells were lysed in RIPA lysis buffer (150 mM NaCl, 50 mM Tris–HCl pH 8, 1% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS) that was added with a protease inhibitor cocktail (Complete, Roche). A total protein amount of 20 μg from each samples was denatured at 95°C for 10 min in Laemmli buffer containing β‐mercaptoethanol before electrophoresis. The primary antibodies that were used are described in Appendix Table S7.
DNA fiber assay
Asynchronous cell cultures were treated with 25 μM IdU (Sigma‐Aldrich, I7125) for 20 min, washed with PBS, and exposed to 250 μM CldU (Sigma‐Aldrich, C6891) for 20 min (−HU) or exposed to 250 μM CldU and 0.2 mM hydroxyurea for 60 min (+HU). After exposure to CldU, cells were washed in PBS and harvested. Cells were then lysed and DNA fibers stretched onto glass slides and fixed as described 93. Fibers were denatured with 2.5 M HCl for 80 min, washed with PBS, and blocked with 2% BSA in phosphate buffer saline for 30 min. The newly synthesized IdU and CldU tracts were visualized with anti‐BrdU antibodies recognizing IdU (1:50 BD Biosciences, 347580) and CldU (1:400, Abcam, ab6326), respectively. Images were taken at 60× magnification using a Leica DM RA2 fluorescence microscope equipped with a Hamamatsu ORCA‐Flash 4.0 V2 (sCMOS‐Monochrome, 4 Mpixel) camera and analyzed using ImageJ software. Statistical analysis was carried out using GraphPad Prism.
Sister chromatid exchange
Cells (1 × 105) were plated on a 10‐cm dish. At 24 h, cells were treated with 5 μM cisplatin. At 3 h post‐cisplatin treatment, cells were washed with fresh medium and treated with 5 mg/ml BrdU (Sigma‐Aldrich, cat. B5002) for 40 h followed by 0.2 μg/ml colchicine for 3 h. Sister chromatid exchange assays were performed as previously described 94.
Statistical analysis
In human tissue samples, the normality of the distribution of MLL3/KMT2C expression in bladder tissue specimens was evaluated by Shapiro–Wilk test. The nonparametric Wilcoxon signed‐rank test was used to analyze MLL3/KMT2C expression between bladder tumor specimens and matched adjacent normal tissues. Animals were randomly assigned into different groups. Group allocation and outcome assessment were not blinded. In two group comparisons, normality of distribution was determined by D'Agostino & Pearson omnibus normality test and Shapiro–Wilk normality test (paired t‐test). For non‐Gaussian sample distribution or small sample size, Mann–Whitney U‐test was employed. Sample sizes met the minimum requirements of the respective statistical test used. A value of P < 0.05 was considered as significant. Animals which did not develop tumors or did not live through the end of the treatment were excluded. Mann–Whitney U‐test was also employed for statistical evaluation of chromosome number differences in karyotyping experiments and tumor volumes in mouse xenograft experiments.
Author contributions
AKl and TR conceived the study and designed all experiments. TR, DK, MA, AKo, ZK, and EKo performed all experiments. AP analyzed expression array data. AS and KS provided the library of human tumor samples. KNM and GEP performed the IR and karyotyping experiments. MT and EKa performed and analyzed CMA experiments. AKl wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
We would like to thank Dr. E. Chavdoula for providing NOD/SCID mice, Dr. M. Roubelaki for help with the cell cycle analysis, Dr. N. Paschalidis for Annexin V FACS analysis and Drs. V. Paraskevopoulou and P. Karakaidos for critically reading the manuscript. This work was supported by a Greek General Secretariat for Research and Technology “Excellence” grant (UTN_1799), a Fondation Santé Grant in Biomedical Sciences, a Worldwide Cancer Research grant (16/1217) and a Horizon 2020 grant (732309) to AK, and a Greek General Secretariat for Research and Technology and the Hellenic Foundation for Research and Innovation (HFRI) grant (472‐EpiNotch) to TR.
EMBO Reports (2019) 20: e46821
References
- 1. Feinberg AP, Koldobskiy MA, Gondor A (2016) Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 17: 284–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, Kandoth C, Payton JE, Baty J, Welch J et al (2010) DNMT3A mutations in acute myeloid leukemia. N Engl J Med 363: 2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T et al (2010) Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet 42: 665–667 [DOI] [PubMed] [Google Scholar]
- 4. Shilatifard A (2012) The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81: 65–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Miller T, Krogan NJ, Dover J, Erdjument‐Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A (2001) COMPASS: a complex of proteins associated with a trithorax‐related SET domain protein. Proc Natl Acad Sci USA 98: 12902–12907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ziemin‐van der Poel S, McCabe NR, Gill HJ, Espinosa R, Patel Y, Harden A, Rubinelli P, Smith SD, LeBeau MM, Rowley JD (1991) Identification of a gene, MLL, that spans the breakpoint in 11q23 translocations associated with human leukemias. Proc Natl Acad Sci USA 88: 10735–10739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cancer Genome Atlas Research Network (2014) Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 507: 315–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gonzalez‐Perez A, Jene‐Sanz A, Lopez‐Bigas N (2013) The mutational landscape of chromatin regulatory factors across 4,623 tumor samples. Genome Biol 14: r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA et al (2013) Mutational landscape and significance across 12 major cancer types. Nature 502: 333–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rao RC, Dou Y (2015) Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer 15: 334–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Robertson AG, Kim J, Al‐Ahmadie H, Bellmunt J, Guo G, Cherniack AD, Hinoue T, Laird PW, Hoadley KA, Akbani R et al (2017) Comprehensive molecular characterization of muscle‐invasive bladder cancer. Cell 171: 540–556 e25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen C, Liu Y, Rappaport Amy R, Kitzing T, Schultz N, Zhao Z, Shroff Aditya S, Dickins Ross A, Vakoc Christopher R, Bradner James E et al (2014) MLL3 is a haploinsufficient 7q tumor suppressor in acute myeloid leukemia. Cancer Cell 25: 652–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee J, Kim DH, Lee S, Yang QH, Lee DK, Lee SK, Roeder RG, Lee JW (2009) A tumor suppressive coactivator complex of p53 containing ASC‐2 and histone H3‐lysine‐4 methyltransferase MLL3 or its paralogue MLL4. Proc Natl Acad Sci USA 106: 8513–8518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kaikkonen MU, Spann NJ, Heinz S, Romanoski CE, Allison KA, Stender JD, Chun HB, Tough DF, Prinjha RK, Benner C et al (2013) Remodeling of the enhancer landscape during macrophage activation is coupled to enhancer transcription. Mol Cell 51: 310–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang C, Lee JE, Lai B, Macfarlan TS, Xu S, Zhuang L, Liu C, Peng W, Ge K (2016) Enhancer priming by H3K4 methyltransferase MLL4 controls cell fate transition. Proc Natl Acad Sci USA 113: 11871–11876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dorighi KM, Swigut T, Henriques T, Bhanu NV, Scruggs BS, Nady N, Still CD II, Garcia BA, Adelman K, Wysocka J (2017) Mll3 and Mll4 facilitate enhancer RNA synthesis and transcription from promoters independently of H3K4 monomethylation. Mol Cell 66: 568–576 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rickels R, Herz HM, Sze CC, Cao K, Morgan MA, Collings CK, Gause M, Takahashi YH, Wang L, Rendleman EJ et al (2017) Histone H3K4 monomethylation catalyzed by Trr and mammalian COMPASS‐like proteins at enhancers is dispensable for development and viability. Nat Genet 49: 1647–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136: E359–E386 [DOI] [PubMed] [Google Scholar]
- 19. Sanli O, Dobruch J, Knowles MA, Burger M, Alemozaffar M, Nielsen ME, Lotan Y (2017) Bladder cancer. Nat Rev Dis Primers 3: 17022 [DOI] [PubMed] [Google Scholar]
- 20. Hurst CD, Alder O, Platt FM, Droop A, Stead LF, Burns JE, Burghel GJ, Jain S, Klimczak LJ, Lindsay H et al (2017) Genomic subtypes of non‐invasive bladder cancer with distinct metabolic profile and female gender bias in KDM6A mutation frequency. Cancer Cell 32: 701–715 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ali M, Hom RA, Blakeslee W, Ikenouye L, Kutateladze TG (2014) Diverse functions of PHD fingers of the MLL/KMT2 subfamily. Biochim Biophys Acta 1843: 366–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rampias T, Vgenopoulou P, Avgeris M, Polyzos A, Stravodimos K, Valavanis C, Scorilas A, Klinakis A (2014) A new tumor suppressor role for the Notch pathway in bladder cancer. Nat Med 20: 1199–1205 [DOI] [PubMed] [Google Scholar]
- 23. Cancer Genome Atlas Network (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487: 330–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cancer Genome Atlas Research Network (2012) Comprehensive genomic characterization of squamous cell lung cancers. Nature 489: 519–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cancer Genome Atlas Network (2015) Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 517: 576–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cancer Genome Atlas Research Network (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511: 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rabello DDA, Ferreira V, Berzoti‐Coelho MG, Burin SM, Magro CL, Cacemiro MDC, Simoes BP, Saldanha‐Araujo F, de Castro FA, Pittella‐Silva F (2018) MLL2/KMT2D and MLL3/KMT2C expression correlates with disease progression and response to imatinib mesylate in chronic myeloid leukemia. Cancer Cell Int 18: 26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z (2017) GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 45: W98–W102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang WY, Hsu SD, Huang HY, Sun YM, Chou CH, Weng SL, Huang HD (2014) MethHC: a database of DNA methylation and gene expression in human cancer. Nucleic Acids Res 43: D856–D861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ibragimova I, Dulaimi E, Slifker MJ, Chen DDY, Uzzo RG, Cairns P (2014) A global profile of gene promoter methylation in treatment‐naïve urothelial cancer. Epigenetics 9: 760–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hanahan D, Weinberg Robert A (2011) Hallmarks of cancer: the next generation. Cell 144: 646–674 [DOI] [PubMed] [Google Scholar]
- 32. Rada‐Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J (2011) A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470: 279–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu G, Guo Z, Chang X, Kim MS, Nagpal JK, Liu J, Maki JM, Kivirikko KI, Ethier SP, Trink B, et al (2007) LOXL1 and LOXL4 are epigenetically silenced and can inhibit ras/extracellular signal‐regulated kinase signaling pathway in human bladder cancer. Cancer Res 67: 4123–4129 [DOI] [PubMed] [Google Scholar]
- 34. Lin‐Shiao E, Lan Y, Coradin M, Anderson A, Donahue G, Simpson CL, Sen P, Saffie R, Busino L, Garcia BA et al (2018) KMT2D regulates p63 target enhancers to coordinate epithelial homeostasis. Genes Dev 32: 181–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boros J, Donaldson IJ, O'Donnell A, Odrowaz ZA, Zeef L, Lupien M, Meyer CA, Liu XS, Brown M, Sharrocks AD (2009) Elucidation of the ELK1 target gene network reveals a role in the coordinate regulation of core components of the gene regulation machinery. Genome Res 19: 1963–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dhar SS, Zhao D, Lin T, Gu B, Pal K, Wu SJ, Alam H, Lv J, Yun K, Gopalakrishnan V et al (2018) MLL4 is required to maintain broad H3K4me3 peaks and super‐enhancers at tumor suppressor genes. Mol Cell 70: 825–841 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yan J, Chen SA, Local A, Liu T, Qiu Y, Dorighi KM, Preissl S, Rivera CM, Wang C, Ye Z et al (2018) Histone H3 lysine 4 monomethylation modulates long‐range chromatin interactions at enhancers. Cell Res 28: 204–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang L, Zhao Z, Ozark PA, Fantini D, Marshall SA, Rendleman EJ, Cozzolino KA, Louis N, He X, Morgan MA et al (2018) Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nat Med 24: 758–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chae YK, Anker JF, Carneiro BA, Chandra S, Kaplan J, Kalyan A, Santa‐Maria CA, Platanias LC, Giles FJ (2016) Genomic landscape of DNA repair genes in cancer. Oncotarget 7: 23312–23321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bernstein C, Bernstein H (2015) Epigenetic reduction of DNA repair in progression to gastrointestinal cancer. World J Gastrointest Oncol 7: 30–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lahtz C, Pfeifer GP (2011) Epigenetic changes of DNA repair genes in cancer. J Mol Cell Biol 3: 51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Aze A, Zhou JC, Costa A, Costanzo V (2013) DNA replication and homologous recombination factors: acting together to maintain genome stability. Chromosoma 122: 401–413 [DOI] [PubMed] [Google Scholar]
- 43. Carr AM, Lambert S (2013) Replication stress‐induced genome instability: the dark side of replication maintenance by homologous recombination. J Mol Biol 425: 4733–4744 [DOI] [PubMed] [Google Scholar]
- 44. Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V (2010) Rad51 protects nascent DNA from Mre11‐dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17: 1305–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lambert S, Mizuno K, Blaisonneau J, Martineau S, Chanet R, Freon K, Murray JM, Carr AM, Baldacci G (2010) Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol Cell 39: 346–359 [DOI] [PubMed] [Google Scholar]
- 46. Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double‐strand break repair‐independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Berti M, Vindigni A (2016) Replication stress: getting back on track. Nat Struct Mol Biol 23: 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Halazonetis TD, Gorgoulis VG, Bartek J (2008) An oncogene‐induced DNA damage model for cancer development. Science 319: 1352–1355 [DOI] [PubMed] [Google Scholar]
- 49. Macheret M, Halazonetis TD (2018) Intragenic origins due to short G1 phases underlie oncogene‐induced DNA replication stress. Nature 555: 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ait Saada A, Teixeira‐Silva A, Iraqui I, Costes A, Hardy J, Paoletti G, Freon K, Lambert SAE (2017) Unprotected replication forks are converted into mitotic sister chromatid bridges. Mol Cell 66: 398–410 e4 [DOI] [PubMed] [Google Scholar]
- 51. Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, Thompson LH, Takeda S (2001) Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol 21: 2858–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bunting SF, Callén E, Wong N, Chen H‐T, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez‐Capetillo O, Cao L et al (2010) 53BP1 inhibits homologous recombination in Brca1‐deficient cells by blocking resection of DNA breaks. Cell 141: 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wilson TE, Grawunder U, Lieber MR (1997) Yeast DNA ligase IV mediates non‐homologous DNA end joining. Nature 388: 495–498 [DOI] [PubMed] [Google Scholar]
- 54. Mansour WY, Rhein T, Dahm‐Daphi J (2010) The alternative end‐joining pathway for repair of DNA double‐strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res 38: 6065–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G (2006) PARP‐1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res 34: 6170–6182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Srivastava M, Nambiar M, Sharma S, Karki Subhas S, Goldsmith G, Hegde M, Kumar S, Pandey M, Singh Ram K, Ray P et al (2012) An inhibitor of nonhomologous end‐joining abrogates double‐strand break repair and impedes cancer progression. Cell 151: 1474–1487 [DOI] [PubMed] [Google Scholar]
- 57. Menear KA, Adcock C, Boulter R, Cockcroft X‐l, Copsey L, Cranston A, Dillon KJ, Drzewiecki J, Garman S, Gomez S et al (2008) 4‐[3‐(4‐Cyclopropanecarbonylpiperazine‐1‐carbonyl)‐4‐fluorobenzyl]‐2H‐phthalazin‐1‐one: a novel bioavailable inhibitor of Poly(ADP‐ribose) polymerase‐1. J Med Chem 51: 6581–6591 [DOI] [PubMed] [Google Scholar]
- 58. Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C et al (2005) Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434: 917–921 [DOI] [PubMed] [Google Scholar]
- 59. Patel AG, Sarkaria JN, Kaufmann SH (2011) Nonhomologous end joining drives poly(ADP‐ribose) polymerase (PARP) inhibitor lethality in homologous recombination‐deficient cells. Proc Natl Acad Sci USA 108: 3406–3411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McCabe N (2006) Deficiency in the repair of DNA damage by homologous recombination and sensitivity to Poly(ADP‐Ribose) polymerase inhibition. Can Res 66: 8109–8115 [DOI] [PubMed] [Google Scholar]
- 61. Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez‐Lopez R, Nava Rodrigues D, Robinson D, Omlin A, Tunariu N et al (2015) DNA‐repair defects and olaparib in metastatic prostate cancer. N Engl J Med 373: 1697–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR et al (2013) Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res 41: D955–D961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Glaser AP, Fantini D, Shilatifard A, Schaeffer EM, Meeks JJ (2017) The evolving genomic landscape of urothelial carcinoma. Nat Rev Urol 14: 215–229 [DOI] [PubMed] [Google Scholar]
- 64. Lee J, Saha PK, Yang QH, Lee S, Park JY, Suh Y, Lee SK, Chan L, Roeder RG, Lee JW (2008) Targeted inactivation of MLL3 histone H3‐Lys‐4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proc Natl Acad Sci USA 105: 19229–19234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sedkov Y, Benes JJ, Berger JR, Riker KM, Tillib S, Jones RS, Mazo A (1999) Molecular genetic analysis of the Drosophila trithorax‐related gene which encodes a novel SET domain protein. Mech Dev 82: 171–179 [DOI] [PubMed] [Google Scholar]
- 66. Jamal‐Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R et al (2017) Tracking the evolution of non‐small‐cell lung cancer. N Engl J Med 376: 2109–2121 [DOI] [PubMed] [Google Scholar]
- 67. Anjanappa M, Hao Y, Simpson ER, Bhat‐Nakshatri P, Nelson JB, Tersey SA, Mirmira RG, Cohen‐Gadol AA, Saadatzadeh MR, Li L et al (2018) A system for detecting high impact‐low frequency mutations in primary tumors and metastases. Oncogene 37: 185–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Herz HM, Mohan M, Garruss AS, Liang K, Takahashi YH, Mickey K, Voets O, Verrijzer CP, Shilatifard A (2012) Enhancer‐associated H3K4 monomethylation by Trithorax‐related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev 26: 2604–2620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hu D, Gao X, Morgan MA, Herz HM, Smith ER, Shilatifard A (2013) The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol 33: 4745–4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sur I, Taipale J (2016) The role of enhancers in cancer. Nat Rev Cancer 16: 483–493 [DOI] [PubMed] [Google Scholar]
- 71. Gonda TJ, Ramsay RG (2015) Directly targeting transcriptional dysregulation in cancer. Nat Rev Cancer 15: 686–694 [DOI] [PubMed] [Google Scholar]
- 72. Herz HM (2016) Enhancer deregulation in cancer and other diseases. BioEssays 38: 1003–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Herz HM, Hu D, Shilatifard A (2014) Enhancer malfunction in cancer. Mol Cell 53: 859–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Denissov S, Hofemeister H, Marks H, Kranz A, Ciotta G, Singh S, Anastassiadis K, Stunnenberg HG, Stewart AF (2014) Mll2 is required for H3K4 trimethylation on bivalent promoters in embryonic stem cells, whereas Mll1 is redundant. Development 141: 526–537 [DOI] [PubMed] [Google Scholar]
- 75. Hu D, Gao X, Cao K, Morgan MA, Mas G, Smith ER, Volk AG, Bartom ET, Crispino JD, Di Croce L et al (2017) Not all H3K4 methylations are created equal: Mll2/COMPASS dependency in primordial germ cell specification. Mol Cell 65: 460–475 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D'Andrea AD (2015) Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov 5: 1137–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lord CJ, Ashworth A (2017) PARP inhibitors: synthetic lethality in the clinic. Science 355: 1152–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, Smith G, Powell JE, Rudzki Z, Kearns P et al (2010) The PARP inhibitor olaparib induces significant killing of ATM‐deficient lymphoid tumor cells in vitro and in vivo . Blood 116: 4578–4587 [DOI] [PubMed] [Google Scholar]
- 79. Ford E, Nikopoulou C, Kokkalis A, Thanos D (2014) A method for generating highly multiplexed ChIP‐seq libraries. BMC Res Notes 7: 312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nussbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ye T, Krebs AR, Choukrallah M‐A, Keime C, Plewniak F, Davidson I, Tora L (2010) seqMINER: an integrated ChIP‐seq data interpretation platform. Nucleic Acids Res 39: e35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Anders S, Pyl PT, Huber W (2014) HTSeq ‐ A Python framework to work with high‐throughput sequencing data. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Huang DW, Sherman BT, Lempicki RA (2008) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- 89. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- 90. Papafotiou G, Paraskevopoulou V, Vasilaki E, Kanaki Z, Paschalidis N, Klinakis A (2016) KRT14 marks a subpopulation of bladder basal cells with pivotal role in regeneration and tumorigenesis. Nat Commun 7: 11914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Pantelias GE (1986) Radiation‐induced cytogenetic damage in relation to changes in interphase chromosome conformation. Radiat Res 105: 341 [PubMed] [Google Scholar]
- 92. Schorl C, Sedivy JM (2007) Analysis of cell cycle phases and progression in cultured mammalian cells. Methods 41: 143–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Nieminuszczy J, Schwab RA, Niedzwiedz W (2016) The DNA fibre technique ‐ tracking helicases at work. Methods 108: 92–98 [DOI] [PubMed] [Google Scholar]
- 94. Bayani J, Squire JA (2005) Sister chromatid exchange. Curr Protoc Cell Biol Chapter 22: Unit 22.7 [DOI] [PubMed] [Google Scholar]
- 95. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3