

Abstract
Members of the GxGD‐type intramembrane aspartyl proteases have emerged as key players not only in fundamental cellular processes such as B‐cell development or protein glycosylation, but also in development of pathologies, such as Alzheimer's disease or hepatitis virus infections. However, one member of this protease family, signal peptide peptidase‐like 2c (SPPL2c), remains orphan and its capability of proteolysis as well as its physiological function is still enigmatic. Here, we demonstrate that SPPL2c is catalytically active and identify a variety of SPPL2c candidate substrates using proteomics. The majority of the SPPL2c candidate substrates cluster to the biological process of vesicular trafficking. Analysis of selected SNARE proteins reveals proteolytic processing by SPPL2c that impairs vesicular transport and causes retention of cargo proteins in the endoplasmic reticulum. As a consequence, the integrity of subcellular compartments, in particular the Golgi, is disturbed. Together with a strikingly high physiological SPPL2c expression in testis, our data suggest involvement of SPPL2c in acrosome formation during spermatogenesis.
Keywords: glycosyltransferases, intramembrane proteases, SPP/SPPL‐family, SNARE, spermatogenesis
Subject Categories: Membrane & Intracellular Transport; Post-translational Modifications, Proteolysis & Proteomics
Introduction
The high degree of compartmentalization in eukaryotic cells creates a need for specific and precise protein trafficking. To meet this need, cells have developed a complex system of vesicle transport that ensures safe sorting of cargo proteins, in particular between the different compartments of the secretory pathway 1, 2, 3. Vesicles originate from a donor membrane, translocate in a targeted manner, and get specifically tethered to the target membrane, before fusing with it. Soluble N‐ethylmaleimide‐sensitive factor attachment protein receptor (SNARE) proteins are known since three decades to mediate specific membrane fusion 4, 5. So far, 38 SNARE proteins have been identified in mammalian cells and in their vast majority they are tail‐anchored type II transmembrane proteins (type IV). Their N‐terminal domains face the cytosol and comprise a 60–70 amino acid‐long conserved “SNARE motif”, which facilitates coiled‐coil formation 3, 6, 7. For two membranes to fuse, a tetrameric SNARE motif complex needs to form with proteins from the opposing membranes. In most cases, one vesicle‐membrane SNARE (v‐SNARE) binds three target‐membrane SNAREs (t‐SNAREs) forming a trans‐complex, which becomes a cis‐complex as soon as the membranes fuse and all proteins locate to one membrane 6. Classification of v‐ and t‐SNAREs is not always consistent with the actual localization of the proteins, so a more robust way to categorize them is based on the conserved glutamine (Q) or arginine (R) residue within their SNARE motif. Each SNARE complex is formed by one R‐SNARE and three Q‐SNAREs (Qa‐, Qb‐, and Qc‐SNAREs). In most cases, the v‐SNARE corresponds to the R‐SNARE and the t‐SNAREs to the Q‐SNAREs. The N‐ethylmaleimide‐sensitive factor (NSF) and the NSF attachment protein (SNAP) are involved in the dissociation of the cis‐SNARE complex. Subsequently, the incoming SNARE proteins are returned to their donor compartment 5. Vesicle‐associated membrane proteins (VAMPs), syntaxins, and the 25‐kD synaptosomal‐associated protein (SNAP25) are the main components of the SNARE complex. SNAP‐25 represents a subgroup of SNAREs, mostly Qb‐ and Qc‐SNAREs, that are the only ones lacking a transmembrane domain and comprising two SNARE motifs. Syntaxins predominantly act as Qa‐SNAREs, and VAMPs as R‐SNAREs 5. Different combinations of SNARE proteins are responsible for different intracellular trafficking routes.
In mammals, the family of GxGD‐type intramembrane aspartyl proteases comprises presenilin 1 and presenilin 2, which form the catalytically active subunit of the Alzheimer disease‐associated γ‐secretase complex, as well as signal peptide peptidase (SPP) and its four homologous, the SPP‐like (SPPL) proteases, SPPL2a, SPPL2b, SPPL2c, and SPPL3 8, 9. Other vertebrates, such as zebrafish, only express one representative of the SPPL2 subfamily suggesting that in particular the individual members of the SPPL2 subfamily have evolved diverse functions 10. Although SPPL2c exhibits a FD and a GFGD motif, which reflect the catalytic signature motifs of GxGD‐type intramembrane aspartyl proteases 11, the proof of SPPL2c being a catalytically active protease is still missing, not least because to date no SPPL2c substrates have been identified. Due to its highly polymorphic and intron‐less structure, the SPPL2C gene is even discussed to be a pseudogene 12.
All members of the SPP/SPPL family are multi‐pass transmembrane proteins that span the membrane with nine transmembrane (TM) domains 13. In contrast to presenilins, which only process type I (Nout)‐oriented TM segments, one mutuality of all SPP/SPPL‐family members is their selectivity for type II (Nin) TM segments 8. Recently, it was shown that SPP is capable of also processing the type II‐oriented tail‐anchored (type IV) proteins cytochrome B5A (CYB5A), ribosome‐associated membrane protein 4 (RAMP4), and heme oxygenase‐1 (HO‐1) 14, 15. Generally, GxGD‐type aspartyl proteases favor substrates with a short luminal domain 16, 17, 18. Only SPPL3, which was recently shown to act as a sheddase on glycan‐modifying enzymes in cells and in mice, constitutes an exception 19, 20. By proteolytic release of the active site‐containing ectodomain of mature glycosyltransferases, such as N‐acetylglucosaminyltransferase V (GnTV), increased SPPL3 expression induces hypoglycosylation of many cellular glycoproteins, while decreased SPPL3 expression results in hyperglycosylated proteins. Thus, changes in SPPL3 expression can provide a switch to adapt glycan structures in reaction to environmental changes 19. While SPPL3 most likely resides in the Golgi 13, 19, SPP is retained in the endoplasmic reticulum (ER) via a KKXX retention signal 13, 21 and was initially reported to mediate turnover of remnant signal peptides released from nascent protein chains by signal peptidase 22, 23. Deglycosylation of ectopically expressed SPPL2c in HEK293 cells indicates that SPPL2c also resides in the ER 13. However, whether SPPL2c is expressed and what physiological functions it has remains enigmatic.
In the present study, we provide first proof that SPPL2c is a catalytically active GxGD‐type intramembrane protease that impacts on vesicular trafficking by interfering with membrane fusion through processing of SNARE proteins. By altering transport of various cargo proteins, SPPL2c affects different cellular processes, among them protein glycosylation through mislocalization of glycan‐modifying enzymes, such as GnTV. This results in changes in the protein composition of cellular compartments and in the glycan structure of cellular proteins. Such changes naturally occur during spermatogenesis, a process characterized by pronounced compartmental reorganization and specific changes in glycan structures that result in the formation of the acrosome and the glycocalyx in the mature spermatozoon 24. Our data indicate that a strikingly high expression of SPPL2c in spermatids supports these processes during spermatogenesis.
Results
SPPL2c is catalytically active and cleaves SNARE proteins
To identify potential candidate substrates of SPPL2c, we subjected membrane preparations from T‐Rex™‐293 (HEK293) cells stably expressing catalytically active SPPL2c under a doxycycline‐inducible promoter and from control cells without SPPL2c expression to protein label‐free quantification (LFQ) mass spectrometry. Under these conditions, 4,841 proteins were identified by at least two peptides, and 3,726 proteins were relatively quantified on the basis of at least three replicates of SPPL2c‐expressing cells and control cells, and were subjected to statistical analysis. Statistical data analysis revealed 451 out of 917 proteins with a P‐value < 0.05 that were significantly altered in SPPL2c‐expressing cells after false discovery rate (FDR) correction for multiple hypotheses (Dataset EV1). The volcano plot summarizes changes in protein abundance from cells ectopically expressing SPPL2c compared to control cells (Fig 1A). As expected, the strongest change was observed for SPPL2c, which was highly enriched in SPPL2c‐expressing HEK293 cells (Fig 1A). Since SPPL2c is not expressed endogenously in HEK293 cells, the very high fold change determined for SPPL2c may be explained by little carryover of SPPL2c peptides from samples of overexpressing cells into control samples. Because of the known substrate selectivity of SPP/SPPL‐family 8 proteases, we only considered type II transmembrane proteins and type IV tail‐anchored proteins that were decreased upon exogenous expression of SPPL2c as potential candidate substrates. Twenty‐five transmembrane proteins fulfilling these criteria were identified (Table 1). Although not annotated as type 2 or 4 transmembrane, two proteins with the strongest decrease, cytochrome b5 types A and B (fold difference: 0.04 and 0.13), are predicted to have a short C‐terminal vesicular domain followed by a transmembrane anchor in agreement with the topology of type 4 transmembrane proteins. Using the STRING 10.5 software (https://string-db.org/), a protein–protein interaction network of these candidate substrates was created (Fig 1B). This analysis presented a compact interaction network consisting of type IV transmembrane proteins involved in SNARE complex formation with an FDR of 1.01e−07 (Fig 1B; Table 2). In addition, some type II candidate SPPL2c substrates clustered to pathways associated with glycosaminoglycan synthesis with an FDR of 2.6e−04 (Fig 1B; Table 2).
Figure 1. Identification of SPPL2c substrates in HEK293 cells.
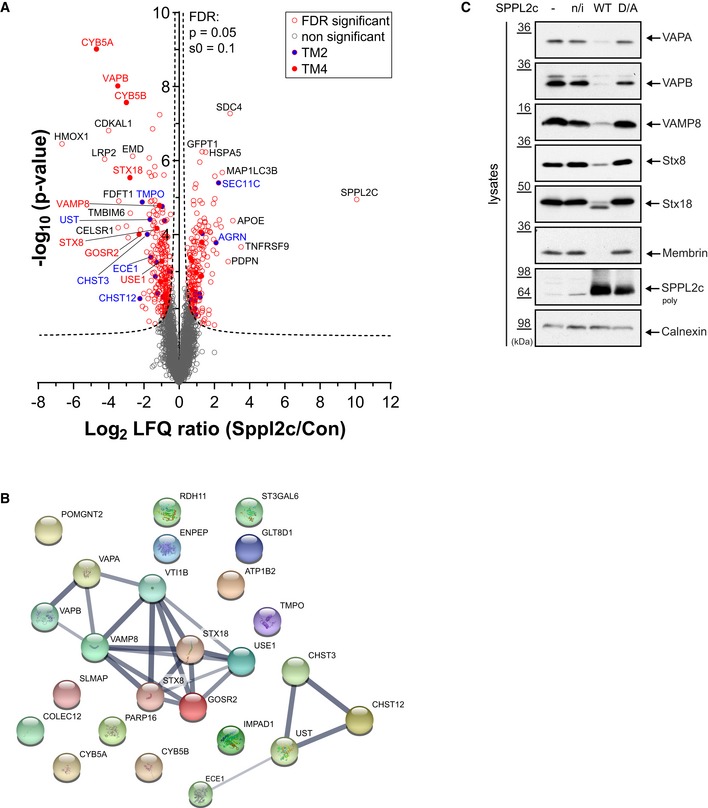
- Volcano plot of proteome analysis. Plotted is the negative log10 of the P‐value (y‐axis) versus the log2 LFQ ratio of SPPL2c‐overexpressing and control cells (x‐axis) of a given identified protein. A permutation‐based FDR correction for multiple hypotheses was applied (P = 0.05; s0 = 0.1; dashed line). Open gray circles indicate proteins not significantly altered in SPPL2c‐expressing cells and red circles significantly altered proteins after FDR correction. Filled red circles indicate type IV membrane proteins and filled blue circles with red line and blue names type II proteins altered in SPPL2c‐expressing cells, n = 6 biological repl. per cell line.
- Pathway analysis of SPPL2c candidate substrates. All type II and type IV membrane proteins significantly decreased in the proteomic analysis following overexpression of SPPL2c (see Table 1) were uploaded on the online STRING 10.5 software (https://string-db.org/). Interaction networks of the candidate substrates are depicted, and the thickness of the lines indicates the degree of confidence in the prediction of the interaction.
- Western blot of selected type IV SPPL2c candidate substrates. Endogenous protein levels of VAPA, VAPB, VAMP8, Syntaxin 8, Syntaxin 18 (Stx8, Stx18), and Membrin (encoded by the gene Golgi SNAP receptor complex member 2, GOSR2) were analyzed in HEK293 cells ectopically expressing wild‐type SPPL2c (SPPL2c WT) or a catalytically inactive SPPL2c (SPPL2c D/A). Non‐transfected cells (−) or stably transfected but non‐induced SPPL2c wt cells (n/i) served as controls. VAPA, VAPB, VAMP8, Stx8, and Membrin protein levels were decreased in SPPL2c‐expressing cells but not in control or SPPL2c D/A‐expressing cells. For Stx18, a cleavage product was detected in SPPL2c WT‐expressing cells. Expression levels of SPPL2c are depicted, and Calnexin serves as loading control.
Source data are available online for this figure.
Table 1.
Significantly decreased type 2 and type 4 transmembrane proteins upon ectopic SPPL2c expression
| UniProt Accession | Protein name | Gene Name | TM2 | TM4 | Ratio (Sppl2c vs. Con) | Log2 (Sppl2c vs. Con) | P‐value |
|---|---|---|---|---|---|---|---|
| P00167 | Cytochrome b5 | CYB5A | 0 | 1* | 0.04 | −4.69 | 9.65E‐10 |
| O95292 | Vesicle‐associated membrane protein‐associated protein B/C | VAPB | 0 | 1 | 0.09 | −3.48 | 9.58E‐09 |
| O43169 | Cytochrome b5 type B | CYB5B | 0 | 1* | 0.13 | −2.99 | 2.67E‐08 |
| Q9P2W9 | Syntaxin 18 | STX18 | 0 | 1 | 0.14 | −2.80 | 2.91E‐06 |
| O14653 | Golgi SNAP receptor complex member 2 | GOSR2 | 0 | 1 | 0.21 | −2.27 | 9.81E‐05 |
| Q9NRB3 | Carbohydrate sulfotransferase 12 | CHST12 | 1 | 0 | 0.21 | −2.23 | 5.36E‐03 |
| P42167 | Lamina‐associated polypeptide 2, isoforms beta/gamma; thymopoietin; thymopentin | TMPO | 1 | 0 | 0.24 | −2.09 | 1.31E‐05 |
| Q7LGC8 | Carbohydrate sulfotransferase 3 | CHST3 | 1 | 0 | 0.28 | −1.81 | 9.84E‐05 |
| Q9Y2C2 | Uronyl 2‐sulfotransferase | UST | 1 | 0 | 0.32 | −1.65 | 3.86E‐05 |
| P42892 | Endothelin‐converting enzyme 1 | ECE1 | 1 | 0 | 0.33 | −1.61 | 4.06E‐04 |
| Q9Y274 | Type 2 lactosamine alpha‐2,3‐sialyltransferase | ST3GAL6 | 1 | 0 | 0.39 | −1.35 | 1.35E‐03 |
| Q9NX62 | Inositol monophosphatase 3 | IMPAD1 | 1 | 0 | 0.41 | −1.28 | 5.60E‐04 |
| Q9UNK0 | Syntaxin 8 | STX8 | 0 | 1 | 0.42 | −1.26 | 6.77E‐05 |
| Q68CQ7 | Glycosyltransferase 8 domain‐containing protein 1 | GLT8D1 | 1 | 0 | 0.43 | −1.22 | 3.90E‐03 |
| Q9BV40 | Vesicle‐associated membrane protein 8 | VAMP8 | 0 | 1 | 0.46 | −1.12 | 1.63E‐05 |
| P14415 | Sodium/potassium‐transporting ATPase subunit beta‐2 | ATP1B2 | 1 | 0 | 0.48 | −1.06 | 1.86E‐03 |
| Q9NZ43 | Vesicle transport protein USE1 | USE1 | 0 | 1 | 0.49 | −1.04 | 1.87E‐03 |
| Q14BN4 | Sarcolemmal membrane‐associated protein | SLMAP | 0 | 1 | 0.51 | −0.97 | 5.22E‐04 |
| Q8TC12 | Retinol dehydrogenase 11 | RDH11 | 1 | 0 | 0.51 | −0.96 | 1.72E‐05 |
| Q5KU26 | Collectin‐12 | COLEC12 | 1 | 0 | 0.53 | −0.92 | 4.86E‐03 |
| Q07075 | Glutamyl aminopeptidase | ENPEP | 1 | 0 | 0.56 | −0.84 | 4.17E‐05 |
| Q9P0L0 | Vesicle‐associated membrane protein‐associated protein A | VAPA | 0 | 1 | 0.58 | −0.77 | 4.15E‐03 |
| Q9UEU0 | Vesicle transport through interaction with t‐SNAREs homolog 1B | VTI1B | 0 | 1 | 0.59 | −0.75 | 1.66E‐03 |
| Q8N5Y8 | Mono [ADP‐ribose] polymerase PARP16 | PARP16 | 0 | 1 | 0.60 | −0.74 | 6.10E‐03 |
| Q8NAT1 | Protein O‐linked‐mannose beta‐1,4‐N‐acetylglucosaminyltransferase 2 | POMGNT2 | 1 | 0 | 0.68 | −0.56 | 9.83E‐04 |
Proteins are sorted by the program according to their fold change (*manually annotated, TM2: transmembrane type 2, TM4: transmembrane type 4). Proteins involved in glycoprotein homeostasis are labeled in italics. Proteins that have been identified as SPPL3 substrates or candidate substrates earlier 19, 20 are marked in bold. Candidate or validated SPPL3 substrates involved in glycoprotein homeostasis are marked in bold italics.
Table 2.
Biological pathways and processes identified by STRING 10.5 analysis of proteins in Table 1
| Pathway description | Observed gene | False discovery rate | Matching proteins in network |
|---|---|---|---|
| KEGG Pathways | |||
| SNARE interactions in vesicular transport | 5 | 1.01e‐07 | STX18, STX8, USE1, VAMP8, VTI1B |
| Glycosaminoglycan biosynthesis—chondroitin sulfate/dermatan sulfate | 3 | 0.00026 | CHST12, CHST3, UST |
| Biological Processes (GO) | |||
| Membrane fusion | 6 | 6.19e‐05 | GOSR2, STX18, STX8, VAMP8, VAPA, VTI1B |
| Vesicle organization | 6 | 0.000645 | GOSR2, STX8, VAMP8, VAPA, VAPB, VTI1B |
| Glycoprotein metabolic process | 7 | 0.000784 | CHST12, CHST3, POMGNT2, IMPAD1, PARP16, ST3GAL6, UST |
| Golgi vesicle transport | 6 | 0.000784 | GOSR2, STX18, VAMP8, VAPA, VAPB, VTI1B |
Proteins were sorted into interaction groups and assigned to different pathways. Listed below are the pathways identified according to either the “Kyoto Encyclopedia of Genes and Genomes” (KEGG) or the Biological Process of the Gene Ontology (GO) project.
To validate the impact of SPPL2c on vesicular transport, selected candidate substrates implicated in this pathway were analyzed in lysates of HEK293 cells either expressing catalytically active SPPL2c (SPPL2c wt) or a catalytically inactive mutant, SPPL2c D448A (SPPL2c D/A; Fig 1C). Levels of the vesicle‐associated membrane protein‐associated proteins A (VAPA) and B (VAPB), the vesicle‐associated membrane protein 8 (VAMP8), Syntaxin 8, and Membrin (encoded by the gene Golgi reassembly‐stacking protein 2, GOSR2) were decreased in cells expressing SPPL2c wt compared to control and SPPL2c D/A‐expressing cells (Fig 1C). This indicates a proteolytic cleavage of the respective substrate by SPPL2c leading to a release of the substrate from the membrane and its subsequent degradation. Syntaxin 18 was also processed by wt SPPL2c resulting in a cleavage product that was readily detected in immunoblot (Fig 1C), indicating a stable cleavage product. In conclusion, these findings demonstrate that SPPL2c is a catalytically active intramembrane aspartyl protease of the GxGD‐type that cleaves protein components of the vesicular transport system in a cellular context.
SPPL2c reduces SPPL3 protein levels and impacts on transport of glycosyltransferases
Proteomic analysis of SPPL2c‐expressing cells also indicated an impact of SPPL2c on glycan‐modifying enzymes (Fig 1A and B, and Table 1). In addition to the glycan‐modifying enzymes with reduced protein levels (Tables 1 and 3), a number of glycosyltransferases accumulated in SPPL2c‐expressing cells (Table 3). Some of these proteins were previously confirmed SPPL3 substrates and accumulated in SPPL3 −/− cells 19, 20. We, thus, analyzed endogenous SPPL3 levels in cells ectopically expressing SPPL2c and observed a reduced SPPL3 expression (Fig 2A), while SPP expression remained unchanged (Fig EV1). To exclude that this observation resulted from clonal variation, we tested three independent SPPL2c‐expressing cell clones for their endogenous SPPL3 expression (Fig EV2A–C). This confirmed that endogenous SPPL3 expression is reduced depending on the amount of ectopically expressed SPPL2c and suggests that this may be the reason for the accumulation of some SPPL3 substrates observed in the mass spectrometric screening.
Table 3.
Type 2 transmembrane proteins increased or decreased upon SPPL2c expression and involved in glycan modification
| UniProt Accession | Protein name | Gene Name | Ratio (Sppl2c vs. Con) | Log2 (Sppl2c vs. Con) | P‐value |
|---|---|---|---|---|---|
| Q10469 | Alpha‐1,6‐mannosyl‐glycoprotein 2‐beta‐N‐acetylglucosaminyltransferase | MGAT2 | 4.74 | 2.24 | 3.98E‐06 |
| O43505 | Beta‐1.4‐glucuronyltransferase 1 | B4GAT1 | 2.23 | 1.16 | 3.97E‐03 |
| O60476 | Mannosyl‐oligosaccharide 1.2‐alpha‐mannosidase IB | MAN1A2 | 1.88 | 0.91 | 1.01E‐02 |
| Q8NCL4 | Polypeptide N‐acetylgalactosaminyltransferase 6 | GALNT6 | 1.88 | 0.91 | 3.51E‐03 |
| Q10471 | Polypeptide N‐acetylgalactosaminyltransferase 2 | GALNT2 | 1.85 | 0.89 | 1.05E‐02 |
| Q10472 | Polypeptide N‐acetylgalactosaminyltransferase 1 | GALNT1 | 1.61 | 0.69 | 1.64E‐02 |
| Q9Y673 | Dolichyl‐phosphate beta‐glucosyltransferase | ALG5 | 1.57 | 0.65 | 2.15E‐02 |
| Q14435 | Polypeptide N‐acetylgalactosaminyltransferase 3 | GALNT3 | 1.55 | 0.63 | 2.09E‐02 |
| Q09328 | Alpha‐1.6‐mannosyl‐glycoprotein 6‐beta‐N‐acetylglucosaminyltransferase A | MGAT5 | 1.53 | 0.61 | 6.03E‐02 |
| Q86SF2 | N‐acetylgalactosaminyltransferase 7 | GALNT7 | 1.49 | 0.58 | 3.40E‐02 |
| Q8WVQ1 | Soluble calcium‐activated nucleotidase 1 | CANT1 | 1.49 | 0.57 | 9.43E‐02 |
| P33908 | Mannosyl‐oligosaccharide 1.2‐alpha‐mannosidase IA | MAN1A1 | 1.47 | 0.56 | 9.26E‐02 |
| Q9UBQ6 | Exostosin‐like 2; processed exostosin‐like 2 | EXTL2 | 1.37 | 0.46 | 1.37E‐01 |
| Q9UKM7 | Endoplasmic reticulum mannosyl‐oligosaccharide 1.2‐alpha‐mannosidase | MAN1B1 | 1.27 | 0.35 | 3.13E‐01 |
| O94766 | Galactosylgalactosylxylosylprotein 3‐beta‐glucuronosyltransferase 3 | B3GAT3 | 1.25 | 0.33 | 1.99E‐01 |
| Q9UBV7 | Beta‐1.4‐galactosyltransferase 7; Xylosylprotein 4‐beta‐galactosyltransferase | B4GALT7 | 1.20 | 0.26 | 3.51E‐01 |
| O43529 | Carbohydrate sulfotransferase 10 | CHST10 | 0.80 | −0.32 | 3.24E‐01 |
| Q8NBI6 | Xyloside xylosyltransferase 1 | XXYLT1 | 0.80 | −0.32 | 1.22E‐01 |
| Q3T906 | N‐acetylglucosamine‐1‐phosphotransferase subunits alpha/beta | GNPTAB | 0.78 | −0.36 | 1.12E‐01 |
| Q7LGA3 | Heparan sulfate 2‐O‐sulfotransferase 1 | HS2ST1 | 0.73 | −0.44 | 1.43E‐01 |
| O94923 | D‐glucuronyl C5‐epimerase | GLCE | 0.71 | −0.50 | 2.66E‐01 |
| Q9NY97 | N‐acetyllactosaminide beta‐1.3‐N‐acetylglucosaminyltransferase 2 | B3GNT2 | 0.69 | −0.53 | 1.40E‐01 |
| P52848 | Bifunctional heparan sulfate N‐deacetylase/N‐sulfotransferase 1 | NDST1 | 0.69 | −0.54 | 3.68E‐02 |
| Q8NAT1 | Protein O‐linked‐mannose beta‐1.4‐N‐acetylglucosaminyltransferase 2 | POMGNT2 | 0.68 | −0.56 | 9.83E‐04 |
| Q4G148 | Glucoside xylosyltransferase 1 | GXYLT1 | 0.67 | −0.59 | 1.56E‐02 |
| Q8NCH0 | Carbohydrate sulfotransferase 14 | CHST14 | 0.65 | −0.63 | 2.15E‐02 |
| P15291 | Beta‐1.4‐galactosyltransferase 1 | B4GALT1 | 0.63 | −0.68 | 3.73E‐02 |
| Q9NS00 | Glycoprotein‐N‐acetylgalactosamine 3‐beta‐galactosyltransferase 1 | C1GALT1 | 0.44 | −1.19 | 4.23E‐02 |
| Q68CQ7 | Glycosyltransferase 8 domain‐containing protein 1 | GLT8D1 | 0.43 | −1.22 | 3.90E‐03 |
| Q9Y274 | Type 2 lactosamine alpha‐2.3‐sialyltransferase | ST3GAL6 | 0.39 | −1.35 | 1.35E‐03 |
| Q9Y2C2 | Uronyl 2‐sulfotransferase | UST | 0.32 | −1.65 | 3.86E‐05 |
| Q7LGC8 | Carbohydrate sulfotransferase 3 | CHST3 | 0.28 | −1.81 | 9.84E‐05 |
| Q9NRB3 | Carbohydrate sulfotransferase 12 | CHST12 | 0.21 | −2.23 | 5.36E‐03 |
Figure 2. SPPL2c downregulates SPPL3 and affects trafficking of membrane proteins in HEK293 cells.
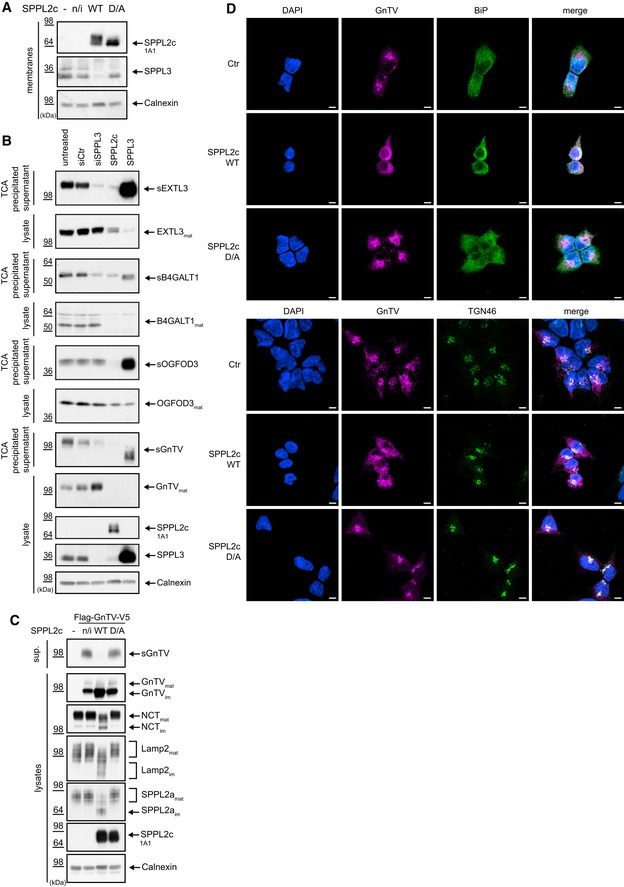
- SPPL2c overexpression causes a decrease in SPPL3 levels. Endogenous levels of SPPL3 were assessed in cells ectopically expressing wild‐type SPPL2c (SPPL2c WT) or catalytically inactive SPPL2c (SPPL2c D/A) by Western blot. Non‐transfected cells (−) or stably transfected but non‐induced SPPL2c wt cells (n/i) served as controls. Note that SPPL3 expression is reduced in cells expressing SPPL2c wt.
- SPPL2c impairs secretion and maturation of glycan‐modifying enzymes. Protein levels of endogenous EXTL3, B4GALT1, OGFOD3, and GnTV were monitored in lysates of cells treated with control siRNA (siCtr) or with siRNA targeting SPPL3 (siSPPL3) and in cells ectopically expressing either SPPL2c or SPPL3 by Western blot. In addition, the secreted forms (sEXTL3, sB4GALT1, sOGFOD3, sGnTV) were analyzed in the corresponding conditioned media using Western blot. Note that SPPL2c reduces secretion of the glycosyltransferases similar to SPPL3 knockdown, but additionally induces a decrease in the mature glycosyltransferases in the cell lysate similar to ectopic expression of SPPL3.
- Processing of ectopically expressed GnTV with an N‐terminal Flag‐ and C‐terminal V5‐tag (Flag‐GnTV‐V5) was analyzed in lysates and conditioned media (sup.) of either SPPL2c wt or SPPL2c D/A‐expressing cells by Western blot. Non‐transfected cells (−) or stably transfected but non‐induced SPPL2c wt cells (n/i) served as controls. In addition, maturation of the glycoproteins Nicastrin (NCT), Lamp2, and SPPL2a was monitored by detection of the respective mature (mat) and immature (im) version. Note that secretion of GnTV is impaired in SPPL2c wt‐expressing cells and immature glycoproteins accumulate.
- SPPL2c induces mislocalization of GnTV. Ectopically expressed GnTV was visualized by immunofluorescence staining using the anti‐V5 antibody either in control cells (Ctr) or in cells co‐expressing either SPPL2c wt or SPPL2c D/A. The ER was stained using the anti‐BiP antibody and the Golgi with the anti‐TGN46 antibody. While in control cells GnTV mainly localizes to the Golgi, its localization shifts to more ER‐like structures in SPPL2c‐expressing cells. Scale bar 5 μm.
Figure EV1. SPP expression is not affected by SPPL2c overexpression.
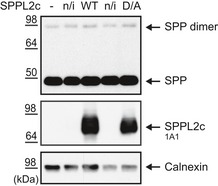
Western blot analysis depicting the SPP expression levels in control HEK293 cells (−), cells overexpressing wild‐type SPPL2c (WT, ind) or catalytically inactive SPPL2c (D/A, ind) and their non‐induced (n/i) controls. Calnexin served as loading control, and ectopic expression of SPPL2c WT and SPPL2c D/A was confirmed using the monoclonal SPPL2c‐specific antibody. Note that neither SPP monomer nor SPP dimer is changed in SPPL2c‐expressing cells compared to control cells.Source data are available online for this figure.
Figure EV2. SPPL3 expression decreases upon SPPL2c overexpression.
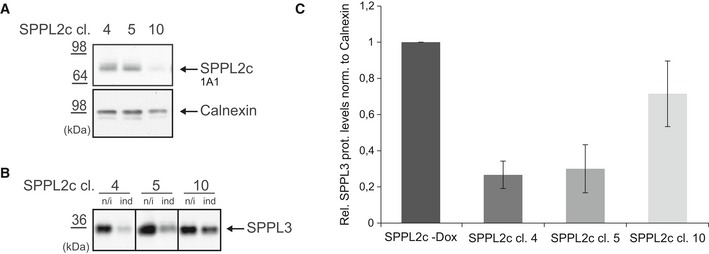
- Western blot analysis depicting the ectopic SPPL2c expression of three independent HEK293 cell clones. Calnexin served as loading control.
- Western blot analysis depicting the SPPL3 expression levels in the three SPPL2c clones under non‐induced (n/i) and induced (ind) conditions.
- Quantification of endogenous SPPL3 protein levels from Fig EV2B normalized to Calnexin (n = 4). Depicted are the ratios of SPPL3 expression before and after induction of SPPL2c overexpression with doxycycline for each SPPL2c‐expressing clone shown in (A). SPPL3 expression of all three non‐induced clones is set to 1 (SPPL2c‐Dox). Mean ± SD. Note that clone number 10 depicts the highest SPPL3 expression and the lowest exogenous SPPL2c expression.
Source data are available online for this figure.
Since some glycosyltransferases that were downregulated in SPPL2c‐expressing cells also have been previously validated as SPPL3 substrates (Table 1, bold letters), we tested whether SPPL2c and SPPL3 substrate spectra overlap. To this end, we analyzed some of the previously characterized SPPL3 substrates 19, 20 for processing by SPPL2c (Fig 2B). Similar to cells with ectopic SPPL3 expression, the mature variants of exostosin‐like 3 (EXTL3), β1,4 galactosyltransferase (B4GALT1), and 2‐oxoglutarate and iron‐dependent oxygenase domain‐containing protein 3 (OGFOD3) were reduced in SPPL2c‐expressing cells (Fig 2B). However, while in SPPL3‐expressing cells secretion of EXTL3, B4GALT1, and OGFOD3 was increased, as indicated by increased amounts of the respective soluble protein fragments in the conditioned media, in SPPL2c‐expressing cells secretion was decreased (Fig 2B). This confirmed that SPPL3 acts as a sheddase on EXTL3, B4GALT1, and OGFOD3 19, 20 and suggested that SPPL2c does not proteolytically process these glycosyltransferases but acts on them differentially. Since the prototype substrate of SPPL3, β1,6‐N‐acetylglucosaminyltransferase V (GnTV), 19 accumulates in the proteomic screening, we also analyzed its processing in SPPL2c‐expressing cells and observed similar effects as for EXTL3, B4GALT1, and OGFOD3 (Fig 2B).
These results suggest that the substrate spectra of SPPL2c and SPPL3 do not overlap. However, if the reduced secretion of SPPL3 substrates in SPPL2c‐expressing cells would be solely caused by downregulation of SPPL3 expression, levels of mature glycosyltransferases in lysates should not be reduced but should rather be stable or accumulate, as observed for GnTV in SPPL3 knockdown conditions (Fig 2B; 19, 20). Thus, SPPL2c may affect protein levels of certain SPPL3 substrates either by reduced transcription/translation or by increased degradation of these proteins. Alternatively, SPPL2c may alter subcellular trafficking of these proteins, resulting in increased amounts of immature glycosyltransferases, which, however, are hardly detected by the antibodies available 19.
Since we have previously shown that immature GnTV is readily detected from ectopically expressed GnTV 19, we co‐expressed N‐terminally Flag‐ and C‐terminally V5‐tagged GnTV with wt SPPL2c or catalytically inactive SPPL2c D/A (Fig 2C). Co‐expression of GnTV with catalytically active SPPL2c revealed a substantial reduction in GnTV secretion (Fig 2C), similar to that observed on endogenous GnTV (Fig 2B), while co‐expression with SPPL2c D/A had no effect (Fig 2C). Immature GnTV was enriched in SPPL2c‐expressing cells, but not in cells expressing in SPPL2c D/A (Fig 2C). This is consistent with the results on endogenous GnTV obtained in the mass spectrometric screening (Table 3;Dataset EV1) and meets our hypothesis that trafficking of GnTV is altered in cells expressing catalytically active SPPL2c. Similarly, endogenous glycoproteins, such as Nicastrin, one component of the γ‐secretase complex, LAMP2, a lysosomal protein, and SPPL2a, another member of the SPP/SPPL family localizing to lysosomes, depicted hypoglycosylation and accumulation of the respective immature protein (Fig 2C). This also depends on the catalytic activity of SPPL2c, since SPPL2c D/A had no effect on protein glycosylation (Fig 2C) and indicates that SPPL2c most likely affects protein transport and not protein expression. To further support this, ectopically expressed GnTV was visualized by immunofluorescence (Fig 2D). While GnTV predominantly resides in the Golgi in control and SPPL2c D/A‐expressing cells, it appears retained in the ER upon co‐expression with wt SPPL2c (Fig 2D).
In summary, our results suggest that catalytically active SPPL2c partially retains glycosyltransferases and most likely also some of their substrates in the ER and, thus, prevents them from reaching their mature state in the Golgi. Subsequently, this leads to reduced catalytically active glycosyltransferases in the Golgi, accounting for the hypoglycosylation of cellular glycoproteins and reduced secretion of the glycosyltransferases.
SPPL2c causes impaired cargo transport by proteolytic processing of SNARE proteins
We next assessed whether cleavage of one of the validated SPPL2c substrates (Fig 1C) might account for the transport defect of GnTV and other cellular proteins. To this end, cells stably expressing tagged GnTV were treated with siRNA directed against the mRNA of the verified SPPL2c substrates. GnTV maturation and cleavage, as well as maturation of endogenous SPPL2a, as a representative endogenous glycoprotein, were compared between the siRNA‐treated cells and cells overexpressing catalytically active SPPL2c (Fig 3A–F). siRNA‐mediated knockdown of the substrates essentially mimicked the reduction in the respective substrate by SPPL2c expression (Fig 3A–F). Although Syntaxin 8 knockdown slightly altered glycosylation of SPPL2a, none of the substrate knockdowns affected maturation of endogenous SPPL2a similarly to SPPL2c expression. In addition, the GnTV maturation and secretion defect in SPPL2c‐overexpressing cells were not entirely mimicked in any of the substrates’ knockdown (Fig 3A–F). Even though Syntaxin 18 (Fig 3E) and Membrin (Fig 3F) slightly reduced secretion of sGnTV, none of the two affected GnTV maturation, indicating that SPPL2c acts additionally on other trafficking factors, which have so far not been validated as SPPL2c substrates. Therefore, we extended our protein–protein interaction network analysis focusing only on the type IV candidate substrates. Using the “More” function of the STRING 10.5 software (https://string-db.org/), we were able to identify further proteins that are involved in membrane fusion during vesicle transport but were not identified in the proteomic screening. Among others, this analysis revealed Syntaxin 5 and Syntaxin 6 as proteins that localize to the Golgi and are closely related to the same functions as the already verified substrates (Fig 4A). Analysis of their protein levels in lysates from cells expressing wt SPPL2c revealed cleavage of Syntaxin 5, while Syntaxin 6 was not processed (Fig 4B). siRNA‐mediated knockdown of Syntaxin 5 in HEK293 cells stably expressing GnTV largely phenocopied the effect of SPPL2c expression on GnTV maturation and cleavage (Fig 4C). While SPPL2a maturation was hardly affected upon Syntaxin 5 knockdown, maturation and glycosylation of Nicastrin were impaired in these cells (Fig 4C), indicating that effects observed in SPPL2c‐expressing cells result from processing of multiple different substrates.
Figure 3. Impact of the validated SPPL2c substrates on GnTV trafficking and glycoprotein maturation in HEK293 cells.
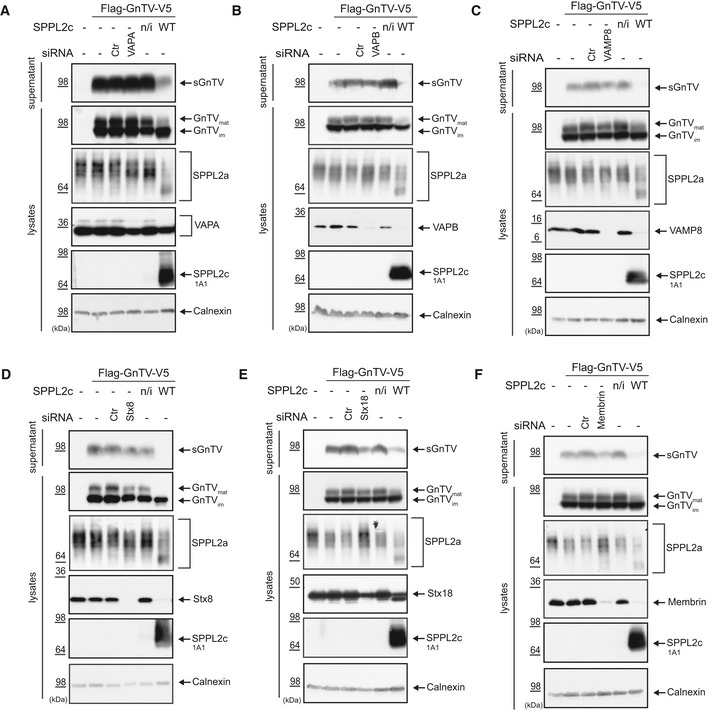
-
A–FProtein levels of the validated SPPL2c substrates VAPA (A), VAPB (B), VAMP8 (C), Syntaxin 8 (Stx8) (D), Syntaxin 18 (Stx18) (E), and Membrin (F) were reduced using specific siRNAs. Non‐targeting control siRNA (Ctr) served as negative control, and overexpression of SPPL2c WT was used as a positive control. Non‐transfected cells (−) or stably transfected but non‐induced SPPL2c wt cells (n/i) served as additional controls for antibody specificity. Processing and maturation of ectopically expressed GnTV and maturation of endogenous SPPL2a were monitored by Western blotting. None of the substrates’ knockdown fully recapitulated the effect on GnTV or on endogenous glycoproteins observed in SPPL2c‐overexpressing cells. Calnexin served as loading control, and ectopic expression of SPPL2c WT and SPPL2c D/A was confirmed using a SPPL2c‐specific antibody.
Source data are available online for this figure.
Figure 4. SPPL2c‐mediated cleavage of Syntaxin 5 impairs GnTV trafficking in HEK293 cells.
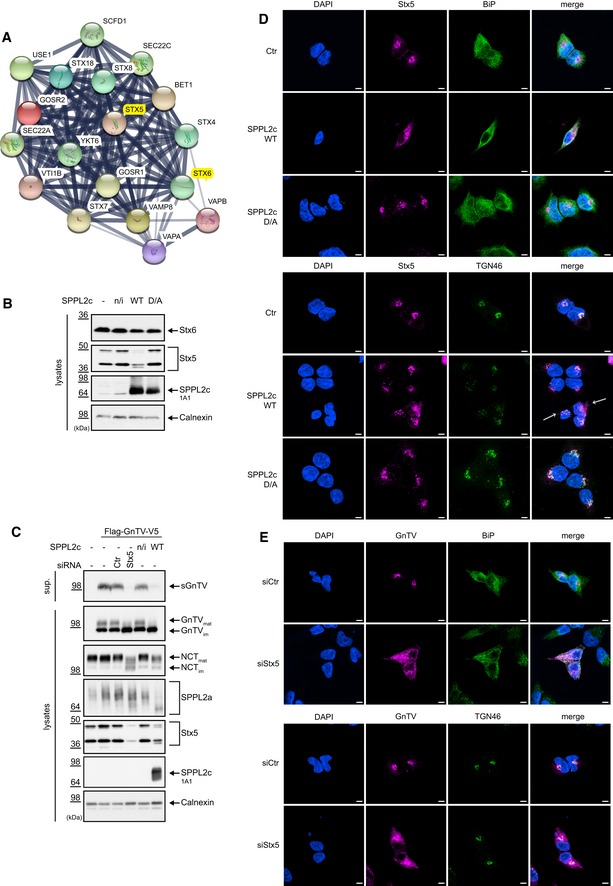
- Additional candidate SPPL2c substrates associated with vesicular trafficking were identified using the function “More” of the STRING 10.5 software. The type IV membrane proteins identified as candidate substrates in the proteomic analysis served as basis for the analysis.
- Processing of endogenous Syntaxin 5 (Stx5) and Syntaxin 6 (Stx6) by SPPL2c wt was assessed as described in Fig 1C by Western blot. Non‐transfected cells (−), stably transfected but non‐induced SPPL2c wt cells (n/i) and induced cells stably expressing SPPL2c D/A served as controls. Results in Figs 1C and 4B are derived from one experiment; thus, expression (SPPL2c) and loading (Calnexin) control are the same. Note, that Syntaxin 5 is cleaved by SPPL2c while Syntaxin 6 is not.
- Syntaxin 5 protein levels were down regulated as described in Fig 3, and processing and maturation of ectopically expressed GnTV, as well maturation of the endogenous glycoproteins Nicastrin (NCT) and SPPL2a, were analyzed in Western blot. Calnexin served as loading control, and ectopic expression of SPPL2c WT was confirmed using a SPPL2c‐specific antibody. While GnTV and Nicastrin maturation were similarly affected by Syntaxin 5 knockdown and SPPL2c overexpression, SPPL2a maturation was hardly impaired in Syntaxin 5 knockdown cells.
- Syntaxin 5 localization is impaired by overexpression of SPPL2c. Endogenous Syntaxin 5 was visualized in control cells without SPPL2c expression (Ctr) and in cells overexpressing SPPL2c wt or SPPL2c D/A using immunofluorescence. BiP was used as a marker protein of the ER and TGN46 as Golgi marker. White arrow indicating cells with disturbed Golgi and Syntaxin 5 staining. Scale bar 5 μm.
- Syntaxin 5 knockdown induces mislocalization of GnTV similar to SPPL2c overexpression (Fig 2D). Cells ectopically expressing GnTV were treated either with non‐specific siRNA (siCtr) or with siRNA targeting Syntaxin 5 (siStx5). GnTV was visualized in immunofluorescence using anti‐V5 antibody targeting its C‐terminal tag and either BIP was co‐stained as ER marker protein or TGN46 as Golgi marker protein. Scale bar 5 μm.
Source data are available online for this figure.
To further support that SPPL2c‐mediated cleavage of Syntaxin 5 results in transport blockage, we carried out immunofluorescence stainings of Syntaxin 5 in cells expressing SPPL2c wt or SPPL2c D/A (Fig 4D). As expected, in control cells and SPPL2c D/A‐expressing cells, Syntaxin 5 mainly localized to the Golgi. In contrast, co‐localization with Golgi markers in cells expressing catalytically active SPPL2c appears to be less pronounced. However, predominant co‐localization with the ER marker protein BiP is also not observed (Fig 4D). Thus, we conclude that SPPL2c induces functional inactivation of Syntaxin 5, which destroys its capability to mediate membrane fusion at the cis‐Golgi.
Since SPPL2c induced mislocalization of GnTV (Fig 2D) and Syntaxin 5 (Fig 4D), we asked to what extent the absence of Syntaxin 5 affects GnTV trafficking. siRNA‐mediated knockdown of Syntaxin 5 induced pronounced retention of GnTV in the ER, as indicated by a strong overlap of BIP and GnTV staining in Syntaxin 5 siRNA‐treated cells, while it mainly localized to the Golgi in control siRNA‐treated cells (Fig 4E).
Taken together, these data indicate that SPPL2c impairs the function of a variety of vesicle‐associated proteins, thus blocking vesicular transport between ER and Golgi, and retaining some cargo proteins in the ER or blocking the fusion of the respective vesicles with the Golgi.
SPPL2c expression disturbs integrity of subcellular compartments
An intact protein transport is a prerequisite for maintaining the integrity of subcellular compartments in particular the Golgi 25. Therefore, we asked whether prolonged expression of SPPL2c impacts on the integrity of cellular compartments, in our HEK293 model. To this end, we induced ectopic expression of SPPL2c for 24 and 48 h, and visualized the ER, the cis/medial‐Golgi, and the trans‐Golgi network using immunofluorescence staining (Figs 5 and EV3). Compared to control cells, the ER marker protein BiP, the cis/medial‐Golgi marker protein giantin, and Cab45, which stains all Golgi subcompartments, were mislocalized in SPPL2c‐expressing cells (Figs 5 and EV3), indicating that the integrity of these subcellular structures was disturbed. Impairment of compartment morphology became more pronounced the longer catalytically active SPPL2c was expressed (Figs 5 and EV3), supporting our hypothesis, that vesicular trafficking is impaired and as a consequence, Golgi integrity is disturbed. Although not as pronounced, also the trans‐Golgi marker protein TGN46 depicted changes upon SPPL2c expression. After 48 h of SPPL2c expression, ER and trans‐Golgi network appeared impaired, probably due to the lack of export from the ER and limited supply to the later compartments. Importantly, changes in compartment morphology were not observed upon ectopic expression of SPPL2c D/A, excluding some alterations of subcellular structures caused solely by overexpression of a polytopic ER membrane protein.
Figure 5. SPPL2c impacts on the integrity of subcellular compartments in HEK293 cells.
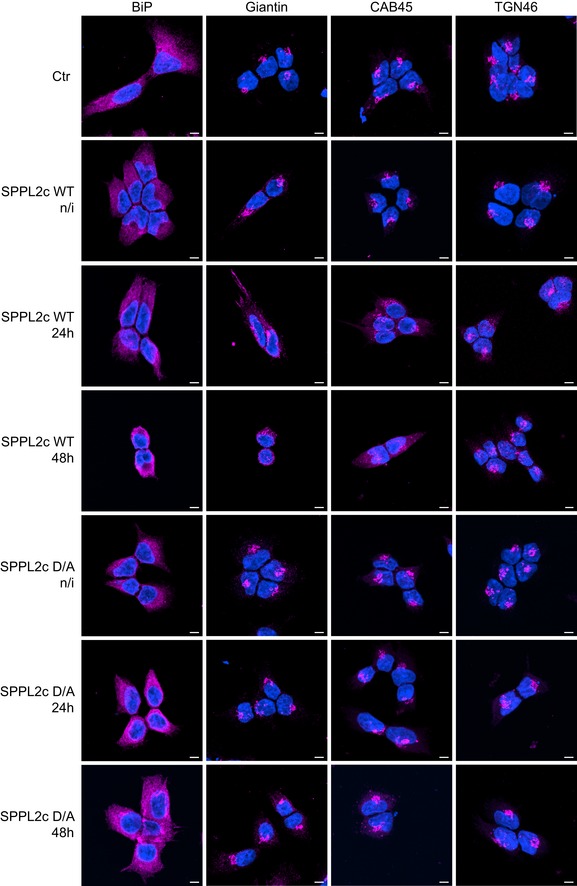
After seeding, cells were cultured for 72 h and expression of catalytically active (wt) or non‐active (D/A) SPPL2c was induced by addition of doxycycline for either 24 or 48 h as indicated. Non‐transfected cells (Ctr) or non‐induced SPPL2c cells (n/i) served as controls. The ER was stained with the anti‐BiP antibody, the cis/medial‐Golgi with the anti‐Giantin, the trans‐Golgi network with the anti‐TGN46 antibody, and all Golgi subcompartments with the CAB45‐specific antibody in immunofluorescence. Note that only upon expression of catalytically active SPPL2c, the morphologies of ER and cis‐Golgi change compared to controls. Scale bar 5 μm.
Figure EV3. ER and Golgi morphology in HEK293 cells with ectopic expression of SPPL2c.
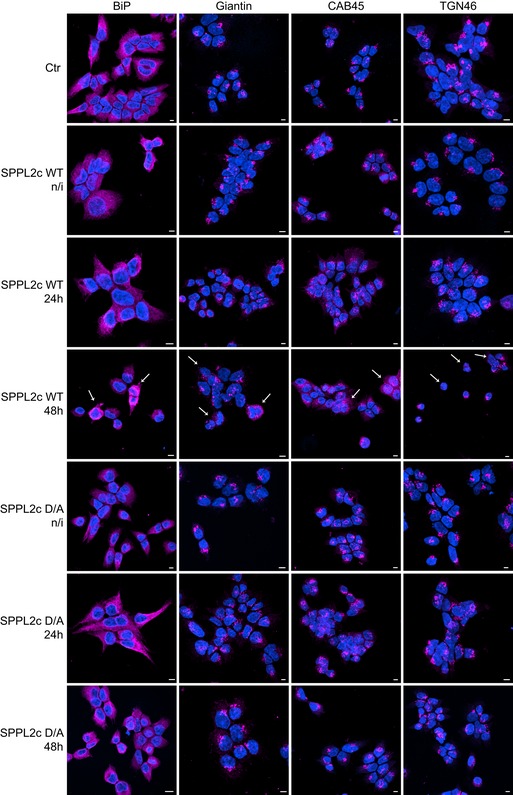
Cells were seeded and cultured for 72 h and expression of catalytically active (WT) or non‐active (D/A) SPPL2c was induced by addition of doxycycline for either 24 or 48 h as indicated. Non‐transfected cells (Ctr) or non‐induced SPPL2c cells (n/i) served as controls. The ER was stained with the anti‐BiP antibody, the cis/medial‐Golgi with the anti‐Giantin, the trans‐Golgi network with the anti‐TGN46 antibody, and all Golgi subcompartments with the CAB45‐specific antibody. Note that only upon expression of catalytically active SPPL2c, the morphologies of ER and cis‐Golgi change compared to controls. The most representative changes are indicated with white arrows at SPPL2c WT 48 h. This figure contains a smaller magnification of the same experimental setup as in Fig 5D. Scale bar 5 μm.
SPPL2c supports acrosome formation in the maturating spermatids
Expression analysis on mRNA and protein level revealed physiological SPPL2c expression in murine and human testis but so far not in any other tested tissue (Fig 6A and B, and accompanying manuscript by Niemeyer et al 26). Proteomic analysis of SPPL2c −/− testis samples identified Syntaxin 8 as a candidate SPPL2c substrate also in vivo 26, suggesting cleavage of SNARE proteins by SPPL2c also in vivo. Since expression of SPPL2c has a pronounced effect on vesicular transport, it is comprehensible why it is expressed in hardly any cell or tissue under normal conditions. Of note, SPPL2c expression in testis is remarkably high, since the amount of SPPL2c detected in only 5 μg protein obtained from membrane preparations of murine or human whole testis is similar to that detected in 0.1 μg membrane protein (1:50 dilution) of HEK293 cells ectopically expressing SPPL2c (Fig 6A). In comparison with other tissues from wt C57BL/6 mice, testis depicted a strong accumulation of immature Nicastrin and SPPL2a (Fig 6B), supporting that protein transport and/or protein maturation in testis differs from that in other tissues. To investigate whether processing of proteins involved in vesicular transport is impaired in testis from SPPL2c‐deficient mice, we analyzed whole testis membrane preparations from these mice with regard to accumulation of the verified SPPL2c substrates involved in vesicular transport. Confirming the data obtained by proteomics (Fig 1A and Ref. 26), a significant accumulation of Syntaxin 8 was observed in SPPL2c −/− mouse testis (Fig 6C and D), while Syntaxin 8 mRNA remained unchanged (Fig 6E), suggesting a lack of Syntaxin 8 processing in SPPL2c −/− mouse testis. However, we were not able to demonstrate the accumulation of other SNARE proteins, most likely because SPPL2c expression in testis was found predominantly in maturating spermatids 26. Thus, accumulation of SNARE proteins in a single subpopulation of cells will not be readily detected if the respective SNARE proteins are expressed in all other testis cells. On the other hand, restricted SPPL2c expression but detection of high SPPL2c protein levels in whole testis lysate (Fig 6A) suggests an even higher expression of SPPL2c in spermatids that may almost achieve levels detected in HEK293 cells with ectopic expression of SPPL2c. Spermatids elongate, and during this final step of spermatogenesis loss of many organelles and parts of the cytosol occurs and, thus, formation of the acrosome is initiated 27. To investigate whether SPPL2c impacts on this fundamental reorganization of subcellular compartments, we analyzed stages VII and VIII of spermatogenesis (according to stage classification, as detailed by Ref. 28) in cross sections of seminiferous tubules of SPPL2c −/− mice and littermate wt controls. To this end Cab45, the Golgi marker protein that was most prominently changed upon SPPL2c expression in our cell culture model, was labeled immunohistochemically in the cross sections of seminiferous tubules. While in wt spermatids Cab45 predominantly localized to compact, dense structures, in SPPL2c −/− mice, it mainly localizes to less condensed and ordered structures (Fig 6F). This indicates that the compartment reorganization that takes place in spermatids is less efficient in SPPL2c −/− spermatids. Quantification revealed a roughly 50% reduction in these compact, dense structures in SPPL2c −/− mice compared to wt controls (Fig EV4A). To further corroborate differences between the two genotypes, 14 images were blindly evaluated. Five independent people assigned the genotypes with an average hit rate of 86% (Fig EV4B) assuring changes in spermatid maturation in the absence of SPPL2c.
Figure 6. SPPL2c cleaves SNARE proteins in testis and is involved in acrosome formation.
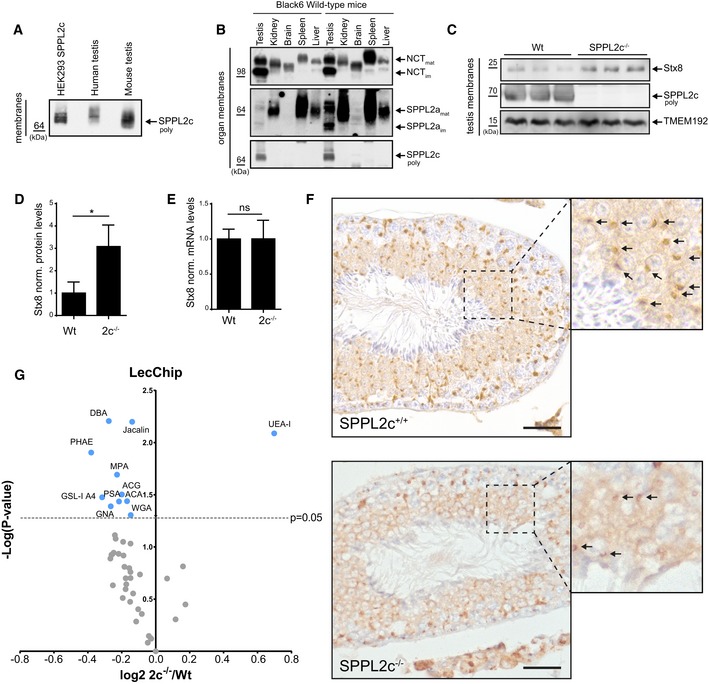
- Western blot analysis of SPPL2c in membrane preparations from HEK293 inducibly overexpressing SPPL2c, and human testis and mouse testis tissue. For human and mouse tissue, 5 μg protein of each total testis preparation is loaded. The membrane fraction obtained from a confluent 6‐cm dish of SPPL2c‐expressing HEK293 cells is diluted 1:50 (0.1 μg total protein).
- Western blot analysis of selected glycoproteins in mouse tissues from two wild‐type C57BL/6J (Black6) mice. Endogenous Nicastrin (NCT) and SPPL2a were analyzed in membrane preparations from testis, kidney, brain, spleen, and liver in littermate male mice. Note that immature proteins (im) accumulate in testis compared to other tissues. SPPL2c protein is only detected in testis tissue.
- Analysis of Syntaxin 8 (Stx8) in SPPL2c −/− mice compared to wild‐type (wt) littermates. Membrane isolates from whole testis were analyzed for Syntaxin 8 (Stx8) and SPPL2c expression. TMEM192 serves as loading control.
- Syntaxin 8 protein levels were quantified from the respective Western blots by densitometric analysis and normalized to those of TMEM192. Bars represent mean values of n = 3 animals ± SD normalized to mean Syntaxin 8 levels of wild‐type mice. Statistical significance was calculated applying an unpaired, two‐sided Student's t‐test. *P ≤ 0.05. Note that Syntaxin 8 is accumulating in SPPL2c −/− mice.
- Real‐time qPCR analysis of Syntaxin 8 (Stx8) mRNA levels in testis of SPPL2c −/− and the wild‐type mice normalized to that of Tuba1a. The diagram depicts mean values of Stx8 mRNA levels normalized to that of wild‐type animals of n = 6 individuals from two independent experiments ± SD. For statistical analysis, an unpaired, two‐sided Student's t‐test was performed. ns = not significant.
- Immunohistochemical stainings of cross‐sectioned seminiferous tubules of SPPL2c +/+ and SPPL2c −/− mice using anti‐Cab45 antibody. Stages VII and VIII are depicted. Higher magnifications are indicated with dashed lines and black arrows indicate pre‐acrosomal structures. Scale bar 50 μm.
- Volcano plot depicting the binding affinities of lectins on sperm lysates from SPPL2c −/− mice compared to SPPL2c +/+ (wt). Each circle represents one lectin. The lectins above the dashed line (light blue) have a significant P‐value < 0.05. Lectins on the left side of the blot demonstrate reduced binding while UEA‐I on the right side has increased binding. n = 4 animals per genotype, two‐sided, unpaired Student's t‐test.
Source data are available online for this figure.
Figure EV4. Quantification of pre‐acrosomal structures in seminiferous tubular cross sections.
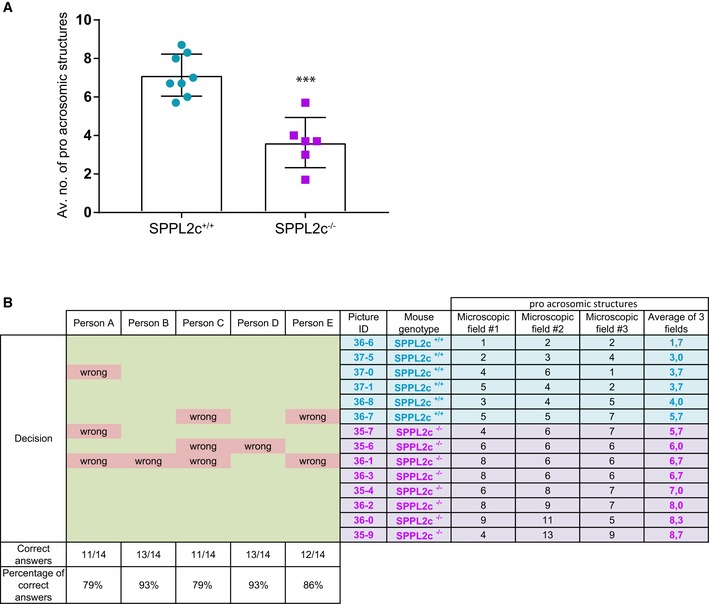
- Pre‐acrosomal resembling structures stained immunohistochemically with Cab45 (Fig 6F, black arrows) were quantified in n = 14 stage VII/VIII seminiferous tubules (n = 6 SPPL2c+/+ (petrol) and n = 8 SPPL2c−/− (lilac)) as described in Materials & Methods. The average number of stainings per tubule is depicted for each genotype, mean ±SD. Unpaired, two‐sided Welch's t‐test. ***P < 0.001 (P = 0.0004).
- Table depicting the stainings counted for each single picture (microscopic fields #1–3) as well as the “average of 3 fields” plotted on Fig EV4A. The genotype of each mouse is indicated, as well as the blinded assignment of the pictures by five independent people, accompanied by the number and percentage of correct evaluations.
Maturation of spermatids and acrosome formation is accompanied by changes in glycan structures, an important prerequisite for fertility 29. Using a lectin chip microarray (LecChip), we analyzed the glycome fingerprint of mature spermatozoa isolated from the cauda epididymis of SPPL2c −/− and control mice. Total lysates of mature spermatozoa were applied to a LecChip containing 45 different lectins. Reactivity with 10 lectins was significantly reduced, while one was increased in sperms from SPPL2c −/− compared to wt controls (Figs 6G and EV5, and Dataset EV2), indicating that limitations in compartmental reorganization during spermatid maturation also lead to manifest changes in the glycan structures of mature spermatozoa. Several glycan structures involved in sperm maturation showed a reduced abundance detected by decreased binding affinity of the lectins Jacalin, MPA, and ACA, as well as WGA, reduction of which is related to teratozoospermia 30, 31. Of note, PSA, a lectin from Pisum sativium that had been previously reported as an acrosome marker 30, was also found to be one of the lectins that depicted reduced binding affinity in SPPL2c −/− sperms (Figs 6G and EV5). Together these data support our hypothesis that catalytic activity of SPPL2c supports compartmental reorganization in maturating spermatids and most likely helps to form the acrosome.
Figure EV5. Glycome fingerprint of mature spermatozoa isolated from the cauda epididymis of SPPL2c−/− and control mice.
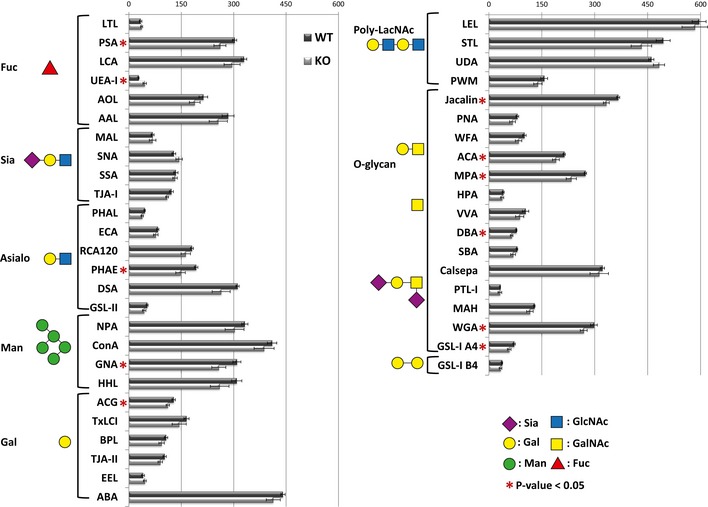
The lectins that were significantly changed are marked with a red asterisk (P‐value < 0.05, unpaired, two‐sided Student's t‐test, n = 4 animals per genotype).
Discussion
In this study, we identified the first substrates for SPPL2c, a so far orphan protease, using systematic proteomic approaches. While analysis of testis tissue from SPPL2c −/− mice revealed significant enrichment of only a few proteins 26, ectopic expression of SPPL2c in HEK293 cells resulted in changes in a variety of proteins that mainly cluster to pathways involved in vesicular trafficking, specifically membrane fusion, and protein glycosylation. Given the restricted expression of SPPL2c to maturating spermatids 26, it is likely that many candidate substrates have been missed in the analysis of whole testis preparations due to dilution effects. Similar to proteome‐wide screens on SPPL3 −/− cells and cells with ectopic SPPL3 expression 20, substrate spectra of SPPL2c overlapped to some extent in the knock out model and the overexpression model, but the majority of candidate substrates differed. However, since testis samples were of murine, while our cell culture system is of human origin, we cannot exclude that also the species may account for differences observed in the two proteomic screenings. Validation of selected substrates in cells expressing either wt SPPL2c or SPPL2c D/A, which carries a mutation that inactivates its potential catalytic center, revealed that SPPL2c indeed is a catalytically active protease with a functional GxGD motif (Figs 1 and 2).
Besides candidates that were validated as SPPL2c substrates, also protein levels of validated SPP substrates, such as Cyb5a, which were not processed by SPPL2c 26, were reduced in SPPL2c‐expressing cells (Fig 1). Since SPP levels were unchanged in SPPL2c‐expressing cells compared to control cells (Fig EV1) and SPPL2c expression results in ER retention of certain secretory proteins (Fig 4), we conclude that SPP likely has more access to some of its substrates due to increased co‐localization, and thus, elevated processing is observed. In addition, this mechanism may also account for increased processing of proteins by other unrelated proteases in the ER or prevent processing of proteins in later secretory compartments, accounting for the pronounced changes observed in the proteome of SPPL2c‐expressing cells (Fig 1).
Pathway analysis of SPPL2c candidate substrates reveals that SNARE‐mediated vesicle fusion is the most significantly affected biological process in cells ectopically expressing SPPL2c (Fig 1). Biochemical analysis of selected proteins implicated in this biological process revealed processing of different SNARE proteins by SPPL2c in HEK cells and analysis of Syntaxin 8 in testis of SPPL2c −/− mice revealed accumulation compared to wild‐type controls (Fig 6C and D), indicating that SPPL2c processes SNARE proteins also in a physiological context.
Although SPPL2c affects a variety of trafficking‐associated proteins, it yet displays some specificity. For instance, Syntaxin 6, which is implicated in a variety of transport processes including transport to the trans‐Golgi network, the early endosomes, and the plasma membrane, is not processed by SPPL2c (Fig 4). This might be because Syntaxin 6 rather belongs to the family of SNAP‐25 proteins and not the syntaxins 32. From this, we conclude that SPPL2c displays certain selectivity in substrate selection as known for other members of the SPP/SPPL family 17, 18, 33. In order to finally determine the substrate requirements of SPPL2c in detail, more in‐depth studies will be required.
Cleavage of SNARE proteins by SPPL2c mostly results in reduction in the substrates protein levels (Fig 1C), suggesting that the SNARE‐cleavage products are rapidly degraded. However, for some SNARE proteins, such as Syntaxin 5 or Syntaxin 18, a stable cleavage product was readily detected (Figs 1C and 4B), indicating different degradation pathways. Although we cannot ultimately exclude an indirect effect of SPPL2c on Syntaxin 5 or Syntaxin 18 via, for instance, a proteolytic cascade, these results strongly suggest proteolytic processing of various SNARE proteins by SPPL2c.
According to deglycosylation experiments on ectopically expressed SPPL2c 13, SPPL2c most likely localizes to the ER and early Golgi compartments, where it gets access to a variety of SNARE proteins that either fulfill their function in these subcellular compartments or pass through during maturation. Based on the impairment of ER and Golgi integrity upon SPPL2c expression (Fig 5), we hypothesize that SPPL2c cleaves its substrates in these early compartments, resulting in a release of the cytosolic SNARE protein domain and, thus, impairing their function in normal vesicle fusion (Figs 7A and B). Consequently, cargo proteins such as GnTV, Nicastrin, SPPL2a, and others remain immature and are likely retained in the ER (Figs 2 and 7C). Accumulation of such cargo proteins in the ER may induce endoplasmic‐reticulum‐associated protein degradation (ERAD), explaining why some of these cargo proteins, such as glycosyltransferases (Table 3), displayed reduced protein abundance in the mass spectrometric analysis of SPPL2c‐overexpressing cells (Fig 1A).
Figure 7. Hypothetical model depicting the effect of SPPL2c overexpression on vesicular trafficking.
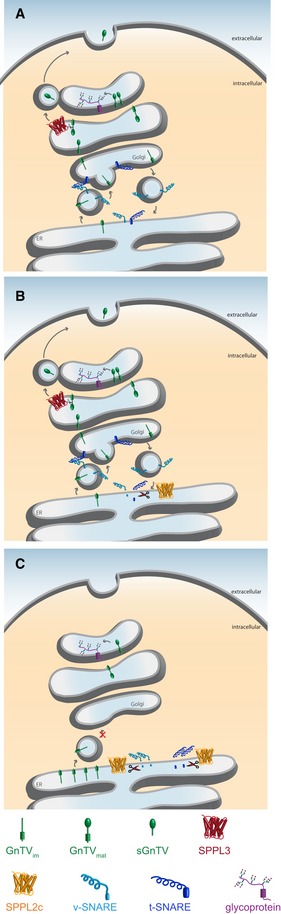
- Membrane‐tethered glycosyltransferases (represented here by GnTV in green) and members of the SNARE family (dark and light blue representing t‐ and v‐SNAREs respectively) are synthesized and inserted in the ER. The glycosyltransferases are transported to the Golgi with the aid of the SNAREs via vesicle transport. Within different compartments of the Golgi, they mature and either meet SPPL3 (red) that can shed and liberate their active site, or they catalyze protein glycosylation (glycoprotein depicted in purple).
- Upon expression of SPPL2c (orange) in the ER, selective SNARE proteins are proteolytically processed.
- Cleavage of SNARE proteins impairs vesicle fusion at the cis‐Golgi and among others, immature glycosyltransferases accumulate in the ER, while the amount of glycosyltransferases in the Golgi decreases. This leads to a decrease in protein glycosylation, and at the same time, SPPL3 (red) probably decreases, most likely to preserve the small amount of glycoproteins that have reached the Golgi.
Other cargo proteins accumulated in cells overexpressing SPPL2c (Fig 1A). Among them, proteins implicated in glycan modification of secreted and membrane proteins (Table 3), which have been identified as SPPL3 substrates earlier 19, 20. SPPL3 has been recently shown to proteolytically release the extracellular domains of glycan‐modifying enzymes, which leads to secretion of the active site‐containing domain of these enzymes and, thus, causes their inactivation 19, 20. Consequently, low levels of SPPL3 preserve more of the active glycan‐modifying enzymes, such as GnTV, in the Golgi where they act on cellular glycoproteins 19. Ectopic expression of SPPL2c induces a reduction in endogenous expression of SPPL3 (Figs 2A and EV2). Therefore, accumulation of certain glycan‐modifying enzymes in SPPL2c‐expressing cells may not be solely due to retention in the ER, but also due to reduced processing by SPPL3. The mechanism inducing the downregulation of SPPL3 remains so far enigmatic. However, since ectopic expression of SPPL2c causes impaired cargo transport from the ER to the Golgi and reduces the number of active glycan‐modifying enzymes in the Golgi, it is tempting to speculate that reduction of SPPL3 expression is a counter reaction to preserve the remaining glycosyltransferase activity in the Golgi (Fig 7C). This suggests that due to mislocalization of cargo, SPPL2c can induce secondary effects with strong impact on cellular processes. At present, it is unclear which signaling cascades trigger these secondary effects and whether they differ in distinct cell types. Further studies will be necessary to unravel these details.
Besides those SNARE proteins that were identified as SPPL2c substrates in the proteomic approaches, also Syntaxin 5 and potentially further members of the SNARE family are processed in a SPPL2c‐dependent manner (Fig 4). In contrast to substrates identified in the proteomic screening, it seems that the total amount of Syntaxin 5 is not reduced upon processing by SPPL2c, since the cleavage product is stable and readily detected in Western blot (Fig 4B and C). Thus, hardly any change in total protein is expected when cells with and without SPPL2c expression are compared by mass spectrometry. However, the localization of Syntaxin 5 was changed upon expression of SPPL2c (Fig 4E). As reported already in 1998, the presence of Syntaxin 5 in the Golgi is essential, not only for transport of cargo to the Golgi, but most probably also for the assembly of pre‐Golgi intermediates 34.
Given the diverse functions of the SNARE proteins affected, SPPL2c expression likely impairs not only ER to Golgi trafficking, which is the dominant phenotype observed in our model system, but also trafficking between other compartments of the secretory pathways. Therefore, SPPL2c expression is apparently highly restricted under healthy, physiological conditions. In contrast to this hypothesis, SPPL2c mRNA was found in a variety of human tissues 13; however, since the SPPL2c gene does not contain any introns, traces of genomic DNA co‐purified with RNA may well reflect a false‐positive detection of mRNA. This is supported by the FANTOM5 CAGE profiles of human and mouse samples, and the genotype‐tissue expression (GTEx) project, which detect SPPL2c mRNA almost exclusively in testis 35, 36.
In fact, SPPL2c protein expression has so far only been unequivocally detected in testes of mice and humans (Fig 6A and B, and 26). In particular, SPPL2c was predominantly found in maturating spermatids, which undergo a comprehensive reorganization of their subcellular compartments to form spermatozoa 27. During this differentiation step, the Golgi is dissolved to form proacrosomic granules, which subsequently fuse with each other to form large acrosomic granules that associate with the nuclear envelope 37. Our data suggest that SPPL2c −/− spermatids depict altered capability of forming these large acrosomic granules (Fig 6F), indicating that either fragmentation of the Golgi and/or fusion of the newly formed granules are impaired. Detection of a surprisingly high amount of SPPL2c in total testis lysate (Fig 6A), together with the very limited number of cells that actually express SPPL2c 26, indicates a massive expression of SPPL2c in spermatids. Consequently, HEK293 cells with ectopic expression reflect the situation in wt spermatids, while the control cell lines represent spermatids of SPPL2c −/− mice. Since we observed a Golgi fragmentation in HEK293 cells overexpressing SPPL2c, it seems likely that due to impaired cleavage of SNARE proteins, the formation of proacrosomic granules in SPPL2c −/− occurs less efficiently than in wt mice. However, also the fusion of these granules requires specialized transport processes that may depend on blockage of regular transport processes in the spermatids. Thus, we cannot exclude that processing of SNARE proteins by SPPL2c is also required for this fusion process.
Maturation of secretory and membrane proteins in testis is different compared to other organs, as demonstrated by a substantial accumulation of immature Nicastrin and SPPL2a in testis compared to brain or other mouse tissues (Fig 6B). Most likely due to these transport defects and the consequent mislocalization of glycan‐modifying enzymes during the maturation process, mature spermatozoa of SPPL2c −/− mice depict profound changes in their glycan fingerprint compared to wt controls, as demonstrated by changes in lectin affinity (Fig 6G). Reduced reactivity of the lectin PSA has been previously associated with a reduction in the acrosome content 30, supporting deficits in acrosome formation in SPPL2c −/− mice. Even more importantly, reduced binding affinity to the lectin WGA is considered an established marker of teratozoospermia 30. Additionally, several of the lectins that depicted reduced binding affinity in SPPL2c −/− spermatozoa, for instance Jacalin, MPA, and ACA, detect glycan structures that are considered important for proper sperm maturation and function 31. Together with alterations in Ca2+ signaling induced by SPPL2c deficiency 26, this may contribute to the significantly reduced litter sizes of homozygous breedings of SPPL2c −/− mice. However, litter sizes were comparable to wt × wt breedings, when SPPL2c −/− male mice were crossed with wt female mice 26. This is in line with the observation that SPPL2c deficiency only causes deficits in spermatid maturation and Ca2+ signaling but not a complete loss of these processes and indicates that the function of SPPL2c in sperm maturation may have a certain redundancy, maybe caused by expression of other SPP/SPPL‐family members. Alternatively, sperm maturation may be additionally supported by other mechanisms that allow successful maturation of a reduced number of spermatids. The latter would put SPPL2c into the role of a booster that increases maturation of functional sperms, which is supported by the increase in SPPL2c expression during onset of sexual maturity 26.
In summary, we identified a variety of vesicular transport‐associated proteins as the first physiological substrates of SPPL2c, a so far orphan member of the SPP/SPPL protease family. Processing of these substrates impairs vesicular transport and leads to missorting of cargo proteins. During maturation of spermatids, alternate transport processes and cargo sorting are required to ensure proper formation of the mature spermatozoa, explaining a strong SPPL2c expression in maturating spermatids. So far, protein expression of SPPL2c has not been detected in any other body cell, but based on our findings, it is tempting to speculate that upregulation of SPPL2c expression may provide a switch to block protein secretion as a reaction to specific environmental stimuli. Based on impaired fertility observed in female SPPL2c−/−, one cell type that may express SPPL2c is the maturing oocyte and it is well imaginable that also other tissues or individual cell populations may express SPPL2c upon certain stimuli to trigger compartment reorganization or changes in Ca2+ signaling.
Materials and Methods
Antibodies
Monoclonal antibodies against SPPL2a (clone 6E9), SPPL3 (clone 7F9), SPP (clone 5H7), and TMEM192 were characterized previously 19, 38, 39, 40, 41, 42 "/>. The monoclonal antibody against SPPL2c (clone L2C3 1A1; IgG2a/k) was produced by immunization of Lou/c rats with a synthetic peptide comprising amino acids 592–612 of human SPPL2c. The polyclonal SPPL2c antibody directed against the C‐Terminus of murine SPPL2c cross‐reacts with human SPPL2c was kindly provided by Dr. Bernd Schröder and is characterized in Niemeyer et al 26. The anti‐Cab45 antibody was generated and purified as described in Ref. 43. Other antibodies used in this study were obtained from commercial sources: anti‐Syntaxin 5 (mIgG1, clone B‐8, sc‐365124, Santa Cruz, Dallas, USA), anti‐Syntaxin 6 (rabbit mAb, C34B2, Cell Signaling, Danvers, USA), anti‐Syntaxin 8 (mIgG1, clone A‐9, sc‐376521, Santa Cruz, Dallas, USA), anti‐Syntaxin 18 (mIgG2b, clone 10, sc‐293067, Santa Cruz, Dallas, USA), anti‐VAPA (rabbit mAb, ab181067, Abcam, Cambridge, UK), anti‐VAPB (rabbit pAb, ab103638, Abcam, Cambridge, UK), anti‐VAMP8 (rabbit mAb, ab76021, Abcam, Cambridge, UK), anti‐GOSR2 (mIgG1, clone 4HAD6, Thermo Fisher, Waltham, USA), anti‐Nicastrin (rabbit pAb, N1660, Sigma‐Aldrich, St. Louis, USA), anti‐hLAMP2 (mouse, mAb, H4B4, Developmental Studies Hybridoma Bank, Iowa, USA), anti‐GnTV (mouse mIgG2A, clone 706824, R&D Systems, Minneapolis, USA), anti‐EXTL3 (mouse, mAb, G‐5, Santa Cruz, Dallas, USA), anti‐B4GALT1 (mouse, mIgG2b, MAB 3609, R&D Minnesota, USA), anti‐OGFOD3 (goat, pAb, F‐19, Santa Cruz, Dallas, USA), anti‐V5 (mIgG2A, clone R960‐25, Life Technologies, Carlsbad, USA), anti‐Calnexin (rabbit pAb, Enzo Life Sciences, Farmingdale, USA), anti‐GM130 (rabbit mAb, EP892Y, ab52649, Abcam, Cambridge, UK), anti‐Giantin (mIgG1, ALX‐804‐600, Enzo Life Sciences, Farmingdale, USA), anti‐GRP78 BiP (rabbit pAb, ab21685, Abcam, Cambridge, UK), and anti‐TGN46 (sheep, pAb, AP32693PU, Acris, Maryland, USA). Horseradish peroxidase (HRP)‐conjugated secondary antibodies were purchased from Promega (Madison, USA), and the fluorescent secondary antibodies goat anti‐mouse 555, goat anti‐rabbit 488, donkey anti‐mouse 555, donkey anti‐sheep 568 were obtained from Thermo Fisher, Waltham, USA.
cDNA constructs, cell culture, transfection, and protein extraction
The cDNAs encoding human SPPL2c isoform A and SPPL2c D448A were kindly provided by Bruno Martoglio. Using PCR, a C‐terminal HA tag (AYPYDVPDYA) was added and both cDNAs were subcloned into pcDNA 4/TO A (Thermo Fisher, Waltham, USA). The cDNA that encodes human MGAT5 (protein name: GnTV) with a N‐terminal Flag (DYKDDDDK) epitope tag after the initiating methionine residue and a C‐terminal in‐frame V5 (GKPIPNPLLGLDST) epitope tag (Flag‐GnTV‐V5) was described before 19. T‐Rex™‐293 (Thermo Fisher, Waltham, USA) cells expressing C‐terminally HA‐tagged SPPL3 under a doxycycline‐inducible promotor have been described earlier 19. SPPL2c and SPPL2c D448A‐expressing cells were generated by stably transfecting the respective cDNAs into doxycycline‐inducible T‐Rex™‐293 cells. All cells were maintained in DMEM GlutaMAX™ media (Thermo Fisher, Waltham, USA) supplemented with L‐glutamine (Life Technologies, Carlsbad, USA), 10% (v/v) fetal calf serum (Sigma‐Aldrich, St. Louis, USA), 1% (v/v) penicillin/streptomycin (Life Technologies, Carlsbad, USA), and 5 μg/ml blasticidin (Life Technologies Carlsbad, USA). Cells stably expressing an SPPL protease were additionally supplemented with 200 μg/ml zeocin (Life Technologies). When plated for experimental use, all additives, except fetal calf serum, were removed from the culture medium. To induce expression of the SPPL proteases, 1 μg/ml of doxycycline (Carl Roth, Karlsruhe, Germany) was added to the culture medium for at least 48 h, unless differently indicated. Cells stably expressing or co‐expressing Flag‐GnTV‐V5, as indicated in the respective experiment, were generated by transfecting the respective plasmid using Lipofectamine™ 2000 according to the manufacturer's instructions (Thermo Fisher, Waltham, USA). Subsequently, cells were selected by addition of 100 μg/ml hygromycin (Life Technologies, Carlsbad, USA) to the culture medium 3 days after transfection and indefinitely. Transient transfections were also achieved using Lipofectamine™ 2000 according to the manufacturer's instructions (Thermo Fisher, Waltham, USA), however, without subsequent antibiotic selection.
For transient knockdown, T‐Rex™‐293 cells were transfected with siGENOME SMARTpool siRNA from Dharmacon (Lafayette, USA) at a final concentration of 20 nM using Lipofectamine™ RNAiMAX according to the manufacturer's instructions (Thermo Fisher, Waltham, USA). The following siGENOME SMARTpool siRNAs were applied: siGENOME Non‐Targeting siRNA Pool #1 (D‐001206‐13), Syntaxin 5 (M‐017768‐01), Syntaxin 8 (M‐019873‐02), Syntaxin 18 (M‐020624‐00), VAPA (M‐021382‐01), VAPB (M‐017795‐00), VAMP8 (M‐013503‐01), GOSR2 (M‐010980‐02), and SPPL3 (M‐006042‐02). For detection of secreted proteins, conditioned media was collected after 24‐ to 48‐h incubation and cleared at 17,000 g, 4°C for 20 min. Subsequently, samples were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) analysis. For detection of endogenous secreted proteins, cells were cultured in the absence of fetal calf serum for 24 h to allow trichloroacetic acid (TCA) precipitation, which has been described earlier in detail 19. To detect intracellular proteins, cells were washed with PBS (140 mM NaCl, 10 mM Na2HPO4, 1.75 mM KH2PO4, pH 7.4), harvested on ice and lysed using ice‐cold STE buffer (150 mM NaCl, 50 mM Tris, pH 7.6, 2 mM EDTA), supplemented just before use with 1:500 protease inhibitor mix (Sigma‐Aldrich, St. Louis, USA). Samples were incubated on ice for 30 min, and the cell debris was removed by centrifuging at 17,000 g, 4°C for 30 min. Total protein concentration in the cell lysates was determined with a BCA assay (Interchim, Montlucon, France), and inconsistencies between sample concentrations were normalized appropriately. For isolation of cellular membranes, cell pellets were resuspended in hypotonic buffer (10 mM Tris, pH 7.6, 1 mM EDTA, 1 mM EGTA, pH 7.6), supplemented just before use with 1:500 protease inhibitor mix (Sigma‐Aldrich, St. Louis, USA), vortexed and cooled on ice for 10 min. Cells were lysed with a 23‐gauge needle and centrifuged at 2,500 g at 4°C for 5 min, to remove cell debris and nuclei. Subsequently, the supernatant was centrifuged at 17,000 g at 4°C for 45 min and the membrane pellet was lysed with buffer containing 40 mM Tris, pH 7.8, 40 mM KCH3COO, 1.6 mM Mg(CH3COO)2, 100 mM sucrose, and 0.8 mM DTT.
Mice and tissue homogenates
The SPPL2c −/− mice, as well as their wild‐type littermates, were kindly provided Dr. Bernd Schröder and are described in detail in Niemeyer et al 26. Testis membrane preparations were produced as described in Niemeyer et al 26 and were directly subjected to SDS–PAGE. For tissue analysis, organs (testis, kidneys, brain, spleen, liver) from three pairs of male C57BL/6J littermate mice at ages of 16, 28, or 36 weeks were removed postmortem and snap‐frozen in liquid nitrogen. Human testis samples were previously described 44. For the scientific use of the samples, the patients had granted written informed consent, and the Ethical Committee (Ethikkommission, Technische Universität München, Fakultät für Medizin, München, project number 5158/11) has approved the study. The experiments were performed in accordance with relevant guidelines and regulations. Thawed organs (100–250 mg) were homogenized in ice‐cold organ‐lysis buffer (5 mM Tris–HCL, pH 7.4, 250 mM sucrose, 5 mM EGTA) supplemented just before use with 1:500 protease inhibitor mix (Sigma‐Aldrich, St. Louis, USA). Samples were centrifuged at 500 g, 4°C for 5 min, and the supernatant was subjected to further centrifugation at 100,000 g, 4°C for 60 min. Pellets were resuspended in organ‐lysis buffer containing 2% (v/v) Triton X‐100 and incubated for 30 min on ice. Cell debris was removed by centrifugation at 17,000 g, 4°C for 30 min, and supernatants were used for protein quantification by BCA and immunoblot analysis.
Immunoblotting
For protein detection lysates, membrane preparations, conditioned media, or TCA‐precipitations were subjected to SDS–PAGE. Depending on the abundance of the protein in question, 5–40 μg was loaded. After separation, proteins were transferred onto PVDF membranes. Primary and secondary antibodies for protein detection were diluted in PBS supplemented with 2% (w/v) Tropix I‐Block™ and 0.5% (v/v) Tween 20. The proteins were visualized using enhanced chemiluminescence technique (ECL) Pierce™ ECL Western Blotting Substrate (Thermo Fisher) or Westar Supernova (Cyanagen, Bologna, Italy) for increased sensitivity and detected on Super RX‐N X‐ray films (Fuji, Tokyo, Japan). For quantification, proteins were detected using the Pierce™ ECL Plus Western Blotting Substrate (Thermo Fisher). The chemiluminescence signals of at least three independent experiments were measured with a CCD camera‐based imaging system (ImageQuant LAS‐4000 Fuji Film).
Immunostaining and confocal imaging
Cells were seeded at a confluency of ~30% on 15‐mm Poly‐L‐Lysine‐coated coverslips and where indicated, treated with siGENOME SMARTpool siRNA during seeding as described above. On day 3 after seeding media was removed, cells were washed once with PBS and fixated for 20 min at room temperature in 4% paraformaldehyde in PBS. Subsequently, cells were washed three times with PBS and were permeabilized for 5 min using 5% (w/v) bovine serum albumin (BSA) supplemented with 0.2% (v/v) Triton X‐100 and 0.5% (w/v) SDS. One‐hour blocking at room temperature with 5% (w/v) BSA was followed by incubation with primary antibodies diluted in 5% (w/v) BSA for 16–20 h at 4°C. After extensive washing, the secondary antibodies diluted 1:400 in 5% (w/v) BSA were added for one hour at room temperature. Again, cells were washed six times with PBS and incubated for 5 min with 1:10,000 4′,6‐Diamidino‐2‐phenylindol (DAPI, 5 μg/ml) solution at room temperature. Coverslips were loaded onto glass plates using Mowiol mounting media (6.0 g glycerol, 2.4 g Mowiol 4‐88 (Carl Roth, Karlsruhe, Germany), 12.0 ml 0.2 M Tris × HCl pH 8.5, 6.0 ml double‐distilled water, 25 mg/ml DABCO (Carl Roth)) and left to dry overnight. Pictures were taken using a LSM 710 Zeiss Observer Z.1 confocal microscope with a 60× oil lens and 2× optical zoom. The intensity of the pictures was increased for visualization purposes with the Zen lite 2011 program.
Histological analysis
Murine testis was fixed by immersion in Bouin's solution and paraffin‐embedded according to standard procedures. Immunohistochemical stainings of paraffin sections were performed utilizing the ABC System (Vector Laboratories) and diaminobenzidine as peroxidase substrate. Cab45 was visualized using the ant‐Cab45 antibody described above in 1:500 dilution. To control for specificity, rabbit polyclonal control IgG (BioLegend) was employed in parallel. After completion of immunostaining, nuclei were visualized with hematoxylin. Images were taken using a Zeiss Axiovert microscope with an Insight Camera (18.2 Color Mosaik) and Spot advanced software 4.6 (SPOT Imaging Solutions, Sterling Heights, MI, USA). For quantification, 14 images each containing one stage VII/VIII seminiferous tubule were used. The stages were chosen because they are characterized by two generations of spermatids and further can be reliably recognized in immuno‐stained sections (e.g., also by the presence of residual bodies; 28). Three squares of the same size were randomly positioned around each seminiferous tubule, and the structures resembling the pre‐acrosomal vesicles within each square were counted. The average of the three squares was used for each tubule. The mean and standard deviation for the two genotypic groups were calculated and depicted in Prism. n = 6 for the SPPL2c +/+ and n = 8 for the SPPL2c −/−.
Mass spectrometry
Cells were harvested and centrifuged 400 g for 10 min at 4°C. Cell pellets were resuspended in STE Buffer (250 mM sucrose, 5 mM Tris, pH 7, 1 mM EGTA, PI mix 1:500) and lysed with a 27‐gauge needle. Samples were centrifuged 10 min at 800 g to remove nuclei, then 10 min at 15,000 g to remove mitochondria, and finally 1 h 100,000 g. The resulting pellets were washed twice with 100 mM Na2CO3 and centrifuged 30 min at 100,000 g after each wash. Pellets from membrane preparations were dissolved in lysis buffer (150 mM NaCl, 50 mM Tris–HCl pH 7.5, 2 mM EDTA, 1% Triton X‐100). The protein concentration was estimated using the Pierce 660 nm protein assay (Thermo Fisher Scientific, US). A protein amount of 15 μg was subjected to proteolytic digestion with trypsin and LysC (Promega, Germany) using the filter‐aided sample preparation (FASP) with Vivacon centrifugal concentrators (30 kDa cut‐off, Sartorius, Germany) according to a standard protocol 45. Peptides were enriched and desalted using stop and go extraction with self‐packed C18 Tips (3M Empore, US) 46. Eluted peptides were dried by vacuum centrifugation and dissolved in 20 μl 0.1% formic acid. Peptides were analyzed on an Easy nLC 1000 nanoHPLC (Thermo Scientific, US) which was coupled online via a Nanospray Flex Ion Source (Thermo Scientific, US) equipped with a PRSO‐V1 column oven (Sonation, Germany) to a Q‐Exactive mass spectrometer (Thermo Scientific, US). An amount of 1.3 μg of peptides was separated on an in‐house packed C18 column (30 cm × 75 μm ID, ReproSil‐Pur 120 C18‐AQ, 1.9 μm, Dr. Maisch GmbH, Germany) using a binary gradient of water (A) and acetonitrile (B) supplemented with 0.1% formic acid (0 min, 2% B; 3:30 min, 5% B; 137:30 min, 25% B; 168:30 min, 35% B; 182:30 min, 60% B) at 50°C column temperature. A data‐dependent acquisition method was used. Full mass spectrometric scans were acquired at a resolution of 70,000 (m/z range: 300–1,400, AGC target: 3E+6). The ten most intense peptide ions per full MS scan were chosen for peptide fragmentation (resolution: 17,500, isolation width: 2.0 m/z, AGC target: 1E+5, NCE: 25%). A dynamic exclusion of 120 s was used for peptide fragmentation.
Data were analyzed with the software Maxquant (maxquant.org, Max Planck Institute Munich) version 1.5.5.1 47. The mass spectrometric data were searched against a reviewed fasta database of Homo sapiens from UniProt including isoforms (download: August 8, 2017, 42,219 entries). Trypsin was defined as protease. Two missed cleavages were allowed for the database search. The option first search was used to recalibrate the peptide masses within a window of 20 ppm. For the main search peptide and peptide fragment, mass tolerances were set to 4.5 and 20 ppm, respectively. Carbamidomethylation of cysteine was defined as static modification. Acetylation of the protein N‐term as well as oxidation of methionine was set as variable modifications. The false discovery rate for both peptides and proteins was adjusted to less than 1%. Label‐free quantification (LFQ) of proteins required at least two ratio counts of razor peptides. Only razor and unique peptides were used for quantification. The software Perseus (version 1.5.8.5) was used for further data analysis. The protein LFQ intensities were log2‐transformed, and two‐sided Student's t‐test was applied to evaluate the significance of proteins with changed abundance. Additionally, a permutation‐based false discovery rate estimation was used 48. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 49 partner repository with the dataset identifier PXD011923.
Pathway analysis
Bioinformatic analysis of the SPPL2c candidate substrates identified through the mass spectrometric approach was performed using the online STRING 10.5 (https://string-db.org/) program. In order to identify potential SPPL2c substrates that were missed by the proteomic analysis, the function “More” of STRING 10.5 was used.
Lectin chip microarray (LecChip)
Mature spermatozoa were recovered from the cauda epididymides of SPPL2c −/− and wild‐type mice (n = 4 per group) as described in Ref. 26. For lectin chip microarray analysis (LecChip), samples were lysed in STEN Buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 2 mM EDTA, 1% NP‐40, PI mix 1:500) incubated on ice for 30 min, centrifuged at 10,000 g for 10 min, and brought to identical protein concentrations (measurement was performed by BCA reagent, Interchim, Montlucon, France) in TBS to obtain a 0.05 mg/ml solution. Cy3 NHS ester reagent (PA13101, GE Healthcare Life Sciences), dissolved in dimethyl sulfoxide, was added (final concentration, 0.05 mg/ml) for 1 h at room temperature. Excess reactive reagent was blocked by inactivation with free lysine (final concentration, 0.05 mM; L5501, Sigma‐Aldrich). LecChip (GlycoTechnica Ltd.) was washed three times with Probing Solution (provided by the manufacturer), and Cy3‐labeled sperm lysates (1 μg/ml) (n = 4 per group) were added to the wells in Probing Solution (100 μl per well). Samples were incubated overnight at 18°C. Subsequently, the LecChip was washed with TBS and double‐distilled water 30 min each. The LecChip was scanned with an InnoScan 710 Microarray scanner (Innopsys), and results were analyzed with CLIQS Array Professional software (TotalLab). Overall, 45 lectin intensities were measured in each sample. An average Pearson correlation coefficient of 0.98 and above was calculated for all samples. Lectin intensities were log2‐transformed, and two‐sided Student's t‐test was applied to evaluate the significance of lectin binding with changed abundance.
Author contributions
SAM and SFL performed the mass spectrometric analysis. TM, JN, and BS provided SPPL2c −/− mouse tissue and performed the analysis of Syntaxin 8 in SPPL2c −/− tissue. JB provided antibodies and helped with analysis of compartment morphology. AM provided human testis samples and performed immunohistological analysis of seminiferous tubular cross sections. RFe established the monoclonal SPPL2c antibody. MH‐K performed experiments shown in Figs 2A and EV2, and provided technical assistance. MDS performed the Lectin Chip microarray. AAP performed all other experiments and wrote parts of the manuscript. RFl conceived the experiments, supervised the project, and wrote the manuscript with input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Acknowledgements
We thank Prof. Marius Lemberg for fruitful discussion. We are grateful to J. Ullrich Schwarzer and Frank‐Michael Köhn for providing human testes samples and Astrid Tiefenbacher as well as Anna Berghofer for technical assistance. We thank Dr. Yifat Merbl for the help with the LecChip scanning. This work was supported by funds from the Deutsche Forschungsgemeinschaft within the research unit FOR 2290 and grants FL 635/2‐1, FL 635/2‐2. AAP was generously supported by IMPRS‐LS. BS received support from the Deutsche Forschungsgemeinschaft within the SFB 877 (project B7) and grant SCHR1284/2‐1. SL was supported by the Breuer Foundation Research Award, the Helmholtz Israel program, and the Centers of Excellence in Neurodegeneration. AM was supported by grants MA 1080/23‐1 and MA 1080/23‐2 from the Deutsche Forschungsgemeinschaft.
EMBO Reports (2019) 20: e46451
See also: J Niemeyer et al (March 2019) and HS Young & MJ Lemieux (March 2019)
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 48 partner repository with the dataset identifier PXD011923 (https://www.ebi.ac.uk/pride/archive/projects/PXD011923).
References
- 1. Lee MC, Miller EA, Goldberg J, Orci L, Schekman R (2004) Bi‐directional protein transport between the ER and Golgi. Annu Rev Cell Dev Biol 20: 87–123 [DOI] [PubMed] [Google Scholar]
- 2. Gomez‐Navarro N, Miller E (2016) Protein sorting at the ER‐Golgi interface. J Cell Biol 215: 769–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang T, Li L, Hong W (2017) SNARE proteins in membrane trafficking. Traffic 18: 767–775 [DOI] [PubMed] [Google Scholar]
- 4. Trimble WS, Cowan DM, Scheller RH (1988) VAMP‐1: a synaptic vesicle‐associated integral membrane protein. Proc Natl Acad Sci USA 85: 4538–4542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jahn R, Scheller RH (2006) SNAREs–engines for membrane fusion. Nat Rev Mol Cell Biol 7: 631–643 [DOI] [PubMed] [Google Scholar]
- 6. Bonifacino JS, Glick BS (2004) The mechanisms of vesicle budding and fusion. Cell 116: 153–166 [DOI] [PubMed] [Google Scholar]
- 7. Teng FY, Wang Y, Tang BL (2001) The syntaxins. Genome Biol 2: REVIEWS3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mentrup T, Fluhrer R, Schroder B (2017) Latest emerging functions of SPP/SPPL intramembrane proteases. Eur J Cell Biol 96: 372–382 [DOI] [PubMed] [Google Scholar]
- 9. Mentrup T, Loock AC, Fluhrer R, Schroder B (2017) Signal peptide peptidase and SPP‐like proteases – Possible therapeutic targets? Biochim Biophys Acta 1864: 2169–2182 [DOI] [PubMed] [Google Scholar]
- 10. Voss M, Schroder B, Fluhrer R (2013) Mechanism, specificity, and physiology of signal peptide peptidase (SPP) and SPP‐like proteases. Biochim Biophys Acta 1828: 2828–2839 [DOI] [PubMed] [Google Scholar]
- 11. Steiner H, Kostka M, Romig H, Basset G, Pesold B, Hardy J, Capell A, Meyn L, Grim MG, Baumeister R et al (2000) Glycine 384 is required for presenilin‐1 function and is conserved in polytopic bacterial aspartyl proteases. Nat Cell Biol 2: 848–851 [DOI] [PubMed] [Google Scholar]
- 12. Golde TE, Wolfe MS, Greenbaum DC (2009) Signal peptide peptidases: a family of intramembrane‐cleaving proteases that cleave type 2 transmembrane proteins. Semin Cell Dev Biol 20: 225–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Friedmann E, Lemberg MK, Weihofen A, Dev KK, Dengler U, Rovelli G, Martoglio B (2004) Consensus analysis of signal peptide peptidase and homologous human aspartic proteases reveals opposite topology of catalytic domains compared with presenilins. J Biol Chem 279: 50790–50798 [DOI] [PubMed] [Google Scholar]
- 14. Boname JM, Bloor S, Wandel MP, Nathan JA, Antrobus R, Dingwell KS, Thurston TL, Smith DL, Smith JC, Randow F et al (2014) Cleavage by signal peptide peptidase is required for the degradation of selected tail‐anchored proteins. J Cell Biol 205: 847–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hsu FF, Yeh CT, Sun YJ, Chiang MT, Lan WM, Li FA, Lee WH, Chau LY (2015) Signal peptide peptidase‐mediated nuclear localization of heme oxygenase‐1 promotes cancer cell proliferation and invasion independent of its enzymatic activity. Oncogene 34: 2360–2370 [DOI] [PubMed] [Google Scholar]
- 16. Struhl G, Adachi A (2000) Requirements for presenilin‐dependent cleavage of Notch and other transmembrane proteins. Mol Cell 6: 625–636 [DOI] [PubMed] [Google Scholar]
- 17. Martin L, Fluhrer R, Haass C (2009) Substrate requirements for SPPL2b‐dependent regulated intramembrane proteolysis. J Biol Chem 284: 5662–5670 [DOI] [PubMed] [Google Scholar]
- 18. Lemberg MK, Martoglio B (2002) Requirements for signal peptide peptidase‐catalyzed intramembrane proteolysis. Mol Cell 10: 735–744 [DOI] [PubMed] [Google Scholar]
- 19. Voss M, Kunzel U, Higel F, Kuhn PH, Colombo A, Fukumori A, Haug‐Kroper M, Klier B, Grammer G, Seidl A et al (2014) Shedding of glycan‐modifying enzymes by signal peptide peptidase‐like 3 (SPPL3) regulates cellular N‐glycosylation. EMBO J, 33: 2890–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kuhn PH, Voss M, Haug‐Kroper M, Schroder B, Schepers U, Brase S, Haass C, Lichtenthaler SF, Fluhrer R (2015) Secretome analysis identifies novel signal Peptide peptidase‐like 3 (Sppl3) substrates and reveals a role of Sppl3 in multiple Golgi glycosylation pathways. Mol Cell Proteomics 14: 1584–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Urny J, Hermans‐Borgmeyer I, Schaller HC (2006) Cell‐surface expression of a new splice variant of the mouse signal peptide peptidase. Biochim Biophys Acta 1759: 159–165 [DOI] [PubMed] [Google Scholar]
- 22. Weihofen A, Lemberg MK, Friedmann E, Rueeger H, Schmitz A, Paganetti P, Rovelli G, Martoglio B (2003) Targeting presenilin‐type aspartic protease signal peptide peptidase with gamma‐secretase inhibitors. J Biol Chem 278: 16528–16533 [DOI] [PubMed] [Google Scholar]
- 23. Weihofen A, Lemberg MK, Ploegh HL, Bogyo M, Martoglio B (2000) Release of signal peptide fragments into the cytosol requires cleavage in the transmembrane region by a protease activity that is specifically blocked by a novel cysteine protease inhibitor. J Biol Chem 275: 30951–30956 [DOI] [PubMed] [Google Scholar]
- 24. Tecle E, Gagneux P (2015) Sugar‐coated sperm: unraveling the functions of the mammalian sperm glycocalyx. Mol Reprod Dev 82: 635–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ward TH, Polishchuk RS, Caplan S, Hirschberg K, Lippincott‐Schwartz J (2001) Maintenance of Golgi structure and function depends on the integrity of ER export. J Cell Biol 155: 557–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Niemeyer J, Mentrup T, Heidasch R, Müller SA, Biswas U, Meyer R, Papadopoulou AA, Dederer V, Haug‐Kröper M, Adamski V, Lüllmann‐Rauch R, Bergmann M, Mayerhofer A, Saftig P, Wennemuth G, Jessberger R, Fluhrer R, Lichtenthaler SF, Lemberg MK, Schröder B (2019) The intramembrane protease SPPL2c promotes male germ cell development by cleaving phospholamban. EMBO Rep 20: e46449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berruti G, Paiardi C (2011) Acrosome biogenesis: revisiting old questions to yield new insights. Spermatogenesis 1: 95–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Meistrich ML, Hess RA (2013) Assessment of spermatogenesis through staging of seminiferous tubules. Methods Mol Biol 927: 299–307 [DOI] [PubMed] [Google Scholar]
- 29. Pang PC, Chiu PC, Lee CL, Chang LY, Panico M, Morris HR, Haslam SM, Khoo KH, Clark GF, Yeung WS et al (2011) Human sperm binding is mediated by the sialyl‐Lewis(x) oligosaccharide on the zona pellucida. Science 333: 1761–1764 [DOI] [PubMed] [Google Scholar]
- 30. Benoff S (1997) Carbohydrates and fertilization: an overview. Mol Hum Reprod 3: 599–637 [DOI] [PubMed] [Google Scholar]
- 31. Xin A, Cheng L, Diao H, Wu Y, Zhou S, Shi C, Sun Y, Wang P, Duan S, Zheng J et al (2016) Lectin binding of human sperm associates with DEFB126 mutation and serves as a potential biomarker for subfertility. Sci Rep 6: 20249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jung JJ, Inamdar SM, Tiwari A, Choudhury A (2012) Regulation of intracellular membrane trafficking and cell dynamics by syntaxin‐6. Biosci Rep 32: 383–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fluhrer R, Martin L, Klier B, Haug‐Kroeper M, Grammer G, Nuscher B, Haass C (2012) The alpha‐helical content of the transmembrane domain of the British dementia protein‐2 (Bri2) determines its processing by Signal Peptide Peptidase‐Like 2b (SPPL2b). J Biol Chem 287: 5156–5163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rowe T, Dascher C, Bannykh S, Plutner H, Balch WE (1998) Role of vesicle‐associated syntaxin 5 in the assembly of pre‐Golgi intermediates. Science 279: 696–700 [DOI] [PubMed] [Google Scholar]
- 35. Consortium GT (2013) The genotype‐tissue expression (GTEx) project. Nat Genet 45: 580–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Noguchi S, Arakawa T, Fukuda S, Furuno M, Hasegawa A, Hori F, Ishikawa‐Kato S, Kaida K, Kaiho A, Kanamori‐Katayama M et al (2017) FANTOM5 CAGE profiles of human and mouse samples. Sci Data 4: 170112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Abou‐Haila A, Tulsiani DR (2000) Mammalian sperm acrosome: formation, contents, and function. Arch Biochem Biophys 379: 173–182 [DOI] [PubMed] [Google Scholar]
- 38. Behnke J, Schneppenheim J, Koch‐Nolte F, Haag F, Saftig P, Schroder B (2011) Signal‐peptide‐peptidase‐like 2a (SPPL2a) is targeted to lysosomes/late endosomes by a tyrosine motif in its C‐terminal tail. FEBS Lett 585: 2951–2957 [DOI] [PubMed] [Google Scholar]
- 39. Voss M, Fukumori A, Kuhn PH, Kunzel U, Klier B, Grammer G, Haug‐Kroper M, Kremmer E, Lichtenthaler SF, Steiner H et al (2012) Foamy virus envelope protein is a substrate for signal peptide peptidase‐like 3 (SPPL3). J Biol Chem 287: 43401–43409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneppenheim J, Dressel R, Huttl S, Lullmann‐Rauch R, Engelke M, Dittmann K, Wienands J, Eskelinen EL, Hermans‐Borgmeyer I, Fluhrer R et al (2013) The intramembrane protease SPPL2a promotes B cell development and controls endosomal traffic by cleavage of the invariant chain. J Exp Med 210: 41–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fleck D, Voss M, Brankatschk B, Giudici C, Hampel H, Schwenk B, Edbauer D, Fukumori A, Steiner H, Kremmer E et al (2016) Proteolytic processing of neuregulin 1 type III by three intramembrane‐cleaving proteases. J Biol Chem 291: 318–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schroder B, Wrocklage C, Hasilik A, Saftig P (2010) Molecular characterisation of ‘transmembrane protein 192’ (TMEM192), a novel protein of the lysosomal membrane. Biol Chem 391: 695–704 [DOI] [PubMed] [Google Scholar]
- 43. Crevenna AH, Blank B, Maiser A, Emin D, Prescher J, Beck G, Kienzle C, Bartnik K, Habermann B, Pakdel M et al (2016) Secretory cargo sorting by Ca2+‐dependent Cab45 oligomerization at the trans‐Golgi network. J Cell Biol 213: 305–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mayer C, Adam M, Glashauser L, Dietrich K, Schwarzer JU, Kohn FM, Strauss L, Welter H, Poutanen M, Mayerhofer A (2016) Sterile inflammation as a factor in human male infertility: involvement of Toll like receptor 2, biglycan and peritubular cells. Sci Rep 6: 37128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wisniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6: 359–362 [DOI] [PubMed] [Google Scholar]
- 46. Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix‐assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75: 663–670 [DOI] [PubMed] [Google Scholar]
- 47. Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M (2014) Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vizcaino JA, Csordas A, del‐Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez‐Riverol Y, Reisinger F, Ternent T et al (2016) 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44: D447–D456 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE 48 partner repository with the dataset identifier PXD011923 (https://www.ebi.ac.uk/pride/archive/projects/PXD011923).