

KSHV LANA mediates episomal persistence of viral genomes. LANA binds the KSHV terminal-repeat (TR) sequence to mediate DNA replication and tethers KSHV DNA to mitotic chromosomes to segregate genomes to daughter cell nuclei. Here, we investigate LANA sequence upstream of the internal repeat elements that contributes to episome segregation and persistence. Mutants with deletions within this sequence maintained the ability to bind mitotic chromosomes or bind and replicate TR DNA. Deletion of upstream sequence within the region reduced segregation of TR DNA to daughter cells, but not episome maintenance. In contrast, mutants with deletions of 80 amino acids immediately downstream were highly deficient for episome persistence yet maintained the ability to replicate and segregate TR DNA, the two principal components of episome persistence, suggesting another role for the residues. In summary, this work identifies adjacent LANA sequence with distinct roles in episome segregation and persistence.
KEYWORDS: KSHV, LANA, episome
ABSTRACT
Kaposi’s sarcoma-associated herpesvirus (KSHV) latency-associated nuclear antigen (LANA) is a 1,162-amino-acid protein that mediates episome persistence of viral genomes. LANA binds the KSHV terminal-repeat (TR) sequence through its carboxy-terminal domain to mediate DNA replication. LANA simultaneously binds mitotic chromosomes and TR DNA to segregate virus genomes to daughter cell nuclei. Amino-terminal LANA attaches to chromosomes by binding histones H2A/H2B, and carboxy-terminal LANA contributes to mitotic-chromosome binding. Although amino- and carboxy-terminal LANA are essential for episome persistence, they are not sufficient, since deletion of all internal LANA sequence renders LANA highly deficient for episome maintenance. Internal LANA sequence upstream of the internal repeat elements contributes to episome segregation and persistence. Here, we investigate this region with a panel of LANA deletion mutants. Mutants retained the ability to associate with mitotic chromosomes and bind TR DNA. In contrast to prior results, deletion of most of this sequence did not reduce LANA’s ability to mediate DNA replication. Deletions of upstream sequence within the region compromised segregation of TR DNA to daughter cells, as assessed by retention of green fluorescent protein (GFP) expression from a replication-deficient TR plasmid. However, deletion of this upstream sequence did not reduce episome maintenance. In contrast, deletions that included an 80-amino-acid sequence immediately downstream resulted in highly deficient episome persistence. LANA with this downstream sequence deleted maintained the ability to replicate and segregate TR DNA, suggesting a unique role for the residues. Therefore, this work identifies adjacent LANA regions with distinct roles in episome segregation and persistence.
IMPORTANCE KSHV LANA mediates episomal persistence of viral genomes. LANA binds the KSHV terminal-repeat (TR) sequence to mediate DNA replication and tethers KSHV DNA to mitotic chromosomes to segregate genomes to daughter cell nuclei. Here, we investigate LANA sequence upstream of the internal repeat elements that contributes to episome segregation and persistence. Mutants with deletions within this sequence maintained the ability to bind mitotic chromosomes or bind and replicate TR DNA. Deletion of upstream sequence within the region reduced segregation of TR DNA to daughter cells, but not episome maintenance. In contrast, mutants with deletions of 80 amino acids immediately downstream were highly deficient for episome persistence yet maintained the ability to replicate and segregate TR DNA, the two principal components of episome persistence, suggesting another role for the residues. In summary, this work identifies adjacent LANA sequence with distinct roles in episome segregation and persistence.
INTRODUCTION
Kaposi’s sarcoma (KS)-associated herpesvirus (KSHV), or human herpesvirus 8 (HHV-8), is a gamma-2 herpesvirus. Like other herpesviruses, KSHV establishes lifelong infection. KSHV has an etiologic role in KS, primary effusion lymphoma (PEL), and multicentric Castleman’s disease (1–4). KSHV infection can alternate between latent and lytic states but predominantly persists in its latent form. During latent infection, KSHV expresses only a small subset of genes, and no virions are produced. The KSHV genome persists in latently infected cells as a multiple-copy, circular, covalently closed, extrachromosomal episome (plasmid) (1, 5). Episome persistence is mediated by KSHV latency-associated nuclear antigen (LANA), encoded by open reading frame 73 (ORF73) (6–8). LANA is necessary and sufficient for KSHV episome persistence in the absence of other viral genes (9, 10).
Episome persistence is comprised of two components—DNA replication and segregation of episomes to progeny cell nuclei following mitosis—and LANA provides both functions. LANA serves as a molecular tether by simultaneously binding terminal-repeat (TR) DNA and mitotic chromosomes to segregate KSHV episomes to daughter cell nuclei. Amino- and carboxy-terminal LANA domains are each essential for episome persistence. LANA associates with mitotic chromosomes through two chromosome binding regions located in amino- and carboxy-terminal LANA (10–18). Carboxy-terminal LANA interacts with the pericentromeric and peri-telomeric regions of mitotic chromosomes, while N-terminal LANA directly binds histones H2A/H2B on the folded portion of the nucleosome and is the dominant chromosome attachment region. Amino-terminal LANA interaction with histones H2A/H2B is essential for episome maintenance and efficient DNA replication (11, 19, 20). The LANA carboxy-terminal domain self-associates to bind three adjacent sites in each KSHV TR element, and this binding is necessary for KSHV to mediate TR-associated DNA replication (9, 20–30).
In addition to amino- and carboxy-terminal LANA, internal LANA sequence is also important for episome persistence. LANA is a 1,162-amino-acid protein and contains a proline-rich region, central repeat elements, and a carboxy-terminal DNA binding domain (Fig. 1). Although amino- and carboxy-terminal LANA are essential for episome persistence, fusion of these two LANA components resulted in episome maintenance that was highly deficient (31). Investigation of LANA internal sequence with deletion mutants identified LANA residues 33 to 273 as critical for episome persistence and LANA residues 33 to 331 as important for LANA segregation of KSHV DNA (32). Here, we performed a detailed investigation of this region. We found that upstream sequence within the region is important for LANA segregation, while residues 80 amino acids immediately downstream are critical for LANA episome persistence.
FIG 1.

Schematic representation of KSHV LANA and deletion mutants. The two vertical black bands indicate the amino-terminal bipartite nuclear localization signal (NLS) at residues 24 to 30 and 41 to 47 (15, 55). The proline-rich (P), aspartate and glutamate (DE), glutamine (Q), and glutamate and glutamine (EQE) regions and the putative leucine zipper (LZ) are indicated. The DE, Q, LZ, and EQE regions each contain imperfect repeat elements. Amino acids 5 to 13 mediate chromosome association through interaction with histones H2A/H2B (11, 19). Amino acids 996 to 1139 bind TR DNA, mediate self-association, and also associate with mitotic chromosomes. Capabilities for chromosome association, TR DNA binding, DNA replication, episome segregation, and episome persistence for LANA and deletion mutants are summarized on the right. For episome persistence, the fractions indicate the number of G418- or puromycin-resistant cell lines containing KSHV episomes divided by the total number of drug-resistant cell lines as assessed by Gardella gel analysis. Percentages are shown in parentheses. Episome maintenance fold deficiencies were determined by dividing the value for each mutant in Tables 1 and 2 by that of WT LANA. NT, not tested. *, data from reference 32.
RESULTS
LANA deletion mutants associate with mitotic chromosomes.
We previously showed that internal LANA residues 33 to 273 are critical for episome persistence (32). To persist in proliferating cells, episomes must replicate with each cell division and segregate to progeny cell nuclei. LANAΔ33–273 was deficient for both LANA-mediated DNA replication and segregation of virus DNA to daughter cells. To further investigate this region, we generated a panel of internal-deletion mutants termed LANAΔ33–273A, LANAΔ74–273, LANAΔ114–273, LANAΔ154–273, LANAΔ194–273, LANAΔ234–273, LANAΔ33–74, LANAΔ33–154, LANAΔ33–194, and LANAΔ33–234 (Fig. 1). LANAΔ33–273A contained four alanine residues in place of the deleted amino acids instead of the GYQ present in the previously described LANAΔ33–273.
Although all the mutants contained the LANA amino- and carboxy-terminal chromosome association regions, we wished to confirm that these mutants associate with mitotic chromosomes, since this function is essential for episome maintenance. BJAB cells stably expressing each of the mutants were arrested in metaphase by overnight incubation with colcemid. We then assessed the subcellular localization of LANA or the LANA mutants by immunofluorescence microscopy. LANA (green) was detected with anti-LANA antibody, and mitotic chromosomes were detected using propidium iodide (red). As previously observed, LANA (Fig. 2A) was distributed broadly over mitotic chromosomes (overlaying green and red generates yellow) with preferential localization near centromeres and telomeres. LANAΔ33–273A, LANAΔ74–273, LANAΔ114–273, LANAΔ154–273, LANAΔ194–273, LANAΔ33–74, LANAΔ33–154, and LANAΔ33–194 (Fig. 2) were distributed similarly to LANA over chromosomes, with preferential localization to pericentromeres (arrowhead) and peri-telomeres (arrow), indicating that the deletions do not affect the ability of the mutants to bind mitotic chromosomes. In contrast, LANAΔ33–234 (Fig. 2K) did not associate with mitotic chromosomes and LANAΔ234–273 (Fig. 2G) only weakly associated with mitotic chromosomes, with much of LANAΔ234–273 accumulating on the peripheries of chromosomes. The two mutants share a junction at LANA residue 234, which may induce a conformation that interferes with chromosome binding; these mutants were not investigated further. The data indicate that, other than LANAΔ33–234 and LANAΔ234–273, all the internal-deletion mutants associated with mitotic chromosomes in a wild-type (WT) fashion.
FIG 2.

LANA and LANA deletion mutant association with mitotic chromosomes. BJAB cell lines stably expressing LANA or LANA deletion mutants were arrested in metaphase with colcemid and assessed for colocalization with mitotic chromosomes. LANA was detected using a monoclonal anti-LANA antibody (green), and the chromosomes were counterstained with propidium iodide (red). Overlaying green and red generates yellow. Brightness and contrast were uniformly adjusted for the panels using ImageJ. Magnification, ×630.
LANA deletion mutants bind TR DNA.
LANA binds KSHV TR DNA through its carboxy-terminal domain. Since none of the deletions involved carboxy-terminal LANA, we expected this interaction to be preserved. However, since TR DNA binding is essential for LANA-mediated DNA replication and episome persistence, we assessed the abilities of the deletion mutants to bind the high-affinity LANA TR DNA binding site by electrophoretic mobility shift assay (EMSA). LANA and the mutants were in vitro translated and incubated with a radiolabeled probe containing the TR binding site. As expected, incubation of the TR probe with rabbit reticulocyte lysate (RRL) did not result in complex formation (Fig. 3, lane 1). Incubation of full-length LANA with the probe resulted in two major complexes (Fig. 3, lane 3; indicated by vertical lines at left), as observed previously. Incubation of antibody that detects the T7 epitope tag fused to N-terminal LANA induced a supershift of the LANA complexes (Fig. 3, lane 2; the supershifted complexes are indicated by symbols), demonstrating the presence of LANA in the complexes. Incubation of the probe with LANAΔ33–273A (Fig. 3, lane 4), LANAΔ74–273 (Fig. 3, lane 5), LANAΔ114–273 (Fig. 3, lane 6), LANAΔ154–273 (Fig. 3, lane 7), LANAΔ194–273 (Fig. 3, lane 8), LANAΔ33–74 (Fig. 3, lane 9), LANAΔ33–154 (Fig. 3, lane 10), or LANAΔ33–194 (Fig. 3, lane 11) resulted in similar complexes. These results indicate that all the deletion mutants retained the ability to bind TR DNA.
FIG 3.
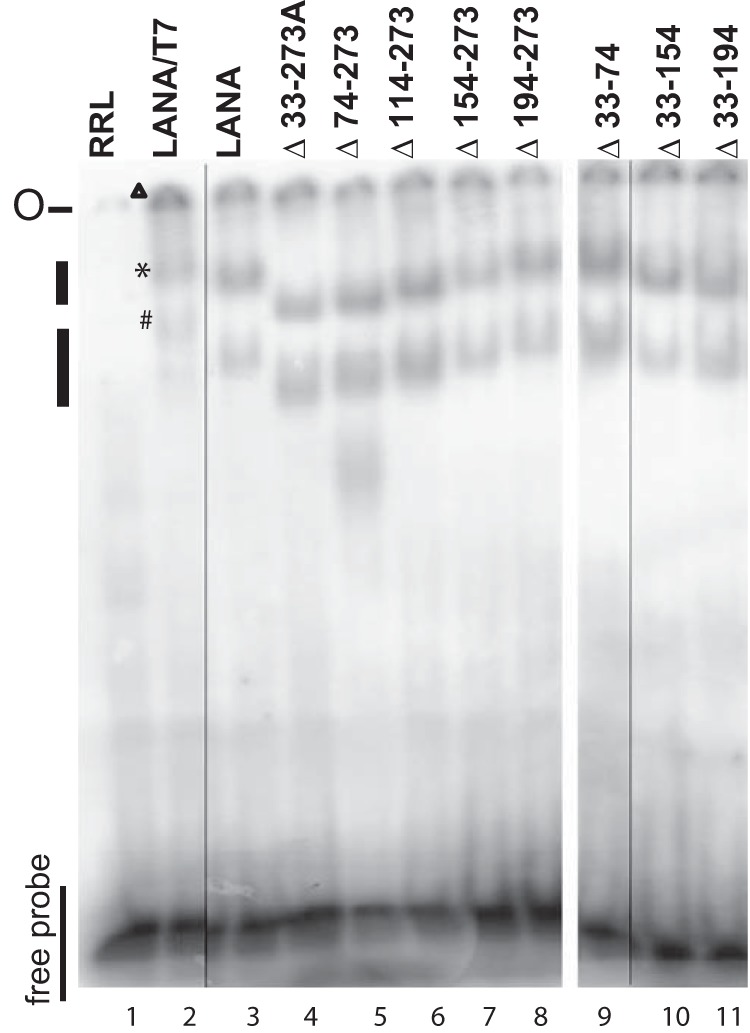
LANA and its deletion mutants maintain the ability to bind KSHV TR DNA, as assessed by EMSA. RRL (lane 1) or similar amounts (as assessed by Western blotting) of in vitro-translated LANA (lane 3) or LANA mutants (lanes 4 to 11) were each incubated with radiolabeled TR DNA probe. The lane 2 incubation included anti-T7 antibody, which detects the amino-terminal LANA T7 epitope. After incubation, complexes were resolved on a nondenaturing polyacrylamide gel. Brightness and contrast were uniformly adjusted using Adobe Photoshop. The results are representative of 3 independent experiments. The data are from two gels (separated by a space between lanes 8 and 9) run simultaneously. The thin lines between lanes 2 and 3 and lanes 9 and 10 indicate where lanes from the same gel were not included. The lines on the left indicate LANA or LANA mutant complexes. ▶, *, and # indicate supershifted LANA complexes in lane 2. O, gel origin.
Deletion of LANA residues 33 to 273 does not reduce TR DNA replication.
LANA-mediated DNA replication is essential for episome persistence. LANA carboxy-terminal DNA binding is necessary for replication, and amino-terminal LANA is also critical for replication. We previously reported that LANAΔ33–273 had reduced DNA replication (32). Since LANAΔ33–273 contains GYQ in place of the deleted LANA residues while LANAΔ33–273A contains four alanines in place of the deletion, we compared the mutants for DNA replication. We cotransfected 293T cells with p8TR-gB (a plasmid containing 8 TRs) alone or with LANAΔ33–273A or LANAΔ33–273. p8TR-gB was purified from Dam methylase-positive bacteria and is therefore susceptible to DpnI digestion. After replication in 293T cells, p8TR-gB is resistant to DpnI digestion, since mammalian cells lack Dam methylase. Twenty-four hours after transfection, a portion of the cells were harvested for Hirt DNA; the DNA was digested with HindIII, which linearizes p8TR-gB; and the linearized p8TR-gB was detected by Southern blotting. All the lanes contained similar levels of linearized p8TR-gB (Fig. 4A, lanes 1 to 3, arrow), indicating similar transfection efficiencies. Seventy-two hours after transfection, replicated DNA was assessed by double digestion with HindIII and DpnI. As expected, in the absence of LANA, no replicated p8TR-gB was detected (Fig. 4B, lane 1). In contrast, replicated DNA was present in both LANAΔ33–273A and LANAΔ33–273 lanes (Fig. 4B, lanes 2 and 3, arrow). However, the level of replicated DNA was substantially higher for LANAΔ33–273A than for LANAΔ33–273. The higher replication was not due to increased LANAΔ33–273 expression compared to LANAΔ33–273A (Fig. 4C). This indicates that GYQ results in reduced LANA replication compared to the use of four alanines in place of the deleted residues. We then assessed LANA, LANAΔ33–273A, LANAΔ33–194, and LANAΔ194–273 for the ability to replicate TR DNA. HindIII digestion of Hirt DNA at 24 h posttransfection indicated similar transfection efficiencies (Fig. 4D). HindIII/DpnI digestion of Hirt DNA at 72 h demonstrated that LANAΔ33–273A, LANAΔ33–194, and LANAΔ194–273 (Fig. 4E, arrow) all replicated p8TR-gB at a level similar to that of LANA. Expression of LANA and the deletion mutants was confirmed by Western blotting, and all were expressed at similar levels (Fig. 4F). Therefore, LANA residues 33 to 273 are dispensable for TR DNA replication.
FIG 4.

Deletions within LANA residues 33 to 273 do not reduce DNA replication. LANA or each LANA mutant was cotransfected with a plasmid containing 8 TR copies (p8TR-gB) into 293T cells. Twenty-four or 72 h after transfection, DNA was extracted by the method of Hirt. (A, B, D, and E) HindIII-digested Hirt DNA harvested at 24 h (A and D) or Hirt DNA digested with HindIII and DpnI at 72 h (B and E) was assessed by Southern blotting after incubation with 32P-radiolabeled TR probe. (C and F) Western blotting for LANA or tubulin 24 h after 293T cell transfection. Brightness and contrast were uniformly adjusted using Adobe Photoshop. The results are representative of at least 3 independent experiments. The arrows in panels A and D indicate linear p8TR-gB plasmid; the arrows in panels B and E indicate replicated DNA.
LANA residues 33 to 194 are critical for retention of green fluorescent protein (GFP) expression from replication-deficient TR episomes.
We wished to assess the episome segregation abilities of the LANA mutants, since segregation of virus DNA to daughter nuclei following mitosis is essential for episome maintenance. In the absence of segregation, episomes are rapidly degraded in the cytoplasm. Amino- and carboxy-terminal LANA tether episomes to mitotic chromosomes. N-terminal LANA binds mitotic chromosomes, and C-terminal LANA simultaneously binds KSHV DNA. In addition to the essential role of N- and C-terminal LANA domains in segregation, we previously showed that LANA residues 33 to 331, comprising the unique internal sequence, are important for episome segregation, despite lacking a role in chromosome binding.
We used a replication-deficient plasmid that contains two TRs, termed p2TR-ΔRE-GFP (33) (a gift from Rolf Renne), to assess segregation. TR elements each contain three adjacent LANA binding sites; one of the three LANA binding sites is deleted in each TR of p2TR-ΔRE-GFP. Deletion of the third LANA binding site (also termed replication element) renders p2TR-ΔRE-GFP incompetent for LANA-mediated DNA replication. LANA binds to the two remaining binding sites in each TR to tether p2TR-ΔRE-GFP to mitotic chromosomes to mediate segregation. We used GFP expression from p2TR-ΔRE-GFP to track the presence of episomes. GFP retention is expected to directly reflect LANA-mediated segregation. To account for any differences in growth rates, the percentages of GFP-positive cells were compared after a constant amount of cell proliferation (e1.5, or 4.48-fold) for each cell line, which generally occurred 3 to 4 days after transfection and was well within the linear growth range for the cell lines. In the absence of selection, TR DNA is expected to be lost from cells, and LANA reduces the rate of loss (33–35). As expected, BJAB cells, which lack LANA, rapidly lost GFP expression (Fig. 5A and C), and only 1.5 to 2.6% of the cells retained GFP expression. In contrast, 20 to 24% of the cells expressing LANA retained GFP. LANAΔ194–273 maintained GFP expression at a level similar to that of LANA (Fig. 5A). In contrast, LANAΔ33–273A, LANAΔ74–273, LANAΔ114–273, and LANAΔ154–273 were highly deficient in the ability to maintain GFP expression, and retention levels were generally similar to that of BJAB cells (Fig. 5A), despite similar or only slightly reduced expression levels compared to LANA (Fig. 5B). We further assessed LANA residues 33 to 194 with LANAΔ33–74, LANAΔ33–154, LANAΔ33–194, and LANAΔ154–194. Each of the mutants was reduced in GFP retention compared to LANA, although each was more efficient than BJAB cells (Fig. 5C). The reduction in GFP retention was not due to reduced LANA expression levels, since each mutant was expressed at levels similar to those of LANA (Fig. 5D). These results indicate LANA residues 33 to 194 play a critical role in LANA segregation ability.
FIG 5.
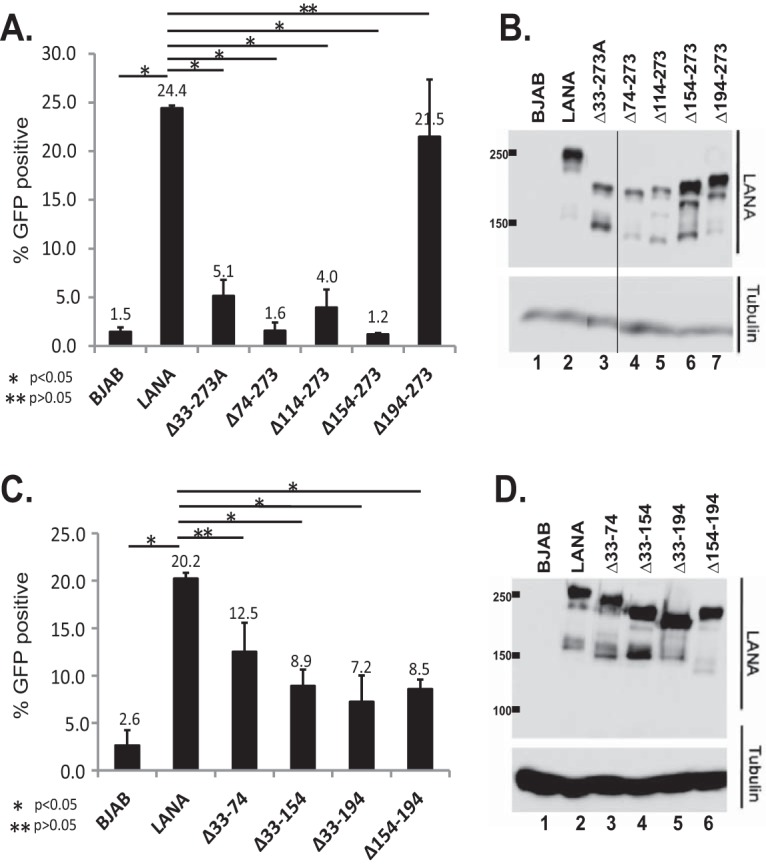
Segregation of TR DNA by LANA or LANA deletion mutants. (A and C) Ten million BJAB cells, BJAB cells stably expressing LANA, or BJAB cells expressing different LANA mutants were transfected with p2TR-ΔRE-GFP (5,000 copies per cell). GFP expression was monitored daily by FACS starting 48 h after transfection (when the maximum percentage of cells expressing GFP was attained). Cell growth was assessed by counting the cells daily. The percentages of cells expressing GFP were compared after the cells had reached a concentration of e1.5 (4.48) times the concentration measured at day 0 of FACS. Each bar is an average of three independent experiments. The error bars indicate standard deviation. (B and D) Western blots from representative experiments for LANA or tubulin of BJAB cells or BJAB cells stably expressing LANA or deletion mutants 7 days after p2TR-ΔRE-GFP transfection. *, P < 0.05; **, P > 0.05, as determined by independent-samples t test.
LANA residues 33 to 194 are dispensable, while residues 194 to 273 are critical, for episome persistence.
We assessed the LANA mutants for the ability to mediate episome persistence. BJAB cells or BJAB cells stably expressing LANA, LANAΔ33–273A, LANAΔ74–273, LANAΔ114–273, LANAΔ154–273, or LANAΔ194–273 were each transfected with p8TR, a plasmid containing 8 copies of the KSHV TRs; seeded at 1,000, 100, 10, or 1 cell per well in microtiter plates; and placed under G418 selection (resistance encoded on the plasmid vector). Since the BJAB cell lines stably expressing LANAΔ33–74, LANAΔ33–154, or LANAΔ33–194 are resistant to G418, these cells were transfected instead with p8TR-P, seeded into microtiter plates, and placed under puromycin resistance (resistance encoded on p8TR-P). Twenty-one days after selection, the number of wells with macroscopically visible outgrowth was determined. As expected, G418 or puromycin-resistant outgrowth of LANA-expressing cells was robust, with outgrowth in an average of 96, 96, 67.2, and 11.5 wells after seeding at 1,000, 100, 10, or 1 cell/well and G418 selection or 96, 79, 24.7, and 1.3 wells after seeding at 1,000, 100, 10, or 1 cell/well and puromycin selection (averaged from the results of three experiments) (Fig. 6). In contrast, BJAB outgrowth was much lower in 81.2, 18.2, 2.5, or 0 wells after seeding at 1,000, 100, 10, or 1 cell/well and G418 selection or 6, 0.3, 0, or 0 wells after seeding at 1,000, 100, 10, or 1 cell/well and puromycin selection. The lower outgrowth of BJAB cells than of LANA-expressing cells is due to the need for integration of p8TR DNA, which is a rare event compared with episome persistence. BJAB cells expressing LANAΔ33–273A, LANAΔ74–273, LANAΔ114–273, LANAΔ154–273, or LANAΔ194–273 were all greatly reduced in G418-resistant outgrowth, with outgrowth levels similar to that of BJAB cells (Fig. 6A and B). In contrast, BJAB cells expressing LANAΔ33–74, LANAΔ33–154, and LANAΔ33–194 demonstrated efficient outgrowth, with levels similar to that of LANA (Fig. 6C and D). These results suggest that LANAΔ33–273A, LANAΔ74–273, LANAΔ114–273, LANAΔ154–273, and LANAΔ194–273 are highly deficient for episome persistence, while LANAΔ33–74, LANAΔ33–154, and LANAΔ33–194 are wild type for episome persistence.
FIG 6.

LANA mutants with deletions including amino acids 194 to 273 are deficient for drug-resistant outgrowth after TR DNA transfection. Limiting-dilution outgrowth assays were performed after p8TR (A and B) or p8TR-P (C and D) transfection into BJAB cells, BJAB cells stably expressing LANA, or BJAB cells stably expressing LANA deletion mutants. Forty-eight hours posttransfection, cells were seeded at an average of 1, 10, 100, or 1,000 cells per well into 96-well microtiter plates and selected for p8TR using G418 (A and B) or puromycin (C and D). Drug-resistant outgrowth was recorded 21 days after cell plating. Averages of the results of three independent experiments are shown in graph form (A and C), or numerical values of the averages are shown (B and D). The error bars indicate standard deviations.
To directly assess for the presence of episomes, Gardella gel assays were performed on G418- or puromycin-resistant cell lines. Cell lines were expanded from microtiter wells from plates that had outgrowth in approximately half or fewer of the wells. In Gardella gels, live cells are loaded into agarose gel wells, and cell lysis occurs in situ upon the start of the gel run due to the presence of SDS and protease (36). During electrophoresis, episomes as large as several hundred kilobases migrate into the gel, while chromosomal DNA remains at the gel origin. Episomes are then detected by Southern blotting. Gardella analysis of BCBL-1 cells, a KSHV-infected PEL cell line, revealed a slowly migrating episomal band (Fig. 7A, lane 1; Fig. 7B, lanes 1 and 2) and a more quickly migrating band of linear DNA due to lytic replication of a subset of cells. As expected, BJAB cells, which lack LANA, lacked episomal DNA (Fig. 7A, lanes 3 and 4, and Fig. 7B, lanes 4 and 5). In contrast, BJAB cells expressing LANA contained episomes in all lanes (Fig. 7A, lanes 5 and 6, and Fig. 7B, lanes 6 to 8). As previously observed, much of the episomal DNA migrated more slowly than the p8TR plasmid covalently closed circular DNA (cccDNA) (Fig. 7A, lane 2, and Fig. 7B, lane 3). The more slowly migrating DNA is due to duplication of TR elements and multimerization of input plasmids (9, 37). In contrast to LANA, BJAB cells expressing LANAΔ33–273A had episomes in only 2 of 4 lanes (Fig. 7A), LANAΔ74–273 had episomes in 2 of 2 lanes, LANAΔ114–273 had episomes in 2 of 4 lanes, LANAΔ154–273 in 2 of 2 lanes, and LANAΔ194–273 in 1 of 2 lanes. In a total of 6 experiments, LANA had episomes in 20/20 (100%) G418- or puromycin-resistant cell lines. In 3 experiments, LANAΔ33–273A had episomes in 34/53 (64.2%), LANAΔ74–273 had episomes in 10/24 (41.7%), LANAΔ114–273 had episomes in 10/37 (27%), LANAΔ154–273 had episomes in 4/16 (25%), and LANAΔ194–273 had episomes in 12/36 (33.3%) G418-resistant cell lines (Fig. 1). When episomal DNA was present for the LANA deletion mutants, it typically migrated more slowly than p8TR cccDNA, and the signal was more intense than that in LANA lanes for some cell lines, such as LANAΔ114–273 (Fig. 7A, lane 15) or LANAΔ154–273 (Fig. 7A, lanes 17 and 18). The deficits in episome persistence were not due to absence of LANA expression as detected by Western blotting (Fig. 7C). Therefore, deletions terminating at residue 273 all induced substantial LANA episome maintenance deficiency.
FIG 7.

LANA mutants with deletions including amino acids 194 to 273 are deficient for episome maintenance after TR DNA transfection, as assessed by Gardella gel analysis. BJAB cells, BJAB cells stably expressing LANA, or BJAB cells stably expressing each deletion mutant were transfected with p8TR. Forty-eight hours after transfection, the cells were seeded into 96-well plates and selected for drug resistance encoded on the p8TR plasmid. Twenty-one days after plating, the cells were expanded from individual wells and analyzed by Gardella gel analysis for the presence of episomes. (A and B) Gardella gels containing LANA or LANA mutants, as indicated. Lanes with p8TR plasmid or with BCBL-1 cells (a KSHV-infected primary effusion lymphoma cell line) are also indicated. O, gel origin; E, BCBL-1 episomes; L, BCBL-1 linear forms (from lytic replication); ccc, p8TR cccDNA. The number of days of drug selection are indicated below each gel. (C and D) Western blot analyses of LANA, LANA mutants, or tubulin. The lanes correspond to those shown in the Gardella gels. Brightness and contrast were uniformly adjusted in each panel using Adobe Photoshop.
In contrast to LANA deletions terminating at residue 273, LANA mutants with deletions between residues 33 and 194 were not reduced in episome persistence. LANAΔ33–74, LANAΔ33–154, and LANAΔ33–194 all had episomes in all lanes, similar to LANA (Fig. 7B). In 3 experiments, LANAΔ33–74 had episomes in 14/14 (100%), LANAΔ33–154 had episomes in 14/14 (100%), and LANAΔ33–194 had episomes in 14/14 (100%) puromycin-resistant cell lines (Fig. 1). These results indicate that LANA residues 33 to 194 are dispensable, but residues 194 to 273 are critical, for LANA episome maintenance.
To facilitate the comparison of episome maintenance functions, the predicted numbers of cells seeded per well required to obtain 63.2% of wells with episome-containing cells were determined by nonlinear regression analyses. Using the Poisson distribution, when the average number of episome-containing cells seeded into a microtiter plate is 1 per well, an average of 36.8% of wells are expected to lack these cells. The estimates of the numbers of cells needed to be seeded per well to yield 63.2% of wells with at least 1 episome-containing cell were 7.4, 381.5, 313.5, 766.1, 699.9, and 1,066.7 for G418-selected LANA, LANAΔ33–273A, LANAΔ74–273, LANAΔ114–273, LANAΔ154–273, and LANAΔ194–273 cell lines, respectively (Table 1). The values obtained for puromycin-selected LANA, LANAΔ33-74, LANAΔ33-154, and LANAΔ33-194 were 32.2, 26.7, 7.1, and 19.7, respectively (Table 2). Compared to LANA, these results indicate deficiencies ranging from 42.5- to 144.8-fold for deletions terminating at amino acid 273; however, the results indicate no deficiencies for deletions between residues 33 and 194 (Fig. 1).
TABLE 1.
Efficiencies of episome maintenance (p8TR-G418 plasmid)
| Cell line | No. of cells per wella |
|---|---|
| LANA | 7.4 |
| LANAΔ33–273A | 381.5 |
| LANAΔ74–273 | 313.5 |
| LANAΔ114–273 | 766.1 |
| LANAΔ154–273 | 699.9 |
| LANAΔ194–273 | 1,066.7 |
Predicted number of cells per well necessary for 63.2% outgrowth of wells with episome-containing cells.
TABLE 2.
Efficiencies of episome maintenance (p8TR-puromycin plasmid)
| Cell line | No. of cells per wella |
|---|---|
| LANA | 32.2 |
| LANAΔ33–74 | 26.7 |
| LANAΔ33–154 | 7.1 |
| LANAΔ33–194 | 19.7 |
Predicted number of cells per well necessary for 63.2% outgrowth of wells with episome-containing cells.
DISCUSSION
We previously identified LANA internal residues upstream of the repeat elements as important for episome persistence and segregation (32). Here, we further investigated this region and found that LANA residues 33 to 194 are important for segregation, as assessed by the ability to retain GFP expression from TR episomes that are not competent for DNA replication, but dispensable for episome maintenance. In contrast, residues 194 to 273 are largely dispensable for segregation but critical for episome persistence. The deficiencies in segregation or episome maintenance were not due to the inability of LANA to associate with mitotic chromosomes or to the inability to bind or replicate KSHV TR DNA.
Simultaneous binding to mitotic chromosomes and KSHV TR DNA results in LANA tethering episomes to mitotic chromosomes to ensure KSHV DNA segregates to progeny cell nuclei. Amino-terminal LANA residues 5 to 13 bind histones H2A/H2B (19), and carboxy-terminal LANA independently binds chromosomes (12, 14). In this work, LANAΔ33–74, LANAΔ33–154, and LANAΔ33–194 demonstrated increasing levels of segregation deficiency, correlating with the increasing sizes of the deletions. In addition, LANAΔ154–194 was also similarly deficient. These results suggest that the entire sequence between amino acids 33 and 194 plays a role in segregation. This sequence does not have a direct role in binding chromosomes, since LANA residues 33 to 331 does not associate with mitotic chromosomes (32). However, it is possible LANA residues 33 to 194 could serve to strengthen amino-terminal LANA binding to nucleosomes through auxiliary contacts with chromatin. Alternatively, the sequence may function during DNA replication, when nucleosomes are removed. For instance, it is possible that residues 33 to 194 could be important to maintain contact with histones H2A/H2B after removal from DNA or to ensure reattachment after nucleosomes are reconstituted into replicated DNA. LANA interacts with SSRP1 (38), a member of the FACT complex, which allows nucleosome reorganization through destabilizing interactions between H2A/H2B dimers and H3/H4 tetramers, and it is intriguing to speculate that the complex could be involved in segregation (39–41). The kinetochore proteins Bub1 and Cenp-F colocalize with LANA at centromeres during mitosis at 55% and 30% of sites, respectively (42). These proteins interact with LANA residues 1 to 340 and with LANA residues 842 to 1162. Since these interactions overlap with residues 33 to 194, they are potential candidates for involvement in episome segregation. Further, cells depleted of Bub1 show a reduction in KSHV episomal DNA. It is possible that these or other cellular proteins interacting with LANA between amino acids 33 and 194 could be important for segregation.
Although deletion of residues 33 to 194 did not result in a deficit for episome maintenance in these experiments, it is possible the sequence plays an important role in vivo. Transfection of p8TR DNA into cells in these experiments is expected to result in a high copy number of plasmids per transfected cell, which may compensate for the observed segregation defect. In contrast, in vivo, virus infection likely usually occurs at a low multiplicity of infection, and in such a setting, the segregation defect could potentially exert a profound effect on virus persistence. Alternatively, since the segregation assay was performed using a plasmid that contains two TR copies, it is possible that the presence of a higher TR copy number compensates for the segregation deficiency. There are 8 TRs in p8TR, and as observed here and previously (9, 31, 32, 37), there is subsequent selection for large recombinant episomes that comigrate near episomal virus DNA (Fig. 7B). These large episomes result from duplication of TR elements and recombined, tandem p8TR plasmids arranged head to tail and likely contain at least 40 TR copies, similar to the KSHV genome. Notably, LANAΔ33–74, LANAΔ33–154, and LANAΔ33–194 each had episomal signal more intense than that of LANA, suggesting selection for increased TR copy numbers for these cell lines (Fig. 7B). Each TR contains three adjacent LANA binding sites. C-terminal LANA forms multimers through dimer-dimer interactions (43–47), and it is possible that a higher-order LANA structure may compensate for the segregation defect induced by deletion of amino acids 33 to 194. Since it is unlikely virus could undergo such TR recombination events and remain viable, these segregation deficiencies could be highly detrimental to KSHV in vivo.
LANA residues 194 to 273 play a critical role in episome persistence; deletions within this sequence resulted in profound deficiencies (Fig. 1). Episome persistence is comprised of two steps. Episomes first replicate prior to cell division, and virus DNA then segregates to daughter cell nuclei following mitosis. It is significant that deletion of residues 194 to 273 did not result in deficiency of either LANA-mediated DNA replication or segregation. It is possible this region may perform a unique function, such as prevention of an inhibitory host cell response. Another possibility could be related to the absence of TR DNA replication in the segregation assay used here. Perhaps this sequence could be important for ensuring reattachment to LANA TR sequence following TR DNA replication. Loss of such reattachment would lead to loss of episomes. This sequence also partially overlaps residues 262 to 320, previously shown to be important for interaction with RFC, the DNA polymerase clamp loader (48, 49). However, LANAΔ194–273’s episome maintenance deficiency is much greater than that of LANAΔ262–320 (144.5- versus 14.4-fold), indicating that the deficiency in LANAΔ194–273 is unlikely to be due to loss of RFC interaction.
LANA residues 194 to 273 contains several notable functional motifs. One of these is a bipartite suppressor of cytokine signaling (SOCS) box-like motif (50). The amino-terminal portion of the SOCS box is located at residues 212 to 222, which interact with Elongin B and C, while the carboxy-terminal portion is located at residues 1085 to 1100 and is the Cullin box. Through these motifs, LANA interacts with the Cul5-Elongin BC complex to serve as an E3 ubiquitin ligase. LANA residues 194 to 273 also contains a SUMO-interacting motif located at residues 244 to 250 and 264 to 270 (51). This region is specific for SUMO-2 rather than SUMO-1 binding. Deletion of these SUMO-interacting motifs led to a deficiency in LANA episome maintenance, as assessed by cotransfection of LANA and TR DNA into 293 cells and selecting for puromycin-resistant colonies at 14 days. The SUMO-interacting motif interacted with the SUMO-2-modified transcriptional repressor KAP1 and was also shown to have a role in blocking lytic replication (51, 52). Hypoxia reduced KAP1 SUMOylation and transcription of the Rta lytic activator. LANA residues 194 to 273 also contain four GSK-3beta phosphorylation sites at residues 219, 250, 261, and 268 (53). GSK-3beta phosphorylates these sites, and the residues also mediate LANA residue interaction with GSK-3beta. Despite GSK-3beta phosphorylation of the sites, GSK-3beta that coprecipitates with LANA is inhibited in its kinase activity. It is interesting to speculate whether any or a combination of these motifs and their functions might be related to LANAΔ194–273’s episome maintenance deficiency.
This work identified a region important for LANA segregation and an independent adjacent region critical for episome persistence. The latter region contains three functional motifs that potentially could play roles in episome maintenance. Future work elucidating the mechanisms through which LANA performs these functions would provide important insight into LANA mediation of viral persistence.
MATERIALS AND METHODS
Plasmids.
pT7LANA, p8TR, p8TR-gB, and p8TR-P have been previously described (31, 32, 37). p2TR-RE-GFP was a kind gift from Rolf Renne (33). To construct a pT7LANA plasmid resistant to G418 (pT7LANA-G418), the kanamycin/G418 resistance gene from pEGFP-C1 was PCR amplified using primers containing AseI-NdeI restriction sites. The fragment was then cloned into the pT7LANA AseI-NdeI sites.
pT7LANAΔ33-273A, pT7LANAΔ74-273, pT7LANAΔ114-273, pT7LANAΔ154-273, pT7LANAΔ194-273, and pT7LANAΔ234-273 were generated as follows. The oligonucleotide NotT7Epitop Fwd was used with the corresponding Ala Rev oligonucleotide (Table 3) to PCR amplify the relevant LANA amino-terminal sequence from pT7LANA using Pfx DNA polymerase (Thermo Fisher). The amplified fragment was digested with XhoI (NEB), which cuts within the N-terminal T7 epitope, and BamHI (NEB). pT7LANAB−, which has the BamHI site in the polylinker disabled, was digested with XhoI and BamHI (which cuts at LANA codon 273), and the XhoI/BamH1-digested PCR fragment was inserted to generate each of the deletion mutants that contain four alanine residues in place of the deleted amino acids.
TABLE 3.
Oligonucleotides used to generate LANA mutants

aLANA sequence is in boldface.
bBamHI restriction sites are indicated by italics.
cAlanine codons are underlined.
Deletions for LANAΔ33–74, LANAΔ33–154, LANAΔ33–194, LANAΔ33–234, and LANAΔ154–194 were generated as follows. pT7LANABam contains the N-terminal 273 LANA codons, without downstream LANA sequence, and was generated by BamHI digestion of pT7LANA, which cuts at codon 273 and in the 3′ polylinker, followed by religation. PCR amplification with PrimeStar (TaKaRa) polymerase of pT7LANABam was performed using oligonucleotides in opposite orientations (Table 3) and with the 5′ ends encoding the margins of each LANA deletion. Oligonucleotides 33 REV and 74 FWD, 154 FWD, 194 FWD, or 234 FWD were used to generate LANAΔ33–74, LANAΔ33–154, LANAΔ33–194, and LANAΔ33–234, respectively, while 154 REV and 194 REV were used to generate LANAΔ154–194. After amplification, DNA was digested with DpnI (NEB) to remove the template, amplified with plasmids circularized using the Quick Ligase (NEB) protocol modified to include T4 polynucleotide kinase (NEB) in the incubation, transformed into bacteria, and plasmid purified. The relevant regions of LANA deletions were then transferred from the pT7LANABam vector to pT7LANA-G418B− by digestion with XhoI/BamHI. All mutations were confirmed by DNA sequencing.
Cell lines.
BJAB and BCBL1 cells were maintained in RPMI medium (Corning) supplemented with 10% bovine growth serum (BGS) (GE Healthcare) and 15 μg/ml gentamicin (Gemini). Stable BJAB cell lines expressing LANA or LANA deletion mutants were obtained by nucleofection with 5 μg of LANA expression plasmid into 10 million cells using Amaxa Nucleofector I (program C-09) and solution V. Forty-eight hours after transfection, cells were seeded at an average of 10, 100, or 1,000 cells per well in microtiter plates in RPMI containing 10% BGS and either 0.2 mg/ml hygromycin (EMD Millipore) or 0.8 mg/ml G418 (Gemini). Cells from plates with the least drug-resistant outgrowth (i.e., at most 40% of wells positive) were screened for LANA expression by Western blotting and immunofluorescence. If fewer than 75% of cells expressed LANA, then the cells were subcloned by limiting dilution and screened again until at least 75% of the cells expressed LANA or LANA mutants. BJAB cells containing p8TR plasmid were maintained using RPMI with 0.8 mg/ml G418 (Gemini) or 0.003 mg/ml puromycin (Invivogen).
Fluorescence microscopy.
BJAB cells, BJAB cells stably expressing LANA, or BJAB cells stably expressing deletion mutants were incubated overnight with 1 μg/ml colcemid (Calbiochem) to induce metaphase arrest. The next day, the cell pellets were resuspended in hypotonic buffer (1% sodium citrate, 10 mM CaCl2, 10 mM MgCl2) for 5 min at room temperature. The cells were spread onto polylysine-coated slides by Cytospin (Thermo Shandon) centrifugation for 10 min at 1,900 rpm. The cells were then fixed for 10 min in 4% paraformaldehyde. LANA or LANA mutants were detected using anti-LANA LN53 antibody (1:300; ABI or EMD Millipore) and secondary Alexa Fluor 488 anti-rat antibody (Invitrogen). DNA was stained with propidium iodide at 1 μg/ml (Invitrogen). Coverslips were mounted using Aqua-Polymount (Polysciences). Microscopy was performed using a Zeiss AxioPlan 2 microscope.
Electrophoretic mobility shift assays.
LANA or LANA deletion mutants were in vitro translated using the TNT Quick Coupled reticulocyte lysate system (Promega). Similar amounts of protein (as determined by Western blotting) were incubated for 30 min at 40°C in DNA binding buffer [20 mM Tris-HCl, pH 7.5, 10% glycerol, 50 mM KCl, 10 mM MgCl2, 0.1 mM dithiothreitol (DTT), 1 mM EDTA, 20 μg/ml poly(dI-dC) (Sigma)] with TR-13 probe, which contains a 20-bp, high-affinity LANA binding site (9) labeled with 32P. For the supershift assay, 1 μg of anti-T7 antibody (Novagen) was included in the incubation for 15 min. Complexes were resolved in a 3.5% nondenaturing polyacrylamide gel, and signal was detected by autoradiography.
DNA replication assays.
293T cells in 6-well plates were cotransfected with 1 μg p8TR-gB and 0.5 μg to 1 μg of LANA or LANA mutant expression vector to obtain similar LANA expression levels by Western blotting or 1 μg of pSG5. Transfections were performed using polyethylenimine (PEI). Twenty-four hours after transfection, half the cells were harvested to assess transfection efficiency, while the other half of the cells were expanded in 10-cm plates. Seventy-two hours after transfection, the remaining cells were harvested. DNA harvested at 24 or 72 h after transfection was extracted by the Hirt method (54). Similar amounts of DNA harvested at 24 h were digested overnight with HindIII (NEB) to linearize p8TR plasmid. Similar amounts of DNA harvested 72 h after transfection were digested overnight with HindIII and DpnI (NEB) to linearize the replicated p8TR plasmids and degrade input, transfected plasmid. DNA was resolved in a 0.8% agarose gel and transferred to a nylon membrane (Bio-Rad), Southern blotting was performed using 32P-labeled TR probe, and signal was detected by autoradiography.
GFP retention assay.
Ten million BJAB cells or BJAB cells stably expressing LANA deletion mutants were transfected with 466.7 ng p2TR-ΔRE-GFP (corresponding to 5,000 copies per cell) using an Amaxa Nucleofector I (program C-09) and solution V and plated into one well of a six-well plate. Twenty-four hours later, the cells were transferred to a T25 flask in 20 ml RPMI. At 48 h after transfection, at which time there was the highest percentage of cells that were GFP positive, the cells were assessed by fluorescence-activated cell sorting (FACS) daily for 5 days with a FACSCalibur (BD Biosciences), and the percentages of GFP-positive cells were plotted over this period. Cell concentrations were measured daily, starting from the day of transfection, by manual counting in trypan blue and plotted for 7 days. The cells were maintained in log-phase growth by splitting them every day to a concentration of about 0.4 × 106 cells/ml. To account for small differences in growth rates between cell lines, percentages of GFP-positive cells were compared when the amount of cell proliferation for each cell line reached e1.5 (4.48) times the concentration measured at the time of the first FACS analysis. e1.5 times the concentration was previously validated as an optimal point to determine differences and is well within the linear part of the growth pattern for the cells (32). GFP percentages and cell concentration data after e1.5 times the cell concentrations were determined by fitting the data to regression curves using GraphPad Prism.
Limiting-dilution outgrowth and Gardella gel analysis.
Ten million BJAB cells or BJAB cells stably expressing the deletion mutants were transfected with 5 μg p8TR or p8TR-P using an Amaxa Nucleofector I (program C-09) and solution V as described above. Twenty-four hours after transfection, the cells were expanded to 20 ml in a T25 flask. Forty-eight hours after transfection, the cells were counted and seeded at an average of 1, 10, 100, or 1,000 cells per well in 96-well microtiter plates with 0.8 mg/ml G418 (Gemini) for cells transfected with p8TR or 0.003 mg/ml puromycin (Invivogen) for cells transfected with p8TR-puro. The cells were fed twice a week with medium containing drug. Twenty-one days after seeding, the number of wells with macroscopically visible outgrowth for each plate was assessed.
Cells were assessed for the presence of episomes. Cells from individual wells of microtiter plates with drug-resistant outgrowth ranging from 10% to about 50% (where the Poisson distribution predicted an average of one drug-resistant outgrowth event per well in most wells) were expanded in 12-well plates for 1 week. Two million cells for each cell line were assessed for episome maintenance by Gardella gels as previously described (32). Briefly, live cells were loaded and lysed in situ in agarose gel wells containing sodium dodecyl sulfate and protease (Sigma), followed by electrophoresis in a Tris-borate-EDTA buffer. DNA was transferred to a nylon membrane, and p8TR DNA was detected using 32P-radiolabeled TR probe. Signal was detected by autoradiography.
Efficiencies of episome-containing cell outgrowth were calculated. The percentages of episome-containing cell lines (Fig. 1) were used to estimate the numbers of episome-containing wells from plates with an average drug-resistant outgrowth of about 10% to 50% of wells (Fig. 6), where most events are expected to be clonal. Using these values, the predicted numbers of episome-containing wells from plates seeded with 10-fold (increment) fewer or greater numbers of cells plated per well were then determined using the Poisson distribution. These numbers were used in nonlinear regression analyses to estimate the predicted number of cells per well necessary to obtain 63.2% outgrowth of wells containing cells maintaining episomes (32).
ACKNOWLEDGMENTS
This work was supported by NIH grants CA082036, DE025208, and DE024971 to K.M.K. and by Fondation ARC pour la Recherche sur le Cancer grant SAE20121206018 to F.J.
REFERENCES
- 1.Cesarman E, Moore PS, Rao PH, Inghirami G, Knowles DM, Chang Y. 1995. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi's sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood 86:2708–2714. [PubMed] [Google Scholar]
- 2.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science 266:1865–1869. [DOI] [PubMed] [Google Scholar]
- 3.Moore PS, Chang Y. 1995. Detection of herpesvirus-like DNA sequences in Kaposi's sarcoma in patients with and without HIV infection. N Engl J Med 332:1181–1185. doi: 10.1056/NEJM199505043321801. [DOI] [PubMed] [Google Scholar]
- 4.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay M-F, Clauvel J-P, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280. [PubMed] [Google Scholar]
- 5.Decker LL, Shankar P, Khan G, Freeman RB, Dezube BJ, Lieberman J, Thorley-Lawson DA. 1996. The Kaposi sarcoma-associated herpesvirus (KSHV) is present as an intact latent genome in KS tissue but replicates in the peripheral blood mononuclear cells of KS patients. J Exp Med 184:283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kedes DH, Lagunoff M, Renne R, Ganem D. 1997. Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi's sarcoma-associated herpesvirus. J Clin Invest 100:2606–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kellam P, Boshoff C, Whitby D, Matthews S, Weiss RA, Talbot SJ. 1997. Identification of a major latent nuclear antigen, LNA-1, in the human herpesvirus 8 genome. J Hum Virol 1:19–29. [PubMed] [Google Scholar]
- 8.Rainbow L, Platt GM, Simpson GR, Sarid R, Gao SJ, Stoiber H, Herrington CS, Moore PS, Schulz TF. 1997. The 222- to 234-kilodalton latent nuclear protein (LNA) of Kaposi's sarcoma-associated herpesvirus is encoded by orf73 and is a component of the latency-associated nuclear antigen. J Virol 71:5915–5921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ballestas ME, Kaye KM. 2001. Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through cis-acting terminal repeat (TR) sequence and specifically binds TR DNA. J Virol 75:3250–3258. doi: 10.1128/JVI.75.7.3250-3258.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ballestas ME, Chatis PA, Kaye KM. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641–644. [DOI] [PubMed] [Google Scholar]
- 11.Barbera AJ, Ballestas ME, Kaye KM. 2004. The Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen 1 N terminus is essential for chromosome association, DNA replication, and episome persistence. J Virol 78:294–301. doi: 10.1128/JVI.78.1.294-301.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelley-Clarke B, Ballestas ME, Komatsu T, Kaye KM. 2007. Kaposi's sarcoma herpesvirus C-terminal LANA concentrates at pericentromeric and peri-telomeric regions of a subset of mitotic chromosomes. Virology 357:149–157. doi: 10.1016/j.virol.2006.07.052. [DOI] [PubMed] [Google Scholar]
- 13.Kelley-Clarke B, Ballestas ME, Srinivasan V, Barbera AJ, Komatsu T, Harris TA, Kazanjian M, Kaye KM. 2007. Determination of Kaposi's sarcoma-associated herpesvirus C-terminal latency-associated nuclear antigen residues mediating chromosome association and DNA binding. J Virol 81:4348–4356. doi: 10.1128/JVI.01289-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krithivas A, Fujimuro M, Weidner M, Young DB, Hayward SD. 2002. Protein interactions targeting the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus to cell chromosomes. J Virol 76:11596–11604. doi: 10.1128/JVI.76.22.11596-11604.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Piolot T, Tramier M, Coppey M, Nicolas JC, Marechal V. 2001. Close but distinct regions of human herpesvirus 8 latency-associated nuclear antigen 1 are responsible for nuclear targeting and binding to human mitotic chromosomes. J Virol 75:3948–3959. doi: 10.1128/JVI.75.8.3948-3959.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Szekely L, Kiss C, Mattsson K, Kashuba E, Pokrovskaja K, Juhasz A, Holmvall P, Klein G. 1999. Human herpesvirus-8-encoded LNA-1 accumulates in heterochromatin-associated nuclear bodies. J Gen Virol 80:2889–2900. doi: 10.1099/0022-1317-80-11-2889. [DOI] [PubMed] [Google Scholar]
- 17.Lim C, Seo T, Jung J, Choe J. 2004. Identification of a virus trans-acting regulatory element on the latent DNA replication of Kaposi's sarcoma-associated herpesvirus. J Gen Virol 85:843–855. doi: 10.1099/vir.0.19510-0. [DOI] [PubMed] [Google Scholar]
- 18.Wong LY, Matchett GA, Wilson AC. 2004. Transcriptional activation by the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen is facilitated by an N-terminal chromatin-binding motif. J Virol 78:10074–10085. doi: 10.1128/JVI.78.18.10074-10085.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barbera AJ, Chodaparambil JV, Kelley CB, Joukov V, Walter JC, Luger K, Kaye KM. 2006. The nucleosomal surface as a docking station for Kaposi's sarcoma herpesvirus LANA. Science 311:856–861. doi: 10.1126/science.1120541. [DOI] [PubMed] [Google Scholar]
- 20.Hu J, Garber AC, Renne R. 2002. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J Virol 76:11677–11687. doi: 10.1128/JVI.76.22.11677-11687.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garber AC, Hu J, Renne R. 2002. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J Biol Chem 277:27401–27411. doi: 10.1074/jbc.M203489200. [DOI] [PubMed] [Google Scholar]
- 22.Cotter MA, Subramanian C, Robertson ES. 2001. The Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen binds to specific sequences at the left end of the viral genome through its carboxy-terminus. Virology 291:241–259. doi: 10.1006/viro.2001.1202. [DOI] [PubMed] [Google Scholar]
- 23.Fejer G, Medveczky MM, Horvath E, Lane B, Chang Y, Medveczky PG. 2003. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus interacts preferentially with the terminal repeats of the genome in vivo and this complex is sufficient for episomal DNA replication. J Gen Virol 84:1451–1462. doi: 10.1099/vir.0.18940-0. [DOI] [PubMed] [Google Scholar]
- 24.Garber AC, Shu MA, Hu J, Renne R. 2001. DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J Virol 75:7882–7892. doi: 10.1128/JVI.75.17.7882-7892.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lagunoff M, Ganem D. 1997. The structure and coding organization of the genomic termini of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8). Virology 236:147–154. doi: 10.1006/viro.1997.8713. [DOI] [PubMed] [Google Scholar]
- 26.Grundhoff A, Ganem D. 2003. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus permits replication of terminal repeat-containing plasmids. J Virol 77:2779–2783. doi: 10.1128/JVI.77.4.2779-2783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Komatsu T, Ballestas ME, Barbera AJ, Kelley-Clarke B, Kaye KM. 2004. KSHV LANA1 binds DNA as an oligomer and residues N-terminal to the oligomerization domain are essential for DNA binding, replication, and episome persistence. Virology 319:225–236. doi: 10.1016/j.virol.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Lim C, Sohn H, Lee D, Gwack Y, Choe J. 2002. Functional dissection of latency-associated nuclear antigen 1 of Kaposi's sarcoma-associated herpesvirus involved in latent DNA replication and transcription of terminal repeats of the viral genome. J Virol 76:10320–10331. doi: 10.1128/JVI.76.20.10320-10331.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verma SC, Lu J, Cai Q, Kosiyatrakul S, McDowell ME, Schildkraut CL, Robertson ES. 2011. Single molecule analysis of replicated DNA reveals the usage of multiple KSHV genome regions for latent replication. PLoS Pathog 7:e1002365. doi: 10.1371/journal.ppat.1002365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madireddy A, Purushothaman P, Loosbroock CP, Robertson ES, Schildkraut CL, Verma SC. 2016. G-quadruplex-interacting compounds alter latent DNA replication and episomal persistence of KSHV. Nucleic Acids Res 44:3675–3694. doi: 10.1093/nar/gkw038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Leon Vazquez E, Kaye KM. 2011. The internal Kaposi's sarcoma-associated herpesvirus LANA regions exert a critical role on episome persistence. J Virol 85:7622–7633. doi: 10.1128/JVI.00304-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Leon Vazquez E, Carey VJ, Kaye KM. 2013. Identification of KSHV LANA regions important for episome segregation, replication and persistence. J Virol 87:12270–12283. doi: 10.1128/JVI.01243-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skalsky RL, Hu J, Renne R. 2007. Analysis of viral cis elements conferring Kaposi's sarcoma-associated herpesvirus episome partitioning and maintenance. J Virol 81:9825–9837. doi: 10.1128/JVI.00842-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De León Vázquez E, Kaye KM. 2011. Rapid and quantitative assessment of KSHV LANA-mediated DNA replication. Arch Virol 156:1323–1333. doi: 10.1007/s00705-011-0985-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grundhoff A, Ganem D. 2004. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J Clin Invest 113:124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gardella T, Medveczky P, Sairenji T, Mulder C. 1984. Detection of circular and linear herpesvirus DNA molecules in mammalian cells by gel electrophoresis. J Virol 50:248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Habison AC, de Miranda MP, Beauchemin C, Tan M, Cerqueira SA, Correia B, Ponnusamy R, Usherwood EJ, McVey CE, Simas JP, Kaye KM. 2017. Cross-species conservation of episome maintenance provides a basis for in vivo investigation of Kaposi's sarcoma herpesvirus LANA. PLoS Pathog 13:e1006555. doi: 10.1371/journal.ppat.1006555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu J, Liu E, Renne R. 2009. Involvement of SSRP1 in latent replication of Kaposi's sarcoma-associated herpesvirus. J Virol 83:11051–11063. doi: 10.1128/JVI.00907-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Orphanides G, LeRoy G, Chang CH, Luse DS, Reinberg D. 1998. FACT, a factor that facilitates transcript elongation through nucleosomes. Cell 92:105–116. [DOI] [PubMed] [Google Scholar]
- 40.Reinberg D, Sims RJ. 2006. De facto nucleosome dynamics. J Biol Chem 281:23297–23301. doi: 10.1074/jbc.R600007200. [DOI] [PubMed] [Google Scholar]
- 41.Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D. 2003. FACT facilitates transcription-dependent nucleosome alteration. Science 301:1090–1093. doi: 10.1126/science.1085703. [DOI] [PubMed] [Google Scholar]
- 42.Xiao B, Verma SC, Cai Q, Kaul R, Lu J, Saha A, Robertson ES. 2010. Bub1 and CENP-F can contribute to Kaposi's sarcoma-associated herpesvirus genome persistence by targeting LANA to kinetochores. J Virol 84:9718–9732. doi: 10.1128/JVI.00713-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Correia B, Cerqueira SA, Beauchemin C, Pires de Miranda M, Li S, Ponnusamy R, Rodrigues L, Schneider TR, Carrondo MA, Kaye KM, Simas JP, McVey CE. 2013. Crystal structure of the gamma-2 herpesvirus LANA DNA binding domain identifies charged surface residues which impact viral latency. PLoS Pathog 9:e1003673. doi: 10.1371/journal.ppat.1003673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ponnusamy R, Petoukhov MV, Correia B, Custodio TF, Juillard F, Tan M, Pires de Miranda M, Carrondo MA, Simas JP, Kaye KM, Svergun DI, McVey CE. 2015. KSHV but not MHV-68 LANA induces a strong bend upon binding to terminal repeat viral DNA. Nucleic Acids Res 43:10039–10054. doi: 10.1093/nar/gkv987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Domsic JF, Chen HS, Lu F, Marmorstein R, Lieberman PM. 2013. Molecular basis for oligomeric-DNA binding and episome maintenance by KSHV LANA. PLoS Pathog 9:e1003672. doi: 10.1371/journal.ppat.1003672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hellert J, Weidner-Glunde M, Krausze J, Richter U, Adler H, Fedorov R, Pietrek M, Rückert J, Ritter C, Schulz TF, Lührs T. 2013. A structural basis for BRD2/4-mediated host chromatin interaction and oligomer assembly of Kaposi sarcoma-associated herpesvirus and murine gammaherpesvirus LANA proteins. PLoS Pathog 9:e1003640. doi: 10.1371/journal.ppat.1003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hellert J, Weidner-Glunde M, Krausze J, Lunsdorf H, Ritter C, Schulz TF, Luhrs T. 2015. The 3D structure of Kaposi sarcoma herpesvirus LANA C-terminal domain bound to DNA. Proc Natl Acad Sci U S A 112:6694–6699. doi: 10.1073/pnas.1421804112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Leon Vazquez E, Juillard F, Rosner B, Kaye KM. 2014. A short sequence immediately upstream of the internal repeat elements is critical for KSHV LANA mediated DNA replication and impacts episome persistence. Virology 448:344–355. doi: 10.1016/j.virol.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun Q, Tsurimoto T, Juillard F, Li L, Li S, De Leon Vazquez E, Chen S, Kaye K. 2014. Kaposi's sarcoma-associated herpesvirus LANA recruits the DNA polymerase clamp loader to mediate efficient replication and virus persistence. Proc Natl Acad Sci U S A 111:11816–11821. doi: 10.1073/pnas.1404219111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cai QL, Knight JS, Verma SC, Zald P, Robertson ES. 2006. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog 2:e116. doi: 10.1371/journal.ppat.0020116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai Q, Cai S, Zhu C, Verma SC, Choi JY, Robertson ES. 2013. A unique SUMO-2-interacting motif within LANA is essential for KSHV latency. PLoS Pathog 9:e1003750. doi: 10.1371/journal.ppat.1003750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sun R, Liang D, Gao Y, Lan K. 2014. Kaposi's sarcoma-associated herpesvirus-encoded LANA interacts with host KAP1 to facilitate establishment of viral latency. J Virol 88:7331–7344. doi: 10.1128/JVI.00596-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fujimuro M, Liu J, Zhu J, Yokosawa H, Hayward SD. 2005. Regulation of the interaction between glycogen synthase kinase 3 and the Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen. J Virol 79:10429–10441. doi: 10.1128/JVI.79.16.10429-10441.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hirt B. 1967. Selective extraction of polyoma DNA from infected mouse cell cultures. J Mol Biol 26:365–369. [DOI] [PubMed] [Google Scholar]
- 55.Cherezova L, Burnside KL, Rose TM. 2011. Conservation of complex nuclear localization signals utilizing classical and non-classical nuclear import pathways in LANA homologs of KSHV and RFHV. PLoS One 6:e18920. doi: 10.1371/journal.pone.0018920. [DOI] [PMC free article] [PubMed] [Google Scholar]