

Abstract
Background
Mycobacterium bovis Bacille Calmette-Guérin (BCG) is not only used as a vaccine against tuberculosis but also protects against leprosy and is used as part of bladder cancer treatment to induce a protective immune response. However, protection by BCG vaccination is not optimal. To improve vaccine efficacy, recombinant BCG expressing heterologous antigens has been put forward to elicit antigen-specific cellular and humoral responses. Cell surface localized or secreted antigens induce better immune responses than their cytosolic counterparts. Optimizing secretion of heterologous proteins or protein fragments holds therefore unexplored potential for improving the efficacy of recombinant BCG vaccine candidates. Secretion of heterologous antigens requires crossing the mycobacterial inner and outer membrane. Mycobacteria have specialized ESX or type VII secretion systems that enable translocation of proteins across both membranes. Probing this secretion system could therefore be a valid approach to surface localize heterologous antigens.
Results
We show that ESX-5 substrate LipY, a lipase, can be used as a carrier for heterologous secretion of an ovalbumin fragment (OVA). LipY contains a PE domain and a lipase domain, separated by a linker region. This linker domain is processed upon secretion. Fusion of the PE and linker domains of LipY to OVA enabled ESX-5-dependent secretion of the fusion construct LipY-OVA in M. marinum, albeit with low efficiency. Subsequent random mutagenesis of LipY-OVA and screening for increased secretion resulted in mutants with improved heterologous secretion. Detailed analysis identified two mutations in OVA that improved secretion, i.e. an L280P mutation and a protein-extending frameshift mutation. Finally, deletion of the linker domain of LipY enhanced secretion of LipY-OVA, although this mutation also reduced surface association. Further analysis in wild type LipY showed that the linker domain is required for surface association.
Conclusion
We show that the ESX-5 system can be used for heterologous secretion. Furthermore, minor mutations in the substrate can enhance secretion. Especially the C-terminal region seems to be important for this. The linker domain of LipY is involved in surface association. These findings show that non-biased screening approaches aid in optimization of heterologous secretion, which can contribute to heterologous vaccine development.
Electronic supplementary material
The online version of this article (10.1186/s12934-019-1093-1) contains supplementary material, which is available to authorized users.
Keywords: Heterologous secretion, Mycobacterium, Vaccine, BCG, LipY, ESX, Type VII secretion
Background
Mycobacterium bovis Bacille Calmette-Guérin (BCG) is well known for its use as a vaccine against the infectious disease tuberculosis, caused by Mycobacterium tuberculosis. One of the major characteristics of BCG vaccination is the induction of the T-helper cell 1 (Th1)-polarized CD4+ T cell responses that confer cellular immune responses against bacterial infection [1, 2]. However, the efficacy of BCG vaccination is known to be suboptimal [3] and many attempts have been made to further improve vaccine efficacy. Particularly, recombinant expression of M. tuberculosis antigens such as 6-kDa early-secreted antigenic target (ESAT-6 or EsxA), Antigen 85B (Ag85B or FbpB) or the complete ESX-1 secretion system (of less pathogenic mycobacteria) in BCG are of great interest, as this has been shown to further enhance cellular immune responses [4–7].
Next to recombinant expression of mycobacterial proteins, expression and secretion of heterologous antigens is also of clinical interest. Expression of antigens from other pathogens in BCG has been shown to induce antigen-specific T-cell and humoral responses and to provide subsequent protection against the corresponding pathogen in vivo [8–15]. Furthermore the heterologous expression of tumor antigens has been advocated as a possible approach to improve BCG for bladder cancer treatment [16].
Heterologous expression of antigens can be accomplished in different manners. First, antigens can be expressed without making alterations to the coding region of the original gene, which usually results in cytosolic expression of the antigen. Second, antigens can be fused to a mycobacterial carrier protein that is known to be secreted by the bacteria, potentially resulting in secreted or cell surface localized antigens. Although this approach is successfully applied for Gram-negative bacteria, heterologous secretion in mycobacteria is still far from established. Carriers that have been used most frequently thus far are lipoproteins, such as the 19 kDa lipoprotein LpqH [13, 17, 18]. Next to LpqH also Ag85B is an often used carrier to secrete proteins [19, 20]. Although results were promising, surface localization and surface accessibility have not been evaluated in most studies investigating heterologous protein expression in mycobacteria, even though this is important when designing new vaccine candidates expressing heterologous antigens. The latter is illustrated by a large body of work suggesting that secreted and cell surface localized antigens evoke a better immune response than their cytosolic counterparts [13, 18, 21–23].
For successful surface localization, heterologous proteins have to cross both the inner membrane and the unique mycobacterial outer membrane. This second membrane has a similar function as the Gram-negative outer membrane, but evolved independently and contains many unique lipids such as mycolic acids. Mycobacteria use specialized secretion systems to secrete proteins across both membranes of the cell envelope. These are known as the type VII secretion systems and are named ESX-1 to ESX-5 [24]. In particular ESX-5 has been shown to secrete a large amount of proteins [25, 26]. Importantly, the type VII secretion systems secrete substrates with T-cell epitopes recognized by the host, and disruption of the ESX-5 system abolished antigen specific CD4+ T-cell responses against its substrates [23, 27]. Furthermore, ESX-5 is, unlike ESX-1, still present and functional in BCG, enabling its use for vaccine purposes.
Previously, we characterized the secretion, processing and surface localization of ESX-5 substrate LipY [28]. LipY is one of the very few ESX-5 substrates with a described function. This protein acts as a triacylglycerol hydrolase that utilizes triacylglycerol under hypoxic conditions and nutrient starvation [29]. LipY is a member of the PE/PPE protein family, named after the conserved proline and glutamic acid residues near their N-terminus [30]. Especially in slow growing pathogenic mycobacteria, there is a large number of PE/PPE proteins [31]. Although PE/PPE proteins lack classic secretion signals, many have been shown to be secreted in an ESX-5 dependent manner [25, 26, 32]. Furthermore, many of these proteins appear to be cell surface localized, making them an interesting carrier for heterologous secretion [33–36]. M. tuberculosis LipY (LipYtub) contains a PE-domain, followed by a linker domain and the functional lipase domain. Based on homology with the alpha/beta hydrolase fold (pfam07859) the lipase domain extends from amino acid (aa) 205 to 443. We have previously shown that the PE domain of LipYtub is required for ESX-5 dependent secretion of LipYtub in M. marinum, which was recently also confirmed by Santucci et al. [37]. Upon secretion the protein is cleaved after amino acid 149 of the linker domain. The processed C-terminal part, including the lipase domain, is then exposed at the cell surface where it carries out its function [28]. Importantly, LipY can be secreted to the cell surface in large amounts, making it a suitable carrier for heterologous secretion.
Here, we fused the PE and linker domains of LipYtub to an ovalbumin fragment (OVA, a synthetic protein containing the major immunogenic epitopes of ovalbumin) to study heterologous secretion. Using mutant analysis we could show that both the LipY carrier domain and the heterologous cargo domain have an effect on secretion efficiency and membrane association. This study shows that heterologous secretion via the ESX-5 secretion system can be achieved in mycobacteria by using LipY as a carrier and provides a method to systematically improve the secretion efficiency of heterologous proteins by mycobacteria.
Methods
Bacterial cultures
Wild type (WT) Mycobacterium marinum E11 strain and ESX-5 deficient strain 7C1 with a transposon in espG5/MMAR_2676 [26] were grown in 7H9 liquid medium (Difco) supplemented with 10% albumin-dextrose-catalase supplement (ADC; BD Bioscience), with or without 0.05% Tween-80, depending on the experiment, while shaking at 90 rpm at 30 °C. Solid cultures were grown on 7H10 agar plates supplemented with OADC at 30 °C. Appropriate antibiotics were used for selection (50 μg/ml hygromycin, 25 μg/ml kanamycin).
Escherichia coli DH5α was used for cloning experiments and was grown in LB broth or LB agar plates at 37 °C with 50 μg/ml hygromycin.
Plasmid construction
The sequence corresponding to amino acid 1–205 from LipY was amplified from pSMT3-lipYtub [28] using primers LipYss Fw and LipYss Rv (primer sequences are listed in Additional file 1: Table S1). OVA was amplified with a sequence coding for a C-terminal Human influenza hemagglutinin (HA)-tag from plasmid pEH3-HbpDL-Glyk-OVAL (personal communications, Wouter Jong) with primers OVA Fw and OVA-HA Rv. Both fragments were digested with BamHI and ligated. The ligated product was amplified with primers LipYss Fw and OVA-HA Rv and subsequently ligated into vector pSMT3 using NheI and EcoRV, resulting in pSMT3-lipY-OVAwt.
pSMT3-LipYtub [28] was used as template for all linker deletion constructs. PCR reactions amplifying the N-terminal flanking sequences of the deletions were performed with LipY fw primer and reverse primers complementary to the respective 5′-deletion flanking sequences and a 3′-tail complementary to the 3′-deletion flanking sequences (Δ100–145 R, Δ158–205 R, Δ158–180 R, Δ181–205 R). For the C-terminal parts, a reverse primer for the HA-tag (LA RP570 (HA_R)) was combined with primers complementary to the specific truncations ((Δ100–145 F, Δ158–205 F, Δ158–180 F, Δ181–205 F). Resulting PCR products were used as template for a second PCR with primers MDTBLipY_F and LA RP570 (HA_R) to create the linker deletion LipY constructs. Products were cloned into pSMT3 by restriction with BamHI and EcoRV, creating vectors pSMT3-lipYΔ100–145, pSMT3-lipYΔ158–205, pSMT3-lipYΔ158–180 and pSMT3-lipYΔ181–205.
pSMT3_lipYΔ98–201 was constructed by overlap PCR. First, the N-terminal part flanking the deletion was amplified using primers MJB_pSMT3_65C_Fw and MJB_LD_Rv. Second, the C-terminal domain flanking the deletion was amplified with primers RU_LipY_LD_Fw and MJB_pSMT3_64C_Rv. PCR products of these PCR reactions were used as template for the overlap PCR using primers MJB_pSMT3_65C_Fw and MD_pSMT3_Fw. The resulting product was cloned into pSMT3 by restriction with BamHI and NheI.
The plasmid pSMT3-hsp60 was cloned from pSMT3-mspA Ates et al. [25]. The plasmid was digested using restriction enzymes EcoRV and NheI. The resulting backbone was isolated, blunt ended using T4DNA polymerase and re-ligated to obtain plasmid pSMT3-hsp60.
All resulting plasmids are summarized in Table 1. All vectors were transformed in E. coli DH5α following standard heat-shock protocol and after plasmid purification electroporated in M. marinum E11.
Table 1.
Plasmids used in this study
| Plasmid name | Plasmid backbone, promotor, selection marker | Protein product | Reference |
|---|---|---|---|
| pSMT3-lipY-OVA | pSMT3, hsp60, hygromycin | Rv3097c aa 1–205, OVA | This study |
| pSMT3-lipY-OVA1 | pSMT3, hsp60, hygromycin | Rv3097c aa 1–205 mut 1, OVA mut 1 | This study |
| pSMT3-lipY-OVA2 | pSMT3, hsp60, hygromycin | Rv3097c aa 1–205 mut 2, OVA mut 2 | This study |
| pSMT3-lipY-OVA3 | pSMT3, hsp60, hygromycin | Rv3097c aa 1–205 mut 3, OVA mut 3 | This study |
| pSMT3-lipYwt-OVA1 | pSMT3, hsp60, hygromycin | Rv3097c aa 1–205, OVA mut 1 | This study |
| pSMT3-lipYwt-OVA3 | pSMT3, hsp60, hygromycin | Rv3097c aa 1–205, OVA mut 3 | This study |
| pSMT3-lipY1-OVAwt | pSMT3, hsp60, hygromycin | Rv3097c mut 1, OVA | This study |
| pSMT3-lipY3-OVAwt | pSMT3, hsp60, hygromycin | Rv3097c mut 3, OVA | This study |
| pSMT3-lipY-OVAL280Pext | pSMT3, hsp60, hygromycin | Rv3097c aa 1–205, OVA L280P with extension caused by framehsift | This study |
| pSMT3-lipY-OVAL280P | pSMT3, hsp60, hygromycin | Rv3097c aa 1–205, OVA L280P | This study |
| pSMT3-lipYtub | pSMT3, hsp60, hygromycin | Rv3097c | Daleke et al. [38] |
| pSMT3-lipY-Δ100–145 | pSMT3, hsp60, hygromycin | Rv3097c Δ100–145 | This study |
| pSMT3-lipY-Δ158–180 | pSMT3, hsp60, hygromycin | Rv3097c Δ158–180 | This study |
| pSMT3-lipY-Δ158–205 | pSMT3, hsp60, hygromycin | Rv3097c Δ158–205 | This study |
| pSMT3-lipY-Δ191–205 | pSMT3, hsp60, hygromycin | Rv3097c Δ191–205 | This study |
| pSMT3-lipY-Δ98–201 | pSMT3, hsp60, hygromycin | Rv3097c Δ98–201 | This study |
All plasmids harbor a C-terminal human influenza hemagglutinin (HA) tag. Mutations found in LipY-OVA mutant constructs can be found in Fig. 2b. OVA ovalbumin aa
Error-prone PCR and screen for LipY-OVA supersecretion
To find LipY-OVA mutants that would show improved secretion, a random mutagenesis library was made. Error-prone polymerase from Pyrococcus furiosus [39] was used for amplification of LipY-OVA with the primers LipYss Fw and OVA-HA Rv. After 30 cycles of amplification, PCR product was isolated and cloned into pSMT3 by using NheI and EcoRV restriction enzymes. Ligation products were transformed in E. coli DH5α and the resulting library was plated on LB plates. About 1000 colonies were harvested and pooled from the plates and plasmids were isolated. The isolated plasmid library was electroporated into M. marinum E11 and cells were plated on nitrocellulose filters (0.45 μm, Merck Millipore) on 7H10 plates. Once colonies were visible, double filter assay (see below) was performed to select for colonies that showed increased secretion of LipY-OVA. Plasmids isolated from supersecreting mutants were sanger sequenced (Macrogen Europe) to detect single nucleotide polymorphisms in the insert.
Double filter assay
To analyze secretion on double filter, bacteria were grown until cultures reached an OD600 of 0.5–1.0 and spotted or homogeneously distributed on nitrocellulose filter (0.45 μm, Merck Millipore) on a plate with appropriate antibiotics. Bacteria were grown at 30 °C and once colonies were visible, the filter was incubated overnight on a second filter on a fresh plate. The second filter was blocked with 5% milk in PBS and subsequently stained with a monoclonal antibody against the HA-tag (HA.11 clone 16B12, Covance) and detected via a peroxidase labelled Goat-anti-Mouse secondary antibody and 4-chloronaphthol/3,3-diaminobenzidine staining.
Secretion analysis and immunoblot
Secretion analysis and immunoblots were performed as previously described [28]. Briefly, bacterial cultures of mid-logarithmic phase were washed with and subsequently grown overnight in 7H9 without ADC supplemented with 0.2% glycerol and 0.2% dextrose. At OD600 = 0.8–1.0 cells were harvested and washed with PBS. Surface-exposed proteins were extracted by 30 min incubation with 0.5% genapol X-080 (v/v; Sigma-Aldrich). For pellet samples, cells were suspended in SD buffer and disrupted by sonication. Supernatant was filtered through 0.2 μm Millipore filters and proteins were subsequently precipitated with tRNA and 10% trichloroacetic acid after overnight incubation. Protein samples were boiled and separated by SDS PAGE. After transferring proteins to nitrocellulose membranes (Merck Millipore) by Western blotting, membranes were blocked with 5% milk in PBS and incubated with antibodies raised against the HA-tag (HA 1.11), GroEL2 (CCs44, J. Belisle, NIH, Bethesda, MD, USA). Staining was performed via Goat-anti-Mouse IgG peroxidase-labelled antibodies (American Qualex Antibodies) and electro-chemi-luminescence Western Blotting Detection Reagent (Amersham Bioscience).
The immunoblots were quantified by generating optical density profiles for each lane with ImageJ. The areas under all peaks per lane were summed and normalized to the lane with the highest summed value of the blot. The colony blots were quantified by measuring the average intensity per spot with ImageJ and correct this value for the average background intensity. Spot intensity was subsequently normalized to the spot with the highest average intensity of the blot.
Flow cytometry
Cell surface localization on whole cells was analyzed by flow cytometry. Bacteria were grown to an OD600 of 0.8–1.2 and were washed with 5% Bovine serum albumin (BSA) in PBS. Subsequently, bacteria were stained with antibodies raised against the HA-tag (HA.11 clone 16B12, Covance) and Goat-anti-Mouse IgG Alexa-488 (Life Technologies). After washing with 5% BSA, cells were analyzed by flow-cytometry (BD Accuri C6). Cells stained with only Goat-anti-Mouse IgG Alexa 488 were taken as control to calculate the fold change in cell surface staining.
Lipase assay
To measure lipase activity, bacteria were grown to an OD600 of 0.8–1.2 in 7H9 liquid medium (Difco) supplemented with 10% ADC (BD Bioscience) and diluted to 0.5 OD600 in 7H9 supplemented with 10% ADC and 0.05% Tween-80. 160 μl of bacterial suspension was added to a 96-well plate together with 25 μl development solution DGGR [10 mg/ml BSA, 120 mM NaCl and 50 mM Tris pH 7.5, 1% Triton X100, 4uM 1,2-Di-O-lauryl-rac-glycero-3-(glutaric acid 6-methyl resorufin ester)]. Fluorescence was measured using a plate reader (excitation 530 nm, emission 600 nm), bottom mode. The lipase inhibitor Paraoxon (POX, 10 μM, Sigma) was used as a control for inhibition of LipY lipase activity.
Structure prediction
To predict the structure of the lipase domain and analyze homology with other proteins aa 158–443 of LipY were analyzed by Phyre2 [40].
Results
LipY as carrier for heterologous secretion of OVA
The opportunities for heterologous secretion in mycobacteria were investigated using the ESX-5 substrate LipY as a carrier. As cargo we used an HA-tagged fragment of ovalbumin (referred to as OVA), containing amino acids 259–357 from ovalbumin, including the major known epitopes, i.e. Major histocompatibility complex (MHC) class I epitope SIINFEKL and MHC class II epitope ISQAVHAAHAEINEAGR [41, 42]. Previously, we have shown that the PE domain of LipY is required for secretion by ESX-5 and that, during or after secretion, the linker domain is cleaved after amino acid 149 [28]. Processed LipY remains associated with the cell surface. Therefore, the PE and linker domain of LipY were fused to OVA, resulting in the fusion protein LipY-OVAwt (Fig. 1a).
Fig. 1.
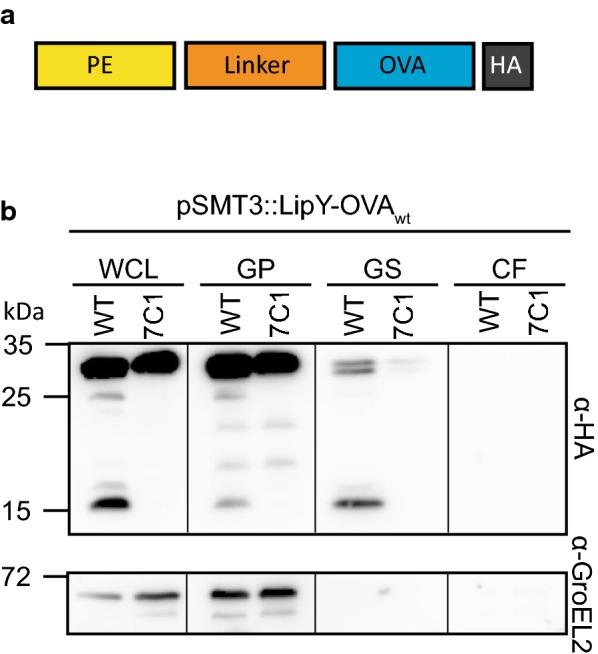
ESX-5 dependent secretion of LipY-OVA in M. marinum. a The PE domain and linker domain of LipYtub are fused to OVA (amino acid 259-357 of ovalbumin) and a C-terminal HA-tag. b Immunoblot of secretion analysis of LipY-OVAwt in M. marinum WT and the ESX-5 mutant espG5::tn (7C1). WCL Whole cell lysate, GP genapol pellet, GS, surface-enriched protein fraction genapol supernatant, CF culture filtrate are shown. Blot was stained with antibodies directed against HA and GroEL2 (cytosolic control)
To study surface localization of LipY-OVAwt, surface proteins were extracted with the mild detergent Genapol X-080. The fusion protein was readily expressed in M. marinum, but only a modest amount was found in the cell surface-enriched and nothing was detected in the supernatant fraction (Fig. 1b). Analysis of LipY-OVAwt in ESX-5 transposon mutant 7C1 (espG5::tn) showed that this modest secretion of LipY-OVAwt was ESX-5 dependent, resembling the secretion of LipYtub. Thus, LipY can serve as a carrier for heterologous secretion of OVA in an ESX-5 dependent manner, although the efficiency is rather low.
Mutations in OVA can enhance secretion of LipY-OVA in M. marinum
Secretion, or surface localization, of mycobacterial antigens is required for the induction of specific CD4+ T-cell responses [27, 43, 44] and is therefore a crucial factor to optimize vaccine design. To investigate whether we could enhance secretion of LipY-OVAwt, we introduced random point mutations in the gene coding for LipY-OVAwt by amplification of the DNA fragment with error-prone DNA polymerase. Subsequently, a library of these error-prone constructs were introduced in M. marinum and colonies were screened for increased secretion of LipY-OVA by double filter screening.
We identified three LipY-OVA clones with increased secretion (Fig. 2a). Sequencing of the mutated constructs isolated from these clones revealed multiple mutations (Fig. 2b). Whereas LipY-OVAwt was not secreted on double filter, mutants 1 (LipY-OVA1), 2 (LipY-OVA2) and 3 (LipY-OVA3) showed secretion. Especially mutant 2 (LipY-OVA2) showed a strong increase in secretion, e.g. twofold higher than LipY-OVA1. Strikingly, both LipY-OVA1 and LipY-OVA3 had independent frame shift mutations in the same codon, resulting in a nineteen amino acid extension after the HA-tag (Table 2). In LipY-OVA1 the extension was caused by a 2 basepair (bp) frameshift deletion in the last two codons before the stop codon. In LipY-OVA3 a 1 bp insertion in the penultimate codon caused a frameshift resulting in a nearly identical extension of the HA-tag. The most striking alteration was located in LipY-OVA2, instead of a single basepair change we observed a deletion that removed aa 98–201 of the gene, which spans almost the complete LipY linker domain, leaving only the PE domain in front of OVA.
Fig. 2.

Error-prone PCR mutants of LipY-OVA show increased secretion in M. marinum. a Secretion of LipY-OVAwt, LipY-OVA1, LipY-OVA2 and LipY-OVA3 was analyzed by double filter assay. Proteins were visualized by anti-HA. b Sequencing of constructs showed different mutations in both LipY and OVA domains. LipY-OVA1 and LipY-OVA3 show an extension of the HA-tag due to frameshift mutations. c Double filter assay showing secretion of LipY-OVA swap constructs by M. marinum. Mutant LipY and OVA domains were swapped with wild type domains. Total amount of HA-labeled protein was calculated using ImageJ Gel Analysis tools and the highest score was set to 100%
Table 2.
Independent polymorphisms lead to a C-terminal extension of LipY-OVA
| Construct | C-terminal amino acid sequence | Deletion/insertion (bp) |
|---|---|---|
| LipY-OVAwt | YPYDVPDYA | – |
| LipY-OVA1 | YPYDVPDYLRYLERPPPRCEPDLVIMS | Δ942T and Δ943G |
| LipY-OVA3 | YPYDVPDYALRYLERPPPRCEPDLVIMS | + 944C |
Depicted is the C-terminal amino acid sequence of LipY-OVAwt containing the HA-tag. Constructs LipY-OVA1 and LipY-OVA3 had independent deletions/insertions (column 3) leading to a similar C-terminal extension after the HA-tag (Column 2, shown in italics). Proline triplet is underlined
To determine which mutation was responsible for the increased secretion we first exchanged either the LipY-encoding domain for the original unmutated version (LipYWT) or the original OVA-encoding domain (OVAWT). Whereas LipYwt-OVA1 and LipYwt-OVA3-showed high secretion levels, similar to LipY-OVA1 and LipY-OVA3, secretion of LipY1-OVAwt and LipY3-OVAwt was not detectable (Fig. 2c). Therefore, we concluded that the mutations in the OVA domain are responsible for the enhanced secretion phenotype. Importantly, expression of the fusion constructs was similar between different mutants (Additional file 2: Figure S1). Notably, LipYwt-OVA1 exhibited a single mutation, i.e. the extension of the OVA domain, indicating that this change is sufficient to enhance the signal on double filter. This is not entirely surprising, as we identified two independent mutations that resulted in exactly the same frameshift.
A notable feature of the extension in LipY-OVA1 and LipY-OVA3 was the presence of three consecutive prolines (underlined in Table 2). Such a proline repeat can cause stalling of the ribosome [45], possibly leading to better folding and enhanced secretion of LipY-OVA. To test this hypothesis we replaced the proline triplet by three alanines, resulting in LipY-OVA1(PPP/AAA). Double filter analysis showed however that LipY-OVA1(PPP/AAA) was secreted to a similar extent as LipY-OVA1, indicating that enhanced secretion is not caused by the proline triplet (Additional file 3: Figure S2).
Mutation L280P enhances secretion and surface localization of LipY-OVA
To analyze the localization of LipY-OVA in more detail, we performed secretion analysis of the different mutants with improved secretion on double filter using liquid cultures. Secreted mycobacterial proteins can be found either in the supernatant or in the cell surface-enriched fraction, which is obtained after washing harvested cells with the mild detergent Genapol X-080. All chimeric proteins were expressed, levels ranging from 0.1 to 2 fold as compared to LipYtub (Fig. 3). LipY-OVA2 was the only fusion protein that could be detected in modestly increased amounts in the culture supernatant. Although the total amount of LipY-OVA2 found in the supernatant was approximately fourfold higher than LipYtub, this modified fusion protein showed various degradation products. This indicates that our mutants are poorly released from the mycobacterial surface into the culture medium, or that they are not stable in culture medium. Evidence for the latter hypothesis is the presence of degradation products for LipY-OVA2 in the supernatant fraction. Instability of released proteins is also observed for EspE [46].
Fig. 3.

Secretion analysis of LipY-OVA mutants. Immunoblots of whole cell lysate (WCL), genapol pellet (GP), genapol supernatant (GS) and culture filtrate (CF) of M. marinum WT, LipYtub-, LipY-OVAwt, LipY-OVA1, LipY-OVA2 and LipY-OVA3. Proteins were visualized with anti-HA and anti-GroEL2 (cytosolic control). Total amount of HA-labeled protein was calculated for each blot separately using ImageJ Gel Analysis tools and the highest score was set to 100%, taking into account all bands in the lane
The picture was different for the cell surface-enriched fraction. In this fraction the amount of LipY-OVA3 was 30 fold higher than LipY-OVAwt (Fig. 3). Interestingly, it was also present in higher amounts than LipY-OVA1, although both mutants showed similar secretion phenotypes on double filter. In addition, LipY-OVA3 showed a band with higher mobility in the surface-enriched fraction, indicating processing during or after the translocation to the cell surface. This processed band resembles the processing seen for full-length LipY, corresponding with cell surface localization of LipY. Hence, these results indicate that LipY-OVA3 is the most efficiently translocated protein.
Next, we aimed to determine which mutation(s) in LipY-OVA3 were responsible for the increased secretion and surface localization. Secretion analysis of the previously constructed pSMT3-lipYwt-OVA3 construct showed that the mutations in the OVA3 domain were responsible for the presence of LipY-OVA3 on the cell surface (Fig. 4). To further elucidate the role of the individual mutations, i.e. N219T, L280P and the extension of the HA-tag present in the OVA-domain of LipY-OVA3, we created LipY-OVA constructs with these polymorphisms. Western blot analysis of LipY-OVAL280P and the same construct with the C-terminal extension (LipY-OVAL280Pext) showed secretion and subsequent surface localization of the processed form, resembling the phenotype found for LipY-OVA3 (Fig. 4, genapol supernatant). Secretion of LipY-OVAL280P and LipY-OVAL280Pext was even a bit more efficient than LipY-OVA3 (respectively 1.3 and 3 fold higher). In contrast, LipY-OVAwt was mainly present in its full length form and was only partially processed (15 kDa band, 7% of total expression) (Fig. 4, whole cell lysate). In conclusion, mutation L280P appeared to be sufficient to markedly enhance secretion and surface localization of LipY-OVA, indicating that modest cargo modification can be sufficient for improving secretion efficiency.
Fig. 4.

Secretion analysis of LipY-OVA L280P mutation. Immunoblot of whole cell lysate (WCL), genapol pellet (GP), genapol supernatant (GS) and culture filtrate (CF) of M. marinum WT, LipY-OVAwt, LipY-OVA3, LipYwt-OVA1, LipY-OVAL280Pext and LipY-OVAL280P. Proteins were visualized by anti-HA-tag and anti-GroEL2 (cytosolic control). Total amount of HA-labeled protein was calculated for each blot separately using ImageJ Gel Analysis tools and the highest score was set to 100%, taking into account all bands in the lane
Since application of heterologous secretion for vaccine strategies would ultimately require expression in BCG, the selected LipY-OVA mutant constructs were also introduced in BCG. All these LipY-OVA constructs were expressed at comparable levels (Additional file 4: Figure S3) in BCG. LipY-OVA could be observed in the surface-enriched fraction (GS), but only after relatively long exposure times. Similar to the observations in M. marinum, the amount of LipY-OVA3 was higher than LipY-OVAwt (twofold), whereas the amount in the pellet fraction was less for this mutant (fivefold). The other two chimeras, LipY-OVA1 and LipY-OVA2 were not increased in the surface-enriched fraction as compared to LipY-OVAwt. As published before [28], LipYtub was not secreted by BCG in vitro, as was shown by the absence of the processed band (35 kDa) in all fractions.
Surface exposure of LipY-OVA on intact cells
Surface localization does not necessarily equal surface accessibility, since proteins that are detected in denatured conditions in cell surface fractions might be shielded by other capsular components and are therefore not exposed to the exterior of the cell. Therefore, we analyzed surface accessibility of LipY-OVA1, LipY-OVA2 and LipY-OVA3 in M. marinum on intact cells by immunolabeling and flow cytometry. All LipY-OVA constructs showed surface labeling, as did full-length LipYtub (Fig. 5). The increase in surface signal suggested that LipYtub (7.5 fold increase) and LipY-OVA3 (4.1 fold increase) have a higher surface labeling as compared to LipY-OVA1 and LipY-OVA2 (2.8 and 2.7 fold increase, respectively). This resembles the subcellular fractionation data, in which LipYtub and LipY-OVA3 were present in significant amounts in the genapol supernatant fraction. Unexpectedly, no clear difference was seen between the surface labeling of LipY-OVAwt (5.0 fold increase) and LipY-OVA3, despite the clearly enhanced presence of LipY-OVA3 in the cell surface-enriched fraction. This might be explained by the observation that LipY-OVA3 is mainly present in its processed form. Due to the orientation and/or length of the processed protein, the HA-tag could be less well accessible on the cell surface for antibody labeling.
Fig. 5.

Flow cytometry results of surface labeling of LipY and LipY-OVA. M. marinum cells were labeled for LipY-HA or LipY-OVA-HA and measured by flow cytometry. Fold change averages of the median fluorescence from wild-type (black line) to LipY-HA or LipY-OVA-HA (red line) and standard deviation over 3 independent experiments are depicted. Plots show results of one representative experiment
Deletion of the linker domain reduces cell surface localization of LipY
LipY-OVA2 showed a deletion of aa 98–201, which is almost completely coinciding with the predicted linker domain (aa 100–205). This mutant showed a higher level of secretion, as was shown by both double filter assay (Fig. 2a) and supernatant precipitation (Fig. 3). These results suggest a role for the linker domain in surface localization. To further investigate this, different parts of the linker domain (aa 100–205) were deleted in the original LipYtub protein and analyzed in M. marinum (Fig. 6a). To retain the known cleavage site (after aa 149) in the partial linker deletions, we constructed LipYΔ100–145 and LipYΔ158–205. In addition, two smaller deletions of the C-terminal part of the linker domain were constructed based on the conservation of amino acids between M. marinum and M. tuberculosis LipY, i.e. either the less conserved amino acids 158–180 were deleted in LipYΔ158–180, or the highly conserved amino acids 181–205 were deleted to obtain LipYΔ181–205. Finally, the deletion found in LipY-OVA2 (aa 98–201) was mimicked in LipYtub (LipYΔ98–201). Expression and secretion of all LipY deletion constructs were analyzed in M. marinum. Deletion of the N-terminal part of the linker domain (LipYΔ100–145) did not result in a stable product and was not observed on double filter nor upon cell fractionation. This finding resembles previous data, which showed that the PE-domain with only the first 25 aa of the linker domain alone is not stable [28]. All other linker domain mutants were reasonably well expressed and were further analyzed by cell fractionation (Fig. 6b). These mutants showed expression of the full length protein in the whole-cell lysate. LipYΔ158–180, LipYΔ158–205 and LipYΔ181–205 also showed smaller bands that correspond to their relative molecular weight after processing after amino acid 149. Surprisingly, processing was also observed for LipYΔ98–201, although this construct lacks the protease cleavage site at amino acid 149 in the linker. This suggests that the PE-domain is still removed, since the length of the smaller band corresponded to the predicted molecular weight of the lipase domain only, which is in line with previous observations [28].
Fig. 6.

The effect of linker domain deletions in LipYtub. a Schematic representation of deletions that were made in the linker domain of LipY. b Immunoblot showing whole cell lysate (WCL), genapol pellet (GP), genapol supernatant (GS) and culture filtrate (CF) of M. marinum expressing LipYtub and LipYtub linker domain deletion mutants. Proteins were visualized with anti-HA and anti-GroEL (cytosolic control). Total amount of HA-labeled protein was calculated for each blot separately using ImageJ Gel Analysis tools and the highest score was set to 100%, taking into account all bands in the lane. c Lipase activity was measured on whole cells M. marinum by using DGGR as a substrate. One representative experiment is shown out of three independent experiments that showed similar results
The processed bands of LipYΔ158–180 and LipYΔ181–205 were mainly found in the genapol X-080 supernatant, indicating that they were processed and retained at the cell surface. In comparison, LipYΔ158–205 was present in much lower amounts in this fraction. Apparently, partial deletion of the C-terminal part of the linker domain (aa 158–180 or aa 181–205) did not affect cell surface localization, while deletion of the whole C-terminal part (aa 158–205) reduced surface localization.
Together these results indicate that in M. marinum the C-terminal part of the linker domain is involved in retention of LipY to the surface. Deletion of the full linker domain reduced surface localization of both LipYΔ98–201 and LipY-OVA2. However, in contrast to LipY-OVA2, culture supernatant levels of LipYΔ98–201 did not increase.
The linker domain is involved in lipase activity of LipY
To examine whether the linker domain is also important in LipY activity, we assessed the lipase activity of the different linker deletion mutants. Lipase activity was measured using a whole cells assay. Lipase activity was almost reduced to zero in a secretion deficient ESX-5 negative background (Fig. 6c). Introduction of a plasmid containing the lipY gene results in significantly increased lipase activity, which can be reduced again by addition of the lipase inhibitor POX. The lack of activity of LipYΔ100–145 is likely due to the instability of the construct. Surprisingly, also most other deletion mutants did not show any lipase activity exceeding the background lipase activity of M. marinum WT even though production levels of these variants were in fairly similar amounts. Only LipYΔ158–180 still showed lipase activity. This was most clear for LipYΔ181–205, which seems in amounts comparable to intact LipY, but which showed no lipase activity above background levels. This means that, even though partial deletions of the linker domain do not affect surface localization of LipY, lipase activity is severely hampered, especially when the C-terminal part of the linker domain is removed.
Discussion
The BCG strain is one of the most widely used vaccines worldwide. Because of the immunogenic properties of BCG that induce clear cellular immunity against many intracellular pathogens, several attempts have been made to enhance vaccine efficacy by recombinant expression of mycobacterial and non-mycobacterial antigens [5]. Since secretion of the antigens increases the adaptive immune response against Mycobacterium tuberculosis antigens [43], we aimed to optimize heterologous secretion in mycobacteria by using the ESX-5 system and assessing both secretion and cell surface localization of a non-mycobacterial protein fragment of OVA.
By using LipY as a carrier we provided a proof of principle that the ESX-5 system could indeed be used for heterologous secretion in M. marinum, although secretion efficiencies were reduced as compared to the wild-type protein. By introducing random mutations in LipY-OVA we were able to identify mutants that showed enhanced secretion and surface localization, indicating that this is a viable approach that could be used for other heterologous constructs. These experiments showed two different mutations in the cargo that improved secretion. The first group of mutations had their stop codon removed by small insertions or deletions. Remarkably, both these alterations gave rise to the same frameshift resulting in an extension containing a proline triplet. Proline triplets can in theory be responsible for enhanced secretion, by securing better folding of the protein due to ribosome stalling [45]. However, further analysis showed that the proline triplet was not responsible for the increased signal. At this point, we cannot exclude that extension of the HA-tag increased detection by the antibody, for example by affecting protein folding.
The second mutation improving secretion was L280P in the OVA-domain. In addition to increased secretion, we found that LipY-OVAL280P induced processing of LipY-OVA in a way similar to the processing seen for LipY. Particularly, this processed form showed increased surface localization. Most likely, this increased surface localization and subsequent processing are the result of enhanced secretion through the ESX-5 system. Prolines are known to induce kinks in peptides [47], which could make the OVA-domain more suitable for secretion through the ESX-5 system. This experiment shows that a single mutation in the OVA-domain can be sufficient for enhanced secretion by the ESX-5 system. This confirms the expectation that there are restrictions on protein size and/or conformation of ESX-5 substrates [48]. On the other hand, our experiments also show that already minor mutations can enable secretion of heterologous proteins through the ESX-5 system. Obviously, these mutations will be specific for each potential substrate and might change substrate structure, but do not necessarily have to change substrate function or immunogenic epitopes. This is illustrated by mutation L280P, which increased processing but is not part of the MHC-I or MHC-II epitopes present in the OVA domain.
Surface localization of LipY-OVA was further analyzed by immunolabeling of whole cells. Whereas surface localization was well visible after secretion analysis on western blot, the differences on whole cells, analyzed with flow cytometry, were relatively small. This might be due to reduced accessibility of the HA-tag in a folded state on the cell surface and putative shielding by the mycobacterial capsule. This could explain why LipY-OVA3 was detected in high amounts in the genapol-extracted fraction in denatured form, but not on the surface of intact cells. Nevertheless, LipY-OVA was clearly present on the cell surface, although cell surface accessibility did not significantly differ between mutants.
The absence of LipY secretion by BCG in culture most likely resembles previous observations in M. tuberculosis [28]. This bacterium only secretes LipY and shows lipase activity upon nutrient starvation or hypoxia, which might also explain the low levels of LipY-OVA in the surface-enriched fraction. Possibly one or more yet unknown proteins required for efficient processing, secretion and surface localization of LipY are not expressed in BCG in nutrient rich in vitro conditions.
The deletion of the linker domain (aa 98–201) enhanced secretion of LipY-OVA. This compelled us to study the functional role of the LipY linker domain in M. marinum. It is likely that surface localization is linked to processing of the linker domain, since LipY-OVA was retained at the cell surface only after processing of the linker domain, as seen in LipY-OVAL280P. These data correspond well with our previously published model of LipYtub secretion, in which the protein is cleaved after Gly-149, after which the C-terminal part stays associated with the cell surface [28]. Further analysis of the linker domain of LipYtub showed that when the major part of the linker domain (aa 98–201) or its C-terminal part (aa 158–205) was deleted, surface localization was severely reduced.
Although surface localization and processing of LipYΔ158–180 and LipYΔ181–205 were only slightly affected, especially LipYΔ181–205 showed strongly reduced lipase activity in whole cells. Previously, it has been shown that the PE domain negatively regulates lipase activity [37, 49, 50]. Our data suggests that on whole cells also the C-terminal part of the linker domain is (in)directly involved in lipase activity. One option is that this domain is important for the folding of the actual lipase domain. More detailed analysis of the lipase domain (Phyre2 [40]) indicated that the lipase domain itself could be extended to aa 192, which would include an extra loop that might be important for proper functioning of the lipase domain. Another option is that the C-terminal aa of the linker domain serve as a platform or stem-like structure that is necessary to make the active site of the lipase domain accessible to the substrate at the cell surface. Since the mycobacterial cell wall is enveloped by a capsular layer of proteins and polysaccharides certain distance might be needed to bring the lipase domain in contact with the extracellular environment [51].
While further research is needed to optimize LipY-mediated heterologous secretion in BCG, this study shows that ESX-5 substrate LipY can act as a carrier for secretion of heterologous proteins both in M. marinum and BCG. This paves the way to improve recombinant expression approaches that already have been taken to improve vaccine efficacy. More importantly, we show a viable and affordable method for optimization of heterologous secretion in mycobacteria by combining error-prone PCR with double filter screening methods. In principle, this method can be adopted to any carrier-cargo sequence to find individual mutations required for enhanced secretion, which in turn may lead to improved performance of recombinant mycobacterial vaccine candidates. If one could combine this screen with Flow Cytometry Cell Sorting many more mutants could be analyzed, although one would miss mutants with improved secretion without increased surface exposure, such as LipY-OVA2.
Conclusions
Here we have shown that the mycobacterial ESX-5 system can serve as a route for heterologous secretion. By fusing the PE domain and linker domain of ESX-5 substrate LipY to a fragment of OVA, OVA was secreted in an ESX-5 dependent manner in M. marinum. Subsequent random mutagenesis of the fusion protein LipY-OVA showed that minor changes in the heterologous substrate can enhance secretion. In addition deletion of the linker domain of LipY-OVA enhanced secretion, but reduced surface association. In agreement with the latter finding, the linker domain was also shown to be important for surface localization of intact LipY. Together, this work is the first to methodologically analyze and optimize heterologous secretion in mycobacteria. This could give new momentum to recombinant BCG development, with implications for recombinant vaccines and BCG bladder cancer treatment.
Additional files
Additional file 1: Table S1. Primers used in this study.
Additional file 2: Figure S1. Expression of LipY-OVA swap constructs in M. marinum. LipY-OVA swap mutants showed similar expression levels, as was analyzed by collecting pellet fractions and staining with anti-HA and anti-GroEL2 (cytosolic control).
Additional file 3: Figure S2. Double filter assay of LipY-OVAPPP/AAA Double filter assay showing LipY-OVA in which the proline triplet in the extension of LipY-OVA1 was replaced by an alanine triplet. HA-tagged proteins were detected by immunoblotting with anti HA.
Additional file 4: Figure S3. Secretion analysis of LipY-OVA in M. bovis BCG. LipYtub and LipY-OVA constructs were expressed in BCG. Whole cell lysate (WCL), genapol pellet (GP), genapol supernatant (GS) and culture filtrate (CF) were collected and analyzed on western blot. Proteins were visualized by anti-HA-tag and anti-GroEL2 (cytosolic control). Proteins in genapol supernatant were only visible after long exposure (exposure time pellet fraction = 0.3 s, surface-enriched fraction = 10 s). Total amount of HA-labeled protein was calculated for each blot separately.
Authors’ contributions
MJB, LSA, CK and KK performed the experiments. AS developed the lipase assay. MJB and LSA analyzed the results and wrote the manuscript. CK and WB supervised the research and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We are grateful to Joen Luirink and Joke den Haan for their scientific advice. We acknowledge Vincent van Winden for purification of the error-prone polymerase and Roy Ummels for making plasmid constructs. We thank Maria Daleke and Wouter Jong for providing plasmids.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This study is funded by CCA_Huijgens program.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Abbreviations
- aa
amino acid
- BCG
Bacille Calmette-Guérin
- bp
basepair
- E. coli
Escherichia coli
- HA
human influenza hemagglutinin
- M. marinum
Mycobacterium marinum
- M. tuberculosis
Mycobacterium tuberculosis
- OVA
amino acid 259–357 of ovalbumin
- Th1
T-helper cell 1
- WT
wild type
Contributor Information
Maroeska J. Burggraaf, Email: m.burggraaf@vumc.nl
Louis S. Ates, Email: l.s.ates@amc.uva.nl
Alexander Speer, Email: a.speer@vumc.nl.
Kim van der Kuij, Email: kimvanderkuij@gmail.com.
Coen Kuijl, Email: c.kuijl@vumc.nl.
Wilbert Bitter, Email: w.bitter@vumc.nl.
References
- 1.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM. Disseminated tuberculosis in interferon gamma gene-disrupted mice. J Exp Med. 1993;178:2243–2247. doi: 10.1084/jem.178.6.2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR. An essential role for interferon 7 in resistance to Mycobacterium tuberculosis infection. J Exp Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fletcher HA, Schrager L. TB vaccine development and the End TB Strategy: importance and current status. Trans R Soc Trop Med Hyg. 2016;110:212–218. doi: 10.1093/trstmh/trw016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Groschel MI, Sayes F, Shin SJ, Frigui W, Pawlik A, Orgeur M, Canetti R, Honore N, Simeone R, van der Werf TS, et al. Recombinant BCG expressing ESX-1 of Mycobacterium marinum combines low virulence with cytosolic immune signaling and improved TB protection. Cell Rep. 2017;18:2752–2765. doi: 10.1016/j.celrep.2017.02.057. [DOI] [PubMed] [Google Scholar]
- 5.Bastos RG, Borsuk S, Seixas FK, Dellagostin OA. Recombinant Mycobacterium bovis BCG. Vaccine. 2009;27:6495–6503. doi: 10.1016/j.vaccine.2009.08.044. [DOI] [PubMed] [Google Scholar]
- 6.Castanon-Arreola M, Lopez-Vidal Y, Espitia-Pinzon C, Hernandez-Pando R. A new vaccine against tuberculosis shows greater protection in a mouse model with progressive pulmonary tuberculosis. Tuberculosis. 2005;85:115–126. doi: 10.1016/j.tube.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Horwitz MA. Recombinant BCG expressing Mycobacterium tuberculosis major extracellular proteins. Microbes Infect. 2005;7:947–954. doi: 10.1016/j.micinf.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 8.Yu Q, Huang X, Gong P, Zhang Q, Li J, Zhang G, Yang J, Li H, Wang N, Zhang X. Protective immunity induced by a recombinant BCG vaccine encoding the cyclophilin gene of Toxoplasma gondii. Vaccine. 2013;31:6065–6071. doi: 10.1016/j.vaccine.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 9.Bueno SM, Gonzalez PA, Cautivo KM, Mora JE, Leiva ED, Tobar HE, Fennelly GJ, Eugenin EA, Jacobs WR, Jr, Riedel CA, Kalergis AM. Protective T cell immunity against respiratory syncytial virus is efficiently induced by recombinant BCG. Proc Natl Acad Sci USA. 2008;105:20822–20827. doi: 10.1073/pnas.0806244105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nascimento IP, Dias WO, Quintilio W, Christ AP, Moraes JF, Vancetto MD, Ribeiro-Dos-Santos G, Raw I, Leite LC. Neonatal immunization with a single dose of recombinant BCG expressing subunit S1 from pertussis toxin induces complete protection against Bordetella pertussis intracerebral challenge. Microbes Infect. 2008;10:198–202. doi: 10.1016/j.micinf.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Mazzantini RP, Miyaji EN, Dias WO, Sakauchi D, Nascimento AL, Raw I, Winter N, Gicquel B, Rappuoli R, Leite LC. Adjuvant activity of Mycobacterium bovis BCG expressing CRM197 on the immune response induced by BCG expressing tetanus toxin fragment C. Vaccine. 2004;22:740–746. doi: 10.1016/j.vaccine.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 12.Varaldo PB, Leite LC, Dias WO, Miyaji EN, Torres FI, Gebara VC, Armoa GR, Campos AS, Matos DC, Winter N, et al. Recombinant Mycobacterium bovis BCG expressing the Sm14 antigen of Schistosoma mansoni protects mice from cercarial challenge. Infect Immun. 2004;72:3336–3343. doi: 10.1128/IAI.72.6.3336-3343.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stover CK, Bansal GP, Hanson MS, Burlein JE, Palaszynski SR, Young JF, Koenig S, Young DB, Sadziene A, Barbour AG. Protective immunity elicited by recombinant Bacille Calmette-Guerin (BCG) expressing outer surface protein A (OspA) lipoprotein: a candidate Lyme disease vaccine. J Exp Med. 1993;178:197–209. doi: 10.1084/jem.178.1.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yasutomi Y, Koenig S, Haun SS, Stover CK, Jackson RK, Conard P, Conley AJ, Emini EA, Fuerst TR, Letvin NL. Immunization with recombinant BCG-SIV elicits SIV-specific cytotoxic T lymphocytes in Rhesus monkeys. J Immunol. 1993;150:3101–3107. [PubMed] [Google Scholar]
- 15.Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, et al. New use of BCG for recombinant vaccines. Nature. 1991;351:456–459. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- 16.Begnini KR, Buss JH, Collares T, Seixas FK. Recombinant Mycobacterium bovis BCG for immunotherapy in nonmuscle invasive bladder cancer. Appl Microbiol Biotechnol. 2015;99:3741–3754. doi: 10.1007/s00253-015-6495-3. [DOI] [PubMed] [Google Scholar]
- 17.Bastos RG, Dellagostin OA, Barletta RG, Doster AR, Nelson E, Osorio FA. Construction and immunogenicity of recombinant Mycobacterium bovis BCG expressing GP5 and M protein of porcine reproductive respiratory syndrome virus. Vaccine. 2002;21:21–29. doi: 10.1016/s0264-410x(02)00443-7. [DOI] [PubMed] [Google Scholar]
- 18.Langermann S, Palaszynski SR, Burlein JE, Koenig S, Hanson MS, Briles DE, Stover CK. Protective humoral response against pneumococcal infection in mice elicited by recombinant Bacille Calmette-Guerin vaccines expressing pneumococcal surface protein A. J Exp Med. 1994;180:2277–2286. doi: 10.1084/jem.180.6.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeuchi A, Eto M, Tatsugami K, Shiota M, Yamada H, Kamiryo Y, Dejima T, Kashiwagi E, Kiyoshima K, Inokuchi J, et al. Antitumor activity of recombinant Bacille Calmette-Guerin secreting interleukin-15-Ag85B fusion protein against bladder cancer. Int Immunopharmacol. 2016;35:327–331. doi: 10.1016/j.intimp.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 20.Tang C, Yamada H, Shibata K, Maeda N, Yoshida S, Wajjwalku W, Ohara N, Yamada T, Kinoshita T, Yoshikai Y. Efficacy of recombinant Bacille Calmette-Guerin vaccine secreting interleukin-15/antigen 85B fusion protein in providing protection against Mycobacterium tuberculosis. J Infect Dis. 2008;197:1263–1274. doi: 10.1086/586902. [DOI] [PubMed] [Google Scholar]
- 21.Yu JS, Peacock JW, Jacobs WR, Jr, Frothingham R, Letvin NL, Liao HX, Haynes BF. Recombinant Mycobacterium bovis bacillus Calmette-Guerin elicits human immunodeficiency virus type 1 envelope-specific T lymphocytes at mucosal sites. Clin Vaccine Immunol. 2007;14:886–893. doi: 10.1128/CVI.00407-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hayward CM, O’Gaora P, Young DB, Griffin GE, Thole J, Hirst TR, Castello-Branco LR, Lewis DJ. Construction and murine immunogenicity of recombinant Bacille Calmette Guerin vaccines expressing the B subunit of Escherichia coli heat labile enterotoxin. Vaccine. 1999;17:1272–1281. doi: 10.1016/s0264-410x(98)00350-8. [DOI] [PubMed] [Google Scholar]
- 23.Sayes F, Blanc C, Ates LS, Deboosere N, Orgeur M, Le Chevalier F, Groschel MI, Frigui W, Song OR, Lo-Man R, et al. Multiplexed quantitation of intraphagocyte Mycobacterium tuberculosis secreted protein effectors. Cell Rep. 2018;23:1072–1084. doi: 10.1016/j.celrep.2018.03.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bitter W, Houben EN, Bottai D, Brodin P, Brown EJ, Cox JS, Derbyshire K, Fortune SM, Gao LY, Liu J, et al. Systematic genetic nomenclature for type VII secretion systems. PLoS Pathog. 2009;5:e1000507. doi: 10.1371/journal.ppat.1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ates LS, Ummels R, Commandeur S, van de Weerd R, Sparrius M, Weerdenburg E, Alber M, Kalscheuer R, Piersma SR, Abdallah AM, et al. Essential role of the ESX-5 secretion system in outer membrane permeability of pathogenic mycobacteria. PLoS Genet. 2015;11:e1005190. doi: 10.1371/journal.pgen.1005190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abdallah AM, Verboom T, Weerdenburg EM, Gey van Pittius NC, Mahasha PW, Jimenez C, Parra M, Cadieux N, Brennan MJ, Appelmelk BJ, Bitter W. PPE and PE_PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol Microbiol. 2009;73:329–340. doi: 10.1111/j.1365-2958.2009.06783.x. [DOI] [PubMed] [Google Scholar]
- 27.Sayes F, Sun L, Di Luca M, Simeone R, Degaiffier N, Fiette L, Esin S, Brosch R, Bottai D, Leclerc C, Majlessi L. Strong immunogenicity and cross-reactivity of Mycobacterium tuberculosis ESX-5 type VII secretion: encoded PE-PPE proteins predicts vaccine potential. Cell Host Microbe. 2012;11:352–363. doi: 10.1016/j.chom.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 28.Daleke MH, Cascioferro A, de Punder K, Ummels R, Abdallah AM, van der Wel N, Peters PJ, Luirink J, Manganelli R, Bitter W. Conserved Pro-Glu (PE) and Pro-Pro-Glu (PPE) protein domains target LipY lipases of pathogenic mycobacteria to the cell surface via the ESX-5 pathway. J Biol Chem. 2011;286:19024–19034. doi: 10.1074/jbc.M110.204966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deb C, Daniel J, Sirakova TD, Abomoelak B, Dubey VS, Kolattukudy PE. A novel lipase belonging to the hormone-sensitive lipase family induced under starvation to utilize stored triacylglycerol in Mycobacterium tuberculosis. J Biol Chem. 2006;281:3866–3875. doi: 10.1074/jbc.M505556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brennan MJ, Delogu G. The PE multigene family: a ‘molecular mantra’ for mycobacteria. Trends Microbiol. 2002;10:246–249. doi: 10.1016/s0966-842x(02)02335-1. [DOI] [PubMed] [Google Scholar]
- 31.Gey van Pittius NC. Sampson SL, Lee H, Kim Y, van Helden PD, Warren RM. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol Biol. 2006;6:95. doi: 10.1186/1471-2148-6-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abdallah AM, Verboom T, Hannes F, Safi M, Strong M, Eisenberg D, Musters RJ, Vandenbroucke-Grauls CM, Appelmelk BJ, Luirink J, Bitter W. A specific secretion system mediates PPE41 transport in pathogenic mycobacteria. Mol Microbiol. 2006;62:667–679. doi: 10.1111/j.1365-2958.2006.05409.x. [DOI] [PubMed] [Google Scholar]
- 33.Cascioferro A, Delogu G, Colone M, Sali M, Stringaro A, Arancia G, Fadda G, Palu G, Manganelli R. PE is a functional domain responsible for protein translocation and localization on mycobacterial cell wall. Mol Microbiol. 2007;66:1536–1547. doi: 10.1111/j.1365-2958.2007.06023.x. [DOI] [PubMed] [Google Scholar]
- 34.Delogu G, Pusceddu C, Bua A, Fadda G, Brennan MJ, Zanetti S. Rv1818c-encoded PE_PGRS protein of Mycobacterium tuberculosis is surface exposed and influences bacterial cell structure. Mol Microbiol. 2004;52:725–733. doi: 10.1111/j.1365-2958.2004.04007.x. [DOI] [PubMed] [Google Scholar]
- 35.Banu S, Honore N, Saint-Joanis B, Philpott D, Prevost MC, Cole ST. Are the PE-PGRS proteins of Mycobacterium tuberculosis variable surface antigens? Mol Microbiol. 2002;44:9–19. doi: 10.1046/j.1365-2958.2002.02813.x. [DOI] [PubMed] [Google Scholar]
- 36.Brennan MJ, Delogu G, Chen Y, Bardarov S, Kriakov J, Alavi M, Jacobs WR., Jr Evidence that mycobacterial PE_PGRS proteins are cell surface constituents that influence interactions with other cells. Infect Immun. 2001;69:7326–7333. doi: 10.1128/IAI.69.12.7326-7333.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santucci P, Diomandé S, Poncin I, Alibaud L, Viljoen A, Kremer L, de Chastellier C, Canaan S. Delineating the physiological roles of the PE and catalytic domain of LipY in lipid consumption in mycobacteria-infected macrophages. Infect Immun. 2018;86:e00394–e00418. doi: 10.1128/IAI.00394-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cascioferro A, Daleke MH, Ventura M, Dona V, Delogu G, Palu G, Bitter W, Manganelli R. Functional dissection of the PE domain responsible for translocation of PE_PGRS33 across the mycobacterial cell wall. PLoS ONE. 2011;6:e27713. doi: 10.1371/journal.pone.0027713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Biles BD, Connolly BA. Low-fidelity Pyrococcus furiosus DNA polymerase mutants useful in error-prone PCR. Nucleic Acids Res. 2004;32:e176. doi: 10.1093/nar/gnh174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnsen G, Elsayed S. Antigenic and allergenic determinants of ovalbumin–III. MHC Ia-binding peptide (OA 323–339) interacts with human and rabbit specific antibodies. Mol Immunol. 1990;27:821–827. doi: 10.1016/0161-5890(90)90147-r. [DOI] [PubMed] [Google Scholar]
- 42.Rötzke O, Falk K, Stevanovic S, Jung G, Walden P, Rammensee H. Exact prediction of a natural T cell epitope. Eur J Immunol. 1991;21:2891–2894. doi: 10.1002/eji.1830211136. [DOI] [PubMed] [Google Scholar]
- 43.Ates LS, Sayes F, Frigui W, Ummels R, Damen MPM, Bottai D, Behr MA, van Heijst JWJ, Bitter W, Majlessi L, Brosch R. RD5-mediated lack of PE_PGRS and PPE-MPTR export in BCG vaccine strains results in strong reduction of antigenic repertoire but little impact on protection. PLoS Pathog. 2018;14:e1007139. doi: 10.1371/journal.ppat.1007139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Majlessi L, Simsova M, Jarvis Z, Brodin P, Rojas M, Bauche C, Nouzé C, Ladant D, Cole ST, Sebo P, Leclerc C. An increase in antimicrobial Th1-cell responses by prime-boost protocols of immunization does not enhance protection against Tuberculosis. Infect Immun. 2006;74:2128–2137. doi: 10.1128/IAI.74.4.2128-2137.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melnikov S, Mailliot J, Rigger L, Neuner S, Shin BS, Yusupova G, Dever TE, Micura R, Yusupov M. Molecular insights into protein synthesis with proline residues. EMBO Rep. 2016;17:1776–1784. doi: 10.15252/embr.201642943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ates LS, van der Woude AD, Bestebroer J, van Stempvoort G, Musters RJ, Garcia-Vallejo JJ, Picavet DI, Weerd R, Maletta M, Kuijl CP, et al. The ESX-5 system of pathogenic mycobacteria is involved in capsule integrity and virulence through its substrate PPE10. PLoS Pathog. 2016;12:e1005696. doi: 10.1371/journal.ppat.1005696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barlow DJT, Thornton JM. Helix geometry in proteins. J Mol Biol. 1988;201:601–619. doi: 10.1016/0022-2836(88)90641-9. [DOI] [PubMed] [Google Scholar]
- 48.Beckham KS, Ciccarelli L, Bunduc CM, Mertens HD, Ummels R, Lugmayr W, Mayr J, Rettel M, Savitski MM, Svergun DI, et al. Structure of the mycobacterial ESX-5 type VII secretion system membrane complex by single-particle analysis. Nat Microbiol. 2017;2:17047. doi: 10.1038/nmicrobiol.2017.47. [DOI] [PubMed] [Google Scholar]
- 49.Garrett CK, Broadwell LJ, Hayne CK, Neher SB. Modulation of the activity of Mycobacterium tuberculosis LipY by its PE domain. PLoS ONE. 2015;10:e0135447. doi: 10.1371/journal.pone.0135447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mishra KC, de Chastellier C, Narayana Y, Bifani P, Brown AK, Besra GS, Katoch VM, Joshi B, Balaji KN, Kremer L. Functional role of the PE domain and immunogenicity of the Mycobacterium tuberculosis triacylglycerol hydrolase LipY. Infect Immun. 2008;76:127–140. doi: 10.1128/IAI.00410-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sani M, Houben EN, Geurtsen J, Pierson J, de Punder K, van Zon M, Wever B, Piersma SR, Jimenez CR, Daffe M, et al. Direct visualization by cryo-EM of the mycobacterial capsular layer: a labile structure containing ESX-1-secreted proteins. PLoS Pathog. 2010;6:e1000794. doi: 10.1371/journal.ppat.1000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Primers used in this study.
Additional file 2: Figure S1. Expression of LipY-OVA swap constructs in M. marinum. LipY-OVA swap mutants showed similar expression levels, as was analyzed by collecting pellet fractions and staining with anti-HA and anti-GroEL2 (cytosolic control).
Additional file 3: Figure S2. Double filter assay of LipY-OVAPPP/AAA Double filter assay showing LipY-OVA in which the proline triplet in the extension of LipY-OVA1 was replaced by an alanine triplet. HA-tagged proteins were detected by immunoblotting with anti HA.
Additional file 4: Figure S3. Secretion analysis of LipY-OVA in M. bovis BCG. LipYtub and LipY-OVA constructs were expressed in BCG. Whole cell lysate (WCL), genapol pellet (GP), genapol supernatant (GS) and culture filtrate (CF) were collected and analyzed on western blot. Proteins were visualized by anti-HA-tag and anti-GroEL2 (cytosolic control). Proteins in genapol supernatant were only visible after long exposure (exposure time pellet fraction = 0.3 s, surface-enriched fraction = 10 s). Total amount of HA-labeled protein was calculated for each blot separately.
Data Availability Statement
Not applicable.