

Abstract
Hypertension is a multifaceted disease that is involved in ~40% of cardiovascular mortalities and is the result of both genetic and environmental factors. Because of its complexity, hypertension has been studied by using various models and approaches, each of which tends to focus on individual organs or tissues to isolate the most critical and treatable causes of hypertension and the related damage to endorgans. Animal models of hypertension have ranged from Goldblatt’s kidney clip models in which the origin of the disease is clearly renal to animals that spontaneously develop hypertension either through targeted genetic manipulations, such as the TGR(mRen2)27, or selective breeding resulting in more enigmatic origins, as exemplified by the spontaneously hypertensive rat (SHR). These two genetically derived models simulate the less-common human primary hypertension in which research has been able to define a Mendelian linkage. Several models are more neurogenic or endocrine in nature and illustrate that crosstalk between the nervous system and hormones can cause a significant rise in blood pressure (BP). This review will examine one of these neurogenic models of hypertension, i.e., the deoxycorticosterone acetate (DOCA), reduced renal mass, and high-salt diet (DOCA-salt) rodent model, one of the most common experimental models used today. Although the DOCA-salt model is mainly believed to be neurogenic and has been shown to impact the central and peripheral nervous systems, it also significantly involves many other body organs.
Keywords: DOCA-salt, Neurogenic hypertension, Neurohormonal, Cardiovascular, Renal, Immune
Introduction
Hypertension is historically a difficult disease to study due to the fact that genetic, dietary, and environmental factors (or more likely a combination of the three) contribute to the chronic rise in BP. Essential hypertension is defined by Oparil and Carretero as “high BP in which secondary causes such as renovascular disease, renal failure, pheochromocytoma, aldosteronism, or other causes of secondary hypertension or Mendelian forms (monogenic) are not present.” Essential hypertension accounts for 95% of all forms of hypertension [1]; however, the etiology of essential hypertension is unknown. A growing body of literature in both human and animal studies indicates that this disease often stems from an overactive sympathetic nervous system [2] and an imbalance in the renin- angiotensin system (RAS). The deoxycorticosterone acetate (DOCA)-salt model is ideal for defining the role of these two major pathways that are critical in essential hypertension pathogenesis.
The DOCA-salt model was first used to study hypertension in the 1970s and has continued to be refined to act as a more translational model since then. In this model, DOCA is administered to the animal, ranging anywhere from 20 to 150 mg/kg in rats (depending on the study). This leads to an imbalance of renal sodium handling where greater amounts of sodium and water are reabsorbed by the kidney resulting in hypervolemia [3–5]. Additionally, the model incorporates a high-salt diet consisting of 0.6–1% NaCl in the drinking water and often accompanied by uninephrectomy to increase the onset of hypertension [4, 6•, 7•]. Salt sensitivity and its involvement in the development of human essential hypertension have been well documented. Salt sensitivity was found in 26% of the normotensive population and 51% of the hypertensive population, indicating this element could be a key indicator in people who are predisposed to the development of hypertension [8]. Interestingly, renin, an enzyme that converts angiotensinogen into angiotensin (Ang)-I, was observed to be low in this salt-sensitive population [9]. This is important because the same phenomenon is observed in DOCA-salt-treated animals, indicating that this model not only incorporates a high-salt diet but also results in a low-renin hypertension, similar to what is observed in the human population [10].
The combination of DOCA, reduced kidney mass, and increased salt intake results in chronic high BP developing in distinct stages. DOCA-salt hypertension is understood to occur in two major phases characterized as an initial rise in BP over the first few days and then a sustained elevated BP for weeks. There are very few studies that followed the animals past 3 weeks and those that have done so show contradicting results on whether BP increases further or decreases after a month of treatment [11, 12].
Neurogenic Component
BP is highly variable throughout the day due to behavior but remains extremely consistent when measured over 24 h. The autonomic nervous system plays a critical role in both sensing and maintaining BP through the complex interactions of sympathetic and parasympathetic reflexes. Put simply, the sympathetic nerves are responsible for innervating the blood vessels, heart, kidneys, and adrenal medulla, all critical to sustaining healthy BP. Irregularities or overactivation in this neural network can result in chronically elevated BP [2, 13•] (Fig. 1).
Fig. 1.
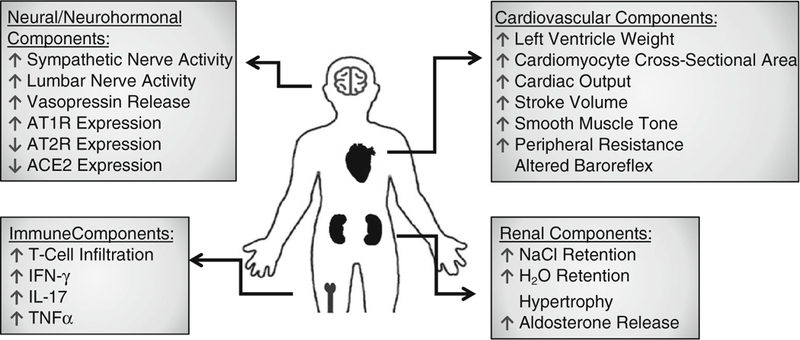
Effects of DOCA-salt treatment on different parts of the body
Takeda et al. showed that elevated sympathetic nerve discharge and altered baroreceptor reflex preceded a rise in BP in DOCA-salt-treated rats [14, 15••]. These were some of the first observations that an overactive sympathetic nervous system could play a major role in the development of hypertension. The same group went on to show that electrical stimulation of the hypothalamus increases splanchnic nerve activity, supporting the hypothesis that central control of the sympathetic nerves plays a role in elevating BP. As time moved forward, more novel and specific techniques were employed and evidence continued to build.
In 2006, O’Donaughy and Brooks measured lumbar nerve activity in conscious rats that had undergone DOCA-salt treatment [16]. Their findings indicate that DOCA amplifies the role of NaCl, leading to the sympatho-excitatory response seen in this hypertensive model. These results built upon previous work done by Scrogin et al. where rats were infused with a hypertonic fluid causing increased lumbar nerve activity and BP [17]. Experiments such as these emphasize that a high-salt diet could be critical to the onset of neurogenic essential hypertension in humans [18].
Secondary to the sensitivity to salt in the central nervous system (CNS), an overactive sympathetic drive has been shown to cause a rise in BP through its effect on venous smooth muscle tone and peripheral vascular resistance. Fink et al. showed that an increase in venous smooth muscle tone was due to increased sympathetic nerve activity in DOCA- salt-treated Sprague-Dawley rats. The effects on venous smooth muscle tone were alleviated in two different ways. First is by utilizing ABT-627, an endothelin subtype A receptor blocker administered intravenously. Secondly, smooth muscle tone was decreased through administration of the ganglionic blocker hexamethonium. Both of these blockers were more effective in lowering BP in hypertensive animals than in normotensive controls [19].
In response to the observations of increased nerve activity, there is an abundance of literature on ablating and/or sympathectomizing different regions of the nervous system in hypertension models. Some examples include lesioning areas such the area postrema, the anterolateral third ventricle, and the paraventricular nucleus of the hypothalamus. Animals that had these areas lesioned and underwent DOCA-salt treatment failed to develop or had attenuated BP relative to their controls [20–24]. These studies have provided evidence of putative candidates responsible for the sympathoexcitation that is observed in hypertensive patients and invaluable insight into potential therapeutic targets.
Neurohormonal Component
The neurohormonal contribution to high BP has been studied for decades but it is only recently that new mechanisms of action have come to light. The DOCA-salt model causes plasma renin and Ang-II to decrease. However, Ang-II levels, and those of its precursor angiotensinogen, increase in the cerebrospinal fluid (CSF) which is highly suggestive of brain RAS activity involvement [25, 26]. Ang-II, by means of its type 1 receptor (AT1R), promotes increased sympathetic activity, salt and water reabsorption, vasoconstriction, and aldosterone and vasopressin release and inflammation, contributing to tissue fibrosis, endothelium dysfunction, and hypertension [27–30] (Fig. 1).
It has been shown that in the DOCA-salt model, there is increased Ang-II binding to AT1R post-treatment in areas of the brain associated with BP regulation. Gutkind et al. quantified binding of Ang-II after a month of DOCA-salt treatment in experimental and control groups using male Wistar-Kyoto rats. Bound Ang-II expression was significantly increased in the median preoptic nucleus and the subfornical organ [31]. Other authors provided evidence supporting a similar increase and confirmed the role of AT 1R in the paraventricular nucleus, the nucleus of the solitary tract, and the area postrema [32–34]. Conversely, parts of the brain not involved in BP regulation such as the olfactory bulb and suprachiasmatic nucleus did not display increased binding to or expression of AT1R.
When losartan, an AT1R antagonist that is frequently used in the clinic, was administered to rats or mice through intra-cerebroventricular (ICV) infusion, hypertension was significantly attenuated [6•, 35, 36]. Notably, when losartan (25 mg/kg/day) was administered to rats in their drinking water, the AT1R antagonist failed to decrease hypertensive symptoms, BP remained high, and endothelium-dependent vessel relaxation did not change [7•, 37]. Other pharmacological approaches have isolated changes in brain RAS as central to hypertension pathology. Captopril, a clinical drug that decreases vasopressin release and acts as an ACE inhibitor, has also been identified as acting almost entirely through brain RAS in the DOCA-salt model [38,39]. Rats that received oral captopril (100 mg/kg/day) did not show reduced symptoms of hypertension after going through the DOCA-salt protocol [40]. When captopril was administered ICV (1.25 mg/h) before and during the DOCA-salt treatment, there was a significant decrease in BP. High BP was also attenuated when captopril was given ICV to animals with established hypertension (rats that had been going through the DOCA-salt hypertension protocol for 4 weeks) [41].
Recently, there has been an abundance of studies on the pro-renin receptor (PRR) and its involvement in the CNS regulation of BP. The presence of Ang-II depends on the precursor renin and/or pro-renin (for details, see 2015 review by A.H. Jan Danser [42]). It has been proposed that when activated, the PRR plays a detrimental role in upregulating the formation of Ang-I that eventually gets converted to Ang-II through ACE, as well as increasing other factors leading to inflammation, increased Erk1/2, and more [42]. When brain PRR is selectively antagonized pharmacologically through the use of PRO20, BP is attenuated in the DOCA-salt model [43]. Additionally, in studies performed by Li et al. (2014), in neuron-specific PRR-knockout mice, DOCA-salt hypertension was completely prevented [44]. The authors attributed this highly significant prevention of hypertension to the PRR role in mediating the formation of Ang-II in the brain.
All pathways of the brain RAS are not always viewed as detrimental when upregulated. The ACE2/Ang-(1–7)/MasR arm of the RAS pathway has shown promise as a counterbalance to the Ang-II/AT1R activation. ACE2 converts Ang-II to Ang-(1–7), a vasodilatory peptide shown by our group and others to have vasodilatory effects and decrease overall BP [6•, 45–47]. However, this beneficial axis ofthe RAS pathway is downregulated in DOCA-salt hypertension, leading to an unbalanced brain RAS where Ang-II levels are no longer regulated and as a result, a chronic elevation of BP occurs. These findings taken together with the pharmacological studies and the increased expression ofthe AT 1R binding after DOCA-salt treatment clearly implicate brain RAS as part of hypertension pathology and therapeutic target.
Peripheral neurohormonal factors contribute to the pathology of hypertension as well. Endothelin and circulating vasopressin, well-characterized peptides involved in blood vessel constriction, are upregulated in DOCA-salt-induced hypertension. Lange et al. and De Champlain et al. observed that endothelin upregulation leads to sympathoadrenal activation through the adrenal medulla and potentiates high BP [48, 49]. The adrenal medulla is necessary to the development of DOCA-salt hypertension [50, 51]. Both authors, utilizing the DOCA-salt model, showed increased catecholamine release from the adrenal medulla dependent on the endothelin. Similarly, it is well established that during the development of hypertension, circulating vasopressin increases, causing vasoconstriction and water retention. Since the late 1970s, authors have observed up to tenfold increases in vasopressin and specific antagonists result in the prevention of high BP associated with persistent and extreme hypernatremia [52–54].
Therefore, it is important to understand that in the DOCA-salt model, peripheral effects are in play and must be considered when deciphering the cause of hypertension.
Cardiovascular Component
The DOCA-salt model results in hypervolemia and circulatory system remodeling. Cardiovascularly, these changes are primarily seen in the heart but the periphral vasculature is altered as well. The combination of increased peripheral resistance and venous return to the heart causes long-term modifications (Fig. 1). In order for the heart to remain an effective pump, it hypertrophies to meet the demand of the changing body. It does this in a number of ways including, but not limited to, increasing left ventricle weight, cardiomyocyte crosssectional area, cardiac output, and stroke volume. Each of these maladaptations could be beneficial in the short-term but contributes to the pathology of hypertension. Indeed, patients that have or have had hypertension are six times as likely to suffer from heart failure as those that have not [55].
Numerous experimental studies have examined hypertrophy during and post-DOCA-salt treatment, identifying alterations in cardiac function [56]. Notably, Reiter et al. have recently (2016) performed a study using the DOCA-salt model on pigs and observed both morphological and functional changes of the heart [57]. In their experiment, pigs that received DOCA-salt treatment and controls underwent 3T cardiovascular magnetic resonance imaging (MRI) at rest and during dobutamine stress. Left ventricular/atrial function, myocardial mass, strain, and torsion were measured along with the use of phase contrast to quantify blood flow and peak velocities. Many of the results mirror what has been seen in human hypertensive patients [58, 59]. To match the force needed to maintain healthy circulation, the DOCA-salt-treated pigs displayed increased left ventricle mass and wall thickening. This resulted in an increased left ventricle ejection fraction and decreased end systolic volume. The authors concluded that the increased left atrial volume was the most remarkable change and the changes seen in the heart could be indicators of early-stage heart failure.
A brief review was recently published by Lee et al. (2015) focusing on cardiovascular changes due to DOCA-salt treatment [56]. In this meta-analysis of numerous studies, it is widely accepted that cardiovascular alterations do occur. Hypertrophy of both the left and right ventricles are seen along with aortic wall thickening, lumen diameter decreasing, and an increase in heart-rate variability. Observations such as these provide evidence that the cardiovascular system is one of the components that contribute to the pathogenesis of hypertension in DOCA-salt hypertension.
Renal Components
The notion that the kidneys play a role in hypertension has been known and studied for almost two centuries [60]. The kidneys have an extreme capacity to handle volume overload through water and salt excretion. As the nephrology community understands it, only through dysfunctional kidneys can the pressure-natriuresis curve (the balance between blood, water, and salt in the body) shift, with the result being the imbalances known as either hypo- or hypertension [61•, 62, 63]. However, with recent work being done on renal nerves, renal arteries, and aldosterone equilibrium, many classic assumptions about sodium retention and fluid expansion are being questioned (Fig. 1).
As stated previously, hypertension is a multifactorial disease and it remains difficult to parse out what part of the pathology occurs first. The DOCA-salt model intentionally disrupts the salt and volume balance in order to cause the animal to develop hypertension [64]. It is still debated as to whether, in the DOCA-salt model and in humans, the autonomic nervous system is sensing a homeostatic change and attempting to right itself and in so doing is resulting in hypertension or it is a feedforward loop initiated by the kidneys.
This debate can be exemplified through work done by Kandlikar and Fink in 2011 [65, 66] and then juxtaposed by more recent findings by Banek et al. in 2016 [67]. Kandlikar and Fink published two studies utilizing a milder form of the DOCA-salt model, implanting a 50 mg/kg DOCA pellet subcutaneously and 1% saline in the rats’ drinking water and opting not to decrease renal mass or perform a uninephrectomy. In their experiments, BP in the rats was monitored through radio-telemetry and norepinephrine (NE) spill over was measured to quantify sympathetic nerve activity. Additionally, they performed renal denervation with appropriate controls. The results showed that there was both no effect of renal denervation on the development of hypertension in DOCA-treated rats and there was also not a significant rise in sympathetic nerve activity. Interestingly, in a second publication, different portions of the sympathetic nervous system were studied due to the heterogeneity of the nerves and the authors did find, unlike the renal nerve, the splanchnic nerve was necessary in the development of hypertension.
Recently, with the more common DOCA-salt model including the uninephrectomy, there is evidence that renal nerve activity does in fact increase [67]. Banek et al. showed that renal afferent and total denervation does attenuate the hypertension induced by DOCA-salt treatment and describes a role that inflammation could also play in the kidney. There is a plethora of studies in both humans, reaching clinical trials [68, 69], and animals manipulating renal nerve activity to influence hypertension. In regard to the neural component of the renal system in the development and maintenance of hypertension, there is clearly still more to understand.
Intrarenal RAS is viewed as another piece of the hypertension pathology. Like the brain, the kidneys develop many of the major players of the RAS intrinsically. It has been shown in hypertensive animals that received an Ang-II infusion the RAS components in the kidney (angiotensinogen, renin, Ang- I, ACE, and Ang-II) contribute to increased BP [70, 71]. The DOCA-salt model and certain forms of human hypertension, unlike the Ang-II hypertension model, induce low levels of circulating renin [8, 9, 71]. Song et al. performed a study in 2016 looking at the kidneys’ (specifically the collecting ducts) role in the low-renin, DOCA-salt hypertension model. When renin was selectively removed from the collecting ducts, there was no amelioration of hypertension, and no improvement of renal injury, observed in animals that went through the DOCA-salt protocol, opposite to what was observed in the Ang-II infusion model [72]. This indicates that while RAS components of the kidneys could play a role in certain types of hypertension (e.g., high-renin forms), the intrarenal RAS may not be involved in the pathology of low-renin forms of hypertension. These studies also emphasize the differences between hypertension models and the potential to isolate effects and variation of hypertension development.
Immune Component
A relatively novel but increasingly important player in the pathology of hypertension is the immune system and the inflammation observed with chronic high BP. The DOCA-salt model among others (e.g., Ang-II infusion) has shown an increase in reactive oxygen species (ROS) and inflammatory markers throughout the body, including the brain, which in turn results in worse end-organ damages [73] (Fig. 1). Experiments utilizing the DOCA-salt model in combination with genetic mouse lines have elucidated likely candidates involved in this inflammation and worsening of the disease state.
Marvar et al., among others, have parsed out a pivotal role T cells play in the pathology of hypertension [74, 75••, 76]. These lymphocytes are necessary to the increase in BP and systemic inflammation observed in DOCA-salt hypertension. This group has run a series of studies examining the many roles that T cells play in hypertension, ranging from increased kidney damage to inflammation of the heart and decreased vascular function. Using Rag1 −/− mice lacking immune cells, the group originally showed that these mice were resistant to or failed to develop hypertension [77]. In follow-up, using an adoptive transfer technique, the group introduced different types of lymphocytes into the mice and challenged them with different hypertension protocols (including DOCA-salt treatment). Interestingly, it was only the addition of T cells, not B cells, which produced hypertension when the animals were challenged. This provides evidence that T cells are a necessary component of hypertension pathology.
The mechanism by which the T cells are inducing increased BP was examined by both knockdown and pharmacological means. When T cells lacking AT 1R, the receptor for Ang-II, were transferred into Rag−/− mice that then went through the hypertension protocol, high BP was severely blunted. Additionally, when etanercept, a TNFα antagonist, was administered subcutaneously, hypertension was prevented and superoxide-induced vascular dysfunction decreased as well [77]. Similar studies go on to show that activated T cells infiltrate the kidneys, vasculature, and heart releasing inflammatory cytokines including IFN-γ, IL-17, and TNFα, which promote sodium retention, vasoconstriction, hypertrophy, and oxidative injury during the development of hypertension in DOCA-salt hypertension [74].
Further supporting the hypothesis that inflammation is pivotal in the progression of DOCA-salt hypertension, Krishnan et al. performed a study examining the role of inflammasomes, “multimeric complexes that facilitate caspase-1-mediated processing ofthe pro-inflammatory cytokines IL-1β and IL-18,” in the development of hypertension. DOCA-salt mice that lacked inflammasomes displayed blunted high BP, along with a decrease in macrophages and pro-inflammatory cytokines in the kidneys when compared to controls [78]. The presence of inflammasomes, lymphocytes, and ROS is clearly advancing the hypertensive state. This path of chronic inflammation is not solely observed in hypertension but other diseases as well (e.g., cardiovascular disease) [79], creating an even larger demand for research to continue in this field.
Concluding Remarks
When studying hypertension, there are many animal models to choose from. Experimenters must be conscious of each model’s strengths and limitations. The questions being asked should dictate what model is used. The phenotypes of hypertension in humans are understood to develop in many different ways and the variety of animal models provide excellent coverage ofthe different types of pathology [80]. The DOCA-salt model produces a low-renin, neurogenic form of hypertension and is better suited to examine neurocentric or high-salt diet hypotheses. On the other hand, this model may not be the correct choice for a genetically inherited form of hypertension that likely develops in a different way.
Although the model does display peripheral effects, ranging from renal to cardiovascular alterations, most of these are predated by CNS and neurohormonal changes. The brain RAS plays an essential role in DOCA-salt hypertension. Increase in Ang-II and its receptor AT1R provides evidence of an increased role of BP upregulation by the brain. When brain regions involved with BP regulation become overactive and sympathetic innervation of the cardiovascular, renal, and hormonal components of the body become unbalanced, baroreflex impairment and hypertension develop. High BP should inhibit sympathetic nerve activity, causing a drop in arterial pressure. In the DOCA-salt model, and in humans, this baroreflex arc becomes blunted and results in high BP (Fig. 1). The DOCA-salt treatment is not perfect but provides a model of hypertension that has and will continue to serve in further understanding the pathology of hypertension.
Footnotes
Compliance with Ethical Standards
Conflict of Interest Drs. Basting and Lazartigues declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently have been highlighted as:
• Of importance,
•• Of major importance
- 1.Carretero OA, Oparil S. Essential hypertension part I: definition and etiology. Circulation. 2000:329–35 [DOI] [PubMed] [Google Scholar]
- 2.Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. 2006;7(5):335–46. [DOI] [PubMed] [Google Scholar]
- 3.Drenjancevic-Peric I, Jelakovic B, Lombard JH, Kunert MP, Kibel A, Gros M. High-salt diet and hypertension: focus on the renin- angiotensin system. Kidney Blood Press Res. 2011;34(1): 1–11. doi: 10.1159/000320387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zicha J, Kunes J, Lebl M, Pohlova I, Slaninova J, Jelinek J. Antidiuretic and pressor actions of vasopressin in age-dependent DOCA-salt hypertension. Am J Phys. 1989;256(1 Pt 2):R138–45. [DOI] [PubMed] [Google Scholar]
- 5.Anderson PG, Bishop SP, Digerness SB. Coronary vascular function and morphology in hydralazine treated DOCA salt rats. J Mol Cell Cardiol. 1988;20(10):955–67. [DOI] [PubMed] [Google Scholar]
- 6.• Xia H, Sriramula S, Chhabra K, Lazartigues E. Brain ACE2 shedding contributes to the development of neurogenic hypertension. Circ Res. 2013;113:1087–96. This study highlights how ACE2 and Ang1–7 are downregulated in hypertension and could be major beneficial players in future therapeutics.
- 7.• Grobe JL, Buehrer BA, Hilzendeger AM, Liu X, Davis DR, Xu D, et al. Angiotensinergic signaling in the brain mediates metabolic effects of deoxycorticosterone (DOCA)-salt in C57 mice. Hypertension. 2011;57(3):600–7. doi: 10.1161/hypertensionaha.110.165829.This emphasizes the critical players in the CNS contributing to DOCA-salt hypertension.
- 8.Weinberger MH, Miller JZ, Luft FC, Grim CE, Fineberg NS. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension. 1986;8(6 Pt 2):II127–34. [DOI] [PubMed] [Google Scholar]
- 9.Poch E, Gonzalez D, Giner V, Bragulat E, Coca A, de La Sierra A. Molecular basis of salt sensitivity in human hypertension. Evaluation of renin-angiotensin-aldosterone system gene polymorphisms. Hypertension. 2001;38(5):1204–9. [DOI] [PubMed] [Google Scholar]
- 10.Funder J, New MI. Low renin hypertension (LRH): shades of John Laragh. Trends Endocrinol Metab. 2008;19(3):83. doi: 10.1016/j.tem.2008.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Turkkan JS, Goldstein DS. Production and reversal of DOCA-salt hypertension in baboons. Clin Exp Hypertens A. 1987;9(1):125–40. [DOI] [PubMed] [Google Scholar]
- 12.Abrams JM, Engeland WC, Osborn JW. Effect of intracerebroven- tricular benzamil on cardiovascular and central autonomic responses to DOCA-salt treatment. Am J Physiol Regul Integr Comp Physiol. 2010;299(6):R1500–10. doi: 10.1152/ajpregu.00431.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.• Dampney RA. Central neural control of the cardiovascular system: current perspectives. Adv Physiol Educ. 2016;40(3):283–96. doi: 10.1152/advan.00027.2016.A detailed review of the cardiovascular and circulatory components involved in the pathogenesis of hypertension.
- 14.Takeda K, Nakamura Y, Hayashi J, Kawasaki S, Nakata T, Oguro M, et al. Effects of salt and DOCA on hypothalamic and baroreflex control of blood pressure. Clin Exp Hypertens A. 1988;10(Suppl 1): 289–99. [DOI] [PubMed] [Google Scholar]
- 15.•• Takeda K, Nakamura Y, Oguro M, Kawasaki S, Hayashi J, Tanabe S, et al. Central attenuation of baroreflex precedes the development of hypertension in DOCA-salt-treated rats. Am J Hypertens. 1988;1(3 Pt 3):23S–5S.An early study showing that changes in the central nervous system predate those in the periphery in the pathogenesis of hypertension.
- 16.O’Donaughy TL, Brooks VL. Deoxycorticosterone acetate-salt rats: hypertension and sympathoexcitation driven by increased NaCl levels. Hypertension. 2006;47(4):680–5. doi: 10.1161/01.HYP.0000214362.18612.6e. [DOI] [PubMed] [Google Scholar]
- 17.Scrogin KE, Grygielko ET, Brooks VL. Osmolality: a physiological long-term regulator of lumbar sympathetic nerve activity and arterial pressure. Am J Phys. 1999;276(6 Pt 2):R1579–86. [DOI] [PubMed] [Google Scholar]
- 18.Weinberger MH, Fineberg NS, Fineberg SE, Weinberger M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension. 2001;37(2 Pt 2):429–32. [DOI] [PubMed] [Google Scholar]
- 19.Fink GD, Johnson RJ, Galligan JJ. Mechanisms of increased venous smooth muscle tone in desoxycorticosterone acetate-salt hypertension. Hypertension. 2000;35(1 Pt2):464–9. [DOI] [PubMed] [Google Scholar]
- 20.Fink GD, Pawloski CM, Blair ML, Mangiapane ML. The area postrema in deoxycorticosterone-salt hypertension in rats. Hypertension. 1987;9(6 Pt2):III206–9. [DOI] [PubMed] [Google Scholar]
- 21.Berecek KH, Barron KW, Webb RL, Brody MJ. Vasopressin central nervous system interactions in the development of DOCA hypertension. Hypertension. 1982;4(3 Pt 2): 131–7. [PubMed] [Google Scholar]
- 22.Ciriello J Contribution of forebrain mechanisms in the maintenance of deoxycorticosterone acetate-salt hypertension. Clin Exp Hypertens A. 1988;10(Suppl 1):169–78. [DOI] [PubMed] [Google Scholar]
- 23.Bruner CA, Mangiapane ML, Fink GD, Webb RC. Area postrema ablation and vascular reactivity in deoxycorticosterone-salt-treated rats. Hypertension. 1988;11(6 Pt 2):668–73. [DOI] [PubMed] [Google Scholar]
- 24.Fink GD, Bruner CA, Mangiapane ML. Area postrema is critical for angiotensin-induced hypertension in rats. Hypertension. 1987;9: 355–61. [DOI] [PubMed] [Google Scholar]
- 25.Ueno Y, Mohara O, Brosnihan KB, Ferrario CM. Characteristics of hormonal and neurogenic mechanisms of deoxycorticosterone- induced hypertension. Hypertension. 1988;11(2 Pt2):I172–7. [DOI] [PubMed] [Google Scholar]
- 26.Hamlyn JM, Blaustein MP. Sodium chloride, extracellular fluid volume, and blood pressure regulation. Am J Phys. 1986;251(4 Pt 2):F563–75. [DOI] [PubMed] [Google Scholar]
- 27.Falcon JC, Phillips MI, Hoffman WE, Brody MJ. Effects of intra-ventricular angiotensin II mediated by the sympathetic nervous system. Am J Phys. 1978;235:H392–H9. [DOI] [PubMed] [Google Scholar]
- 28.Esler M, Kaye D. Sympathetic nervous system activation in essential hypertension, cardiac failure and psychosomatic heart disease. J Cardiovasc Pharmacol. 2000;35(7 Suppl. 4):S1–7. [DOI] [PubMed] [Google Scholar]
- 29.Albrecht vBuHOD. The CNS renin-angiotensin system 2006. [DOI] [PubMed]
- 30.Chapleau MW, Abboud FM. Neuro-cardiovascular regulation: from molecules to man. Ann NY Acad Sci. 2001:1 New York. [PubMed] [Google Scholar]
- 31.Gutkind JS, Kurihara M, Saavedra JM. Increased angiotensin II receptors in brain nuclei of DOCA-salt hypertensive rats. Am J Hypertens. 1988;255(3 Pt 2):H646–H50. [DOI] [PubMed] [Google Scholar]
- 32.Mangiapane ML, Simpson JB. Subfornical organ lesions reduce the pressor effect of systemic angiotensin II. Neuroendocrinology. 1980;31(6):380–4. [DOI] [PubMed] [Google Scholar]
- 33.Li W, Liu J, Hammond SL, Tjalkens RB, Saifudeen Z, Feng Y. Angiotensin II regulates brain (pro)renin receptor expression through activation of cAMP response element-binding protein. Am J Physiol Regul Integr Comp Physiol. 2015;309(2):R138–47. doi: 10.1152/ajpregu.00319.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan JY, Wang LL, Lee HY, Chan SH. Augmented upregulation by cfos of angiotensin subtype 1 receptor in nucleus tractus solitarii of spontaneously hypertensive rats. Hypertension. 2002;40(3):335–41. [DOI] [PubMed] [Google Scholar]
- 35.Hilzendeger AM, Morgan DA, Brooks L, Dellsperger D, Liu X, Grobe JL, et al. A brain leptin-renin angiotensin system interaction in the regulation of sympathetic nerve activity. Am J Physiol Heart Circ Physiol. 2012;303(2):H197–206. doi: 10.1152/ajpheart.00974.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park CG, Leenen FH. Effects of centrally administered losartan on deoxycorticosterone-salt hypertension rats. J Korean Med Sci. 2001;16(5):553–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Somers MJ, Mavromatis K, Galis ZS, Harrison DG. Vascular superoxide production and vasomotor function in hypertension induced by deoxycorticosterone acetate-salt. Circulation. 2000;101(14):1722–8. [DOI] [PubMed] [Google Scholar]
- 38.Basso N, Ruiz P, Kurnjek ML, Cannata MA, Taquini AC. The brain renin-angiotensin system and the development of DOC-salt hypertension. Clin Exp Hypertens A. 1985;7(9):1259–68. [DOI] [PubMed] [Google Scholar]
- 39.Tada Y, Wada K, Shimada K, Makino H, Liang EI, Murakami S, et al. Roles of hypertension in the rupture of intracranial aneurysms. Stroke. 2014;45(2):579–86. doi: 10.1161/STROKEAHA.113.003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Basso N, Ruiz P, Mangiarua E, Taquini AC. Renin-like activity in the rat brain during the development of DOC-salt hypertension. Hypertension. 1981;3(6 Pt 2):II-14–7. [DOI] [PubMed] [Google Scholar]
- 41.Itaya Y, Suzuki H, Matsukawa S, Kondo K, Saruta T. Central renin- angiotensin system and the pathogenesis of DOCA-salt hypertension in rats. Am J Phys. 1986;251(2 Pt 2):H261–H8. [DOI] [PubMed] [Google Scholar]
- 42.Danser AH. The role of the (pro)renin receptor in hypertensive disease. Am J Hypertens. 2015;28(10):1187–96. doi: 10.1093/ajh/hpv045. [DOI] [PubMed] [Google Scholar]
- 43.Li W, Sullivan MN, Zhang S, Worker CJ, Xiong Z, Speth RC, et al. Intracerebroventricular infusion of the (pro)renin receptor antagonist PRO20 attenuates deoxycorticosterone acetate-salt-induced hypertension. Hypertension. 2015;65(2):352–61. doi: 10.1161/HYPERTENSIONAHA.114.04458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li W, Peng H, Mehaffey EP, Kimball CD, Grobe JL, van Gool JM, et al. Neuron-specific (pro)renin receptor knockout prevents the development of salt-sensitive hypertension. Hypertension. 2014;63(2):316–23 doi: 10.1161/HYPERTENSIONAHA.113.02041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Feng Y, Xia H, Cai Y, Halabi CM, Becker LK, Santos RAS, et al. Brain-selective overexpression of human angiotensin-converting enzyme type 2 attenuates neurogenic hypertension. Circ Res. 2010;106(2):373–82. doi: 10.1161/circresaha.109.208645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu P, Sriramula S, Lazartigues E. ACE2/Ang-(1–7)/Mas pathway in the brain: the axis of good. Am J Physiology-Regul Integr Comp Physiol. 2011;300(4):R804–17. doi: 10.1152/ajpregu.00222.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferrario CM. ACE2: more of Ang-(1–7) or less Ang II? Curr Opin Nephrol Hypertens. 2011;20(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lange DL, Haywood JR, Hinojosa-Laborde C. Endothelin enhances and inhibits adrenal catecholamine release in deoxycorticosterone acetate-salt hypertensive rats. Hypertension. 2000;35(1 Pt 2):385–90. [DOI] [PubMed] [Google Scholar]
- 49.de Champlain J, Eid H, Papin D. Potentiated endothelin-1-induced phosphoinositide hydrolysis in atria and mesenteric artery of DOCA-salt hypertensive rats. J Hypertens Suppl. 1989;7(6): S136–7. [DOI] [PubMed] [Google Scholar]
- 50.Lange DL, Haywood JR, Hinojosa-Laborde C. Role of the adrenal medullae in male and female DOCA-salt hypertensive rats. Hypertension. 1998;31(1 Pt2):403–8. [DOI] [PubMed] [Google Scholar]
- 51.Moreau P, Drolet G, Yamaguchi N, de Champlain J. Role of pre- synaptic beta 2-adrenergic facilitation in the development and maintenance of DOCA-salt hypertension. Am J Hypertens. 1993;6(12): 1016–24. [DOI] [PubMed] [Google Scholar]
- 52.Hofbauer KG, Studer W, Mah SC, Michel JB, Wood JM, Stalder R. The significance of vasopressin as a pressor agent. J Cardiovasc Pharmacol. 1984;6(Suppl 2):S429–38. [DOI] [PubMed] [Google Scholar]
- 53.Mohring J, Mohring B, Petri M, Haack D. Vasopressor role of ADH in the pathogenesis of malignant DOC hypertension. Am J Phys. 1977;232(3):F260–9. [DOI] [PubMed] [Google Scholar]
- 54.Mimura Y, Ogura T, Yamauchi T, Otsuka F, Oishi T, Harada K, et al. Effect of vasopressin V1- and V2-receptor stimulation on blood pressure in DOCA-salt hypertensive rats. Acta Med Okayama. 1995;49(4):187–94. [DOI] [PubMed] [Google Scholar]
- 55.Rao MR. Effects oftetrandrine on cardiac and vascular remodeling. Acta Pharmacol Sin. 2002;23(12):1075–85. [PubMed] [Google Scholar]
- 56.Lee LK, Kim MY, Kim JH, Lee JU, Park BS, Yang SM, et al. A review of deoxycorticosterone acetate-salt hypertension and its relevance for cardiovascular physiotherapy research. J Phys Ther Sci. 2015;27(1):303–7. doi: 10.1589/jpts.27.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reiter U, Reiter G, Manninger M, Adelsmayr G, Schipke J, Alogna A, et al. Early-stage heart failure with preserved ejection fraction in the pig: a cardiovascular magnetic resonance study. J Cardiovasc Magn Reson. 2016;18(1):63. doi: 10.1186/s12968-016-0283-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moreno MU, Eiros R, Gavira JJ, Gallego C, Gonzalez A, Ravassa S, et al. The hypertensive myocardium: from microscopic lesions to clinical complications and outcomes. Med Clin North Am. 2017;101(1):43–52. doi: 10.1016/j.mcna.2016.08.002. [DOI] [PubMed] [Google Scholar]
- 59.Koito H, Yutaka H. CT and MRI findings of pulmonary hypertension. Nihon Rinsho. 2001;59(6):1107–12. [PubMed] [Google Scholar]
- 60.Bright R. Observations on the treatment of fever. Case of simple fever, protracted by irritation of the bowels, and attended by relapse. Guy’s Hospital Reports. 1836;1:1–8. [Google Scholar]
- 61.• Coffman TM. The inextricable role ofthe kidney in hypertension. J Clin Invest. 2014;124(6):2341–7. doi: 10.1172/JCI72274.A brief review that touches upon multiple alterations of the kidney during hypertension, including some mechanistic and physiological findings.
- 62.Mullins LJ, Conway BR, Menzies RI, Denby L, Mullins JJ. Renal disease pathophysiology and treatment: contributions from the rat. Dis Model Mech. 2016;9(12):1419–33. doi: 10.1242/dmm.027276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pavlov TS, Staruschenko A. Involvement of ENaC in the development of salt-sensitive hypertension. Am J Physiol Renal Physiol. 2016; doi: 10.1152/ajprenal.00427.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yemane H, Busauskas M, Burris SK, Knuepfer MM. Neurohumoral mechanisms in deoxycorticosterone acetate (DOCA)-salt hypertension in rats. Exp Physiol. 2010;95(1):51–5. doi: 10.1113/expphysiol.2008.046334. [DOI] [PubMed] [Google Scholar]
- 65.Kandlikar SS, Fink GD. Splanchnic sympathetic nerves in the development of mild DOCA-salt hypertension. Am J Physiol Heart Circ Physiol. 2011;301(5):H1965–73. doi: 10.1152/ajpheart.00086.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kandlikar SS, Fink GD. Mild DOCA-salt hypertension: sympathetic system and role of renal nerves. Am J Physiol Heart Circ Physiol. 2011;300(5):H1781–7. doi: 10.1152/ajpheart.00972.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Banek CT, Knuepfer MM, Foss JD, Fiege JK, Asirvatham-Jeyaraj N, Van Helden D, et al. Resting afferent renal nerve discharge and renal inflammation: elucidating the role of afferent and efferent renal nerves in deoxycorticosterone acetate salt hypertension. Hypertension. 2016;68(6):1415–23. doi: 10.1161/HYPERTENSIONAHA.116.07850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mahfoud F, Brilakis N, Bohm M, Narkiewicz K, Ruilope L, Schlaich M, et al. TCT-761 long-term (3-year) safety and effectiveness from the Global SYMPLICITY Registry of renal denervation in a real world patient population with uncontrolled hypertension. J Am Coll Cardiol. 2016;68(18S):B308. doi: 10.1016/j.jacc.2016.09.791. [DOI] [Google Scholar]
- 69.Warchol-Celinska E, Januszewicz A, Prejbisz A, Kadziela J. Renal denervation after the symplicity HTN-3 trial. Postepy Kardiol Interwencyjnej. 2014;10(2):75–7. doi: 10.5114/pwki.2014.43509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rohrwasser A, Morgan T, Dillon HF, Zhao L, Callaway CW, Hillas E, et al. Elements of a paracrine tubular renin-angiotensin system along the entire nephron. Hypertension. 1999;34(6):1265–74. [DOI] [PubMed] [Google Scholar]
- 71.Liu L, Gonzalez AA, McCormack M, Seth DM, Kobori H, Navar LG, et al. Increased renin excretion is associated with augmented urinary angiotensin II levels in chronic angiotensin II-infused hypertensive rats. Am J Physiol Renal Physiol. 2011;301(6):F1195—201. doi: 10.1152/ajprenal.00339.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Song K, Stuart D, Abraham N, Wang F, Wang S, Yang T, et al. Collecting duct renin does not mediate DOCA-salt hypertension or renal injury. PLoSOne. 2016;11(7):e0159872. doi: 10.1371/journal.pone.0159872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Malik KU, Jennings BL, Yaghini FA, Sahan-Firat S, Song CY, Estes AM, et al. Contribution of cytochrome P450 1B1 to hypertension and associated pathophysiology: a novel target for antihypertensive agents. Prostaglandins Other Lipid Mediat. 2012;98(3– 4):69–74. doi: 10.1016/j.prostaglandins.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Harrison D. Sy 17–2 inflammation, immunity and hypertension. J Hypertens. 2016;34(Suppl 1- ISH 2016 Abstract Book):e535. doi: 10.1097/01.hjh.0000501473.77203.33. [DOI] [Google Scholar]
- 75.•• Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, et al. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res. 2010;107(2):263–70. doi: 10.1161/CIRCRESAHA.110.217299.This study examines the role of the immune system in the development of hypertension and the necessity of T cells to disease pathogenesis
- 76.Dikalov SI, Nazarewicz RR, Bikineyeva A, Hilenski L, Lassegue B, Griendling KK, et al. Nox2-induced production of mitochondrial superoxide in angiotensin II-mediated endothelial oxidative stress and hypertension. Antioxid Redox Signal. 2014;20(2):281–94. doi: 10.1089/ars.2012.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II-induced hypertension and vascular dysfunction. J Exp Med. 2007;204(10): 2449–60. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Krishnan SM, Dowling JK, Ling YH, Diep H, Chan CT, Ferens D, et al. Inflammasome activity is essential for one kidney/ deoxycorticosterone acetate/salt-induced hypertension in mice. Br J Pharmacol. 2016;173(4):752–65. doi: 10.1111/bph.13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Youn JC. Sy 17–3 role of cmv induced T cell senescence in the pathophysiology of cardiovascular disease. J Hypertens. 2016;34(Suppl 1- ISH 2016 Abstract Book):e535. doi: 10.1097/01.hjh.0000501474.77203.fd. [DOI] [Google Scholar]
- 80.Pinto YM, Paul M, Ganten D. Lessons from rat models of hypertension: from Goldblatt to genetic engineering. Cardiovasc Res. 1998;39(1):77–88. [DOI] [PubMed] [Google Scholar]