

Abstract
The maintenance of genome integrity involves multiple independent DNA damage avoidance and repair mechanisms. Yet, the origin and pathways of the focal chromosomal reshuffling phenomena collectively referred to as chromothripsis remain mechanistically obscure. Here, we discuss the role, mechanisms, and regulation of HR in the formation of simple and complex chromosomal rearrangements. We emphasize features of the recently characterized Multi- invasions Induced Rearrangement (MIR) pathway, which uniquely amplifies the initial DNA damage. HR intermediates and cellular contexts at risk for genomic stability are discussed along with the emerging roles of various classes of nucleases in the formation of genome rearrangements. Long-read sequencing and improved mapping of repeats should enable better appreciation of the significance of recombination in generating genomic rearrangements.
Keywords: chromothripsis, copy number variation, multi-invasion, non-allelic homologous recombination, structure-selective endonuclease, structural variant
Complex chromosomal rearrangements and chromothripsis
Chromosomal rearrangements encompass any structural variation (SV) of the genome, regardless of its association with copy number variation (CNV). The advent of high throughput sequencing technologies revealed massive and complex clustered structural variations [1], which have been proposed to constitute a novel genomic instability phenomenon found in cancer genomes, congenital diseases, as well as in asymptomatic individuals [1–4]. Although the terminology varies with the precise nature of the alterations and possibly its etiology, this phenomenon is widely referred to as chromothripsis, and we will use this umbrella term here (Box: Mutational Phenomena). Formal criteria based on the CNV pattern and the physically confined nature of the rearrangement junctions have been proposed to define chromothripsis ([1, 5] but see also [6] for a different perspective). The key underlying feature of chromothripsis is the abrupt acquisition of the associated rearrangements. The number of junctions in localized rearrangements suggests a continuum of complexity, with chromothripsis being potentially an extreme expression of mechanism(s) also responsible for simpler SVs [7].
Pathways for complex chromosomal rearrangements as studied in S. cerevisiae
A number of experimental systems have been developed in tractable model organisms to decipher the origin of both simple and complex SVs as well as the pathways promoting and preventing their occurrence in various sequence contexts. Here, we will focus on work performed in Saccharomyces cerevisiae, in which the conserved double-strand break (DSB) repair mechanisms and the consequences of their defect have been best understood thanks to the exquisite genetic and molecular tools in that organism [8].
Most strikingly, and despite their relatively low abundance in the S. cerevisiae genome, repeated DNA elements were the predominant mediators of SVs [9–12]. Beyond obvious pathological consequences, these repeat-mediated CNVs also contributed to rapid adaptation upon artificial gene dosage imbalances [13, 14] or nutrient–limiting conditions [15, 16].
Genetic screens revealed the complex networks of proteins involved in genome maintenance in yeast with implications for human cancer [17]. Kolodner and co-workers identified an astounding 182 genes (3%) playing a primary role and 438 genes (7%) playing a supporting role in suppressing genomic instability in the absence of exogenous DNA damage [17]. These genes participate in two broad functions whose simultaneous inactivation synergizes to destabilize the genome: 1) the prevention or removal of structural DNA damage or aberrant structures, and 2) the promotion of accurate repair of the damage, with certain functions intersecting both categories (e.g. mismatch repair). Structural DNA lesions at risk for genomic stability are varied in nature and origin. In unchallenged cells they are believed to mainly originate from replication errors in the form of a persistent ssDNA gaps or broken replication fork, i.e. a single-ended DSB (see Glossary) [8, 18, 19]. These two types of lesion are recombinogenic substrates. For simplicity we will focus here on DSBs, which have been best studied in their double-ended form upon site-specific induction [20].
Homologous recombination pathways and their associated risks to genomic stability
The DSB repair strategies can be broadly separated based on their homology requirements and sub-categorized based on their genetic dependencies, as (i) a homology-independent end- joining (NHEJ) mechanism, (ii) single strand annealing mechanisms relying either on micro- homology (Alt-EJ/MMEJ) or extensive homology (SSA), and (iii) homology-dependent DNA strand invasion mechanisms collectively referred to as HR (Fig. 1). While SSA is a homology- dependent process, it does not involve DNA strand invasion and may be responsible for homology-directed repair independent of Rad51 [21]. We briefly review here the risks to genomic stability inherent to the HR pathway, and refer the reader interested in the role of EJ and annealing mechanisms to other recent reviews [22–24].
Figure 1: Overview of the DSB repair pathways and their associated risk for genomic stability.

The repair products are boxed. The box color (from green to red) indicates the threat to genomic stability posed by the product of each pathway and sub-pathway. DNA synthesis is indicated by an arrow and newly-synthesized DNA by a dotted line. The long ssDNA associated with BIR has the potential to undergo MIR. More detailed mechanisms for D-loop cleavage and MIR are provided in Figures 2A and 2C, respectively. The reversibility of DSB resection provided by the Shieldin complex with fill-in by the DNA polymerase α-primase complex provides an unanticipated degree of flexibility in the choice of DSB repair between HR and EJ mechanisms [130]. Hence, EJ pathways are available not only on unresected or minimally resected DSB, they can also be engaged after extensive resection and subsequent fill-in.
HR templates DSB repair by locating and copying an extensive identical (homologous) or near- identical (homeologous) sequence present in intact duplex DNA (Fig. 1). Hence, HR uniquely entails homology search and DNA strand invasion of the broken molecule to identify and invade a homologous template. The DNA strand invasion reaction results in a D-loop (Fig. 1) containing heteroduplex DNA (hDNA). These reactions are catalyzed by a helical filament of Rad51 (RecA in bacteria) and associated proteins assembled on the resected ssDNA flanking the DSB [25]. Upon pairing of the 3’ extremity of the broken molecule, DNA synthesis restores the sequence information disrupted by the DSB. The HR sub-pathways branch based on the differential processing of this extended D-loop intermediate (Fig. 1). Two features of this pathway have important consequences for genomic stability: the unstable nature of the DNA synthesis occurring in the context of the D-loop and the potential of various types of DNA joint molecules generated throughout HR to be aberrantly processed by structure-selective endonuclease (SSE) (see below). Additionally, HR-mediated rearrangements can lead to dicentric chromosome formation and subsequent chronic instability by Breakage-Fusion- Bridge cycles [26, 27].
Disruption of the extended D-loop funnels the pathway towards Synthesis-Dependent Strand Annealing (SDSA), which leads to a non-crossover outcome with minimal associated gene conversion. As such, it is the most conservative sub-pathway of HR, even when using a donor at an ectopic locus.
Alternatively, the displaced strand in the extended D-loop can anneal to the second end of the DSB leading to the formation of a double Holliday Junction (dHJ), as part of the pathway historically coined Double-Strand Break Repair (DSBR) [28]. This step may depend on more extensive DNA synthesis than SDSA [29]. This covalently linked intermediate, detected physically both in somatic and meiotic cells [30, 31], can either be topologically dissolved [32] or resolved endonucleolytically [33]. While dissolution always yields a non-crossover outcome with minimal gene conversion, resolution can lead to a crossover outcome. If occurring at an allelic locus, crossovers will cause a loss of heterozygosity at the next cell division in half of the cases. If occurring between non-allelic loci, a crossover will lead to a reciprocal translocation (Fig. 1).
Alternatively to this second end annealing scenario, a defect in extended D-loop disruption or the absence of a second end to anneal to (e.g. in the case of a single-ended DSB) leads to a long- range displacement DNA synthesis mechanism known as Break-Induced Replication (BIR) until stabilization of the broken chromosome by capture of a telomeric sequence or the merging with a convergent replication fork [34, 35]. BIR is a physiological pathway of broken fork recovery also prone to generate various types of rearrangements (reviewed in ref. [36]). Replication forks can break through direct cleavage by structure-selective endonucleases, such as MUS81-EME1, or by replicating through an existing single-stranded nick in the template [19]. Indeed, the massive gene conversion resulting from its conservative nature [37, 38] will lead either to loss of heterozygosity upon repair templated by the homologous chromosome or an unbalanced SV if initiated at an ectopic site. Ectopic BIR is also associated with copy number (CN) gain of the template molecule. The overall mutagenic potential of BIR depends on the extent of synthesis it can achieve, which can be several hundreds of kilobases [37]. Finally, the long-lived single-stranded DNA associated with BIR [38, 39] is at risk for increased mutagenesis and kataegis [40].
A consequence of the unstable displacement DNA synthesis during HR is the occasional occurrence of template switches at regions of extensive homo- and homeologies over rounds of extended D-loop dissociation and re-invasion, both as part of BIR and SDSA [41–45]. Template switches can also occur at nearby micro-homologies when D-loop extension during BIR is impaired in a PIF1 mutant [46]. These switches thus provide opportunities for multiple CN gain-associated SVs from a single DSB repair event. This propensity to switch decreases at a distance from the invasion point, as the migrating synthesis bubble is converted by structure- selective endonucleases (SSEs) to a stable replication fork [35, 44, 47, 48]. Hence, BIR- mediated SVs are predicted to be found clustered at one end of the gained region, where the initial invasion and synthesis have occurred. Moreover, several HR intermediates are substrates for SSEs, and a combination of strand cleavage and ligation can convert a D-loop to a half- crossover (Figs. 1, 2A) [44, 49–51]. It is predicted to transfer a co-oriented 3’-protruding single- ended DSB on the donor, thus maintaining the initial amount and orientation of DSB extremities (Fig. 2A). Such DSB transfer on the donor have been physically detected in a related mechanism (MIR, see below) and shown to be dependent on both Rad51 (i.e. DNA strand invasion) and SSEs [52].
Figure 2: Single and multi-invasion joint molecules are at-risk HR intermediates for SSE- mediated genomic instability.

(A) Model for half-crossover formation from cleavage of a single D-loop. (B) Multi-invasions (MI) joint molecules are formed when a Rad51-ssDNA filament invades two independent donors along its length. It features an internal and a terminal D-loop. (C) Model for multi-invasion-induced rearrangement (MIR). (D) Several activities inhibit MIR in a reversible (Sgs1-Top2-Rmi1 (STR), Mph1, Srs2) or irreversible (Rad1-Rad10) fashion. The specificity of STR, Mph1 and Srs2 as well as the precise nature of their substrates has not been established.
Multi-invasions-Induced Rearrangement (MIR): an HR-based mechanism for the formation of complex rearrangements
Biochemical work revealed that a single presynaptic filament can pair and invade (i.e. form hDNA with) multiple dsDNA donors at once, resulting in a multi-invasion intermediate (MI; Fig. 2B) [52, 53]. MI joint molecules are readily formed by long presynaptic filaments made with bacterial RecA, yeast Rad51-Rad54, and human RAD51-RAD54, and are stimulated by increasing homology length ([52, 53] and Wright & Heyer, unpublished). These results suggest that MIs are byproducts of basic activities of the presynaptic filament: the inter-segmental homology sampling process [54] and hDNA formation.
It is noteworthy that terminal homologies are not required for MI formation [52, 53]. It suggests that ssDNA regions, such as replication-associated gaps, distant from or devoid of a freely rotating end could form MI joint molecules, despite the topological constraints for strand intertwining [55]. This possibility greatly expands the pathological and physiological contexts conducive to MI beyond the DSB-induced and -proximal situation studied so far [52] (see below and Fig. 3).
Figure 3: Nuclease involvement in MIR.
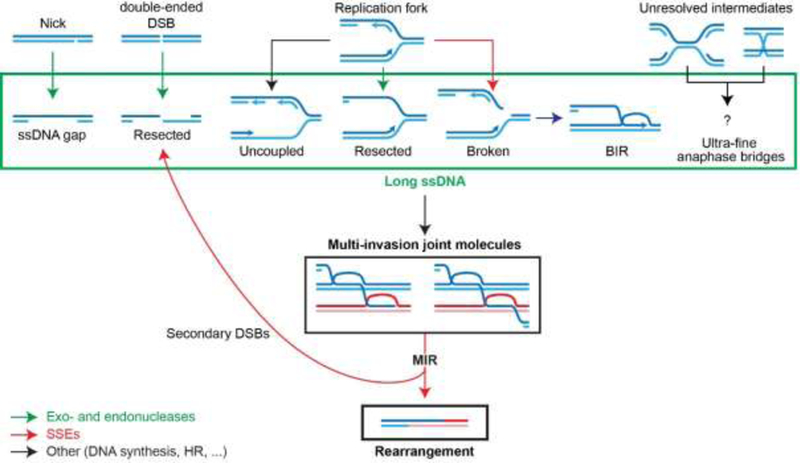
Exo- and endonucleases involved in resection (green) generate long recombinogenic substrates, while SSEs (red) process MI joint molecules into rearrangements and additional resected secondary single-ended DSBs (indicated by backwards arrow to resected DSBs).
MI joint molecules form in S. cerevisiae cells and induce the formation of chromosomal rearrangements (Fig. 2C) [52]. Specifically, the endonucleolytic processing of MI by SSEs leads to a translocation of the donors that inserts the intervening sequence of the invading molecule between the two invaded regions, in a mechanism referred to as MI-Induced Rearrangement (MIR; Fig. 2B) [52]. In addition to the translocation, MIR generates additional single-ended DSBs on each donor (Fig. 2C; mechanisms detailed in ref. [56]). Moreover, the initiating DSB is not repaired during the MIR process (Fig. 2C). Hence, while D-loop cleavage leading to half-crossover keeps the total amount of DSB ends constant, MIR generates two additional single-ended DSBs (compare Figs. 2A and 2C). In both cases, the propensity to generate additional and sometimes complex rearrangements upon attempted repair of the remaining DSBs (initial and newly formed) depends on their respective sequences [52, 56]. Hence, MIR is a HR pathway that uniquely amplifies the initial damage, and is thus at risk of runaway cascades of rearrangements.
Reversibility of HR intermediates by HR regulators guards against MIR and repeat- mediated genomic instability
HR accuracy relies in part on kinetic proof-reading, which is enforced by the reversal of several non-covalent intermediates of the pathway as a safeguard against HR-mediated rearrangements [57]. Distinct but possibly overlapping reversal activities in yeast are supported by a diverse set of HR regulators including the Srs2 helicase (putative functional human homologs are FBH1, RTEL, PARI and RECQ5), the Mph1 helicase (human FANCM), the Sgs1-Top3-Rmi1 helicase-topoisomerase complex (STR; human BLM-TOPO3α-RMI1/2), and mismatch repair (MMR) factors (reviewed in [57]). These regulators operate at multiple possible steps during the HR pathway [57]. Srs2 dissociates Rad51 filaments [58, 59] likely formed on ssDNA gaps generated during replication that are otherwise toxic [60]. Nascent and extended D-loops are nodes for two radically different decisions: anti-recombination and anti-crossover, respectively (Fig. 1). Srs2, Mph1, and STR disrupt D-loops in reconstituted biochemical reactions with Rad51, Rad54 and RPA [61–63]. In cells, they inhibit homeologous recombination in coordination with MMR [9], promote and/or bias the HR repair outcome towards non-crossover [61, 64–66], and in the case of Mph1 and Srs2 promote template-switch during BIR [43]. Additionally, the STR complex uniquely catalyzes dHJ dissolution [32, 67]. Hence, Srs2, Mph1 and STR are believed to operate throughout the HR pathway at the presynaptic (resection and Rad51 filament), synaptic (nascent D-loop formation and stability) and post-synaptic (extended D-loop stability and dHJ processing) steps, which results in conservative repair products and mitigates the risk of HR-induced genomic instability.
Consistent with their proposed roles in D-loop disruption, Srs2, Mph1 and STR also suppress MIR [52]. Additionally, the Rad1-Rad10 endonuclease (human XPF-ERRC1) suppresses MIR by clipping the 3’-flap of the internal invasion, thus preempting formation of the MI intermediate (Fig. 2D) [52]. Analysis of combinations between these MIR suppressive pathways suggested that these inhibitory activities are exerted at different steps or substrates [52]. Conversely, physical analysis of BIR intermediates suggested that Srs2 inhibits MI formation presumably by preventing Rad51 filament assembly on and/or by disrupting internal D-loops formed by the trailing ssDNA upstream of the extending D-loop [39]. Hence, multiple independent activities prevent the formation and/or accumulation of HR byproducts at risk for genomic instability. Their precise substrate(s), mechanisms and interactions have not yet been deciphered.
Long single-stranded DNAs and nucleases are instrumental in MIR
Various endo- and exonucleases are instrumental for both the early and late steps of MIR, by generating the recombinogenic substrates and by processing the subsequent MI joint molecules, respectively (Fig. 3). Long ssDNAs are key precursors of MIR: the amount of MI species in reconstituted D-loop reaction in vitro and the frequency of MIR in vivo are greatly stimulated by increasing ssDNA and homology length, respectively [52, 53]. By exposing long ssDNAs, extensive resection presumably enable the concomitant encounter of independent donors by the Rad51-ssDNA homology search engine [52–54] and exposes repeated sequences located at a distance from the DSB site, thus enabling SSA [12, 68]. Consequently, various cellular processes that generate long ssDNA are potential instigators of repeat-mediated genomic instability by MIR (Fig. 3). DSB resection is a regulated process that involves several endo- and/or exonucleases [69], and which has recently been shown to be reversible [70]. Briefly, the Sae2-MRX (human CtIP-MRN) endo- and exonuclease complex initiates DSB resection at short range (~1 kb), while the Exo1 exonuclease (human EXO1) and Sgs1/Dna2 (human BLM/DNA2) redundantly generate ssDNA of up to several tens of kilobases [69]. Single-strand breaks as well as gaps on the lagging strand of replication forks can also be exonucleolytically processed in a similar fashion [71, 72] (Fig. 3). Long ssDNAs can also be generated in a nuclease-independent fashion at the replication fork, such as upon uncoupling between the leading strand DNA polymerase and the replicative helicase [73–75] or when DNA primase becomes limiting [74, 76]. The endonucleolytic processing of replication forks by SSEs is also expected to yield single-ended DSBs exhibiting varying degrees of ssDNA, especially upon uncoupling (Fig. 3). Broken forks can be rescued by BIR, which generates persistent, kilobases- long newly synthesized ssDNA trailing behind the extending D-loop [38], which was physically shown to invade in cis upstream of the extending D-loop [39]. Previously observed BIR- induced recombination events between donor chromosomes may result from the endonucleolytic processing of such MIs [43].
As discussed earlier, DNA strand invasion intermediates generated during HR exhibit a variety of branch points that can be recognized and cleaved by SSEs with varying efficiency [33]. Consistently, three SSEs in S. cerevisiae (Mus81-Mms4, Slx1-Slx4, and Yen1) are required in a redundant fashion for MIR [52]. Functional redundancy between theses SSEs in cleaving other DNA structures had been previously reported [44]. Importantly, while moving the pathway towards the completion of the donor translocation, MI cleavage by SSEs propagates additional single-ended DSBs onto the donors (Fig. 2C).
In conclusion, nucleases contribute to HR-mediated genomic stability and specifically play a two-fold role in MIR by: 1) generating the initiating recombinogenic substrate and 2) processing DNA joint molecules into both genomic rearrangements and additional recombinogenic damage [44, 52].
Chromothripsis as a consequence of a spatial and temporal failure to separate nucleases from their potential substrate?
Recent inducible experimental systems of chromothripsis in mammalian cells started to shed light on cellular contexts prone to generate massive and localized structural DNA damage and their commonalities, and the repair mechanisms involved in the formation of the rearrangement.
Nuclear compartmentalization defects are associated with DNA damage formation and chromothripsis
Cellular compartmentalization and cell cycle regulation ensure that mutually exclusive metabolic activities are separated spatially and/or temporally [77]. For instance, failure to temporally separate DNA replication and mitotic entry results in mild chromosomal fragility up to full chromosome shattering [78, 79]. The breakage results from replication fork collapse triggered by the activation of SSEs [80–82], which normally act at mitosis as a last resort mechanism for disentangling various types of DNA joint molecules that dissolution and decatenation have failed to process [83, 84]. From a spatial point of view, one role of the nuclear envelope (NE) is to isolate the genomic DNA from the nucleases that patrol the cytosol as part of the cellular immune system (Fig. 4) [85]. In fact, NE disruption and/or programmed entry of endo- and exonucleases (endoG, TREX1 and NM23-H1) into the nucleus are integral to the genomic DNA elimination program of apoptosis induced during viral infection or in tumor cells [86–88]. The cytosol also contains Yen1/GEN1, an SSE involved in the elimination of various persistent replication and HR intermediates upon NE breakdown at mitosis [89]. Its activity is subjected to a dual inhibitory control in human: a spatial exclusion from its potential substrates and a cell-cycle regulated inactivation of the enzyme by phosphorylation [90, 91]. Consequently, NE rupture will accidentally expose genomic DNA to various nucleases, thus fulfilling one of the prerequisite for chromothripsis: the formation of massive DNA damage (Fig. 4).
Figure 4: S-phase-specific DNA damage from defective nucleoplasm isolation and DNA metabolism in micronuclei.
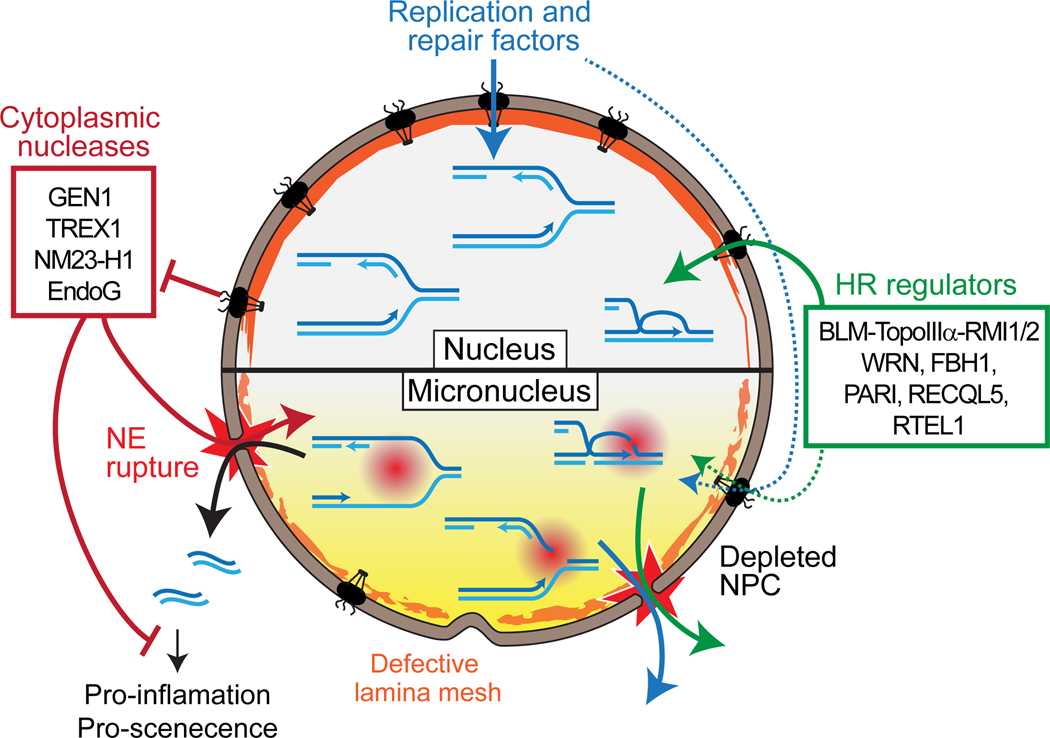
The nuclear envelope (NE) of micronuclei is depleted for nuclear pore complex and features abnormal lamina deposition. These dysregulations lead to abnormal nuclear protein import and frequent NE rupture, respectively. NE rupture leads to the penetration of cytoplasmic components to the micronucleus and the leakage of soluble nuclear components and DNA.
Two independent chromothripsis-inducing contexts that both entail nuclear compartmentalization defects have recently been investigated (Fig. 5) [92–94]. In two studies, chromothripsis arose from the isolation of whole or pieces of chromosome within micronuclei consecutive to their mis-segregation at the previous mitosis (Fig. 5A) [92, 93]. A consequence of this disrupted nuclear homeostasis in micronuclei is the massive formation of DNA damage containing, or coinciding with, long ssDNA (as revealed by colocalized phosphorylated RPA and γH2AX staining) specifically during or following S-phase, but not in G1 (Fig. 5A) [95]. Their S-phase-dependent nature suggested that defective replication itself or the exposure of DNA structures generated during replication to cytoplasmic components such as SSEs leads to DNA damage (Fig. 4).
Figure 5: Cellular models for chromothripsis.

(A) Chromothripsis by isolation isolation and breakage of chromosomes in micronuclei [92, 93]. (i-ii) Mis-segregation and physical separation of a large chromatin fragment leads to micronucleus formation. (ii-iii) Micronuclei are defective for the nuclear import and barrier function (Fig. 4). (iii) Upon S-phase entry, NE rupture leads to the formation of massive DNA damage, which features co-localizing DSBs (green) and γH2AX foci [95] (iv-v) The micronuclear DNA reaches mitosis under-replicated and fragmented. (v-vi) Micronuclear DNA fragments can be re-incorporated into the main nucleus at subsequent mitosis where it may undergo final repair. (B) Chromothripsis originating from attempted segregation of dicentric chromosomes [94]. (i) Spindle-exerted tensions can stretch but not break the central portion of a dicentric chromosome. (ii) NE forms around the daughter nuclei and the intervening chromatin bridge, depleted for lamin and nuclear pore complex. Right inset: the bridge is partially depleted for nucleosomes. (iii) Tensions exerted on the bridge cause NE rupture at the base of the bridge in G1/S phase. NE rupture leads to leaking of nuclear components and penetration of cytoplasmic proteins such as the TREX1 exonuclease (right inset). (iv) TREX1 exploits nicks or ssDNA gaps present in the bridge or generated by other cytoplasmic endonuclease to resect the chromatin bridge (right inset). This leads to accumulation of RPA foci (green) and (v) culminates in the resolution of the bridge. In addition to RPA, the resected DNA causes the formation of 53BP1, γH2AX and Mre11 foci (yellow) in the daughter nuclei.
In another study, chromothripsis was initiated by the attempted segregation of a dicentric chromosome (Fig. 5B) [94]. The force exerted by the spindle at metaphase (≈ 1 nN) [96] is expected to cause structural transitions and partial melting of protein-free dsDNA [97, 98], but remains insufficient to break the molecules, as rupture of covalent bonds occurs only above 2 nN [99]. Instead, resolution of various types of DNA bridges involved the action of nucleases, either at the mid-body during cytokinesis or upon breakdown or rupture of the NE [83, 84, 94]. In the case of chromatin bridges, the NE reformed around the segregated chromosomes as well as the bridge that persisted between the two daughter cells, which remains subjected to robust antagonistic forces (Fig. 5B) [94]. These forces ultimately lead to localized NE rupture at the base of the bridge, exposing the intervening chromatin to cytoplasmic components. The TREX1 exonuclease, either by exploiting pre-existing nicks or aided by an uncharacterized endonuclease, resects the stretched bridge until its resolution, causing the snapback of massive amounts of RPA-coated ssDNA into the daughter cells (Fig. 5B). This damaged DNA prompted the formation of 53BP1, γH2AX and Mre11 foci in the daughter cells, where it induced chromothriptic rearrangements [94]. Notably, these micronuclei-independent chromothripsis events were uniquely found associated with kataegis, consistent with long-lived ssDNA as an initiating substrate for chromothripsis (see below).
Hence, the commonality of these two experimentally distinct chromothripsis-inducing contexts is a defect in nuclear compartmentalization, and in one case the demonstrated involvement of a cytoplasmic nuclease and long ssDNAs.
Repair mechanisms generating chromothriptic rearrangements
Initial examination of the genomic characteristics of chromothriptic rearrangements, such as the SV junction sequences and the limited and oscillating copy number variation, suggested that chromothripsis resulted either from the EJ-mediating stitching of numerous DSBs and/or of microhomology-mediated template-switching during replicative or repair processes [1, 4]. Experimental evidence indicated that the DNA damage generated in both micronuclei during S-phase and upon bridge resolution in G1 or S phase stains with RPA antibodies, and certain chromothripsis events were found associated with kataegis over kilobases-long regions [94, 100, 101]. These observations suggested the involvement of long ssDNAs in chromothripsis, a recombinogenic substrate that is not readily processed by EJ mechanisms. Furthermore, HR- proficient breast cancer cells exhibit clustered rearrangements of various types while HR- deficient cells do not [102]. Moreover, chromothripsis was found mutually exclusive with biallelic BRCA2 mutations in metastatic prostate cancers [103]. Finally, p53-deficient mice inactivated for EJ (XRCC4 or LIG4) or HR factors (BRCA2) still exhibited complex rearrangements consistent with chromothripsis or chromoanasynthesis, suggesting the involvement of both pathways in chromothripsis etiology [104].
Ly and colleagues experimentally addressed whether DNA damage, in addition to be generated in micronuclei, could also be repaired there [92]. Depletion of canonical NHEJ factors (LIG4 and DNA-PKcs), but not HR (BRCA2 and RAD51) or MMEJ factors (LIG3 and PARP1), led to an increase of the fragmentation of the micronuclei-contained Y chromosome observed at the subsequent mitosis. It suggests that a significant fraction of the micronuclei-induced damages are substrate for the NHEJ machinery, and that this type of repair is active in the context of the micronuclei while HR repair is not. The remaining damages are subsequently repaired upon reincorporation of the micronuclear DNA into the main nucleus at mitosis (Fig. 5A) [93], the nature of which remains unknown. Although not yet demonstrated by sequencing, this NHEJ-dependent minimization of micronuclear DNA fragmentation suggests that the mechanisms generating chromothriptic rearrangements could occur in the micronuclei in addition to the main nucleus. The involvement of NHEJ is consistent with the analysis of junction sequences in chromothripitic genomes [1].
In contrast to the Y-chromosome system, bridge-induced chromothripsis originated from extensive ssDNA and did not undergo partial repair in micronuclei [94]. How could a single long ssDNA or multiple ssDNA gaps that are not readily substrate for EJ repair trigger chromothripsis in the daughter cell? Since long ssDNAs are suitable for Rad51 filament assembly, bridge-induced chromothripsis potentially resulted from attempted HR repair. This repair is expected to be independent of factors required for resection initiation and extension, thus bypassing an important (although not unique [105]) G1-specific block to HR, at the resection initiation level (reviewed in ref. [69]). Defects in p53 in the strain used may also have potentiated HR [106] by failing to repress RAD51 expression [107]. This situation is nonetheless relevant as p53 is inactivated in the majority of human cancers [108]. Furthermore, the prevalence of chromothripsis is higher in p53-deficient tumors [109]. Given the concomitant NE integrity defect, we suspect that HR intermediates generated along this ssDNA may lead to damage transfer and amplification similar to what is observed during half-crossover and MIR, respectively (Fig. 2).
Hence, the etiology of chromothripsis is complex and likely influenced by the damage-inducing context, with different DSB and/or ssDNA gap repair pathways involved in the formation of the rearrangements.
Why is there no extensive homology at the chromothriptic SV junctions? The need for long-read genome assemblies.
It is striking that prior to the advent of high-throughput DNA sequencing the finely-mapped SV junctions observed in human genomes involved repeated elements [110, 111], in agreement with yeast studies [8]. Then how is it that the vast majority of the SV junctions documented since then exhibit significant micro-homology (1–10 nt) but rarely longer homologies [112], despite the massive repeat content of the human genome? Instead of a biological reality, we suspect that this bias originates from technical limitations of the dominant paired-end sequencing technology and analysis pipelines. First, reads ambiguously mapped (i.e. in repeats) are usually discarded. Second, paired-end sequencing of <400 bp fragments used to detect SV poses an absolute upper threshold to the repeat size that can be detected at a SV junction. Consistently, germline and somatic cancer rearrangements in humans identified by such methods are found enriched near or at short repeats (SINEs) but strongly depleted at long repeats (LINEs) [113]. Accordingly, the exhaustive establishment of mutational profiles (particularly SVs) in S. cerevisiae required the use of additional molecular techniques in addition to paired-end sequencing [114]. We anticipate that the improved mappability of long repeated regions thanks to long-read technologies, optical mapping or Hi-C approaches will reveal more SV formed at repeats [115–118]. The recent application of long-read sequencing and across-platform comparisons reported a dramatic increase in the identification of SVs [115, 119–121], and improved SV calling algorithms could detect significant enrichment of junctions within LINEs [112]. These technical advances will produce a more comprehensive picture of the patterns of SVs in genomic rearrangements and will enable to test the involvement of HR and especially MIR in chromothripsis.
Concluding remarks
Far from an error-free pathway, HR can generate a variety of rearrangements due to the repetitive nature of the genome. The HR pathway entails the formation of DNA joint molecules at risk to induce rearrangements when cleaved inappropriately. Consequently, dysregulation of SSEs and other nucleases puts the cell at risk of a HR-mediated runaway cascade of damage and rearrangement, as demonstrated with the MIR pathway. Given that the integrity of the NE is compromised in experimental models for chromothripsis, we suspect that a key aspect of both SV and damage formation of chromothripsis reside in the accidental exposure of replication and HR intermediates to various nucleases. Finally, we highlight the need for genomes assembled from long-read DNA sequencing techniques to allow evaluating the contributions of HR between repeated DNA in the formation of genomic rearrangements and chromothripsis.
Outstanding Questions.
What is the role of SSEs and cytoplasmic nucleases in generating massive DNA damage and the complex rearrangements of chromothripsis?
What are the DNA repair pathways involved in different damage-inducing contexts? Especially for the repair of long ssDNAs?
Are HR regulators involved in suppressing chromothripsis?
Does MIR operates in human cells? In which context? Is there a mutational signature unique to MIR?
What additional types of SVs and junction sequences will be uncovered by long-read assemblies of genomes with simple and complex rearrangements?
Highlights.
Homologous recombination generates genome rearrangements involving repeated DNA elements with identical (homologous) or near identical (homeologous) sequences that can be located anywhere in the genome.
Multi-invasions are recombination byproducts that physically bridge two copies of a repeated DNA element that can be processed by structure-selective endonucleases into genome rearrangements and additional DNA double-stranded breaks.
Different chromothripsis-inducing contexts in mammalian cells feature defective isolation of genomic DNA from cytoplasmic components, including various types of nucleases.
Long DNA sequence read assemblies paired with additional approaches and improved bioinformatic pipelines are needed to fully evaluate the contributions of homologous recombination between repeated DNA to generate genomic rearrangements.
Mutational Phenomena.
Chromothripsis was originally described by Campbell and colleagues as massive focal genome rearrangements in a patient with Chronic Lymphocytic Leukemia [1] and is associated with a significant and variable proportion of cancer genomes depending on cancer type [2]. Independently, chromothripsis was identified as a constitutional genetic change in a child with severe congenital abnormalities [125]. The term derives from Greek (chromos for chromosome; thripsis, shattering into pieces) describing the interpretation that chromothripsis results from catastrophic chromosomal breakage into many individual DNA fragments.
Chromoanasynthesis is a constitutionally acquired complex genome arrangement phenomenon that was discovered in individuals with developmental delay and cognitive anomalies [126]. The patterns of CNVs and breakpoint junctions differ from chromothripsis, and the breakpoint sequence analysis suggested repeated template switching at microhomologies as the process leading to the observed rearrangements.
Chromoplexy describes yet another pattern of complex genome rearrangements involving a connected chain of translocations which first discovered in prostate tumors [127].
Kataegis describes localized hypermutation of single nucleotide changes identified in mice and in some cancer genomes [101, 128]. Kataegis is the C-to-T mutagenic signature of APOBEC deaminases that act on single-stranded DNA, suggesting that kataegis signals regions of long-lived single-stranded DNA intermediates [40, 101, 129].
Acknowledgements
We are grateful to present and former members of the Heyer, Hunter and Kowalczykowski laboratories for stimulating discussions, Steven Gore, Thomas Guérin and Stéphane Marcand for helpful insights and comments, and Jim Haber, John McPherson, Richard Kolodner and Markus Löbrich for their critical reading of the manuscript. We apologize to all authors whose primary work could not be cited due to tight constraints in the allowed number of references. AP was supported by fellowships from the Fondation ARC pour la Recherche sur le Cancer (PDF20171206726), the EMBO (ALTF-238–2013), the Framework Project 7 of the European Union (Marie Curie International Outgoing Fellowship 628355), and received financial support from the Philippe Foundation. Research in the WDH laboratory is supported by NIH grants GM58015 and CA92276 and the France-Berkeley Fund.
Abbreviations:
- BIR
break-induced replication
- CNV
copy number variant
- dsDNA
double-stranded DNA
- DSB
double-strand break
- EJ
endjoining
- hDNA
heteroduplex DNA
- HR
homologous recombination
- MI
multi-invasion
- MIR
MI-induced rearrangement
- MMEJ
microhomology-mediated end-joining
- NE
nuclear envelope
- NHEJ
non-homologous end-joining
- ssDNA
single-strand DNA
- SSE
structure-selective endonuclease
- SV
structural variant
Glossary
- Breakage-Fusion-Bridge cycle
Breakage-Fusion-Bridge cycle denotes a genetic phenomenon first described by Barbara McClintock of telomere fusions generating a dicentric chromosome that cannot be properly segregated during anaphase leading to breakage and a new cycle of fusions [122]. Any genome rearrangements between sister chromatids, homologs or different chromosomes resulting in a dicentric chromosomes will lead to chronic genomic instability until stabilization of the broken fragments.
- Conversion/Crossover/half-crossover/non-crossover
Conversion is a non-reciprocal exchange of genetic information. Crossover is the reciprocal exchange of genetic markers during HR, where both reciprocal products are recovered. In somatic cells, crossover is actively avoided by using the SDSA pathway and dissolution of double Holliday junctions, leading to non-crossovers which often involve conversion (Fig.1). Half-crossover is an event where one of the expected reciprocal products is lost
- D-loop (Displacement loop)
D-loops are a central HR intermediate, whose processing determines the HR subpathway (SDSA, DSBR, or BIR) and crossover/non-crossover outcome (Fig.1). D-loops are formed by Rad51-ssDNA filament through DNA strand invasion. Nascent and extended D-loop denotes D-loops prior and after extension of the 3’-OH of the invading DNA strand by DNA polymerase, respectively
- Double-strand break (DSB)
A DSB consists in the interruption of the phospho-diester backbone of two complementary DNA strands at the same or nearby position. DSBs come in two flavors: either single-ended such as upon replication fork breakage or frank (two-sided) (Fig.1). Single-ended DSBs can be accurately repaired only by HR
- Micronucleus
Micronuclei are a classic hallmark of genotoxic stress and form when a chromosome or chromosome fragment is not incorporated into the two daughter nuclei after anaphase. Micronuclei are nuclear-like structures that features defective isolation of the nucleoplasm from the cytoplasm (Fig.4) [93, 95, 123]. Micronuclei are depleted for nuclear pore complexes and other NE proteins, which is suspected to dysregulate nuclear protein homeostasis and underlie micronuclei-specific NE fragility (Fig.4) [124]
- Structure-selective endonucleases (SSE)
Non-linear DNA structures including intermediates generated during HR (flaps, D-loops or HJs) and stalled replications forks are substrates for SSEs. Mostly studied for their role in genome maintenance are Mus81-Mms4, Slx1-Slx4, Rad1- Rad10 and Yen1 in S. cerevisiae, respectively MUS81-EME1/2, SLX1-SLX4, XPF-ERCC1 and GEN1 in humans
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stephens PJ et al. (2011) Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cortes-Ciriano I et al. (2018) Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. bioRxiv [DOI] [PMC free article] [PubMed]
- 3.Kloosterman WP and Cuppen E (2013) Chromothripsis in congenital disorders and cancer: similarities and differences. Curr Opin Cell Biol 25, 341–8. [DOI] [PubMed] [Google Scholar]
- 4.Liu P et al. (2011) Chromosome catastrophes involve replication mechanisms generating complex genomic rearrangements. Cell 146, 889–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Korbel JO and Campbell PJ (2013) Criteria for inference of chromothripsis in cancer genomes. Cell 152, 1226–36. [DOI] [PubMed] [Google Scholar]
- 6.Kinsella M et al. (2014) The elusive evidence for chromothripsis. Nucleic Acids Res 42, 8231–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Storchova Z and Kloosterman WP (2016) The genomic characteristics and cellular origin of chromothripsis. Curr Opin Cell Biol 40, 106–13. [DOI] [PubMed] [Google Scholar]
- 8.Putnam CD and Kolodner RD (2017) Pathways and mechanisms that prevent genome instability in Saccharomyces cerevisiae. Genetics 206, 1187–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Putnam CD et al. (2009) Specific pathways prevent duplication-mediated genome rearrangements. Nature 460, 984–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan JE and Kolodner RD (2011) A genetic and structural study of genome rearrangements mediated by high copy repeat Ty1 elements. PLoS Genet 7, e1002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Argueso JL et al. (2008) Double-strand breaks associated with repetitive DNA can reshape the genome. Proc Natl Acad Sci USA 105, 11845–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoang ML et al. (2010) Competitive repair by naturally dispersed repetitive DNA during non- allelic homologous recombination. PLoS Genet 6, e1002633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koszul R et al. (2004) Eucaryotic genome evolution through the spontaneous duplication of large chromosomal segments. EMBO J 23, 234–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Payen C et al. (2008) Segmental duplications arise from Pol32-dependent repair of broken forks through two alternative replication-based mechanisms. PLoS Genet 4, e1000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown CJ et al. (1998) Multiple duplications of yeast hexose transport genes in response to selection in a glucose-limited environment. Mol Biol Evol 15, 931–42. [DOI] [PubMed] [Google Scholar]
- 16.Dunham MJ et al. (2002) Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 99, 16144–16149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Putnam CD et al. (2016) A genetic network that suppresses genome rearrangements in Saccharomyces cerevisiae and contains defects in cancers. Nat Commun 7, 11256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lisby M et al. (2001) Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci USA 98, 8276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Branzei D and Foiani M (2010) Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11, 208–219. [DOI] [PubMed] [Google Scholar]
- 20.Haber JE (2012) Mating-yype genes and MAT switching in Saccharomyces cerevisiae. Genetics 191, 33–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verma P and Greenberg RA (2016) Noncanonical views of homology-directed DNA repair. Genes Dev 30, 1138–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pannunzio NR et al. (2018) Nonhomologous DNA end-joining for repair of DNA double-strand breaks. J Biol Chem 293, 10512–10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sallmyr A and Tomkinson AE (2018) Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J Biol Chem 293, 10536–10546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bhargava R et al. (2016) Regulation of single-strand annealing and its role in genome maintenance. Trends Genet 32, 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bell JC and Kowalczykowski SC (2016) RecA: regulation and mechanism of a molecular search engine. Trends Biochem Sci 41, 491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mizuno K et al. (2013) Recombination-restarted replication makes inverted chromosome fusions at inverted repeats. Nature 493, 246–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lambert S et al. (2010) Homologous recombination restarts blocked replication forks at the expense of genome rearrangements by template exchange. Mol Cell 39, 346–59. [DOI] [PubMed] [Google Scholar]
- 28.Szostak JW et al. (1983) The double-strand-break repair model for recombination. Cell 33, 25–35. [DOI] [PubMed] [Google Scholar]
- 29.Juhasz S et al. (2018) ATRX Promotes DNA Repair Synthesis and Sister Chromatid Exchange during Homologous Recombination. Molecular Cell 71, 11–24. [DOI] [PubMed] [Google Scholar]
- 30.Bzymek M et al. (2010) Double Holliday junctions are intermediates of DNA break repair. Nature 464, 937–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwacha A and Kleckner N (1995) Identification of double Holliday junctions as intermediates in meiotic recombination. Cell 83, 783–791. [DOI] [PubMed] [Google Scholar]
- 32.Wu LJ and Hickson ID (2003) The Bloom’s syndrome helicase suppresses crossing-over during homologous recombination. Nature 426, 870–874. [DOI] [PubMed] [Google Scholar]
- 33.Schwartz EK and Heyer WD (2011) Processing of joint molecule intermediates by structure- selective endonucleases during homologous recombination in eukaryotes. Chromosoma 120, 109–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malkova A et al. (1996) Double-strand break repair in the absence of RAD51 in yeast: a possible role for break-induced DNA replication. Proc Natl Acad Sci USA 93, 7131–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mayle R et al. (2015) Mus81 and converging forks limit the mutagenicity of replication fork breakage. Science 349, 742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Anand RP et al. (2013) Break-induced DNA replication. Cold Spring Harbor Persp Biol 5, a010397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Donnianni RA and Symington LS (2013) Break-induced replication occurs by conservative DNA synthesis. Proc Natl Acad Sci USA 110, 13475–13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saini N et al. (2013) Migrating bubble during break-induced replication drives conservative DNA synthesis. Nature 502, 389–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elango R et al. (2017) Break-induced replication promotes formation of lethal joint molecules dissolved by Srs2. Nat Commun 8, 1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakofsky CJ et al. (2014) Break-induced replication is a source of mutation clusters underlying kataegis. Cell Rep 7, 1640–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith CE et al. (2007) Template switching during break-induced replication. Nature 447, 102–105. [DOI] [PubMed] [Google Scholar]
- 42.Anand RP et al. (2014) Chromosome rearrangements via template switching between diverged repeated sequences. Genes Dev 28, 2394–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stafa A et al. (2014) Template switching during break-induced replication is promoted by the Mph1 helicase in Saccharomyces cerevisiae. Genetics 196, 1017–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pardo B and Aguilera A (2012) Complex chromosomal rearrangements mediated by break- induced replication involve structure-selective endonucleases. PLoS Genet 8 e1002979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hicks WM et al. (2010) Increased mutagenesis and unique mutation signature associated with mitotic gene conversion. Science 329, 82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilson MA et al. (2013) Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature 502, 393–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lydeard JR et al. (2007) Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448, 820–823. [DOI] [PubMed] [Google Scholar]
- 48.Lydeard JR et al. (2010) Break-induced replication requires all essential DNA replication factors except those specific for pre-RC assembly. Genes Dev 24, 1133–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deem A et al. (2008) Defective break-induced replication leads to half-crossovers in Saccharomyces cerevisiae. Genetics 179, 1845–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Smith CE et al. (2009) aberrant double-strand break repair resulting in half crossovers in mutants defective for Rad51 or the DNA polymerase delta complex. Mol Cell Biol 29, 1432–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hum YF and Jinks-Robertson S (2018) DNA strand-exchange patterns associated with double- strand break-induced and spontaneous mitotic crossovers in Saccharomyces cerevisiae. PLoS Genet 14, e1007302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piazza A et al. (2017) Multi-invasions are recombination byproducts that induce chromosomal rearrangements. Cell 170, 760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wright WD and Heyer WD (2014) Rad54 functions as a heteroduplex DNA pump modulated by its DNA substrates and Rad51 during D-loop formation. Mol Cell 53, 420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Forget AL and Kowalczykowski SC (2012) Single-molecule imaging of DNA pairing by RecA reveals a three-dimensional homology search. Nature 482, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wright WD et al. (2018) Homologous recombination and the repair of DNA double-strand breaks. J Biol Chem 293, 10524–10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Piazza A and Heyer WD (2018) Multi-invasion-induced rearrangements as a pathway for physiological and pathological recombination. Bioessays 40, e1700249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heyer WD (2015) Regulation of recombination and genomic maintenance. Cold Spring Harb Perspect Biol 7, a016501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Veaute X et al. (2003) The Srs2 helicase prevents recombination by disrupting Rad51 nucleoprotein filaments. Nature 423, 309–312. [DOI] [PubMed] [Google Scholar]
- 59.Kaniecki K et al. (2017) Dissociation of Rad51 presynaptic complexes and heteroduplex DNA joints by tandem assemblies of Srs2. Cell Rep 21, 3166–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fabre F et al. (2002) Alternate pathways involving Sgs1/Top3, Mus81/Mms4, and Srs2 prevent formation of toxic recombination intermediates from single-stranded gaps created by DNA replication. Proc Natl Acad Sci USA 99, 16887–16892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Prakash R et al. (2009) Yeast Mph1 helicase dissociates Rad51-made D-loops: implications for crossover control in mitotic recombination. Genes Dev 23, 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Liu J et al. (2017) Srs2 promotes synthesis-dependent strand annealing by disrupting DNA polymerase delta-extending D-loops. Elife 6 e22195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fasching CL et al. (2015) Top3-Rmi1 dissolve Rad51-mediated D-loops by a topoisomerase- based mechanism. Mol Cell 57, 595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ira G et al. (2003) Srs2 and Sgs1-Top3 suppress crossovers during double-strand break repair in yeast. Cell 115, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mazon G and Symington LS (2013) Mph1 and Mus81-Mms4 prevent aberrant processing of mitotic recombination imtermediates. Mol Cell 52, 63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mitchel K et al. (2013) Heteroduplex DNA position defines the roles of the Sgs1, Srs2, and Mph1 helicases in promoting distinct recombination outcomes. PLoS Genet 9 e1003340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cejka P et al. (2010) Rmi1 stimulates decatenation of double Holliday junctions during dissolution by Sgs1-Top3. Nature Struct.l Mol. Biol 17, 1377–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mimitou EP and Symington LS (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455, 770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Symington LS (2016) Mechanism and regulation of DNA end resection in eukaryotes. Crit Rev Biochem Mol Biol 51, 195–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mirman Z et al. (2018) 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polalpha-dependent fill-in. Nature 560, 112–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hashimoto Y et al. (2010) Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17, 1305–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schlacher K et al. (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sogo JM et al. (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297, 599–602. [DOI] [PubMed] [Google Scholar]
- 74.Graham JE et al. (2017) Independent and stochastic action of DNA polymerases in the replisome. Cell 169, 1201–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yeeles JT and Marians KJ (2013) Dynamics of leading-strand lesion skipping by the replisome. Mol Cell 52, 855–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lemoine FJ et al. (2005) Chromosomal translocations in yeast induced by low levels of DNA polymerase a model for chromosome fragile sites. Cell 120, 587–98. [DOI] [PubMed] [Google Scholar]
- 77.Hartwell LH and Weinert TA (1989) Checkpoints: controls that ensure order of cell cycle events. Science 246, 629–634. [DOI] [PubMed] [Google Scholar]
- 78.Kato H and Sandberg AA (1968) Chromosome pulverization in human cells with micronuclei. J Natl Cancer Inst 40, 165–79. [PubMed] [Google Scholar]
- 79.Letessier A et al. (2011) Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470, 120–3. [DOI] [PubMed] [Google Scholar]
- 80.Ying S et al. (2013) MUS81 promotes common fragile site expression. Nat Cell Biol 15, 1001–1007. [DOI] [PubMed] [Google Scholar]
- 81.Naim V et al. (2013) ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat Cell Biol 15, 1008–1015. [DOI] [PubMed] [Google Scholar]
- 82.Techer H et al. (2016) Signaling from Mus81-Eme2-dependent DNA damage elicited by Chk1 deficiency modulates replication fork speed and origin usage. Cell Rep 14, 1114–1127. [DOI] [PubMed] [Google Scholar]
- 83.Szakal B and Branzei D (2013) Premature Cdk1/Cdc5/Mus81 pathway activation induces aberrant replication and deleterious crossover. EMBO J 32, 1155–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Matos J et al. (2013) Cell-cycle kinases coordinate the resolution of recombination intermediates with chromosome segregation. Cell Rep 4, 76–86. [DOI] [PubMed] [Google Scholar]
- 85.Atianand MK and Fitzgerald KA (2013) Molecular basis of DNA recognition in the immune system. J Immunol 190, 1911–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chowdhury D et al. (2006) The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell 23, 133–42. [DOI] [PubMed] [Google Scholar]
- 87.Fan Z et al. (2003) Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL- mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell 112, 659–72. [DOI] [PubMed] [Google Scholar]
- 88.Li LY et al. (2001) Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 412, 95–9. [DOI] [PubMed] [Google Scholar]
- 89.West SC et al. (2015) Resolution of recombination intermediates: Mechanisms and regulation. Cold Spring Harb Symp Quant Biol 80, 103–9. [DOI] [PubMed] [Google Scholar]
- 90.Blanco MG et al. (2014) Dual control of Yen1 nuclease activity and cellular localization by Cdk and Cdc14 prevents genome instability. Mol Cell 54, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eissler CL et al. (2014) The Cdk/Cdc14 module controls activation of the Yen1 Holliday junction resolvase to promote genome stability. Mol Cell 54, 80–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ly P et al. (2017) Selective Y centromere inactivation triggers chromosome shattering in micronuclei and repair by non-homologous end joining. Nat Cell Biol 19, 68–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang CZ et al. (2015) Chromothripsis from DNA damage in micronuclei. Nature 522, 179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maciejowski J et al. (2015) Chromothripsis and kataegis induced by telomere crisis. Cell 163, 1641–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Crasta K et al. (2012) DNA breaks and chromosome pulverization from errors in mitosis. Nature 482, 53–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Houchmandzadeh B et al. (1997) Elasticity and structure of eukaryote chromosomes studied by micromanipulation and micropipette aspiration. J Cell Biol 139, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cluzel P et al. (1996) DNA: an extensible molecule. Science 271, 792–4. [DOI] [PubMed] [Google Scholar]
- 98.Smith SB et al. (1996) Overstretching B-DNA: the elastic response of individual double-stranded and single-stranded DNA molecules. Science 271, 795–9. [DOI] [PubMed] [Google Scholar]
- 99.Grandbois M et al. (1999) How strong is a covalent bond? Science 283, 1727–30. [DOI] [PubMed] [Google Scholar]
- 100.Nik-Zainal S et al. (2012) Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chan K and Gordenin DA (2015) Clusters of multiple mutations: incidence and molecular mechanisms. Annu Rev Genet 49, 243–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nik-Zainal S et al. (2016) Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Quigley DA et al. (2018) Genomic hallmarks and structural variation in metastatic prostate cancer. Cell 174, 758–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ratnaparkhe M et al. (2018) Defective DNA damage repair leads to frequent catastrophic genomic events in murine and human tumors. bioRxiv [DOI] [PMC free article] [PubMed]
- 105.Orthwein A et al. (2015) A mechanism for the suppression of homologous recombination in G1 cells. Nature 528, 422–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mekeel KL et al. (1997) Inactivation of p53 results in high rates of homologous recombination. Oncogene 14, 1847–57. [DOI] [PubMed] [Google Scholar]
- 107.Arias-Lopez C et al. (2006) p53 modulates homologous recombination by transcriptional regulation of the RAD51 gene. EMBO Rep 7, 219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Hollstein M et al. (1991) p53 mutations in human cancers. Science 253, 49–53. [DOI] [PubMed] [Google Scholar]
- 109.Rausch T et al. (2012) Genome sequencing of pediatric medulloblastoma links catastrophic DNA rearrangements with TP53 mutations. Cell 148, 59–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Carvalho CM and Lupski JR (2016) Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 17, 224–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sasaki M et al. (2010) Genome destabilization by homologous recombination in the germ line. Nat Rev Mol Cell Biol 11, 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wala JA et al. (2017) Selective and mechanistic sources of recurrent rearrangements across the cancer genome. bioRxiv
- 113.van Heesch S et al. (2014) Genomic and functional overlap between somatic and germline chromosomal rearrangements. Cell Rep 9, 2001–10. [DOI] [PubMed] [Google Scholar]
- 114.Serero A et al. (2014) Mutational landscape of yeast mutator strains. Proc Natl Acad Sci U S A 111, 1897–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cretu Stancu M et al. (2017) Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat Commun 8, 1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Harewood L et al. (2017) Hi-C as a tool for precise detection and characterisation of chromosomal rearrangements and copy number variation in human tumours. Genome Biol 18, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Monlong J et al. (2018) Human copy number variants are enriched in regions of low mappability. bioRxiv [DOI] [PMC free article] [PubMed]
- 118.Dixon JR et al. (2018) Integrative detection and analysis of structural variation in cancer genomes. Nat Genet 50, 1388–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Nattestad M et al. (2018) Complex rearrangements and oncogene amplifications revealed by long-read DNA and RNA sequencing of a breast cancer cell line. Genome Res 28, 1126–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sedlazeck FJ et al. (2018) Accurate detection of complex structural variations using single- molecule sequencing. Nat Methods 15, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chaisson MJP et al. (2018) Multi-platform discovery of haplotype-resolved structural variation in human genomes. bioRxiv [DOI] [PMC free article] [PubMed]
- 122.McClintock B (1941) The stability of broken ends of chromosomes in Zea Mays. Genetics 26, 234–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hatch EM et al. (2013) Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell 154, 47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Liu S et al. (2018) Nuclear envelope assembly defects link mitotic errors to chromothripsis. bioRxiv [DOI] [PMC free article] [PubMed]
- 125.Kloosterman WP et al. (2011) Chromothripsis as a mechanism driving complex de novo structural rearrangements in the germline. Hum Mol Genet 20, 1916–24. [DOI] [PubMed] [Google Scholar]
- 126.Liu P et al. (2012) Mechanisms for recurrent and complex human genomic rearrangements. Curr Opin Genet Dev 22, 211–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Berger MF et al. (2011) The genomic complexity of primary human prostate cancer. Nature 470, 214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wang J et al. (2007) Evidence for mutation showers. Proc Natl Acad Sci USA 104, 8403–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Roberts SA et al. (2012) Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol Cell 46, 424–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mirman Z et al. (2018) 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Pol alpha-dependent fill-in. Nature 560, 112–6. [DOI] [PMC free article] [PubMed] [Google Scholar]