

Abstract
The chemistry of Fe2(μ-SH)2(CO)4(PPh3)2 (2HH) is described with attention to S-S coupling reactions. Produced by reduction of 2 (Fe2(μ-S2)(CO)4(PPh3)2), 2HH is an analogue of Fe2(μ-SH)2(CO)6 (1HH) that exhibits well-behaved S-centered redox. Both 2HH and the related 2MeH exist as isomers that differ with respect to the stereochemistry of the μ-SR ligands (R = H, Me). Compounds 2HH, 2MeH, and 2 protonate to give rare examples of Fe-SH-hydrides and Fe-S2-hydrides. Salts of [H2]+, [H2HH]+, and [H2MeH]+ were characterized crystallographically. Complex 2HH reduces O2, H2O2, (PhCO2)2, and Ph2N2 giving 2. Related reactions involving 1HH gave uncharacterizable polymers. The differing behaviors of 2HH vs 1HH reflects the stabilization of the ferrous intermediates by the PPh3 ligands. When independently generated by reaction of 2HH with TEMPO, 2* quantitatively converts to 2 or, in the presence of C2H4, is trapped as the ethanedithiolate Fe2(μ-S2C2H4)(CO)4(PPh3)2. Evidence is presented that the Hieber-Gruber synthesis of 1 involves polysulfido intermediates [Fe2(μ-Sn)2(CO)6]2- (n > 1). Two relevant experiments are: (i) protonation of [Fe4(μ-S)2(μ-S2)CO)12]2- gives 1 and 1HH and (ii) oxidation of 1HH by sulfur gives 1.
Graphical Abstract
Introduction
The S-H bond of organic thiols is known to be relatively weak, with bond dissociation energies near 70 kcal/mol.1 Indeed this weakness is widely exploited since thiols serve as antioxidants and inhibit radical reactions. The thiol-ene reaction is a route to useful polymers that exploits the addition of thiyl radicals to alkenes.2 Given this rich background, the corresponding homolysis of the S-H bond of metallothiols (LnMSH) is a likely reaction, especially since electropositive metal centers might be expected to stabilize S-centered radicals, leading to even weaker S-H than those quoted for organosulfur compounds.3
Of the many metallothiol complexes,4,5 those of iron are of greatest interest because of their pervasiveness. The reactivity of FeS-H bonds is relevant to contemporary questions in bioinorganic chemistry. It is fairly certain that Fe-SH intermediates are involved in the biosynthesis of all Fe-S cofactors.6 This assertion rests on the fact that the second pKa of H2S is near or above 14.7 Consequently, all M-S forming reactions attributed to “sulfide” are in fact effected by SH−. A specific functional role of an SH− cofactor has recently been implicated in nitrogenase.8,9 Finally, the FeSH/FeSSFe couple is invoked in the Iron-Sulfur Theory of evolution.10
To examine the radical properties of metallothiols, we selected Fe2(μ-SH)2(CO)6 (1HH).11 Although only one of many Fe-SH complexes (Figure 1), Fe2(μ-SH)2(CO)6 is is the most studied metallothiol.12–15
Figure 1.

Structures of selected crystallographically characterized Fe-SH complexes.16−19
While this project began with an intended focus on Fe2(μ-SH)2(CO)6, it quickly became apparent that this complex oxidizes (dehydrogenates) to uncharacterizable insoluble solids. Fortunately, the closely related derivative Fe2(μ-SH)2(CO)4(PPh3)2 exhibits the reversible S-centered reactivity that we sought. Of specific interest is the possibility that Fe2(μ-SH)2 species might possess antioxidant behavior, akin to but extending that of glutathione and thioredoxin. In the present context, “antioxidant’ is a chemical agent that reduces reactive oxygen species (ROS).20,21 A well behaved antioxidant is one that generates a well-defined, ideally recyclable oxidized product upon exposure to the ROS. As demonstrated in this work, the Fe2(SH)2 group is a well-behaved antioxidant (Scheme 1).
Scheme 1.

Dithiol-Disulfide Antioxidant Reaction.
Results
I. Characterization of Fe2(μ-S2)(CO)4(PPh3)2 and Related Compounds.
Fe2(μ-S2)(CO)6-x(PPh3)x (x = 1, 2).
The disubstituted complex Fe2(μ-S 2)(CO)4(PPh3)2 (2) was prepared in 15–40% yield by substitution of the CO ligand in Fe2(μ-S2)(CO)6 (1) by PPh3. The monophosphine complex Fe2(μ-S2)(CO)5(PPh3) (3) has been prepared previously.22
Compound 2 is well-behaved in solution. A single isomer was observed by 31P NMR spectroscopy. Its structure was confirmed by X-ray crystallography (Figure 2). The structure of 2 has also been reported by Gao et al.23 Compared to 1,24 the Fe-CO bonds in 2 are shorter by 0.05 Å, consistent with strengthened π-bonding. Curiously a valence isomer of 3, the diferrous sulfide Fe2(μ-S)2(CO)4(PPh3)2 has also been claimed on the basis of X-ray crystallography,25 but we see no evidence for such a species.
Figure 2.

Structure of Fe2(μ-S2)(CO)4(PPh3)2 (2). Selected distances (Å): Fe(1)-Fe(1)’, 2.5557(5); Fe(1)-S(1), 2.2391(4); Fe(1)-S(1)’, 2.2585(5); Fe(1)’-S(1), 2.2585(5); Fe(1)’-S(1)’, 2.2391(4); Fe(1)-P(1), 2.2210(5); Fe(1)’-P(1)’, 2.2210(5); S(1)-S(1)’, 2.0315(6).
[HFe2(μ-S2)(CO)4(PPh3)2]+.
Protonation of the simplest diiron dithiolates of Fe2(μ-SR)2(CO)4(PR3)2 is well studied for many phosphine derivatives.26,27 Usually protonation causes changes in stereochemistry. Two scenarios have been elucidated:
Protonation of chelating dithiolates Fe2(μ-xdt)(CO)4(PR’3)2 causes the phosphine ligands to shift to basal sites (xdt = edt and pdt).26,28
Protonation of bis(alkylthiolate)s Fe2(μ-SR)2(CO)4(PR’3)2 proceeds with no repositioning of the phosphines but a change in stereochemistry of the thiolate substituents R. In Fe2(μ-SR)2(CO)4(PR’3)2 (R = Me,29 Et28), the SR groups are diequatorial.
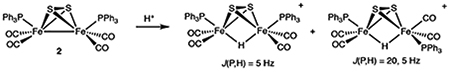
The Fe2(S2)(CO)4(PPh3)2 case follows neither pattern (eq 1).
 |
(1) |
Treatment of CH2Cl2 solutions of 2 with H(OEt2)2BArF4 resulted in a lightening of the solution color indicating protonation to give [H2]+. Excess acid had no effect. In the FT-IR spectrum, the vCO bands exhibited the expected shift of ca. 60–90 cm−1 toward higher energy. The magnitude of ΔvCO indicate that protonation occurs at the Fe-Fe bond, not at sulfur. For comparison, ΔvCO of 90 cm−1 is observed for the protonation of Fe2(μ-pdt)(CO)4(PMe3)2 to give [(μ-H)Fe2(μ-pdt)(CO)4(PMe3)2]+.27,28
Two isomers of [H2]+ are indicated by both 1H and 31P NMR spectroscopy. Two sets of hydride signals appear in a 3:2 ratio, at −19.8 (dd, J = 20, 5 Hz) and −20.6 (t, J = 5 Hz). Accordingly, two sets of 31P NMR signals are observed, and the HMBC experiment indicates the hydrides couple to distinct 31P signals (Figure S6). The stereochemistry of the two isomers can be assigned by the splitting of the hydride signals: small values of J(31P,1H) are typically observed for phosphines trans (apical sites) to μ-hydride, and larger values (~20 Hz) are observed for cis (basal sites).30
Persulfido hydrido complexes are rare, the precedents being Cp*2Ta(S2)H and its Cr(CO)5 adduct.31 The cation [H2]+ is the first example of an iron persulfido hydride. Its valence isomer [Fe2(μ-S)(μ-SH)(CO)4(PPh3)2]+ would be an analogue of our recently reported [Fe2(μ-S)(μ-SCH2Ph)(CO)2(dppv)2]+.32 X-ray crystallographic analysis confirmed the structure of [H2]+ (Figure 3). Compared to its conjugate base Fe2(μ-S 2)(CO)4(PPh3)2 (Supporting Information), the Fe-Fe distance in the hydride cation is elongated by almost 0.1 Å, from 2.5558(5) to 2.6326(5) Å. Compared to 2, the Fe-CO bond distances are elongated by ~0.03 Å.
Figure 3.

Structure of the cation in [(μ-H)Fe (μ-S2 2)(CO)4(PPh3)2]BArF4. Selected distances (Å): Fe(1)-Fe(2), 2.6327(6); Fe(1)-S(1), 2.2349(7); Fe(1)-S(2), 2.2565(8); Fe(2)-S(1), 2.2364(7); Fe(2)-S(2), 2.2527(7); Fe(1)-P(1), 2.2570(7); Fe(2)-P(2), 2.2518(8); Fe-CO: 1.788(3)-1.783(3); Fe(1)-H(1): 1.65(4); Fe(2)-H(1): 1.65(3); S(1)-S(2), 2.0201(9).
II. Fe2(μ-SH)2(CO)4(PPh3)2 and Related Compounds.
Fe2(μ-SH)2(CO)4(PPh3)2 and Fe2(μ-SH)2(CO)5(PPh3).
Compounds 2 and 3 undergo reductive conversion to the respective (SH)2-containing derivatives, Fe2(μ-SH)2(CO)6-x(PPh3)x, 2HH and 3HH. The conversion followed Seyferth’s protocol (LiBHEt3 reduction followed by trifluoroacetic acid).11 Of course, 2 and 3 are unreactive toward H2 (1 atm).
Both 2HH and 3HH exist of three isomers, which differ in terms of the stereochemistry of the SH groups (Table 1).33 The rationale for the assignment of these isomers is presented in a subsequent section of this paper. The triplet at δ−0.79 is assigned to the ee isomer, the singlet at δ −0.16 and the triplet at δ −3.84 (J = 5 Hz) can be assigned respectively to the axial-equatorial isomer ae, and the triplet at d-4.55 (J = 5 Hz) is assigned to aa. Related to 3HH, the complex Fe2(μ-SH)2(CO)5(PhP(CH2OH)2) also consists of three isomers, with a ratio similar to that of 2HH.33
Table 1.
Selected NMR Data for 1HH, 3HH, 2HH, and 2MeH.
| Complex | NMR Chemical shifts (ppm) | Isomer Ratio (ee:ae:aa) | |
|---|---|---|---|
| 1H NMR SH Signals (multiplicity, J in Hz) | 31P NMR | ||
| Fe2(μ-SH)2(CO)6 (1HH) | 0.3 (s) | - | 0.2: 0.7: 0.1 |
| −0.27 (s), −2.14 (s) | |||
| −2.25 (s) | |||
| Fe2(μ-SH)2(CO)5(PPh3)(3HH) | 0.05 (d, 1.25) | 61.2(s) | 0.1: 0.8: 0.1 |
| −0.56 (d, 1.25), −3.01 (d, 5.5) | 58.3(s) | ||
| −3.33 (d, 5.5) | 55.8(s) | ||
| Fe2(μ-SH)2(CO)4(PPh3)2(2HH) | −0.16 (s) | 59.2(s) | 0.1: 0.8: 0.1 |
| −0.79 (s), −3.84 (t, 10) | 55.2(s) | ||
| −4.55 (t, 10) | 52.2(s) | ||
| Fe2(μ-SH)(SMe)(CO)4(PPh3)2(2MeH) | −0.73 (br s) | 57.3 (s) | 0.1: 0.8: 0.1 |
| −0.79 (br s) | 55.4 (s) | ||
| −3.73 (t, 5) | 53.1 (s) | ||
[HFe2(μ-SH)2(CO)4(PPh3)2]+.
Solutions of 2HH react with one equiv of H(OEt2)2BArF4 at −80°C. The initial product is the hydride [HFe2(μ-SH)2(CO)4(PPh3)2]+ ([H2HH]+). The closest precedent to [H2HH]+ is the radical (μ-H)Fe2(μ-SH)2(CO)6, one of several products obtained by UV-irradiation of pentane solution of Fe(CO)5 and H2S at low-temperatures.34 The structure of [(μ-H)Fe2(μ-SH)2(CO)4(PPh3)2]BArF4 was confirmed by X-ray crystallography (Figure 4). The ae and ee isomers cocrystallized.
Figure 4.

Structure of the cation in [(μ-H)Fe2(μ-SH)2(CO)4(PPh3)2]BArF4. Selected distances (Å): Fe(1)-Fe(2), 2.5893(6); Fe(1)-S(1), 2.2861(9); Fe(1)-S(2), 2.283(1); Fe(2)-S(1), 2.2780(8); Fe(2)-S(2), 2.2679(9); Fe(1)-P(1), 2.2588(9); Fe(2)-P(2), 2.272(1); Fe(1)-H(1): 1.76(4); Fe(2)-H(1), 1.67(4); S(1)-H(1A), 1.21(4); S(2)-H(2), 1.22(3).
Two isomers of [H2HH]+ are indicted by the hydride signals at δ−17.0 and −17.6 as well as 31P NMR singlets at δ 59.5 and 56.5 (−20°C). The isomer ratio is 1:1. The small value of J(H,P) indicates that the phosphines are diapical in each isomer. HMBC measurements indicate that the isomers differ with respect to the relative orientation of the SH groups (Figure S14), consisting of an unsymmetrical ae and a symmetrical isomer (Scheme 2). For the symmetrical isomer, only one SH signal correlated with one 31P NMR signal whereas for the unsymmetrical isomer, two SH signals correlate with the second 31P NMR signal. The symmetrical isomer is probably ee, the favored stereochemistry in other complexes of the type [HFe2(μ-SR)2(CO)4(PR’3)2]+ (R = Me, Et).28,29 The stabilization of the ee isomer is possibly associated with the shortened S ⋯S distances (by 0.1 Å) induced by ~0.05 Å protonation-induced elongation of the Fe⋯Fe distances.
Scheme 2.

Effect of Protonation on Isomerism of the Fe2(SH)2 Center in 2HH and [H2HH]+.
Upon warming the solution to near 0°C, decomposition was evident by the evolution of gas. Gas chromatographic analysis showed that the gas was a mixture of H2 and CO. The main organometallic product of the decomposition is 2HH, which forms in ~50% yield.
Fe2(μ-SH)(μ-SMe)(CO)4(PPh3)2 and [HFe2(μ-SH)(μ-SMe)(CO)4(PPh3)2]+.
A mixed SH-SMe complex was prepared in the form of Fe2(μ-SH)(μ-SMe)(CO)4(PPh3)2 (2MeH). The complex was obtained by the addition of an excess of MeLi to 2 followed by acidification. The presumed intermediate in this reaction is [Fe2(μ-SLi)(μ-SMe)(CO)4(PPh3)2].
The NMR properties of 2MeH are sufficiently definitive to allow not only the assignment of these isomers to structures, but also the assignments for Fe2(μ-SH)2(CO)6-x(PPh3)x, for which consistent, logical assignments are lacking.12 According to 1H and 31P NMR analysis, 2MeH consists of three isomers (Figure 5).
Figure 5.

1H NMR (500 MHz, CD2Cl2) spectrum of Fe2(μ-SMe)(μ-SH)(CO)4(PPh3)2 at 20°C, depicting expansion of SMe and SH signals. Three isomers, ae, ee and ea are observed.
Four isomers are possible: aHeMe, eHeMe, and eHaMe, aHaMe. The missing isomer must be aHaMe. The aa isomer is minor in 1HH and 2HH, and completely absent in Fe2(μ-SMe)2(CO)6,35–38 which exists only as eMeeMe and aMeeMe (Scheme 3).39,40 Apparently only the miniscule H substituent is compatible with the diaxial configuration.41 We conclude that for 2MeH the aa isomer is destabilized by a steric clash. The relative chemical shifts of the SH signals and the magnitude of the 31P-1H couplings are consistent with the previous assignments.
Scheme 3.

Four isomers of 2MeH. The aHaMe isomer is not observed.
Protonation of 2MeH gave an isolable hydride salt [H2MeH]BF4, which was characterized by X-ray crystallography (Figure 6). Crystallographic analysis of [H2MeH]BF4 revealed a single isomer with equatorial SMe and axial SH. As seen for [HFe2(μ-SR)2(CO)4(PR’3)2]+ (R = Me,29 Et28), the two phosphine ligands remain apical, reflecting the steric protection afforded by the single equatorial methyl group.
Figure 6.

Structure of the cation in [(μ-H)Fe2(μ-SH)(μ-SMe)(CO)4(PPh3)2]BF4. Selected distances (Å): Fe(1)-Fe(2), 2.5797(9); Fe(1)-S(1), 2.268(1); Fe(1)-S(2), 2.284(1); Fe(2)-S(1), 2.251(1); Fe(2)-S(2), 2.289(1); Fe(1)-P(1), 2.271(1); Fe(2)-P(2), 2.255(1); Fe(1)-H(1), 1.58(4); Fe(2)-H(1), 1.61(5); S(1)-C(5), 1.818(4); S(2)-H(2), 1.24(4).
Compared to [H2HH]+, [H2MeH]+ is noticeably more stable in solution at room temperature. This enhanced stability is attributed in part to the greater electron releasing properties of the SMe− vs SH− groups. This trend is indicated by the relative values of vCO for 2HH vs 2MeH, which are 2000, 1955, 1938 and 1995, 1948, 1930 cm−1, respectively.
III. Oxidation of Fe2(μ-SH)2(CO)4(PPh3)2.
Although 1HH has been described as “air-sensitive,” little is known about the products of air oxidation. When left open to air in THF solution, 1HH degrades over the course of hours to a black solid and ca. 50% yield of 1. Under these conditions 1 is very stable in THF solution, so the black solid does not result from degradation of 1. In solution, 2HH is more air-stable than 1, but converts to 2 over the course of days. The conversion of 2HH into 2 proceeds quantitatively according to 31P NMR spectroscopy.
Given the efficiency of the 2HH + O2 reaction, other oxidants were examined. Azobenzene was found to react over the course of several hours at room temperature to generate diphenylhydrazine and 2. The hydrogenation of azobenzene by iron sulfido complexes has not been reported, but the reaction is catalyzed by (CH3C5H4)2Mo2S2(S2CH2).42
The O2 + 2HH reaction is proposed to involve H-atom abstraction by O2, leading sequentially to hydroperoxide (HO2), H2O2, and H2O. Using aqueous H2O2 in place of air, the conversion 2HH → 2 proceeds in minutes (vs days, eq 2).
| (2) |
Benzoyl peroxide ((PhCO2)2) is a very fast oxidant, the conversion being complete in the time of mixing.
IV. Mechanistic Studies on the Oxidation of Fe2(μ-SH)2(CO)4(PPh3)2.
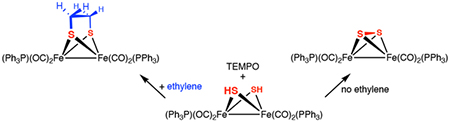
To learn more about the O2 + 2HH reaction, the reagent 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) was employed. Since it abstracts H atoms from weak X-H bonds,43 TEMPO serves as a weighable surrogate of O2 with enhanced reactivity. Within seconds of being treated with TEMPO, 2HH quantitatively converted into 2. A deficiency of TEMPO resulted in partial conversion to 2, leaving unreacted 2HH. This result indicates that mixed valence species Fe2(μ-SH)(μ-S)CO)4(PPh3)2 (2H) is a superior H-atom donor relative to 2HH (Scheme 4).
Scheme 4.

Reaction of TEMPO with 2HH and 2H.
These results are consistent with DFT-calculated bond dissociation free energies for Fe2(μ-SH)2-x(μ-S)x(CO)6. Those S-H bond energies are calculated to be 72 and 45 kcal/mol, respectively for x = 0 and x = 1.44 Oxidation of 3HH with two equiv of TEMPO cleanly gave 3. On the other hand, oxidation of 1HH with 0.5–2 equiv of TEMPO resulted in nearly full conversion to a black solid.
Trapping experiments implicate unsaturated intermediates in the TEMPO-induced dehydrogenation of Fe2(μ-SH)2(CO)6-x(PPh3)x. Addition of 2 equiv of TEMPO to an C2H4-saturated solution of 2HH gave a 1:3 mixture of 2 and the ethanedithiolate Fe2(μ-S2C2H4)(CO)4(PPh3)2, respectively. The identity of Fe2(μ-S2C2H4)(CO)4(PPh3)2 was verified by independent synthesis.45 In a control experiment, C2H4 was shown to be unreactive toward 2, i.e. no thermal reaction occurs (Scheme 5).
Scheme 5.

Effect of Ethylene on Oxidation of Fe2(SH)2(CO)4(PPh3)2.
The TEMPO-C2H4-trapping experiments were extended to 3HH and 1HH. Addition of two equiv of TEMPO to a C2H4-saturated solution of the monophosphine 3HH mainly produced black solids together with small amounts of the ethanedithiolate Fe2(μ- S2C2H4)(CO)5(PPh3) and 3 in a ratio of 1:9, based on 31P NMR analysis. This ethanedithiolate was identified by independent synthesis (Fe2(μ-S2C2H4)(CO)6 + PPh3). Its 1H NMR spectrum is distinctive in the CH2 region showing two multiplets, consistent with an AA’BB’ coupling pattern where AA’ and BB’ correspond to H’s facing toward or away from the PPh3 ligand.
Trapping agents other than ethylene were tested without success. Treatment of solutions of 2HH with two equiv TEMPO under an atmosphere of D2, in the presence of 10 equiv of PhMe2SiH, phenylacetylene, or 1-hexene gave only 2. Substituted alkenes are known to be unreactive toward UV-irradiated solutions of 1.46,47
V. Synthetic Pathway Leading to Fe2(μ-S2)(CO)6.
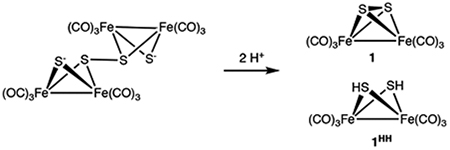
The results described above are relevant to an old mystery: the pathway for the synthesis of 1. Since it was reported in 1958 by Hieber and Gruber (H-G), the synthesis of 1 has remained unaddressed.48 This oversight contrasts with the hundreds of studies that use 1. The H-G synthesis involves the formation of an Fe-S-CO anion by the reaction of [HFe(CO)4]− and sodium polysulfide (or elemental sulfur) followed by acidification. A key fact is that 12- is not a precursor to 1, it simply protonates to give 1HH. The requirement of the H-G synthesis for excess sulfur provides the main clue to the pathway (Table 2). Although the H-G synthesis reaction does not involve 12-, this dianion can be used to generate intermediates in the pathway of the H-G synthesis. Indeed, treating a THF solution of Li2[1] with excess S (as S8) followed by acid efficiently gave 1.
Table 2.
Effect of Fe:xS Ratio on the Yields of 1 and Fe3S2(CO)9 by the H-G Synthesis Method.
| Equiv S | 2.5 | 4.5 | 5.5 | 6.0 | 6.5 | 7.5 | 8.5 | 10 |
|---|---|---|---|---|---|---|---|---|
| Fe3S2(CO)9 | 37% | 0.4% |
The role of polysulfide intermediate(s) in the conversion of 12- to 1 is supported by 13C NMR studies. 13C NMR spectroscopy allows the identification of 1HH and 1, which are otherwise difficult to distinguish by FT-IR spectroscopy (Figure S26). The 13C NMR spectrum of 1HH consists of three singlets (δ209.4, 209.2, 209.0), consistent with the presence of aa, ee, and ae isomers, respectively. The center signal, corresponding to a minor isomer, overlaps with the singlet for 1. According to 13C NMR spectroscopy, a THF solution of 1HH converts to 1 in the presence of elemental sulfur. Unlike the 1HH + O2 reaction, the 1HH + S reaction is more efficient (no black solids). The 1HH + S → 1 conversion does not proceed in the presence of acid, so it is probably not relevant to the H-G synthesis.
Further insights were obtained using salts of the persulfide [Fe4S4(CO)12]2-.49 In terms of their IR spectra, the species generated by the reaction of Li2[1] with S is almost identical to (BnNMe3)2[Fe4S4(CO)12] (Figure 7). This salt was obtained by the addition of BnNMe3+ to a H-G synthesis solution. Treatment of MeCN solution of (BnNMe3)2[Fe4S4(CO)12] with excess p-toluene sulfonic acid (HOTs) gave an
 |
(3) |
Figure 7.

IR spectra of THF solutions of Li2Fe2S2(CO)6 (a) before and (b) after addition 4 equiv S, (c) the salt precipitated by addition of BnNMe3Cl to a Hieber-Gruber solution, and (d) (BnNMe3)2[Fe4S4(CO)12]
approximate 1:1 mixture of 1HH and 1 (Figure 8, eq 3). This finding is consistent with the hypothesis that polysulfides derived from [1]2- undergo ring-closure upon protonation to give 1.
Figure 8.

13C NMR spectra for 1HH (top, showing the three isomers), a 1:1 mixture of 1 and 1HH (middle), and the pentane-soluble products (bottom) from treatment of acetonitrile solution of (BnNMe3)2[Fe4S4(CO)12] with 15 equiv of HOTs. All solutions are in CD2Cl2 solvent.
Summary and Conclusions
The work mainly examines the diiron dithiol Fe2(μ-SH)2(CO)4(PPh3)2 (2HH) as a well-behaved, readily interrogated analogue of Fe2(μ-SH)2(CO)6. Complex 2HH exhibits unusual redox properties resulting in hydrogen atom transfer, which regenerates the disulfide 2. In this regard, the diiron dithiol exhibits well-behaved antioxidant behavior.
Why is 2HH a superior and better-behaved reductant relative to 1HH? We propose that the PPh3 ligands stabilize the dehydrogenated intermediate Fe2(μ-S)2(CO)4(PPh3)2 (2*), which resists the addition of further equiv of Fe2(μ-SH)2(CO)4(PPh3)2. In contrast, Fe2(μ-S)2(CO)6 (1*) consumes 1HH, giving polymers competitive with conversion to 1.
Both electronic and steric arguments can be used to rationalize the stability of 2*. Featuring ferrous centers, 2* would be stabilized strongly by presence of phosphine ligands. Indicative of the effect of phosphine substitution, the reduction potentials for [Fe2(μ-SR)2(CO)6]+ and [Fe2(μ-SR)2(CO)4(PPh3)2]+ differ by ~0.8 V.50 Computational experiments indicate that the diferrous species Fe2(μ-S)2(CO)6, analogous to 2*, exists as a singlet ground state.51 It is also possible that the stabilizing influence of the PPh3 ligands is steric in origin, preventing 2* from attacking 2HH.
The trapping results using 2HH/TEMPO/C2H4 reaction are reminiscent of the photochemical reactivity of 1. UV-irradiation of 1 in presence of alkynes, ethylene, and CO gives Fe2(μ-S2C2R2)(CO)6,52 Fe2(μ-S2C2H4)(CO)6, and Fe2(μ-S2CO)(CO)6, respectively.22,46,47,53,54 The intermediate generated in the 1HH/TEMPO and 1HH/O2 reactions appears to react with 1HH to give intractable solids. In contrast the intermediate 2* generated in the corresponding oxidations of Fe2(μ-SH)2(CO)4(PPh3)2 does not add 2HH but cleanly converts to 2.
Finally, the experiments on 1HH were extended to partially illuminate the final stages in the Hieber-Gruber synthesis of 1. Acid-labile per- or polysulfides of the type [Fe2(μ-Sn)2(CO)6]2- or [Fe2(μ-S)(μ-Sn)(CO)6]2- are implicated (n > 1, Scheme 6). The salient findings are (i) 1HH + S8 gives 1, while 1HH + O2 does not and (ii) Fe4(μ-S)2(μ-S 2)(CO)12]2- + H+ gives 1, while 12- + H+ + does not.
Scheme 6.

Simplified View of the Proposed Final Steps of the Hieber-Gruber Synthesis of 1.
Experimental
Materials and methods have been described recently.32
Fe2(S2)(CO)4(PPh3)2 (2).
A 100-mL Schlenk flask was charged with 0.80 g (2.33 mmol) of 1 and 50 mL of THF to give a red-orange solution. At −78°C, addition of 1.22 g (4.66 mmol) of PPh3 and 0.35 g (4.66 mmol) of Me3NO. The solution was allowed to warm to room temperature over the course of several min. After 28 h, solvent was evaporated. The dark red residue was purified by chromatography on silica gel eluting with 1:10 CH2Cl2-hexanes. Three red bands eluted, the first being 1, the second being 3, and the third band was 2 (further unidentified products could be collected by eluting with CH2Cl2). Yield: 0.65 g (15–35%). IR (CH2Cl2): ʋCO 2000, 1950, 1935 cm−1. 1H NMR (500 MHz, CD2Cl2): δ 7.44–7.40, (m, 30H, PPh3). 31P NMR (CD2Cl2): δ 57.69 (s). Single X-ray crystals were grown from a concentrated pentane solution at −20°C. Anal. Calcd for C40H30P2Fe2O4S2: C, 59.13; H, 3.72. Found: C, 58.73; H, 3.69.
[HFe2(μ-S2)(CO)4(PPh3)2]BArF4 ([H2]BArF4).
A solution of 50 mg (0.06 mmol) of 2 in 2 mL of CH2Cl2 was treated with H(OEt2)2BArF4 (62.3 mg, 0.06 mmol). Pentane was added, and the mixture was maintained at −20°C overnight. The resulting solid precipitate was washed with 10 mL of pentane. Yield: 87 mg (86%). IR (CH2Cl2): ʋCO 2061, 2041, 2008 cm−1. 1H NMR (500 MHz, CD2Cl2): δ 7.73–7.28 (m, 42 H), −19.80 (dd, J = 20 and 5 Hz, 0.37H, μ-H isomer A), −20.56 (t, J = 5Hz, 0.26 H, μ-H isomer B). 31P NMR (CD2Cl2): δ 61.52, 61.55, 61.63, 61.66 (isomer B) and 55.5 (isomer A). Single X-ray crystals were grown from a concentrated CH2Cl2 solution layered with pentane at −20°C.
Fe2(μ-SH)2(CO)4(PPh3)2 (2HH).
A stirred solution of 0.2 g (0.25 mmol) of 2 in 30 mL of THF at −78°C was treated dropwise with 0.62 mL (0.62 mmol) of LiHBEt3. The initially red solution became green, indicating the formation of (μ-LiS)2Fe2(CO)4(PPh3)2. After the reaction mixture had stirred for an additional 15 min., 68 μL (0.90 mmol) of CF3CO2H was added, causing an immediate color change from green to red, indicating the formation of 2HH. After stirring for an additional 15 min at −78°C, the mixture was concentrated in vacuo. The red gummy residue was extracted with ca. 15 mL of CH2Cl2, and this extract was passed through a plug of Celite. The solvent was evaporated under vacuum to afford a red solid. Yield: 0.18 g (90%). IR (CH2Cl2): ʋCO = 2000, 1955, 1938 cm−1. 1H NMR (500 MHz, CD2Cl2): δ 7.47–7.39 (m, 30H, PPh3), −0.16 (s, e-H of ae isomer), −0.79 (s, e-H of ee isomer), −3.84 (s, a-H of ae isomer), −4.55 (s, a-H of aa isomer). 31P NMR (CD2Cl2): δ 59.2 (s, aa), 55.2 (s, ae), 52.2 (s, ee). Single X-ray crystals were grown from a concentrated CH2Cl2 solution layered with pentane at −20°C. Anal. Calcd for C 40H32Fe2O4P2S2 CH2Cl2: C, 54.74; H, 3.81 Found: C, 55.14; H, 3.59.
[HFe2(μ-SH)2(CO)4(PPh3)2]BArF4 ([H2HH]BArF4).
To a stirred solution of 30 mg (0.04 mmol) of 2HH in 1 mL of CD2Cl2 at - 80°C was added H(OEt2)2BArF4 (37 mg, 0.04 mmol). IR (CH2Cl2): ʋCO 2061, 2041, 2008 cm−1. 1H NMR (500 MHz, CD2Cl2): δ δ 7.72–7.43 (m), −0.01 (s, e-H of ae isomer), −3.51 (s, a-H of ae isomer), −3.81 (s, a-H of aa isomer), −17.02 (br s, μ-H of aa isomer) and −17.63 (br s, μ-H of ae isomer). 31P NMR (CD2Cl2): δ 59.5 (s, aa), 56.5 (s, ae).
Fe2(μ-SMe)(μ-SH)(CO)4(PPh3)2 (2MeH).
A stirred solution of 0.0812 g (0.1 mmol) of 2 in 5 mL of THF at −78°C was treated with 0.19 mL (0.3 mmol, 1.6 M) of MeLi. The mixture assumed a dark green color attributed to Fe2(μ-LiS)(μ-SMe)(CO)4(PPh3)2. After being maintained at −78°C for 15 min, the reaction mixture was treated with 23 μL (0.3 mmol) of CF3CO2H, causing an immediate color change from green to red, consistent with the formation of 2MeH. After an additional 15 min at −78°C, the mixture was allowed to warm to room temperature, and solvent was removed. The red gummy residue was extracted into ca. 2 mL of CH2Cl2, and this extract was filtered through a plug of Celite. The solvent was evaporated under vacuum to afford a dark red solid. Recrystallization of this solid was achieved by addition of pentane to a CH2Cl2 solution, followed by cooling to −20°C. Yield: 0.074 g (90%). Anal. Calcd for C41H34Fe2O4P2S2•0.5 CH2Cl2: C, 57.23; H, 4.05. Found: C, 57.01; H, 3.98. IR (CH2Cl2): ʋCO 1995, 1948, 1930 cm−1. 1H NMR (500 MHz, CD2Cl2): δ 7.53–7.38 (m, 45H, PPh3), 1.35 (s, ae, e-CH3), 1.25 (s, ee, e-CH3), 0.07 (s, ea, a-CH3), −0.73 (s, ee, e-H), −0.79 (s, ea, e-H), −3.73 (t, ae, a-H, J = 5Hz). 31P NMR (CD2Cl2): δ 57.3 (s, ea, e-CH3, a-H), 55.4 (s, ae, a-H, e-CH3), 53.1 (s, ee, e-H, e-CH3). Single X-ray crystals were grown by layering a concentrated CH2Cl2 solution with pentane at −20°C.
[HFe2(μ-SMe)(μ-SH)(CO)4(PPh3)2]BArF4 ([H2MeH]BArF4).
A stirred solution of 0.0828 g (0.1 mmol) of 2MeH in 2 mL of CH2Cl2 was treated with 0.1012 g of HBArF4.2Et2O, causing an immediate color change from dark read to light red. The product was purified by layering a concentrated CH2Cl2 solution (~0.5 mL) with 20 mL of pentane at −20°C. Anal. Calcd for C73H47Fe2O4P2S2BF24•0.5 CH~ 2Cl2: C, 50.88; H, 2.79. Found: C, 50.92; H, 2.52. IR (CH2Cl2): ʋCO 2099, 2055, 2040, 2003 cm−1. 1H NMR (500 MHz, CD2Cl2): δ 7.51–7.75 (m, 2 PPh3 and BArF4), 1.42, 1.38, 1.09, 0.99, 0.65, 0.14 (CH3), −0.59, −0.70, −1.30, −1.53, −1.85, −2.97 (S-H), −15.51, −16.26, −16.35, −17.35 (m, μ-H). 31P NMR (CD2Cl2): δ 56.6 (m), 51.9 (m), 51.2 (m), 50.2 (t), 50.5 (m), 43.1 (m). Single X-ray crystals were grown from a concentrated CH2Cl2 solution layered with pentane at −20°C.
Ethylene Trapping Reactions.
A solution of 2HH (50 mg, 0.06 mmol) in THF (15 mL) was cooled to 0°C and saturated with ethylene. A THF solution of TEMPO (0.61 M, 0.2 mL) was injected, and the mixture was stirred for an additional 30 min., maintaining the ethylene purge. Solvent was removed under vacuum, and the flask was taken into a glove box. The residue was dissolved in a minimum amount of DCM, and the solution was filtered to remove a small amount of insoluble material. Addition of pentane to the filtrate yielded a red-pink powder consisting of Fe2(S2C2H4)(CO)4(PPh3)2 and 2.45 1H-NMR (CD2Cl2):δ 7.57 (m, 14H), 7.40 (m, 34H), 0.65 (s, 4H). 31P NMR (CD2Cl2): δ 60.25 (s), 57.69 (s). When a solution of 2HH (50 mg, 0.06 mmol) in THF (15 mL) was treated with TEMPO (19 mg, 0.12 mmol), 2 was the exclusive product as demonstrated by 31P NMR analysis and thin-layer chromatography.
(BnNMe3)2[Fe2S2(CO)6]2.
The modified procedure was based on the literature.45 A solution of 1 (5.0 mmol, 1.72 g) in THF (10 mL) was treated dropwise with LiBHEt3 (5.0 mmol, 5 mL, 1 M in THF) at −78°C. The solution turned to greenish dark immediately. After 30 min, a solution of PhCH2NMe3Cl (5.0 mmol, 925 mg) in a mixture of MeCN (8 mL) and MeOH (2 mL) was added to the above solution. After stirring the mixture at −20°C for 1 h, solvents were removed under vacuum. The residue was washed with cold THF and Et2O and dried under vacuum. Yield: 2.67 g (54%). IR (MeCN): ʋCO 2041, 2029, 2002, 1954 cm−1. 13C NMR (125 MHz, CD3CN): δ 53.4, 70.2, 128.6, 130.1, 131.7, 133.8, 214.5.
Effect of S/Fe Ratio on Hieber-Gruber Synthesis.
A 500-mL flask was charged with Fe(CO)5 (1.0 equiv, 14.8 mmol, 2 mL) and MeOH (12 mL) followed by aqueous KOH (50% w/w, 7.2 equiv, 6.0 g). After 10 min., the orange solution was cooled to 0°C. Elemental sulfur (x equiv, see Table 2) was added, producing a dark brown dark solution. Caution: gas evolution! After stirring for 1 h at 0°C, the mixture was treated with water (75 mL), pentane (100 mL), followed by solid NH4Cl (10 equiv, 148 mmol, 8.0 g) in one portion. After being kept at 0°C for ca. 3 h, the solution was allowed to warm to room temperature overnight. The mixture was extracted exhaustively with pentane (in cases where phase separation was unclear, ~10 mL of acetone was added). The combined pentane extracts were filtered through a pad of silica gel and evaporated using a rotary evaporator. For purification by chromatography on silica gel, pentane alone was used. The first orange band was 1, which was further purified by repeated evaporation-extraction into pentane to remove sulfur.
X-ray Crystallographic Determinations.
Crystallographic Data were collected on a Bruker D8 Venture instrument equipped with a four-circle kappa diffractometer and Photon 100 detector. An Iμs microfocus Mo (λ = 0.71073 Å) source was supplied the multi-mirror monochromated incident beam. The samples were mounted on a 0.3 mm loop with the minimal amount of Paratone-N oil. Data were collected as a series of φ and/or ω scans. Data were collected at 100K and integrated and filtered for statistical outliers using SAINT,55 and corrected for absorption by multi-scan methods using SADABS56 v2014/7. The structures were phased using direct methods57 or intrinsic phasing methods58 and then refined with the SHELX software package SHELX-2014–7.57
Supplementary Material
Synopsis.
In contrast with Fe2(μ-SH)2(CO)6, the complex Fe2(μ-SH)2(CO)4(PPh3)2 exhibits well-behaved anti-oxidant properties resulting from S-centered redox. It reduces TEMPO, O 2, H2O2, (PhCO2)2 and Ph2N2 giving Fe2(μ-S2)(CO)4(PPh3)2. Oxidation of Fe2(μ-SH)2(CO)4(PPh3)2 with TEMPO in the presence of C2H4 gives the ethanedithiolate Fe2(μ-S2C2H4)(CO)4(PPh3)2. Related studies show that the original synthesis of Fe2(μ-S 2)(CO)6 involves protonation of polysulfides [Fe2(μ-Sn)2(CO)6]2- (n > 1) with elimination of H2Sx.
ACKNOWLEDGMENTS
This work was supported by GM-61153 from the National Institutes of Health.
Footnotes
Supporting Information
Spectroscopic data, selected reaction schemes, selected procedures.
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website at DOI: _
Accession Codes
CCDC 1877173, 1877177, 1877175, 1877176 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
REFERENCES
- 1.Luo YR Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, 2007. [Google Scholar]
- 2.Dénès F; Pichowicz M; Povie G; Renaud P, Thiyl Radicals in Organic Synthesis. Chem. Rev 2014, 114, 2587–2693. [DOI] [PubMed] [Google Scholar]
- 3.Appel AM; Lee S-J; Franz JA; DuBois DL; Rakowski DuBois M, Free Energy Landscapes for S−H Bonds in Cp*2Mo2S4 Complexes. J. Am. Chem. Soc 2009, 131, 5224–5232. [DOI] [PubMed] [Google Scholar]
- 4.Kuwata S; Hidai M, Hydrosulfido Complexes of Transition Metals. Coord. Chem. Rev 2001, 213, 211–305. [Google Scholar]
- 5.Peruzzini M; de los Rios I; Romerosa A, Coordination Chemistry of Transition Metals with Hydrogen Chalcogenide and Hydrogen Chalcogenido Ligands. Prog. Inorg. Chem 2001, 49, 169–543. [Google Scholar]
- 6.Lill R, Function and Biogenesis of Iron-Sulfur Proteins. Nature 2009, 460, 831–838. [DOI] [PubMed] [Google Scholar]
- 7.Meyer B; Ward K; Koshlap K; Peter L, Second Dissociation Constant of Hydrogen Sulfide. Inorg. Chem 1983, 22, 2345–2346. [Google Scholar]
- 8.Hoffman BM; Lukoyanov D; Dean DR; Seefeldt LC, Nitrogenase: A Draft Mechanism. Acc. Chem. Res 2013, 46, 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khadka N; Milton RD; Shaw S; Lukoyanov D; Dean DR; Minteer SD; Raugei S; Hoffman BM; Seefeldt LC, Mechanism of Nitrogenase H2 Formation by Metal-Hydride Protonation Probed by Mediated Electrocatalysis and H/D Isotope Effects. J. Am. Chem. Soc 2017, 139, 13518–13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wächtershäuser G, From Chemical Invariance to Genetic Variability In Bioinspired Catalysis; Weigand Wand Schollhammer P, 2014, Wiley-VCH, Weinheim. [Google Scholar]
- 11.Seyferth D; Henderson RS, Di-μ-Thiolbis(tricarbonyliron), (μ-HS)2Fe2(CO)6: An Inorganic Mimic of Organic Thiols. J. Organomet. Chem 1981, 218, C34–C36. [Google Scholar]
- 12.Crouthers DJ; Ding S; Denny JA; Bethel RD; Hsieh C-H; Hall MB; Darensbourg MY, A Reduced 2Fe2S Cluster Probe of Sulfur-Hydrogen versus Sulfur-Gold Interactions. Angew. Chem., Int. Ed 2015, 54, 11102–11106. [DOI] [PubMed] [Google Scholar]
- 13.Seyferth D; Womack GB; Henderson RS; Cowie M; Hames BW, Michael-Type Addition Reactions of Bis(μ-mercapto)bis(tricarbonyliron): Proximity-Induced Formation of Bidentate Organosulfur Ligands. Organometallics 1986, 5, 1568–1575. [Google Scholar]
- 14.Rauchfuss TB, Diiron Azadithiolates as Models for the [FeFe]-Hydrogenase Active Site and Paradigm for the Role of the Second Coordination Sphere. Acc. Chem. Res 2015, 48, 2107–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y; Rauchfuss TB, Synthesis of Diiron(I) Dithiolato Carbonyl Complexes. Chem. Rev 2016, 116, 7043–7077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arif AM; Hefner JG; Jones RA; Koschmieder SU, Mononuclear Complexes of Cr(II) and Fe(II) with Terminal -SH Groups. Synthesis and X-ray Crystal Structures of trans-M(SH)2(dmpe)2 (M = Cr, Fe; dmpe = 1,2-Bis(dimethylphosphino)ethane). J. Coord. Chem 1991, 23, 13–19. [Google Scholar]
- 17.Segal BM; Hoveyda HR; Holm RH, Terminal Ligand Assignments Based on Trends in Metal−Ligand Bond Lengths of Cubane-Type [Fe4S4]2+,+Clusters. Inorg. Chem 1998, 37, 3440–3443. [Google Scholar]
- 18.Di Vaira M; Midollini S; Sacconi L, Synthesis, Properties, and X-Ray Characterization of 3d Metal-Mercapto and -Methylthio Complexes with the Poly(tertiary phosphines) Tris(2-diphenylphosphinoethyl)amine and Tris(2-diphenylphosphinoethyl)phosphine. Inorg. Chem 1977, 16, 1518–1524. [Google Scholar]
- 19.Tsou C-C; Chiu W-C; Ke C-H; Tsai J-C; Wang Y-M; Chiang M-H; Liaw W-F, Iron(III) Bound by Hydrosulfide Anion Ligands: NO-Promoted Stabilization of the [FeIII–SH] Motif. J. Am. Chem. Soc 2014, 136, 9424–9433. [DOI] [PubMed] [Google Scholar]
- 20.Deponte M, Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochimi. Biophys. Acta 2013, 1830, 3217–3266. [DOI] [PubMed] [Google Scholar]
- 21.Powis G; Montfort WR, Properties and Biological Activities of Thioredoxins. Ann. Rev. Biophys. Biomolec. Struct 2001, 30, 421–455. [DOI] [PubMed] [Google Scholar]
- 22.Westmeyer MD; Rauchfuss TB; Verma AK, Iron Sulfido Derivatives of the Fullerenes C60 and C70. Inorg. Chem 1996, 35, 7140–7147. [DOI] [PubMed] [Google Scholar]
- 23.Gao W; Yang JYJ, Synthesis and crystal structure of complex (μ-S2)Fe2(CO)4(PPh3)2. Asian J. Chem 2013, 25, 9755–9757. [Google Scholar]
- 24.Farrugia LJ; Evans C; Senn HM; Hanninen MM; Sillanpaa R, QTAIM View of Metal-Metal Bonding in Di- and Trinuclear Disulfido Carbonyl Clusters. Organometallics 2012, 31, 2559–2570. [Google Scholar]
- 25.Zhuang B; Chen J; He L; Chen J; Zhou Z; Wu K, Synthesis, Structure and Formation Pathways of New Fe–S Complexes Containing [Fe2S2]-Units in Different Valences, [Fe2S2(CO)4(PPh3)2], [Fe3S2(CO)6(PPh3)3] and [Fe4S2(CO)10]2− and the Origin of the [Fe2S2]-unit in Metal–[Fe2S2(CO)6] Complexes. J. Organomet. Chem 2004, 689, 2674–2683. [Google Scholar]
- 26.Liu C; Peck JNT; Wright JA; Pickett CJ; Hall MB, Density Functional Calculations on Protonation of the [FeFe]-Hydrogenase Model Complex Fe2(μ-pdt)(CO)4(PMe3)2 and Subsequent Isomerization Pathways. Eur. J. Inorg. Chem 2011, 1080–1093. [Google Scholar]
- 27.Jablonskyte A; Webster LR; Simmons TR; Wright JA; Pickett CJ, Electronic Control of the Protonation Rates of Fe-Fe Bonds. J. Am. Chem. Soc 2014, 136, 13038–13044. [DOI] [PubMed] [Google Scholar]
- 28.Zhao X; Georgakaki IP; Miller ML; Mejia-Rodriguez R; Chiang C-Y; Darensbourg MY, Catalysis of H2/D2 Scrambling and Other H/D Exchange Processes by [Fe]-Hydrogenase Model Complexes. Inorg. Chem 2002, 41, 3917–3928. [DOI] [PubMed] [Google Scholar]
- 29.Savariault J-M; Bonnet J-J; Mathieu R; Galy J, Étude Cristallographique du Composé {[(SCH3)Fe(CO)2P(CH3)2(C6H5)]2H}+PF6-. C. R. Acad. Sci. Paris 1977, 284, 663–667. [Google Scholar]
- 30.Barton BE; Zampella G; Justice AK; De Gioia L; Rauchfuss TB; Wilson SR, Isomerization of the Hydride Complexes [HFe2(SR)2(PR3)x(CO)6-x]+ (x = 2, 3, 4) Relevant to the Active Site Models for the [FeFe]-Hydrogenases. Dalton Trans 2010, 39, 3011–3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brunner H; Wachter J; Gehart G; Leblanc J-C; Moïse C, Preparation and Reactivity of Peralkylated Tantalocene Sulfur Complexes Having a Fulvenoid Substructure. Organometallics 1996, 15, 1327–1330. [Google Scholar]
- 32.Li Q; Lalaoui N; Woods TJ; Rauchfuss TB; Arrigoni F; Zampella G, Electron-Rich, Diiron Bis(monothiolato) Carbonyls: C-S Bond Homolysis in a Mixed Valence Diiron Dithiolate. Inorg. Chem 2018, 57, 4409–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song L-C; Zhao P-H; Du Z-Q; Tang M-Y; Hu Q-M, Unexpected Synthesis of Tetrahedral Fe/S Clusters via Highly Reactive Butterfly Intermediates (μ-HS)2Fe2(CO)5[RP(CH2OH)2]. Organometallics 2010, 29, 5751–5753. [Google Scholar]
- 34.Keizer PN; Krusic PJ; Morton JR; Preston KF, Thiolato- and Selenato-Bridged Dinuclear Iron Carbonyl Radicals. J. Am. Chem. Soc 1991, 113, 5454–5456. [Google Scholar]
- 35.King RB, Organosulfur Derivatives of Metal Carbonyls. I. The Isolation of Two Isomeric Products in the Reaction of Triiron Dodecacarbonyl with Dimethyl Disulfide. J. Am. Chem. Soc 1962, 84, 2460. [Google Scholar]
- 36.Mueting A; Mattson BM, Kinetics and Thermodynamics of the Intramolecular Isomerization of [Fe(SCH3)(CO)3]2. J. Inorg. Nucl. Chem 1981, 43, 749–751. [Google Scholar]
- 37.Seyferth D; Henderson RS; Song L-C, Chemistry of μ-Dithio-bis(tricarbonyliron), a Mimic of Inorganic Disulfides. 1. Formation of Di-μ-thiolato-bis(tricarbonyliron) Dianion. Organometallics 1982, 1, 125–133. [Google Scholar]
- 38.Dahl LF; Wei C-H, Structure and Nature of Bonding of [C2H5SFe(CO)3]2. Inorg. Chem 1963, 2, 328–333. [Google Scholar]
- 39.Ortega-Alfaro MC; Hernandez N; Cerna I; Lopez-Cortes JG; Gomez E; Toscano RA; Alvarez-Toledano C, Novel Dinuclear Iron(0) Complexes from a,b-Unsaturated Ketones b-Positioned with Sulfide and Sulfoxide Groups. J. Organomet. Chem 2004, 689, 885–893. [Google Scholar]
- 40.Shi Y-C; Wu Z-D; Hou X-L; Li Z-W; Wang Y, Syntheses, Crystal Structures, and Electrochemical Studies of Dinuclear Coordination Compounds with the Fe2(CO)6 Core. J. Coord. Chem 2016, 69, 3603–3618. [Google Scholar]
- 41.Song L-C; Zhao Z-Y; Wang J-T, Synthesis and Conformational Analysis of α-Ethoxycarbonylmethyl-Substittuted Sulfur-Bridged Iron Carbonyl Complexes (μ-RS)(μ-EtOC(O)CH2S)Fe2(CO)6. Acta Chim. Sin 1987, 1, 80–84 (Chinese edition 1987, 1945, 1467─1471). [Google Scholar]
- 42.Casewit CJ; Coons DE; Wright LL; Miller WK; Rakowski DuBois M, Homogeneous Reductions of Nitrogen-Containing Substrates Catalyzed by Molybdenum(IV) Complexes with μ-Sulfido Ligands. Organometallics 1986, 5, 951–955. [Google Scholar]
- 43.Warren JJ; Tronic TA; Mayer JM, Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev 2010, 110, 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Franz JA; Lee S-J; Bowden TA; Alnajjar MS; Appel AM; Birnbaum JC; Bitterwolf TE; Dupuis M, Activation of the S-H Group in Fe(μ2-SH)Fe Clusters: S-H Bond Strengths and Free Radical Reactivity of the Fe(μ2-SH)Fe Cluster. J. Am. Chem. Soc 2009, 131, 15212–15224. [DOI] [PubMed] [Google Scholar]
- 45.Gao W; Zhang T-T; Sun Y-J, Synthesis and Structural Characterization of Diiron Ethanedithiolate Complex [(μ-SCH2)2Fe2(CO)4](PPh3)2 Related to the Active Site of [Fe-Fe]-Hydrogenases. Asian J. Chem 2014, 26, 6687–6688. [Google Scholar]
- 46.Kramer A; Lingnau R; Lorenz IP; Mayer HA, Komplexchemischer Aufbau cyclischer 1,2-Dithiolato-Liganden mit dem nido Cluster [(CO)3FeS]2. - Molekülstruktur und partielle S-Oxidation von Hexacarbonyl(mu2-cis-1,2-cyclohexandithiolato-S,S}dieisen. Chem. Ber 1990, 123, 1821–1826. [Google Scholar]
- 47.Kramer A; Lorenz IP, Photochemisch induzierte [2 + 2]-Cycloadditionen von Alkenen und Dienen mit der S-S-Bindung des nido-Clusters [(CO)3FeS]2. J. Organomet. Chem 1990, 388, 187–193. [Google Scholar]
- 48.Hieber W; Gruber J, Zur Kenntnis der Eisencarbonylchalkogenide. Z. anorg. allg. Chem 1958, 296, 91–103. [Google Scholar]
- 49.Bose KS; Sinn E; Averill BA, Synthesis and X-Ray Structure of the [Fe4S4(CO)12]2- Ion: an Example of intermolecular Sisulfide Formation by the (μ-S)2Fe2(CO)6 Unit. Organometallics 1984, 3, 1126–1128. [Google Scholar]
- 50.Felton GAN; Mebi CA; Petro BJ; Vannucci AK; Evans DH; Glass RS; Lichtenberger DL, Review of Electrochemical Studies of Complexes Containing the Fe2S2 Core Characteristic of [FeFe]-Hydrogenases Including Catalysis by These Complexes of the Reduction of Acids to Form Dihydrogen. J. Organomet. Chem 2009, 694, 2681–2699. [Google Scholar]
- 51.Silaghi-Dumitrescu I; Bitterwolf TE; King RB, Butterfly Diradical Intermediates in Photochemical Reactions of Fe2(CO)6(μ-S2). J. Am. Chem. Soc 2006, 128, 5342–5343. [DOI] [PubMed] [Google Scholar]
- 52.Seyferth D; Henderson RS, Photochemically Induced Insertion of Acetylenes into μ-Dithiobis(tricarbonyliron). J. Organomet. Chem 1979, 182, C39–C42. [Google Scholar]
- 53.Messelhäuser J; Gutensohn KU; Lorenz IP; Hiller W, Insertionreaktionen von Ethen und Kohlenmonoxid in die S-S Bindung des [(CO)3FeS]2 nido-Clusters und Synthese und Struktur des 1,3-Ethanesulfenatothiolato Complex [(CO)3Fe]2SC2H4S(O). J. Organomet. Chem 1987, 321, 377–388. [Google Scholar]
- 54.Zhao P; Gray DL; Rauchfuss TB, Rational Synthesis of the Carbonyl(perthiolato)diiron [Fe2(S3CPh2)(CO)6] and Related Complexes. Eur. J. Inorg. Chem 2016, 2016, 2681–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.SAINT SHELXTL, XCIF XPREP, Bruker AXS, Inc, Madison, Wisconsin, 2014. [Google Scholar]
- 56.Krause L; Herbst-Irmer R; Sheldrick GM; Stalke D, Comparison of Silver and Molybdenum Microfocus X-Ray Sources for Single-Crystal Structure Determination. J. Appl. Cryst, 2015, 48, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sheldrick GM, Crystal Structure Refinement with SHELXL. Acta Cryst 2015, C71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sheldrick GM, SHELXT - Integrated Space-Group and Crystal-Structure Determination. Acta Cryst 2015, A71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.