

Abstract
Enantioenriched molecules bearing indole-substituted stereocenters form a class of privileged compounds in biological, medicinal, and organic chemistry. Thus, the development of methods for asymmetric indole alkylation is highly valuable in organic synthesis. Traditionally, achieving N-selectivity in indole alkylation reactions is a significant challenge, since there is an intrinsic preference for alkylation at C3, the most nucleophilic position. Furthermore, selective and predictable access to either N- or C3-alkylated chiral indoles using catalyst control has been a long-standing goal in indole functionalization. Herein, we report a ligand-controlled regiodivergent synthesis of N- and C3-alkylated chiral indoles that relies on a polarity reversal strategy. In contrast to conventional alkylation reactions in which indoles are employed as nucleophiles, this transformation employs electrophilic indole derivatives, N-(benzoyloxy)indoles, as coupling partners. N- or C3-alkylated indoles are prepared with high levels of regio- and enantioselectivity using a copper hydride catalyst. The regioselectivity is governed by the use of either DTBM-SEGPHOS or Ph-BPE as the supporting ligand. Density functional theory (DFT) calculations are conducted to elucidate the origin of the ligand-controlled regiodivergence.
Graphical Abstract
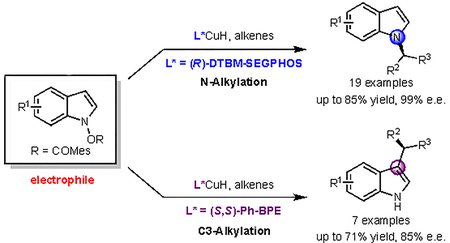
Introduction
Enantiomerically enriched indole derivatives are ubiquitous in biologically active natural products, and widely recognized as privileged components in pharmacologically relevant compounds (Figure 1a).1,2 Therefore, the development of techniques for the efficient enantioselective synthesis of indoles is a prominent objective in organic synthesis. A few powerful methods have been developed recently to access chiral alkylated indoles. In these transformations, the indole derivatives generally serve as the nucleophilic coupling fragments, reacting with a variety of electrophiles, including activated olefins, ketones, imines, allylic alcohol derivatives, and alkynes.3–5 Most alkylation reactions take place largely or entirely at C3, due to the higher nucleophilicity at this position (Figure 1b).6,7
Figure 1. Design of a CuH-catalyzed indole alkylation reaction using electrophilic indole reagents.

a) Representative biologically active N- and C3-substituted α-chiral indoles. b) Regioselectivity in conventional enantioselective indole alkylation reactions. c) Proposed mechanistic pathway of enantioselective alkylation of electrophilic indoles. d) Ligand-controlled regiodivergent alkylation of electrophilic indoles.
The development of methods that generate enantioenriched N-alkylated indoles has been an active area of research for the past few years. The majority of these processes involve the enantioselective N-allylation of indoles with electron-withdrawing groups or with substituents blocking the C3 position. In addition, two-step procedures including N-allylation/oxidation of indolines and N-allylation/Fischer indolization of aryl hydrazines have been developed.8–13 Despite these notable advances, methods for highly enantioselective N-alkylation (non-allylic alkylation) of indoles with broad substrate scope remain rare.14,15 The difficulty in controlling regioselectivity in indole alkylation reactions originates from the nucleophilic character of the indole, with the C3 position being much more nucleophilic than N or other positions. We hypothesized that if the indole could be employed as an electrophile instead of as a nucleophile, alkylation reactions could potentially occur at positions other than C3.
During the past few years, CuH catalysis16–18 has emerged as an efficient method for the enantioselective formation of C–N and C–C bonds. In these reactions, enantioenriched alkylcopper(I) intermediates such as II (Figure 1c) are generated from the reaction of alkenes and a CuH catalyst ligated by a chiral phosphine ligand.19,20 The alkylcopper(I) species can then be efficiently trapped by electrophiles such as electrophilic amines,21–25 ketones,26–28 imines,29,30 allylic phosphate,31–33and other reagents.34–38 We felt that these alkylcopper(I) species could act as nucleophiles toward appropriate indole electrophiles, providing alkylated indoles potentially with high levels of enantioselectivity and chemoselectivity. Among a variety of possible classes of electrophilic indoles,39 we considered N-hydroxyindole derivatives to be promising reagents for our proposed transformation (Figure 1c, electrophilic indole). N-hydroxyindole derivatives with a variety of leaving groups (e.g., OH, OMe, OBz, OTs) have been prepared and their nucleophilic substitution reactions have been studied.40,41 The ability of these to react with nucleophiles at N-,42 C2-,43 and C3-positions44 of indoles has been demonstrated, although not in a catalytic, enantioselective manner. In order to be useful, the chosen electrophilic indole reagents need to be stable under reductive reaction conditions in the presence of LCuH, but reactive enough to productively interact with the alkylcopper(I) intermediate. By analogy to previous CuH-catalyzed hydroamination reactions that use (benzoyloxy)amine derivatives as aminating reagents,21 we postulated that N-(benzoyloxy)indole derivatives (e.g., Figure 1c, electrophilic indoles, R = Bz) might be suitable coupling partners.
Herein, we report a CuH-catalyzed enantioselective alkylation of indoles using a polarity reversal (umpolung) strategy.45 In this method, electrophilic indole derivatives (N-(2,4,6-trimethylbenzoyloxy)indoles) are employed as starting materials. With a DTBM-SEGPHOS-modified CuH catalyst, N-alkylated indoles, which are difficult to access by other methods, can be efficiently synthesized with high levels of regio- and enantioselectivity (Figure 1d, blue). During the course of this study, we unexpectedly found that the regioselectivity (N/C3) of this process is ligand-controlled. By switching the ligand from DTBM-SEGPHOS to Ph-BPE, enantioenriched C3-alkylated indoles could be selectively accessed (Figure 1d, purple). Using DFT calculations, we have proposed a model for the regioselectivity based on the structure of the ligands in the regiochemistry-determining transition states.
Results and discussion
Reaction development and optimization
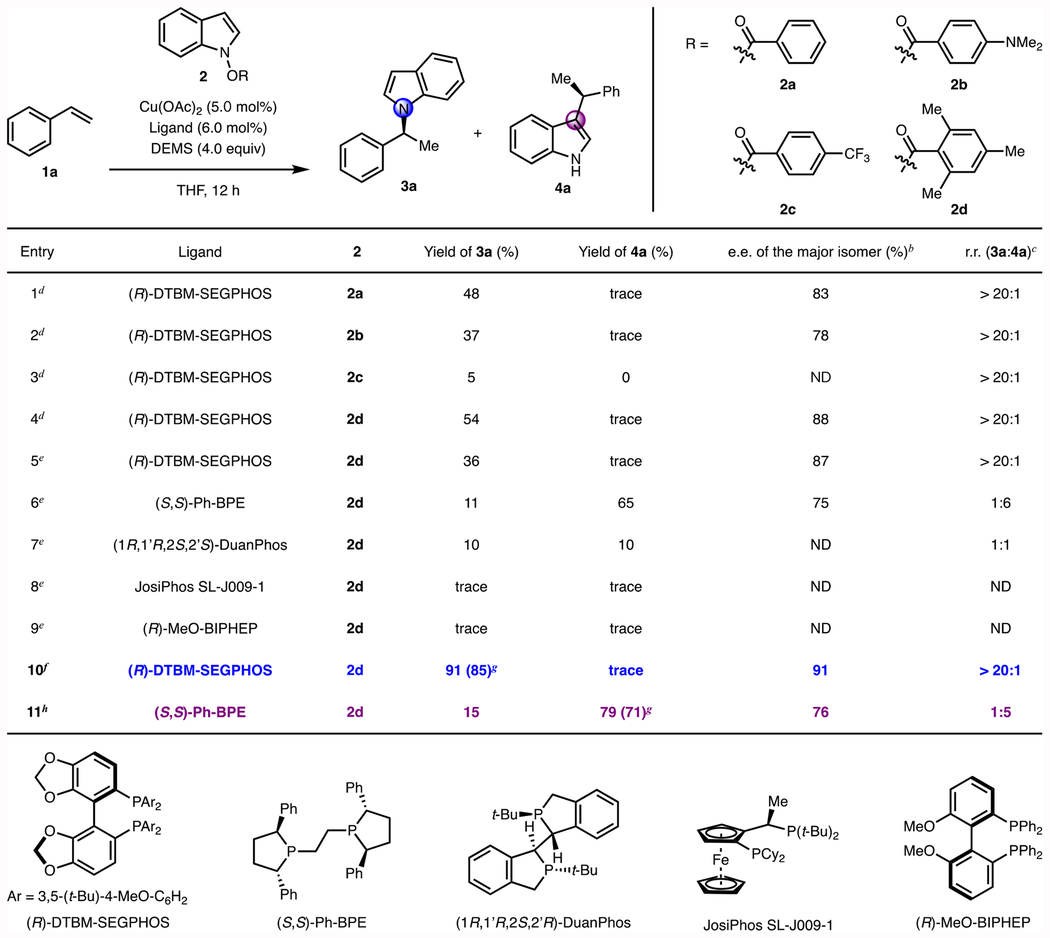
We reasoned that the stability and reactivity of the electrophilic indole reagent could be modulated by tuning the leaving group as we had seen before.46 Thus, N-(benzoyloxy)indole derivatives with different benzoate substituents were prepared (Table 1, 2a, 2b, 2c, 2d). The feasibility of the alkylation process was then investigated using styrene as the pronucleophile, copper(II) acetate as the precatalyst, and DTBM-SEGPHOS as the ligand. For all four electrophiles evaluated, the corresponding N-alkylated indole was generated with excellent regioselectivity (>20:1 N:C3) and with good enantioselectivity (Table 1, entries 1–4). Among the indole electrophiles tested, the use of N-(2,4,6-trimethylbenzoyl)indole (2d) provided the best yield and the highest enantiometric excess. We hypothesized that the steric hindrance provided by the two ortho-methyl groups on the benzoate slows direct reductive cleavage of the N–O bond by LCuH, but still allows the reagent to undergo the desired C–N bond formation at elevated temperatures.
Table 1.
Optimization of CuH-catalyzed enantioselective N- and C3-alkylation of indole derivatives.a
|
Reactions were conducted on 0.10 mmol scale. Yields were determined by gas chromatography using dodecane as internal standard.
The ee was determined by SFC analysis.
The regioisomeric ratio (rr) was determined by GC analysis of the crude reaction mixture.
Conditions: 1 (0.10 mmol), 2 (0.10 mmol), Cu(OAc)2 (5.0 mol%), (R)-DTBM-SEGPHOS (6.0 mol%), DEMS (4.0 equiv), THF (0.1 M), 90 °C, 12 h.
Conditions: 1 (0.15 mmol), 2 (0.10 mmol), Cu(OAc)2 (5.0 mol%), ligand (6.0 mol%), DEMS (4.0 equiv), THF (0.5 M), 50 °C, 12 h.
Conditions: 1 (0.10 mmol), 2 (0.15 mmol), Et3COH (0.02 mmol), Cu(OAc)2 (5.0 mol%), (R)-DTBM-SEGPHOS (6.0 mol%), DEMS (4.0 equiv), THF (0.1 M), 90 °C, 20 h.
Isolated yield on a 0.50 mmol scale.
Conditions: 1 (0.20 mmol), 2d (0.10 mmol), Et3COH (0.02 mmol), Cu(OAc)2 (1.0 mol%), (S,S)-Ph-BPE (1.2 mol%), DMMS (4.0 equiv), THF (0.5 M), 40 °C, 24 h. ND = Not determined. DEMS = (EtO)2MeSiH. DMMS = (MeO)2MeSiH.
We found that the regioselectivity of this reaction was sensitive to the choice of ligand. When DTBM-SEGPHOS was replaced with Ph-BPE, instead of, 3a, which arises from carbon-nitrogen bond formation, 4a, resulting from carbon-carbon bond formation at C3 was the predominant product. (Table 1, entries 5 and 6). Catalysts based on DuanPhos provided the alkylated indoles in diminished yields and with poor selectivity (Table 1, entry 7). Other phosphine ligands, such as a Josiphos derivative and MeO-BIPHEP, showed limited ability to facilitate the reaction (Table 1, entries 8 and 9).
After further optimization of the reaction parameters (Table S1 and S2 in the Supporting Information), chiral N-alkylated indole 3a could be accessed in 85% yield with 91% ee and >20:1 regioselectivity using DTBM-SEGPHOS as the supporting ligand (Table 1, entry 10). C3-alkylated indole 4a could be isolated in 71% yield with 76% ee and >5:1 regioselectivity using Ph-BPE as the ligand (Table 1, entry 11). As we have previously seen, the inclusion of an alcohol as an additive was found to improve the efficiency of both transformations.29
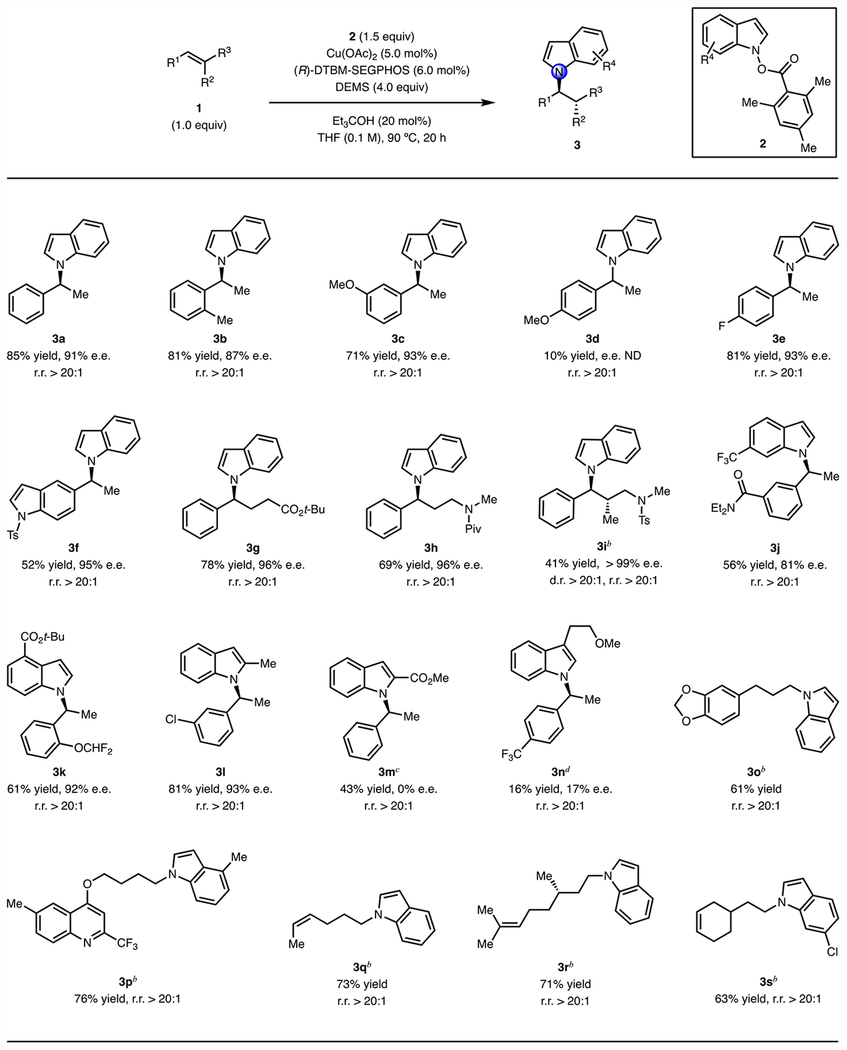
Substrate scope of the N-alkylation reaction.
Using the optimized reaction conditions described above, we evaluated a range of styrenes as substrates (Table 2). In all cases with DTBM-SEGPHOS as the supporting ligand, we observed that the N-to-C3 selectivity was greater than 20:1. Styrenes bearing ortho- (3b, 3k), meta- (3c, 3j, 3l), and para-substituents (3e) were all suitable, yielding the desired N-alkylated indoles with high efficiency and high levels of enantioselectivity. Electron-withdrawing groups on the aryl ring of the styrenes are tolerated (3c, 3j, 3k, 3l), while an electron-donating group (3d) slowed down the reaction.47a trans-β-Substituted styrenes were also successfully transformed using this protocol (3g, 3h). In particular, 3h, the derivative of an important serotonin reuptake inhibitor, was efficiently prepared in a reaction that proceeded with excellent levels of regio- and enantioselectivity. A more sterically hindered β,β-substituted styrene was transformed to the desired product (3i) in moderate yield as a single diastereomer.
Table 2.
Substrate scope of CuH-catalyzed enantioselective N-alkylation.a
|
Conditions: 1 (0.50 mmol), 2 (0.75 mmol), Et3COH (0.10 mmol), Cu(OAc)2 (5.0 mol%), (R)-DTBM-SEGPHOS (6.0 mol%), DEMS (4.0 equiv), THF (0.1 M), 90 °C, 20 h. The ee was determined by SFC analysis. The regioisomeric ratio (rr) was determined by GC analysis of the crude reaction mixture.
Conditions: 1 (0.75 mmol), 2 (0.50 mmol), KF (0.10 mmol), Cu(OAc)2 (5.0 mol%), (R)-DTBM-SEGPHOS (6.0 mol%), DEMS (4.0 equiv), THF (0.1 M), 70 °C, 20 h.
Using 1,4-dioxane instead of THF.
Conditions: 1 (0.50 mmol), 2 (1.5 mmol), Et3COH (0.10 mmol), Cu(OAc)2 (5.0 mol%), (R)-DTBM-SEGPHOS (6.0 mol%), DEMS (4.0 equiv), 1,4-dioxane (0.1 M), 90 °C, 20 h. ND = Not determined. DEMS = (EtO)2MeSiH.
Terminal aliphatic alkenes could also be employed as substrates (3o, 3p). In these cases, the reaction exhibited a nearly complete change in regioisomeric preference toward the anti-Markovnikov product. Monosubstituted C=C double bonds underwent the transformation selectively (3q, 3r, 3s), in the presence of cis-disubstituted or trisubstituted olefins.
We next surveyed the scope of indole electrophiles that could be used in the N-alkylation reaction. It was found that a variety of functional groups were accommodated at different positions on the benzene ring of the indole, including a 6-trifluoromethyl (3j), a 4-tert-butyl ester (3k), and a 6-chloro (3s) substituent. Alkyl groups at the 2- and 4-position of the indole electrophile were tolerated and the corresponding products were synthesized in good yield (3l, 3p) with excellent enantioselectivity in the case of (3l). An indole electrophile with a 2-carbomethoxy substituent exhibited low reactivity under the standard reaction conditions (10% conversion). However, by using an excess of the styrene substrate and employing dioxane as solvent, the desired product was obtained in moderate yield (3m) although in racemic form.47b Indole electrophiles bearing substituents at the 3-position were generally poor substrates in this N-alkylation reaction. In the case shown in Table 2, the desired product was generated in low yield with a low level of enantioselectivity (3n).48
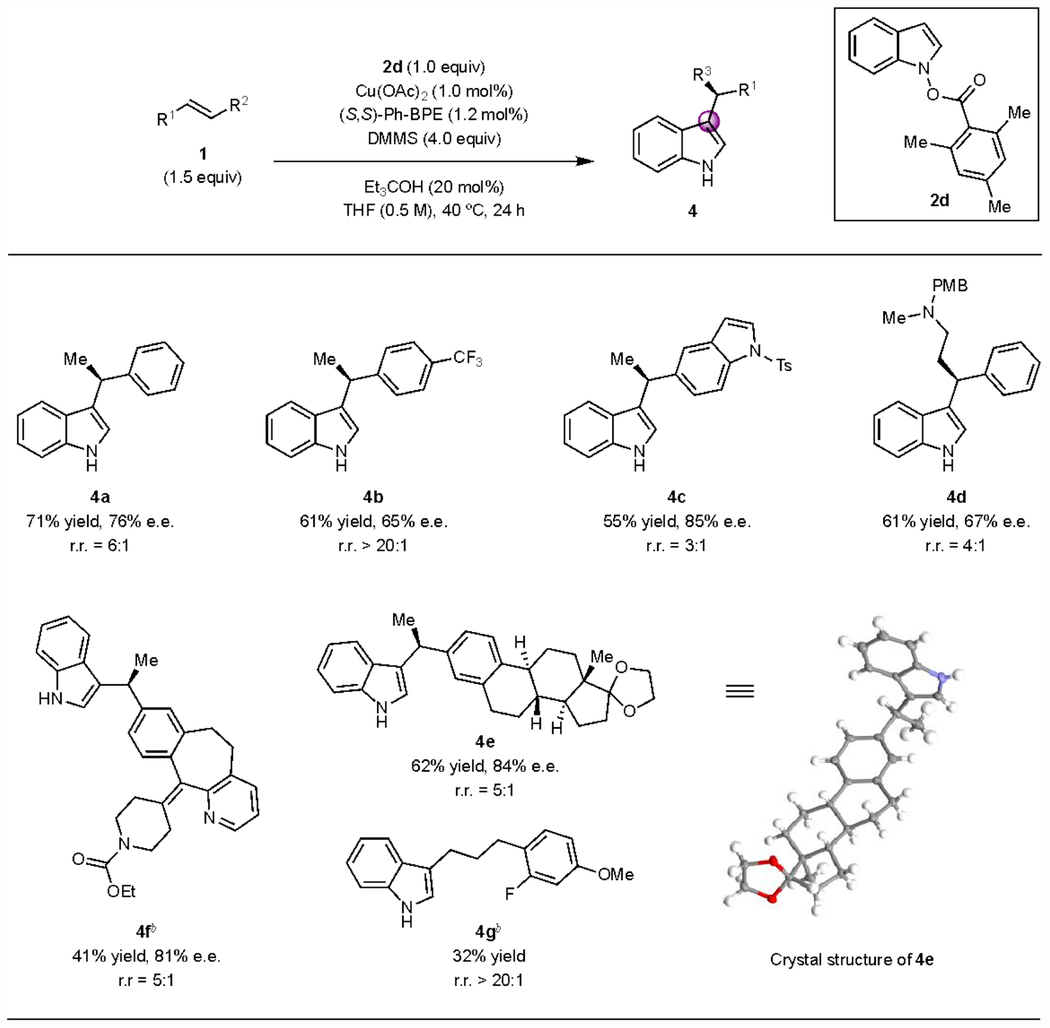
Substrate scope of the C3-alkylation reaction.
Using the reaction conditions that favor carbon-carbon bond formation, (i.e., with Ph-BPE) a number of C3-alkylated indoles were prepared in moderate-to-good yields and with useful levels of enantiomeric excess (Table 3). Styrene bearing a 4-trifluoromethyl substituent was found to be a suitable substrate in this transformation, providing the desired indole product (4b) in moderate yield with a high level of regioselectivity. The lower enantioselectivity observed compared to 4a can reasonably be ascribed to the relatively fast epimerization of the electron-deficient alkylcopper species.20,49 In contrast, 1-tosyl-5-vinylindole, a relatively electron-rich alkene, underwent the transformation with lower regioselectivity but higher enantioselectivity (4c). A trans-β-substituted styrene was also effectively transformed by this protocol (4d). To further demonstrate the synthetic utility of this method, chiral C3-alkylated indoles derived from estrone and loratadine (a common antihistamine agent) were prepared with good regio- and enantioselectivities (4e, 4f). In addition to styrenes, a terminal aliphatic olefin was used as the coupling partner, generating the corresponding product in a moderate yield with excellent regioselectivity (4g, >20:1).
Table 3.
Substrate scope of CuH-catalyzed enantioselective C3-alkylation.a
|
Conditions: 1 (1.0 mmol), 2d (0.50 mmol), Et3COH (0.10 mmol), Cu(OAc)2 (1.0 mol%), (S,S)-Ph-BPE (1.2 mol%), DMMS (4.0 equiv), THF (0.5 M), 40 °C, 24 h. The ee was determined by SFC analysis. The regioisomeric ratio (rr) was determined by 1H NMR analysis of the crude reaction mixture. DMMS = (MeO)2MeSiH.
Using 5.0 mol% Cu(OAc)2 and 6.0 mol% (S,S)-Ph-BPE instead of 1.0 mol% Cu(OAc)2 and 1.2 mol% (S,S)-Ph-BPE.
Mechanistic discussion
DFT calculations were performed to construct a mechanistic model that explains the regiodivergence observed in the reaction. Since the SEGPHOS ligand displays the same regioselectivity as DTBM-SEGPHOS (>20:1 N:C3), we used SEGPHOS in the calculations to reduce the computational cost. In addition, styrene and 2d were selected as model substrates. The olefin insertion step, which is responsible for determining the enantioselectivity, is well-understood24,27 and thus, we focused on subsequent steps in the mechanism to explain the regioselectivity.50
The alkylcopper(I) complex II can form either the N-indoyl copper(III) complex IIIN (Figure 2a, N-oxidative addition) or the C3-indoyl copper(III) complex IIIC (Figure 2a, C3-oxidative addition) through the oxidative addition transition states II-TS and II-TS’, respectively. Subsequent reductive elimination affords the products 3a and 4a, respectively; in the latter case after tautomerization of the initially formed intermediate 4a’. An intramolecular interconversion between IIIN and IIIC through intermediate IV, which leads to decreased regioselectivity, was also considered.
Figure 2.

Proposed regiochemistry-determining reaction pathway. The mesitoate anion is omitted for clarity in the many of the structures.
Figure 2b illustrates the most important portion of the reaction energy profiles for the SEGPHOS and Ph-BPE ligands (subscripts S and P indicate SEGPHOS and Ph-BPE as the supporting ligand, respectively). In good agreement with the experimental observation of excellent regioselectivity of greater than 20:1 (N:C3), the alkylcopper(I) complex IIS bearing the SEGPHOS ligand is predicted to prefer oxidative addition at N rather than at C3. These processes proceed through the transition states IIS-TS and IIS-TS’ at 23.4 and 26.3 kcal/mol, respectively (Figure 2b, SEGPHOS). In addition, the intramolecular interconversion from IIISN to IIISC is predicted to be insignificant, since the barrier of 1,3-migration (from IIISN through the transition states IIISN-TS’) was found to be 17.8 kcal/mol higher in energy than the reductive elimination barrier (from IIISN through the transition states IIISN-TS).
When the SEGPHOS ligand is replaced with Ph-BPE, oxidative addition is instead slightly preferred at C3 rather than N: the transition state IIP-TS’ at 19.9 kcal/mol is lower in energy than IIP-TS at 20.9 kcal/mol (Figure 2b, Ph-BPE). In this case, the barrier of 1,3-migration from IIIPC to IIIPN is comparable with the reductive elimination barrier to generate 4a. The small preference for the C3-alkylation regioisomer, and the competitive 1,3-migration are in qualitative agreement with experimental results, which showed moderate levels of regioselectivity (~5:1 C3:N).
We hoped to identify the origins of this ligand effect at a more fundamental level. Therefore, we compared the structures of the alkylcopper(I) intermediates supported by Ph-BPE (IIP) and SEGPHOS (IIS), respectively. As shown in Figure 2c, the phenyl group in the Ph-BPE ligand, highlighted in green, rests above an open coordination site of the copper in IIP, whereas the metal center in the intermediate IIS is relatively unhindered and freely accessible. During the N-oxidative addition with Ph-BPE ligand (Figure 2c, IIP-TS), when catalyst IIP engages the indole substrate at the N-position (blue), the benzoate ligand (red) is in close proximity to the ligand phenyl group (green), which gives rise to unfavorable steric interactions. In contrast, C3-oxidative addition can be accomplished without such steric crowding, because the benzoate fragment can point away from the Ph-BPE ligand, as shown in the computed structure of IIP-TS’.
These steric considerations are less important in the SEGPHOS case. Thus the N–O oxidative insertion, which appears to be favored by the intrinsic electrophilicity of the indole electrophile, takes place (Figure 2c, IIS-TS vs. IIS-TS’). To confirm our hypotheses that the N-oxidative addition transition state is destabilized by steric interactions when Ph-BPE is used rather than SEGPHOS, an energy decomposition analysis (see the Supporting Information for details) was used to compare transition states IIP-TS and IIP-TS’. The distortion energy on the phosphine fragment is estimated to be ~5 kcal/mol higher in IIP-TS than in IIP-TS’. This difference is more significant than the intrinsic energetic advantages of N-oxidative addition over C3-oxidative addition, and steric effects are able to reverse this preference.
Conclusion
In summary, we have developed an enantioselective CuH-catalyzed process to access either N- or C3-alkylated indoles depending on the choice of ligand. In contrast to conventional indole functionalizations in which indoles are used as nucleophiles, N-(benzoyloxy)indole derivatives are employed as electrophiles in this method. When the DTBM-SEGPHOS is used as the supporting ligand, N-alkylated indoles with a variety of functional groups are prepared in good yields with high levels of regio- and enantioselectivity. When the Ph-BPE ligand is employed, chiral C3-alkylated indoles can be accessed as well. We anticipate that the application of this polarity reversal strategy is not limited to indole alkylation but may be further extended to the catalytic functionalization of other important electron-rich nitrogen heterocycles.
Supplementary Material
Figure 3.

Energy profiles of the oxidative addition and reductive elimination steps with 2d and alkylcopper(I) complex II supported by SEGPHOS and Ph-BPE, respectively. (Subscripts S and P indicate SEGPHOS and Ph-BPE as the supporting ligand, respectively).
Figure 4.

The optimized structures of II, II-TS, and II-TS’ with Ph-BPE and SEGPHOS as the ligand, respectively. The phenyl group in the Ph-BPE ligand obscuring the copper center is displayed in green. (Subscripts S and P indicate SEGPHOS and Ph-BPE as the supporting ligand, respectively)
Acknowledgements
The authors are grateful to Solvias AG for donations of ligands used in this project. We are grateful to the National Institutes of Health (R35-GM122483), and the Institute for Basic Science (IBS-R010-D1) in Korea. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Health. We thank Mr. Richard Liu, Drs. Scott McCann, Christine Nguyen, and Andy Thomas for their advice on the preparation of this manuscript. We acknowledge Drs. Peter Müller (MIT) and Charlene Tsay (MIT) for X-ray crystallographic analysis of 4e. We thank Mr. Marek Domin (Boston College), and Dr. Zhenxing Liu (Boston College) for the help with the High Resolution Mass Spectrometry analysis. We thank National Institutes of Health for a supplemental grant for the purchase of supercritical fluid chromatography (SFC) equipment (GM058160–17S1).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental details and computational data (PDF)
Spectroscopic Data (PDF)
The authors declare no competing financial interests.
References
- (1).Gul W; Hamann MT, Indole Alkaloid Marine Natural Products: An Established Source of Cancer Drug Leads with Considerable Promise for the Control of Parasitic, Neurological and Other Diseases. Life Sci. 2005, 78, 442–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sravanthi TV; Manju SL, Indoles–A Promising Scaffold for Drug Development. Eur. J. Pharm. Sci 2016, 91, 1–10. [DOI] [PubMed] [Google Scholar]
- (3).Chen JB; Jia YX, Recent Progress in Transition-Metal-Catalyzed Enantioselective Indole Functionalizations. Org. Biomol. Chem 2017, 15, 3550–3567. [DOI] [PubMed] [Google Scholar]
- (4).Bandini M; Eichholzer A, Catalytic Functionalization of Indoles in a New Dimension. Angew. Chem. Int. Ed 2009, 48, 9608–9644. [DOI] [PubMed] [Google Scholar]
- (5).Cruz FA; Zhu Y; Tercenio QD; Shen Z; Dong VM, Alkyne Hydroheteroarylation: Enantioselective Coupling of Indoles and Alkynes via Rh-Hydride Catalysis. J. Am. Chem. Soc 2017, 139, 10641–10644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Lakhdar S; Westermaier M; Terrier F; Goumont R; Boubaker T; Ofial AR; Mayr H, Nucleophilic Reactivities of Indoles. J. Org. Chem 2006, 71, 9088–9095. [DOI] [PubMed] [Google Scholar]
- (7).Otero N; Mandado M; Mosquera RA, Nucleophilicity of Indole Derivatives: Activating and Deactivating Effects Based on Proton Affinities and Electron Density Properties. J. Phys. Chem. A 2007, 111, 5557–5562. [DOI] [PubMed] [Google Scholar]
- (8).Stanley LM; Hartwig JF, Iridium-Catalyzed Regio- and Enantioselective N-allylation of Indoles. Angew. Chem. Int. Ed 2009, 48, 7841–7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Trost BM; Krische MJ; Berl V; Grenzer EM, Chemo-, Regio-, and Enantioselective Pd-Catalyzed Allylic Alkylation of Indolocarbazole Pro-Aglycons. Org. Lett 2002, 4, 2005–2008. [DOI] [PubMed] [Google Scholar]
- (10).Trost BM; Osipov M; Dong G, Palladium-Catalyzed Dynamic Kinetic Asymmetric Transformation of Vinyl Aziridines with Nitrogen Heterocycles: Rapid Access to Biologically Active Pyrroles and Indoles. J. Am. Chem. Soc 2010, 132, 15800–15807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cui H-L; Feng X; Peng J; Lei J; Jiang K; Chen Y-C, Chemoselective Asymmetric N-allylic Alkylation of Indoles with Morita-Baylis-Hillman Carbonates. Angew. Chem. Int. Ed 2009, 48, 5737–5740. [DOI] [PubMed] [Google Scholar]
- (12).Liu W; Zhang X; Dai L; You S, Asymmetric N-Allylation of Indoles through the Iridium-Catalyzed Allylic Alkylation/Oxidation of Indolines. Angew. Chem. Int. Ed 2009, 51, 5183–5187. [DOI] [PubMed] [Google Scholar]
- (13).Xu K; Gilles T; Breit B, Asymmetric Synthesis of N-Allylic Indoles via Regio- and Enantioselective Allylation of Aryl Hydrazines. Nat. Commun 2015, 6, 7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Sevov CS; Zhou J; Hartwig JF, Iridium-Catalyzed, Intermolecular Hydroamination of Unactivated Alkenes with Indoles. J. Am. Chem. Soc 2014, 136, 3200–3207. [DOI] [PubMed] [Google Scholar]
- (15).Bandini M; Eichholzer A; Tragni M; Umani-Ronchi A, Enantioselective Phase-Transfer-Catalyzed Intramolecular Aza-Michael Reaction: Effective Route to Pyrazino-Indole Compounds. Angew. Chem. Int. Ed 2008, 47, 3238–3241. [DOI] [PubMed] [Google Scholar]
- (16).Deutsch C; Krause N; Lipshutz BH, CuH-Catalyzed Reactions. Chem. Rev 2008, 108, 2916–2927. [DOI] [PubMed] [Google Scholar]
- (17).Lipshutz BH, Rediscovering Organocopper Chemistry Through Copper Hydride. It’s All About the Ligand. Synlett 2009, 509–524. [Google Scholar]
- (18).Jordan AJ; Lalic G; Sadighi JP, Coinage Metal Hydrides: Synthesis, Characterization, and Reactivity. Chem. Rev 2016, 116, 8318–8372. [DOI] [PubMed] [Google Scholar]
- (19).Bandar JS; Pirnot MT; Buchwald SL, Mechanistic Studies Lead to Dramatically Improved Reaction Conditions for the Cu-Catalyzed Asymmetric Hydroamination of Olefins. J. Am. Chem. Soc 2015, 137, 14812–14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Xi Y; Hartwig JF, Mechanistic Studies of Copper-Catalyzed Asymmetric Hydroboration of Alkenes. J. Am. Chem. Soc 2017, 139, 12758–12772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Pirnot M; Wang Y-M; Buchwald SL, Copper Hydride Catalyzed Hydroamination of Alkenes and Alkynes. Angew. Chem. Int. Ed 2016, 55, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Miki Y; Hirano K; Satoh T; Miura M, Copper-Catalyzed Intermolecular Regioselective Hydroamination of Styrenes with Polymethylhydrosiloxane and Hydroxylamines. Angew. Chem. Int. Ed 2013, 52, 10830–10834. [DOI] [PubMed] [Google Scholar]
- (23).Zhu S; Niljianskul N; Buchwald SL, Enantio- and Regioselective CuH-Catalyzed Hydroamination of Alkenes. J. Am. Chem. Soc 2013, 135, 15746–15749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Yang Y; Shi S-L; Niu D; Liu P; Buchwald SL, Catalytic Asymmetric Hydroamination of Unactivated Internal Olefins to Aliphatic Amines. Science 2015, 349, 62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Xi Y; Butcher TW; Zhang J; Hartwig JF, Regioselective, Asymmetric Formal Hydroamination of Unactivated Internal Alkenes. Angew. Chem. Int. Ed 2016, 55, 776–780. [DOI] [PubMed] [Google Scholar]
- (26).Saxena A; Choi B; Lam HW, Enantioselective Copper-Catalyzed Reductive Coupling of Alkenylazaarenes with Ketones. J. Am. Chem. Soc 2012, 134, 8428–8431. [DOI] [PubMed] [Google Scholar]
- (27).Yang Y; Perry IB; Lu G; Liu P; Buchwald SL, Copper-Catalyzed Asymmetric Addition of Olefin-Derived Nucleophiles to Ketones. Science 2016, 353, 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Li K; Shao X; Tseng L; Malcolmson SJ, 2-Azadienes as Reagents for Preparing Chiral Amines: Synthesis of 1,2-Amino Tertiary Alcohols by Cu-Catalyzed Enantioselective Reductive Couplings with Ketones. J. Am. Chem. Soc 2018, 140, 598–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yang Y; Perry IB; Buchwald SL, Copper-Catalyzed Enantioselective Addition of Styrene-Derived Nucleophiles to Imines Enabled by Ligand-Controlled Chemoselective Hydrocupration. J. Am. Chem. Soc 2016, 138, 9787–9790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Shao X; Li K; Malcolmson SJ, Enantioselective Synthesis of Anti-1,2-Diamines by Cu-Catalyzed Reductive Couplings of Azadienes with Aldimines and Ketimines. J. Am. Chem. Soc 2018, 140, 7083–7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wang Y-M; Buchwald SL, Enantioselective CuH-Catalyzed Hydroallylation of Vinylarenes. J. Am. Chem. Soc 2016, 138, 5024–5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lee J; Torker S; Hoveyda AH, Versatile Homoallylic Boronates by Chemo-, SN2’- Diastereo- and Enantioselective Catalytic Sequence of Cu–H Addition to Vinyl-B(pin)/Allylic Substitution. Angew. Chem. Int. Ed 2007, 56, 821–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Xu G; Zhao H; Fu B; Gang A; Zhang G; Zhang Q; Xiong T; Zhang Q, Ligand-Controlled Regiodivergent and Enantioselective Copper-Catalyzed Hydroallylation of Alkynes. Angew. Chem. Int. Ed 2017, 56, 13130–13134. [DOI] [PubMed] [Google Scholar]
- (34).Bandar JS; Ascic E; Buchwald SL, Enantioselective CuH-Catalyzed Reductive Coupling of Aryl Alkenes and Activated Carboxylic Acids. J. Am. Chem. Soc 2016, 138, 5821–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Gui Y-Y; Hu N; Chen X-W; Liao L-L; Ju T; Ye J-H; Zhang Z; Li J; Yu D-G, Highly Regio- and Enantioselective Copper-Catalyzed Reductive Hydroxymethylation of Styrenes and 1,3-Dienes with CO2. J. Am. Chem. Soc 2017, 139, 17011–17014. [DOI] [PubMed] [Google Scholar]
- (36).Gribble MW Jr; Guo S; Buchwald SL, Asymmetric Cu-Catalyzed 1,4-Dearomatization of Pyridines and Pyridazines without Preactivation of the Heterocycle or Nucleophile. J. Am. Chem. Soc 2018, 140, 5057–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Yu S; Sang HL; Ge S, Enantioselective Copper-Catalyzed Alkylation of Quinolone N-Oxides with Vinylarenes. Angew. Chem. Int. Ed 2017, 56, 15896–15900. [DOI] [PubMed] [Google Scholar]
- (38).Wang Y-M; Bruno NC; Placeres ÁL; Zhu S; Buchwald SL, Enantioselective Synthesis of Carbo- and Heterocycles Through a CuH-Catalyzed Hydroalkylation Approach. J. Am. Chem. Soc 2015, 137, 10524–10527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bandini M, Electrophilicity: the “Dark-Side” of Indole Chemistry. Org. Biomol. Chem 2013, 11, 5206–5212. [DOI] [PubMed] [Google Scholar]
- (40).Somei M, 1-Hydroxylindoles. Heterocycles 1990, 50, 1157–1211. [Google Scholar]
- (41).Somei M, 1-Hydroxyindoles, 1-Hydroxytryptamines, and 1-Hydroxytryptophans. Top. Heterocycl. Chem 2006, 6, 77–111. [Google Scholar]
- (42).Yamada F; Goto A; Peng W; Hayashi T; Saga Y; Somei M, Nucleophilic Substitution Reaction at the 1-Position of 1-Hydroxytryptamine and -Tryptophan Derivatives. Heterocycles 2003, 61, 163–172. [Google Scholar]
- (43).Yamada K; Yamada F; Shiraishi T; Tomioka S; Somei M, Nucleophilic Substitution Reactions of 1-Methoxy-6-Nitroindole-3-Carbaldehyde. Heterocycles 2002, 58, 53–56. [Google Scholar]
- (44).Yamada F; Goto A; Somei M, Synthesis of 3a-(Indol-3-yl)-1,2,3,3a,8,8a-Hexahydropyrrolo-[2,3-b]indole Core of Leptosins D–F Based on Nucleophilic Substitution Reaction on Indole Nucleus. Heterocycles 2000, 53, 1255–1258. [Google Scholar]
- (45).Seebach D, Methods of Reactivity Umpolung. Angew. Chem. Int. Ed 1979, 18, 239–258. [Google Scholar]
- (46).Niu D; Buchwald SL, Design of Modified Amine Transfer Reagents Allows the Synthesis of α-Chiral Secondary Amines via CuH-Catalyzed Hydroamination. J. Am. Chem. Soc 2015, 137, 9716–9721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).(a) We speculate that the para-OMe group of 1d might stabilize a benzylic radical/cation and in turn facilitate the dissociation of the Cu–C bond of the corresponding benzyl copper intermediate at high temperature (90°C). This would result in catalyst decomposition and might be responsible for the low yield of 3d. (b) We speculate that the lower yield seen for 3m is due to substrate chelation.
- (48).In the case of 3-substituted indole electrophiles, we mainly observed the direct reduction of the electrophile to the corresponding indole. Computational studies indicate that this process begins with oxidative insertion of LCuH into the N–O bond of the indole electrophile, rather than hydrocupration of the styrene double bond. See Figure S4 in the Supporting Information for details.
- (49).(a) Lee J; Radomkit S; Torker S; del Pozo J; Hoveyda AH, Mechanism-Based Enhancement of Scope and Enantioselectivity for Reactions Involving a Copper-Substituted Stereogenic Carbon Centre. Nat. Chem 2017, 10, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Huang Y; del Pozo J; Torker S; Hoveyda AH, Enantioselective Synthesis of Trisubstituted Allenyl–B(pin) Compounds by Phosphine–Cu-Catalyzed 1,3-Enyne Hydroboration. Insights Regarding Stereochemical Integrity of Cu–Allenyl Intermediates. J. Am. Chem. Soc 2018, 140, 2643–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).The absolute configurations of the products were assigned to be S in both C–N and C–C formation reactions (see the Supporting Information for details). By comparison with previous reactions involving irreversible, stereoselective hydrocupration of styrenes by DTBM-SEGPHOS- and Ph-BPE-ligated copper hydride species, we conclude that the configuration of the benzylic stereogenic center is most likely retained during the subsequent reaction with the electrophilic indole reagent.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.