

Abstract
p53 is a tumor suppressor protein that maintains genome stability, but its Δ133p53β and Δ160p53β isoforms promote breast cancer cell invasion. The sequence truncations in the p53 core domain raise key questions related to their physicochemical properties, including structural stabilities, interaction mechanisms, and DNA-binding abilities. Herein, we investigated the conformational dynamics of Δ133p53β and Δ160p53β with and without binding to p53-specific DNA by using molecular dynamics simulations. We observed that the core domains of the 2 truncated isoforms are much less stable than wild-type (wt) p53β, and the increased solvent exposure of their aggregation-triggering segment indicates their higher aggregation propensities than wt p53. We also found that Δ133p53β stability is modulable by peptide or DNA interactions. Adding a p53 peptide (derived from truncated p53 sequence 107–129) may help stabilize Δ133p53. Most importantly, our simulations of p53 isomer-DNA complexes indicate that Δ133p53β dimer, but not Δ160p53β dimer, could form a stable complex with p53-specific DNA, which is consistent with recent experiments. This study provides physicochemical insight into Δ133p53β, Δ133p53β-DNA complexes, Δ133p53β’s pathologic mechanism, and peptide-based inhibitor design against p53-related cancers.—Lei, J., Qi, R., Tang, Y., Wang, W., Wei, G., Nussinov, R., Ma, B. Conformational stability and dynamics of the cancer-associated isoform Δ133p53β are modulated by p53 peptides and p53-specific DNA.
Keywords: Δ160p53β cancer, p53 aggregation, p53 response element
The transcription factor p53 is at the center of a complex signaling network that controls cell fate (1, 2). Cellular stress, such as DNA damage and oncogene activation, activates p53, promoting expression of p53 target genes involved in cell-cycle arrest, DNA repair, apoptosis, senescence, angiogenesis, and autophagy (3, 4). p53 regulates hundreds of genes and is a guardian maintaining genome stability (5, 6). p53 mutants lose their tumor suppressive functions in over 50% of human cancers (7, 8). One reason may be a prion-like aggregation behavior, resulting in dominant-negative and gain-of-function effects (9–12).
The human p53 gene encodes at least 12 p53 isoforms (p53α, p53β, p53γ, Δ40p53α, Δ40p53β, Δ40p53γ, Δ133p53α, Δ133p53β, Δ133p53γ, Δ160p53α, Δ160p53β, and Δ160p53γ) through alternative promoters, alternative initiation of translation, and alternative splicing (13–15). Canonical p53α has 393 aa and 3 major functional domains, including transactivation (TA) domain, DNA-binding domain (DBD), and oligomerization domain (13, 16, 17). The β and γ isoforms of p53 contain different C-terminal amino acid sequences, with the oligomerization and negative-regulation domains missing in both isoforms. p53 isoforms are differentially expressed in human tissues and have cell-type–dependent activities (13). They play roles in programmed cell death, cell-cycle progression, senescence, inflammation, stem-cell renewal and differentiation, aging, neurodegeneration, glucose metabolism, angiogenesis, and embryo development (18–32). p53 isoforms can be found in various cancers. Clinical studies also suggest that specific expression patterns of p53 isoforms are associated with tumor development, cancer progression, clinical response, and prognoses (33).
The Δ40, Δ133, and Δ160 isoforms of p53 protein lack the first 39, 132, and 159 aa, respectively. Translation of the Δ40p53 protein, also known as p44, is regulated by the intracellular domain of the amyloid precursor protein (34), a polypyrimidine tract binding protein, as well as miR-1285 (35) and 20S proteasome-mediated cleavage (36). Δ133p53 transcript generates 2 different p53 isoforms, Δ133p53 and Δ160p53 (37). Specific p53 mutants can induce translation of Δ160p53, promoting tumorigenesis (38). Though the functional roles of Δ160p53 are less clear, studies revealed cancer-promoting effects of Δ133p53. Recent studies show that the p53 isoform Δ133p53β regulates cancer cell deterioration, and it is expressed at higher levels in highly invasive breast cancer cells than other isoforms (2) and promotes breast cancer cell invasion (39). Cancer stem cells with Δ133p53β-containing tumors are more likely to develop secondary tumors regardless of p53 mutations (32). Δ133p53β also prevents cancer cells from undergoing apoptosis in a RhoB-dependent manner (40). It has been suggested that the Δ133p53 isoform and its mouse analog Δ122p53 promote invasion and metastasis in a manner similar to gain-of-function of mutant p53 proteins (41). Therefore, it is important to understand the physicochemical properties of Δ133p53β compared with p53 protein with normal DBD.
The perturbations of the DBD by Δ133 and Δ160 deletions question their structural stability, DNA-binding abilities, and function. The Δ40p53 still contains a part of the p53 TA domain and the whole DBD, and it can activate gene expression after transfection through a second TA domain (42). The Δ133 p53 isoforms lack both TA domains and a small part of the first conserved Cys box of the DBD, whereas the Δ160p53 isoforms lack both TA domains and the entire first conserved Cys box of the DBD, retaining the 3 other conserved Cys boxes of the DBD. We ask 1) what are structural stabilities of Δ133 and Δ160 p53 isoforms, 2) how are the Δ133 and Δ160 p53 isoforms able to bind DNA, and 3) whether adding the peptide with the truncated p53 sequence can affect Δ133p53β’s stability. To address these questions, we used molecular dynamic (MD) simulations. We investigated the structural properties, conformational dynamics, and DNA interactions of human 94-341p53β [as a model of wild-type (wt) p53], Δ133p53β, and Δ160p53β. We compared the structural stability of Δ133p53β with that of wt p53β and Δ160p53β. Though the DBD of wt p53 is very stable, deletion of the N-terminal domains destabilizes the structure of both isoforms. Compared with Δ160p53β, Δ133p53β still partially maintains the folded structure of wt p53 and is still able to form stable structure with p53 DNA-binding motif, helped by the K139 motif. Our simulations and experiments suggest that Δ133p53β has a higher aggregation propensity than wt p53β. In considering whether peptide with the same amino acid sequence as fragments from the truncated DBD region can restore Δ133p53β to the wt p53 conformation, our simulation indicated that a peptide consisting of p53 107–129 fragment could be a good candidate to rescue Δ133p53β.
MATERIALS AND METHODS
The wt p53 protein and its Δ133p53β and Δ160p53β isoforms
In this work, we investigated structural properties and aggregation propensity of the Δ133p53β isoform in the absence and presence of DNA or peptides. For comparison, we also performed MD simulations on the isolated 94-341p53β protein, the Δ160p53β isoform, 94-341p53-DNA complex, and Δ160p53β-DNA complex (Supplemental Table S1). The structure of 94-341p53β consists of DBD from solution NMR structure (Protein Data Bank identification 2FEJ, residues 94–297) (43) and C-terminal random structure [residues 298–331 and DQTSFQKENC (i.e., β)]. The 94-341p53β is used to model the wt p53. The initial structures of Δ133p53β and Δ160p53β are taken from p53 structure, but they lack residues 94–132 and 94–159, respectively. In the case of the p53-DNA complex, a dimer of 94-341p53β, Δ133p53β, or Δ160p53β is considered. The p53-binding DNA sequence (response element) used in our simulations is 5′-GCGTGAGCATGCTCAC-3′, corresponding to a half-site palindrome in a full p53 response element (44). The underlined sequence indicates the direct binding sitE of the p53 protein. Using the X-ray structure of the p53 dimer-DNA complex (2GEQ) (45) as a template, we aligned the structure of 94-341p53β, Δ133p53β, or Δ160p53β to obtain the initial structure of 94-341p53β-, Δ133p53β-, or Δ160p53β- dimer-DNA complex. Finally, we examined possible restoration of Δ133p53β to the conformation of wt p53 by adding a peptide that has the same amino acid sequence as the fragment from the missing N-terminal region of DBD. We selected the following 4 peptides from p53 DBD region: peptide-1, residues 102–128; peptide-2, residues 107–129; peptide-3, residues 107–130; and peptide-4, residues 107–131 of p53.
Three Cys residues (C176, C238, and C242), which are coordinated by a Zn ion, were deprotonated. Following our earlier simulation settings (46), the lengths of the 3 Zn-S bonds (between Zn and Cys) and the Zn-N bond (between Zn and His179) were fixed using the distance restraint method implemented in Gromacs (47). The partial charges and van der Waals parameters of the Zn ion and deprotonated Cys were taken from the work by Maynard and Covell (48).
Details of MD simulations and analysis methods
All simulations were performed in the isothermal-isobaric ensemble using the Gromacs package v.4.5.3 (47) with Charmm27 force field (49). Each p53 isoform system is placed in a box filled with transferable interatomic potential with 3 points water. The pressure was kept at 1 bar using the Parrinello–Rahman method (50) with a coupling time constant of 1.0 ps. The temperature of the system was maintained at 310 K by separately coupling the protein and nonprotein groups to an external heat bath using a V-Rescaling coupling method (with a relaxation time of 0.1 ps) (51). Constraints were applied to all-bond lengths using the Settle algorithm (52) for water molecules and the LINCS method (53) for the protein and DNA. Electrostatic interactions were treated with the Particle Mesh Ewald method (54) with a real space cutoff of 1.0 nm. Van der Waals interactions were calculated using a cutoff of 1.4 nm. The integration time step is 2 fs and the simulation time is 500 ns for each MD run. All simulations were conducted using periodic boundary conditions.
Trajectory analysis was carried out using the facilities implemented in the Gromacs v.4.5.3 software package (47) and our in-house codes. The DSSP program (55) was used to calculate the secondary structure. A salt bridge is considered if the minimum distance between the Nζ atom of Lys/Arg and the O atom in the side chain COO− group of Asp/Glu is smaller than 0.4 nm. We used the community network method (56–58) to analyze the dynamical correlations in each isoform. In the analysis, residues 96–300, 135–300, and 162–300 are considered for 94-341p53β, Δ133p53β, and Δ160p53β systems, respectively. We assigned each amino acid as a node using a Cα atom and drew an edge between each of the 2 nodes whose corresponding residues have atomic contacts (58). A contact is defined if the distance between 2 atoms is within 0.5 nm for at least 70% of the MD trajectory. Visual molecular dynamics is used to display the structures of all isoforms and p53-DNA complexes.
The protein’s root mean square deviation (RMSD) between 2 conformations (v, w) was calculated by a rigid superposition, which minimizes the following value:
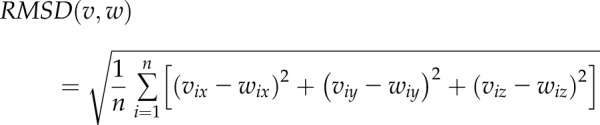 |
Where n is the total number of protein atoms, and x, y, and z are Cartesian coordinates of i’s atom.
The root mean square fluctuation (RMSF) of the protein was calculated as
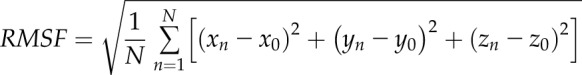 |
where N is the number of conformations used in the calculation, and xn, yn, and zn and x0, y0, and z0 are the Cartesian coordinates of n’s conformation and reference conformation, respectively. We used the positions of Cα in both RMSD and RMSF calculations. Thus, the Cα RMSD of p53 core domain (residues 96–300 for 94-341p53β, residues 135–300 for Δ133p53β, and residues 162–300 for Δ160p53β) in the 3 systems were calculated following structural alignment. The Cα RMSF of 94-341p53β, Δ133p53β, and Δ160p53β were calculated with respect to their MD generated average structures in the last 250 ns.
Details of protein expression experiments
The p53(94-312) and p53(133-312) fragments were individually cloned in a pET-M3C vector. Each of the resulting proteins contains a His6 tag in its N termini. Escherichia coli BL21 (DE3) were grown in Lysogeny broth medium at 37°C to an OD600 of 0.5 followed by overnight induction at 16°C with 0.4 mM isopropyl-b-d-thiogalactoside. After induction, cells were harvested by centrifugation and resuspended in buffer 1 [25 mM Na3PO4 (pH 7.0), 0.15 M KCl, 5% glycerol, 2 mM 2-ME], buffer 2 [25 mM HEPES-NaOH (pH 7.6), 5 mM DTT, 15% glycerol, 1% Triton X-100], or buffer 3 [50 mM Tri-HCl (pH 7.5), 5 mM EDTA, 5 mM DTT, 1% NP40, 1 mM PMSF, and 0.15 mM KCl], and then disrupted by high-pressure dispersion. After 25 min at 18,000 rpm centrifugation, the pellets were resuspended in the above-mentioned 3 buffers and subjected to SDS-PAGE together with all supernatants.
RESULTS
Δ133p53β and Δ160p53β are destabilized and display higher aggregation propensity because of truncation of core domain residues
The sequence of the wt p53 core domain and its structure are shown in Fig. 1A, B. The p53 core domain consists of 11 β strands (B1–B11) and 2 helices [H1 and H2]. The first and the 4th–11th β-sheets form a β-sandwich structure. It was reported that the thermodynamic stability of the p53 core domain is similar to that of full-length p53 (59). To compare the structural stability of 94-341p53β, Δ133p53β, and Δ160p53β, we first calculated the Cα RMSD of the p53 core domain (residues 96–300 for 94-341p53β, residues 135–300 for Δ133p53β, and residues 162–300 for Δ160p53β) in the 3 systems as a function of simulation time. It can be seen in Fig. 1C that the Cα RMSDs of the core domain in 94-341p53β, Δ133p53β, and Δ160p53β systems increase within the first 250 ns and fluctuate, respectively, around 0.45, 0.5, and 0.9 nm during the remaining 250 ns. Both Δ133p53β and Δ160p53β have higher RMSD values than 94-341p53β, indicating that truncation of core domain residues destabilizes the p53 core domain structure. The equilibrated Cα RMSD value of Δ133p53β is close to that of 94-341p53β, but the value of Δ160p53β is much larger than that of 94-341p53β, indicating that the structure of Δ133p53β is more similar to the native p53 structure than that of Δ160p53β.
Figure 1.

∆133p53β and Δ160p53β are destabilized because of truncation of core domain residues, whereas the effect on Δ133p53β is much smaller than that on Δ160p53β system. A) The sequence of the wt p53 DBD. β-strands and α-helices are represented by yellow arrows and blue cylinders, respectively. B) The 3D structure of the p53 DBD (residues 94–300). C) Time evolution of Cα RMSD of the p53 core domain (residues 96–300 for 94-341p53β, residues 135–300 for Δ133p53β, and residues 162–300 for Δ160p53β) in the 3 systems. D) The single site residue mutation frequencies of cancer-associated p53 and average Cα RMSF of 94-341p53β, Δ133p53β and Δ160p53β.
To measure the flexibilities of 94-341p53β and its 2 isoforms, we calculated their Cα RMSF using the last 250 ns trajectories (Fig. 1D). The N- and C-terminal domains have highest RMSF, indicative of a high flexibility of these 2 domains. In the DBD, 4 peaks appear in the regions of residues 165–169, 183–187, 208–213, and 223–227, corresponding to the fluctuating loop regions. The 2 isoforms show overall higher flexibility than 94-341p53β, especially for the residues 250–291 region containing B10, B11, and H2, indicating strong p53 core domain perturbation effects upon truncation.
To check whether the residue truncations affect the flexibilities of residues with high-mutational frequencies (59–62), we examined a possible relationship between mutational frequencies and the Cα RMSF values using the TP53 mutant database (http://p53.iarc.fr) (Fig. 1D). We found that the top 10 high-frequency mutation sites have relatively low RMSF values in 94-341p53β systems; however, increased RMSFs are observed in 1 position (residue R282) in the Δ133p53β system but in 4 positions (residues C176, R213, R273, and R282) in the Δ160p53β system. This suggests that residue deletions can influence the flexibilities of those residues in both Δ133p53β and Δ160p53β systems, whereas the effect on Δ133p53β is much smaller than that on Δ160p53β system.
The changes in the secondary structures and salt bridge interactions are responsible for the larger structural fluctuations following deletions of the core domain residues. Figure 2A–C shows the time evolution of the secondary structure profile of the 3 systems. Δ133p53β lacks the first 2 β-strands (B1 and B2) and Δ160p53β loses the first 5 β-strands (B1–B5) because of truncation of the N-terminal region. In the Δ133p53β system, although most β-strands (B3–B10) are maintained as those in wt p53, the 11th β-strand (B11) and the second helix (H2) become shorter. In contrast, 3 β-strands (B7, B8, and B9) of Δ160p53β disappear gradually with simulation time. It can be seen from Fig. 2D–F that helix H2 in both Δ133p53β and Δ160p53β systems deviates from its initial orientation, whereas the H2 orientation in the 94-341p53β system has minor changes. The larger deviation in the 2 isoforms is likely because of missing the 2 nearest neighboring β-strands (B2 and B3) of H2 and the loop connecting the 2 β-strands (B1 and B2). In addition, all β-strands (B3–B11) in Δ133p53β superimpose well with their initial positions, but the β-strands (B6–B11) in Δ160p53β deviate. These results indicate that different truncations of the N-terminal domains can cause different extents of conformational changes in the 2 isoforms.
Figure 2.

A–C) The changes in the secondary structures are responsible for the larger structural fluctuations following deletions of the core domains residues, evidenced by the time evolution of secondary structure profiles. D–F) The superposed core domain structures of the final conformations (blue) of 94-341p53β (D), Δ133p53β (E), and Δ160p53β (F) with respect to their initial structure (red) revealed a larger deviation of DNA-binding motif helix H2.
Salt bridges can be important stabilizers of protein structures. Table 1 displays the residue pairs with salt bridge formation probability ≥10% (during the last 250 ns trajectory) in wt p53 and the β-sheets or helices where these residue pairs are located. For comparison, the results of Δ133p53β and Δ160p53β are also given. The salt bridges K132-E271 between B3 and B11, R156-E258, R158-E258, and K164-E271 between B5 and B10 in 94-341p53β have high probabilities (94.6, 53.1, 99.7, and 99.5%). These salt bridges can stabilize the 2 β-sheets of B3–B11 and B5–B10. In the Δ133p53β system, the K132-E271 salt bridge between B3 and B11 disappears, which is correlated with the β-strand B11 shortening. In 94-341p53β, 4 salt bridges (R282-E286, R283-E287, R290-E287, and K291-E287) exist within helix H2, though with different populations. However, in Δ133p53β, 3 of the 4 salt bridges have greatly reduced probabilities (from 100, 16.5 and 21.2–15.0, and 1.9 and 0.2% for R282-E286, R283-E287, and R290-E287, respectively). In Δ160p53β, almost all salt bridges are lost except for K164-E271 and R273-D281. These results indicate that residue deletions in N-terminal region of DBD disrupt salt bridges in B3–B11 and B5–B10 β-sheets and H2 helix, and thus destabilize Δ133p53β and Δ160p53β.
TABLE 1.
Formation probabilities of salt bridges in β-sheets and helices in 94-341p53β, Δ133p53β, and Δ160p53β systems
| Salt bridge pair | K132-E271 | R156-E258 | R158-E258 | K164-E271 | R273-D281 | R282-E286 | R283-E287 | R290-E287 | K291-E287 |
|---|---|---|---|---|---|---|---|---|---|
| Location | B3-B11 | B5-B10 | B5-B10 | B5-B10 | B11-H2 | H2-H2 | H2-H2 | H2-H2 | H2-H2 |
| 94-341p53β | 94.6% | 53.1% | 99.7% | 99.5% | 74.4% | 100% | 16.5% | 21.2% | 26.9% |
| Δ133p53β | 0% | 52.6% | 100% | 97.5% | 64.9% | 15.0% | 1.9% | 0.2% | 86.3% |
| Δ160p53β | 0% | 0% | 0% | 96.8% | 64.1% | 2.4% | 0.1% | 0% | 0% |
Even with the larger structural flexibilities in the 2 isoforms, Δ133p53β is able to retain a similar global residue–residue dynamic correlation as the 94-341p53β with unperturbed core domain. In Fig. 3A–C, the 3 most fluctuating loops (residues 165–169, residues 183–187, and residues 223–227) display the strongest negative correlation with all the other residues. We can see in Fig. 3A that residues 100–132 correlate well with residues 275–300, which are important for DNA binding. Indeed, through dynamic network analyses (as shown in Fig. 3D), we also found that residues 125–132 and the DNA-binding helix fall into 1 community. The core domain of 94-341p53β is divided into 13 communities. The top 3 largest communities are colored in blue, red, and gray. They contain part of B8, B10, B11; part of B8, B10, B11, and H1; and part of B2, B3, and H2. Residue truncations change the global residue dynamics correlations. Though the correlation pattern of Δ133p53β maintains most 94-341p53β features (Fig. 3B), that of Δ160p53β differs (Fig. 3C). Δ133p53β has the same number of communities (13 in total) with 94-341p53β, but their sizes decrease (Fig. 3E), as shown in the blue and gray communities, whereas Δ160p53β only has 9 communities (Fig. 3F). These analyses suggest that unlike Δ160p53β, Δ133p53β could preserve 94-341p53β’s dynamic communication network.
Figure 3.

Covariance matrix (A–C) and dynamical network (D–F) analyses indicate that Δ133p53β is able to retain similar global residue–residue dynamic correlation as 94-341p53β with unperturbed core domain. D–F) In the dynamical network plots [94-341p53β (D), Δ133p53β (E), and Δ160p53β systems (F)], residues belonging to the same community groups have the same color.
The destabilized p53 is likely to increase the solvent exposure of the hydrophobic core region, thus making it prone to aggregation (63). Experiments suggest that p53 aggregation can cause cancers (10, 33, 64, 65). Recent experiments show that the hydrophobic region 251ILTIITL257 can trigger the aggregation of p53 (66). To examine the aggregation propensity of Δ133p53β, we calculated the solvent accessible surface area (SASA) of this aggregation-triggering segment. As seen in Fig. 4, the SASA value of 251ILTIITL257 in Δ133p53β (∼1.5 nm2) is much larger than that in wt p53 (∼0.9 nm2), suggesting that Δ133p53β has higher aggregation propensity than wt p53. This prediction was confirmed indirectly by protein expression experiments in Fig. 4, showing that a fraction of soluble p53(94-312) can be obtained in the supernatant, but p53(133-312) are all in pellet.
Figure 4.

∆133p53β has a higher aggregation propensity because of the deletion of core domain residues. A) The SASA analyses of the p53 aggregation core fragment 251ILTIITL257 in the 94-341p53β and Δ133p53β systems. B) Protein expression of p53 (94–312) and p53 (133–312). A total of 3 buffer conditions were tested for expression of both p53 fragments. A fraction of soluble p53 (94–312) can be obtained in the supernatant (S), but p53 (133–312) are all in pellet (P).
p53 peptide (107–129) may stabilize the structure of Δ133p53β near the native conformation of wt p53
Δ133p53β promotes cancer cell invasion (39), and 1 possible underlying mechanism is the destabilization by the deletion of B1 and B2. We asked whether a peptide from the truncated p53 DBD sequence could stabilize the conformation toward the native structure of p53. If so, it may prevent the abnormal function of Δ133p53β. We selected 4 peptides containing B1 and B2 from the N-terminal region of p53 DBD (Supplemental Fig. S1) and labeled them peptide-1 (residues 102–128), peptide-2 (residues 107–129), peptide-3 (residues 107–130), and peptide-4 (residues 107–131). We aim to find the shortest peptide that can stabilize Δ133p53β’s structure.
To examine the effect of the 4 peptides on strand B11 and helix H2, we calculated the β-sheet probability of each residue in B11 and the helix probability of each residue in H2. As shown in Fig. 5A, all 4 peptides bound to Δ133p53β and can restore residues 273–275 to the β-sheet states with ∼100% probability. Figure 5B shows that both peptide-1 and -2 can increase the helix probabilities of residues 288–291 from 62, 48, 48, and 48% in isolated Δ133p53β to ∼100, ∼100, ∼100, and 72%, and peptide-2 further improves the helix probability of residue 292 from 44 to 94%. Although peptide-3 and -4 stabilize the helical structure of residues 288–290, they cannot recover the helix states of residues 291 and 292. We also checked the orientation of H2 by monitoring the tilt angle of H2 relative to its initial orientation as a function of simulation time (Fig. 5C). Compared with the angle value in isolated Δ133p53β, the tilt angle in peptide-bound Δ133p53β decreases and becomes close to the wt state. These results indicate that all 4 peptides have a capacity to restore B11 and H2 to the native state and peptide-2 is the most efficient.
Figure 5.

The effect of the 4 peptides (peptide-1, -2, -3, -4) on the structural stabilities of strand B11 and helix H2 indicates that peptide-2 has the potential to stabilize p63 core domain secondary structure. A) The β-sheet probability of each residue in B11. B) The helix probability of each residue in H2. C) The time evolution of tilt angle of H2 with respect to the initial orientation of H2.
A comparison of the Cα RMSDs of the core domain of peptide-bound Δ133p53β with those of isolated Δ133p53β and 94-341p53β indicates that these 4 peptides have different effects on the global conformation of Δ133p53β (Fig. 6). When binding with peptide-1, -2, and -4, Δ133p53β has a smaller Cα RMSD value than the isolated Δ133p53β, whereas peptide-3–bound Δ133p53β has a larger RMSD value than the isolated Δ133p53β. The RMSD value of peptide-2–bound Δ133p53β is smaller than that of the wt 94-341p53β. Qualitatively similar results are obtained in the secondary structure analyses (Supplemental Fig. S2) and the superimposed structures of peptide-bound Δ133p53β (Supplemental Fig. S3). These data suggest that peptide-2 may even restore the global structure of Δ133p53β toward the wt conformation of p53.
Figure 6.

The effect of the 4 peptides (peptide-1, -2, -3, -4) on the conformation of ∆133p53β suggests that peptide-1 and peptide-2 could enhance structural stability of ∆133p53β. Time evolution of Cα-RMSD of 94-341p53β, isolated Δ133p53β, and Δ133p53β bound with peptide-1 (A), peptide-2 (B), peptide-3 (C), and peptide-4 (D).
To examine whether each of the 4 peptides can change the residue–residue correlation, we calculated the covariance matrix of the core domains of 94-341p53β, isolated Δ133p53β, and peptide-bound Δ133p53β. As shown in Fig. 7, only peptide-2–bound Δ133p53β has similar residue–residue correlation pattern with that of the wt p53, indicating that the peptide-2–bound Δ133p53β and wt p53 may have similar biologic functions. This observation, together with the increased structural stability (Figs. 5 and 6) of B11, H2, and the whole conformation of Δ133p53β, suggests that introducing peptide-2 to Δ133p53β may be an effective way to shift the conformation and possibly the function of Δ133p53β toward the wt p53 (Supplemental Fig. S3).
Figure 7.

Covariance matrix analyses of Δ133p53β in the isolated Δ133p53β system and the peptide-bound Δ133p53β indicate that peptide-2–bound ∆133p53β has similar residue–residue correlation pattern with wt p53. A) 94-341p53β; B) ∆133p53β+pep-1; C) ∆133p53β+pep-2; D) ∆133p53β+pep-3; E) ∆133p53β+pep-4.
Δ133p53β dimer could form a relatively stable complex with p53-specific DNA, and DNA binding alters the correlation dynamics of Δ133p53β
p53 tetramer binds to 2 half-sites (each with 10 bp) of p53-specific DNA, and p53 dimer binds to 1 half-site of p53-specific DNA. If the Δ133p53β is still able to interact with p53 response elements, it may interfere with the normal function of wt p53. We simulated the 94-341p53β-dimer-DNA complex, Δ133p53β-dimer-DNA complex, and Δ160p53β-dimer-DNA complex to examine the structural stabilities of the 3 complexes. We first calculated the Cα RMSD of p53 core domain in DNA-bound 94-341p53β, Δ133p53β, and Δ160p53β dimers. It can be seen from Fig. 8A that the Cα RMSD value of the Δ133p53β dimer-DNA complex quickly increases to 0.75 nm within the first 50 ns and decreases to a value of 0.48 nm, which is similar to that of 94-341p53β-dimer-DNA complex, but the RMSD value of Δ160p53β-dimer-DNA complex is always larger than those of Δ133p53β-dimer-DNA complex and 94-341p53β-dimer-DNA complex. Qualitatively similar results are obtained for the RMSD of each monomer of 94-341p53β, Δ133p53β, and Δ160p53β in the 3 complexes. Structural alignments between the final and initial snapshots of each complex (Fig. 8D–F) show that the Δ133p53β-dimer-DNA complex and the 94-341p53β-dimer-DNA complex fit well with their initial states, but Δ160p53β-dimer-DNA complex does not. These results indicate that Δ133p53β could form stable complex with p53-specific DNA and the structure of the complex could be similar to wt p53-DNA complex, whereas Δ160p53β could not.
Figure 8.

Structural stability analyses of 94-341p53β-dimer-, Δ133p53β-dimer-, and Δ160p53β-dimer-DNA complex suggest that Δ133p53β-dimer could form stable complex with p53 response element. A) The time evolution of Cα RMSD of p53 core domain in 94-341p53β-dimer-DNA complex, Δ133p53β-dimer-DNA complex, and Δ160p53β-dimer-DNA complex system. B, C) The RMSD of each 94-341p53β monomer, Δ133p53β monomer (B), and Δ160p53β monomer (C) in the DNA-bound complex. D–F) The superposed structures of 94-341p53β-dimer-DNA complex (D), Δ133p53β-dimer-DNA complex (E), and Δ160p53β-dimer-DNA complex (F). The initial and final snapshots are colored in red and blue, respectively. G–I) Contact probabilities of each amino acid with DNA in the following 3 complexes: 94-341p53β-dimer-DNA complex (G), Δ133p53β-dimer-DNA complex (H), and Δ160p53β-dimer-DNA complex (I).
Three regions containing positively charged residues of p53 have relatively strong interactions with DNA: loop 1 around residue K120, loop 2 around R248, and helix H2 containing residues R273 and R280. Loop 1 around residue K120 is very important for p53-DNA interaction (46, 67, 68). However, loop 1 is truncated in both Δ133p53β and Δ160p53β. In order to examine the effect of residue truncation on p53-DNA interaction (Fig. 8G–I), we calculated the contact probabilities of each amino acid with DNA in the 3 complexes. In the Δ133p53β-dimer-DNA complex, residues 134–141, which are adjacent to loop 1 and contain K139, have increased contact probabilities compared with those in the 94-341p53β-dimer-DNA complex. As seen in Fig. 1D, residues 134–151 have much higher flexibility in Δ133p53β than in 94-341p53β. The higher flexibility of this N-terminal segment of Δ133p53β helps residues 134–141 move toward DNA to make up for the lost contact of loop 1. As a result, Δ133p53β has 3 similar p53-DNA interacting regions with wt p53, but Δ160p53β has only 2, as both loop 1 and residues 134–141 were removed in Δ160p53β. These results suggest that Δ133p53β and wt p53 may have similar interaction patterns with DNA.
We further examined the residue–residue dynamics correlation of DNA-bound 94-341p53β, Δ133p53β, and Δ160p53β by calculating the covariance matrix. As seen in Supplemental Figure S4, the 2 monomers of Δ133p53β and Δ160p53β in the p53-DNA complex have different covariance matrix than 94-341p53β monomers. This indicates that the residue–residue correlation dynamics of DNA-bound Δ133p53β differs from that of the wt p53, although the interaction mode of Δ133p53β with DNA and the conformation of Δ133p53β are similar to that of wt p53, which explains why Δ133p53β exhibits opposite biologic effects with respect to wt p53.
DISCUSSION
As a transcription factor, wt p53 functions mainly through recognizing p53 DNA response elements. Experimental (69) and computational (61) studies revealed that p53 has evolved to become a conformationally sensitive protein, and vertebrate p53 has a low thermodynamic stability. A p53 mutant can induce the aggregation of wt p53 because of the reduced structural stabilities of the core domain (70) via residues 251–257 acting as an aggregation nucleus (66). p53 isoforms can interfere with p53’s normal functions (2), such as transcriptional activity (13), apoptosis (30), senescence, and cell death (71), possibly because of their decreased structural stabilities. Using all-atom MD simulations, we systematically investigated the structural properties, conformational dynamics, and DNA interactions of 94-341p53β, Δ133p53β, and Δ160p53β. Our results revealed that Δ133p53β is aggregation-prone but is still able to form a relatively stable complex with p53-specific DNA.
The prion-like aggregating behavior of p53 mutants can lead to dominant-negative and gain-of-function effects that increase cancer aggressiveness and progression (9–12). Longer truncations of the N-terminal region can cause larger destabilization. It is known that solvent exposure of residues 251–257 can trigger p53 aggregation (66). Our simulations showed that these residues in Δ133p53β have larger SASA than those in wt p53, indicating that Δ133p53β has a higher aggregation propensity than wt p53β. This prediction is in line with our protein expression experiments. The higher aggregation capability of destabilized Δ133p53β may cause the abnormal function. Unlike Δ160p53β, Δ133p53β still shares similar local structural properties and correlation dynamics with wt p53 but with a tunable stability. We propose that p53 fragment peptide 107–129 could shift Δ133p53β toward the native conformation and restore p53 properties (Fig. 9). However, further experimental study is required to validate the potential, for example, by more in-depth functional analyses (e.g., regarding the DNA binding or biologic impact of the N-terminally truncated p53 forms). Previous experiments found that a 17-residue peptide inhibitor can prevent p53 mutant aggregation (72) by restoring the variants to a wt conformation (73). If in vivo experiments can confirm the stabilization effects of p53 fragment peptide 107–129, our results will be a crucial clue to developing a new therapeutic strategy.
Figure 9.

Schematic diagram of hypothetical mechanisms leading to Δ133p53β abnormal function and a potential therapeutic strategy. A) Destabilized ∆133p53β has a strong aggregation capability. B) ∆133p53β could prevent p53’s normal function by competing with wt p53 in recognizing p53 response elements. C) The peptide with residues 107–129 of wt p53 could inhibit the aggregation of ∆133p53β and restore ∆133p53β to a wt conformation of p53, and thus potentially re-establish tumor suppressor function.
Our results show that Δ133p53β dimer could form relatively stable complex with p53-specific DNA, whereas Δ160p53β cannot. Although DNA binding facilitates Δ133p53β adopting wt conformation, it’s residue–residue correlation dynamic differs from that of wt p53. There are 2 possible consequences of the Δ133p53β–DNA interaction. First, when binding to p53 target genes, Δ133p53β could compete with wt p53 in recognizing p53 response elements. Second, Δ133p53β may form complex with p53 family proteins to bind to p53 response element (Fig. 9B). Such a scenario has been reported recently just after completion of this work. Gong et al. (74) found that Δ133p53 binds to ΔNp63 and utilizes its TA domain to promote glycolytic metabolism, leading to cancer cell proliferation. Additionally, p73 and Δ133p53 act synergistically to promote the expression of RAD51, LIG4, and RAD52 by binding to a region containing a Δ133p53-responsive element and a p73-responsive element in the promoters of all 3 DNA double-strand break repair genes (75).
Supplementary Material
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
ACKNOWLEDGMENTS
This work has been supported by the Ministry of Science and Technology of China (Grant 2016YFA0501702) and the National Natural Science Foundation of China (Grant 11674065). This work has also been funded in whole, or in part, by the U.S. National Institutes of Health (NIH) Frederick National Laboratory for Cancer Research under contract HHSN261200800001E. This research was supported, in part, by the Intramural Research Program of the NIH, Frederick National Lab, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. All simulations reported in this work were performed using the high-performance computational facilities at the NIH Biowulf Cluster and the National High Performance Computing Center of Fudan University. The authors declare no conflicts of interest.
Glossary
- DBD
DNA-binding domain
- MD
molecular dynamic
- RMSD
root mean square deviation
- RMSF
root mean square fluctuation
- SASA
solvent accessible surface area
- TA
transactivation
- wt
wild type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
J. Lei, R. Qi, and Y. Tang performed experiments; J. Lei, R. Nussinov, W. Wang, G. Wei, and B. Ma wrote the paper; and W. Wang, G. Wei, and B. Ma conceived and coordinated the study.
REFERENCES
- 1.Joerger A. C., Fersht A. R. (2016) The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu. Rev. Biochem. 85, 375–404 [DOI] [PubMed] [Google Scholar]
- 2.Joruiz S. M., Bourdon J. C. (2016) p53 isoforms: key regulators of the cell fate decision. Cold Spring Harb. Perspect. Med. 6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vogelstein B., Lane D., Levine A. J. (2000) Surfing the p53 network. Nature 408, 307–310 [DOI] [PubMed] [Google Scholar]
- 4.Vousden K. H., Prives C. (2009) Blinded by the light: the growing complexity of p53. Cell 137, 413–431 [DOI] [PubMed] [Google Scholar]
- 5.Riley T., Sontag E., Chen P., Levine A. (2008) Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 9, 402–412 [DOI] [PubMed] [Google Scholar]
- 6.Stracquadanio G., Wang X., Wallace M. D., Grawenda A. M., Zhang P., Hewitt J., Zeron-Medina J., Castro-Giner F., Tomlinson I. P., Goding C. R., Cygan K. J., Fairbrother W. G., Thomas L. F., Sætrom P., Gemignani F., Landi S., Schuster-Böckler B., Bell D. A., Bond G. L. (2016) The importance of p53 pathway genetics in inherited and somatic cancer genomes. Nat. Rev. Cancer 16, 251–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olivier M., Eeles R., Hollstein M., Khan M. A., Harris C. C., Hainaut P. (2002) The IARC TP53 database: new online mutation analysis and recommendations to users. Hum. Mutat. 19, 607–614 [DOI] [PubMed] [Google Scholar]
- 8.Béroud C., Soussi T. (2003) The UMD-p53 database: new mutations and analysis tools. Hum. Mutat. 21, 176–181 [DOI] [PubMed] [Google Scholar]
- 9.Silva J. L., De Moura Gallo C. V., Costa D. C., Rangel L. P. (2014) Prion-like aggregation of mutant p53 in cancer. Trends Biochem. Sci. 39, 260–267 [DOI] [PubMed] [Google Scholar]
- 10.Ano Bom A. P., Rangel L. P., Costa D. C., de Oliveira G. A., Sanches D., Braga C. A., Gava L. M., Ramos C. H., Cepeda A. O., Stumbo A. C., De Moura Gallo C. V., Cordeiro Y., Silva J. L. (2012) Mutant p53 aggregates into prion-like amyloid oligomers and fibrils: implications for cancer. J. Biol. Chem. 287, 28152–28162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Muller P. A., Vousden K. H. (2014) Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25, 304–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muller P. A., Vousden K. H. (2013) p53 mutations in cancer. Nat. Cell Biol. 15, 2–8 [DOI] [PubMed] [Google Scholar]
- 13.Bourdon J. C., Fernandes K., Murray-Zmijewski F., Liu G., Diot A., Xirodimas D. P., Saville M. K., Lane D. P. (2005) p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 19, 2122–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khoury M. P., Bourdon J. C. (2010) The isoforms of the p53 protein. Cold Spring Harb. Perspect. Biol. 2, a000927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khoury M. P., Bourdon J. C. (2011) p53 isoforms: an intracellular microprocessor? Genes Cancer 2, 453–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bourdon J. C. (2007) p53 and its isoforms in cancer. Br. J. Cancer 97, 277–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marcel V., Hainaut P. (2009) p53 isoforms - a conspiracy to kidnap p53 tumor suppressor activity? Cell. Mol. Life Sci. 66, 391–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maier B., Gluba W., Bernier B., Turner T., Mohammad K., Guise T., Sutherland A., Thorner M., Scrable H. (2004) Modulation of mammalian life span by the short isoform of p53. Genes Dev. 18, 306–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J., Ruan H., Ng S. M., Gao C., Soo H. M., Wu W., Zhang Z., Wen Z., Lane D. P., Peng J. (2005) Loss of function of def selectively up-regulates Delta113p53 expression to arrest expansion growth of digestive organs in zebrafish. Genes Dev. 19, 2900–2911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujita K., Mondal A. M., Horikawa I., Nguyen G. H., Kumamoto K., Sohn J. J., Bowman E. D., Mathe E. A., Schetter A. J., Pine S. R., Ji H., Vojtesek B., Bourdon J. C., Lane D. P., Harris C. C. (2009) p53 isoforms Delta133p53 and p53beta are endogenous regulators of replicative cellular senescence. Nat. Cell Biol. 11, 1135–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davidson W. R., Kari C., Ren Q., Daroczi B., Dicker A. P., Rodeck U. (2010) Differential regulation of p53 function by the N-terminal ΔNp53 and Δ113p53 isoforms in zebrafish embryos. BMC Dev. Biol. 10, 102–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pehar M., O’Riordan K. J., Burns-Cusato M., Andrzejewski M. E., del Alcazar C. G., Burger C., Scrable H., Puglielli L. (2010) Altered longevity-assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death. Aging Cell 9, 174–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pehar M., Ko M. H., Li M., Scrable H., Puglielli L. (2014) P44, the ‘longevity-assurance’ isoform of P53, regulates tau phosphorylation and is activated in an age-dependent fashion. Aging Cell 13, 449–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ungewitter E., Scrable H. (2010) Delta40p53 controls the switch from pluripotency to differentiation by regulating IGF signaling in ESCs. Genes Dev. 24, 2408–2419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aoubala M., Murray-Zmijewski F., Khoury M. P., Fernandes K., Perrier S., Bernard H., Prats A. C., Lane D. P., Bourdon J. C. (2011) p53 directly transactivates Δ133p53α, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 18, 248–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinault C., Kawamori D., Liew C. W., Maier B., Hu J., Keller S. R., Mirmira R. G., Scrable H., Kulkarni R. N. (2011) Δ40 Isoform of p53 controls β-cell proliferation and glucose homeostasis in mice. Diabetes 60, 1210–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Slatter T. L., Hung N., Campbell H., Rubio C., Mehta R., Renshaw P., Williams G., Wilson M., Engelmann A., Jeffs A., Royds J. A., Baird M. A., Braithwaite A. W. (2011) Hyperproliferation, cancer, and inflammation in mice expressing a Δ133p53-like isoform. Blood 117, 5166–5177 [DOI] [PubMed] [Google Scholar]
- 28.Terrier O., Marcel V., Cartet G., Lane D. P., Lina B., Rosa-Calatrava M., Bourdon J. C. (2012) Influenza A viruses control expression of proviral human p53 isoforms p53β and Delta133p53α. J. Virol. 86, 8452–8460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bernard H., Garmy-Susini B., Ainaoui N., Van Den Berghe L., Peurichard A., Javerzat S., Bikfalvi A., Lane D. P., Bourdon J. C., Prats A. C. (2013) The p53 isoform, Δ133p53α, stimulates angiogenesis and tumour progression. Oncogene 32, 2150–2160 [DOI] [PubMed] [Google Scholar]
- 30.Dichtel-Danjoy M. L., Ma D., Dourlen P., Chatelain G., Napoletano F., Robin M., Corbet M., Levet C., Hafsi H., Hainaut P., Ryoo H. D., Bourdon J. C., Mollereau B. (2013) Drosophila p53 isoforms differentially regulate apoptosis and apoptosis-induced proliferation. Cell Death Differ. 20, 108–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marcel V., Fernandes K., Terrier O., Lane D. P., Bourdon J. C. (2014) Modulation of p53β and p53γ expression by regulating the alternative splicing of TP53 gene modifies cellular response. Cell Death Differ. 21, 1377–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arsic N., Gadea G., Lagerqvist E. L., Busson M., Cahuzac N., Brock C., Hollande F., Gire V., Pannequin J., Roux P. (2015) The p53 isoform Δ133p53β promotes cancer stem cell potential. Stem Cell Reports 4, 531–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim S., An S. S. (2016) Role of p53 isoforms and aggregations in cancer. Medicine (Baltimore) 95, e3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li M., Pehar M., Liu Y., Bhattacharyya A., Zhang S. C., O’Riordan K. J., Burger C., D’Adamio L., Puglielli L. (2015) The amyloid precursor protein (APP) intracellular domain regulates translation of p44, a short isoform of p53, through an IRES-dependent mechanism. Neurobiol. Aging 36, 2725–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Katoch A., George B., Iyyappan A., Khan D., Das S. (2017) Interplay between PTB and miR-1285 at the p53 3'UTR modulates the levels of p53 and its isoform Δ40p53α. Nucleic Acids Res. 45, 10206–10217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Solomon H., Bräuning B., Fainer I., Ben-Nissan G., Rabani S., Goldfinger N., Moscovitz O., Shakked Z., Rotter V., Sharon M. (2017) Post-translational regulation of p53 function through 20S proteasome-mediated cleavage. Cell Death Differ. 24, 2187–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marcel V., Perrier S., Aoubala M., Ageorges S., Groves M. J., Diot A., Fernandes K., Tauro S., Bourdon J. C. (2010) Δ160p53 is a novel N-terminal p53 isoform encoded by Δ133p53 transcript. FEBS Lett. 584, 4463–4468 [DOI] [PubMed] [Google Scholar]
- 38.Candeias M. M., Hagiwara M., Matsuda M. (2016) Cancer-specific mutations in p53 induce the translation of Δ160p53 promoting tumorigenesis. EMBO Rep. 17, 1542–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gadea G., Arsic N., Fernandes K., Diot A., Joruiz S. M., Abdallah S., Meuray V., Vinot S., Anguille C., Remenyi J., Khoury M. P., Quinlan P. R., Purdie C. A., Jordan L. B., Fuller-Pace F. V., de Toledo M., Cren M., Thompson A. M., Bourdon J. C., Roux P. (2016) TP53 drives invasion through expression of its Δ133p53β variant. eLife 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Arsic N., Ho-Pun-Cheung A., Evelyne C., Assenat E., Jarlier M., Anguille C., Colard M., Pezet M., Roux P., Gadea G. (2017) The p53 isoform delta133p53ß regulates cancer cell apoptosis in a RhoB-dependent manner. PLoS One 12, e0172125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roth I., Campbell H., Rubio C., Vennin C., Wilson M., Wiles A., Williams G., Woolley A., Timpson P., Berridge M. V., Fleming N., Baird M., Braithwaite A. W. (2016) The Δ133p53 isoform and its mouse analogue Δ122p53 promote invasion and metastasis involving pro-inflammatory molecules interleukin-6 and CCL2. Oncogene 35, 4981–4989 [DOI] [PubMed] [Google Scholar]
- 42.Harms K. L., Chen X. (2005) The C terminus of p53 family proteins is a cell fate determinant. Mol. Cell. Biol. 25, 2014–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cañadillas J. M., Tidow H., Freund S. M., Rutherford T. J., Ang H. C., Fersht A. R. (2006) Solution structure of p53 core domain: structural basis for its instability. Proc. Natl. Acad. Sci. USA 103, 2109–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ma B., Pan Y., Zheng J., Levine A. J., Nussinov R. (2007) Sequence analysis of p53 response-elements suggests multiple binding modes of the p53 tetramer to DNA targets. Nucleic Acids Res. 35, 2986–3001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ho W. C., Fitzgerald M. X., Marmorstein R. (2006) Structure of the p53 core domain dimer bound to DNA. J. Biol. Chem. 281, 20494–20502 [DOI] [PubMed] [Google Scholar]
- 46.Ma B., Pan Y., Gunasekaran K., Venkataraghavan R. B., Levine A. J., Nussinov R. (2005) Comparison of the protein-protein interfaces in the p53-DNA crystal structures: towards elucidation of the biological interface. Proc. Natl. Acad. Sci. USA 102, 3988–3993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., Berendsen H. J. (2005) GROMACS: fast, flexible, and free. J. Comput. Chem. 26, 1701–1718 [DOI] [PubMed] [Google Scholar]
- 48.Maynard A. T., Covell D. G. (2001) Reactivity of zinc finger cores: analysis of protein packing and electrostatic screening. J. Am. Chem. Soc. 123, 1047–1058 [DOI] [PubMed] [Google Scholar]
- 49.Bjelkmar P., Larsson P., Cuendet M. A., Hess B., Lindahl E. (2010) Implementation of the CHARMM force field in GROMACS: analysis of protein stability effects from correction maps, virtual interaction sites, and water models. J. Chem. Theory Comput. 6, 459–466 [DOI] [PubMed] [Google Scholar]
- 50.Nosé S., Klein M. L. (1983) Constant pressure molecular dynamics for molecular systems. Mol. Phys. 50, 1055–1076 [Google Scholar]
- 51.Bussi G., Donadio D., Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 [DOI] [PubMed] [Google Scholar]
- 52.Miyamoto S., Kollman P. A. (1992) Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 13, 952–962 [Google Scholar]
- 53.Hess B. B. H., Berendsen H. J., Fraaije J. G. (1997) Lincs: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 [Google Scholar]
- 54.Darden T., York D., Pedersen L. (1993) Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 55.Kabsch W., Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 56.Alexander R. W., Eargle J., Luthey-Schulten Z. (2010) Experimental and computational determination of tRNA dynamics. FEBS Lett. 584, 376–386 [DOI] [PubMed] [Google Scholar]
- 57.Black Pyrkosz A., Eargle J., Sethi A., Luthey-Schulten Z. (2010) Exit strategies for charged tRNA from GluRS. J. Mol. Biol. 397, 1350–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sethi A., Eargle J., Black A. A., Luthey-Schulten Z. (2009) Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA 106, 6620–6625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ang H. C., Joerger A. C., Mayer S., Fersht A. R. (2006) Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J. Biol. Chem. 281, 21934–21941 [DOI] [PubMed] [Google Scholar]
- 60.Bullock A. N., Henckel J., DeDecker B. S., Johnson C. M., Nikolova P. V., Proctor M. R., Lane D. P., Fersht A. R. (1997) Thermodynamic stability of wild-type and mutant p53 core domain. Proc. Natl. Acad. Sci. USA 94, 14338–14342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ji X., Huang Q., Yu L., Nussinov R., Ma B. (2014) Bioinformatics study of cancer-related mutations within p53 phosphorylation site motifs. Int. J. Mol. Sci. 15, 13275–13298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang Q., Yu L., Levine A. J., Nussinov R., Ma B. (2014) Dipeptide analysis of p53 mutations and evolution of p53 family proteins. Biochim. Biophys. Acta 1844(1 Pt B), 198–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bullock A. N., Fersht A. R. (2001) Rescuing the function of mutant p53. Nat. Rev. Cancer 1, 68–76 [DOI] [PubMed] [Google Scholar]
- 64.Levy C. B., Stumbo A. C., Ano Bom A. P., Portari E. A., Cordeiro Y., Silva J. L., De Moura-Gallo C. V. (2011) Co-localization of mutant p53 and amyloid-like protein aggregates in breast tumors. Int. J. Biochem. Cell Biol. 43, 60–64; erratum: 946 [DOI] [PubMed] [Google Scholar]
- 65.Silva J. L., Cino E. A., Soares I. N., Ferreira V. F., A P de Oliveira G. (2018) Targeting the prion-like aggregation of mutant p53 to combat cancer. Acc. Chem. Res. 51, 181–190 [DOI] [PubMed] [Google Scholar]
- 66.Xu J., Reumers J., Couceiro J. R., De Smet F., Gallardo R., Rudyak S., Cornelis A., Rozenski J., Zwolinska A., Marine J. C., Lambrechts D., Suh Y. A., Rousseau F., Schymkowitz J. (2011) Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 7, 285–295 [DOI] [PubMed] [Google Scholar]
- 67.Pan Y., Ma B., Venkataraghavan R. B., Levine A. J., Nussinov R. (2005) In the quest for stable rescuing mutants of p53: computational mutagenesis of flexible loop L1. Biochemistry 44, 1423–1432 [DOI] [PubMed] [Google Scholar]
- 68.Lukman S., Lane D. P., Verma C. S. (2013) Mapping the structural and dynamical features of multiple p53 DNA binding domains: insights into loop 1 intrinsic dynamics. PLoS One 8, e80221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khoo K. H., Andreeva A., Fersht A. R. (2009) Adaptive evolution of p53 thermodynamic stability. J. Mol. Biol. 393, 161–175 [DOI] [PubMed] [Google Scholar]
- 70.Wang G., Fersht A. R. (2015) Propagation of aggregated p53: cross-reaction and coaggregation vs. seeding. Proc. Natl. Acad. Sci. USA 112, 2443–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gong L., Gong H., Pan X., Chang C., Ou Z., Ye S., Yin L., Yang L., Tao T., Zhang Z., Liu C., Lane D. P., Peng J., Chen J. (2015) p53 isoform Δ113p53/Δ133p53 promotes DNA double-strand break repair to protect cell from death and senescence in response to DNA damage. Cell Res. 25, 351–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Soragni A., Janzen D. M., Johnson L. M., Lindgren A. G., Thai-Quynh Nguyen A., Tiourin E., Soriaga A. B., Lu J., Jiang L., Faull K. F., Pellegrini M., Memarzadeh S., Eisenberg D. S. (2016) A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell 29, 90–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Blandino G., Di Agostino S. (2018) New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 37, 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gong L., Pan X., Lim C. B., de Polo A., Little J. B., Yuan Z. M. (2018) A functional interplay between Δ133p53 and ΔNp63 in promoting glycolytic metabolism to fuel cancer cell proliferation. Oncogene 37, 2150–2164 [DOI] [PubMed] [Google Scholar]
- 75.Gong H., Zhang Y., Jiang K., Ye S., Chen S., Zhang Q., Peng J., Chen J. (2018) p73 coordinates with Δ133p53 to promote DNA double-strand break repair. Cell Death Differ. 25, 1063–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.