

Abstract
Background
This study aimed at investigating whether NLRP3 (the Nod like receptor family, pyrin domain‐containing 3 protein) inflammasome activation induced HMGB1 (high mobility group box‐1 protein) secretion and foam cell formation in human vascular smooth muscle cells (VSMCs) and atherosclerosis in ApoE−/− mice.
Methods and Results
VSMCs or ApoE−/− mice were treated with lipopolysaccharides (LPS) and/or ATP or LPS and high‐fat diet to induce NLRP3 inflammasome activation. HMGB1 distribution and foam cell formation in VSMCs were characterized. Liver X receptor α and ATP‐binding cassette transporter expression were determined. The impact of NLRP3 or receptor for advanced glycation end product silencing, ZYVAD‐FMK (caspase‐1 inhibitor), glycyrrhizin (HMGB1 inhibitor) or receptor for advanced glycation end product antagonist peptide on HMGB1 secretion, foam cell formation, liver X receptor α and ATP‐binding cassette transporter expression was examined. Expression level of HMGB1 in human atherosclerosis obliterans arterial tissues was characterized. Our results found that NLRP3 inflammasome activation promoted foam cell formation and HMGB1 secretion in VSMCs. Extracellular HMGB1 was a key signal molecule in inflammasome activation‐mediated foam cell formation. Furthermore, inflammasome activation‐induced HMGB1 activity and foam cell formation were achieved by receptor for advanced glycation end product/liver X receptor α /ATP‐binding cassette transporter glycyrrhizin. Experiments in vivo found glycyrrhizin significantly attenuated the LPS/high‐fat diet‐induced atherosclerosis and serum HMGB1 levels in mice. Finally, levels of HMGB1 and NLRP3 were increased in tunica media adjacent to intima of atherosclerosis obliteran arteries.
Conclusions
Our results revealed that HMGB1 is a key downstream signal molecule of NLRP3 inflammasome activation and plays an important role in VSMCs foam cell formation and atherogenesis by downregulating liver X receptor α and ATP‐binding cassette transporter expression through receptor for advanced glycation end product.
Keywords: ATP‐binding cassette transporter; foam cells; Lipid metabolites; Nod like receptor family, pyrin domain‐containing 3 protein; receptor for advanced glycation end product
Subject Categories: Inflammation, Lipids and Cholesterol, Vascular Biology, Vascular Disease, Atherosclerosis
Clinical Perspective
What Is New?
The study employed a cellular model of foam cell formation and mouse model of atherosclerosis to test the effect of NLRP3 inflammasome activation on HMGB1 secretion, foam cell formation and atherosclerosis development.
The results revealed that HMGB1 is a key downstream signal molecule of NLRP3 inflammasome activation, which may play an important role in the foam cell formation and atherogenesis by downregulating liver X receptor α and ATP‐binding cassette transporter expression through its receptor for advanced glycation end product.
What Are the Clinical Implications?
NLRP3 inflammasome activation‐dependent HMGB1 release and its downstream signal could represent a potential therapeutic target to modulate foam cell formation and atherosclerosis.
Introduction
Atherosclerosis is a pathological basis of many vascular disorders. Inhibition of the atherosclerosis process benefits patients with vascular diseases, such as aneurysm, atherosclerosis obliterans (ASO) and carotid arterial disease (CAD).1 During the atherosclerosis process, Lipid accumulates in macrophages and VSMCs to form foam cells that deposit in the artery wall.2, 3 However, the contribution of VSMC‐based foam cells to the development of atherosclerosis has been undervalued. Recent studies have demonstrated that vascular smooth muscle cells (VSMCs) can form foam cells after exposure to cholesterol or lipoprotein and VSMC‐based foam cells account for about 45% to 90%.2, 3, 4, 5 However, the mechanisms underlying the formation of VSMC‐based foam cells have not been clarified.
Inflammation is another key factor for the development of atherosclerosis. Innate immune cells can secrete inflammatory mediators, contributing to early stage of the atherosclerosis process.6, 7 Innate immune cells can recognize and be activated by pathogen‐associated molecular patterns or damage‐associated molecular patterns through pattern recognition receptors. The NLR family, pyrin domain‐containing 3 protein (NLRP3) is a member of the nucleotide‐binding oligomerization domain‐like receptors (NLRs) family which is one of the most important pattern recognition receptors. It is well known that danger signals, such as reactive oxygen species, ox‐LDL, and cholesterol crystals can activate the NLRP3, which recruits caspase‐1 through adaptor protein apoptosis‐associated speck‐like protein containing a caspase recruitment domain and assembles a protein platform called NLRP3 inflammasome, leading to caspase‐1 activation, and interleukin‐1β (IL‐1β and IL‐18 maturation and secretion.8, 9, 10, 11 Furthermore, the NLRP3 inflammation activation can also promote the cytoplasmic transportation and secretion of nuclear high mobility group box‐1 protein (HMGB1).12, 13, 14 HMGB1, which acts as an architectural chromatin‐binding factor located in the nucleus of most cell types in physiologic conditions, can be released by necrotic or damaged cells or secreted by activated immunocompetent cells.15, 16, 17, 18 The extracellular HMGB1 bind to receptor for advanced glycation end‐products (Receptor for Advanced Glycation End Product [RAGE] or Advanced Glycosylation End Product Specific Receptor [AGER]), Toll‐like receptor‐2, and Toll‐like receptor‐4 to induce signal transduction.19
Many studies have reported that NLRP3 inflammasome activation promotes the formation of macrophage foam cells through IL‐1β.20, 21, 22 In addition, it is well known that HMGB1 and its receptor RAGE contribute to the development of atherosclerosis.23, 24, 25, 26 However, whether NLRP3 inflammasome activation can induce HMGB1 secretion in VSMCs and whether HMGB1 can promote foam cell formation as well as the potential mechanism are still unclear.
In this study, we employed a cellular model of foam cell formation and mouse model of atherosclerosis to test the effect of NLRP3 inflammasome activation on HMGB1 secretion, foam cell formation, and atherosclerosis development. Our findings indicate that NLRP3 inflammasome activation promotes HMGB1 secretion from VSMCs and HMGB1 induces VSMC‐based foam cell formation by downregulating LXRα and ATP‐binding cassette transporter (ABCA1) expression through its RAGE for the first time.
Materials and Methods
The data and analytic methods are available from the corresponding author on reasonable request. All materials used in this study are available commercially from the indicated vendors.
Human Arterial Samples
The experimental protocols were approved by the Research Ethics Committee of the First Affiliated Hospital of Sun Yat‐sen University. Informed consent was obtained from individual patients and healthy subjects. ASO arterial samples were femoral arteries obtained from 6 patients, who received lower limb amputation. Healthy femoral arterial samples were acquired from 6 healthy organ donors. All arterial samples were fixed with 4% paraformaldehyde overnight and embedded in paraffin.
Primary VSMC Culture and Stimulation
Primary human VSMCs were isolated from walls of femoral artery tissues from healthy organ donors and the cells were identified by anti‐α‐SM actin staining, according to the protocols described previously.27 The cells were maintained in Dulbecco's modified Eagle’ medium (DMEM; Gibco, Karlsruhe, Germany) supplemented with 10% fetal bovine serum (FBS, Gibco), 100 units/mL penicillin and 100 μg/mL streptomycin at 37°C in a humidified incubator with 5% CO2. The cells at passages 3 to 9 were used for experiments.
To activate NLRP3 inflammasome, VSMCs were pretreated with, or without, different concentrations (100–1000 ng/mL) LPS for 16 hours and 5 mmol/L ATP (Sigma‐Aldrich, Missouri) for 1 hour. Furthermore, some cells were first treated with vehicle DMSO or 2 μmol/L Z‐YVAD‐FMK for 24 hours to inactivate caspase activity and then treated with LPS and/or ATP. Similarly, some cells were pretreated with vehicle PBS or 2 mmol/L glycyrrhizin (Sigma‐Aldrich, Missouri) for 24 hours to inhibit HMGB1 release and then stimulated with LPS and/or ATP. In addition, some cells were treated with different concentrations (0, 5, 10, 20, 50, 100 ng/mL) recombinant human HMGB1 (Sigma‐Aldrich) for 24 hours to determine the effect of extracellular HMGB1 on cell function.
Transfection Small Interfering RNA (siRNA)
To knock down NLRP3 or RAGE expression, VSMCs were cultured in 24 or 6‐well plates and when they reached at 80% confluency, the cells were transfected for 24 hours with 100 nmol/L NLRP3‐, RAGE‐specific siRNA or control siRNA (RiboBio, Guangzhou, China) using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA), according to manufacturer's instructions. The sequences were siRNA 1 5′‐GAAAUGGAUUGAAGUGAAAdTdT‐3′ (sense) and 5′‐UUUCACUUCAAUCCAUUUCdTdT‐3′ (antisense) and siRNA 2 5′‐GGAUCAAACUACUCUGUGAdTdT‐3′ (sense) and 5′‐UCACAGAGUAGUUUGAUCCdTdT‐3′ (antisense) for NLRP3; siRNA 1 5′‐GGCAGUAGUAGGUGCUCAAdTdT‐3′ (sense) and 5′‐UUGAGCACCUACUACUGCCdTdT‐3′ (antisense) and siRNA 25′‐CACUGCAGUCGGAGCUAAUdTdT‐3′ (sense) and 5′‐AUUAGCUCCGACUGCAGUGdTdT‐3′ (antisense) for RAGE; and negative control siRNA (NC group) were synthesized by Guangzhou RiboBio (Guangzhou, China).
Quantitative Real‐Time Polymerase Chain Reaction
Individual groups of VSMCs were harvested and their total RNA was extracted using Trizol reagent (Invitrogen). Individual RNA samples were reversely transcribed into cDNA using Transcriptor First Strand cDNA Synthesis Kit (Roche Applied Sciences). The relative levels of target gene mRNA transcripts were determined by quantitative real‐time polymerase chain reaction (qRT‐PCR) using the FastStart Universal SYBR Green Master (Roche Applied Sciences) and specific primers on a Roche LightCycler 480 Real Time PCR System. The sequences of primers were forward 5′‐GCACCGTCAAGGCTGAGAAC‐3′ and revere 5′ ‐TGGTGAAGACGCCAGTGGA‐3′ for GAPDH (138 bp); forward 5′‐AGCCTGGGCAACAATGACCT‐3′ and reverse 5′‐CACTCCTACCAAGAAGGCTCAAAGA‐3′ for NLRP3 (194 bp); forward 5′‐GCCGAGTTTGCCTTGCTCA‐3′ and reverse 5′‐TCCGGAGGCTCACCAGTTTC‐3′ for LXRα (187 bp); forward 5′‐GGATCATGGGCCTGGACAA‐3′ and reverse 5′‐GGGATCACTGTAGGGCAGCA‐3′ for ABCA1 (131 bp). The PCR was performed in triplicate and the data were determined by the 2−ΔΔCt.
Western Blotting
Individual groups of VSMCs were harvested and lysed in RIPA (Radio Immunoprecipitation Assay) buffer containing protease inhibitor cocktail (Roche, Basel, Switzerland), followed by centrifuging. Furthermore, the nuclear and cytoplasmic cellular proteins were extracted using the nuclear and cytoplasmic protein extraction kit (Beyotime, Nantong, China). After quantification of protein concentrations, the cell lysate or protein samples (20 μg/lane) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) on 8% to12% gels and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA). The membranes were blocked with 5% non‐fat dry milk in TBST and incubated with primary antibodies overnight at 4°C. The primary antibodies included rabbit anti‐β‐actin, anti‐LAMIN B1 (Cell Signaling Technologies, Beverly, MA), rabbit anti‐NLRP3, anti‐caspase‐1, anti‐LXRα, anti‐ABCA1 (Sigma‐Aldrich), rabbit anti‐HMGB1 (Abcam, MA) and rabbit anti‐CD36 (Abcam, MA). After being washed, the bound antibodies were detected with horseradish peroxidase (HRP)‐conjugated anti‐rabbit IgG and visualized using the enhanced chemiluminescent reagents (Millipore). The relative levels of target protein to the control β‐actin or LAMIN B1 expression were determined by densitometric analysis using a GE ImageQuant Las 4000 mini.
Enzyme‐Linked Immunosorbent Assay
The levels of HMGB1 in the supernatants of cultured VSMCs or serum sample from each mouse were measured using the HMGB1 ELISA kit (Shino‐Test Corporation, Kanagawa, Japan), according to the manufacturer's instruction.
Oil Red O Staining And Intracellular Cholesterol Measurement
To induce foam cell formation, VSMCs were treated with, or without, siRNA, ZYVAD‐FMK or glycyrrhizin and stimulated with LPS and /or ATP or recombinant human HMGB1. The cells were treated with Cholesterol–methyl‐β‐cyclodextrin (Chol:MβCD) (10 μg/mL, Sigma‐Aldrich) in 0.2% BSA for 72 hours. Chol:MβCD is the abbreviation of methyl‐beta‐cyclodextrin encapsulated cholesterol which was labelled in the Sigma catalogue as “water‐soluble cholesterol”. Chol:MβCD contains about 50 mg of cholesterol/g solid (molar ratio, 1:6 cholesterol/MβCD). Subsequently, the cells were fixed with 4% paraformaldehyde and stained with 0.3% Oil Red O in 60% isopropanol (Sigma‐Aldrich) for 15 minutes. After being washed with 60% isopropanol once and PBS 3 times, the cells were stained with hematoxylin. The stained cells were photoimaged under a microscope (Zeiss Axio Observer Z1, Jena, Germany). The contents of intracellular cholesterol in individual groups of cells were measured by fluorometric assay using the Cholesterol Quantitation Kit, according to manufacturer's instructions.
Cholesterol Efflux Assay
Cholesterol efflux were detected by Cholesterol Efflux Fluorometric Assay Kit (Biovision, Milpitas) according to the manufacturer's instruction. Briefly, VSMCs were cultured in 96 well plates and stimulated as above. After labelling in a 37°C incubator containing 5% CO2 for 16 hours, cells were treated with 50 μg/mL apoA1 as cholesterol acceptors in DMEM media for 6 hours. At the end of incubation, transfer supernatant to a new 96 well plate and measure the fluorescence (Ex/Em=482/515 nm). Add 100 μL of Cell Lysis Buffer and shaking on a plate shaker for 30 minutes at room temperature to solubilize the cells. Pipette up and down to dissolve any cell debris. Measure the fluorescence (Ex/Em=482/515 nm). % Cholesterol Efflux=Fluorescence intensity of the media/(Fluorescence intensity of the cell lysate+media).
Mice
Male ApoE−/− mice at 6 weeks of age were obtained from Charles River (Beijing, China) and housed in a specific pathogen‐free facility with a 12:12 hours light‐dark cycle and free access to food and water. 32 mice were fed with high fat diet (HFD, 1.2% cholesterol, 15% fat and 0.2% sodium cholate, Guangdong Medical Laboratory Animal Center, Guangzhou, China). The mice were randomized and injected intraperitoneally with 0.8 mg/kg LPS alone 3 times per week (HFD+LPS group, n=8), LPS+PBS (HFD+LPS+PBS group, n=8) or LPS+glycyrrhizin (20 mg/kg, injected at 16 hours post LPS injection, HFD+LPS+Glycyrrhizin group,n=8). Other 16 mice were fed with normal chow diet (Xietong Organism, Jiangsu, China). 8 mice were only fed with normal chow diet as the control (control group,n=8) and the other 8 mice were injected with LPS (0.8 mg/kg, the LPS group, n=8). Individual groups of mice (n=8 per group) were euthanized at 16 weeks post HFD and LPS injection. The aorta from aortic arch to the abdominal aorta of individual mice was dissected out and stained with 0.3% Oil Red O in 60% isopropanol (Sigma‐Aldrich) for 5 minutes, followed by imaged. The lesion areas of individual samples were calculated in a masked manner.
Immunofluorescence
The distribution and contents of HMGB1 and NLRP3 in individual healthy arteries and ASO arterial samples were examined by immunofluorescence. Briefly, 3 consecutive tissue sections (5 μm) were deparaffinized, rehydrated, and subjected to antigen retrieval in 0.01 mol/L citrate buffer (pH 6.0) in a pressure cooker for 5 minutes, followed by treatment with 3% hydrogen peroxide for 5 minutes. One section from individual samples was stained with hematoxylin and eosin and the remaining 2 sections were blocked with 5% BSA and stained with rabbit anti‐NLRP3 (1:200) or rabbit anti‐HMGB1 (1:250) and mouse anti‐α‐SM actin (1:500) or mouse anti‐CD68 (1:500) overnight at 4°C. After being washed, the sections were incubated with Alexa488‐conjugated goat anti‐rabbit (1:1000) or Alexa594‐conjugated donkey anti‐mouse (1:1000, Thermo Fisher Scientific, Waltham, MA) for 2 hours at room temperature and counterstained with 40,6‐diamidino‐2‐phenylindole (DAPI, Thermo Fisher Scientific, Waltham, MA). The sections were examined and imaged under an inverted fluorescence microscope (Zeiss Axio Observer Z1, Jena, Germany). The fluorescent signals were measured using the ImageJ software.
Statistical Analysis
All data are expressed as the mean±SD. The difference between 2 groups was analyzed by independent sample t test and the difference among groups was determined by one‐way analysis of variance (ANOVA) and post hoc Bonferroni test using the SPSS 19.0 (SPSS, Chicago, IL). A P<0.05 was considered statistically significant.
Results
LPS and ATP Induce the Activation of NLRP3 Inflammasomes and Promote HMGB1 Secretion in Human Primary VSMCs
Engagement of pattern recognition receptors by damage‐associated molecular patterns and pathogen‐associated molecular patterns can activate NLRP3 inflammasome in many types of cells by inducing caspase‐1 activation. To activate NLRP3 inflammasome in primary VSMCs, human primary arterial VSMCs were treated with different concentrations of LPS for 16 hours, and/or 5 mmol/L ATP for another 1 hour. The relative levels of NLRP3 mRNA transcripts and protein expression as well as cleaved caspase‐1 were determined by qRT‐PCR and Western blot. In comparison with the unmanipulated controls, stimulation with LPS, but not ATP, alone significantly upregulated NLRP3 mRNA transcripts and protein expression but did not significantly alter the levels of cleaved caspase‐1 (Figure 1A through 1C). Treatment with different doses of LPS, together with ATP increased the levels of NLRP3 expression and cleaved caspase‐1 in VSMCs in a dose‐dependent manner (Figure 1C), indicating that ATP enhanced the activation of NLRP3 inflammasomes induced by LPS in VSMCs.
Figure 1.
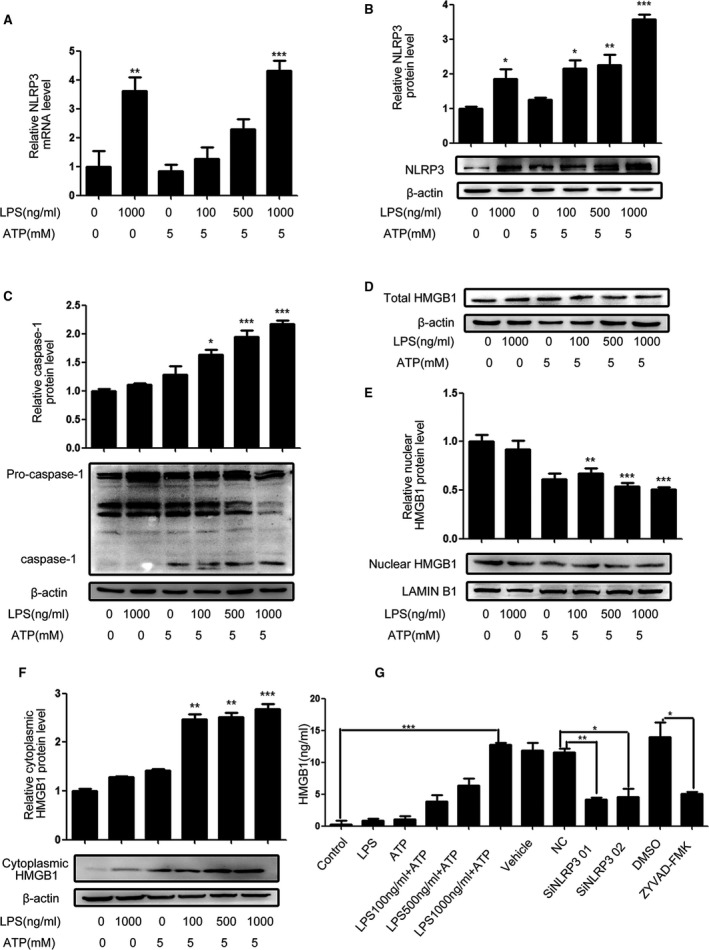
LPS/ATP activate NLRP3 inflammasomes in VSMCs. Human primary VSMCs were treated with vehicle or the indicated concentrations of LPS for 16 hours and/or ATP for 1 hour. Some cells were transfected with control or NLRP3‐specific siRNA or treated with vehicle DMSO or ZYVAD‐FMK, followed by treatment with LPS and/or ATP. The relative levels of NLRP3 and cleaved caspase‐1 expression were determined by qRT‐PCR and Western blot. The distribution of HMGB1 in the different groups of cells and their cultured supernatants were determined by Western blot and ELISA. Data are representative images or expressed as the mean±SD of each group from 4 separated experiments (Western blot) or 3 separated experiments with 4 duplicated wells (qRT‐PCR and ELISA). A, The levels of NLRP3 mRNA transcripts. B, The levels of NLRP3 proteins. C, The levels of cleaved caspase‐1. D, The levels of total HMGB1. E, The levels of nuclear HMGB1. F, The levels of cytoplasmic HMGB1. G, The levels of HMGB1 in the supernatants. *P<0.05, † P<0.01, ‡ P<0.001 vs the control. ATP indicates adenosine triphosphate; DMSO, dimethyl sulfoxide; ELISA, enzyme linked immunosorbent assay; HMGB1, high mobility group box‐1 protein; LPS, lipopolysaccharides; mRNA, messenger RNA; NLRP3, The Nod like receptor family, pyrin domain‐containing 3 protein; qRT‐PCR, quantitative real‐time polymerase chain reaction; siRNA, small interfering RNA; VSMC, vascular smooth muscle cell.
Next, we investigated whether NLRP3 activation by LPS and ATP could induce the cytoplasmic transportation and secretion of nuclear HMGB1 in VSMCs. As shown in Figure 1D through 1F, treatment with LPS or ATP alone did not significantly change the levels of total, nuclear, and cytoplasmic HMGB1 in VSMCs while treatment with both LPS and ATP significantly reduced the levels of total and nuclear HMGB1 but increased the levels of cytoplasmic HMGB1 in VSMCs in a dose‐dependent manner. Further ELISA revealed a similar pattern of HMGB1 levels in the supernatants of different groups of cultured cells (Figure 1G). Moreover, knockdown of NLRP3 by specific siRNAs or inhibition of caspase‐1 activity by ZYVAD‐FMK dramatically reduced the levels of HMGB1 in the supernatants of cultured cells that had been treated with LPS and ATP. Hence, activation of NLRP3 inflammasomes by LPS/ATP promoted the cytoplasmic transportation of nuclear HMGB1 and secretion in VSMCs, dependent on the caspase‐1 activity.
NLRP3 Inflammasome Activation Increased Lipid Accumulation in VSMCs, Which is Related to HMGB1
We further examined whether NLRP3 inflammasome‐dependent HMGB1 secretion could induce foam cell formation in VSMCs. Following treatment with LPS and/or ATP, the cells were treated with Chol:MβCD for 72 hours and the contents of intracellular lipids in VSMCs were characterized by Oil Red O staining and Cholesterol Quantitation Kit. NLRP3 inflammasome activation induced by LPS and ATP significantly increased the levels of cholesterol in VSMCs (Figure 2A and 2E), which was attenuated by NLRP3 silencing or treatment with Z‐YVAD‐FMK (Figure 2B, 2C, and 2E). Glycyrrhizin is an inhibitor of HMGB1 and can inhibit HMGB1 nuclear release and extracellular HMGB1 signaling. Treatment with glycyrrhizin significantly reduced the levels of intracellular cholesterol in the LPS/ATP‐treated VSMCs (Figure 2D). In contrast, treatment with recombinant human HMGB1 increased the levels of intracellular cholesterol in the Chol:MβCD‐treated VSMCs in a dose‐dependent manner. These results indicated that NLRP3 inflammasome activation and HMGB1 secretion promoted the cholesterol accumulation in VSMCs.
Figure 2.
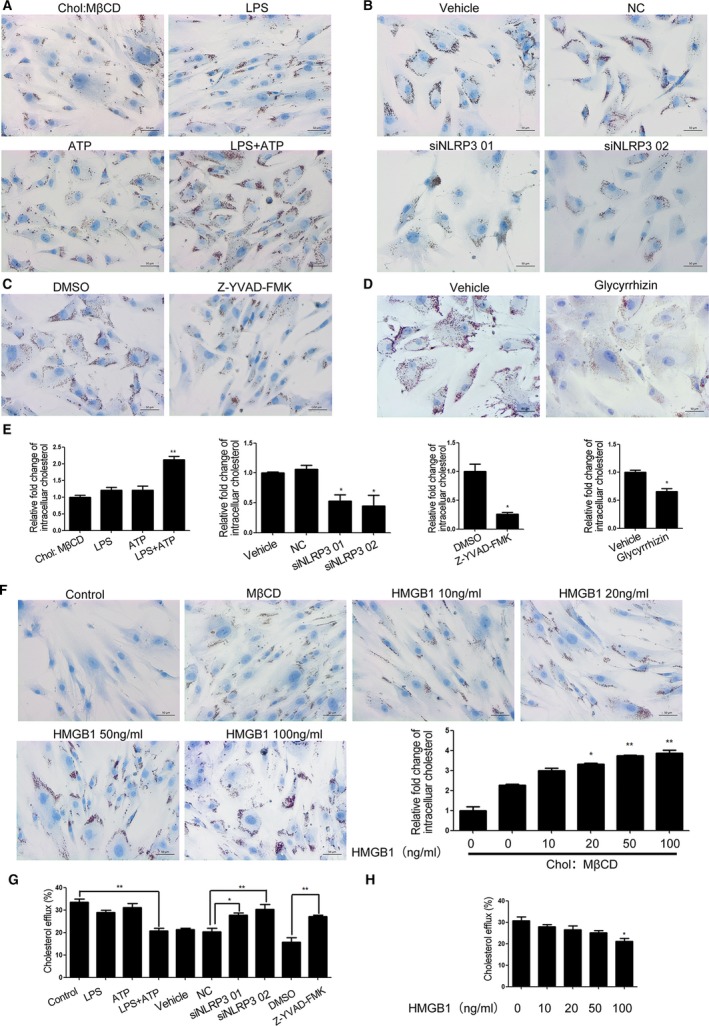
NLRP3 inflammasome activation and HMGB1 promote the cholesterol accumulation and reduce cholesterol efflux in VSMCs. VSMCs were treated with, or without, LPS and/or ATP in the presence of Chol:MβCD (10 μg/mL) for 72 hours. Some cells were transfected with control or NLRP3‐specific siRNA or pretreated with ZYVAD‐FMK or Glycyrrhizin and challenged with LPS and/or ATP in the presence of Chol:MβCD. Other cells were treated with, or without, the indicated concentrations of HMGB1 in the presence of Chol:MβCD. The intracellular cholesterol was stained by Oil Red O, and imaged and the contents of cellular cholesterol were determined by a fluorometric assay. Cholesterol efflux were detected by Cholesterol Efflux Fluorometric Assay Kit. Data are representative images or expressed as the mean±SD of each group from 3 separated Cholesterol Efflux Fluorometric Assay experiment with 4 to 6 duplicated wells. A, NLRP3 inflamasome activation promoted cholesterol accumulation in VSMCs. B through E, Inhibition of caspase‐1 or HMGB1 or NLRP3‐sielncing mitigated the NLRP3 inflammasome activation‐promoted cholesterol accumulation in VSMCs. F, HMGB1 promoted cholesterol accumulation in VSMCS in a dose‐dependent manner. G, NLRP3 inflamasome activation reduced cholesterol efflux which could be mitigated by NLRP3‐sielncing and caspase‐1 inhibitor. H, HMGB1 inhibited cholesterol efflux in VSMCs. *P<0.05, † P<0.01, vs the control. ATP indicates adenosine triphosphate; Chol:MβCD, cholesterol–methyl‐β‐cyclodextrin; HMGB1, high mobility group box‐1 protein; LPS, lipopolysaccharides; NC, negative control; NLRP3, The Nod like receptor family, pyrin domain‐containing 3 protein; siRNA, small interfering RNA; VSMC, vascular smooth muscle cell.
NLRP3 Inflammasome‐Dependent HMGB1 Secretion Inhibit Cholesterol Efflux in VSMCs
Lipid accumulation is an imbalance between cholesterol influx and efflux. To see whether NLRP3 inflammasome activation induced lipid accumulation was dependent on influx of lipids, we analyzed expression of scavenger receptor CD36 with the stimulation of LPS and/or ATP as well as HMGB1. We found that LPS alone are able to upregulated CD36 in both mRNA and protein level in VSMCs (Figure S1A). However, treatment VSMCs with recombinant human HMGB1 did not change CD36 significantly (Figure S1B). Thus, upregulation of CD36‐dependent lipid uptake was not a major cause of HMGB1‐dependent foam cell formation.
Next, we investigated the effects of NLRP3 inflammasome activation and extracellular HMGB1 on cholesterol efflux from VSMCs. VSMCs were treated with LPS and/or ATP or HMGB1 for 24 hours. Cholesterol efflux was initiated by the addition of apoA1. The results revealed that cholesterol efflux to apoA1 was decreased after NLRP3 inflammasome activation or HMGB1 stimulation. Transfection with NLRP3 specific siRNA as well as ZYVAD‐FMK rescued cholesterol efflux in the LPS/ATP‐treated VSMCs (Figure 2G).
The results above indicated that NLRP3 inflammasome activation and HMGB1 secretion inhibited the cholesterol efflux in VSMCs.
NLRP3 Inflammasome Activation Downregulates ABCA1 Expression by Inhibiting the LXRα Expression in VSMCs
Dysregulation of ABCA1 and ABCG1‐dependent cholesterol efflux is crucial for foam cell formation, and both ABCA1 and ABCG1 expression are regulated by liver X receptor (LXRs). LXRs are ligand activated transcription factors, which contain 2 homologies (LXRα and LXRβ). LXRα is expressed primarily in the liver, intestine, adipose tissues, macrophages and VSMCs while LXRβ is expressed ubiquitously. Though expression of LXRα is not as broad as LXRβ, it is much more crucial for lipid metabolism in VSMCs. To understand how NLRP3 inflammasome activation affected the cholesterol accumulation in VSMCs, we determined the relative levels of LXRα, ABCA1 and ABCG1 expression in the LPS/ATP‐treated VSMCs. We found that treatment with LPS or ATP alone did not change the relative levels of LXRα, ABCA1 and ABCG1 in VSMCs while treatment with both significantly reduced the relative levels of LXRα and ABCA1, but not ABCG1 (Data of ABCG1 have not been shown), mRNA transcripts and protein expression in VSMCs (Figure 3A and 3B). In contrast, treatment with ZYVAD‐FMK or NLRP3 silencing rescued LXRα and ABCA1 in the LPS/ATP‐treated VSMCs (Figure 3C through 3F). Furthermore, pre‐treatment with T0901317, an agonist of LXRα, significantly increased the levels of ABCA1 expression in VSMCs and mitigated the NLRP3 inflammasome activation‐reduced ABCA1 expression in VSMCs (Figure 3G and 3H). Together, these data indicated that NLRP3 inflammasome activation induced cholesterol accumulation in VSMCs was achieved by inhibiting the LXRα‐mediated ABCA1 expression.
Figure 3.
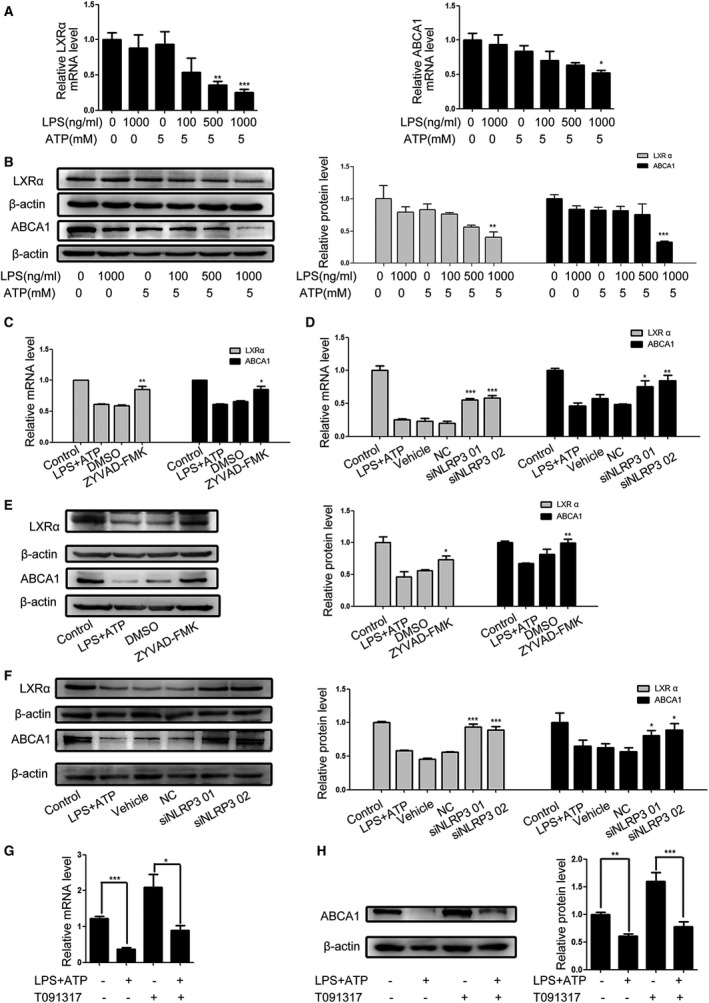
NLRP3 inflammasome activation reduces LXRα and ABCA1 expression in VSMCs. Following treatment with the indicated drugs and LPS/ATP stimulation, the relative levels of LXRα and ABCA1 expression were determined qRT‐PCR and Western blot. Data are representative images or expressed as the mean±SD of each group from 3 separated experiments (Western blot) or 3 separated experiments with 3 duplicated wells (qRT‐PCR). A, qRT‐PCR analysis of LXRα and ABCA1 mRNA transcripts. B, Western blot analysis of LXRα and ABCA1 proteins. C through F, Treatment with ZYVAD‐FMK or NLRP3 silencing mitigated the LPS/ATP‐reduced LXRα and ABCA1 expression. G and H, Treatment with T091317, the LXRα agonist enhanced ABCA1 expression and mitigated the LPS/ATP‐reduced ABCA1 expression. *P<0.05, † P<0.01, ‡ P<0.001 the control. ABCA1 indicates ATP‐binding cassette transporter 1; ATP, adenosine triphosphate; LPS, lipopolysaccharides; LXRα, liver X receptor α; mRNA, messenger RNA; NC, negative control; NLRP3, The Nod like receptor family, pyrin domain‐containing 3 protein; qRT‐PCR, Quantitative real‐time polymerase chain reaction; VSMC, vascular smooth muscle cell.
HMGB1 Downregulates LXRα and ABCA1 Expression in VSMCs
Given that HMGB1 was crucial for the foam cell formation induced by ATP and LPS treatment in VSMCs, we examined the importance of HMGB1 in LPS and ATP reduced LXRα and ABCA1 expression in VSMCs. We found that treatment with glycyrrhizin to inhibit HMGB1 activity significantly mitigated the LPS/ATP‐reduced LXRα and ABCA1 expression in VSMCs (Figure 4A and 4B). Furthermore, treatment with HMGB1 alone reduced the levels of LXRα and ABCA1 expression in VSMCs in a dose‐dependent manner (Figure 4C and 4D). Pre‐treatment with T091317 alone enhanced the levels of ABCA1 expression while combination of it with HMGB1 mitigated the HMGB1‐reduced ABCA1 expression in VSMCs. These data demonstrated that HMGB1 inhibited the LXRα and ABCA1 expression in VSMCs.
Figure 4.
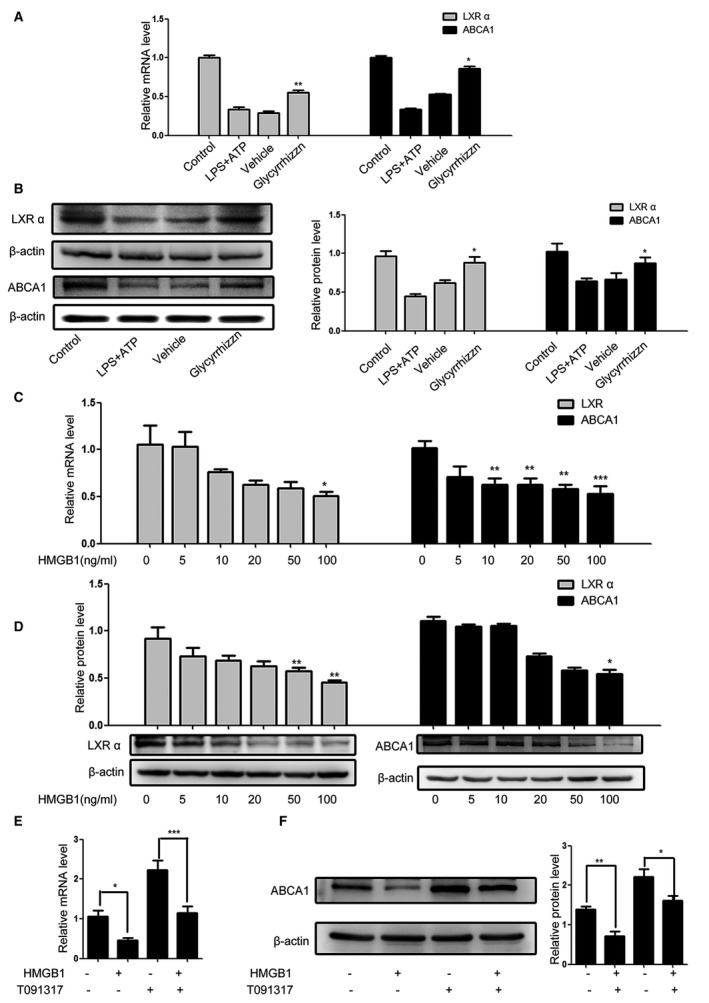
HMGB1 reduces LXRα and ABCA1 expression in VSMCs. Following pretreatment with HMGB1 and/or glycyrrhizin the HMGB1 inhibitor and LPS/ATP stimulation, the relative levels of LXRα and ABCA1 expression were determined by qRT‐PCR and Western blot. Data are representative images or expressed as the mean±SD of each group from 3 separated experiments (Western blot) or 3 separated experiments with 3 duplicated wells (qRT‐PCR). A and B, inhibition of HMGB1 mitigated the LPS/ATP‐reduced LXRα and ABCA1 expression. C and D, HMGB1 reduces the levels of LXRα and ABCA1 expression in a dose‐dependent manner. E and F, Treatment with T091317 mitigated the HMGB1‐reduced ABCA1 expression. *P<0.05, † P<0.01, ‡ P<0.001 vs the control. ABCA1 indicates ATP‐binding cassette transporter 1; ATP, adenosine triphosphate; HMGB1, high mobility group box‐1 protein; LPS, lipopolysaccharides; LXRα, Liver X receptor α; NC, negative control; qRT‐PCR, Quantitative real‐time polymerase chain reaction; VSMC, vascular smooth muscle cell.
NLRP3 Inflammasome‐Dependent HMGB1 Secretion Downregulates LXRα and ABCA1 Expression by Binding to RAGE
Extracellular HMGB1 exerts its function by binding to its receptors, such as RAGE. We further examined the importance of RAGE in the HMGB1‐mediated cholesterol accumulation in VSMCs. Transfection with RAGE‐specific siRNA, but not negative control (NC) significantly reduced the levels of intracellular cholesterol and increased cholesterol efflux in VSMCs. Treatment with RAGE antagonist peptide, but not vehicle (DMSO), also decreased the levels of intracellular cholesterol and promoted cholesterol efflux in VSMCs (Figure 5A through 5D). Similarly, knockdown of RAGE or treatment with RAGE antagonist peptide significantly mitigated the LPS/ATP‐reduced LXRα and ABCA1 expression in VSMCs (Figure 5E through 5H). The results indicated that the foam cell formation induced by LPS/ATP‐mediated NLRP3 inflammasome activation was dependent on the binding of HMGB1 to RAGE in VSMCs.
Figure 5.
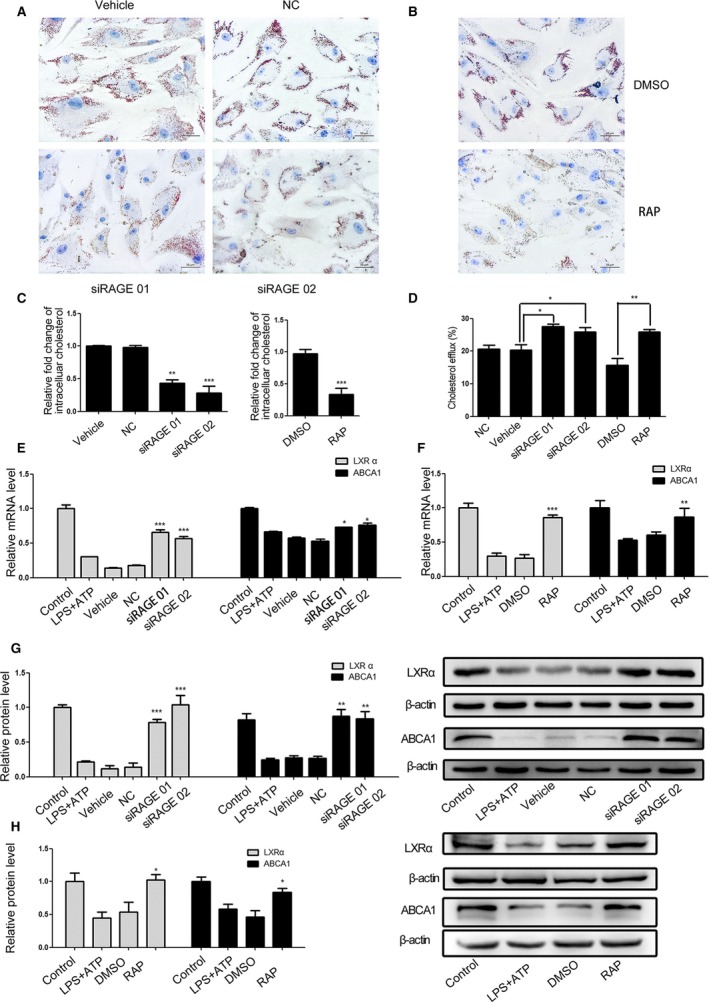
NLRP3 inflammasome‐dependent HMGB1 secretion reduces LXRα and ABCA1 expression through binding to RAGE in VSMCs. Following NLRP3 silencing, pretreatment with RAP, a RAGE antagonist peptide and LPS/ATP stimulation in the presence or absence of Chol:MβCD, the cholesterol accumulation in VSMCs was characterized and the relative levels of LXRα and ABCA1 expression were determined by qRT‐PCR and Western blot. Data are representative images or expressed as the mean±SD of each group from 3 separated experiments (Western blot) or 3 separated experiments with 3 duplicated wells (qRT‐PCR and Cholesterol Efflux Fluorometric Assay). A through C, The cholesterol accumulation in VSMCs. D, The cholesterol efflux in VSMCs. E through H, The relative levels of LXRα and ABCA1 expression in the different groups of VSMCs. *P<0.05, † P<0.01, ‡ P<0.001 vs the control. ABCA1 indicates ATP‐binding cassette transporter 1; HMGB1, high mobility group box‐1 protein; LPS, lipopolysaccharide; LXRα, Liver X receptor α; NC, negative control; NLRP3, The Nod like receptor family, pyrin domain‐containing 3 protein; qRT‐PCR, Quantitative real‐time polymerase chain reaction; RAGE, receptor for advanced glycation end product; RAP, receptor for advanced glycation end product antagonist peptide; Chol:MβCD, cholesterol–methyl‐β‐cyclodextrin; VSMC, vascular smooth muscle cell.
Glycyrrhizin Reduces Atherosclerotic Plaque Formation and Serum HMGB1 Levels In Vivo
It has been reported that LPS can induce NLRP3 inflammasome activation in mice.28, 29 To evaluate the importance of NLRP3 inflammasome activation and HMGB1 secretion in the development of atherosclerosis, ApoE−/− mice were injected intraperitoneally with LPS and/or fed with HFD to induce atherosclerosis. We found that injection with LPS alone or feeding with HFD induced mild or moderate levels of arterial plaque formation while combination of LPS injection and HFD feeding promoted severe levels of arterial plaque formation in mice (Figure 6A). Treatment with glycyrrhizin significantly mitigated the arterial plaque formation in the mice injected with LPS and fed with HFD. Similarly, feeding with HFD, but not injection with LPS significantly increased the levels of serum HMGB1 and combination of both treatments further increased the levels of serum HMGB1 in mice (Figure 6B). In contrast, treatment with glycyrrhizin significantly reduced the levels of serum HMGB1 in the mice, relative to that of the vehicle (PBS) ‐treated mice that had been injected with LPS and fed with HFD. These demonstrated that LPS and HFD feeding induced arterial plaque formation and promoted HMGB1 secretion in mice, which was attenuated by inhibition of HMGB1.
Figure 6.
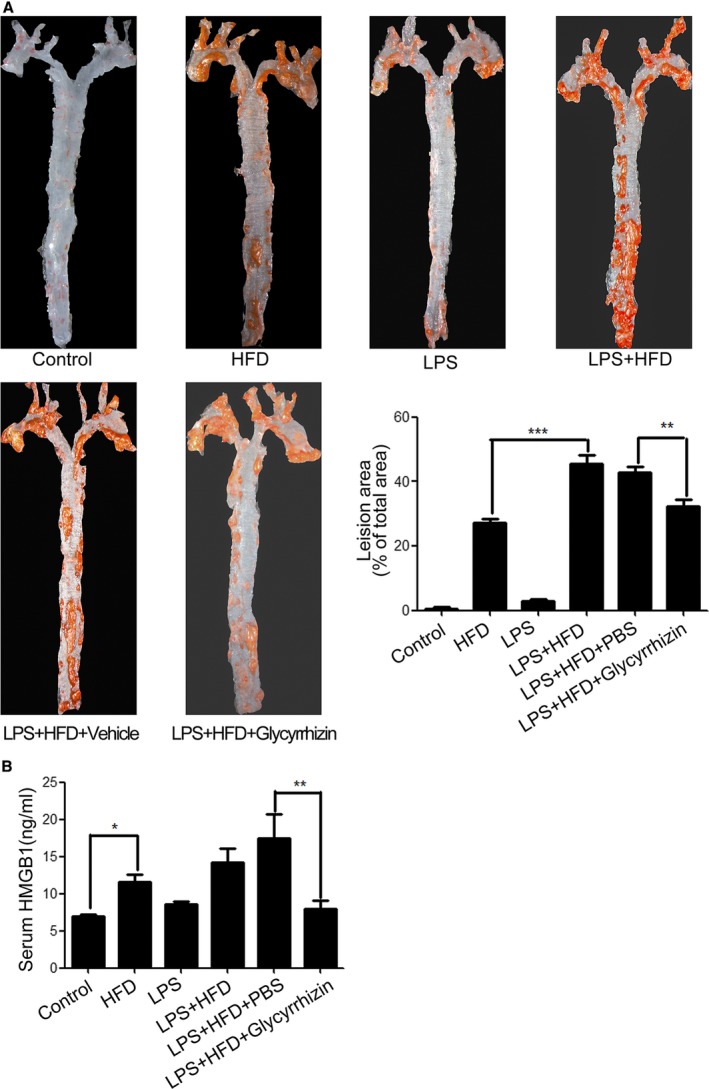
NLRP3 inflammasome‐dependent HMGB1 secretion promotes atherosclerosis development in mice. ApoE−/− mice were fed with HFD and/or injected with LPS for 16 weeks. Some mice were pre‐treated with Glycyrrhizin and stimulated by HFD and LPS injection (n=8 per group). At the end, the mice were euthanized, and their arteries were dissected and imaged. The atherosclerotic plaque areas and the levels of serum HMGB1 in individual mice were measured in a masked manner. A, The atherosclerotic plaque formation in mice. B, The levels of serum HMGB1. *P<0.05, † P<0.01, ‡ P<0.001 vs the control. ApoE−/− mice indicates apolipoprotein E knock out mice; HFD, high‐fat diet; HMGB1, high mobility group box‐1 protein; LPS, lipopolysaccharides; NLRP3, The Nod like receptor family, pyrin domain‐containing 3 protein.
Finally, we characterized the expression level and distribution of HMGB1 in human healthy and ASO arteries tissues by immunofluorescence (Figures 7 and 8). HMGB1 protein expression levels were increased in ASO arteries compared with normal arteries. Moreover, HMGB1 was only detected in the nucleus of VSMCs from healthy arteries and morphologically normal tunica media of ASO arterial tissues (Arrow) but observed in both nucleus and extranuclear area in tunica media closed to neointima (asterisk in Figure 7) and in neointima (triangle in Figure 7) of ASO arteries. In addition, lower levels of α‐SM actin and higher levels of NLRP3 expression were observed in the same area (asterisk in Figure 7), indicating that higher levels of NLRP3 expression were associated with extranuclear HMGB1 expression. To identify distribution relationship between NLRP3, HMGB1 and macrophage, the co‐staining of NLRP3 and HMGB1 with macrophage marker CD68 also been detected. As shown in Figure 8, there were few or no CD68‐positive cells in normal arteries or vascular wall of ASO arteries. In contrast, CD68‐positive cells were observed within plaques of ASO arteries and part of these cells was positive for HMGB1 and NLRP3 (Figure 8B). This results indicated that in artery plaque, NLRP3 inflammasome was activated in macrophage as well as other cell types (such as VSMCs). Moreover, macrophage was a source of HMGB1 in arterial plaque, but it was not the only source.
Figure 7.
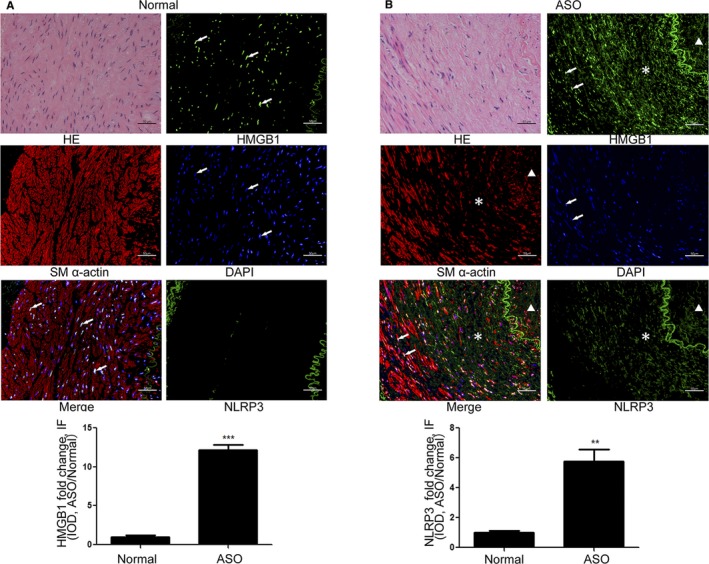
NLRP3 and HMGB1 expression were upregulated in human ASO arterial samples. The expression of NLRP3, HMGB1, and α‐SM actin in the healthy arterial and ASO tissues (n=6 per group) was characterized by immunofluorescence using anti‐NLRP3, anti‐HMGB1, and anti‐α‐SM actin as well as DAPI staining. Data are representative images or fluorescent signals expressed as the mean±SD of each group (6 sections of each group and every section was chosen 3 views randomly). A, Normal arteries. B, ASO arterial samples. † P<0.01, ‡ P<0.001 vs the control. ASO indicates atherosclerosis obliterans; HE, hematoxylin‐eosin staining; HMGB1, high mobility group box‐1 protein; NLRP3, The Nod like receptor family, pyrin domain‐containing 3 protein; α‐SM actin, α smooth muscle actin, DAPI, 4′,6‐diamidino‐2‐phenylindole.
Figure 8.
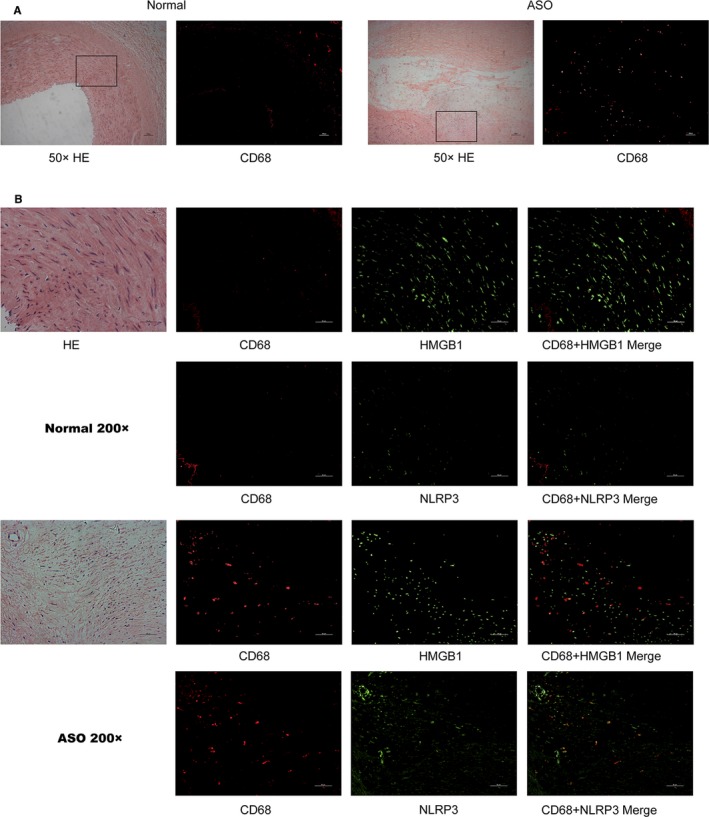
The co‐staining of NLRP3 and HMGB1 with macrophage marker CD68. The expression of NLRP3, HMGB1 and CD68 in the healthy arterial and ASO tissues (n=6 per group) was characterized by immunofluorescence using anti‐NLRP3, anti‐HMGB1, and anti‐CD68 as well as DAPI staining. A, CD68 expression in ×50 magnification. B, CD68 expression in ×200 magnification. ASO indicates atherosclerosis obliterans; HE, hematoxylin‐eosin staining; HMGB1, high mobility group box‐1 protein; NLRP3, The Nod like receptor family, pyrin domain‐containing 3 protein.
Discussion
Both NLRP3 inflammasome activation‐induced innate immunity and foam cell formation are crucial for the development of atherosclerosis. NLRP3 inflammasome activation facilitate HMGB1 transporting from the nucleus to the cytoplasm and secretion in macrophages.13, 30 In this study, we found that NLRP3 inflammasome activation by LPS and ATP promoted HMGB1 secretion in the cultured human primary VSMCs. Extracellular HMGB1 through its RAGE induced cholesterol accumulation and foam cell formation by downregulating LXRα‐ABCA1 pathway in VSMCs. Given that foamed VSMCs contribute to the development of atherosclerosis our novel findings may provide new insights into the pathogenesis of atherosclerosis.
Pathogen‐associated molecular patterns and damage‐associated molecular patterns, such as microbial LPS, can activate the nuclear factor‐κB signaling to induce pro‐IL‐1β and NLRP3 expression in innate immune cells.11 These, together with other factors, such as ATP, triggers NLRP3 inflammasome activation that activates caspase‐1 and induces IL‐1β maturation by inducing potassium (K+) efflux, pore formation in cell membranes, lysosomes damage, elevation of reactive oxygen species or damage in the mitochondria.31, 32 In recent years, it has been reported that activation of NLRP3 inflammasome induced HMGB1 secretion in macrophage in a caspase‐1 dependent manner. Evidentially, our data indicated stimulation with both LPS and ATP, but not either alone reduced the levels of nuclear HMGB1 but increased the levels of cytoplasmic HMGB1 and it in the supernatants of cultured VSMCs. These novel data extended previous observation that NLRP3 inflammasome activation promoted HMGB1 secretion and support the notion that HMGB1 can come from human VSMCs, the potential therapeutic targets during the process of atherosclerotic vascular diseases.23
Foam cell formation in macrophages is a key event in the process of atherosclerosis. Indeed, VSMC is also a non‐negligible source of foam cells3, 4 and VSMCs‐based foam cells account for 45% of total foam cells in an advanced atherosclerosis leision.33 Previous studies have shown that NLRP3 inflammasome activation induces the formation of macrophage foam cells22, 34 and higher levels of HMGB1 are also associated with foam cell formation and atherosclerosis.23, 24 In our study, we found that LPS and ATP triggered foam cell formation in human primary VSMCs, which was attenuated by pre‐treatment with the HMGB1 inhibitor of glycyrrhizin, indicating that the importance of HMGB1 in NLRP3 inflammasome activation‐induced foam cell formation in VSMCs. More importantly, treatment with recombinant human HMGB1 promoted the formation of foamed VSMCs by enhancing the accumulation of cholesterol in VSMCs. In addition, high levels of HMGB1 were associated with NLRP3 inflammasome‐induced atherosclerosis, which was mitigated by treatment with glycyrrhizin in mice. These further indicated that HMGB1 was crucial for the formation of atherosclerotic plaques in the arterial tissues of mice. Moreover, immunofluorescent analysis revealed that high levels of HMGB1 and NLRP3 were detected in human ASO arterial tissues. These independent lines of evidence demonstrated that HMGB1 secretion induced by NLRP3 inflammasome activation was responsible for mediating foam VSMC formation and subsequent atherosclerosis. These novel findings suggest that HMGB1 may be a therapeutic target for intervention of atherosclerosis.
Foam cell formation depends on intracellular cholesterol homeostasis and is regulated by cholesterol uptake and efflux in macrophages and VSMCs. CD36 is one of the main receptors that mediate cholesterol uptake. Furthermore, it is well known that ABCA1 and ABCG1 can facilitate cholesterol efflux.35 ABCA1 and ABCG1 expression are dependent on LXRs, the nuclear receptors, which act as intracellular sensors of cholesterol and regulating cholesterol metabolism.36 LXRα is important in maintaining cholesterol homeostasis and LXRα−/− mice are prone to development of atherosclerosis.37 In this study, we found that LPS alone was able to upregulating CD36 expression in VSMC but recombinant human HMGB1 did not change CD36 significantly. In contrast, NLRP3 inflammasome activation by LPS/ATP reduced the levels of LXRα and ABCA1 expression in VSMCs, which was mitigated by pre‐treatment with glycyrrhizin. Similarly, exogenous HMGB1 decreased the levels of LXRα and ABCA1 expression in VSMCs in a dose‐dependent manner while pre‐treatment with T091317, the LXRα agonist prevented the HMGB1 or NLRP3 inflammasome activation‐reduced ABCA1 expression in VSMCs. These indicated that HMGB1 secretion induced by NLRP3 inflammasome activation acted as an inhibitor of LXRα to inhibit ABCA1 expression in VSMCs. It is well known that HMGB1 can bind to its receptors, such as RAGE, Toll‐like receptor‐2, and Toll‐like receptor‐4 and contribute to inflammatory process.19, 38, 39 Engagement of RAGE by S100B and AGEs can disturb the LXRα/ABCA1 pathway and inhibiting of the RAGE signaling by ursodeoxycholic acid attenuates the process of atherosclerosis.26, 40, 41 Indeed, we found that treatment RAGE antagonist peptide, an antagonist of RAGE or RAGE silencing mitigated the HMGB1 or NLRP3 inflammasome activation‐induced cholesterol accumulation and the reduction of LXRα and ABCA1 expression in VSMCs. Our data uncovered that NLRP3 inflammasome activation by LPS/ATP promoted HMGB1 secretion, which through its RAGE to downregulate LXRα and ABCA1 expression, leading to cholesterol accumulation in VSMCs and foamed VSMC formation as well as atherosclerosis development. These findings may provide new explanation why inhibition of the RAGE signaling by ursodeoxycholic acid attenuates the process of atherosclerosis.
As another vital source of foam cells, we also examined NLRP3‐HMGB1 pathway in macrophages. We found lower concentration of LPS (50–100 ng/mL) was able to downregulate LXRα and ABCA1 in both mRNA and protein levels. However, recombinant human HMGB1 could not reduce LXRα and ABCA1 drastically as LPS did (Figure S2). In addition, in artery plaque of ASO samples, only scattered HMGB1 or NLRP3‐positive cells were positive for CD68 indicating macrophage was not the only source of NLRP3 and HMGB1 in atherosclerosis. Therefore, macrophage play an important role in inflammation‐related foam cell formation, but in HMGB1‐mediated foam cell formation, macrophage may not have an advantage over VSMCs.
In summary, our data indicate that HMGB1 secretion is a new mechanism by which NLRP3 inflammasome activation induced foam VSMC formation and atherosclerosis. Extracellular HMGB1 through its RAGE disturbed the LXRα‐ABCA1 pathway to promote cholesterol accumulation in VSMCs.
Sources of Funding
The research was supported by the National Natural Science Foundation of China (No. 81270378, 81070258, 81370368, 81300237, 81670439, 81600336), and was supported in part by grants from Guangdong Province Industry‐Academia‐Research Program (2011B090400117), the Guangdong Department of Science & Medicine Center grant (2011A080300002), Guangzhou Science and Technology Plan Projects (2011Y2‐00022), Guangdong Department of Finance grant (2014SC104, 20160901), Science and Technology Program of Guangzhou (201510010040) and Guangdong Engineering Laboratory for Diagnosis and Treatment of Vascular Disease grant (2015B090903064), and Pearl River S&T Nova Program of Guangzhou (201806010006).
Disclosures
None.
Supporting information
Figure S1. NLRP3 inflammasome activation and HMGB1 in regulating CD36 expression in VSMCs. Treatment with LPS/ATP stimulation or HMGB1, the relative levels of CD36 expression were determined qRT‐PCR and Western blot. Data are representative images or expressed as the mean ± SD of each group from 3 separated experiments (Western blot) or 3 separated experiments with 3 duplicated wells (qRT‐PCR). A, qRT‐PCR analysis of CD36 mRNA transcripts. (B) Western blot analysis of CD36 proteins. *P<0.05, † P<0.01, ‡ P<0.001 the control. HMGB1 indicates high mobility group box‐1 protein; mRNA, messenger RNA; NLRP3, The Nod like receptor family, pyrin domain‐containing 3 protein; qRT‐PCR, quantitative real‐time polymerase chain reaction; VSMC, vascular smooth muscle cell
Figure S2. NLRP3 inflammasome activation and HMGB1 in regulating LXRα and ABCA1 expression in macrophages. Treatment with LPS/ATP stimulation or HMGB1, the relative levels of LXRα and ABCA1 expression were determined qRT‐PCR and Western blot. Data are representative images or expressed as the mean ± SD of each group from 3 separated experiments (Western blot) or 3 separated experiments with 3 duplicated wells (qRT‐PCR). A, qRT‐PCR analysis of LXRα and ABCA1 mRNA transcripts. B, Western blot analysis of LXRα and ABCA1 proteins. *P<0.05, † P<0.01, ‡ P<0.001 the control. ABCA1 indicates ATP‐binding cassette transporter 1; ATP, adenosine triphosphate; HMGB1, high mobility group box‐1 protein; LPS, Lipopolysaccharides; LXRα, Liver X receptor α; NLRP3, The Nod like receptor family, pyrin domain‐containing 3 protein; qRT‐PCR, quantitative real‐time polymerase chain reaction.
(J Am Heart Assoc. 2018;7:e008596 DOI: 10.1161/JAHA.118.008596.)
Contributor Information
Wen Li, Email: wenli28@163.com.
Shenming Wang, Email: shenmingwang@hotmail.com.
References
- 1. Liapis CD, Avgerinos ED, Kadoglou NP, Kakisis JD. What a vascular surgeon should know and do about atherosclerotic risk factors. J Vasc Surg. 2009;49:1348–1354. [DOI] [PubMed] [Google Scholar]
- 2. Dubland JA, Francis GA. So much cholesterol: the unrecognized importance of smooth muscle cells in atherosclerotic foam cell formation. Curr Opin Lipidol. 2016;27:155–161. [DOI] [PubMed] [Google Scholar]
- 3. Fisher EA, Miano JM. Don't judge books by their covers: vascular smooth muscle cells in arterial pathologies. Circulation. 2014;129:1545–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage‐like cells in human atherosclerosis. Circulation. 2014;129:1551–1559. [DOI] [PubMed] [Google Scholar]
- 5. Orekhov AN, Andreeva ER, Mikhailova IA, Gordon D. Cell proliferation in normal and atherosclerotic human aorta: proliferative splash in lipid‐rich lesions. Atherosclerosis. 1998;139:41–48. [DOI] [PubMed] [Google Scholar]
- 6. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. [DOI] [PubMed] [Google Scholar]
- 7. Karasawa T, Takahashi M. Role of NLRP3 inflammasomes in atherosclerosis. J Atheroscler Thromb. 2017;24:443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jiang Y, Wang M, Huang K, Zhang Z, Shao N, Zhang Y, Wang W, Wang S. Oxidized low‐density lipoprotein induces secretion of interleukin‐1beta by macrophages via reactive oxygen species‐dependent NLRP3 inflammasome activation. Biochem Biophys Res Comm. 2012;425:121–126. [DOI] [PubMed] [Google Scholar]
- 10. Sorci‐Thomas MG, Thomas MJ. Microdomains, inflammation, and atherosclerosis. Circ Res. 2016;118:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wen H, Miao EA, Ting JP. Mechanisms of nod‐like receptor‐associated inflammasome activation. Immunity. 2013;39:432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vande Walle L, Kanneganti TD, Lamkanfi M. Hmgb1 release by inflammasomes. Virulence. 2011;2:162–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lamkanfi M, Sarkar A, Vande Walle L, Vitari AC, Amer AO, Wewers MD, Tracey KJ, Kanneganti TD, Dixit VM. Inflammasome‐dependent release of the alarmin hmgb1 in endotoxemia. J Immunol. 2010;185:4385–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lu B, Wang H, Andersson U, Tracey KJ. Regulation of HMGB1 release by inflammasomes. Protein Cell. 2013;4:163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. [DOI] [PubMed] [Google Scholar]
- 16. Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, Czura CJ, Wang H, Roth J, Warren HS, Fink MP, Fenton MJ, Andersson U, Tracey KJ. Reversing established sepsis with antagonists of endogenous high‐mobility group box 1. Proc Natl Acad Sci USA. 2004;101:296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Frank MG, Weber MD, Watkins LR, Maier SF. Stress sounds the alarmin: the role of the danger‐associated molecular pattern HMGB1 in stress‐induced neuroinflammatory priming. Brain Behav Immun. 2015;48:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG‐1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. [DOI] [PubMed] [Google Scholar]
- 19. Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan XG, Yan Z, Sun X, Wang H, Wang Q, Tsung A, Billiar TR, Zeh HJ III, Lotze MT, Tang D. Hmgb1 in health and disease. Mol Aspects Med. 2014;40:1–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yin YW, Liao SQ, Zhang MJ, Liu Y, Li BH, Zhou Y, Chen L, Gao CY, Li JC, Zhang LL. TLR4‐mediated inflammation promotes foam cell formation of vascular smooth muscle cell by upregulating ACAT1 expression. Cell Death Dis. 2014;5:e1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Eun SY, Ko YS, Park SW, Chang KC, Kim HJ. IL‐1beta enhances vascular smooth muscle cell proliferation and migration via P2Y2 receptor‐mediated rage expression and HMGB1 release. Vascul Pharmacol. 2015;72:108–117. [DOI] [PubMed] [Google Scholar]
- 22. Li X, Zhang Y, Xia M, Gulbins E, Boini KM, Li PL. Activation of NLRP3 inflammasomes enhances macrophage lipid‐deposition and migration: implication of a novel role of inflammasome in atherogenesis. PLoS ONE. 2014;9:e87552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Porto A, Palumbo R, Pieroni M, Aprigliano G, Chiesa R, Sanvito F, Maseri A, Bianchi ME. Smooth muscle cells in human atherosclerotic plaques secrete and proliferate in response to high mobility group box 1 protein. FASEB J. 2006;20:2565–2566. [DOI] [PubMed] [Google Scholar]
- 24. Umahara T, Uchihara T, Koyama S, Hashimoto T, Akimoto J, Haraoka J, Iwamoto T. Local extension of HMGB1 in atherosclerotic lesions of human main cerebral and carotid arteries. Histol Histopathol. 2014;29:235–242. [DOI] [PubMed] [Google Scholar]
- 25. Fu Y, Zhou E, Wei Z, Song X, Liu Z, Wang T, Wang W, Zhang N, Liu G, Yang Z. Glycyrrhizin inhibits lipopolysaccharide‐induced inflammatory response by reducing TLR4 recruitment into lipid rafts in RAW264.7 cells. Biochem Biophys Acta. 2014;1840:1755–1764. [DOI] [PubMed] [Google Scholar]
- 26. Chung J, An SH, Kang SW, Kwon K. Ursodeoxycholic acid (UDCA) exerts anti‐atherogenic effects by inhibiting rage signaling in diabetic atherosclerosis. PLoS ONE. 2016;11:e0147839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang M, Li W, Chang GQ, Ye CS, Ou JS, Li XX, Liu Y, Cheang TY, Huang XL, Wang SM. Microrna‐21 regulates vascular smooth muscle cell function via targeting tropomyosin 1 in arteriosclerosis obliterans of lower extremities. Arterioscler Thromb Vasc Biol. 2011;31:2044–2053. [DOI] [PubMed] [Google Scholar]
- 28. Diao L, Marshall AH, Dai X, Bogdanovic E, Abdullahi A, Amini‐Nik S, Jeschke MG. Burn plus lipopolysaccharide augments endoplasmic reticulum stress and NLRP3 inflammasome activation and reduces PGC‐1alpha in liver. Shock. 2014;41:138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mao Z, Liu C, Ji S, Yang Q, Ye H, Han H, Xue Z. The NLRP3 inflammasome is involved in the pathogenesis of parkinson's disease in rats. Neurochem Res. 2017;42:1104–1115. [DOI] [PubMed] [Google Scholar]
- 30. Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, Duncan JA, Ting JP. NLRP3 (NALP3, cryopyrin) facilitates in vivo caspase‐1 activation, necrosis, and HMGB1 release via inflammasome‐dependent and ‐independent pathways. J Immunol. 2009;183:2008–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Haneklaus M, O'Neill LA, Coll RC. Modulatory mechanisms controlling the NLRP3 inflammasome in inflammation: recent developments. Curr Opin Immunol. 2013;25:40–45. [DOI] [PubMed] [Google Scholar]
- 32. Chen S, Sun B. Negative regulation of NLRP3 inflammasome signaling. Protein Cell. 2013;4:251–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rosenfeld ME, Ross R. Macrophage and smooth muscle cell proliferation in atherosclerotic lesions of WHHL and comparably hypercholesterolemic fat‐fed rabbits. Arteriosclerosis. 1990;10:680–687. [DOI] [PubMed] [Google Scholar]
- 34. Dai Y, Cao Y, Zhang Z, Vallurupalli S, Mehta JL. Xanthine oxidase induces foam cell formation through LOX‐1 and NLRP3 activation. Cardiovasc Drugs Ther. 2017;31:19–27. [DOI] [PubMed] [Google Scholar]
- 35. Calkin AC, Tontonoz P. Liver X receptor signaling pathways and atherosclerosis. Arterioscler Thromb Vasc Biol. 2010;30:1513–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee SD, Tontonoz P. Liver X receptors at the intersection of lipid metabolism and atherogenesis. Atherosclerosis. 2015;242:29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bischoff ED, Daige CL, Petrowski M, Dedman H, Pattison J, Juliano J, Li AC, Schulman IG. Non‐redundant roles for LXRalpha and lxrbeta in atherosclerosis susceptibility in low density lipoprotein receptor knockout mice. J Lipid Res. 2010;51:900–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cai J, Wen J, Bauer E, Zhong H, Yuan H, Chen AF. The role of HMGB1 in cardiovascular biology: danger signals. Antioxid Redox Signal. 2015;23:1351–1369. [DOI] [PubMed] [Google Scholar]
- 39. Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll‐like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7377. [DOI] [PubMed] [Google Scholar]
- 40. Kumar P, Raghavan S, Shanmugam G, Shanmugam N. Ligation of rage with ligand S100B attenuates ABCA1 expression in monocytes. Metabolism. 2013;62:1149–1158. [DOI] [PubMed] [Google Scholar]
- 41. Sun H, Yuan Y, Sun Z. Update on mechanisms of renal tubule injury caused by advanced glycation end products. Biomed Res Int. 2016;2016:5475120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. NLRP3 inflammasome activation and HMGB1 in regulating CD36 expression in VSMCs. Treatment with LPS/ATP stimulation or HMGB1, the relative levels of CD36 expression were determined qRT‐PCR and Western blot. Data are representative images or expressed as the mean ± SD of each group from 3 separated experiments (Western blot) or 3 separated experiments with 3 duplicated wells (qRT‐PCR). A, qRT‐PCR analysis of CD36 mRNA transcripts. (B) Western blot analysis of CD36 proteins. *P<0.05, † P<0.01, ‡ P<0.001 the control. HMGB1 indicates high mobility group box‐1 protein; mRNA, messenger RNA; NLRP3, The Nod like receptor family, pyrin domain‐containing 3 protein; qRT‐PCR, quantitative real‐time polymerase chain reaction; VSMC, vascular smooth muscle cell
Figure S2. NLRP3 inflammasome activation and HMGB1 in regulating LXRα and ABCA1 expression in macrophages. Treatment with LPS/ATP stimulation or HMGB1, the relative levels of LXRα and ABCA1 expression were determined qRT‐PCR and Western blot. Data are representative images or expressed as the mean ± SD of each group from 3 separated experiments (Western blot) or 3 separated experiments with 3 duplicated wells (qRT‐PCR). A, qRT‐PCR analysis of LXRα and ABCA1 mRNA transcripts. B, Western blot analysis of LXRα and ABCA1 proteins. *P<0.05, † P<0.01, ‡ P<0.001 the control. ABCA1 indicates ATP‐binding cassette transporter 1; ATP, adenosine triphosphate; HMGB1, high mobility group box‐1 protein; LPS, Lipopolysaccharides; LXRα, Liver X receptor α; NLRP3, The Nod like receptor family, pyrin domain‐containing 3 protein; qRT‐PCR, quantitative real‐time polymerase chain reaction.