

Abstract
Purpose
Patients with Lynch syndrome (LS) have a significantly increased risk of developing upper-tract urothelial carcinoma (UTUC). Here, we sought to identify differences in the patterns of mutational changes in LS-associated versus sporadic UTUCs.
Patients and Methods
We performed targeted sequencing of 17 UTUCs from patients with documented LS-associated germline mutations (LS-UTUCs) using the Memorial Sloan Kettering Integrated Molecular Profiling of Actionable Cancer Targets targeted exon capture assay and compared the results with those from a recently characterized cohort of 82 patients with sporadic UTUC.
Results
Patients with LS-UTUC were significantly younger, had had less exposure to tobacco, and more often presented with a ureteral primary site compared with patients with sporadic UTUC. The median number of mutations per tumor was significantly greater in LS-UTUC tumors than in tumors from the sporadic cohort (58; interquartile range [IQR], 47-101 v 6; IQR, 4-10; P < .001), as was the MSIsensor score (median, 25.1; IQR, 17.9-31.2 v 0.03; IQR, 0-0.44; P < .001). Differences in the genetic landscape were observed between sporadic and LS-associated tumors. Alterations in KMT2D, CREBBP, or ARID1A or in DNA damage response and repair genes were present at a significantly higher frequency in LS-UTUC. CIC, NOTCH1, NOTCH3, RB1, and CDKN1B alterations were almost exclusive to LS-UTUC. Although FGFR3 mutations were identified in both cohorts, the R248C hotspot mutation was highly enriched in LS-UTUC.
Conclusion
LS- and sporadic UTUCs have overlapping but distinct genetic signatures. LS-UTUC is associated with hypermutation and a significantly higher prevalence of FGFR3 R248C mutation. Prospective molecular characterization of patients to identify those with LS-UTUC may help guide treatment.
INTRODUCTION
Lynch syndrome (LS) is an autosomal dominant cancer predisposition syndrome caused by germline mutations in the mismatch repair (MMR) genes MLH1, MSH2, MSH6, or PMS2. Patients with LS have an increased risk of developing a variety of tumors, particularly those arising in the colon, but also extracolonic cancers, including urothelial carcinomas (UCs).1-3 UC is the third most frequent malignancy in LS, occurring in approximately 5% of patients.3,4 Patients with LS have up to a 22-fold greater risk of developing upper-tract urothelial carcinoma (LS-UTUC) over the general population and a median age of onset 10 to 15 years earlier than patients with sporadic UTUC.2-5
Although both UC of the bladder (UCB) and UTUC are believed to arise from a common precursor cell population within the urothelium, increasing evidence suggests that these two malignancies represent different disease entities from both clinicopathologic and genetic perspectives.6-10 In support of this concept, tumor genomic sequencing of UCB and UTUC has identified distinct mutational profiles between these two urothelial malignancies.11,12
Here, we sought to determine whether patients with UTUC with known germline mutations uniformly harbored loss of heterozygosity of the wild-type allele of the gene that was the basis of their LS diagnosis and whether their tumors exhibited a pattern of hypermutation consistent with MMR deficiency. We also sought to identify differences in the patterns of mutational changes in LS-UTUC versus sporadic UTUC that may represent potential therapeutic targets.
PATIENTS AND METHODS
Study Samples
Upper-tract tumors (n = 21) and normal samples were collected from patients with a known germline mutation in an LS-associated gene through a collaborative effort between the Colon Cancer Family Registry (CCFR) and Memorial Sloan Kettering Cancer Center. The CCFR activities are conducted across six multiple–principal investigator sites and six subaward sites: University of Melbourne (Melbourne, Victoria, Australia), Fred Hutchinson Cancer Research Center (Seattle, WA), Sinai Health System (comprising Mount Sinai Hospital and Lunenfeld-Tanenbaum Research Institute [Toronto, Ontario, Canada]), Mayo Clinic (Mayo Clinic Arizona [Scottsdale, AZ] with a subaward to Mayo Clinic Minnesota [Rochester, MN]), Cedars-Sinai Consortium (comprising Cedars-Sinai Medical Center [Los Angeles, CA] with subawards to Cleveland Clinic [Cleveland, OH], University of Minnesota [Minneapolis, MN], Dartmouth College [Hanover, NH], and University of Virginia [Charlottesville, VA]), and University of Hawaii (University of Hawaii Medical Center [Honolulu, HI] with a subaward to Cancer Prevention Institute of California [Fremont, CA]). This was formed as a resource to support studies on the etiology, prevention, and clinical management of colorectal cancer. The resource comprises data and biospecimens from approximately 40,000 participants from 14,000 families recruited from 1998 to 2016.13 For this study, the inclusion criteria were proven pathogenic germline mutation in one of the DNA MMR genes MLH1, MSH2, MSH6, or PMS2; diagnosis of UTUC confirmed by expert pathologic examination; and availability of archival tissue blocks for genetic analysis. Study approval was obtained from the institutional review board or human research ethics committee of each participating institution. A previously characterized cohort of 82 patients with presumed sporadic UTUC treated with radical nephroureterectomy was used as a comparison group.11
Sample Preparation
Germline DNA was extracted from peripheral blood lymphocytes provided by the CCFR for each patient. Paraffin-embedded tumor blocks were obtained. Hematoxylin and eosin–stained sections were prepared from each block and reviewed by a board-certified pathologist to confirm the histologic diagnosis and tumor grade. For four tumors obtained from the Mount Sinai Hospital, previously extracted tumor genomic DNA was provided. For these cases, pathologic review was performed on high-resolution digital images of the hematoxylin and eosin–stained sections. No patient had a dominant variant histologic subtype. When not provided by the CCFR, DNA was extracted from paraffin sections as previously described.12 Clinical and demographic information were obtained from the prospectively maintained registry of the CCFR.
Targeted Sequencing
All protein-coding exons of 341 cancer-associated genes were sequenced using the Memorial Sloan Kettering Integrated Molecular Profiling of Actionable Cancer Targets (MSK-IMPACT) assay as previously described (Data Supplement).14 All candidate mutations and indels were reviewed manually. The accumulated sequence coverage for each exon was compared in tumor and matched germline samples. Coverage ratios ≥ 3× were defined as amplifications, and ratios ≤ 0.3× were defined as deletions, whereas coverage ratios ≥ 2× and ≤ 0.5× were defined as gains and losses, respectively.
To determine allelic configurations, total and allele-specific copy-number states were inferred for all tumor samples using FACETS (version 0.5.6).15 The proportions of unstable microsatellites were quantitated using MSIsensor (version 0.2).16 Microsatellite instability (MSI) score was defined as the percentage of unstable microsatellite sites divided by total number of microsatellite sites surveyed. Signature decomposition analysis17,18 was performed for all tumor samples with ≥ 10 single-nucleotide somatic mutations. From the somatic mutations in an individual tumor sample, contributions were inferred from known mutational signatures, which are probability distributions over the nucleotide change and flanking 5′ and 3′ nucleotide context of each mutation. The quasi P value for a particular signature was calculated as the fraction of samples with a signature proportion greater than a precalculated noise threshold (1 × 10−5). Signatures representing ≥ 15% mutations and with a significant quasi P value (< .05) were plotted.
Statistical Analysis
Pearson’s χ2 test or Fisher’s exact test were used to test statistical significance of differences between clinical and demographic variables. Bivariable comparisons of individual mutation frequencies by cohort were performed using Fisher’s exact test. Counts of gains, losses, and total copy-number alterations were analyzed using negative binomial regression. P values < .05 were considered statistically significant. All analyses were conducted using R software (version 2.13.1; R Foundation, Vienna, Austria).
RESULTS
Genomic Characterization of LS-UTUC
Of the 21 tumors analyzed, four were reclassified as renal cell carcinoma (RCC) on central pathologic review. Genomic DNA from all 21 sample tumors (LS-UTUC, n = 17; RCC, n = 4) and matched normal peripheral blood lymphocytes were analyzed for alterations in 341 cancer-associated genes. The average sequencing coverage was 315× for all targeted exons across all 21 patients, with average coverage of 251× per tumor (Data Supplement). In all 21 patient cases, we confirmed the presence of a deleterious germline alteration in an LS-associated gene. In patients with LS-UTUC, MSH2 was the most common germline alteration (13 [76%] of 17), followed by MSH6 (three [18%] of 17) and PMS2 (one [6%] of 17). No germline alterations were identified in MLH1 or PMS1. Thirteen (76%) of 17 patients had loss of heterozygosity in the DNA repair genes that corresponded to the germline alterations identified in their germline DNA (Fig 1A; Appendix Fig A1).
Fig 1.
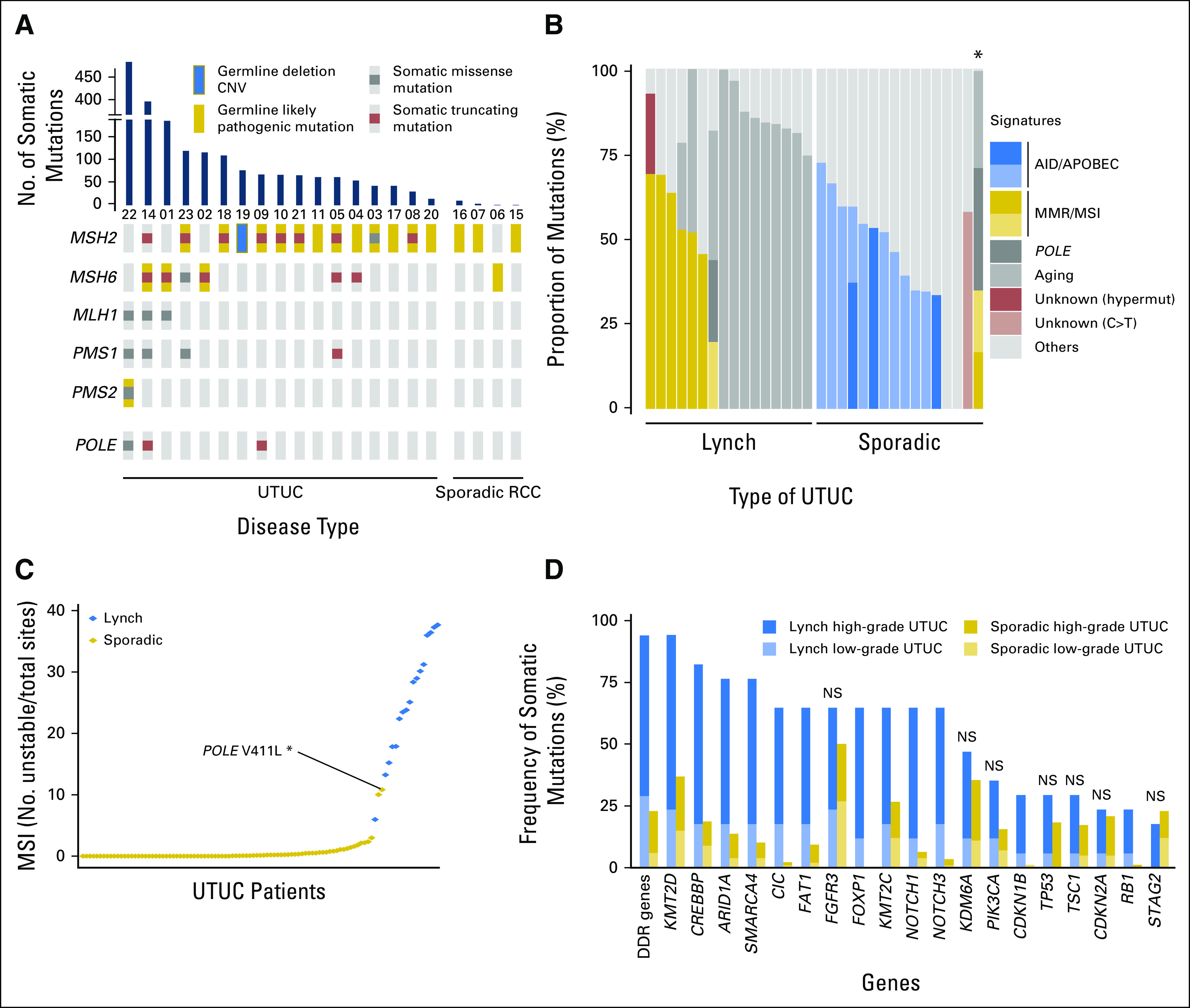
(A) Oncoprint of germline and somatic mutations in mismatch repair (MMR) genes and in POLE in the Lynch cohort. (B) Mutational decomposition analysis of the samples with ≥ 10 somatic mutations. (*) Hypermutated sporadic upper-tract urothelial carcinoma (UTUC) tumor with a hotspot mutation in POLE. (C) Microsatellite instability (MSI) sensor scores across the two cohorts. (D) Frequency of somatic alterations in Lynch syndrome–associated (n = 17) and sporadic UTUC (n = 82) cohorts. AID/APOBEC, activation-induced cytidine deaminase/apolipoprotein B mRNA-editing enzyme catalytic polypeptide; CNV, copy-number variation; DDR, DNA damage repair; NS, non significant; RCC, renal cell carcinoma.
A total of 1,834 somatic coding alterations were identified across all patients with LS-UTUC, involving 300 different genes (Data Supplement). The median number of somatically altered genes per tumor was 47 (range, nine to 204). Characteristic of MMR-deficient neoplasms, the mean MSIsensor score was high (median, 25.1; range, 6 to 37.7), with an excess of frameshift mutations (142 [8%] in 1,834), whereas copy-number alterations were uncommon.19 Using mutational decomposition analysis in samples with ≥ 10 somatic mutations, we identified evidence of MSI/MMR signatures in seven tumors, whereas the mitotic clock/aging signature was predominant in nine (Fig 1B). Notably, the four RCCs found to arise in the patients with LS had low mutation rates (median, two; range, one to nine), low MSIsensor scores (median, 4.7; range, 3 to 11.6), and no evidence of loss of heterozygosity of their LS-associated germline mutation, suggesting that the pathogenesis of these tumors was biologically independent of their LS diagnosis.
The most frequently mutated genes in the 17 LS-UTUC tumors (present in > 10 patients) in decreasing frequency were KMT2D, CREBBP, ARID1A, SMARCA4, CIC, FAT1, FGFR3, FOXP1, KMT2C, NOTCH1, and NOTCH3 (Appendix Fig A2). With the exception of CIC, FOXP1, and KMT2C, these genes were also those with the highest mutation rate (> 0.002 mutations per nucleotide; Data Supplement). TP53 was mutated in a smaller subset of carcinomas (five [29%] of 17), and in four of these, the TP53 mutation co-occurred with FGFR3 mutations. RB1 alteration was found in four (24%) of 17. Of interest, although only 11 focal amplifications were identified within the 17 LS-UTUCs, five of these 11 amplification events involved CDKN1B. Moreover, CDKN1B amplification showed a tendency toward mutual exclusivity with both FGFR3 and TP53 mutations.
Comparison of Genomic Profiles of LS-UTUC and Sporadic UTUC
To identify potential differences in the mutational landscape between LS-UTUC and sporadic UTUC, we compared the results of the LS-UTUC cohort with those from a previously reported analysis by our group of 82 patients with sporadic UTUC. One patient in this prior cohort had an ultramutated phenotype (422 mutations and no focal copy-number alterations) attributable to a hotspot mutation in POLE.11 Patient demographic and clinicopathologic characteristics of the two groups are listed in Table 1. Compared with those with sporadic UTUC, patients with LS were significantly younger (median age, 61 years; interquartile range [IQR], 53–66 years v 68.5 years; IQR, 63-75 years; P = .005), had had less exposure to tobacco (47% v 73%; P = .035), and had a different distribution of primary tumor location (47% ureteral tumor in LS v 18% in the sporadic cohort; P = .011).
Table 1.
Population Demographics and Clinical Characteristics

As expected, the median number of mutations per sample for LS-UTUC was significantly greater than that seen in the sporadic cohort (58; IQR, 47-101 v six; IQR, 4–10; P < .001), and the MSIsensor score was also significantly higher (median, 25.1; IQR, 17.9-31.2 v 0.03; IQR, 0-0.44; P < .001; Fig 1C). Most of the sporadic samples with ≥ 10 somatic mutations had an AID/APOBEC (activation-induced cytidine deaminase/apolipoprotein B mRNA-editing enzyme catalytic polypeptide) signature (12 of 16; Fig 1B).
We next compared the frequencies of genetic alterations in the most commonly altered genes (Table 2; Fig 1D) between the two tumor cohorts. This revealed that, although the mutational landscapes overlapped, some genes were targets of somatic alteration in both cohorts; however, the frequency of alteration was significantly higher in LS-UTUC (ie, KMT2D, CREBBP, ARID1A, and SMARCA4). Moreover, some genes were somatic targets almost exclusively in the LS cohort. Examples of this include CIC, FOXP1, NOTCH1, NOTCH3, or RB1, each of which harbored somatic alterations in nonoverlapping subsets in < 6% of sporadic UTUCs versus 24% to 65% in LS-UTUCs (P < .001). Copy-number alterations such as CDKN1B amplification were also unique to LS-UTUCs (five of 17 v zero of 82; P < .001). Of note, alteration in at least one of the DNA damage response and repair genes (ERCC2, ERCC3, ERCC4, ERCC5, BRCA1, BRCA2, RAD50, RAD51, RAD51B, RAD51C, RAD51D, RAD52, RAD54L, NBN, MRE11A, ATM, ATR, MDC1, CHEK1, CHEK2, PALB2, BRIP1, FANCA, FANCC, BLM, MUTYH, RECQL4, PARP1, and POLE) was found in 94% of the patients with LS, compared with 23% in the sporadic cohort (P < .001). When adjusting for grade, most of the differences between the two cohorts remained significant, although the frequency of gene alterations was different between low- and high-grade tumors (Table 2).
Table 2.
Comparison of Frequency of Gene Alterations in Both Cohorts

Finally, we noted that both cohorts had similar frequencies of FGFR3 mutations (LS-UTUC, 11 [65%] of 17 v sporadic UTUC, 41 [50%] of 82; P = .269; Appendix Fig A3). Not surprisingly, FGFR3 mutations were more frequent in low-grade tumors (80% of low-grade LS-UTUCs v 94% of low-grade sporadic UTUCs; P = .218). However, whereas the most prevalent FGFR3 hotspot mutation in sporadic UTUC was S249C (26 [63%] of 41 mutations; 13 of 22 and 13 of 19 for low- and high-grade tumors, respectively), in LS-UTUC, the FGFR3 mutations noted were predominantly R248C (nine [82%] of 11 mutations; four of four and five of seven for low- and high-grade tumors, respectively) and to a lesser extent G380R (four [36%] of 11 mutations; two of four and two of seven for low- and high-grade tumors, respectively; Fig 2A).
Fig 2.
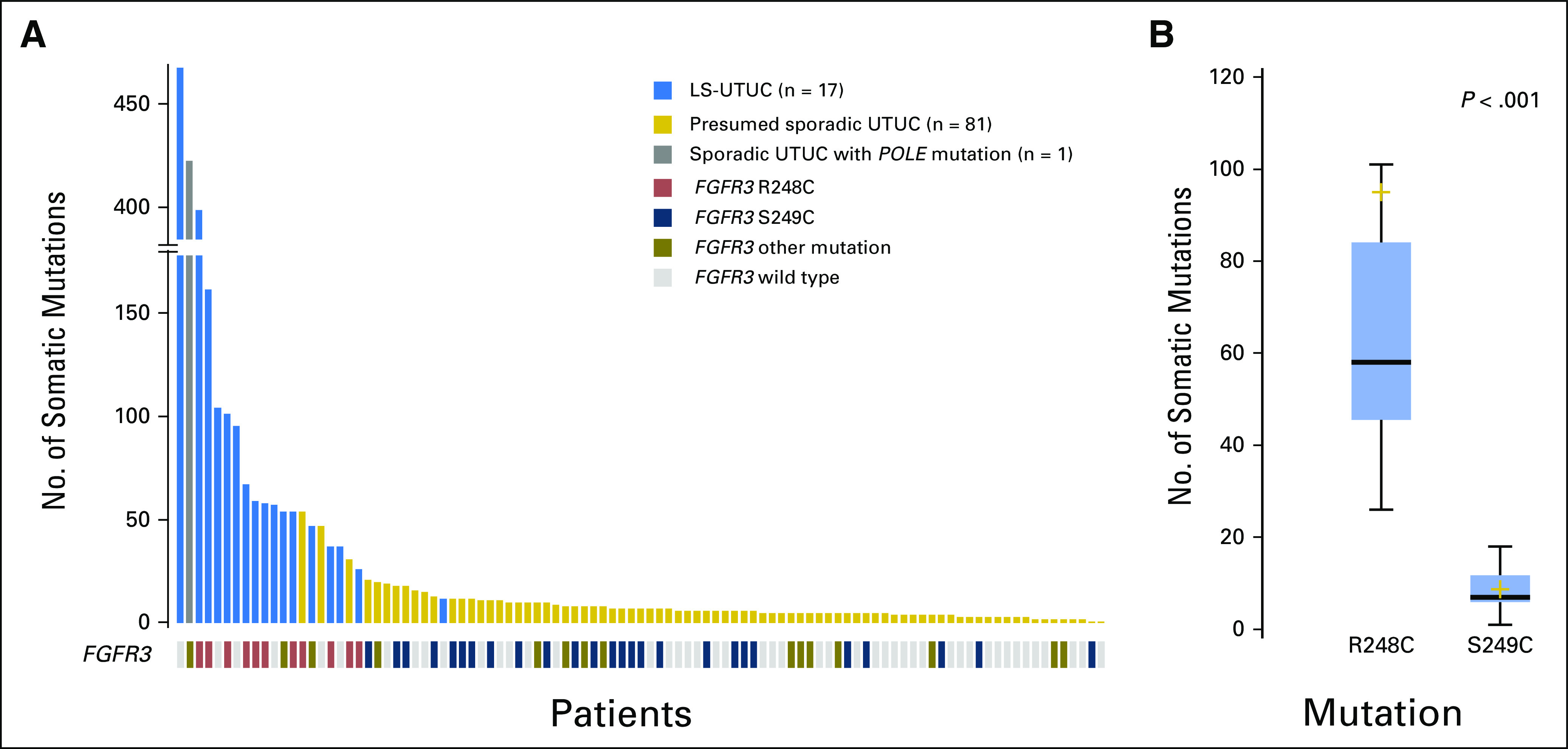
(A) Number of somatic alterations in Lynch syndrome–associated upper-tract urothelial carcinoma (LS-UTUC) and sporadic UTUC tumors and their association with FGFR3 mutation status. (B) Total mutation count in patients with somatic FGFR3 R248C and FGFR3 S249C mutations.
Given this unexpected finding, we rereviewed all 41 FGFR3 mutations in our sporadic cohort of 82 UTUCs and identified two carcinomas with an FGFR3 R248C mutation. These UTUCs had 31 and 54 total somatic mutations, respectively, by MSK-IMPACT, suggesting the possibility of an MMR defect. However, their MSIsensor scores were low (0 and 0.11), and the mutational decomposition analysis revealed an AID/APOBEC signature.
Validation of FGFR3 R248C as Marker of LS-UTUC
To determine the extent to which the presence of an FGFR3 R248C mutation may be associated with LS or an LS-like hypermutator phenotype, we queried 14,800 tumors prospectively sequenced as part of clinical care at Memorial Sloan Kettering Cancer Center using the MSK-IMPACT assay. Twenty-three patients had an FGFR3 R248C mutation, corresponding to 11 UTUCs, nine UCBs, and three tumors with other primary sites (one squamous cell carcinoma of the lung, one lung metastasis from breast carcinoma, and one squamous cell carcinoma of the tongue; Fig 3). The 11 patients with UTUC had a median number of mutations of 41 (range, one to 414 mutations). Consent for germline genomic analysis was lacking for two patients. Of the nine evaluable patients with presumed sporadic UTUC and an FGFR3 R248C hotspot mutation, seven displayed germline evidence of LS (six germline mutations in MSH2 and one in MSH6). One of the patients without evidence of a germline mutation had a hypermutator tumor (41 mutations) with absence of MSH2 and MSH6 expression by immunohistochemistry consistent with a somatic MSI-high, LS-like phenotype.
Fig 3.

Flow chart of identification of the patients with FGFR3 R248C mutation in the clinical Memorial Sloan Kettering Integrated Molecular Profiling of Actionable Cancer Targets (MSK-IMPACT) cohort. LS, Lynch syndrome; SCC, squamous cell carconima; UC, urothelial carcinoma; UCB, UC of the bladder; UTUC, upper tract UC.
In our study, the presence of an FGFR3 R248C hotspot mutation was associated with a higher median number of somatic mutations compared with tumors with an FGFR3 S249C mutation (58; IQR, 45.5-84 v seven; IQR, 6-11.8; P < .001; Fig 2B). Similarly, of the 14,800 tumors prospectively sequenced at our institution, the 23 samples with FGFR3 R248C mutations, including 20 UCs and three other tumors, had a significantly higher median number of somatic mutations compared with that in the 84 patients with FGFR3 S249C mutations across all tumor types (22; IQR, 8-53 v nine; IQR, 6-11.8; P < .006).
DISCUSSION
LS is an autosomal dominant cancer predisposition syndrome attributable to germline mutations in an MMR gene. Upwards of 80% of patients with LS will develop a malignancy, with colorectal cancer being the most common.20 UC is the third most common site of extracolonic malignancy in the LS spectrum and occurs in approximately 5% of patients. Few studies to date have sought to define the somatic mutational profile of MMR-deficient urothelial tumors, in part because of the perception that the high mutational load would limit the ability to identify true driver events.19 In this study, we performed targeted sequencing of UTUC from 17 patients with known germline mutations in an LS-associated gene to characterize the genomic landscape of these tumors and compared the results with those from a cohort of 82 patients with clinically presumed sporadic UTUC.
Next-generation sequencing (NGS) can be used to discover novel, targetable, or pathogenic somatic alterations and identify patients with mutations for which therapies already exist. Although many of the driver genes identified in the LS and sporadic UTUC cohorts tended to be similar (FGFR3, KDM6A, PIC3CA, and TP53), differences in the genomic profiles were observed. As would be expected from an MSI-associated malignancy, the median number of mutations per tumor was significantly higher in LS-UTUC than sporadic UTUC (58 v six; P < .001). There was an excess of frameshift mutations (142 [8%] of 1,834), and copy-number alterations were relatively uncommon. Mutation of some genes was almost exclusive to LS-UTUC (CIC, NOTCH1, NOTCH3, and RB1). Of note, however, hypermutation or loss of heterozygosity of the LS germline mutation was not noted in the four RCC samples collected from patients with LS, suggesting that the pathogenesis of these tumors was unrelated to the LS diagnosis.
Although we were able to identify somatic alterations in most patients driving loss of heterozygosity in the germline LS genes relevant to each corresponding patient, four patients with LS had germline mutations without evidence of loss of heterozygosity as detectable by tumor DNA sequencing. All four, however, had genomic signatures consistent with MSI, suggesting that these patients may have had transcriptional silencing by promoter hypermethylation, as previously reported.21 Regarding the mutational decomposition analysis, only seven LS-UTUC tumors had an MMR/MSI signature. This mutational decomposition analysis only uses single-nucleotide polymorphisms, and these are not the most prevalent type of mutation present in MSI/MMR cases.19 This may partially explain why the decomposition analysis did not identify a strong MSI/MMR signature in some patient cases, including many with high MSIsensor scores.
Among the most striking findings in our study was the identification of an R248C hotspot mutation in FGFR3 that may serve as a potential biomarker for LS in patients with UTUC. FGFR3 is one of four members of the fibroblast growth factor receptor family of receptor tyrosine kinases that promote cell growth and proliferation. Activating mutations of FGFR3 are particularly common in low-grade UCs.22,23 FGFR3 R248C leads to increased FGFR3 dimer stability and constitutive receptor activation in the absence of ligand, resulting in activation of the phosphatidylinositol 3-kinase–AKT and mitogen-activated protein kinase signaling pathways.24,25 This mutation has already been described in UCB,26 but its presence in UTUC was highly associated with a significantly increased median number of somatic mutations.
Some hereditary cancers are misclassified as sporadic, although their identification might have consequences for both patients and their family members, because this information would prompt germline testing and, if positive, screening for LS-related malignancies.27 Our data suggest that the finding of an FGFR3 R248C somatic mutation is a potential biomarker for LS and its identification in a patient should prompt germline testing for LS. Furthermore, recent data suggest that tumors with MMR deficiency are more responsive to immune checkpoint blockade than MMR-proficient tumors.28 It is possible that patients with LS-UTUC may derive greater benefit from anti–PD-1 therapy, suggesting an immediate clinical implication for screening and potential personalization of therapy in these patients.
One potential limitation of our study was the use of a targeted sequencing approach focusing on a panel of 341 cancer-related genes. A broader sequencing approach, such as whole-exome sequencing, may have identified additional genes differentially altered in sporadic and LS-UTUCs. Recently, the analysis of 28 samples of sporadic UTUC identified four RNA expression profiles with different clinical characteristics, and it would be biologically important to study how LS-UTUCs cluster within this classification.29 Additionally, although ours is the largest series to date to our knowledge profiling LS-UTUC using NGS, the sample size remains small, and a larger study may have revealed other significant patterns of comutated genes. Consequently, there is a need for a larger cohort with external validation.
In conclusion, we performed targeted NGS of 17 UTUCs that arose in patients with LS to compare the genetic characteristics of such tumors with those of sporadic UTUC tumors. Although the spectrums of mutated genes showed some similarities, LS-UTUCs were characterized by a unique genetic signature, consisting of hypermutation and frequent hotspot mutations at FGFR3 R248C. The results of this study may help to differentiate between presumed sporadic UTUC and UTUC with an underlying MSI-associated germline defect, which could be used to guide specific screening and treatment recommendations.
Appendix
Fig A1.
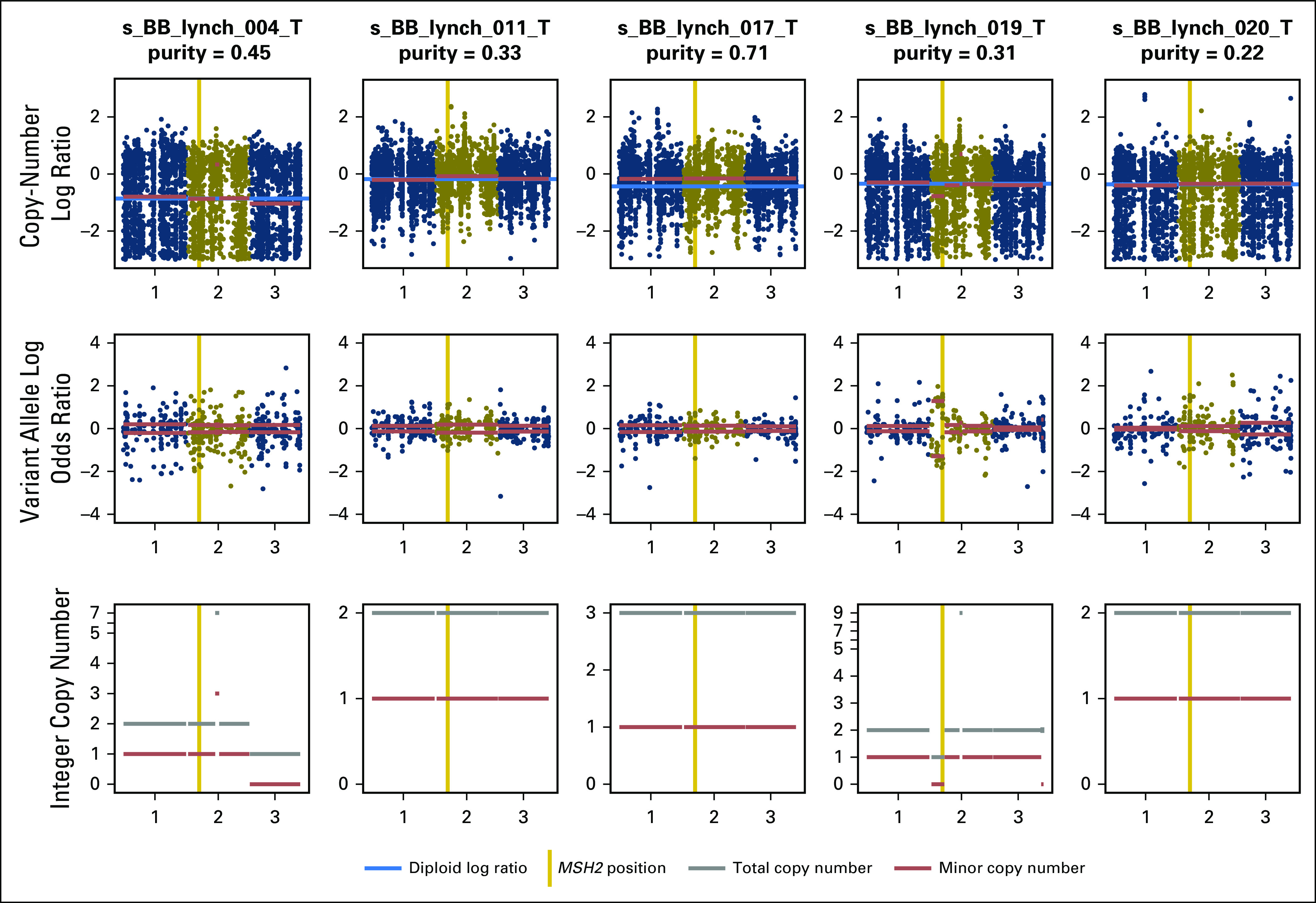
FACETS analysis of five samples without somatic alterations corresponding to germline mutations identified in mismatch repair genes. Sample s_BB_lynch_019_T was found to present loss of heterozygosity in MSH2.
Fig A2.

Oncoprint of the most frequent genes altered in the Lynch cohort.
Fig A3.

Lollipop plots of FGFR3 mutations in the sporadic and Lynch cohorts.
Footnotes
The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Colon Cancer Family Registry (CCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or CCFR.
Supported in part by the Pin Down Bladder Cancer Foundation; Cycle for Survival; National Cancer Institute (NCI) Cancer Center Support Grant No. P30 CA008748; NCI Grant No. UM1 CA167551; and cooperative agreements with the following Colon Cancer Family Registry centers: Australasian Colorectal Cancer Family Registry (Grants No. U01 CA074778 and U01/U24 CA097735), Mayo Clinic Cooperative Family Registry for Colon Cancer Studies (Grant No. U01/U24 CA074800), and Ontario Familial Colorectal Cancer Registry (Grant No. U01/U24 CA074783).
AUTHOR CONTRIBUTIONS
Conception and design: Timothy F. Donahue, Aditya Bagrodia, Hikmat Al-Ahmadie, Mark Jenkins, Barry Taylor, Jonathan Coleman, David B. Solit, Bernard H. Bochner
Financial support: Timothy F. Donahue
Administrative support: Timothy F. Donahue
Provision of study materials or patients: Timothy F. Donahue, Mark Jenkins, Allyson S. Templeton, Noralane M. Lindor
Collection and assembly of data: Timothy F. Donahue, François Audenet, Eugene K. Cha, John P. Sfakianos, Dahlia Sperling, Hikmat Al-Ahmadie, Mark Clendennig, Christophe Rosty, Daniel D. Buchanan, Mark Jenkins, John Hopper, Ingrid Winship, Allyson S. Templeton, Noralane M. Lindor
Data analysis and interpretation: Timothy F. Donahue, Aditya Bagrodia, François Audenet, Mark T.A. Donoghue, Eugene K. Cha, Hikmat Al-Ahmadie, Daniel D. Buchanan, Mark Jenkins, Michael F. Walsh, Zsofia K. Stadler, Gopa Iyer, Barry Taylor, Jonathan Coleman, Noralane M. Lindor, David B. Solit, Bernard H. Bochner
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Timothy F. Donahue
No relationship to disclose
Aditya Bagrodia
No relationship to disclose
François Audenet
No relationship to disclose
Mark T.A. Donoghue
No relationship to disclose
Eugene K. Cha
No relationship to disclose
John P. Sfakianos
Consulting or Advisory Role: EMD Serono
Speakers’ Bureau: Astellas Medivation
Dahlia Sperling
Employment: Next Generation
Stock and Other Ownership Interests: Pharma (I)
Consulting or Advisory Role: Next Generation
Travel, Accommodations, Expenses: Next Generation
Hikmat Al-Ahmadie
Consulting or Advisory Role: Bristol-Myers Squibb, EMD Serono
Mark Clendenning
No relationship to disclose
Christophe Rosty
No relationship to disclose
Daniel D. Buchanan
No relationship to disclose
Mark Jenkins
No relationship to disclose
John Hopper
No relationship to disclose
Ingrid Winship
No relationship to disclose
Allyson S. Templeton
No relationship to disclose
Michael F. Walsh
No relationship to disclose
Zsofia K. Stadler
Consulting or Advisory Role: Allergan (I), Genentech/Roche (I), Regeneron (I), Optos, (I), Adverum (I)
Gopa Iyer
No relationship to disclose
Barry Taylor
No relationship to disclose
Jonathan Coleman
No relationship to disclose
Noralane M. Lindor
Consulting or Advisory Role: UpToDate
Patents, Royalties, Other Intellectual Property: Royalties as editor for section in UpToDate; royalties as editor for section in UpToDate (I)
Travel, Accommodations, Expenses: Intercept Pharmaceuticals (I)
David B. Solit
Honoraria: Loxo, Pfizer
Consulting or Advisory Role: Pfizer, Loxo
Bernard H. Bochner
Honoraria: Genentech/Roche
Consulting or Advisory Role: Genentech
REFERENCES
- 1.Watson P, Lynch HT. Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer. 1993;71:677–685. doi: 10.1002/1097-0142(19930201)71:3<677::aid-cncr2820710305>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 2.Lynch HT, Smyrk TC, Watson P, et al. Genetics, natural history, tumor spectrum, and pathology of hereditary nonpolyposis colorectal cancer: An updated review. Gastroenterology. 1993;104:1535–1549. doi: 10.1016/0016-5085(93)90368-m. [DOI] [PubMed] [Google Scholar]
- 3.Watson P, Riley B. The tumor spectrum in the Lynch syndrome. Fam Cancer. 2005;4:245–248. doi: 10.1007/s10689-004-7994-z. [DOI] [PubMed] [Google Scholar]
- 4.Watson P, Lynch HT. The tumor spectrum in HNPCC. Anticancer Res. 1994;14:1635–1639. [PubMed] [Google Scholar]
- 5.Sijmons RH, Kiemeney LA, Witjes JA, et al. Urinary tract cancer and hereditary nonpolyposis colorectal cancer: Risks and screening options. J Urol. 1998;160:466–470. [PubMed] [Google Scholar]
- 6.Olgac S, Mazumdar M, Dalbagni G, et al. Urothelial carcinoma of the renal pelvis: A clinicopathologic study of 130 cases. Am J Surg Pathol. 2004;28:1545–1552. doi: 10.1097/00000478-200412000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Stewart GD, Bariol SV, Grigor KM, et al. A comparison of the pathology of transitional cell carcinoma of the bladder and upper urinary tract. BJU Int. 2005;95:791–793. doi: 10.1111/j.1464-410X.2005.05402.x. [DOI] [PubMed] [Google Scholar]
- 8.Margulis V, Shariat SF, Matin SF, et al. Outcomes of radical nephroureterectomy: A series from the Upper Tract Urothelial Carcinoma Collaboration. Cancer. 2009;115:1224–1233. doi: 10.1002/cncr.24135. [DOI] [PubMed] [Google Scholar]
- 9.Cha EK, Shariat SF, Kormaksson M, et al. Predicting clinical outcomes after radical nephroureterectomy for upper tract urothelial carcinoma. Eur Urol. 2012;61:818–825. doi: 10.1016/j.eururo.2012.01.021. [DOI] [PubMed] [Google Scholar]
- 10.Rink M, Ehdaie B, Cha EK, et al. Stage-specific impact of tumor location on oncologic outcomes in patients with upper and lower tract urothelial carcinoma following radical surgery. Eur Urol. 2012;62:677–684. doi: 10.1016/j.eururo.2012.02.018. [DOI] [PubMed] [Google Scholar]
- 11.Sfakianos JP, Cha EK, Iyer G, et al. Genomic characterization of upper tract urothelial carcinoma. Eur Urol. 2015;68:970–977. doi: 10.1016/j.eururo.2015.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim PH, Cha EK, Sfakianos JP, et al. Genomic predictors of survival in patients with high-grade urothelial carcinoma of the bladder. Eur Urol. 2015;67:198–201. doi: 10.1016/j.eururo.2014.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Colon Cancer Family Registry. http://coloncfr.org.
- 14.Wagle N, Berger MF, Davis MJ, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2:82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen R, Seshan VE. FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Niu B, Ye K, Zhang Q, et al. MSIsensor: Microsatellite instability detection using paired tumor-normal sequence data. Bioinformatics. 2014;30:1015–1016. doi: 10.1093/bioinformatics/btt755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. Erratum: Nature 502:258, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Deciphering signatures of mutational processes operative in human cancer. Cell Reports. 2013;3:246–259. doi: 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim T-M, Laird PW, Park PJ. The landscape of microsatellite instability in colorectal and endometrial cancer genomes. Cell. 2013;155:858–868. doi: 10.1016/j.cell.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lynch HT, de la Chapelle A. Hereditary colorectal cancer. N Engl J Med. 2003;348:919–932. doi: 10.1056/NEJMra012242. [DOI] [PubMed] [Google Scholar]
- 21.Lahtz C, Pfeifer GP. Epigenetic changes of DNA repair genes in cancer. J Mol Cell Biol. 2011;3:51–58. doi: 10.1093/jmcb/mjq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Billerey C, Chopin D, Aubriot-Lorton MH, et al. Frequent FGFR3 mutations in papillary non-invasive bladder (pTa) tumors. Am J Pathol. 2001;158:1955–1959. doi: 10.1016/S0002-9440(10)64665-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hernández S, López-Knowles E, Lloreta J, et al. Prospective study of FGFR3 mutations as a prognostic factor in nonmuscle invasive urothelial bladder carcinomas. J Clin Oncol. 2006;24:3664–3671. doi: 10.1200/JCO.2005.05.1771. [DOI] [PubMed] [Google Scholar]
- 24.Duperret EK, Oh SJ, McNeal A, et al. Activating FGFR3 mutations cause mild hyperplasia in human skin, but are insufficient to drive benign or malignant skin tumors. Cell Cycle. 2014;13:1551–1559. doi: 10.4161/cc.28492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juanpere N, Agell L, Lorenzo M, et al. Mutations in FGFR3 and PIK3CA, singly or combined with RAS and AKT1, are associated with AKT but not with MAPK pathway activation in urothelial bladder cancer. Hum Pathol. 2012;43:1573–1582. doi: 10.1016/j.humpath.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 26.Al-Ahmadie HA, Iyer G, Janakiraman M, et al. Somatic mutation of fibroblast growth factor receptor-3 (FGFR3) defines a distinct morphological subtype of high-grade urothelial carcinoma. J Pathol. 2011;224:270–279. doi: 10.1002/path.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Audenet F, Colin P, Yates DR, et al. A proportion of hereditary upper urinary tract urothelial carcinomas are misclassified as sporadic according to a multi-institutional database analysis: proposal of patient-specific risk identification tool. BJU Int. 2012;110:E583–E589. doi: 10.1111/j.1464-410X.2012.11298.x. [DOI] [PubMed] [Google Scholar]
- 28.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372:2509–2520. doi: 10.1056/NEJMoa1500596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moss TJ, Qi Y, Xi L, et al. Comprehensive genomic characterization of upper tract urothelial carcinoma. Eur Urol. 2017;72:641–649. doi: 10.1016/j.eururo.2017.05.048. [DOI] [PubMed] [Google Scholar]