

Abstract
Background
Particulate matter (particles < 2.5 μm [PM 2.5]) exposure during the in utero and postnatal developmental periods causes cardiac dysfunction during adulthood. Here, we investigated the potential priming effects of preconception exposure of PM 2.5 on cardiac function in adult offspring.
Methods and Results
Male and female friend leukemia virus b (FVB) mice were exposed to either filtered air (FA) or PM 2.5 at an average concentration of 38.58 μg/m3 for 6 hours/day, 5 days/week for 3 months. Mice were then crossbred into 2 groups: (1) FA male×FA female (both parents were exposed to FA preconception) and, (2) PM 2.5male×PM 2.5female (both parents were exposed to PM 2.5 preconception). Male offspring were divided: (1) preconception FA (offspring born to FA exposed parents) and, (2) preconception PM 2.5 (offspring born to PM 2.5 exposed parents) and analyzed at 3 months of age. Echocardiography identified increased left ventricular end systolic volume and reduced posterior wall thickness, reduced %fractional shortening and %ejection fraction in preconception PM 2.5 offspring. Cardiomyocytes isolated from preconception PM 2.5 offspring showed reduced %peak shortening, −dL/dT, TPS90 and slower calcium reuptake (tau). Gene and protein expression revealed modifications in markers of inflammation (IL‐6, IL‐15, TNFα, NFқB, CRP, CD26E, CD26P, intercellular adhesion molecule 1, and monocyte chemoattractant protein‐1) profibrosis (collagen type III alpha 1 chain), oxidative stress (NOS2), antioxidants (Nrf2, SOD, catalase), Ca2+ regulatory proteins (SERCA2a, p‐PLN, NCX), and epigenetic regulators (Dnmt1, Dnmt3a, Dnmt3b, Sirt1, and Sirt2) in preconception PM 2.5 offspring.
Conclusions
Preconception exposure to PM 2.5 results in global cardiac dysfunction in adult offspring, suggesting that abnormalities during development are not limited to the prenatal or postnatal periods but can also be determined before conception.
Keywords: calcium, cardiovascular function, inflammation, myocyte, particulate matter, preconception
Subject Categories: Calcium Cycling/Excitation-Contraction Coupling, Animal Models of Human Disease, Contractile function, Myocardial Biology, Pathophysiology
Short abstract
See Editorial by Stapleton
Clinical Perspective
What Is New?
The study used an in vivo mouse model of preconception exposure to investigate the adverse cardiac effects on male offspring.
The results revealed that the preconception period represents a “critical window” for the development of cardiac dysfunction in the offspring at adulthood. These data indicate that environmental exposures may cause adaptations to the germ cells that could provide the mechanistic basis for the cardiovascular dysfunction observed.
What Are the Clinical Implications?
Current findings may help in designing future clinical and regulatory guidelines addressing environmental exposures and human health specifically parental exposures in relation to cardiovascular diseases in offspring.
Introduction
Exposure to environmental pollution has significant effects on human health and the progression of various diseases. More specifically, ambient particulate matter (PM) pollution is commonly recognized as one of the top global health burdens following its association with 3.2 million deaths per year.1 An escalating amount of clinical and epidemiologic studies have established a relationship between cardiovascular disease (CVD) and exposure to ambient fine particulate matter (<2.5 μm in diameter; PM2.5)2 specifically arrhythmias, hypertension, myocardial infarction and cardiac remodeling.3, 4, 5 Despite much research focusing on the effects of air pollution on preexisting heart conditions, whether PM exposure can be a primary determinant of cardiovascular disease directly or by predisposing the organ to worse outcomes has become a heightened area of interest. In fact, exposures during “sensitive/critical” windows of development (both prenatal and perinatal exposure) can have lasting impacts on health. These exposures can influence the developing organs, tissues, and cells, and can lead to impairments in adult cardiac phenotypes. Previous work in our laboratory has shown that PM2.5 exposure during the in‐utero period can have detrimental effects on cardiac function of neonatal6 and adult mice.7
A severe outcome may also result from changes occurring in the unfertilized male and female germ cells eg, through epigenetic changes.8, 9, 10 It is increasingly appreciated that parental exposure to environmental stressors can have significant effects on offspring phenotypes and exposure to PM2.5 both before conception and during pregnancy impairing fetal development and resulting in adverse birth outcomes.11, 12, 13 Whether part of the phenotypic changes induced by PM2.5 exposure during the critical preconception period on offspring include a predisposition to or set pathological changes in cardiac function remain unknown. In the present study, we examined the effects of parental preconception exposure on cardiac function in adult offspring and elucidated the potential molecular mechanisms involved.
Materials and Methods
The data and analytic methods are available from the corresponding author for purposes of reproducing the results or replicating the procedures. All materials used in this study are available commercially from the indicated vendors.
Animals and Exposure
All animal procedures were performed according to guidelines provided by the National Institutes of Health and approved by the Institutional Animal Care and Use Committee (IACUC) at The Ohio State University, Columbus, Ohio. Friend leukemia virus B (FVB) mice of both sexes were housed for 1 week in our housing facility before PM exposure (animal numbers are listed in the figure legends). We then exposed male and female mice, separately, to either filtered air (FA) or PM2.5 at an average concentration of 38.58 μg/m3 for 6 hours/day, 5 days/week for 3 months before conception. The aerosol concentration system located at The Ohio State University7 was used to deliver concentrated PM2.5 exposures from the Columbus, OH region. Mice in the FA group were exposed using an identical system with the exception of implementing a HEPA filter at the inlet that successfully removed all ambient particles. After 3 months of exposure, the mice were crossbred into 2 groups: (1) FAmale×FAfemale (both parents were FA exposed before conception) and, (2) PM2.5male×PM2.5female (both parents were PM2.5 exposed before conception). We observed successful vaginal plugs within a week of mating. Male offspring born to these crosses were divided into 2 groups: (1) preconception FA (offspring born to FA exposed parents) and, (2) preconception PM2.5 (offspring born to PM2.5 exposed parents) and were analyzed at 3 months of age for the various experiments as described below. We included 2 mice from each litter for echocardiography and cardiomyocyte functional analyses and 1 mouse per litter was used for molecular studies. A detailed outline of the preconception exposure model is provided in Figure S1.
Physiological Parameters
All mice were euthanized at 3 months following echocardiography. Body weight, heart weight, heart weight/body weight, heart weight/tibial length and tibia length were measured. Body surface area (BSA) was calculated using the formula BSA=K mass0.667; where K is known as the Meeh constant=9.82. This equation is widely accepted for estimating BSA in animals.14 Assessment of fat and lean mass was determined in awake animals using the echo magnetic resonance imaging (EchoMRI) 3‐in‐1 (EchoMRI, Houston, TX).
Assessment by Echocardiography
Cardiac function was assessed via echocardiography (40 MHz transducer, Vevo 2100; Visualsonics [Toronto, ON, Canada]). Three‐month‐old mice were kept sedated with 1.5% isoflurane (in 100% O2) anesthesia supplied through a nose cone, while the body temperature was maintained at 37°C. Following successful anesthesia, the chests were cleared of fur using hair removal cream. Next, pre‐warmed ultrasound gel was applied to the chest, along with a 15‐MHz probe (optimized for mice) that was placed in the parasternal, short‐axis orientation. Cine loops collected from M mode views were analyzed for LV systolic and diastolic internal dimensions (LVESd and LVEDd), as well as systolic and diastolic posterior wall thickness (PWTs and PWTd). The following measurements were calculated: fractional shortening percentage (%FS=[(LVEDd−LVESd)/(LVEDd)×100), LV end diastolic volume (LVEDV; 7/(2.4+LVESd)×LVEDd), LV end systolic volume (LVESV; 7/(2.4+LVESd)×LVESd), ejection fraction (ejection fraction(LVEDV−LVESV)/LVEDV×100), stroke volume (LVEDV−LVESV), and cardiac output (SV×HR).
Cardiomyocyte Isolation and Functional Assessment
Three‐month‐old mice were euthanized, and cardiomyocytes were isolated as previously described.15 In brief, following cardioectomy, isolations were completed by enzymatic digestion (Liberase and Trypsin) via coronary retrograde perfusion through the aorta. Isolated cardiomyocytes were then plated on laminin‐coated glass‐bottom inserts (Cell MicroControls, Norfolk, VA) for functional analyses. Glass inserts were perfused with prewarmed contractile buffer in a chamber mounted on an Olympus IX‐71 microscope. Cells were stimulated (1 Hz, 3‐ms duration) with a Myopacer Field‐Stimulator system (IonOptix, Milton, MA) and functional properties of the cells were evaluated using the Sarclen Sarcomere Length Acquisition Module with the Myocam‐S Digital CCD camera video imaging system (IonOptix, Milton, MA). Sarcomere percent peak shortening (normalized to baseline sarcomere length, %PS; cellular corollary of %FS), sarcomere maximal departure and return velocities (+dL/dT, −dL/dT), and sarcomere time to 90% peak shortening (TPS90) and time to 90% relengthening (TR90) were measured to assess inotropic and lusitropic function using 8 to 12 cells/heart.
Intracellular Ca2+ Transients Measurements
We performed Ca2+ measurements using Fura‐2AM, a Ca2+ sensitive fluorimetric dye. Briefly, cardiomyocytes in glass‐bottom dishes were loaded with Fura‐2‐AM at a concentration of 5 μmol/L for 20 minutes at 25°C. Cells were then washed and treated with normal culture media for 20 minutes at 25°C. Fluorescence was recorded in stimulated cardiomyocytes at 37°C using a dual‐excitation, single‐emission system (IonOptix). Transients were used to monitor basal Ca2+, the amplitude of the Ca2+ transient (∆340/380), and fluorescence decay rate (tau) as an indication of intracellular Ca2+ clearing rate.
Quantitative Real‐Time Polymerase Chain Reaction
Frozen heart tissues of 3‐month‐old mice were used for the isolation of total RNA. Tissues were homogenized with a TissueLyser (Qiagen, Boston, MA) using Trizol buffer followed by transferring to a chloroform containing tube and centrifuge. The supernatant was mixed with equal volume of 70% ethanol and loaded onto a Qiagen RNA processing column from the Qiagen RNeasy Min‐Kit (Qiagen, Boston, MA). All steps were followed according to the manufacturer's instructions. RNA quality and concentration was determined via NanoDrop 2000c (ThermoScientific, Wilmington, DE). A known amount of RNA was reverse transcribed to generate cDNA using the iScript Supermix kit (Bio‐Rad, Hercules, CA) on a CFX96 Thermocycler (BioRad, Hercules, CA). Primers were used at a final concentration of 0.25 to 0.5 μmol/L for target genes and normalized to GAPDH expression. Relative gene expression levels were quantified using the formula 2−ΔΔCt. A 3‐step amplification protocol was implemented: 10 minutes of denaturation at 95°C, followed by 45 cycles of denaturation (95°C, 1 second), annealing (65°C, 10 seconds), and extension (72°C, 20 seconds). Gene‐specific primer sequences are presented in Table 1.
Table 1.
Primer Sequences Used for PCR Amplification
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Gapdh | CTCACTCAAGATTGTCAGCAATG | GAGGGAGATGCTCAGTGTTGG |
| IL‐6 | GAAATGCCACCTTTTGACAGTG | TGGATGCTCTCATCAGGACAG |
| IL‐15 | ACATCCATCTCGTGCTAC‐TTGT | GCCTCTGTTTTAGGGAGA‐CCT |
| TNF‐α | CTGAACTTCGGGGTGATCGG | GGCTTGTCACTCGAATTTTGAGA |
| NF‐κB | GGAGCCTGGGAAATGAAAGAA | GCCAGAGCCCTACCTGATTG |
| CRP | GTCTGCTACGGGGATTGTAGA | CACCGCCATACGAGTCCTG |
| CD62P | CATCTGGTTCAGTGCTTTGATCT | ACCCGTGAGTTATTCCATGAGT |
| CD62E | ATGAAGCCAGTGCATACTGTC | CGGTGAATGTTTCAGATTGGAGT |
| TGCCTCTGAAGCTCGGAT‐ATAC | TCTGTCGAACTCCTCAGT‐CAC | |
| COL‐3 | AGCTTTGTGCAAAGTGGA‐ACCTGG | CAAGGTGGCTGCATCCCA‐ATTCAT |
| Dnmt1 | ATCCTGTGAAAGAGAACCCTGT | GTGGAAACCACCGAGAACAC |
| Dnmt3a | CTGTCAGTCTGTCAACCTCAC | CATAACTCCACACGTGGTTCTCAG |
| Dnmt3b | AGCGGGTATGAGGAGTGCAT | GGGAGCATCCTTCGTGTCTG |
| Sirt1 | TGATTGGCACCGATCCTCG | CCACAGCGTCATATCATCCAG |
| Sirt2 | GCGGGTATCCCTGACTTCC | CGTGTCTATGTTCTGCGTGTAG |
PCR indicates polymerase chain reaction.
Western Blotting
Frozen heart tissues from 3‐month‐old mice were homogenized in lysis buffer as described previously.6 Protein concentration was then quantified using a bicinchinonic acid assay (ThermoScientific, Wilmington, DE). The proteins were separated on SDS‐PAGE gels (20 g) and transferred to polyvinylidene fluoride or polyvinylidene difluoride (PVDF) membranes. Membranes were probed with antibodies to sarco/endoplasmic reticulum Ca2+‐ATPase (SERCA2a) (Thermo Fisher Scientific, MA3‐919, Waltham, MA), phosphorylated phospholamban (p‐PLN) (Santa Cruz Biotechnology, sc‐12963, Dallas, TX), Na+/Ca2+ exchanger (NCX) (Thermo Fisher Scientific, MA3‐926, Waltham, MA), Nitric oxide synthase 2 (NOS2) (Santa Cruz Biotechnology, sc‐651, Dallas, TX), DNA methyltransferase 1 (DNMT1) (Abcam, ab‐13537, Cambridge, MA), DNA methyltransferase 3A (DNMT3A) (Abcam, ab‐13888, Cambridge, MA), monocyte chemoattractant protein‐1 (MCP‐1) (Santa Cruz Biotechnology, sc‐136750, Dallas, TX), and collagen type III alpha 1 chain (Santa Cruz Biotechnology, sc‐8780‐R, Dallas, TX). The densities of all protein bands were normalized to β‐actin (Sigma, A1978, St. Louis, MO). Band signals were detected via the enhanced chemiluminescence method (PIERCE, SuperSignal West Pico, Chemiluminescent Substrate, CA). X‐ray films were scanned using Image Lab software (Bio‐Rad, Hercules, CA, version 4.1) and later assessed.
Statistical Analyses
All data were analyzed for statistical significance using GraphPad Prism 7 (GraphPad Software, Inc, San Diego, CA). Differences between 2 groups were determined using a Student t test (2‐tailed) with an α‐value of 0.05. All error bars in the figures represent standard error of the mean; *, †, ‡ represent significant differences of P<0.05, <0.01, and <0.001, respectively. Additionally, for echocardiography and cardiomyocyte functional analyses, we performed mixed model analysis with a restricted maximum likelihood test to assess correlation between littermates and to test group difference.
Results
Parental Preconception PM2.5 Exposure Results in Altered Body Weight Parameters
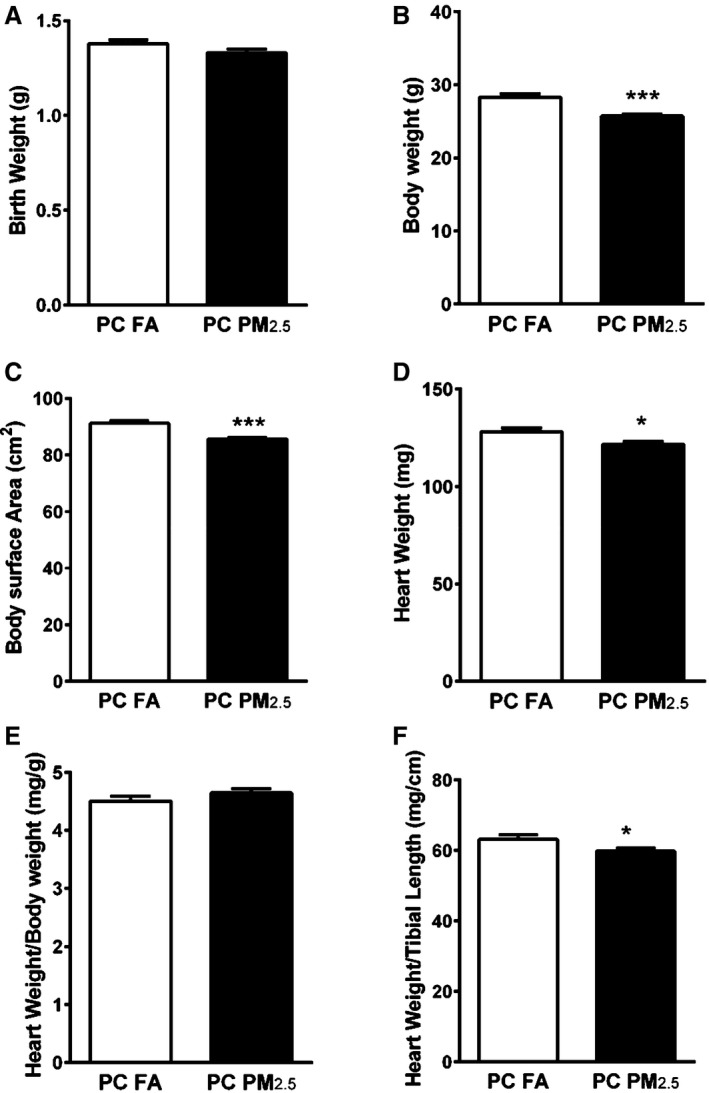
Litter size or sex did not vary significantly between preconception FA (offspring born to preconception FA exposed parents) and preconception PM2.5 (offspring born to preconception PM2.5 exposed parents) groups. At birth, there was no difference in body weights of the pups born to preconception PM2.5 or FA exposed parents (Figure 1A). To measure the impact of preconception PM2.5 exposure on growth and body size, we measured body weight at adulthood (3 months). Body weights of 3‐month‐old preconception PM2.5 offspring were significantly lower compared with preconception FA offspring (28.30±0.45 g preconception FA; 25.73±0.25 g preconception PM2.5; P<0.001) (Figure 1B). Since the body weights were different, we also measured body surface area (BSA), absolute heart weight (HW), heart weight normalized to body weight (HW/BW) and heart weight normalized to tibia length (HW/tibia length). Preconception PM2.5 exposure was associated with significantly lower BSA (91.27±0.97 cm2 preconception FA; 85.68±0.57 cm2 preconception PM2.5; P<0.001) (Figure 1C) and heart weights (128.2±1.92 mg preconception FA; 121.6±1.55 mg preconception PM2.5; P<0.05) at 3 months of age (Figure 1D). HW/BW remained unchanged (4.51±0.08 mg/g preconception FA; 4.65±0.07 mg/g preconception PM2.5; P=0.2), however HW/tibia length was decreased in preconception PM2.5 offspring compared with preconception FA offspring (63.28±1.17 mg/cm preconception FA; 59.89±0.79 mg/cm preconception PM2.5; P<0.05) (Figure 1E and 1F). To perform detailed body composition analyses, we also investigated EchoMRI in 3‐month old offspring. Parental preconception exposure to PM2.5 did not alter fat mass but lean mass was significantly lower in preconception PM2.5 offspring compared with preconception FA offspring (Figure S2).
Figure 1.
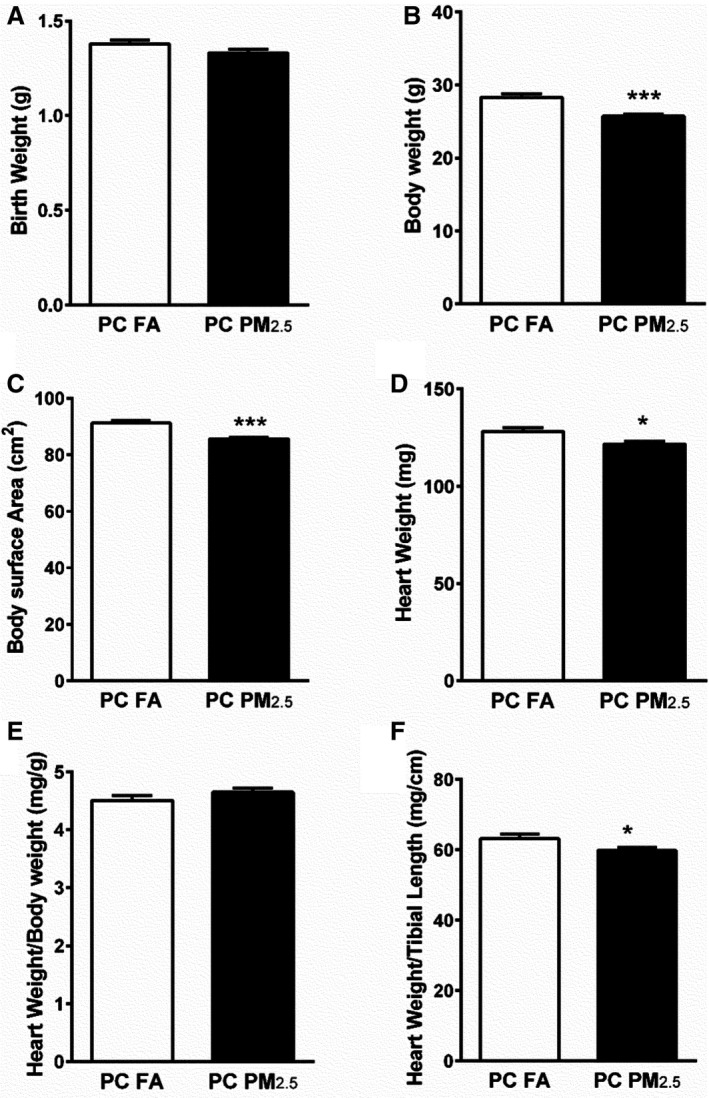
Body weight parameters from mice born to preconception FA or PM 2.5‐exposed parents. A, Birth weight (n=42–49 pups/group), (B) body weight (n=12–13 mice/group), (C) body surface area (BSA) (n=12–13 mice/group), (D) heart weight (n=12–13 mice/group), (E) heart weight/body weight (HW/BW) (n=12–13 mice/group), (F) heart weight/tibial length (HW/TL) (n=12–13). Data are expressed as ±SEM. *P<0.05, ***P<0.001 vs preconception FA controls. A, Represents birth weight of all offspring (irrespective of sex). B through F, Represent parameters that were analyzed in 3‐month‐old male offspring only. PC FA indicates preconception filtered air; PC PM2.5, preconception particulate matter (<2.5 μm in diameter).
Parental PM2.5 Exposure Leads to LV Systolic Dysfunction
Echocardiographic analyses of 3‐month‐old preconception PM2.5 offspring revealed no change in LVEDs (data not shown) and slightly increased LVESd (2.51±0.07 mm preconception FA; 2.74±0.10 mm preconception PM2.5; P=0.06). Posterior wall thickness was not different in diastole (PWTd), but was significantly decreased during systole (PWTs) (1.70±0.06 mm preconception FA; 1.44±0.08 mm preconception PM2.5; P<0.05) (Figure 2A and 2B). These changes were associated with reduced systolic function as evidenced by reduced %FS (35.09±1.34 preconception FA; 29.05±1.25 preconception PM2.5; P<0.01) and %ejection fraction (64.86±1.76 preconception FA; 56.27±1.95 preconception PM2.5; P<0.01) in preconception PM2.5 offspring compared with preconception FA offspring (Figure 2C and 2D). Further, we observed significant increases in LVESV (Table 2). Other important hemodynamic parameters including cardiac output, stroke volume, cardiac index, and heart rate were not different between groups (Table 2). When compared using mix model analysis with restricted maximum likelihood test, our results showed a significant change in %FS and ejection fraction (P<0.05) and a significant trend (P=0.07) in PWTs between both groups.
Figure 2.
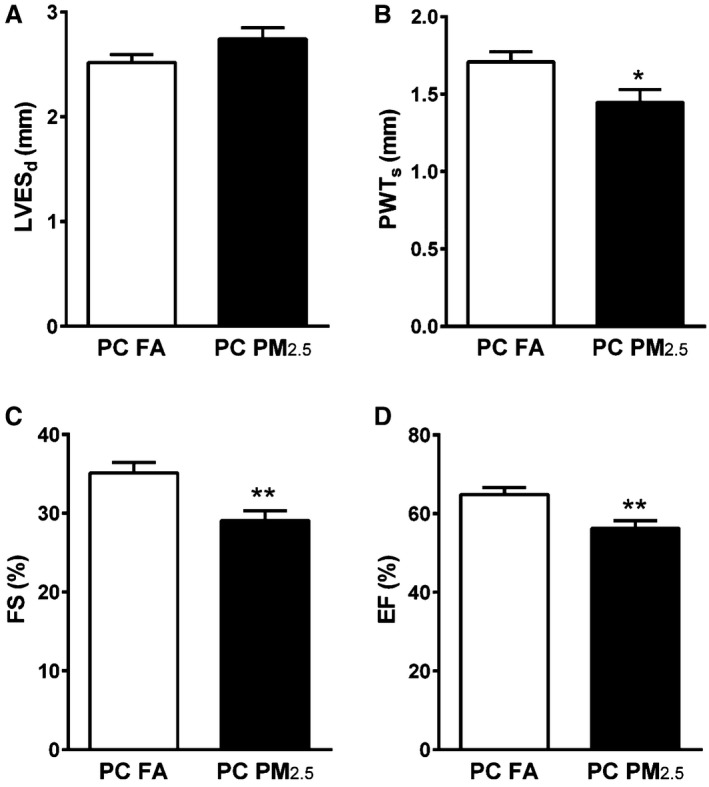
Transthoracic echocardiography to assess cardiac function of 3‐month‐old mice that were born to preconception FA or PM 2.5 exposed parents. A, LVESd, (B) PWTs, (C) %FS, (D) %EF. Data were collected from 10 to 12 mice in each group. Five beat cycles were captured and 3 loops averaged per assessment. Data are expressed as ±SEM. %EF indicates %ejection fraction; %FS, %fractional shortening; LVESd, left ventricular end‐diastolic dimension; PC FA, preconception filtered air; PC PM2.5, preconception particulate matter (<2.5 μm in diameter); PWTs, posterior wall thickness during systole. *P<0.05, **P<0.01 vs PC FA controls.
Table 2.
Echocardiographic Parameters of 3‐Month‐Old Preconception FA and Preconception PM2.5 Mice
| Preconception FA (N=10) | Preconception PM2.5 (N=12) | P Value | |
|---|---|---|---|
| IVSD, mm | 1.12±0.14 | 1.15±0.15 | 0.359 |
| IVSS, mm | 1.65±0.18 | 1.60±0.25 | 0.305 |
| LVEDV, μL | 256.65±58.89 | 257.57±88.61 | 0.712 |
| LVESV, μL | 59.19±19.82 | 82.08±35.11 | 0.029* |
| Stroke volume, μL | 197.46±45.36 | 175.49±59.57 | 0.475 |
| Cardiac output, mL/min | 90.20±19.11 | 83.55±31.11 | 0.686 |
| Cardiac index, L/min per m2 | 0.99±0.06 | 1.00±0.12 | 0.920 |
| Heart rate, bpm | 448.25±3.21 | 442.2±3.24 | 0.370 |
Data are expressed as mean±SEM. FA indicates filtered air; IVSd, interventricular septal end diastole diameter; IVSs, interventricular septal end systole diameter; LVEDV, left ventricular end diastolic volume; LVESV, left ventricular end systolic volume; PM2.5, particulate matter (<2.5 μm diameter).
Parental Preconception Exposure to PM2.5 Impairs Offspring Cardiomyocyte Function
To validate cardiac‐specific effects of preconception PM2.5 exposure, we performed functional analyses in isolated single cardiomyocytes from preconception PM2.5 and preconception FA adult hearts. Our in vitro data demonstrated alterations in sarcomere function as indicated by a reduction in %PS (12.93±0.42 preconception FA; 11.10±0.41 preconception PM2.5; P<0.01), −dL/dT (−10.60±0.22 μm/s preconception FA; −8.50±0.60 μm/s preconception PM2.5; P<0.05) and TPS90 (0.081±0.003 seconds preconception FA; 0.071±0.002 seconds preconception PM2.5; P<0.05) (Figure 3A, 3B, and 3D) in cardiomyocytes isolated from preconception PM2.5 offspring compared with preconception FA offspring. +dL/dT and TR90 were not different between groups (data not shown). These results further corroborate our echocardiography findings. When compared using mix model analysis with restricted maximum likelihood test, our results showed no significant difference in %PS, a significant change in TPS90 and Tau (P<0.05), and a significant trend (P=0.07) in −dL/dT.
Figure 3.
In vitro cardiomyocyte functional and calcium signaling parameters obtained from 3‐month‐old mice that were born to preconception filtered air or particulate matter (<2.5 μm in diameter) exposed parents. Graphs depicted are representative of (A) %peak shortening (%PS), (B) negative velocity (‐dL/dT), (C) time‐to‐peak shortening (TPS90), (D) fluorescence decay rate (τ), (E) calcium transient amplitude (Δ340/380). Data were collected from 70 to 80 cardiomyocytes isolated from 9 to 10 mice per group taken from individual litters. Data are expressed as mean±SEM. %PS indicates %peak shortening; ‐dL/dT, negative velocity; PC FA indicates preconception filtered air; PC PM2.5, preconception particulate matter (<2.5 μm in diameter); TPS90, time‐to‐peak shortening. *P<0.05, **P<0.01 vs PC FA controls.
Parental Preconception Exposure to PM2.5 Impairs Fluorescence Decay Rate Without Affecting Calcium Transient Amplitude
Ca2+ reuptake was delayed as indicated by increased duration of Tau (Ʈ) in preconception PM2.5 offspring compared with preconception FA offspring (0.42±0.06 s preconception FA; 0.67±0.09 s preconception PM2.5; P<0.05). Ca2+ transient amplitude (∆340/380), however, remained unchanged between both groups (Figure 3C and 3E).
Parental Preconception Exposure to PM2.5 Alters Expression of Ca2+ Regulatory Proteins
The normal cardiac cycle (contraction/relaxation) is dependent on regulated movement of calcium between the sarco‐endoplasmic reticulum and the cytosol. To elucidate the role of calcium pathways in cardiac dysfunction, we examined cardiac genes and protein expression of important cellular calcium regulators. While no change was observed in mRNA expression of SERCA2a, PLN, and NCX (Figure S3), a marked increase in SERCA2a and p‐PLN and modest increase in NCX was observed in preconception PM2.5 offspring compared with preconception FA offspring (Figure 4A through 4C).
Figure 4.
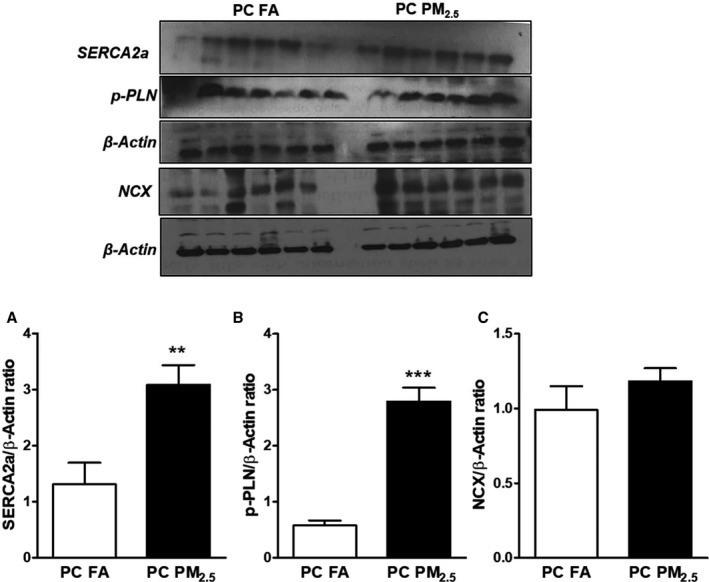
Western blot analysis showing expression Ca2+ handling proteins (A) SERCA‐2A, (B) p‐PLN; (C) NCX. Representative Western blot shown above and quantification below. Data were obtained from 3‐month‐old mouse hearts (n=6 mice/group) that were born to preconception filtered air or particulate matter (<2.5 μm in diameter) exposed parents and expressed as mean±SEM. NCX indicates Na+/Ca2+ exchanger; PC FA indicates preconception filtered air; PC PM2.5, preconception particulate matter (<2.5 μm in diameter); p‐PLN, phosphorylated phospholamban; SERCA‐2A, sarco/endoplasmic reticulum Ca2+‐ATPase. **P<0.01, ***P<0.001 vs PC FA controls.
Parental Preconception Exposure to PM2.5 Activates Oxidative Stress Pathways
To better define the mechanisms of cardiac dysfunction, we studied the expression of oxidative stress markers in the myocardium of 3‐month‐old preconception PM2.5 offspring. Myocardial protein expression of nitric oxide synthase 2 (NOS2) was also increased in preconception PM2.5 offspring compared with preconception FA offspring (Figure 5A). Quantitative polymerase chain reaction data showed increased expression of nuclear factor (erythroid‐derived 2)‐like 2 (Nrf2) and superoxide dismutase (SOD) with no change in the expression of catalase (Figure 5B through 5D).
Figure 5.
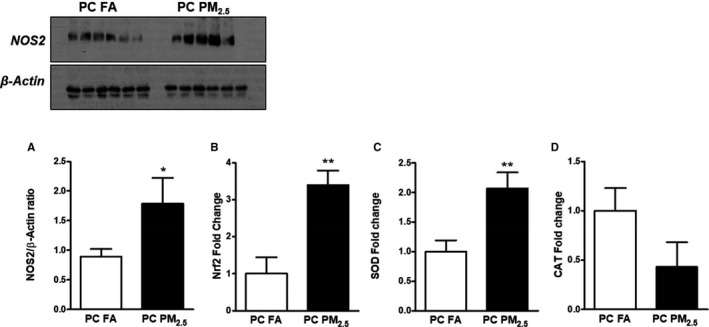
A, Western blot analysis showing expression of NOS‐2. Representative Western blot is shown on the top. Quantitative polymerase chain reaction analysis showing expression of (B) Nrf2, (C) SOD, (D) CAT. Data were obtained from 3‐month‐old mouse hearts (n=6 mice/group) that were born to preconception FA or PM 2.5 exposed parents and expressed as mean±SEM. CAT indicates catalase; NOS‐2, nitric oxide synthase 2; Nrf2, nuclear factor (erythroid‐derived 2)‐like 2; SOD, superoxide dismutase; PC FA, preconception filtered air; PC PM2.5, preconception particulate matter (<2.5 μm in diameter). *P<0.05, **P<0.01 vs preconception FA controls.
Parental Preconception Exposure to PM2.5 Leads to Activation of an Inflammatory Response
Oxidative stress activates inflammatory pathways and therefore we studied the expression of proinflammatory genes. Myocardial mRNA expression of proinflammatory markers such as interleukin‐6 and ‐15 (IL‐6 and IL‐15), tumor necrosis factor alpha (TNF‐α), nuclear factor kappa‐light‐chain‐enhancer of activated B cells (NF‐κB), C‐reactive protein (CRP), E‐selectin (CD26E), P‐selectin (CD26P) and intercellular adhesion molecule 1 (ICAM‐1) were increased in preconception PM2.5 offspring compared with preconception FA offspring (Figure 6A through 6H). Overlapping pathways involving inflammatory mediators (NF‐κB) induce chemotactic cytokine/chemokines (CC) in myocardial tissue. To elucidate the role of chemokine pathways in the present study, we examined protein expression of MCP‐1, a potent chemoattractant for monocytes, T cells, and NK cells. We observed a marked upregulation of MCP‐1 protein levels in preconception PM2.5 mouse hearts compared with preconception FA offspring (Figure 6I).
Figure 6.
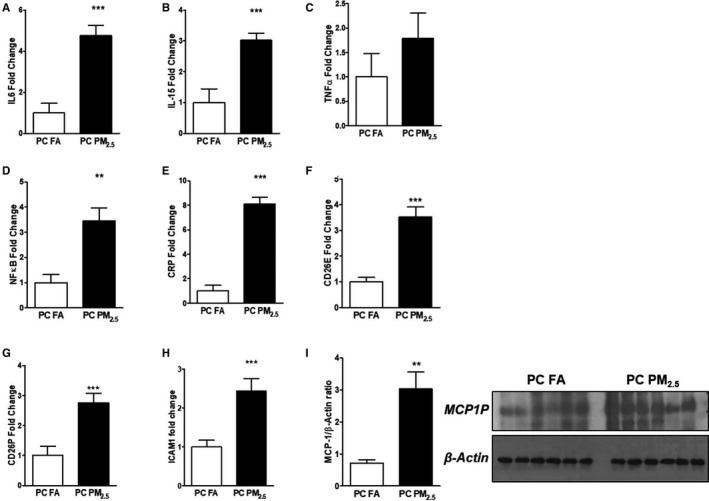
Quantitative polymerase chain reaction expression of (A) IL‐6, (B) IL‐15, (C) TNF‐α, (D) NF‐κB, (E) CRP, (F) E‐selectin (CD26E), (G) P‐selectin (CD26P), (H) ICAM‐1. I, Western blot analysis showing expression of MCP‐1. Representative Western blot was shown on the right and quantification on the left. Data were obtained from 3‐month‐old mouse hearts (n=6 mice/group) that were born to preconception FA or PM 2.5 exposed parents and expressed as mean±SEM. CRP indicates C‐reactive protein; ICAM1, intercellular adhesion molecule 1; IL‐6, interleukin 6; IL‐15, interleukin 15; MCP‐1, monocyte chemoattractant protein‐1; NF‐κB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; PC FA, preconception filtered air; PC PM2.5, preconception particulate matter (<2.5 μm in diameter); TNF‐α, tumor necrosis factor‐alpha. **P<0.01, ***P<0.001 vs PC FA controls.
Parental Preconception Exposure to PM2.5 Increases Cardiac Collagen3a1 Expression
Increased levels of inflammatory mediators were also found to be associated with upregulation of fibrogenic mediators such as structural extracellular matrix protein. Collagen3a1 expression was found to be significantly upregulated both at transcriptional and translational levels (Figure 7A and 7B).
Figure 7.
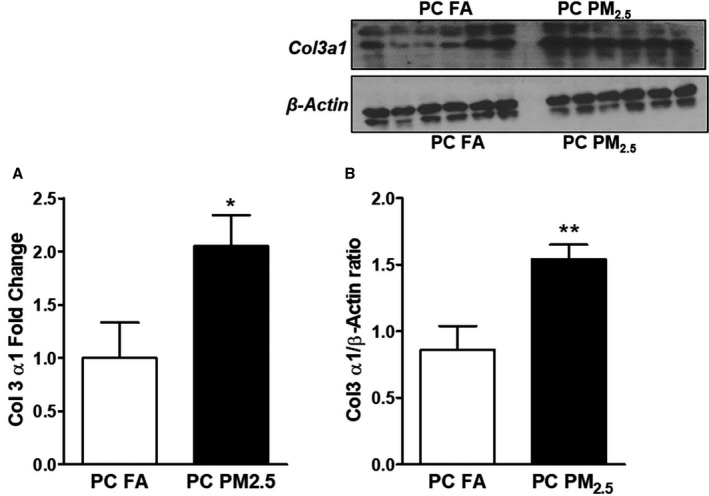
A, Quantitative polymerase chain reaction expression of collagen3a1 (Col3a1) and (B) protein expression of Col3a1 via Western blot analysis. Representative Western blot is shown above and quantification below. Data were obtained from 3‐month‐old mouse hearts (n=6 mice/group) that were born to preconception FA or PM 2.5‐exposed parents and expressed as mean±SEM. PC FA indicates preconception filtered air; PC PM2.5, preconception particulate matter (<2.5 μm in diameter). *P<0.05, **P<0.01 vs PC FA controls.
Parental Preconception Exposure to PM2.5 Alters Epigenetic Mediators
To further elucidate the potential epigenetic mechanisms responsible for the cardiac dysfunction observed in 3‐month‐old mice born to preconception PM2.5‐exposed parents, we examined the expression of epigenetic mediators in myocardial tissue both at mRNA and protein levels. While our quantitative polymerase chain reaction data demonstrated no change in the expression of DNA methyltransferases (Figure S4), we observed significant downregulation in protein expression of Dnmt1a and no change in Dnmt3a. (Figure 8A and 8B). Further, mRNA expression of sirtuins (Sirt1 and Sirt2) was significantly increased in preconception PM2.5 offspring compared with preconception FA offspring (Figure 8C and 8D).
Figure 8.
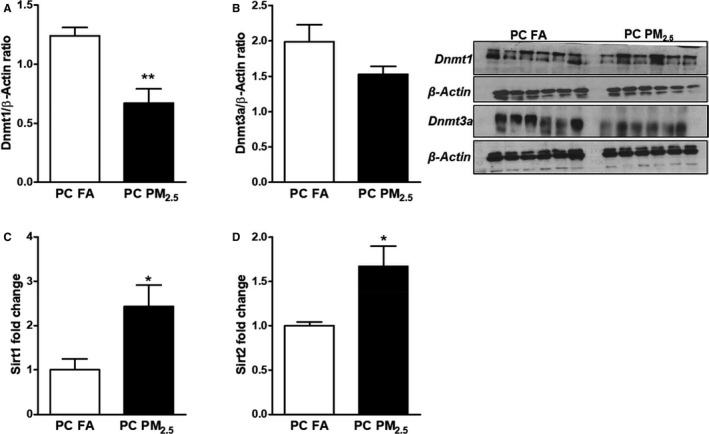
Western blot analysis showing expression of (A) DNA methyltransferases‐1 (Dnmt1) and (B) DNA methyltransferases‐3a (Dnmt3a). Representative Western blots are shown on the left and quantification on the right. Quantitative polymerase chain reaction analysis showing expression of (C) Sirt1 and (D) Sirt2. Data were obtained from 3‐month‐old mouse hearts (n=6 mice/group) that were born to preconception FA or PM 2.5‐exposed parents and expressed as mean±SEM. Dnmt1 indicates DNA methyltransferases‐1; Dnmt3a, DNA methyltransferases; PC FA, preconception filtered air; PC PM2.5, preconception particulate matter (<2.5 μm in diameter); Sirt1, sirtuin 1; Sirt2, sirtuin 2. *P<0.05, **P<0.01 vs PC FA controls.
Discussion
Our study shows for the first time that exposure to PM2.5 exclusively during the preconception period is sufficient to program the developing fetus and cause significant changes in cardiac function, gene and protein expression. While the primary stimulus was preconception PM2.5 exposure, altered Ca2+ regulatory proteins, elevated levels of oxidative stress markers, inflammatory and fibrogenic mediators suggest the involvement of long‐lasting stress responses. Our results are consistent with previous investigations using other models of preconception perturbations such as maternal nutritional status,16 alcohol consumption,17 and exposure to organic pollutants.11 All of these studies underscore the importance of the periods around the time of conception as critical windows of development.
Several epidemiological studies have confirmed an association between PM exposure and other stressful preconception events with low birth weight.13, 18, 19 We did not observe any significant differences in offspring birth weight in response to parental preconception PM2.5 exposure. Therefore, our model is not confounded by low birth weight, a common finding of many other preconception models. Interestingly, when analyzed at 3 months of age, the body weight and heart weight of preconception PM2.5 offspring were significantly less. Furthermore, a detailed body composition profile demonstrated reduced lean mass with fat mass in preconception PM2.5 offspring.
The primary finding of the present study is the development of cardiac dysfunction in adult offspring born to preconception PM2.5‐exposed parents as suggested by the in vivo and in vitro functional data. Preconception PM2.5 offspring also showed a significant increase in LVESV. An increase in LVESV is associated with increased cardiac events in patients with LV systolic dysfunction20 and is considered as an independent predictor of mortality.21 Isolated cardiomyocyte measurements further corroborate the echocardiography data suggestive of cardiomyocyte dysfunction. The understanding of molecular and cellular pathways underlying CVDs may help to define our observed cardiac effects.
The synchronized process of contraction and relaxation is regulated by several mediators, but most important among them is Ca2+. Alterations in Ca2+ signaling and Ca2+ handling proteins can affect cardiac function, potentially leading to impaired myocardial contractility. Thus, we investigated the involvement of the Ca2+ handling proteins. Our results demonstrated no change in the expression of these proteins at the mRNA level, but protein expression was significantly upregulated. The upregulation of SERCA‐2A and simultaneous increased phosphorylated phospholamban in preconception PM2.5 offspring suggests increased Ca2+ cycling to maintain optimal cardiomyocyte function as a protective/adaptive mechanism in response to preconception PM2.5‐associated adverse cardiac effects. Similar results were also observed by our group in a model of maternal inflammation and neonatal hyperoxia22 and in 14‐day‐old mice following in utero PM2.5 exposure.6 Of note, preconception PM2.5 exposure could affect the myofilaments and therefore future studies are warranted to investigate isolated myofilaments.
Another important inference drawn from the present study is the altered expression of oxidative stress markers in our preconception PM2.5 exposure model. Elevated NOS2 leads to a toxic amount of nitric oxide (NO) production which is associated with decreased ventricular contractility and cardiovascular dysfunction.23, 24 Increased NO also contributes to the production of reactive oxygen species (such as superoxide radical [])25 and reactive nitrogen species,26 which further imparts deleterious effects. Moreover, the expression of Nrf2, a key transcription factor involved in regulating various antioxidant genes was also found to be upregulated. We speculate that increased expression of Nfr2 is attributable to simultaneous activation of the body's defense mechanism to counteract inflammation‐associated oxidative insult. Similar results were observed previously in the setting of long‐term air pollution exposure.27 Among several other antioxidant genes, SOD is also regulated by Nrf2 and the simultaneous increased expression of SOD further suggests the antioxidant function of Nrf2. Surprisingly, we did not observe differences in catalase expression between both groups. This could indicate that its expression is not solely regulated by Nrf2 but also by other transcription factors and signaling molecules.28, 29 Based on our results, we speculate increased accumulation of H2O2 because of increased SOD (SOD catalyzes dismutation of into O2 and H2O2) and not simultaneous availability of equal amounts of catalase (catalase converts H2O2 into H2O and O2). The increased H2O2 directly and/or indirectly through the production of highly reactive hydroxyl radicals can cause myocardial injury.30 Taken together, our results suggest suboptimal antioxidant capacity and nitrosative stress and/or nitroso–redox imbalance, both of which can impair LV function.26
In addition to oxidative stress we also observed involvement of inflammatory pathway in our study. Previously, PM2.5 exposure was shown to increase expression of genes associated with inflammation, oxidative stress, and coagulation,31, 32 suggesting that these mechanisms are clinically relevant. The increased expression of IL‐6, IL‐15, and TNF‐α in our study may provide an explanation behind cardiac dysfunction observed in preconception PM2.5 offspring. In fact, previous evidence indicated that chronically elevated levels of IL‐6 induce maladaptive hypertrophy and decreased contractile function.33, 34 Also, myocyte specific production of IL‐6 in response to injury causes depressed basal contractility and leads to decreased contractile function.35 Besides playing a role as an inducer of cardiac dysfunction36 and mediator of cardiac remodeling,37 TNF‐α also activates NF‐κB,38 a key inflammatory mediator known to play a role in attenuated contractility,39 hypertrophy40 and cardiomyopathy.41 The increased expression of NF‐κB in our study was thus directly correlated with increased TNF‐α expression.
As demonstrated previously,42, 43 we also observed nearly an 8‐fold increase in CRP expression in preconception PM2.5 offspring supporting the activation of inflammatory pathways. Combined clinical findings and experimental observations suggest that CRP is not merely a CVD risk (inflammatory) biomarker but is a mediator of various CVDs.44 It has also been demonstrated that CRP upregulates the expression of adhesion molecules on endothelial cells which might serve as an explanation for increased expression of CD26E, CD26P, and ICAM‐1 in our study. These adhesion molecules are expressed in response to PM exposure and independently and jointly known to be associated with an increased risk of CVDs.45, 46 Inflammation is preceded by recruitment of monocytes and MCP‐1 is the chief chemokine responsible for monocyte infiltration to sites of inflammation.47 Functional significance of MCP‐1 has been reported in various cardiovascular diseases48, 49 and its expression has been shown to be upregulated by PM exposure.50, 51 We have also demonstrated the role of MCP‐1 in PM‐induced reduced contractility and Ca2+ handling of cardiomyocytes in our previously published work.52 In the present study, increased expression of MCP‐1 in preconception PM2.5 offspring provides additional evidence for the involvement of an inflammatory cascade. Cytokines augment release of local profibrotic mediators in response to injury.53 Increase in collagen type III alpha 1 chain expression in preconception PM2.5 indicates activation of profibrotic mechanisms to maintain optimum cardiac functioning.
Lastly, to answer the question of how the effects of preconception PM2.5 are transmitted to the offspring, we studied the expression of DNA methyltransferases. The process of DNA methylation (regulated by DNMTs) ensures correct gene expression, maintains genetic stability and may provide the link between parental and offspring phenotype/genotype transgenerationally. It has been shown that under the stressful preconception period epigenetic modifications can affect individuals adversely.54 The marked decrease in protein expression of DNMT1 (and modest decrease in DNMT3a) suggests altered epigenetic regulation. Studies have shown that PM exposure causes DNA methylation and can regulate various biological processes including inflammation leading to underlying CVD.55, 56, 57 In addition, we have also observed increased levels of Sirt1 and Sirt2 in our study; sirtuins are a family of deacetylases that play crucial roles in maintaining the crosstalk between genetic integrity and environment. Sirtuins are known to regulate DNMTs by deacetylating them at various sites58 thereby altering their activity.59 Our results are supported by a recent in vitro study showing antagonistic effects of Sirt1 on DNMTs.60 Taken together, our results established epigenetic links in preconception PM2.5 exposure model.
Limitations of the Study
One major concern with air pollution research is the complex nature of the natural environment. The characteristics of particulate matter and their interaction with other environmental agents/pollutants (such as other gaseous components), could be a potential contributor to the observed epidemiological effects of particulate exposure. In our study, we did not investigate the contributions of these potential confounders that could have more closely mimicked the clinical scenario. Because of such complex environmental interactions, there are still many research questions to be addressed by future air pollution experimental and clinical studies.
Conclusion and Future Directions
The U.S. Environmental Protection Agency (U.S. EPA) has set national ambient air quality standard for PM2.5 as 35 μg/m3 per day and 12 μg/m3 per year. PM2.5 concentrations achieved in this study were close to the daily allowable limit but significantly more than the annual limit. Based on our results, it is clear that this PM2.5 concentration is sufficient to trigger adverse cardiac effects in male offspring born to preconception PM2.5 exposed parents. Results from the present study are of high clinical importance as they demonstrate that parental exposures to PM2.5 before conception can have long‐lasting adverse cardiac effects on offspring. Altered germs cells from parents because of PM exposure can modify or worsen the developing cardiovascular system of the fetus. Future work should focus on the timing of environmental insults and their impact on the developing cardiovascular system which may prove beneficial in treating and preventing CVDs. Additionally, we will also examine the effects of preconception exposure on adult female offspring in our future work. Our findings thus underscore the importance of designing epidemiological studies that ascertain preconception parental exposures in relation to CVDs. Results from our study were unable to identify sole maternal or paternal contribution in the offspring phenotype. Hence, future studies should also focus on paternal versus maternal contribution in response to preconception exposure on the offspring.
Sources of Funding
This work was supported in part by The Ohio State University College of Medicine Roessler Research Scholarship (to Sugar and Katapadi), United States Veterans Affairs grants 1I01CX001329 and 1I01CX001515 (to Falvo), National Institutes of Health grants HL138738 to Stanford and AG057046, HL139348, NR012618 and ES019923 to Wold.
Disclosures
None.
Supporting information
Figure S1. Preconception exposure model.
Figure S2. Body weight parameters from mice born to preconception FA or PM2.5 exposed parents.
Figure S3. Quantitative PCR analysis showing expression of (A) sarco/endoplasmic reticulum Ca2+‐ATPase (SERCA‐2A), (B) Phospholamban (PLN) and (C) Na+/Ca2+ exchanger (NCX).
Figure S4. Quantitative PCR analysis showing expression of (A) DNA methyltransferases‐1 (Dnmt1), (B) DNA methyltransferases‐3a (Dnmt3a), (C) DNA methyltransferases‐3b (Dnmt3b).
Acknowledgments
We are grateful to Dr Federica del Monte for critical evaluation and discussion of the study and Dr Diego Hernandez‐Saavedra for statistical consultation and analysis.
(J Am Heart Assoc. 2018;7:e010797 DOI: 10.1161/JAHA.118.010797.)
References
- 1. Thurston G, Lippmann M. Ambient particulate matter air pollution and cardiopulmonary diseases. Semin Respir Crit Care Med. 2015;36:422–432. [DOI] [PubMed] [Google Scholar]
- 2. Franklin BA, Brook R, Arden Pope C III. Air pollution and cardiovascular disease. Curr Probl Cardiol. 2015;40:207–238. [DOI] [PubMed] [Google Scholar]
- 3. Wang T, Lang GD, Moreno‐Vinasco L, Huang Y, Goonewardena SN, Peng YJ, Svensson EC, Natarajan V, Lang RM, Linares JD, Breysse PN, Geyh AS, Samet JM, Lussier YA, Dudley S, Prabhakar NR, Garcia JG. Particulate matter induces cardiac arrhythmias via dysregulation of carotid body sensitivity and cardiac sodium channels. Am J Respir Cell Mol Biol. 2012;46:524–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Byrd JB, Morishita M, Bard RL, Das R, Wang L, Sun Z, Spino C, Harkema J, Dvonch JT, Rajagopalan S, Brook RD. Acute increase in blood pressure during inhalation of coarse particulate matter air pollution from an urban location. J Am Soc Hypertens. 2016;10:133–139.e4. [DOI] [PubMed] [Google Scholar]
- 5. Argacha JF, Collart P, Wauters A, Kayaert P, Lochy S, Schoors D, Sonck J, de Vos T, Forton M, Brasseur O, Beauloye C, Gevaert S, Evrard P, Coppieters Y, Sinnaeve P, Claeys MJ. Air pollution and ST‐elevation myocardial infarction: a case‐crossover study of the Belgian STEMI registry 2009–2013. Int J Cardiol. 2016;223:300–305. [DOI] [PubMed] [Google Scholar]
- 6. Tanwar V, Adelstein JM, Grimmer JA, Youtz DJ, Sugar BP, Wold LE. PM2.5 exposure in utero contributes to neonatal cardiac dysfunction in mice. Environ Pollut. 2017;230:116–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Tanwar V, Gorr MW, Velten M, Eichenseer CM, Long VP III, Bonilla IM, Shettigar V, Ziolo MT, Davis JP, Baine SH, Carnes CA, Wold LE. In utero particulate matter exposure produces heart failure, electrical remodeling, and epigenetic changes at adulthood. J Am Heart Assoc. 2017;6:e005796 DOI: 10.1161/JAHA.117.005796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guerrero‐Bosagna C, Settles M, Lucker B, Skinner MK. Epigenetic transgenerational actions of vinclozolin on promoter regions of the sperm epigenome. PLoS One. 2010;5:e13100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zama AM, Uzumcu M. Fetal and neonatal exposure to the endocrine disruptor methoxychlor causes epigenetic alterations in adult ovarian genes. Endocrinology. 2009;150:4681–4691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shi L, Wu J. Epigenetic regulation in mammalian preimplantation embryo development. Reprod Biol Endocrinol. 2009;7:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Robledo CA, Yeung E, Mendola P, Sundaram R, Maisog J, Sweeney AM, Barr DB, Louis GM. Preconception maternal and paternal exposure to persistent organic pollutants and birth size: the LIFE study. Environ Health Perspect. 2015;123:88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rappazzo KM, Daniels JL, Messer LC, Poole C, Lobdell DT. Exposure to fine particulate matter during pregnancy and risk of preterm birth among women in New Jersey, Ohio, and Pennsylvania, 2000–2005. Environ Health Perspect. 2014;122:992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dadvand P, Parker J, Bell ML, Bonzini M, Brauer M, Darrow LA, Gehring U, Glinianaia SV, Gouveia N, Ha EH, Leem JH, van den Hooven EH, Jalaludin B, Jesdale BM, Lepeule J, Morello‐Frosch R, Morgan GG, Pesatori AC, Pierik FH, Pless‐Mulloli T, Rich DQ, Sathyanarayana S, Seo J, Slama R, Strickland M, Tamburic L, Wartenberg D, Nieuwenhuijsen MJ, Woodruff TJ. Maternal exposure to particulate air pollution and term birth weight: a multi‐country evaluation of effect and heterogeneity. Environ Health Perspect. 2013;121:267–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cheung MC, Spalding PB, Gutierrez JC, Balkan W, Namias N, Koniaris LG, Zimmers TA. Body surface area prediction in normal, hypermuscular, and obese mice. J Surg Res. 2009;153:326–331. [DOI] [PubMed] [Google Scholar]
- 15. Wold LE, Ying Z, Hutchinson KR, Velten M, Gorr MW, Velten C, Youtz DJ, Wang A, Lucchesi PA, Sun Q, Rajagopalan S. Cardiovascular remodeling in response to long‐term exposure to fine particulate matter air pollution. Circ Heart Fail. 2012;5:452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Watkins AJ, Lucas ES, Wilkins A, Cagampang FR, Fleming TP. Maternal periconceptional and gestational low protein diet affects mouse offspring growth, cardiovascular and adipose phenotype at 1 year of age. PLoS One. 2011;6:e28745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gardebjer EM, Cuffe JS, Pantaleon M, Wlodek ME, Moritz KM. Periconceptional alcohol consumption causes fetal growth restriction and increases glycogen accumulation in the late gestation rat placenta. Placenta. 2014;35:50–57. [DOI] [PubMed] [Google Scholar]
- 18. Witt WP, Mandell KC, Wisk LE, Cheng ER, Chatterjee D, Wakeel F, Park H, Zarak D. Infant birthweight in the US: the role of preconception stressful life events and substance use. Arch Womens Ment Health. 2016;19:529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bell ML, Belanger K, Ebisu K, Gent JF, Lee HJ, Koutrakis P, Leaderer BP. Prenatal exposure to fine particulate matter and birth weight: variations by particulate constituents and sources. Epidemiology. 2010;21:884–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kjoller‐Hansen L, Steffensen R, Grande P. Beneficial effects of ramipril on left ventricular end‐diastolic and end‐systolic volume indexes after uncomplicated invasive revascularization are associated with a reduction in cardiac events in patients with moderately impaired left ventricular function and no clinical heart failure. J Am Coll Cardiol. 2001;37:1214–1220. [DOI] [PubMed] [Google Scholar]
- 21. Migrino RQ, Young JB, Ellis SG, White HD, Lundergan CF, Miller DP, Granger CB, Ross AM, Califf RM, Topol EJ. End‐systolic volume index at 90 to 180 minutes into reperfusion therapy for acute myocardial infarction is a strong predictor of early and late mortality. The Global Utilization of Streptokinase and t‐PA for Occluded Coronary Arteries (GUSTO)‐I Angiographic Investigators. Circulation. 1997;96:116–121. [DOI] [PubMed] [Google Scholar]
- 22. Velten M, Gorr MW, Youtz DJ, Velten C, Rogers LK, Wold LE. Adverse perinatal environment contributes to altered cardiac development and function. Am J Physiol Heart Circ Physiol. 2014;306:H1334–H1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yu X, Kennedy RH, Liu SJ. JAK2/STAT3, not ERK1/2, mediates interleukin‐6‐induced activation of inducible nitric‐oxide synthase and decrease in contractility of adult ventricular myocytes. J Biol Chem. 2003;278:16304–16309. [DOI] [PubMed] [Google Scholar]
- 24. Feng Q, Lu X, Jones DL, Shen J, Arnold JM. Increased inducible nitric oxide synthase expression contributes to myocardial dysfunction and higher mortality after myocardial infarction in mice. Circulation. 2001;104:700–704. [DOI] [PubMed] [Google Scholar]
- 25. Umar S, van der Laarse A. Nitric oxide and nitric oxide synthase isoforms in the normal, hypertrophic, and failing heart. Mol Cell Biochem. 2010;333:191–201. [DOI] [PubMed] [Google Scholar]
- 26. Zimmet JM, Hare JM. Nitroso‐redox interactions in the cardiovascular system. Circulation. 2006;114:1531–1544. [DOI] [PubMed] [Google Scholar]
- 27. Xu X, Liu C, Xu Z, Tzan K, Zhong M, Wang A, Lippmann M, Chen LC, Rajagopalan S, Sun Q. Long‐term exposure to ambient fine particulate pollution induces insulin resistance and mitochondrial alteration in adipose tissue. Toxicol Sci. 2011;124:88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Glorieux C, Zamocky M, Sandoval JM, Verrax J, Calderon PB. Regulation of catalase expression in healthy and cancerous cells. Free Radic Biol Med. 2015;87:84–97. [DOI] [PubMed] [Google Scholar]
- 29. Yano S, Yano N. Regulation of catalase enzyme activity by cell signaling molecules. Mol Cell Biochem. 2002;240:119–130. [DOI] [PubMed] [Google Scholar]
- 30. Slezak J, Tribulova N, Pristacova J, Uhrik B, Thomas T, Khaper N, Kaul N, Singal PK. Hydrogen peroxide changes in ischemic and reperfused heart. Cytochemistry and biochemical and X‐ray microanalysis. Am J Pathol. 1995;147:772–781. [PMC free article] [PubMed] [Google Scholar]
- 31. Peretz A, Peck EC, Bammler TK, Beyer RP, Sullivan JH, Trenga CA, Srinouanprachnah S, Farin FM, Kaufman JD. Diesel exhaust inhalation and assessment of peripheral blood mononuclear cell gene transcription effects: an exploratory study of healthy human volunteers. Inhal Toxicol. 2007;19:1107–1119. [DOI] [PubMed] [Google Scholar]
- 32. Tsai DH, Amyai N, Marques‐Vidal P, Wang JL, Riediker M, Mooser V, Paccaud F, Waeber G, Vollenweider P, Bochud M. Effects of particulate matter on inflammatory markers in the general adult population. Part Fibre Toxicol. 2012;9:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wollert KC, Taga T, Saito M, Narazaki M, Kishimoto T, Glembotski CC, Vernallis AB, Heath JK, Pennica D, Wood WI, Chien KR. Cardiotrophin‐1 activates a distinct form of cardiac muscle cell hypertrophy. Assembly of sarcomeric units in series VIA gp130/leukemia inhibitory factor receptor‐dependent pathways. J Biol Chem. 1996;271:9535–9545. [DOI] [PubMed] [Google Scholar]
- 34. Terrell AM, Crisostomo PR, Wairiuko GM, Wang M, Morrell ED, Meldrum DR. Jak/STAT/SOCS signaling circuits and associated cytokine‐mediated inflammation and hypertrophy in the heart. Shock. 2006;26:226–234. [DOI] [PubMed] [Google Scholar]
- 35. Prabhu SD. Cytokine‐induced modulation of cardiac function. Circ Res. 2004;95:1140–1153. [DOI] [PubMed] [Google Scholar]
- 36. Yang YY, Liu H, Nam SW, Kunos G, Lee SS. Mechanisms of TNFalpha‐induced cardiac dysfunction in cholestatic bile duct‐ligated mice: interaction between TNFalpha and endocannabinoids. J Hepatol. 2010;53:298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, Kassiri Z, Kirshenbaum LA, Arnold M, Khokha R, Liu PP. Tumor necrosis factor‐alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. 2007;115:1398–1407. [DOI] [PubMed] [Google Scholar]
- 38. Moe KT, Khairunnisa K, Yin NO, Chin‐Dusting J, Wong P, Wong MC. Tumor necrosis factor‐alpha‐induced nuclear factor‐kappaB activation in human cardiomyocytes is mediated by NADPH oxidase. J Physiol Biochem. 2014;70:769–779. [DOI] [PubMed] [Google Scholar]
- 39. Liu H, Lee SS. Nuclear factor‐kappaB inhibition improves myocardial contractility in rats with cirrhotic cardiomyopathy. Liver Int. 2008;28:640–648. [DOI] [PubMed] [Google Scholar]
- 40. Gupta S, Young D, Sen S. Inhibition of NF‐kappaB induces regression of cardiac hypertrophy, independent of blood pressure control, in spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2005;289:H20–H29. [DOI] [PubMed] [Google Scholar]
- 41. Kawamura N, Kubota T, Kawano S, Monden Y, Feldman AM, Tsutsui H, Takeshita A, Sunagawa K. Blockade of NF‐kappaB improves cardiac function and survival without affecting inflammation in TNF‐alpha‐induced cardiomyopathy. Cardiovasc Res. 2005;66:520–529. [DOI] [PubMed] [Google Scholar]
- 42. Vogel CF, Sciullo E, Wong P, Kuzmicky P, Kado N, Matsumura F. Induction of proinflammatory cytokines and C‐reactive protein in human macrophage cell line U937 exposed to air pollution particulates. Environ Health Perspect. 2005;113:1536–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Niwa Y, Hiura Y, Sawamura H, Iwai N. Inhalation exposure to carbon black induces inflammatory response in rats. Circ J. 2008;72:144–149. [DOI] [PubMed] [Google Scholar]
- 44. Munkhaugen J, Otterstad JE, Dammen T, Gjertsen E, Moum T, Husebye E, Gullestad L. The prevalence and predictors of elevated C‐reactive protein after a coronary heart disease event. Eur J Prev Cardiol. 2018;25:923–931. DOI: 10.1177/2047487318768940. [DOI] [PubMed] [Google Scholar]
- 45. Pradhan AD, Rifai N, Ridker PM. Soluble intercellular adhesion molecule‐1, soluble vascular adhesion molecule‐1, and the development of symptomatic peripheral arterial disease in men. Circulation. 2002;106:820–825. [DOI] [PubMed] [Google Scholar]
- 46. Rana JS, Arsenault BJ, Despres JP, Cote M, Talmud PJ, Ninio E, Wouter Jukema J, Wareham NJ, Kastelein JJ, Khaw KT, Boekholdt SM. Inflammatory biomarkers, physical activity, waist circumference, and risk of future coronary heart disease in healthy men and women. Eur Heart J. 2011;32:336–344. [DOI] [PubMed] [Google Scholar]
- 47. Dawson J, Miltz W, Mir AK, Wiessner C. Targeting monocyte chemoattractant protein‐1 signalling in disease. Expert Opin Ther Targets. 2003;7:35–48. [DOI] [PubMed] [Google Scholar]
- 48. Sheikine Y, Hansson GK. Chemokines and atherosclerosis. Ann Med. 2004;36:98–118. [DOI] [PubMed] [Google Scholar]
- 49. Becker LC. Yin and yang of MCP‐1. Circ Res. 2005;96:812–814. [DOI] [PubMed] [Google Scholar]
- 50. Pope CA III, Bhatnagar A, McCracken JP, Abplanalp W, Conklin DJ, O'Toole T. Exposure to fine particulate air pollution is associated with endothelial injury and systemic inflammation. Circ Res. 2016;119:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kampfrath T, Maiseyeu A, Ying Z, Shah Z, Deiuliis JA, Xu X, Kherada N, Brook RD, Reddy KM, Padture NP, Parthasarathy S, Chen LC, Moffatt‐Bruce S, Sun Q, Morawietz H, Rajagopalan S. Chronic fine particulate matter exposure induces systemic vascular dysfunction via NADPH oxidase and TLR4 pathways. Circ Res. 2011;108:716–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gorr MW, Youtz DJ, Eichenseer CM, Smith KE, Nelin TD, Cormet‐Boyaka E, Wold LE. In vitro particulate matter exposure causes direct and lung‐mediated indirect effects on cardiomyocyte function. Am J Physiol Heart Circ Physiol. 2015;309:H53–H62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mack M. Inflammation and fibrosis. Matrix Biol. 2018;68–69:106–121. [DOI] [PubMed] [Google Scholar]
- 54. Day J, Savani S, Krempley BD, Nguyen M, Kitlinska JB. Influence of paternal preconception exposures on their offspring: through epigenetics to phenotype. Am J Stem Cells. 2016;5:11–18. [PMC free article] [PubMed] [Google Scholar]
- 55. Baccarelli A, Rienstra M, Benjamin EJ. Cardiovascular epigenetics: basic concepts and results from animal and human studies. Circ Cardiovasc Genet. 2010;3:567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Turunen MP, Aavik E, Yla‐Herttuala S. Epigenetics and atherosclerosis. Biochim Biophys Acta. 2009;1790:886–891. [DOI] [PubMed] [Google Scholar]
- 57. Cantone L, Iodice S, Tarantini L, Albetti B, Restelli I, Vigna L, Bonzini M, Pesatori AC, Bollati V. Particulate matter exposure is associated with inflammatory gene methylation in obese subjects. Environ Res. 2017;152:478–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bosch‐Presegue L, Vaquero A. Sirtuin‐dependent epigenetic regulation in the maintenance of genome integrity. FEBS J. 2015;282:1745–1767. [DOI] [PubMed] [Google Scholar]
- 59. Peng L, Yuan Z, Ling H, Fukasawa K, Robertson K, Olashaw N, Koomen J, Chen J, Lane WS, Seto E. SIRT1 deacetylates the DNA methyltransferase 1 (DNMT1) protein and alters its activities. Mol Cell Biol. 2011;31:4720–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Heo J, Lim J, Lee S, Jeong J, Kang H, Kim Y, Kang JW, Yu HY, Jeong EM, Kim K, Kucia M, Waigel SJ, Zacharias W, Chen Y, Kim IG, Ratajczak MZ, Shin DM. Sirt1 regulates DNA methylation and differentiation potential of embryonic stem cells by antagonizing Dnmt3 l. Cell Rep. 2017;18:1930–1945. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Preconception exposure model.
Figure S2. Body weight parameters from mice born to preconception FA or PM2.5 exposed parents.
Figure S3. Quantitative PCR analysis showing expression of (A) sarco/endoplasmic reticulum Ca2+‐ATPase (SERCA‐2A), (B) Phospholamban (PLN) and (C) Na+/Ca2+ exchanger (NCX).
Figure S4. Quantitative PCR analysis showing expression of (A) DNA methyltransferases‐1 (Dnmt1), (B) DNA methyltransferases‐3a (Dnmt3a), (C) DNA methyltransferases‐3b (Dnmt3b).