

Abstract
Background
In randomized trials (SHARP [Study of Heart and Renal Protection], IMPROVE‐IT [Improved Reduction of Outcomes: Vytorin Efficacy International Trial]), combination of statin and ezetimibe resulted in additional reduction of cardiovascular events. The reduction was greater in patients with type 2 diabetes mellitus (T2DM), where elevated remnant cholesterol and high cardiovascular disease risk is characteristic. To evaluate possible causes behind these results, 40 patients eligible for cholecystectomy, randomized to simvastatin, ezetimibe, combined treatment (simvastatin+ezetimibe), or placebo treatment during 4 weeks before surgery, were studied.
Methods and Results
Fasting blood samples were taken before treatment start and at the end (just before surgery). Bile samples and liver biopsies were collected during surgery. Hepatic gene expression levels were assessed with qPCR. Lipoprotein, apolipoprotein levels, and content of cholesterol, cholesteryl ester, and triglycerides were measured after lipoprotein fractionation. Lipoprotein subclasses were analyzed by nuclear magnetic resonance. Apolipoprotein affinity for human arterial proteoglycans (PG) was measured. Biomarkers of cholesterol biosynthesis and intestinal absorption and bile lipid composition were analyzed using mass spectrometry. Combined treatment caused a statistically significant decrease in plasma remnant particles and apolipoprotein B (ApoB)/lipoprotein content of cholesterol, cholesteryl esters, and triglycerides. All treatments reduced ApoB‐lipoprotein PG binding. Simvastatin and combined treatment modified the composition of lipoproteins. Changes in biomarkers of cholesterol synthesis and absorption and bile acid synthesis were as expected. No adverse events were found.
Conclusions
Combined treatment caused atheroprotective changes on ApoB‐lipoproteins, remnant particles, bile components, and in ApoB‐lipoprotein affinity for arterial PG. These effects might explain the decrease of cardiovascular events seen in the SHARP and IMPROVE‐IT trials.
Clinical Trial Registration
URL: www.clinicaltrialsregister.eu. Unique identifier: 2006‐004839‐30).
Keywords: ApoB‐lipoproteins, bile, proteoglycan binding, statin therapy, T2DM
Subject Categories: Clinical Studies, Lipids and Cholesterol, Mechanisms, Metabolism
Clinical Perspective
What Is New?
This study demonstrated that combined treatment of simvastatin and ezetimibe caused a significant decrease of plasma cholesterol, cholesteryl esters, and triglycerides in remnant particles without changing the lipid composition of bile.
Simvastatin, ezetimibe, and combined treatment reduced the binding of apolipoprotein B (ApoB)‐containing lipoprotein to human arterial proteoglycans.
The expected increase in biliary cholesterol following treatment with ezetimibe alone did not occur in patients with gallstone disease, yet we found evidence that ezetimibe was able to block hepatic Niemann‐Pick C1‐like 1 (NPC1L1) in humans.
Addition of ezetimibe to simvastatin did not affect the hepatic mRNA levels of the sterol regulatory element binding transcription factor 2 (SREBF2) system.
What Are the Clinical Implications?
Combined therapy of simvastatin and ezetimibe seems to be an optimal treatment for lipid disorders characterized by elevated remnant cholesterol (ie, type 2 diabetes mellitus) to reduce cardiovascular events.
Addition of ezetimibe to simvastatin does not increase biliary cholesterol, and this should not lead to an increased risk for cholesterol gallstone disease.
Introduction
Statins are the main treatment for primary and secondary prevention of cardiovascular disease (CVD). Efforts to reach guideline goal levels for low‐density lipoprotein cholesterol (LDL‐C), when statins are not sufficient or poorly tolerated, have led to studies where lipid‐lowering drugs have been used in combination with statins.1 One such drug is ezetimibe, an inhibitor of cholesterol absorption that blocks the intestinal Niemann‐Pick C1‐like 1 (NPC1L1) protein transporter.2 Consistent with previous studies,3 the IMPROVE‐IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial) trial recently reported that adding ezetimibe to simvastatin therapy after acute coronary syndrome resulted in additional reduction of plasma LDL‐C levels by ≈22%.4 This resulted in a 2% absolute risk reduction of death from CVD, major coronary events (nonfatal myocardial infarction, unstable angina, or coronary revascularization), or nonfatal stroke.4 This benefit was more pronounced in the subgroup of patients with type 2 diabetes mellitus (T2DM), with 5.5% absolute risk reduction.5 No additional adverse muscle, gallbladder, or hepatic effects were reported in the IMPROVE‐IT trial. Similar results were observed in the SHARP (Study of Heart and Renal Protection), where the safety and efficacy of plasma LDL‐C reduction in patients with chronic kidney disease were assessed. Treatment with simvastatin and ezetimibe in combination resulted in an average plasma LDL‐C lowering of 0.85 mmol/L and a 17% reduction in major atherosclerotic events (nonfatal myocardial infarction, coronary death, nonhemorrhagic stroke, or arterial revascularization), without any excess risk of persistent elevation of hepatic transaminases, hepatitis, gallstone disease, or pancreatitis compared with placebo.6 In this trial, 22% of the patients, equally distributed in the treatment and placebo group, had T2DM.7
Remnant cholesterol in plasma consists of free cholesterol and cholesteryl esters (CEs) carried in triglyceride‐rich lipoproteins, chylomicron remnants, very low‐density lipoproteins (VLDLs) and intermediate‐density lipoproteins (IDLs). All these particles contain apoliprotein B (ApoB) as their main apolipoprotein. Remnant cholesterol can be estimated by subtracting LDL‐C and high‐density lipoprotein (HDL) cholesterol from total plasma cholesterol.8 The European Atherosclerosis Society recommends that nonfasting lipid profiling, including calculation of remnant cholesterol, can be used routinely in clinics given that both fasting and nonfasting conditions have been shown to be good predictors for CVD and for the evaluation of treatment response. Nonfasting remnant cholesterol ≥0.9 mmol/L and fasting remnant cholesterol ≥0.8 mmol/L can be considered as abnormal.9
Both elevated LDL‐C and remnant cholesterol increase the risk for CVD. In contrast to LDL‐C, elevated remnant cholesterol is also associated with a systemic low‐grade inflammation.8 The residual CVD risk observed after lowering of LDL‐C may partly be related to remnant particles that can be retained in the arterial intima by the interaction of the apoB‐100 with extracellular intima proteoglycans.10, 11 Therefore, the ability of a lipid‐lowering therapeutic regimen to reduce circulating levels of all ApoB particles that can deposit cholesterol and CEs in the arteries (ie, remnants, IDL, and LDL) could be just as important as its ability to lower LDL‐C.10, 11
Three enzymes are responsible for cholesterol esterification. Lecithin‐cholesterol acyltransferase (LCAT) esterifies cholesterol only in the plasma, whereas acyl‐coenzyme A: cholesterol acyltransferase (ACAT) 1 and 2 both acts intracellularly.12 Intestinal and hepatic ACAT2 synthetizes the CEs used for assembly and secretion of nascent chylomicrons and VLDL, respectively.12, 13 CEs synthesized by LCAT is transferred to VLDL.14 Atorvastatin has been shown to reduce the expression and activity of hepatic ACAT2, thereby lowering CEs in VLDL.15 Ezetimibe inhibits intestinal NPC1L1 and reduces the amount of cholesterol that can be esterified by intestinal ACAT2 and secreted with chylomicrons.16 Large and small dense LDLs (sdLDLs) are remnants of secreted VLDL, and their CEs are produced by hepatic ACAT2 and plasma LCAT.12, 17 Concentration of sdLDL can be used to predict the risk of developing CVD in patients with T2DM.18, 19 In these patients, the combination of rosuvastatin and ezetimibe reduced sdLDL‐C more efficiently than doubling the rosuvastatin dose.20 sdLDL particles are more prone to oxidation,21 glycation22 and have a higher binding affinity to arterial proteoglycans (PG).23 This last property is associated with cholesterol deposition in the intima and CVD and can be reduced by lipid‐lowering treatments.24, 25, 26 Plasma ApoB (ApoB‐100)‐containing lipoprotein binding to human intima PG causes lipoprotein deposition in the arterial wall. This is one of the initial steps in intimal deposition of cholesterol and consequent atherogenesis and is a central concept in the current “response to retention hypothesis” of atherosclerosis.11, 23, 24, 25, 26, 27, 28, 29, 30, 31
In contrast to mice, hepatic NPC1L1 is highly expressed in humans.32 The role and effects of ezetimibe on human hepatic NPC1L1 remain uncertain given that most of the available data are based on either in vitro models33 or mouse models overexpressing human NPC1L1.34, 35 The results from recent studies performed so far in humans are not conclusive because of very small sample size in addition to limitations of duodenal bile sampling methods.36, 37
In the present study, we evaluated whether ezetimibe in association with simvastatin has the ability to decrease cholesterol and CEs in remnant particles and LDL, and whether this translates into a reduced PG binding. We assessed the effects of ezetimibe on biliary lipid composition to gain more insight into the function of hepatic NPC1L1. In addition, the effects on mRNA levels of genes involved in the hepatic cholesterol metabolism were analyzed.
Methods
The data, analytical methods, and study materials will be made available on request to other researchers for the purposes of reproducing the results or replicating the procedure. All data are electronically stored at the division of Clinical Chemistry, Department of Laboratory Medicine, Karolinska Institutet and can be available to other researchers by contacting the coordinator (Osman Ahmed) or the principle investigators (Paolo Parini or Mats Eriksson) of the Stockholm study.
Patients, Treatments, and Specimen Collection
Forty patients (14 males, 13 fertile, and 13 postmenopausal females) with uncomplicated cholesterol gallstone disease, eligible for elective cholecystectomy at the Department of Surgery, Danderyd Hospital, Danderyd, Sweden, were enrolled in this study. The study was approved by the Stockholm Region Ethical Board (2006/1204‐31/1), the Swedish Medical Agency, and registered at the EU clinical trial register (EudraCT number: 2006‐004839‐30). All patients gave their written informed consent to participate in this study.
Inclusion Criteria
Patients with gallstone disease, eligible to cholecystectomy, were aged between 25 and 80 years.
Exclusion Criteria
Patients with obesity (body mass index, >30), known familial phytosterolemia, active liver disease, or hepatic dysfunction (aspartate aminotransferase [S‐AST] or alanine aminotransferase [S‐ALT] ≥3 times the upper limit of reference), elevated creatinine phosphokinase (S‐CPK), partial ileal bypass, metabolic diseases (ie, T2DM), inflammatory bowel disease, or other serious disease were not enrolled. Patients with known hypersensitivity to 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitors, ezetimibe, or treatment with drugs known to be associated with rhabdomyolysis in combination with 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitors were excluded.
Study Procedure
After a first screening‐visit, patients qualifying for inclusion were randomized into 4 different treatment groups: simvastatin 80 mg daily (S), ezetimibe 10 mg daily (E), simvastatin 80 mg and ezetimibe 10 mg daily (S+E), or placebo (P). Treatment started 4 weeks (25–30 days) before the surgery and ended at the end‐of study visit, preceding the surgical intervention. Investigators were aware of the treatment received, because this was a randomized single‐blind study.
Fasting blood samples were collected at the first screening visit and at the end‐of study visit. Liver biopsies and bile were collected after an overnight fast during the surgical intervention. All samples were rapidly frozen and stored at −70°C until analysis.
Statistical Analysis
Sample‐size determination
The primary end points of the study were the relative reduction of LDL‐C and the relative changes in hepatic gene expression. No data on the variation of gene expression in humans in a Swedish population were available. Therefore, we used LDL‐C as the variable on which sample size and power for reaching significance in a 2‐sided test at the 5% level (F test; 1‐way ANOVA; fully randomized design) were measured. Mean LDL‐C concentration in our Stockholm reference group was estimated to be 3.68 mmol/L, with an SD equal to 1.21. Considering an LDL‐C lowering effect of 15%, 30%, and 50% by treatment with ezetimibe, simvastatin, and ezetimibe plus simvastatin, respectively (effect‐size “f”: 0.6355), the calculated power was 0.8 if there were 10 patients in each treatment group for the study.
All data are presented as mean±SEM, unless otherwise stated. When appropriate, data were log transformed before parametric analysis when homoscedasticity was not present (unequal variance of different groups). Significance was tested by multiway ANOVA. Post hoc comparisons were done according to the Fisher's least significant difference test. Outlier analysis was performed for all variables and rejection was done if outliers were present.
All statistical analyses were performed using Statistica (version 12.0; StatSoft, Tulsa, OK; Data S1).
Results
Effects on Total Body Cholesterol Synthesis and Intestinal Cholesterol Absorption
In this study, patients were randomized to treatment with placebo, simvastatin 80 mg/day, ezetimibe 10 mg/day, or combined simvastatin 80 mg/day and ezetimibe 10 mg/day (Table 1). Cholesterol synthesis (assessed by the total plasma lathosterol to cholesterol ratio) was reduced by simvastatin (−56%; P<0.001) and combined treatment (−29%; P<0.001), but increased by ezetimibe as a monotherapy (40%; P<0.01; Figure 1). As expected, simvastatin increased intestinal cholesterol absorption, assessed by the plasma campesterol to cholesterol ratio (21%; P<0.05), whereas ezetimibe reduced it as a monotherapy (−52%; P<0.001) and in combination with simvastatin (−41%; P<0.01; Figure 1).
Table 1.
Baseline Clinical Characteristics and Plasma Lipid Levels
| Placebo (n=10) | Simvastatin (n=10) | Ezetimibe (n=10) | Simvastatin+Ezetimibe (n=10) | P Value | |
|---|---|---|---|---|---|
| Sex (M/FF/PF) | 4/3/3 | 3/4/3 | 4/3/3 | 3/3/4 | NS |
| Age, y | 57.7±3.40 | 57.4±6.20 | 54.1±5.70 | 59.4±4.60 | NS |
| BMI | 25.8±0.70 | 26.4±0.80 | 25.5±0.80 | 25.2±0.90 | NS |
| Plasma total cholesterol, mmol/L | 5.80±0.40 | 5.80±0.30 | 5.50±0.40 | 5.50±0.30 | NS |
| Plasma triglycerides, mmol/L | 1.10±0.20 | 1.70±0.60 | 1.10±0.10 | 1.00±0.60 | NS |
| Remnant cholesterol (non‐LDL, non‐HDL), mmol/L | 0.90±0.20 | 1.10±0.30 | 0.80±0.10 | 1.00±0.10 | NS |
| LDL cholesterol, mmol/L | 3.10±0.30 | 3.20±0.20 | 3.20±0.20 | 3.00±0.20 | NS |
| HDL cholesterol, mmol/L | 1.80±0.20 | 1.50±0.10 | 1.60±0.10 | 1.50±0.20 | NS |
| Apolipoprotein B, g/L | 0.87±0.70 | 0.98±0.12 | 0.78±0.04 | 0.84±0.05 | NS |
| Apolipoprotein A1, g/L | 1.60±0.05 | 1.57±0.03 | 1.41±0.04 | 1.41±0.03 | <0.05 |
| Apolipoprotein E, g/L | 0.05±0.00 | 0.05±0.00 | 0.05±0.00 | 0.04±0.00 | <0.05 |
| Apolipoprotein CIII, g/L | 0.95±0.04 | 0.73±0.15 | 0.69±0.12 | 0.95±0.09 | NS |
| Lathosterol/cholesterol, mg/mmol cholesterol | 430±30.00 | 433±62.00 | 559±43.00 | 473±24.00 | <0.05 |
| Campesterol/cholesterol, mg/mmol cholesterol | 568±74.00 | 697±108.0 | 511±58.00 | 489±39.00 | NS |
Data show mean±SEM. Lipoprotein profiling was determined after separation by size‐exclusion chromatography. Multiway ANOVA followed by LSD test. BMI indicates body mass index; FF, fertile female; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; LSD, least significant difference; M, male; NS, not significant; PF, postmenopausal female.
Figure 1.
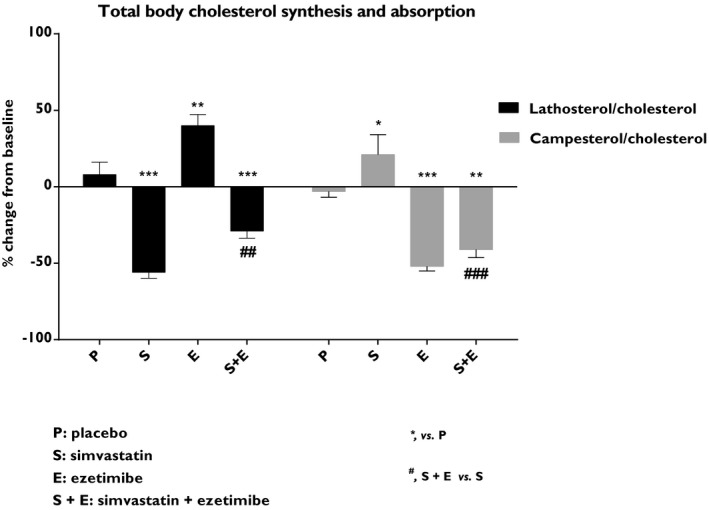
Effects on total body cholesterol synthesis and intestinal cholesterol absorption. The plasma lathosterol/cholesterol ratio is an indirect measurement of total body cholesterol synthesis whereas the plasma campesterol/cholesterol ratio reflects intestinal cholesterol absorption. Data show the % change from baseline and are expressed as mean±SEM. Baseline values are given in Table 1. Multiway ANOVA followed by LSD test. *vs placebo, P<0.05; **vs placebo, P<0.01; ***vs placebo, P<0.001; #simvastatin+ezetimibe vs simvastatin, P<0.05; ##simvastatin+ezetimibe vs simvastatin, P<0.01; and ###simvastatin+ezetimibe vs simvastatin, P<0.001 (n=8–10/treatment group). LSD indicates least significant difference.
Effects on Lipoproteins and Lipoprotein Subclasses
In order to determine in more detail the lipoprotein composition, plasma samples were subjected to analysis by nuclear magnetic resonance. Remnant cholesterol and LDL‐C were reduced by both simvastatin (−51% and −52%; P<0.001) and ezetimibe (−18%; P<0.001 and −14%; P<0.05). Combined treatment with simvastatin and ezetimibe led to an even greater reduction of both remnant cholesterol (−65%; P<0.001) and LDL‐C (−64%; P<0.001; Figure 2A). Simvastatin, ezetimibe, and the combined treatment also led to a reduction of remnant CEs (−53%, −20%, and −68%, respectively; P<0.001; Figure 2B). Ezetimibe added to simvastatin treatment showed an additional reduction (−15%; P<0.01) of remnant CEs compared with simvastatin alone (Figure 2B).
Figure 2.
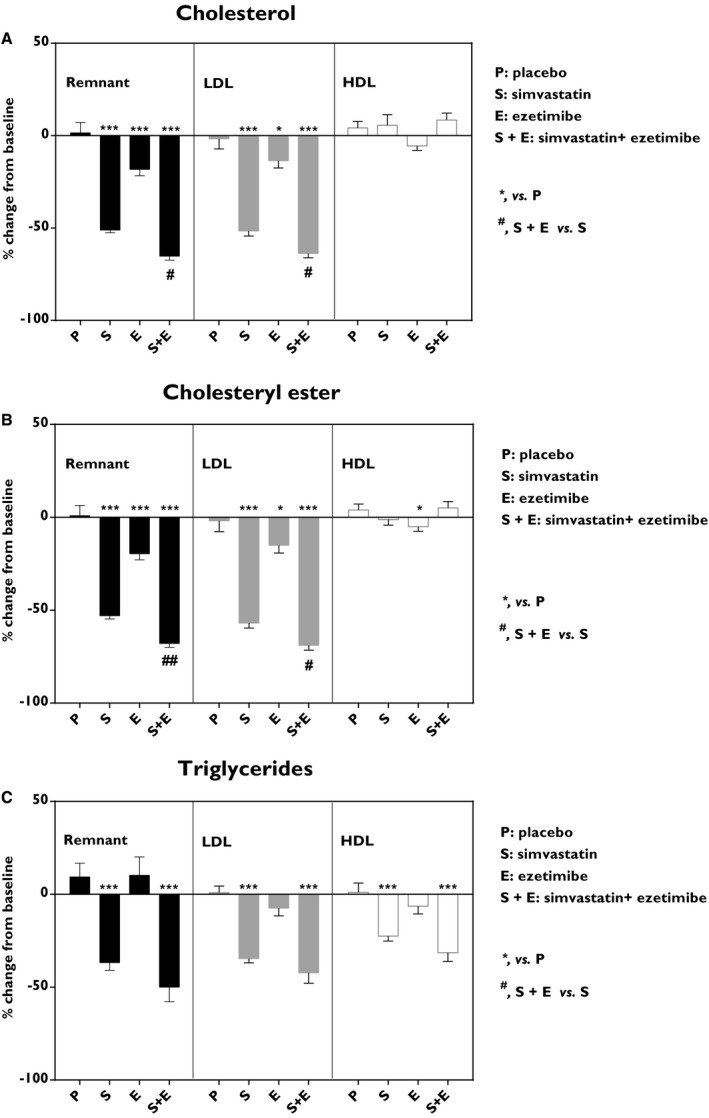
Effects on plasma lipoproteins. A, Cholesterol, (B) cholysteryl ester, and (C) triglycerides. Remnant particles, that is, non‐high‐density lipoproteins (non‐HDL) and non‐low‐density lipoproteins (non‐LDL; black), LDL particles (gray) and HDL particles (white). Data show the % change from baseline and are expressed as mean±SEM. Fasted plasma lipoproteins were subjected to analysis by nuclear magnetic resonance. Lipid contribution in lipoprotein fractions are given in Table S1. Multiway ANOVA followed by LSD test. *vs placebo, P<0.05; ***vs placebo, P<0.001; #simvastatin+ezetimibe vs simvastatin, P<0.05; and ##simvastatin+ezetimibe vs simvastatin, P<0.01 (n=8–10/treatment group). LSD indicates least significant difference.
Simvastatin and combined treatment with ezetimibe showed reduction (−37%; P<0.01 and −50%; P<0.001) of remnant triglyceride (Figure 2C). Similarly, simvastatin and combined treatment with ezetimibe reduced LDL‐triglyceride (−35% and −42%, respectively; P<0.001) and HDL‐triglyceride (−23%; P<0.01 and −32%; P<0.001; Figure 2C). Ezetimibe in monotherapy had no significant effect on triglyceride levels. The lipid contribution in lipoprotein fractions is given in Table S1.
Lipoprotein fractions were divided into subclasses by nuclear magnetic resonance. The contribution of the different subclasses to their respective fraction (ie, remnant, LDL, and HDL) baseline is presented in Table S2. Detailed analysis of the 7 different non‐LDL, non‐HDL lipoprotein subclasses (diameter size range down to 28.6 nm) showed that simvastatin alone or in combination with ezetimibe reduced particle number (P<0.001), triglyceride (P<0.001), and CE (P<0.001) in all subclasses (Figure 3). Ezetimibe treatment alone reduced the particle number in 3 subclasses (XXL‐VLDL/chylomicron, XS‐VLDL, and IDL; P<0.05), triglyceride in 1 subclass (XXL‐VLDL/chylomicron; P<0.05), and CEs in all subclasses except XL‐VLDL (P<0.05; Figure 3).
Figure 3.
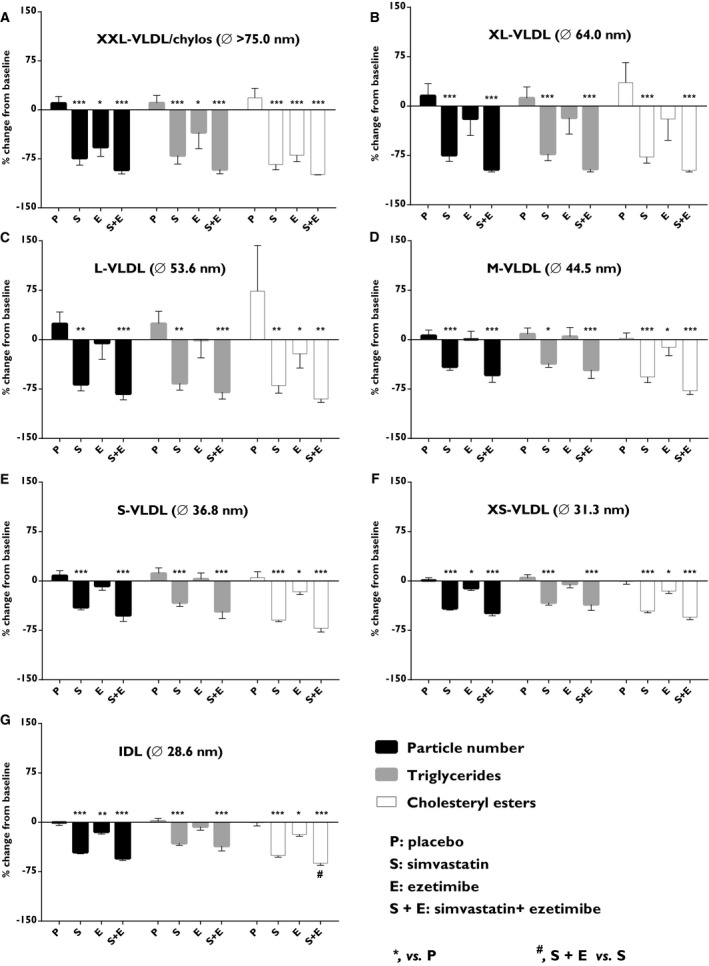
Effects on subclasses of non‐LDL, non‐HDL particles. Particle numbers (black), cholesteryl esters (white), and triglycerides (gray) were quantified in (A) extra‐large very‐low‐density lipoproteins (XXL‐VLDL)/chylomicrons, (B) extra‐large‐VLDL (XL‐VLDL), (C) large‐VLDL (L‐VLDL), (D) medium‐VLDL (M‐VLDL), (E) small‐VLDL (S‐VLDL), (F) extra‐small‐VLDL (XS‐VLDL), and (G) intermediate‐density lipoprotein (IDL). Data show the % change from baseline and are expressed as mean±SEM. Fasted plasma lipoproteins were subjected to analysis by nuclear magnetic resonance. Multiway ANOVA followed by LSD test. *vs placebo, P<0.05; **vs placebo, P<0.01; ***vs placebo, P<0.001; and #simvastatin+ezetimibe vs simvastatin, P<0.05 (n=8–10/treatment group). LSD indicates least significant difference.
Analyses of LDL subclasses following treatment with simvastatin alone or in combination with ezetimibe showed reduced particle number (P<0.001), triglyceride (P<0.001), and CEs (P<0.001) in all 3 LDL subclasses (diameter size range, 18.7–25.5 nm; Figure 4A through 4C). Ezetimibe as monotherapy reduced particle number (P<0.05), triglyceride (P<0.01) in S‐LDL (diameter size, 18.7 nm), and CEs (P<0.05) in L‐LDL particles (diameter size, 25.5 nm; Figure 4A and 4C).
Figure 4.
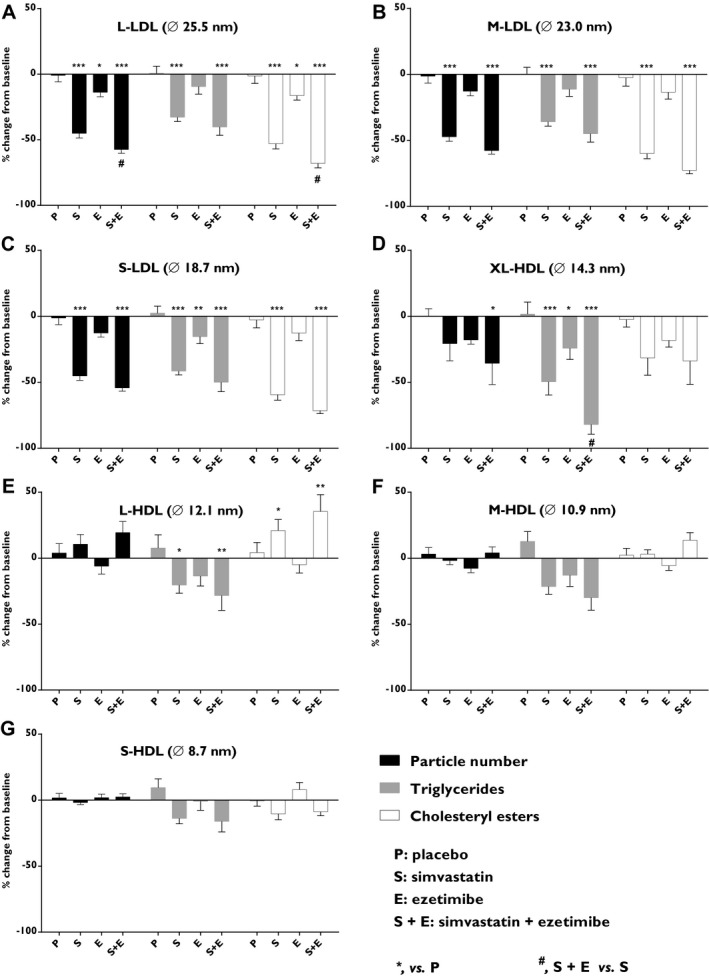
Effects on subclasses of LDL and HDL particles. Particle numbers (black), cholesteryl esters (white) and triglycerides (gray) were quantified in (A) large low‐density lipoproteins (L‐LDL), (B) medium‐LDL (M‐LDL), (C) small‐LDL (S‐LDL), (D) extra‐large high‐density lipoproteins (XL‐HDL), (E) large‐HDL (L‐HDL), (F) medium‐HDL (M‐HDL), and (G) small‐HDL (S‐HDL). Data show the % change from baseline and are expressed as mean±SEM. Fasted plasma lipoproteins were subjected to analysis by nuclear magnetic resonance. Multiway ANOVA followed by LSD test. *vs placebo, P<0.05; **vs placebo, P<0.01; ***vs placebo, P<0.001; and #simvastatin+ezetimibe vs simvastatin, P<0.05 (n=8–10/treatment group). LSD indicates least significant difference.
Evaluation of the effects on the 4 different HDL subclasses (diameter size range, 8.7–14.3 nm) showed that the different treatments only affected the larger HDL subclasses (L‐HDL and XL‐HDL; Figure 4D and 4E). Combined treatment reduced the particle number (P<0.05) in the XL‐HDL subclass (diameter size, 14.3 nm). Triglycerides were reduced by simvastatin (P<0.001), ezetimibe (P<0.05), and by combined treatment (P<0.001), with a clear additive effect when ezetimibe was combined with simvastatin (P<0.05) in the XL‐HDL subclass. Simvastatin and combined treatment reduced triglyceride (P<0.05) and increased CEs (P<0.05) in the L‐HDL subclass (diameter size, 12.1 nm; Figure 4D and 4E).
Effects on the Major Apolipoproteins
ApoB levels were reduced by simvastatin (−38%; P<0.001), ezetimibe (−13%; P<0.01), and combined treatment (−48%; P<0.001). Ezetimibe had an additive effect in combination with simvastatin compared with simvastatin treatment alone (P<0.05; Figure 5A). No significant differences in plasma ApoA1 levels were observed. Thus, the ApoB to ApoA1 ratio was reduced following simvastatin, ezetimibe, or combined treatment (−34%; P<0.001, −10%; P<0.05 and −45%; P<0.001, respectively; Figure 5A). Ezetimibe had an additive effect on the ApoB to ApoA1 ratio in combination with simvastatin compared with simvastatin alone (P<0.01; Figure 5A). Similar decreases in ApoE levels were observed in the simvastatin (−24%; P<0.01) and combined treatment groups (−24%; P<0.01), whereas ezetimibe alone had no significant effect (Figure 5B). In addition, no significant effects were observed in ApoCIII levels (Figure 5B).
Figure 5.
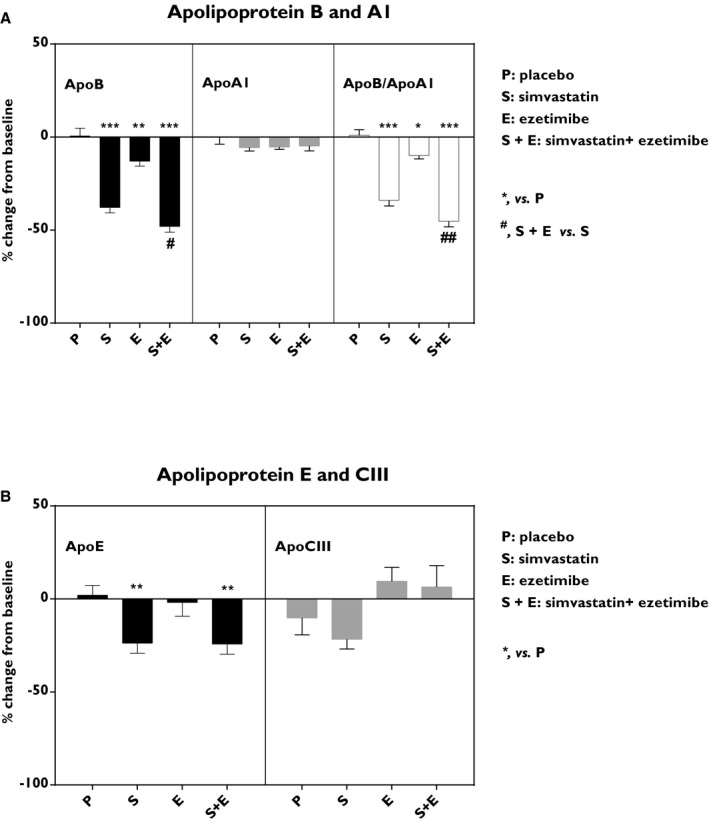
Effects on apolipoproteins. Levels of: (A) apolipoprotein B (ApoB) and apolipoprotein A1 (ApoA1) and the calculated ApoB/ApoA1 ratio, and (B) apolipoprotein E (ApoE) and apolipoprotein CIII (ApoCIII). Data show the % change from the baseline and are expressed as mean±SEM. Multiway ANOVA followed by LSD test. *vs placebo, P<0.05; **vs placebo, P<0.01; ***vs placebo, P<0.001; #simvastatin+ezetimibe vs simvastatin, P<0.05; and ##simvastatin+ezetimibe vs simvastatin, P<0.01 (n=8–10/treatment group). LSD indicates least significant difference.
Effects on Plasma Binding to Human Aortic PG
Simvastatin, ezetimibe, and the combined treatment reduced plasma binding to arterial PG (−53%; P<0.001, −17%; P<0.01, and −57%; P<0.001, respectively) compared with placebo (Figure 6A). When plasma binding was divided by total cholesterol, simvastatin and combined treatment showed (−40%; P<0.001 and −28%; P<0.01) reduction, respectively, whereas the effects by ezetimibe were lost (Figure 6B). After correction for ApoB content, binding to human arterial PG was reduced by simvastatin (−27%; P<0.01), by ezetimibe (−11%; P<0.05), and by the combined treatment (−21%; P<0.01; Figure 6C).
Figure 6.
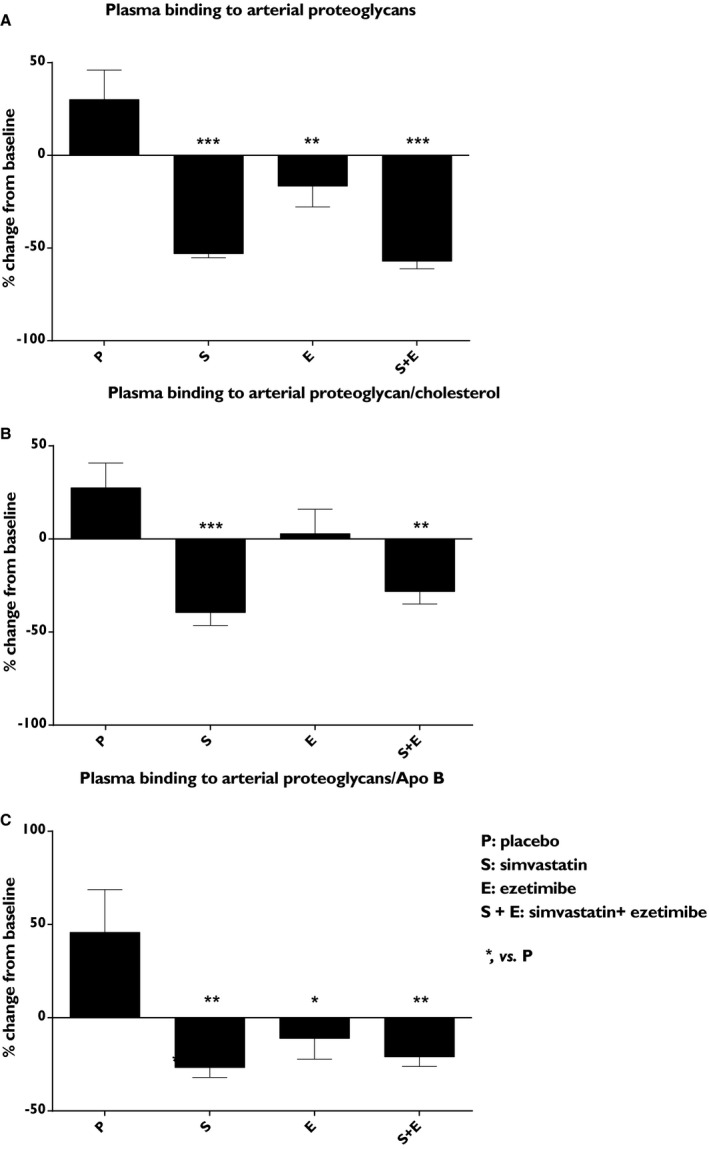
Effects on plasma binding to the human aortic PG. (A), plasma binding to the human arterial proteoglycans (PG), (B) plasma binding to the arterial PG/total cholesterol ratio, and (C) plasma binding to the arterial PG/apolipoprotein B (ApoB) ratio. Data show the % change from baseline and are expressed as mean±SEM. Multiway ANOVA followed by LSD test. *vs placebo, P<0.05; **vs placebo, P<0.01; and ***vs placebo, P<0.001 (n=8–10/treatment group). LSD indicates least significant difference.
When the percentage change from baseline in plasma PG binding was correlated with the percentage change from baseline in each lipoprotein subclass (Table S3), it was clear that the reduction in particle number and lipid composition (CEs and triglyceride) of all ApoB‐lipoprotein contributed to the reduction of PG binding and especially the reduction of the remnant CEs.
Effects on Biliary Lipid Composition and on Bile Acids Synthesis
The effect of intestinal cholesterol absorption and/or cholesterol synthesis inhibition on biliary lipid composition in gallbladder bile after an overnight fast was assessed. As expected,15 simvastatin treatment reduced the absolute biliary cholesterol concentration (−51%; P<0.01) when compared with placebo (Table 2). This reduction was still present when the % molar concentration of biliary cholesterol was calculated (−21%; P<0.05; Figure 7A). In contrast, ezetimibe alone had no significant effect on the absolute biliary cholesterol or on the % molar biliary cholesterol concentrations (Table 2 and Figure 7A). Addition of ezetimibe to simvastatin treatment increased the absolute concentration of all biliary lipids when compared with simvastatin alone (P<0.05 for all; Table 2). The new ratios between bile acids, phospholipids, and cholesterol translated to a loss of significant reduction in the % molar biliary cholesterol concentration when compared with placebo (Figure 7A). Hence, only significant reduction of cholesterol saturation index was observed following treatment with simvastatin (−15%; P<0.05; Figure 7D).
Table 2.
Bile Composition, Plasma Plant Sterols and 7α‐Hydroxy‐4‐Cholesten‐3‐One (C4)
| Placebo (n=10) | Simvastatin (n=10) | Ezetimibe (n=10) | Simvastatin+Ezetimibe (n=10) | |
|---|---|---|---|---|
| Biliary cholesterol, mmol/L | 17.7±2.57 | 8.60±1.13** | 15.6±1.37 | 16.5±2.35# |
| Biliary bile acids, mmol/L | 110±9.80 | 74.8±8.30 | 97.1±12.8 | 139±33.9# |
| Biliary phospholipids, mmol/L | 52.2±7.00 | 30.5±4.20 | 43.2±6.70 | 65.5±11.9# |
| Total biliary lipid, mmol/L | 179±18.60 | 114±13.10 | 156±20.30 | 213±49.00# |
| Cholesterol, % molar | 9.10±0.52 | 7.20±0.20* | 10.4±0.71 | 7.60±0.48 |
| Bile acid, % molar | 61.7±1.49 | 66.2±1.59 | 62.4±0.96 | 64.7±0.82 |
| Phospholipids, % molar | 28.5±1.20 | 26.2±1.31 | 27.2±1.19 | 27.8±0.71 |
| Saturation index, % | 123±8.60 | 104±4.70* | 138±12.5 | 94.0±8.70 |
| Cholic acid, % | 24.0±1.49 | 21.5±1.53 | 21.5±2.16 | 22.7±1.42 |
| Chenodeoxycholic acid, % | 41.4±1.56 | 48.1±4.48 | 47.5±3.80 | 43.1±2.18 |
| Deoxycholic acid, % | 34.5±2.95 | 24.8±6.98 | 25.2±5.15 | 33.7±4.53 |
| Ursodeoxycholic acid, % | 1.50±0.28 | 1.92±0.39 | 1.45±0.27 | 1.46±0.31 |
| Lithocholic acid, % | 0.87±0.31 | 0.62±0.11 | 0.50±0.10 | 0.63±0.13 |
| Bile campesterol, μg/mL | 29.7±3.60 | 16.8±3.9** | 14.5±2.7** | 13.7±1.78** |
| Plasma lathosterol/cholesterol, % variation from baseline | 8.16±8.09 | −56±3.1*** | 40±7.21** | −29±4.6*** , ## |
| Plasma campesterol/cholesterol, % variation from baseline | −3.5±3.90 | 21.2±13.1* | −52±3.1*** | −41±5.3** , ### |
| Plasma C4/cholesterol, ng/mmol/L | 26.8±18.4 | 21.2±17.9 | 13±17.6 | 14±10.3 |
Data show mean±SEM. Multiway ANOVA followed by LSD test. LSD indicates least significant difference.
*vs placebo, P<0.05; **vs placebo, P<0.01; ***vs placebo, P<0.001; #simvastatin + ezetimibe vs simvastatin, P<0.05; ##simvastatin + ezetimibe vs simvastatin, P<0.01; and ###simvastatin+ezetimibe vs simvastatin, P<0.001.
Figure 7.
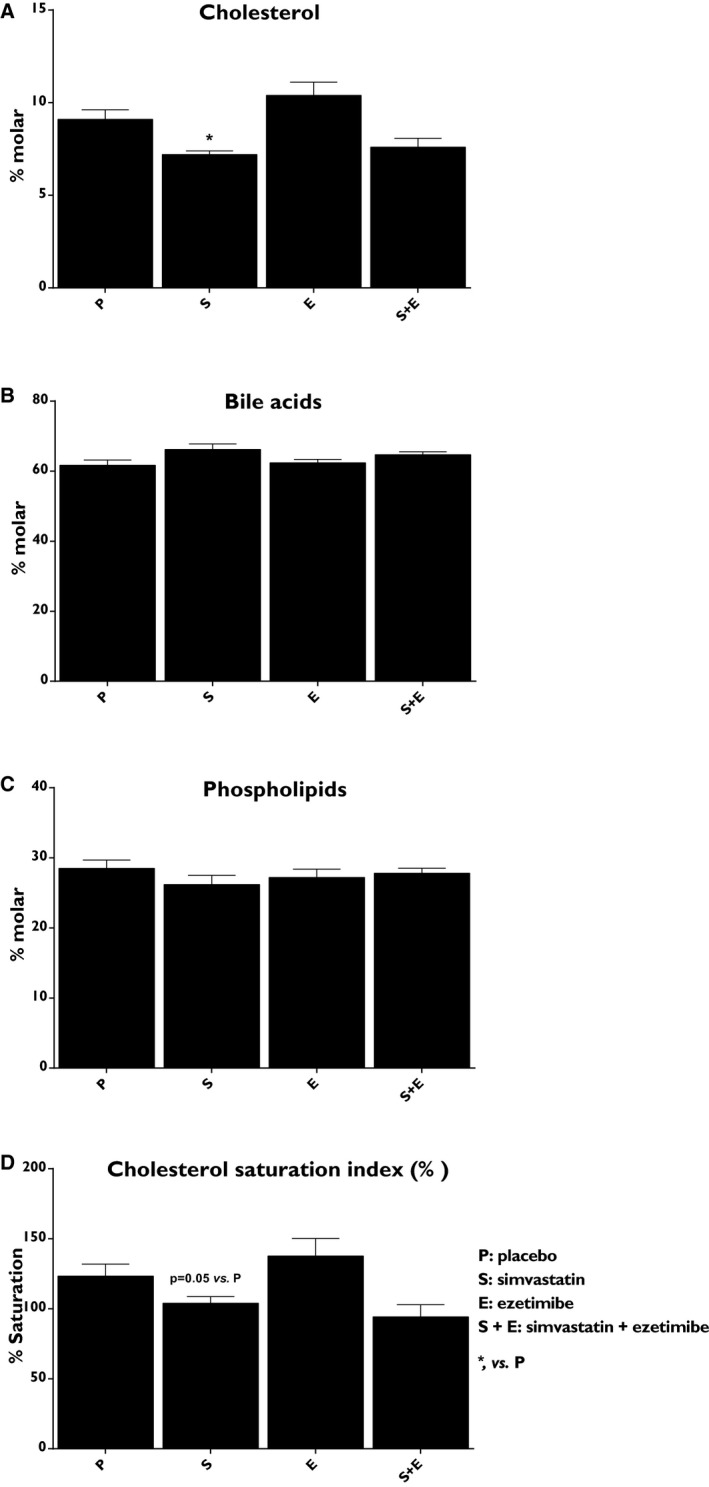
Effects on biliary lipid composition. (A), Percentage molar cholesterol, (B) percentage molar bile acids, (C) percentage molar phospholipids, and (D) cholesterol saturation index. Data show the % change from the baseline and are expressed as mean±SEM. Multiway ANOVA followed by LSD test. *vs placebo, P<0.05 (n=8–10/treatment group). LSD indicates least significant difference.
Simvastatin, ezetimibe, and combined therapy reduced the absolute concentration of campesterol in bile compared with placebo (−43%, −51%, and −54% respectively; P<0.01; Table 2). No significant effects were observed on the composition of individual bile acids or on bile acid synthesis (assayed by C4 to cholesterol ratio; Table 2).
Effects on Genes Involved in Hepatic Cholesterol Metabolism
The mRNA levels of key genes in hepatic cholesterol metabolism were analyzed to investigate the effect of intestinal cholesterol absorption and/or cholesterol synthesis inhibition. Simvastatin and combined treatment significantly increased hepatic mRNA expression of sterol regulatory element binding transcription factor 2 (SREBF2) by 2‐fold, 3‐hydroxy‐3‐methylglutaryl‐CoA reductase (HMGCR; 3.5‐ and 4‐fold, respectively), 3‐hydroxy‐3‐methylglutaryl‐CoA synthase 1 (HMGCS1; 5.5‐ and 6.5‐fold, respectively), low‐density lipoprotein receptor (LDLR; 3.5‐ and 2‐fold, respectively), and proprotein convertase subtilisin/kexin type 9 (PCSK9; 3.5‐ and 2‐fold, respectively; Figure 8A and Table 3). Simvastatin increased hepatic expression of NPC1L1 2‐fold and microsomal triglyceride transfer protein (MTTP) 2‐fold. Ezetimibe added to simvastatin treatment caused 2‐fold reduction in hepatic expression of MTTP and cholesteryl ester transfer protein (CETP), compared with simvastatin monotherapy (Figure 8B and Table 3).
Figure 8.
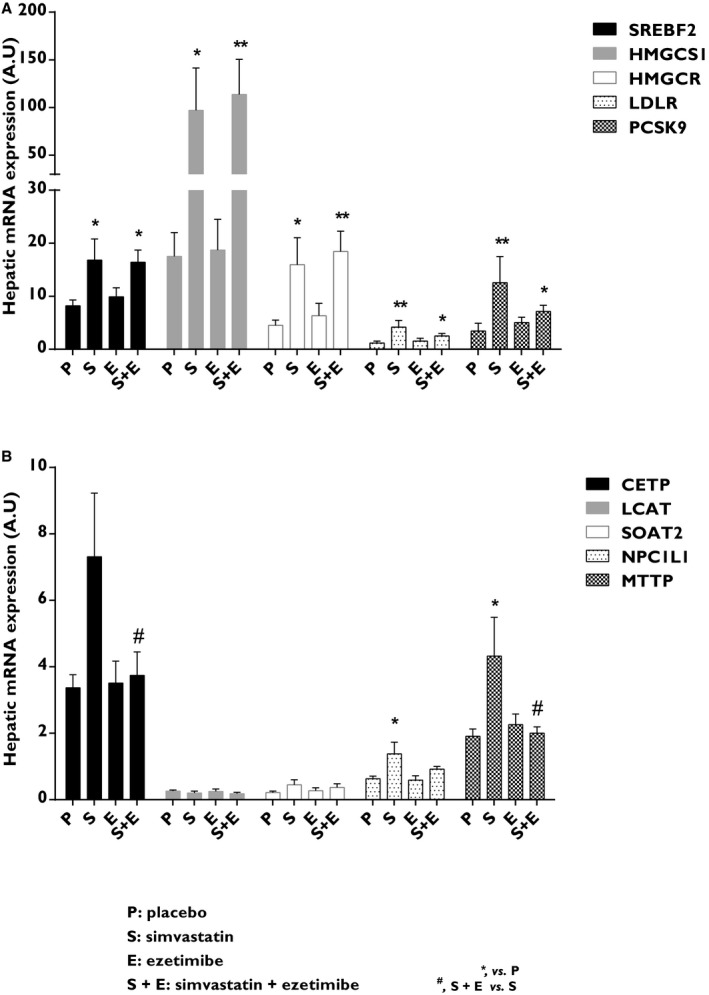
Effects on genes involved in hepatic cholesterol metabolism. A and B, Key genes regulating hepatic cholesterol metabolism. CETP indicates cholesteryl ester transfer protein; HMGCR, 3‐hydroxy‐3‐methylglutaryl‐CoA reductase; HMGCS1, 3‐hydroxy‐3‐methylglutaryl‐CoA synthase 1; LCAT, lecithin‐cholesterol acyltransferase; LDLR, low‐density lipoprotein receptor; LSD, least significant difference; MTTP, microsomal triglyceride transfer protein; NPC1L1, NPC1 like intracellular cholesterol transporter 1; PCSK9, proprotein convertase subtilisin/kexin type 9; SOAT2, sterol O‐acyltransferase 2; SREBF2, sterol regulatory element binding transcription factor 2. Data are expressed as mean±SEM. Primers sequences are given in Table 3. Multiway ANOVA followed by LSD test. *vs placebo, P<0.05; **vs placebo, P<0.01; and #simvastatin+ezetimibe vs simvastatin, P<0.05 (n=5–9/treatment group). A.U. indicates arbitrary units; LSD, least significant difference.
Table 3.
Primer Sequences for Real‐Time PCR
| Gene | Primer Sequence (5′ to 3′) | Final Conc. |
|---|---|---|
| HuSREBF2 | Fwd CAG CTG CAC ATC ACA GGG AA | 700 nmol/L |
| Rev GTA CAT CGG AAC AGG CGG AT | 700 nmol/L | |
| HuHMGCS1 | Fwd GGC ACA GCT GCT GTC TTC AAT | 200 nmol/L |
| Rev ACC AGG GCA TAC CGT CCA T | 200 nmol/L | |
| HuHMGCR | Fwd ATA GGA GGC TAC AAC GCC CAT | 200 nmol/L |
| Rev TTC TGT GCT GCA TCC TGT CC | 200 nmol/L | |
| HuLDLR | Fwd CAG ATA TCA TCA ACG AAG C | 700 nmol/L |
| Rev CCT CTC ACA CCA GTT CAC TCC | 700 nmol/L | |
| HuPCSK9 | Fwd CCA AGA TCC TGC ATG TCT TCC | 200 nmol/L |
| Rev AAC TTC AAG GCC AGC TCC AG | 200 nmol/L | |
| HuCETP | Fwd GAG ACG AGT TCA AGG CAG TGC | 200 nmol/L |
| Rev TCC TGG TTG GTG TTG AAG CC | 200 nmol/L | |
| HuLCAT | Fwd TCC AAC GCC CCT GGT GTC CA | 100 nmol/L |
| Rev TCT CGT CCC GCA CGT AGC CA | 100 nmol/L | |
| HuSOAT2 | Fwd GAG ACT TAC CCT AGG ACG CCC T | 200 nmol/L |
| Rev AGT TCT TGG CCA CAT AAT TCC AC | 200 nmol/L | |
| HuNPC1L1 | Fwd CTT CAG ATG GCC AGG TTT TAG C | 200 nmol/L |
| Rev TGT AAT CCT GTG AGT TTT TCA GGG | 200 nmol/L | |
| HuMTTP | Fwd TTC ACG GTA GCC AGG TGG TT | 100 nmol/L |
| Rev GCG ATT AAG GCT TCC AGT CCT | 100 nmol/L | |
| HuGAPDH | Fwd TGA CAA CTT TGG TAT CGT GGA AGG | 200 nmol/L |
| Rev AGG CAG GGA TGA TGT TCT GGA GAG | 200 nmol/L |
All primer pairs were based on human sequences. CETP indicates cholesteryl ester transfer protein; Conc., concentration; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; HMGCR, 3‐hydroxy‐3‐methylglutaryl‐CoA reductase; HMGCS1, 3‐hydroxy‐3‐methylglutaryl‐CoA synthase 1; LCAT, lecithin‐cholesterol acyltransferase; LDLR, low‐density lipoprotein receptor; MTTP, microsomal triglyceride transfer protein; NPC1L1, NPC1 like intracellular cholesterol transporter 1; PCSK9, proprotein convertase subtilisin/kexin type 9; SOAT2, sterol O‐acyltransferase 2; SREBF2, sterol regulatory element binding transcription factor 2.
Safety Parameters
No significant between‐group differences were observed in the percentage change from baseline of S‐ALT, S‐CPK, or gamma‐glutamyltransferas (Table S4). No adverse events were reported during or after the treatment period.
Discussion
In this randomized, single‐blind, placebo‐controlled study, we found previously unreported effects on plasma lipoprotein particle size, composition, apolipoprotein content, and binding to human arterial proteoglycan following simvastatin, ezetimibe, or combined treatment. In addition, we identified differences in hepatic gene expression of some key genes regulating cholesterol metabolism in the liver and different circulating lipoproteins.
In contrast to mice, humans express NPC1L1 in the liver and its role for hepatic cholesterol and lipoprotein metabolisms has not been fully elucidated. Using a human transgenic mouse model, Temel et al34 suggested that hepatic NPC1L1 is responsible for biliary cholesterol reuptake and is a target for ezetimibe. Our study confirms that this hypothesis is also true in humans. We found that inhibition of cholesterol synthesis with simvastatin leads to a compensatory reduction of cholesterol secretion into bile and to a consequent decrease in biliary cholesterol saturation index. Inhibition of cholesterol synthesis activates the SREBF2 pathway, which also control hepatic NPC1L1 expression in humans.38, 39 Thus, the decreased biliary cholesterol that follows inhibition of cholesterol synthesis seems to be secondary to the concomitant 2‐fold increase in hepatic NPC1L1 expression and to a decrease of the intracellular cholesterol pool available for biliary secretion. The observation that ezetimibe in monotherapy did not affect biliary cholesterol in humans argues against the hypotheis that hepatic NPC1L1 is also a target of this drug, given that Temel et al34 observed an increase in biliary cholesterol after treating transgenic mice with ezetimibe. This discrepancy may have different explanations. In gallstone disease a low hepatic expression of NPC1L1 has been reported,40 and this may reduce the magnitude of the effect of ezetimibe on hepatic NPC1L1. Alternatively, inhibition of intestinal cholesterol absorption by ezetimibe may lead to a decrease in the hepatic cholesterol pool available to biliary secretion counterbalancing the expected increase in biliary cholesterol that ezetimibe would have induced. When ezetimibe was added to statin treatment, a new condition was created as depicted by the analysis of the plasma lathosterol to cholesterol ratio: The liver cannot synthetize cholesterol in response to lower input of cholesterol from the intestine. In this new condition, biliary cholesterol levels were similar to placebo, suggesting that hepatic NPC1L1 in humans is likely a target for ezetimibe. If not, significantly lower levels of biliary cholesterol should have been observed. Our conclusions are in line with the observations that: (1) In pigs,41 ezetimibe added to simvastatin increased hepatic NPC1L1 expression following SREBF2 induction; (2) statin therapy has been associated with a reduced risk of gallstone formation in an epidemiological study42; and (3) genetic variation of NPC1L1 leading to lower activity are associated with an increased risk of gallstone disease.43
Large genetic studies have recently recognized elevated remnant cholesterol, defined as non‐LDL‐C, non‐HDL‐C, as a causal risk factor for CVD and low‐grade inflammation, whereas elevated LDL‐C is associated causally only with CVD.8 Thus, the residual risk of CVD after LDL‐C lowering can partly be attributed to remnant cholesterol.10 In the present study, all treatments, and especially combined therapy, were associated with decreased plasma CEs in almost all 7 subclasses of remnant particles. ACAT2 determines the content of atherogenic CEs secreted into nascent VLDL and chylomicrons12 whereas LCAT adds CEs to VLDL in plasma.14 High‐dose statin treatment decreases the activity of hepatic ACAT215 and CETP44 whereas increasing the activity of LCAT44, 45, 46 in humans. As a net effect, plasma VLDL cholesterol level is decreased. Ezetimibe seems to decrease CETP and LCAT activity in plasma,47 whereas it blocks the NPC1L1‐mediated cholesterol availability to intestinal ACAT2. Consequently, ezetimibe reduces the chylomicron, CE, as shown in mice.16 In this study, ezetimibe reduced CE in almost all remnant lipoprotein subclasses, but not in all LDL subclasses. Thus, the additional reduction of CE in remnant particles by combined treatment compared with simvastatin monotherapy support the complementary and synergistic role of ezetimibe in combination with statin. In line with these findings, it has been shown that the addition of ezetimibe to ongoing statin therapy decreases remnant cholesterol to a greater extent than just doubling the statin dose.48
The primary quantitative dyslipidemia in T2DM is characterized by high plasma triglyceride and low HDL‐cholesterol levels while qualitatively by an increase in L‐VLDL and sdLDL fractions.49 The combined treatment showed consistent significant effects in all remnant lipoprotein subclasses both qualitatively (CE and triglyceride) and quantitatively (particle numbers). These findings suggest that ezetimibe in combination with simvastatin is more effective than simvastatin monotherapy in T2DM patients, who are characterized by high levels of triglyceride‐rich remnant particles. Hence, our study may, in part, explain why the combination therapy of simvastatin and ezetimibe was more efficient in reducing CVD events in patients with T2DM in the IMPROVE‐IT5 and SHARP6 trials.
LDL particles from patients with coronary heart disease have a significantly higher affinity for arterial PG compared with healthy individuals.24, 25 This affinity is reduced by statin treatment in patients with moderate hypercholesterolemia.26 Here, we evaluated the effect of simvastatin and ezetimibe alone or in combination on the binding of lipoproteins in plasma to human arterial PG. The solid‐phase analysis used mainly LDL particles and the triglyceride‐rich VLDL and IDL particles, which have a lower affinity for arterial PG than LDL particles. Simvastatin and ezetimibe monotherapy treatments reduced the amount of plasma ApoB‐lipoprotein cholesterol bound to PG compared with placebo, where simvastatin was more effective. However, addition of ezetimibe did not have any significant synergistic effect compared with simvastatin alone. It has been shown that large LDL particles have a lower affinity for PG than sdLDL particles.11, 25 Simvastatin reduced CE in all LDL subclasses whereas ezetimibe only reduced CE in L‐LDL particles, with no effects on smaller and most atherogenic sdLDL subclasses. This might explain the greater effect of simvastatin and the lack of synergistic effect of ezetimibe on arterial PG binding. To assess further, whether the reduction of lipoprotein binding to PG was attributed to the lowering effect of the cholesterol and particle number, we corrected the PG binding for total cholesterol and ApoB, respectively. In contrast to ezetimibe, simvastatin was still able to reduce the binding to arterial PG. Thus, simvastatin seems to modify the characteristics of the particles and not only their number and cholesterol load.
Simvastatin monotherapy increased the hepatic mRNA levels for genes under the control of SREBF2, as expected. For the majority of these genes, addition of ezetimibe in combination with simvastatin treatment did not enhance this effect, suggesting that inhibition of cholesterol synthesis is per se sufficient to fully activate the SREBF2 pathway. Adding ezetimibe to simvastatin treatment blunted the induction of hepatic expression of MTTP. This effect may, in part, explain why the combined therapy modifies the lipid content of remnant particles. Differently from our previous study with atorvastatin,15 we did not observe a decrease in Soat2 mRNA levels. Unfortunately, the size of the biopsies did not allow for measurement of ACAT2 activity, which is a limitation of this study.
In conclusion, the expected increase in biliary cholesterol following treatment with ezetimibe monotherapy does not occur in gallstone‐diseased patients, yet we found evidence that ezetimibe is able to block hepatic NPC1L1 in humans. In addition, our results suggest that ezetimibe and simvastatin reduces atherogenic CE in remnant cholesterol particles. Moreover, the dramatic reduction of remnant cholesterol and the apparent atheroprotective changes in ApoB‐lipoprotein plasma levels, their composition, size distribution, and possible reduction of affinity for human arterial proteoglycans may all explain why ezetimibe combined with simvastatin was effective in reducing cardiovascular events in the IMPROVE‐IT and SHARP trials. Therefore, it seems reasonable to treat the lipid disorder in those conditions characterized by increased levels of remnant cholesterol, that is, patients with T2DM with a combination of simvastatin and ezetimibe.
Sources of Funding
This work was supported by grants from the Swedish Research Council, the Swedish Heart‐Lung Foundation, the Stockholm City Council, and the Karolinska Institutet.
Disclosures
Eriksson and Parini received jointly an independent research grant from MERCK. The remaining authors have no disclosures to report.
Supporting information
Data S1. Supplemental methods.
Table S1. Lipid Contribution in Lipoprotein Fractions Used in Figure 2
Table S2. Contribution of Different Subclasses for Their Respective Fraction Baseline
Table S3. Correlation of Percentage Changes of Proteoglycan (PG) Binding and Lipoprotein Particle Number and Composition
Table S4. Safety Assessment Analyses
Acknowledgments
We thank Anita Lövgren‐Sandblom for her technical assistance and Maura Heverin for language revision.
(J Am Heart Assoc. 2018;7:e009876 DOI: 10.1161/JAHA.118.009876.)
References
- 1. Mancini GB, Tashakkor AY, Baker S, Bergeron J, Fitchett D, Frohlich J, Genest J, Gupta M, Hegele RA, Ng DS, Pearson GJ, Pope J. Diagnosis, prevention, and management of statin adverse effects and intolerance: Canadian Working Group Consensus update. Can J Cardiol. 2013;29:1553–1568. [DOI] [PubMed] [Google Scholar]
- 2. Clader JW. The discovery of ezetimibe: a view from outside the receptor. J Med Chem. 2004;47:1–9. [DOI] [PubMed] [Google Scholar]
- 3. Couture P, Lamarche B. Ezetimibe and bile acid sequestrants: impact on lipoprotein metabolism and beyond. Curr Opin Lipidol. 2013;24:227–232. [DOI] [PubMed] [Google Scholar]
- 4. Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW, De Ferrari GM, Ruzyllo W, De Lucca P, Im K, Bohula EA, Reist C, Wiviott SD, Tershakovec AM, Musliner TA, Braunwald E, Califf RM. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–2397. [DOI] [PubMed] [Google Scholar]
- 5. Giugliano RP, Cannon CP, Blazing MA, Nicolau JC, Corbalan R, Spinar J, Park JG, White JA, Bohula EA, Braunwald E. Benefit of adding ezetimibe to statin therapy on cardiovascular outcomes and safety in patients with versus without diabetes mellitus: results from IMPROVE‐IT (Improved Reduction of Outcomes: Vytorin Efficacy International Trial). Circulation. 2018;137:1571–1582. [DOI] [PubMed] [Google Scholar]
- 6. Baigent C, Landray MJ, Reith C, Emberson J, Wheeler DC, Tomson C, Wanner C, Krane V, Cass A, Craig J, Neal B, Jiang L, Hooi LS, Levin A, Agodoa L, Gaziano M, Kasiske B, Walker R, Massy ZA, Feldt‐Rasmussen B, Krairittichai U, Ophascharoensuk V, Fellstrom B, Holdaas H, Tesar V, Wiecek A, Grobbee D, de Zeeuw D, Gronhagen‐Riska C, Dasgupta T, Lewis D, Herrington W, Mafham M, Majoni W, Wallendszus K, Grimm R, Pedersen T, Tobert J, Armitage J, Baxter A, Bray C, Chen Y, Chen Z, Hill M, Knott C, Parish S, Simpson D, Sleight P, Young A, Collins R. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo‐controlled trial. Lancet. 2011;377:2181–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sharp Collaborative G . Study of Heart and Renal Protection (SHARP): randomized trial to assess the effects of lowering low‐density lipoprotein cholesterol among 9,438 patients with chronic kidney disease. Am Heart J. 2010;160:785–794.e710. [DOI] [PubMed] [Google Scholar]
- 8. Varbo A, Benn M, Tybjaerg‐Hansen A, Nordestgaard BG. Elevated remnant cholesterol causes both low‐grade inflammation and ischemic heart disease, whereas elevated low‐density lipoprotein cholesterol causes ischemic heart disease without inflammation. Circulation. 2013;128:1298–1309. [DOI] [PubMed] [Google Scholar]
- 9. Nordestgaard BG, Langsted A, Mora S, Kolovou G, Baum H, Bruckert E, Watts GF, Sypniewska G, Wiklund O, Boren J, Chapman MJ, Cobbaert C, Descamps OS, von Eckardstein A, Kamstrup PR, Pulkki K, Kronenberg F, Remaley AT, Rifai N, Ros E, Langlois M. Fasting is not routinely required for determination of a lipid profile: clinical and laboratory implications including flagging at desirable concentration cut‐points‐a joint consensus statement from the European Atherosclerosis Society and European Federation of Clinical Chemistry and Laboratory Medicine. Eur Heart J. 2016;37:1944–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Varbo A, Nordestgaard BG. Remnant cholesterol and ischemic heart disease. Curr Opin Lipidol. 2014;25:266–273. [DOI] [PubMed] [Google Scholar]
- 11. Boren J, Williams KJ. The central role of arterial retention of cholesterol‐rich apolipoprotein‐B‐containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27:473–483. [DOI] [PubMed] [Google Scholar]
- 12. Pramfalk C, Eriksson M, Parini P. Cholesteryl esters and ACAT. Eur J Lipid Sci Technol. 2012;114:624–633. [Google Scholar]
- 13. Parini P, Davis M, Lada AT, Erickson SK, Wright TL, Gustafsson U, Sahlin S, Einarsson C, Eriksson M, Angelin B, Tomoda H, Omura S, Willingham MC, Rudel LL. ACAT2 is localized to hepatocytes and is the major cholesterol‐esterifying enzyme in human liver. Circulation. 2004;110:2017–2023. [DOI] [PubMed] [Google Scholar]
- 14. Eisenberg S. Preferential enrichment of large‐sized very low density lipoprotein populations with transferred cholesteryl esters. J Lipid Res. 1985;26:487–494. [PubMed] [Google Scholar]
- 15. Parini P, Gustafsson U, Davis MA, Larsson L, Einarsson C, Wilson M, Rudling M, Tomoda H, Omura S, Sahlin S, Angelin B, Rudel LL, Eriksson M. Cholesterol synthesis inhibition elicits an integrated molecular response in human livers including decreased ACAT2. Arterioscler Thromb Vasc Biol. 2008;28:1200–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xie C, Zhou ZS, Li N, Bian Y, Wang YJ, Wang LJ, Li BL, Song BL. Ezetimibe blocks the internalization of NPC1L1 and cholesterol in mouse small intestine. J Lipid Res. 2012;53:2092–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee RG, Shah R, Sawyer JK, Hamilton RL, Parks JS, Rudel LL. ACAT2 contributes cholesteryl esters to newly secreted VLDL, whereas LCAT adds cholesteryl ester to LDL in mice. J Lipid Res. 2005;46:1205–1212. [DOI] [PubMed] [Google Scholar]
- 18. Hoogeveen RC, Gaubatz JW, Sun W, Dodge RC, Crosby JR, Jiang J, Couper D, Virani SS, Kathiresan S, Boerwinkle E, Ballantyne CM. Small dense low‐density lipoprotein‐cholesterol concentrations predict risk for coronary heart disease: the Atherosclerosis Risk in Communities (ARIC) study. Arterioscler Thromb Vasc Biol. 2014;34:1069–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang YC, Chang PY, Hwang JS, Ning HC. Association of small dense lowdensity lipoprotein cholesterol in type 2 diabetics with coronary artery disease. Biomed J. 2014;37:375–379. [DOI] [PubMed] [Google Scholar]
- 20. Torimoto K, Okada Y, Mori H, Hajime M, Tanaka K, Kurozumi A, Narisawa M, Yamamoto S, Arao T, Matsuoka H, Inokuchi N, Tanaka Y. Efficacy of combination of ezetimibe 10 mg and rosuvastatin 2.5 mg versus rosuvastatin 5 mg monotherapy for hypercholesterolemia in patients with type 2 diabetes. Lipids Health Dis. 2013;12:137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tribble DL, Rizzo M, Chait A, Lewis DM, Blanche PJ, Krauss RM. Enhanced oxidative susceptibility and reduced antioxidant content of metabolic precursors of small, dense low‐density lipoproteins. Am J Med. 2001;110:103–110. [DOI] [PubMed] [Google Scholar]
- 22. Younis NN, Soran H, Pemberton P, Charlton‐Menys V, Elseweidy MM, Durrington PN. Small dense LDL is more susceptible to glycation than more buoyant LDL in type 2 diabetes. Clin Sci (Lond). 2013;124:343–349. [DOI] [PubMed] [Google Scholar]
- 23. Hurt‐Camejo E, Camejo G, Rosengren B, Lopez F, Wiklund O, Bondjers G. Differential uptake of proteoglycan‐selected subfractions of low density lipoprotein by human macrophages. J Lipid Res. 1990;31:1387–1398. [PubMed] [Google Scholar]
- 24. Linden T, Bondjers G, Camejo G, Bergstrand R, Wilhelmsen L, Wiklund O. Affinity of LDL to a human arterial proteoglycan among male survivors of myocardial infarction. Eur J Clin Invest. 1989;19:38–44. [DOI] [PubMed] [Google Scholar]
- 25. Camejo G, Hurt‐Camejo E, Wiklund O, Bondjers G. Association of apo B lipoproteins with arterial proteoglycans: pathological significance and molecular basis. Atherosclerosis. 1998;139:205–222. [DOI] [PubMed] [Google Scholar]
- 26. Wiklund O, Bondjers G, Wright I, Camejo G. Insoluble complex formation between LDL and arterial proteoglycans in relation to serum lipid levels and effects of lipid lowering drugs. Atherosclerosis. 1996;119:57–67. [DOI] [PubMed] [Google Scholar]
- 27. Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL, Boren J. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002;417:750–754. [DOI] [PubMed] [Google Scholar]
- 28. Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116:1832–1844. [DOI] [PubMed] [Google Scholar]
- 29. Bancells C, Benitez S, Jauhiainen M, Ordonez‐Llanos J, Kovanen PT, Villegas S, Sanchez‐Quesada JL, Oorni K. High binding affinity of electronegative LDL to human aortic proteoglycans depends on its aggregation level. J Lipid Res. 2009;50:446–455. [DOI] [PubMed] [Google Scholar]
- 30. Boren J, Olin K, Lee I, Chait A, Wight TN, Innerarity TL. Identification of the principal proteoglycan‐binding site in LDL. A single‐point mutation in apo‐B100 severely affects proteoglycan interaction without affecting LDL receptor binding. J Clin Invest. 1998;101:2658–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hurt‐Camejo E, Camejo G. ApoB‐100 lipoprotein complex formation with intima proteoglycans as a cause of atherosclerosis and its possible ex vivo evaluation as a disease biomarker. J Cardiovasc Dev Dis. 2018;5:E36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pramfalk C, Jiang ZY, Parini P. Hepatic Niemann‐Pick C1‐like 1. Curr Opin Lipidol. 2011;22:225–230. [DOI] [PubMed] [Google Scholar]
- 33. Yu L, Bharadwaj S, Brown JM, Ma Y, Du W, Davis MA, Michaely P, Liu P, Willingham MC, Rudel LL. Cholesterol‐regulated translocation of NPC1L1 to the cell surface facilitates free cholesterol uptake. J Biol Chem. 2006;281:6616–6624. [DOI] [PubMed] [Google Scholar]
- 34. Temel RE, Tang W, Ma Y, Rudel LL, Willingham MC, Ioannou YA, Davies JP, Nilsson LM, Yu L. Hepatic Niemann‐Pick C1‐like 1 regulates biliary cholesterol concentration and is a target of ezetimibe. J Clin Invest. 2007;117:1968–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tang W, Jia L, Ma Y, Xie P, Haywood J, Dawson PA, Li J, Yu L. Ezetimibe restores biliary cholesterol excretion in mice expressing Niemann‐Pick C1‐like 1 only in liver. Biochim Biophys Acta. 2011;1811:549–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kishikawa N, Kanno K, Sugiyama A, Yokobayashi K, Mizooka M, Tazuma S. Long‐term administration of a Niemann‐Pick C1‐like 1 inhibitor, ezetimibe, does not worsen bile lithogenicity in dyslipidemic patients with hepatobiliary diseases. J Hepatobiliary Pancreat Sci. 2016;23:125–131. [DOI] [PubMed] [Google Scholar]
- 37. Kishikawa N, Kanno K, Sugiyama A, Yokobayashi K, Mizooka M, Tazuma S. Clinical evaluation of ezetimibe on bile lithogenicity in humans: use of transnasal endoscopy for bile sampling. Hepatol Res. 2015;45:693–697. [DOI] [PubMed] [Google Scholar]
- 38. Pramfalk C, Jiang ZY, Cai Q, Hu H, Zhang SD, Han TQ, Eriksson M, Parini P. HNF1alpha and SREBP2 are important regulators of NPC1L1 in human liver. J Lipid Res. 2010;51:1354–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tremblay AJ, Lamarche B, Lemelin V, Hoos L, Benjannet S, Seidah NG, Davis HR Jr, Couture P. Atorvastatin increases intestinal expression of NPC1L1 in hyperlipidemic men. J Lipid Res. 2011;52:558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cui W, Jiang ZY, Cai Q, Zhang RY, Wu WZ, Wang JC, Fei J, Zhang SD, Han TQ. Decreased NPC1L1 expression in the liver from Chinese female gallstone patients. Lipids Health Dis. 2010;9:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Telford DE, Sutherland BG, Edwards JY, Andrews JD, Barrett PH, Huff MW. The molecular mechanisms underlying the reduction of LDL apoB‐100 by ezetimibe plus simvastatin. J Lipid Res. 2007;48:699–708. [DOI] [PubMed] [Google Scholar]
- 42. Bodmer M, Brauchli YB, Krahenbuhl S, Jick SS, Meier CR. Statin use and risk of gallstone disease followed by cholecystectomy. JAMA. 2009;302:2001–2007. [DOI] [PubMed] [Google Scholar]
- 43. Lauridsen BK, Stender S, Frikke‐Schmidt R, Nordestgaard BG, Tybjaerg‐Hansen A. Genetic variation in the cholesterol transporter NPC1L1, ischaemic vascular disease, and gallstone disease. Eur Heart J. 2015;36:1601–1608. [DOI] [PubMed] [Google Scholar]
- 44. Kassai A, Illyes L, Mirdamadi HZ, Seres I, Kalmar T, Audikovszky M, Paragh G. The effect of atorvastatin therapy on lecithin: cholesterol acyltransferase, cholesteryl ester transfer protein and the antioxidant paraoxonase. Clin Biochem. 2007;40:1–5. [DOI] [PubMed] [Google Scholar]
- 45. Weisweiler P. Simvastatin and bezafibrate: effects on serum lipoproteins and lecithin: cholesterol acyltransferase activity in familial hypercholesterolaemia. Eur J Clin Pharmacol. 1988;35:579–583. [DOI] [PubMed] [Google Scholar]
- 46. Adekunle AS, Fatoki JO, Adelusi TI. Antihyperlipidemic and antiatherogenic activity of simvastatin may involve modulation of the expression of lecithin: cholesterol acyl transferase. Acta Biochim Pol. 2013;60:579–583. [PubMed] [Google Scholar]
- 47. Yagyu H, Nagashima S, Takahashi M, Miyamoto M, Okada K, Osuga J, Ishibashi S. Ezetimibe, an inhibitor of Niemann‐Pick C1‐like 1 protein, decreases cholesteryl ester transfer protein in type 2 diabetes mellitus. Endocr J. 2012;59:1077–1084. [DOI] [PubMed] [Google Scholar]
- 48. Nakamura T, Hirano M, Kitta Y, Fujioka D, Saito Y, Kawabata K, Obata JE, Watanabe Y, Watanabe K, Kugiyama K. A comparison of the efficacy of combined ezetimibe and statin therapy with doubling of statin dose in patients with remnant lipoproteinemia on previous statin therapy. J Cardiol. 2012;60:12–17. [DOI] [PubMed] [Google Scholar]
- 49. Verges B. Pathophysiology of diabetic dyslipidaemia: where are we? Diabetologia. 2015;58:886–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental methods.
Table S1. Lipid Contribution in Lipoprotein Fractions Used in Figure 2
Table S2. Contribution of Different Subclasses for Their Respective Fraction Baseline
Table S3. Correlation of Percentage Changes of Proteoglycan (PG) Binding and Lipoprotein Particle Number and Composition
Table S4. Safety Assessment Analyses