

Abstract
Abnormal expression of dermatan sulfate epimerase (DSE) has been found in many types of cancer, while its expression and biological functions in hepatocellular carcinoma (HCC) progression remains obscure. Here we report that DSE, the enzyme that catalyzes the conversion of chondroitin sulfate (CS) to dermatan sulfate (DS), is a critical mediator of malignant character in HCC, through regulation of CCL5 signaling. DSE mRNA and protein were downregulated frequently in HCC tumors, where these events were associated with advanced tumor stages, metastases, and poor survival. DSE-mediated tumor growth was evaluated in immune-deficient and immune-complement mice models. Restoring DSE expression in HCC cells suppressed tumor growth, as well as decreased IL-1β and CCL5 levels in transplanted tumor tissue. Mechanistic investigations revealed that the expression of DSE altered CCL5 signaling and cell surface binding in HCC cells. Accordingly, DSE suppressed CCL5-induced cell growth, migration, and invasion, whereas silencing of DSE enhanced CCL5-triggered malignant phenotypes. Inhibiting CCR1 activity with BX471 decreased CCL5-induced malignant characters caused by siRNA-mediated knockdown of DSE in HCC cells, establishing the critical role of the CCL5/CCR1 axis in mediating the effects of DSE expression. Taken together, our results suggest that DSE dysregulation contributes to the malignant behavior of HCC cells. This provides novel insight into the significance of DSE in CCL5 signaling and HCC pathogenesis.
Keywords: Dermatan sulfate, DS epimerase, hepatocellular carcinoma, tumor infiltrated lymphocyte, CCL5
Introduction
Hepatocellular carcinoma (HCC) is the most common primary liver malignancy and is the third leading cause of cancer-related death worldwide [1]. Recently, mortality rates for HCC have continued to increase, mainly due to late diagnosis and a lack of therapeutic options [2]. Curative surgery or liver transplantation may provide an opportunity for a cure, but only when patients are diagnosed at an early stage [3]. For patients with advanced HCC, the introduction of sorafenib, regorafenib, or immune checkpoint inhibitors offer limited survival benefits [4-6]. Therefore, in order to develop new therapies and new risk stratification approaches, a full understanding of the molecular mechanisms underlying HCC progression is necessary.
Alteration of the tumor microenvironment, especially changes to extracellular matrix (ECM) composition, modulates the progression of established tumors. Glycosaminoglycan (GAG), a family of unbranched polysaccharide chains, is a major component of the extracellular matrix in both normal organs and in tumor tissue. Abnormal GAG expression and modification are often observed in HCC, which may not only be associated with disease progression, but specific glycan structures could also be used as biomarkers for disease diagnosis and pharmacological targets [7-10]. Although aberrant expression of GAG chains in HCC has been proposed in some reports, the biological functions of GAG in HCC cells remain largely unknown.
Chondroitin sulfate (CS) is one of the major types of GAG, composed of repeating glucuronic acid/N-acetylgalactosamine (GlcA-GalNAc) blocks with complex sulfation and epimerization. In certain tissues, dermatan sulfate epimerase (DSE), a C5 epimerase, plays a pivotal role in converting GlcA into iduronic acid (IdoA) within CS chains. DSE works in tandem with dermatan 4-O-sulfotransferase 1 (D4ST1), and increases structural diversity as a CS/DS hybrid polysaccharide composite [11]. CS/DS chains are crucial in shaping the functions of growth factors, proteases, cytokines, and chemokines by regulating their movement and activity. This property of CS/DS results in modulation of various cell behaviors during development [12-14]. For example, CS/DS hybrid chains with specific compositions can modulate their affinity to FGF2 and Heparin Cofactor-II, which regulate migration of neural crest cells [15-17]. Loss-of-function mutation in DSE can cause musculocontractural Ehlers-Danlos syndrome, a developmental disorder characterized by connective tissue fragility and multiple organ disorder in humans [18].
Previous reports indicate that DSE is often up-regulated during carcinogenesis in certain types of cancer, such as glioma and squamous cell carcinoma, and that it could regulate growth factor signaling in cancer cells [19,20]. Notably, recent reports indicate that enzymatic degradation of CS/DS chains could inhibit malignant phenotypes of cancer cells and enhance effectiveness of anti-tumor agents [21-23]. These studies suggest important roles of DSE in tumorgenesis. However, its expression and function in human HCC remain obscure. This study aims to investigate the correlation of DSE expression in HCC progression. Furthermore, we explore possible mechanisms of DSE in regulating HCC malignant phenotypes in vivo and in vitro.
Material and methods
Human tissue samples
Post-surgery frozen HCC tissues for western blots were obtained from the Chung Shan Medical University Hospital (Taichung, Taiwan). This study was approved by the Ethical Committees of Chung Shan Medical University Hospital, and all patients gave informed consent to have their tissues before collection. For immunohistochemistry, two paraffin-embedded human HCC tissue microarrays (CS4 and CSA4) including follow-up survival information were purchased from SuperBioChips.
Immunohistochemistry
UltraVision Quanto Detection System (Thermo Fisher Scientific Inc.) was used for immunohistochemistry. Tissue arrays were incubated with anti-DSE antibody (Sigma-Aldrich) in 1:100 dilutions for 16 hours at 4°C. The specific immunostaining was visualized with 3,3-diaminobenzidine (DAB) and counterstained with hematoxylin for 1 minute (Sigma). Images were obtained by Tissue FAX Plus Cytometer.
Cell culture
Liver cancer cell lines, HA59T, HA22T, HepG2, and Hepa1-6 were purchased from Bioresource Collection and Research Center (Hsinchu, Taiwan). HCC36 cells were kindly provided by Prof. Lei Wan (China Medical University, Taichung, Taiwan). Cells were cultured in DMEM containing 10% FBS in 5% CO2 at 37°C. CCL5 blocking antibody (MAB678) was purchased from R&D SystemsTM, and 2 μg/ml was used for treating cells. BX471 was purchased from CAYMAN CHEMICAL, and 20 μM was used for treating cells in culture medium. For CS/DS enzymatic digestion, cultured cells were pretreated with chondroitinase ABC (0.5 unit/ml; Sigma-Aldrich) in serum free DMEM for 3 hours.
Transfection and RNA interference
To overexpress DSE, cells were transfected with pcDNA3.1/DSE/mycHis plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. Empty pcDNA3.1/mycHis plasmid was used as mock transfectant. The transfected cells were selected with 600 μg/mL of G418 for 14 days, and selected clones were pooled for further studies.
Transient knockdown of DSE by siRNA was performed as a previous report [20]. Overexpression and knockdown of DSE in HCC cell lines were confirmed by western blotting.
Western blotting
Western blotting was carried out as reported previously [24]. For measuring cell signaling, recombinant CCL5 was purchased from PepoTech. Antibodies against p-AKT, p-ERK1/2, p-P38, AKT, ERK1/2, and P38 were purchased from Cell Signaling Technology. Antibodies against Actin were purchased from GeneTex. To analyze CCL5 binding and signaling, CCL5 (50 ng/ml) were added to cells for 5 and 15 minutes. Cells were washed once in ice cold PBS for 10 seconds, and subjected to western blotting using 8% or 13% SDS-PAGE, and 0.22 μm PVDF membrane. Anti-CCL5 antibody (710001) and anti-CCR1 antibody (PA1-41062) was purchased from Thermo Fisher Scientific Inc.
CCK-8 assay
Cells (1 × 103) were seeded into 96-well plates with culture medium 16 hours before the experiments. Cell viability was analyzed by CCK-8 reagent at 0, 24, 48 and 72 hours. Four wells per group for each time point were measured following manufacturer’s protocol. For CCL5 stimulation assays, 2 or 20 ng/ml of CCL5 was added in DMEM medium with 5% FBS for 48 or 72 hours. These experiments were repeated for three times, and relative fold changes of OD 450 nm were shown.
In vivo tumor growth and isolation of tumor infiltrating cells
Male non-obese diabetic/severe combined immnodeficient (NOD/SCID) mice and C57BL/6 mice, 6 weeks of age, were purchased from National Laboratory Animal Center, Taiwan. 5 × 106 of Hepa1-6 transfectants were subcutaneously injected into right flank (n = 7 for each group). Tumor volumes were monitored for 20 days.
For analyzing tumor infiltrated cells, 5 × 106 of Hepa1-6 transfectants were subcutaneously injected, and tumor tissues were excised at day 10 (n = 7 for each group). Specimens were divided into fragments and minced with razor blades. Sterile 50 µm medicons (BD Biosciences) were prepared with culture medium then loaded with tumor tissue. Disaggregation cell suspension was filtered through cell strainer to remove cell clumps and briefly treated with ACK buffer to remove red blood cells. Cells were spin down and analyzed by flow cytometry.
All animal experiments in this study were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Chung Shan Medical University Experimental Animal Center.
Flow cytometry
Tumor cell suspension was stained with Pacific-Blue anti-CD4, Violet 510 anti-Gr1, PE anti-CD11b, PerCP anti-CD45, PE-Cy7 anti-CD3, APC anti-NK1.1, and APC-Cy7 anti-CD8 antibodies on ice for 30 minutes (all form Biolegend). Following staining of surface markers, cells were washed, and applied to FITC anti-Foxp3 staining kit (eBioscience) according to manufacturer’s protocol. For CCL5 staining, above cell surface markers were stained (PE anti-CD11b was replaced by FITC anti-CD11b), cells were then resuspended in Fix/Perm buffer and stained with PE anti-mouse CCL5 (2E9, Biolegend) (n = 5 for each group). Non-specific PE mouse IgG2b (Biolegend) was used as iso-type control. Antibodies against chondroitin sulfate (CS-56) was purchased from GeneTex. FITC-anti-mouse IgM and Alexa488-anti-rabbit IgG was purchased from Invitrogen. Flow cytometry was performed on a BD FACSVerse flow cytometer. Data was analyzed with the FlowJo software version 10.
Multiplex immunoassays
The Bio-Plex Pro Mouse Cytokine 23-Plex immunoassay kit was used to detect IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6I, L-9I, L-10I, L-12 (p40), IL-12 (p70), IL-13, IL-17A, CCL11, CCL2, CCL3, CCL4, CCL5, CXCL1, TNF-α, GM-CSF, G-CSF, and IFN-γ. Assays were performed by Bio-Plex 200 system. 100 μg of mouse tumor tissue lysate was used for each reaction (n = 5 for each group).
Cell migration and invasion assay
Transwell inserts with uncoated porous filters (pore size 8 µm) were used to evaluate cell migration, and Matrigel (BD Biosciences) coated inserts were used to measure cell invasion. 2 × 104 cells in serum-free DMEM were seeded into inserts, DMEM containing 5% FBS was added with or without CCL5 (20 ng/ml) in lower part of the inserts. Hepa1-6 cell were incubated for 24 hours and HA22T cell were incubated for 16 hours. Independent experiments were repeated for three times. Average number of cells per microscopic field was shown.
Immunofluorescence microscopy
For CCL5 staining, frozen sections of mouse tumor tissue were stained with anti-CCL5 antibody. DAPI was used for nuclear staining. Images were captured by ZEISS Axio Imager A2 microscope.
Statistical analysis
All data analysis was performed using GraphPad Prism 6. Student t test was used for statistical analyses. Two-sided Fisher exact test was used for comparisons between DSE expression and clinicopathologic features of HCC tissue array. P < 0.05 was considered statistically significant difference.
Results
Down-regulation of DSE is associated with late tumor stage and worse prognosis of HCC
To explore DSE expression in human liver and HCC, we analyzed the ONCOMINE database [25]. Two independent microarray datasets indicated that DSE is significantly down-regulated in HCC compared to normal liver tissue (Figure 1A). We further measured protein expression of DSE in paired HCC tissues and adjacent non-tumor liver tissues by western blotting and immunohistochemistry (IHC). Consistently, western blotting showed that DSE protein was down-regulated in 75% (9/12) of paired HCC tissues (Figure 1B). Immunohistochemistry revealed dot-like precipitates of DSE mainly expressed in the cytoplasm of adjacent non-tumor hepatocytes, but downregulated in tumor cells (Figure 1C). Additionally, expression of DSE was barely observed in surrounding stromal cells under our experimental conditions. To explore the relationship between DSE expression and clinicopathologic features in patients with HCC, we conducted immunohistochemistry in a tissue array containing 98 primary HCC tissues and 9 non-tumor liver samples. The intensity of staining was scored according to the percentage of DSE-positive parenchymal cells in each sample (0, negative; +1, < 20%; +2, 20%-50%; +3, > 50%). Our data revealed that 78% of non-tumor liver tissues expressed high levels (+2 and +3) of DSE, whereas DSE remained highly expressed in only 27% of HCC tumors (Mann-Whitney U Test, P = 0.004; Figure 1D and 1E). We found that decreased DSE expression was correlated with advanced tumor stage (Fisher exact test, P = 0.0032) and metastasis (Fisher exact test, P = 0.0223) of HCC tumors (Table 1). A Kaplan-Meier survival analysis showed that the survival rate of patients with HCC with low DSE expression was significantly lower than those with high DSE expression. (log-rank test, P = 0.0153; Figure 1F). Collectively, these data suggest that DSE is frequently down-regulated in HCC, and its down-regulation is associated with advanced tumor stage, metastasis, and poor survival in HCC patients.
Figure 1.
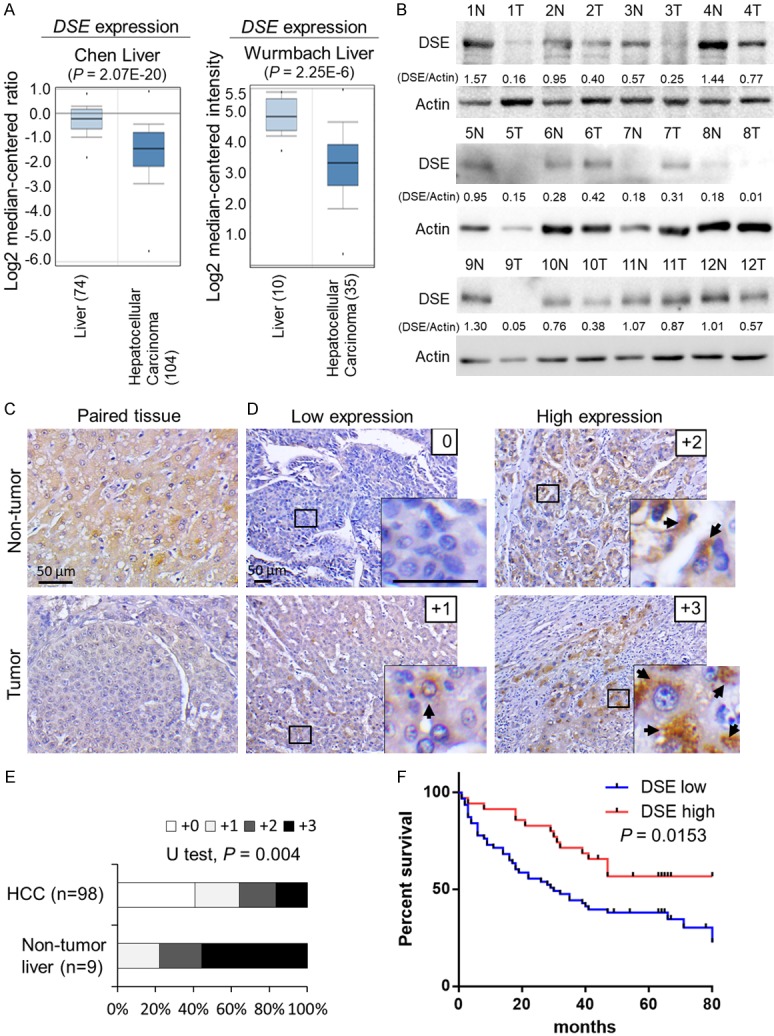
DSE is frequently down-regulated in human HCC and associated with poor overall survival. A. Expression of DSE in the ONCOMINE cancer microarray database. Two independent datasets showed that DSE gene expression is significantly down-regulated in HCC tissue, compared to normal liver tissue. B. Protein expression of DSE in paired HCC tissue. Western blots of DSE using paired non-tumor (N) and HCC tumor tissue (T). Twelve paired samples were tested, and Actin was taken as loading control. Relative quantities are shown. C. Immunohistochemistry of DSE on paired HCC tissue. The staining was visualized in brown color with a 3,3-diaminobenzidine liquid substrate system, and all sections were counterstained with hematoxylin. Representative images of adjacent non-tumor liver (upper) and HCC tumor area (bottom) are shown. Scale bars, 50 μm. D. Intensity of DSE staining on a tissue array comprising 98 primary HCC samples and 9 non-tumor tissue samples. Amplified images are shown at the bottom right. Arrows indicate positive stained HCC cells. Scale bars, 50 μm. E. Statistical analysis of immunohistochemistry in HCC tissue array. Mann-Whitney U Test was used, P = 0.004. F. Kaplan-Meier analysis of overall survival for HCC patients. The analyses were conducted according to the immunohistochemistry of DSE on tissue array. Probability of overall survival was analyzed according supplier’s information. Log-rank test, P = 0.0153.
Table 1.
Correlation of DSE expression with clinicopathological features of HCC tissue array
| Factor | DSE expression | P value (Two-sided Fisher’s exact test) | ||
|---|---|---|---|---|
|
| ||||
| Low | High | |||
| Tissue types | Non-tumor | 2 | 7 | 0.0268* |
| Tumor | 63 | 35 | ||
| Sex | Male | 52 | 24 | 0.1336 |
| Female | 11 | 11 | ||
| Age | < 55 years | 33 | 13 | 0.2050 |
| ≥ 55 years | 30 | 22 | ||
| Histology grade | Well | 10 | 8 | 0.4230 |
| Moderately and pooly | 53 | 27 | ||
| Tumor stage | T1 + T2 | 25 | 25 | 0.0032* |
| T3 + T3 | 38 | 10 | ||
| Metastasis | No | 45 | 32 | 0.0223* |
| Yes | 18 | 3 | ||
P < 0.05 was considered as statistically significant.
DSE suppresses tumor growth in vitro and in vivo
By measuring DSE expression in liver tissue and HCC cell lines by western blotting, we found that HA59T and HA22T expressed DSE, while it was not detectable in HepG2, HCC36, and Hepa1-6 cells (Figure 2A). Because Hepa1-6 cells are tumorigenic in mice, we re-expressed DSE in this cell line for further experiments (Figure 2B). We found that DSE suppressed cell viability in vitro (Figure 2C). To analyze the effect of DSE on tumor growth in vivo, mock and DSE-expressed Hepa1-6 clones were subcutaneously injected to SCID mice. The results showed that overexpression of DSE significantly suppressed the volume of tumors in immunodeficient mice. We further measured tumor growth in immune complement C57BL/6 mice. Interestedly, the volume of DSE-expressed tumors started to decrease from day 10, and 57% (4/7) of tumors had completely disappeared by day 19 (Figure 2D and 2E). These results suggest that DSE inhibited HCC cell growth in vitro and in vivo, and the immune-related response may strengthen this suppressive effect.
Figure 2.
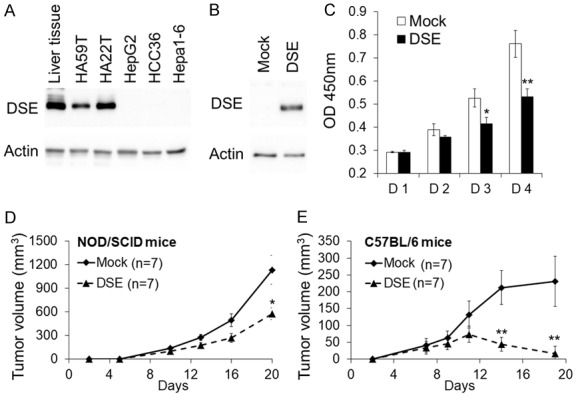
Overexpression of DSE suppresses HCC cell growth in vitro and in vivo. (A) Expression of DSE in HCC cell lines liver tissue. Protein expression was analyzed by western blotting. Actin was used as an internal control. (B) Stable overexpressed DSE in Hepa1-6 cells. Cells were stably transfected with empty vectors (mock) or DSE-expressing plasmids (DSE). G418 selected clones were pooled and the DSE expression were analyzed by western blotting. (C) DSE modulates cell viability in vitro. The cell viability of Hepa1-6 stable clones was measured using a CCK-8 assay at the indicted time-points. Data represent means ± SD from three independent experiments. *P < 0.05; **P < 0.01. DSE suppressed tumor growth in NOD/SCID mice (D) and C57BL/6 mice (E). Hepa1-6 transfectants were subcutaneously injected to mice. The size of the tumors was measured at the indicated time-points. Data is represented as the mean ± SD. *P < 0.05; **P < 0.01, n = 7 for each group.
DSE decreases CCL5 accumulation in mice tumor tissue
We next analyzed different populations of infiltrated immune cells in tumor tissue 10 days after cell injection by multicolor flow cytometry (Figure 3A). We found that populations of leucocytes are not altered in DSE-overexpressed tumors. The percentages of NK cells and NKT cells were significantly decreased in DSE-overexpressed tumors, which contrasted with the decreases in tumor volume. In addition, we found that regulatory T cells (Treg) were slightly decreased in DSE tumors, but neither Th cells nor Tc cells were correspondingly increased (Figure 3B). Therefore, we suppose that changes to the population of infiltrated immune cells may not be directly responsible for DSE-mediated growth suppression.
Figure 3.

Analysis of tumor infiltrated immune cells and cytokines in C57BL/6 mice tumor model. (A) Representative gating strategy for identifying different cell population in tumor tissue. (B) Percentage of tumor infiltrated immune cell population 10 days after cell transplantation. Data is represented as the mean ± SD. **P < 0.01, n = 7 for each group. (C) Expression of cytokines in transplanted tumor tissue, and (D) expression of CCL2, CCL3, CCL4, CCL5, CCL11, and CXCL1 in transplanted tumor tissue. **P < 0.01, n = 5 for each group. (E) Identifying CCL5 expressing cells in tumor tissue. Flow cytometry plots staining for CCL5 PE in mock or DSE Hepa1-6 tumors, gated on live CD45+/CD11b-/Gr1low cells. Iso-type PE was taken as negative stain control. Representative data was shown from 5 mice for each group. (F) Percentage of CCL5+ immune cell population in tumor tissue 10 days after transplantation. Data is represented as the mean ± SD. *P < 0.05, n = 5 for each group.
Aberrant secretion of cytokines and chemokines may promote HCC cell progression, and accumulating evidence suggests that CS/DS chains can modulate binding of these factors to the cell surface. Thus, we measured the levels of certain important inflammatory factors, cytokines, and chemokines in dissected tumor tissue by multiplex immunoassay. Our data revealed that IL-1β and CCL5 were significantly decreased in DSE-overexpressed tumors, while no differences were found in other factors (Figures 3C, 3D and S1). Flow cytometry was used to identify CCL5-producing cells in the tumor tissues. We found that CCL5 was mainly expressed by tumor infiltrated NK cells and Tc cells (Figures 3E and S2). Additionally, population of CCL5-expressing NK cells and Tc cells were decreased in DSE overexpressed tumor tissue (Figure 3F).
DSE modulates CCL5-induced phenotypes in HCC cells
Due to the known influence of CCL5 on the carcinogenesis of many types of cancer [26-28], and its known interactions with CS/DS chains [29,30], we next investigated whether CCL5 is involved in DSE-regulated phenotypes. CCK8 assay revealed that cell viability was dramatically enhanced by CCL5 in Hepa1-6 cells, while overexpression of DSE attenuated the effects of CCL5 (Figure 4A). In contrast, in HA22T cells, where endogenous expression of DSE is high, CCL5 only slightly increased cell viability in control siRNA-transfected cells. Knockdown of DSE by siRNA significantly enhanced CCL5-induced cell viability in this cell line (Figure 4B). In addition, overexpression of DSE inhibited CCL5-induced cell migration and invasion, while knockdown of DSE significantly enhanced CCL5-induced cell migration and invasion (Figure 4C and 4D). These data suggest that CCL5 may have a direct effect on HCC cells, and expression of DSE can regulate CCL5-induced phenotypes.
Figure 4.
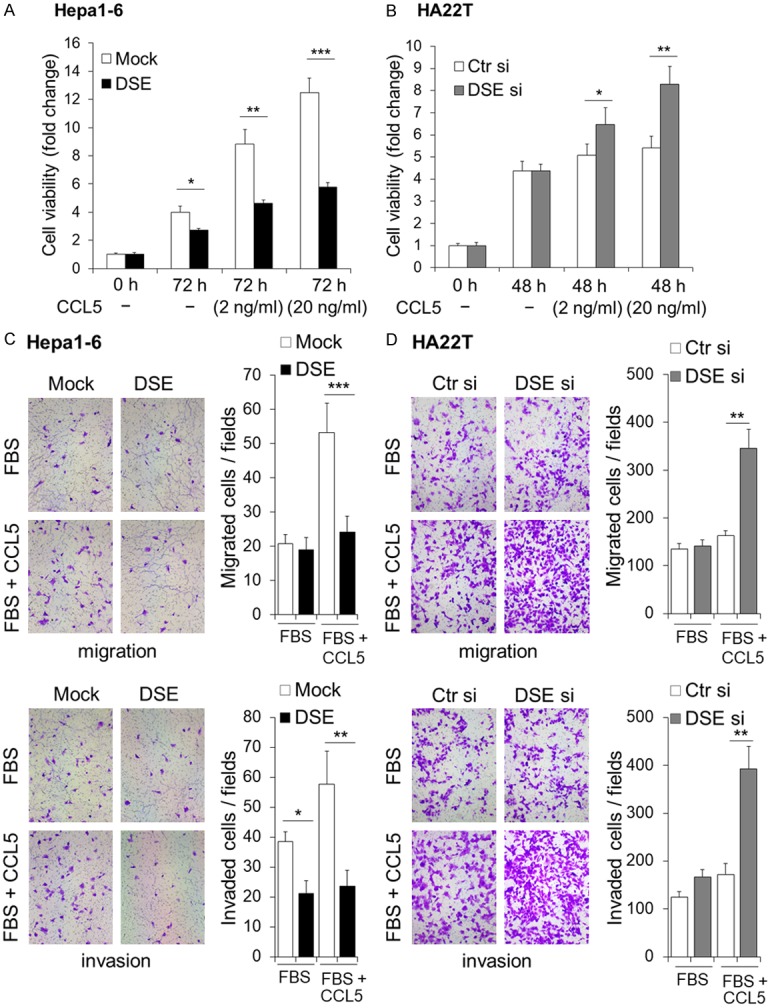
DSE modulates CCL5-induced malignant phenotypes in HCC cells. (A) Overexpression of DSE decreases CCL5-induced cell viability in Hepa1-6 cells. (B) Knockdown of DSE enhances CCL5-triggered cell viability in HA22T cells. Cells were culture with 5% FBS with or without CCL5 for indicated duration. Cell viability was determined using a CCK-8 assay. Fold changes of 450 nm absorbance were shown. Data is represented as the means ± SD from three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001. (C and D) DSE regulates CCL5-induced migration and invasion of HCC cells. Mock and DSE overexpressed hepa1-6 cells (C), and control-siRNA (Ctr si) as well as DSE-siRNA (DSE si) cells (D) were subjected to transwell migration assay and Matrigel invasion assay. Representative images are shown at left. Statistic results are represented as means ± SD from three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001.
DSE regulates CCL5 signaling and binding in HCC cells through CCR1
We further investigated CCL5-triggered downstream signaling in HCC cells. Results of western blots indicate that overexpression of DSE suppressed CCL5-induced phosphorylation of AKT and P38 in Hepa1-6 cells (Figure 5A). In contrast, knockdown of DSE enhanced CCL5-induced activation of AKT and P38 in HA22T cells (Figure 5B).
Figure 5.

DSE modulates CCL5-induced signaling and binding in HCC cells. A. Overexpression of DSE decreases CCL5-induced cell signaling in Hepa1-6 cells. B. Knockdown of DSE enhances CCL5-triggered cell signaling in HA22T cells. Cultured cells were serum free starved for 3 hours and then treated with or without (-) CCL5 (50 ng/mL) for indicated time points. Cell lysates (20 μg) were analyzed by western blotting with various antibodies, as indicated. Actin was using as loading control. Representative blots were shown at left. Relative signals were quantified by Image J, and represented as means ± SD from three independent experiments. *P < 0.05; **P < 0.01. C. CCR1 expresses on Hepa1-6 and HA22T cell surface. Transfected cells were stained by CCR1 and anti-rabbit IgG-Alexa488. Non-specific rabbit IgG with anti-rabbit IgG-Alexa488 () was used as iso-type control. D. DSE mediates CCL5 binding on HCC cells. Transfected cells were treated with or without (-) CCL5 (50 ng/mL) for indicated time points. Cells were washed once with PBS and then lysis for western blots with indicated antibodies. Actin was using as loading control. E. Confocal microscopy analysis of CCL5 subcellular localization on HA22T cells. Recombinant CCL5 were treated to control and DSE knockdowned cells for 15 minutes. Cells were stained with CCL5 (red) and nuclei were counter stain with DAPI (blue). Representative images are shown. Scale bars, 20 μm. F. Knockdown of DSE increases CS on HA22T cells. Flow cytometry with anti-CS56 antibody was used. Non-specific mouse IgM was used as iso-type control. G. Enzyme digestion of CS/DS decreases CCL5 binding on HCC cells. Control and DSE knockdowned cells were incubated with or without chondroitinase ABC (ChasABC, 0.5 units/ml) for 3 hours in serum free DMEM. CCL5 (50 ng/mL) were treated for 15 minutes and analyzed by Western blots.
Through DSE modification of cellular CS/DS chains, it could possibly influenced CCL5 binding to cells. We next examined whether DSE regulated CCL5 binding by western blots and confocal microscopy, and found that endogenous CCL5 expression is undetectable by western blots in cultured HCC cells without treatment. After adding CCL5 and a brief wash with PBS, DSE-overexpressed cells revealed lower of CCL5 levels than mock cells; whereas knockdown of DSE increased CCL5 levels in HA22T cells (Figure 5D). In addition, immunostaining of CCL5 revealed that knockdown of DSE facilitated CCL5 accumulation in the cytoplasm (Figure 5E). Moreover, immunostaining of mouse tumor sections showed that CCL5 accumulated in tumor tissue and the extracellular matrix in mock tumor tissue, while CCL5 was decreased in DSE-overexpressed tumor tissue (Figure S3). To prove CS/DS chains were involved in DSE-mediated CCL5 accumulation in HCC cells, we first analyzed cell surface CS changes on HCC cells. Flow cytometry with CS-56 antibody revealed that knockdown of DSE increased cell surface CS (Figure 5F). Chondroitinase ABC was treated in cultured cells to degrade CS/DS chains, from which results showed that enzyme digestion of CS/DS decreased CCL5 accumulation caused by knockdown of DSE in HCC cells (Figure 5G).
CCL5 activity is mediated through its binding to CCR5, CCR3, and CCR1; while only CCR1 was constitutively expressed in HCC cells, and associated with worse prognosis in HCC patients [31-33]. Our data reveal that CCR1 was also expressed on surface of Hepa1-6 cells and HA22T cells (Figure 5C), and its expression decreased with CCL5 stimulation (Figure 5D). Blocking CCL5 inhibited CCL5-promoted cell viability in DSE knockdowned cells (Figure 6A). Importantly, using a CCR1 antagonist, BX471, CCL5-enhanced cell viability, and cell migration were successfully suppressed in DSE knockdowned cells (Figure 6B and 6C). Additionally, both BX471 and chondroitinase ABC can suppress CCL5-induced AKT and P38 activation in DSE-knockdown cells (Figure 6D). Collectively, these data suggest that DSE may regulate HCC cells receiving or accumulating CCL5, and modulate CCL5 signaling through CCR1 on HCC cells.
Figure 6.
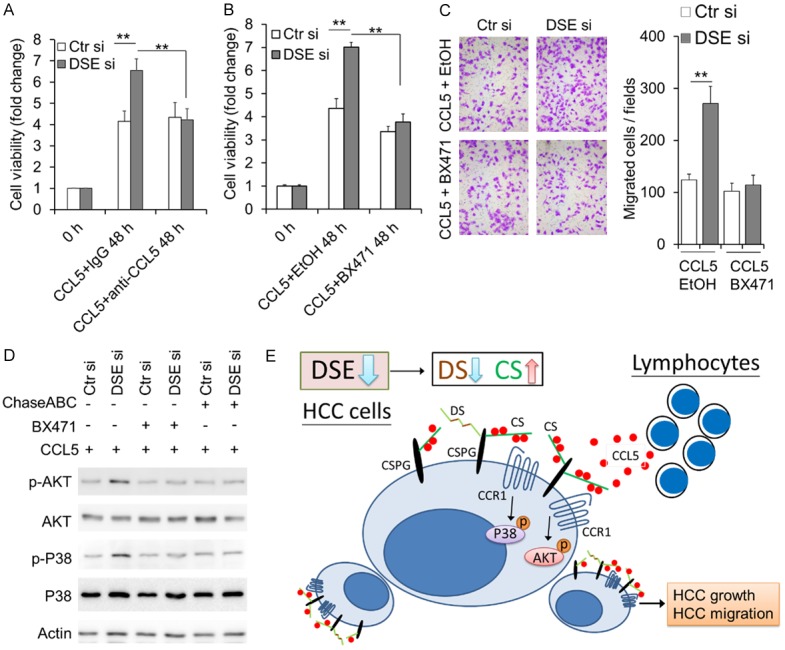
CCR1 antagonist suppresses DSE-mediated CCL5 phenotypes and signaling. A. Anti-CCL5 blocking antibody suppresses CCL5-enhanced cell viability in DSE-knockdowned cells. Control and DSE-knockdowned HA22T cells were cultured with CCL5 (20 ng/ml) for 48 hours. CCL5 antibody (2 µg/ml) or non-specific IgG (2 µg/ml) were used. Cell viability was determined using a CCK-8 assay. Fold changes of 450 nm absorbance were shown. Data is represented as the mean ± SD from three independent experiments. **P < 0.01. B. BX471 suppresses CCL5-enhanced cell viability in DSE-knockdowned cells. Control and DSE-knockdowned HA22T cells were cultured with CCL5 (20 ng/ml) for 48 hours. BX471 (20 μM) or 0.1% of ethanol (EtOH, solvent control) were added to culture medium. Cell viability was determined using a CCK-8 assay. Fold changes of 450 nm absorbance were shown. Data is represented as the mean ± SD from three independent experiments. **P < 0.01. C. BX471 suppresses CCL5-enhanced cell migration in DSE-knockdowned cells. BX471 (20 μM) or EtOH were added to culture medium in lower part of the inserts. D. BX471 and chondroitinase ABC (ChasABC) inhibit CCL5-induced signaling. Cells were preincubated with BX471 (20 μM) or ChaseABC (0.5 units/ml). CCL5 (50 ng/mL) were added to medium for 15 minutes and analyzed by western blotting with indicated antibodies. E. A proposed model illustrating down-regulation of DSE in HCC cells modulates CCL5 binding on HCC cells, P38 and AKT signaling, as well as HCC promotion.
Discussion
In this study, we demonstrated for the first time that DSE is frequently down-regulated in human HCC at both the mRNA and the protein level. Down-regulation of DSE is associated with advanced tumor stage, metastasis, and poor overall survival. Interestingly, re-expressed DSE in HCC cells suppressed cell growth, and the suppressive effects were augmented in the immune complement animal model. Although the changes in infiltrated immune cell populations may not directly explain this tumor suppressive effect, we suggest that the decrease of CCL5 in tumor tissue may be involved in DSE-regulated malignant phenotypes. Indeed, expression of DSE in HCC cells significantly modulated CCL5-induced cell growth, migration, and invasion. We propose that DSE-modulation of CS/DS could mediate HCC cells receiving CCL5, and consequently regulate CCL5-triggered signaling through CCR1 in HCC cells (Figure 6E).
DSE was found to be highly expressed in squamous cell carcinoma from different origins, and was therefore considered as a cancer cell marker, which was named SART2 (squamous cell carcinoma antigen recognized by T-cells 2) [34]. It was suggested to be a potential immunotherapeutic target for many types of cancer, including HCC. It has been reported that DSE peptide-specific cytotoxic T cells demonstrated cytotoxicity to HCC cells [34-36]. However, the therapeutic response of DSE peptide-specific cytotoxic T cells is only limited to certain patients [36,37]. Nevertheless, we searched a public database and found that average DSE gene expression was significantly decreased in HCC tissue in two independent cohorts, compared to its expression in normal liver. In addition, a paired HCC tissue study from our institute also revealed DSE proteins to be constantly expressed in non-tumor liver tissue, but frequently downregulated in HCC tissue. Moreover, IHC on another independent cohort of HCC tissue and non-tumor liver indicated that DSE was significantly down-regulated in HCC tissue. Although our data indicated that a subpopulation of HCC patients highly expressed DSE in cancer tissue, these patients with high DSE expression were associated with benign pathological features and better survival. Therefore, we suggest that it is important to pre-evaluate DSE expression in HCC patients and carefully monitor adverse side effects when using DSE as an immunotherapeutic target.
HCC is a well-known inflammation-induced cancer. The chronic inflammation microenvironment promotes carcinogenesis and progression of HCC [38]. Various chemokines recruit inflammatory cells to the tumor site, which is the main feature of the inflammatory tumor microenvironment. CCL5 is a crucial inflammatory mediator in HCC, which is mainly secreted from infiltrated lymphocytes and hepatic stellate cell [27,39]. In our animal model, multiplex immune assay revealed that CCL5 was significantly decreased in DSE-overexpressed tumor tissue. Decline of CCL5-producing NK cell and Tc cell populations may contribute to the decrease of CCL5. Although these potential antitumor cells decreased, tumor growth was dramatically suppressed in contrast.
Many studies indicate that these CC chemokines, such as CCL5 and CCL15, may not only mediate trafficking and activity of immune cells, but are also directly utilized for promoting malignant phenotypes of tumor cells [28,33,40,41]. Additionally, previous studies have demonstrated that the CC chemokine receptor, CCR1, is overexpressed on HCC cells, and its expression is associated with malignant cell behaviors that worsen prognoses [33,42,43]. As CCR1 is a receptor of CCL5, we thus investigated the direct influence of CCL5 on cancer cells, instead of its effects on immune cells. Our data reveal that CCR1 is expressed in our tested HCC cell lines, a result which is similar to previous studies showing that CCR1 is generally expressed in HCC cells [32,33]. Additionally, we provide evidence that overexpression of DSE suppressed CCL5-induced cell viability and mobility, while knockdown of DSE enhanced CCL5-triggered malignant phenotypes in cultured HCC cells. Moreover, CCR1 antagonist can suppress CCL5-induced malignant phenotypes and cellular signaling caused by DSE silencing in HCC cells. These results suggest that DSE regulated the protein level of CCL5 in the HCC tumor microenvironment, and modulated CCL5 derived effects on cancer cells through CCR1.
Both CS and DS have demonstrated affinity to CCL5. Previous studies indicate that chondroitin-4,6-sulfate and chondroitin-6-sulfate may have higher affinity to CCL5 than DS [29,44]. However, these studies used highly purified GAG fragments for analysis. Our cellular experiments reveal that knockdown of DSE caused CS accumulation on the cell surface, as well as enhanced CCL5 binding and cellular signaling. Meanwhile, overexpression of DSE decreased CCL5 binding and signaling, suggesting that changes in CS/DS composition could regulate the cellular response to CCL5 stimulation. It has been reported that enzyme degradation of cell surface CS/DS can diminish CCL5 binding to cells, and that blocking GAG formation can deplete CCL5 induced phenotypes in HCC cells [40,45]. In agreement with previous reports, our data indicate that enzyme digestion of CS/DS on HCC cells decreased both CCL5 binding and signaling, which was caused by knockdown of DSE in HCC cells. These data suggest that DSE is an important modulator for CCL5 signaling.
In conclusion, the results obtained in this study suggest that DSE could regulate CCL5/CCR1 signaling, which is involved in DSE-induced malignant behaviors of HCC cells. Although other unknown mechanisms may also be involved in DSE-mediated malignant phenotypes of HCC, this study proves that the CCL5/CCR1 axis has direct effects on tumor cells. This study not only shows a pathophysiological role of DSE in HCC cells, but also sheds light on the biological function of aberrant CS/DS expression in HCC progression.
Acknowledgements
Tissue FAX Plus Cytometer, immunofluorescence microscopy (ZEISS Axio Imager A2), and confocal microscopy (Zeiss LSM 510 META) were performed in the Instrument Center of Chung Shan Medical University, which is supported by Ministry of Science and Technology, Ministry of Education, and Chung Shan Medical University. This study was supported by the grants from the Ministry of Science and Technology, Taiwan, MOST-105-2320-B-040-033-MY2 and MOST-106-2320-B-040-009-MY3 (Chiung-Hui Liu), MOST-105-2320-B-040-029-MY3 (Wen-Chieh Liao), MOST-106-2320-B-040-028-MY2 (Chih-Kai Liao), and Chinese Medicine Research Center, China Medical University, from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan, CMRC-CHM-2 (Hung-Rong Yen).
Disclosure of conflict of interest
None.
Abbreviations
- HCC
hepatocellular carcinoma
- ECM
extracellular matrix
- GAGs
Glycosaminoglycans
- PG
proteoglycans
- CS
Chondroitin sulfate
- DS
Dermatan sulfate
Supporting Information
References
- 1.Jou JH, Muir AJ. Hepatocellular carcinoma surveillance. Clin Gastroenterol Hepatol. 2018;16:19–20. doi: 10.1016/j.cgh.2017.06.049. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Ward EM, Johnson CJ, Cronin KA, Ma J, Ryerson B, Mariotto A, Lake AJ, Wilson R, Sherman RL, Anderson RN, Henley SJ, Kohler BA, Penberthy L, Feuer EJ, Weir HK. Annual report to the nation on the status of cancer, 1975-2014, featuring survival. J Natl Cancer Inst. 2017;109 doi: 10.1093/jnci/djx030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Forner A, Llovet JM, Bruix J. Hepatocellular carcinoma. Lancet. 2012;379:1245–1255. doi: 10.1016/S0140-6736(11)61347-0. [DOI] [PubMed] [Google Scholar]
- 4.Worns MA, Galle PR. HCC therapies-lessons learned. Nat Rev Gastroenterol Hepatol. 2014;11:447–52. doi: 10.1038/nrgastro.2014.10. [DOI] [PubMed] [Google Scholar]
- 5.Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, Gerolami R, Masi G, Ross PJ, Song T, Bronowicki JP, Ollivier-Hourmand I, Kudo M, Cheng AL, Llovet JM, Finn RS, LeBerre MA, Baumhauer A, Meinhardt G, Han G RESORCE Investigators. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56–66. doi: 10.1016/S0140-6736(16)32453-9. [DOI] [PubMed] [Google Scholar]
- 6.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, Kim TY, Choo SP, Trojan J, Welling THR, Meyer T, Kang YK, Yeo W, Chopra A, Anderson J, Dela Cruz C, Lang L, Neely J, Tang H, Dastani HB, Melero I. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet. 2017;389:2492–2502. doi: 10.1016/S0140-6736(17)31046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jia XL, Li SY, Dang SS, Cheng YA, Zhang X, Wang WJ, Hughes CE, Caterson B. Increased expression of chondroitin sulphate proteoglycans in rat hepatocellular carcinoma tissues. World J Gastroenterol. 2012;18:3962–3976. doi: 10.3748/wjg.v18.i30.3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lv H, Yu G, Sun L, Zhang Z, Zhao X, Chai W. Elevate level of glycosaminoglycans and altered sulfation pattern of chondroitin sulfate are associated with differentiation status and histological type of human primary hepatic carcinoma. Oncology. 2007;72:347–356. doi: 10.1159/000113145. [DOI] [PubMed] [Google Scholar]
- 9.Salanti A, Clausen TM, Agerbaek MO, Al Nakouzi N, Dahlback M, Oo HZ, Lee S, Gustavsson T, Rich JR, Hedberg BJ, Mao Y, Barington L, Pereira MA, LoBello J, Endo M, Fazli L, Soden J, Wang CK, Sander AF, Dagil R, Thrane S, Holst PJ, Meng L, Favero F, Weiss GJ, Nielsen MA, Freeth J, Nielsen TO, Zaia J, Tran NL, Trent J, Babcook JS, Theander TG, Sorensen PH, Daugaard M. Targeting human cancer by a glycosaminoglycan binding malaria protein. Cancer Cell. 2015;28:500–514. doi: 10.1016/j.ccell.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu CH, Lan CT, Chou JF, Tseng TJ, Liao WC. CHSY1 promotes aggressive phenotypes of hepatocellular carcinoma cells via activation of the hedgehog signaling pathway. Cancer Lett. 2017;403:280–288. doi: 10.1016/j.canlet.2017.06.023. [DOI] [PubMed] [Google Scholar]
- 11.Tykesson E, Hassinen A, Zielinska K, Thelin MA, Frati G, Ellervik U, Westergren-Thorsson G, Malmstrom A, Kellokumpu S, Maccarana M. Dermatan sulfate epimerase 1 and dermatan 4-O-sulfotransferase 1 form complexes that generate long epimerized 4-O-sulfated blocks. J Biol Chem. 2018;293:13725–13735. doi: 10.1074/jbc.RA118.003875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mizumoto S, Yamada S, Sugahara K. Human genetic disorders and knockout mice deficient in glycosaminoglycan. Biomed Res Int. 2014;2014:495764. doi: 10.1155/2014/495764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sugahara K, Mikami T, Uyama T, Mizuguchi S, Nomura K, Kitagawa H. Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr Opin Struct Biol. 2003;13:612–620. doi: 10.1016/j.sbi.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Yamada S, Sugahara K. Potential therapeutic application of chondroitin sulfate/dermatan sulfate. Curr Drug Discov Technol. 2008;5:289–301. doi: 10.2174/157016308786733564. [DOI] [PubMed] [Google Scholar]
- 15.Maimone MM, Tollefsen DM. Structure of a dermatan sulfate hexasaccharide that binds to heparin cofactor-ii with high-affinity. J Biol Chem. 1990;265:18263–18271. [PubMed] [Google Scholar]
- 16.Taylor KR, Rudisill JA, Gallo RL. Structural and sequence motifs in dermatan sulfate for promoting fibroblast growth factor-2 (FGF-2) and FGF-7 activity. J Biol Chem. 2005;280:5300–6. doi: 10.1074/jbc.M410412200. [DOI] [PubMed] [Google Scholar]
- 17.Sugahara K, Mikami T. Chondroitin/dermatan sulfate in the central nervous system. Curr Opin Struct Biol. 2007;17:536–545. doi: 10.1016/j.sbi.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 18.Muller T, Mizumoto S, Suresh I, Komatsu Y, Vodopiutz J, Dundar M, Straub V, Lingenhel A, Melmer A, Lechner S, Zschocke J, Sugahara K, Janecke AR. Loss of dermatan sulfate epimerase (DSE) function results in musculocontractural Ehlers-Danlos syndrome. Hum Mol Genet. 2013;22:3761–3772. doi: 10.1093/hmg/ddt227. [DOI] [PubMed] [Google Scholar]
- 19.Thelin MA, Svensson KJ, Shi XF, Bagher M, Axelsson J, Isinger-Ekstrand A, van Kuppevelt TH, Johansson J, Nilbert M, Zaia J, Belting M, Maccarana M, Malmstrom A. Dermatan sulfate is involved in the tumorigenic properties of esophagus squamous cell carcinoma. Cancer Res. 2012;72:1943–52. doi: 10.1158/0008-5472.CAN-11-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao WC, Liao CK, Tsai YH, Tseng TJ, Chuang LC, Lan CT, Chang HM, Liu CH. DSE promotes aggressive glioma cell phenotypes by enhancing HB-EGF/ErbB signaling. PLoS One. 2018;13:e0198364. doi: 10.1371/journal.pone.0198364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dmitrieva N, Yu L, Viapiano M, Cripe TP, Chiocca EA, Glorioso JC, Kaur B. Chondroitinase ABC I-mediated enhancement of oncolytic virus spread and antitumor efficacy. Clin Cancer Res. 2011;17:1362–1372. doi: 10.1158/1078-0432.CCR-10-2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaime-Ramirez AC, Dmitrieva N, Yoo JY, Banasavadi-Siddegowda Y, Zhang J, Relation T, Bolyard C, Wojton J, Kaur B. Humanized chondroitinase ABC sensitizes glioblastoma cells to temozolomide. J Gene Med. 2017;19 doi: 10.1002/jgm.2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denholm EM, Lin YQ, Silver PJ. Anti-tumor activities of chondroitinase AC and chondroitinase B: inhibition of angiogenesis, proliferation and invasion. Eur J Pharmacol. 2001;416:213–221. doi: 10.1016/s0014-2999(01)00884-6. [DOI] [PubMed] [Google Scholar]
- 24.Liu CH, Chang HM, Wu TH, Chen LY, Yang YS, Tseng TJ, Liao WC. Rearrangement of potassium ions and Kv1.1/Kv1.2 potassium channels in regenerating axons following end-to-end neurorrhaphy: ionic images from TOF-SIMS. Histochem Cell Biol. 2017;148:407–416. doi: 10.1007/s00418-017-1570-8. [DOI] [PubMed] [Google Scholar]
- 25.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 2004;6:1–6. doi: 10.1016/s1476-5586(04)80047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aldinucci D, Colombatti A. The inflammatory chemokine CCL5 and cancer progression. Mediators Inflamm. 2014;2014:292376. doi: 10.1155/2014/292376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mohs A, Kuttkat N, Reissing J, Zimmermann HW, Sonntag R, Proudfoot A, Youssef SA, de Bruin A, Cubero FJ, Trautwein C. Functional role of CCL5/RANTES for HCC progression during chronic liver disease. J Hepatol. 2017;66:743–753. doi: 10.1016/j.jhep.2016.12.011. [DOI] [PubMed] [Google Scholar]
- 28.Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, Suetterlin T, Brand K, Krauss J, Lasitschka F, Lerchl T, Luckner-Minden C, Ulrich A, Koch M, Weitz J, Schneider M, Buechler MW, Zitvogel L, Herrmann T, Benner A, Kunz C, Luecke S, Springfeld C, Grabe N, Falk CS, Jaeger D. Tumoral immune cell exploitation in colorectal cancer metastases can be targeted effectively by Anti-CCR5 therapy in cancer patients. Cancer Cell. 2016;29:587–601. doi: 10.1016/j.ccell.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 29.Martin L, Blanpain C, Garnier P, Wittamer V, Parmentier M, Vita C. Structural and functional analysis of the RANTES-glycosaminoglycans interactions. Biochemistry. 2001;40:6303–6318. doi: 10.1021/bi002670n. [DOI] [PubMed] [Google Scholar]
- 30.Deshauer C, Morgan AM, Ryan EO, Handel TM, Prestegard JH, Wang X. Interactions of the chemokine CCL5/RANTES with medium-sized chondroitin sulfate ligands. Structure. 2015;23:1066–1077. doi: 10.1016/j.str.2015.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Appay V, Rowland-Jones SL. RANTES: a versatile and controversial chemokine. Trends Immunol. 2001;22:83–87. doi: 10.1016/s1471-4906(00)01812-3. [DOI] [PubMed] [Google Scholar]
- 32.Lu P, Nakamoto Y, Nemoto-Sasaki Y, Fujii C, Wang H, Hashii M, Ohmoto Y, Kaneko S, Kobayashi K, Mukaida N. Potential interaction between CCR1 and its ligand, CCL3, induced by endogenously produced interleukin-1 in human hepatomas. Am J Pathol. 2003;162:1249–1258. doi: 10.1016/S0002-9440(10)63921-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu Y, Gao XM, Yang J, Xu D, Zhang Y, Lu M, Zhang Z, Sheng YY, Li JH, Yu XX, Zheng Y, Dong QZ, Qin LX. C-C chemokine receptor type 1 mediates osteopontin-promoted metastasis in hepatocellular carcinoma. Cancer Sci. 2018;109:710–723. doi: 10.1111/cas.13487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakao M, Shichijo S, Imaizumi T, Inoue Y, Matsunaga K, Yamada A, Kikuchi M, Tsuda N, Ohta K, Takamori S, Yamana H, Fujita H, Itoh K. Identification of a gene coding for a new squamous cell carcinoma antigen recognized by the CTL. J Immunol. 2000;164:2565–2574. doi: 10.4049/jimmunol.164.5.2565. [DOI] [PubMed] [Google Scholar]
- 35.Noguchi M, Yao A, Harada M, Nakashima O, Komohara Y, Yamada S, Itoh K, Matsuoka K. Immunological evaluation of neoadjuvant peptide vaccination before radical prostatectomy for patients with localized prostate cancer. Prostate. 2007;67:933–942. doi: 10.1002/pros.20572. [DOI] [PubMed] [Google Scholar]
- 36.Mizukoshi E, Nakamoto Y, Arai K, Yamashita T, Sakai A, Sakai Y, Kagaya T, Yamashita T, Honda M, Kaneko S. Comparative analysis of various tumor-associated antigen-specific t-cell responses in patients with hepatocellular carcinoma. Hepatology. 2011;53:1206–1216. doi: 10.1002/hep.24149. [DOI] [PubMed] [Google Scholar]
- 37.Mizukoshi E, Fushimi K, Arai K, Yamashita T, Honda M, Kaneko S. Expression of chondroitin-glucuronate C5-epimerase and cellular immune responses in patients with hepatocellular carcinoma. Liver Int. 2012;32:1516–1526. doi: 10.1111/j.1478-3231.2012.02853.x. [DOI] [PubMed] [Google Scholar]
- 38.Giannelli G, Rani B, Dituri F, Cao Y, Palasciano G. Moving towards personalised therapy in patients with hepatocellular carcinoma: the role of the microenvironment. Gut. 2014;63:1668–1676. doi: 10.1136/gutjnl-2014-307323. [DOI] [PubMed] [Google Scholar]
- 39.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sutton A, Friand V, Papy-Garcia D, Dagouassat M, Martin L, Vassy R, Haddad O, Sainte-Catherine O, Kraemer M, Saffar L, Perret GY, Courty J, Gattegno L, Charnaux N. Glycosaminoglycans and their synthetic mimetics inhibit RANTES-induced migration and invasion of human hepatoma cells. Mol Cancer Ther. 2007;6:2948–2958. doi: 10.1158/1535-7163.MCT-07-0114. [DOI] [PubMed] [Google Scholar]
- 41.Liu LZ, Zhang Z, Zheng BH, Shi Y, Duan M, Ma LJ, Wang ZC, Dong LQ, Dong PP, Shi JY, Zhang S, Ding ZB, Ke AW, Cao Y, Zhang XM, Xi R, Zhou J, Fan J, Wang XY, Gao Q. CCL15 recruits suppressive monocytes to facilitate immune escape and disease progression in hepatocellular carcinoma. Hepatology. 2019;69:143–159. doi: 10.1002/hep.30134. [DOI] [PubMed] [Google Scholar]
- 42.Yang X, Lu P, Fujii C, Nakamoto Y, Gao JL, Kaneko S, Murphy PM, Mukaida N. Essential contribution of a chemokine, CCL3, and its receptor, CCR1, to hepatocellular carcinoma progression. Int J Cancer. 2006;118:1869–1876. doi: 10.1002/ijc.21596. [DOI] [PubMed] [Google Scholar]
- 43.Wu X, Fan J, Wang X, Zhou J, Qiu S, Yu Y, Liu Y, Tang Z. Downregulation of CCR1 inhibits human hepatocellular carcinoma cell invasion. Biochem Biophys Res Commun. 2007;355:866–871. doi: 10.1016/j.bbrc.2007.01.199. [DOI] [PubMed] [Google Scholar]
- 44.Mizumoto S, Fongmoon D, Sugahara K. Interaction of chondroitin sulfate and dermatan sulfate from various biological sources with heparin-binding growth factors and cytokines. Glycoconj J. 2013;30:619–632. doi: 10.1007/s10719-012-9463-5. [DOI] [PubMed] [Google Scholar]
- 45.Murooka TT, Wong MM, Rahbar R, Majchrzak-Kita B, Proudfoot AE, Fish EN. CCL5-CCR5-mediated apoptosis in T cells: requirement for glycosaminoglycan binding and CCL5 aggregation. J Biol Chem. 2006;281:25184–25194. doi: 10.1074/jbc.M603912200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.