

Abstract
P-glycoprotein (P-gp) is a multidrug transporter, which harnesses the chemical energy of ATP to power the efflux of diverse chemotherapeutics out of cells and thus contributes to the development of multidrug resistance (MDR) in cancer. It has been proved that the ligand-binding pocket of P-gp is located at the transmembrane domains (TMDs). However, the access of ligands into the binding pocket remains to be elucidated, which definitely hinder the development of P-gp inhibitors. Herein, the access pathways of a well-known substrate rhodamine-123 and a cyclopeptide inhibitor QZ-Leu were characterized by time-independent partial nudged elastic band (PNEB) simulations. The decreasing free energies along the PNEB-optimized access pathway indicated that TM4/6 cleft may be an energetically favorable entrance gate for ligand entry into the binding pocket of P-gp. The results can be reconciled with a range of experimental studies, further corroborating the reliability of the gate revealed by computational simulations. Our atomic level description of the ligand access pathway provides valuable insights into the gating mechanism for drug uptake and transport by P-gp and other multidrug transporters.
Keywords: P-glycoprotein, Multidrug resistance, Entrance gate, Partial nudged elastic band, Access pathway
Abbreviations: P-gp, P-Glycoprotein; MDR, Multidrug resistance; TMDs, Transmembrane domains; PNEB, Partial nudged elastic band; ABC, ATP-Binding cassette; QSAR, Quantitative structure-activity relationship; MD, Molecular dynamics; RMSDs, Root-mean-square deviations
Graphical Abstract

Highlights
-
•
P-gp contributes to the development of multidrug resistance in cancer.
-
•
The entrance of drugs into P-gp binding pocket has yet to be elucidated.
-
•
An energetically favorable entrance gate was revealed by PNEB simulations.
-
•
The computational results were reconciled with the experimental data.
-
•
The atomic simulations provide insights into the gating mechanism of P-gp.
Multidrug resistance (MDR) is one of the major clinical impediments in cancer chemotherapy, wherein cells are resistant to the cytotoxic effect of multiple chemotherapeutic drugs. The predominant mechanism of MDR is the drug efflux promoted by the overexpression of membrane-embedded ATP-binding cassette (ABC) multidrug transporters [1]. P-glycoprotein (P-gp), also known as ABCB1 or MDR1, is one of the best characterized multidrug transporters. P-gp can confer MDR to cancer cells by catalyzing efflux of a broad spectrum of chemically and structurally unrelated anticancer drugs in an ATP-dependent manner [2,3]. Therefore, inhibition of drug efflux by P-gp is considered a feasible approach to reverse MDR in cancer treatment. Thus far, a number of compounds have been shown the abilities to inhibit drug efflux of P-gp by in vitro and in vivo experiments [4]. However, the development of effective inhibitors of P-gp and other multidrug transporters for clinical practice has been hampered, largely due to the unsolved molecular mechanism of drug entrance and binding.
The binding nature of P-gp has been extensively investigated since it was discovered in 1976 by Juliano and Ling [5]. Competitive and non-competitive assays have shown that P-gp can bind more than one compounds simultaneously [[6], [7], [8], [9], [10], [11]], which indicates the existence of multiple binding sites, e.g. the H-site (Hoechst-33342 binding site) and R-site (Rhodamine-123 binding site) proposed by Shapiro and Ling [12]. Extensive mutation studies demonstrated that a list of hydrophobic and aromatic residues at the transmembrane domains (TMDs) were essential for substrate binding and transportation [9,[13], [14], [15], [16], [17], [18], [19], [20], [21]]. Accordingly, an intrinsically large, flexible and hydrophobic pocket harboring multiple binding sites was proposed to account for substrate promiscuity and poly-specificity of P-gp [22,23].
In 2009, the first crystal structure of murine P-gp showed a large pocket of approximately 6000 Å3 in TMDs [24]. Three distinct but overlapping ligand-binding sites were identified from the location of co-crystallized QZ59-SSS and QZ59-RRR [[24], [25], [26]]. However, the original structures with lower resolution (4.4 and 4.35 Å) contained registry shifts in TMDs that limited the interpretation of drug interactions with P-gp. Most recently, many higher resolution (3.4–3.8 Å) structures of P-gp in complex with a series of cyclopeptide ligands (QZ-Ala, QZ-Val, QZ-Leu, and QZ-Phe) have been released, which revealed more binding sites with shared residues dispersed in the hydrophobic pocket [27,28]. Additionally, the binding profiles of P-gp substrates and inhibitors were also investigated by ligand-based quantitative structure-activity relationship (QSAR) and receptor-based molecular docking techniques in our previous researches [29,30]. Despite great advances in the characterization of binding sites and binding profiles of P-gp, the access pathway for the binding of ligands remains elusive.
The lipid membrane plays an important regulation role in the binding of ligands to P-gp [31]. Most P-gp substrates are hydrophobic and consequently concentrated in the membrane. So, it is widely accepted that P-gp is likely to function as a flippase [32] or hydrophobic vacuum cleaner [33] to take up hydrophobic molecules directly from the inner leaflet of the lipid bilayer [34,35]. The higher drug sensitivity of P-gp in the membrane than that in detergents also supports this point [36]. Recently, Xu et al. demonstrated that the binding of P-gp substrates is a membrane-mediated process, starting with a lipid-water partitioning step followed by a transporter binding step in the lipid membrane [37]. For that matter, at least one intramembranous entrance gate is indispensable for ligand entry into the binding pocket of P-gp. Structural inspection of P-gp suggests that the ligand-binding pocket is laterally accessible to the inner leaflet of the lipid bilayer through two clefts or gates formed by two pairs of transmembrane helices (TM4/6 and TM10/12), respectively. Unfortunately, there is still no direct experimental proof to support this view.
Intriguingly, a prominent conformational difference between TM4/6 and TM10/12 clefts was observed in the substantially improved structure of apo P-gp with the highest resolution (PDB code: 4Q9H) so far. In comparison with TM10/12 cleft, the TM4/6 cleft seems more accessible to the inner leaflet of the membrane (Fig. S1). Szewczyk et al. also suggested that TM4/6 cleft may be preferable for the uptake of molecules owing to the flexibility of TM4 [27]. Thus, it is assumed that the relatively open TM4/6 cleft is an entrance gate for the access of ligands into the binding pocket from the lipid membrane.
To test the hypothesis above, in this paper, the access pathways of a well-known substrate rhodamine-123 and a cyclopeptide inhibitor QZ-Leu were investigated by molecular dynamics (MD) simulations in explicit lipid bilayer and water. Herein, the time-independent partial nudged elastic band (PNEB) method [38] implementation in Amber12 [39] was used to generate the access pathway from the lipid membrane to the binding pocket of P-gp. In PNEB, a predefined reaction coordinate is not required to guide the access processes, and that the pathways are continuous in all solute degrees of freedom. In each case, the equilibrated P-gp systems in ligand bound and unbound states were selected as two endpoint structures (Fig. S2). The initial access pathway was constructed by 8 copies of the unbound endpoint and 8 copies of the bound endpoint. The 16 structures were connected together by springs, and minimized simultaneously by a simulated annealing protocol (Table S1), with the two copies at endpoints fixed. Based on 20 independent PNEB simulations, the binding free energy profile along the PNEB-optimized pathway was calculated by using MM/GBSA (Molecular Mechanics/Generalized Born Surface Area) method. Structural preparation and computational details are available in Supporting Information.
The PNEB-optimized access pathways for the rhodamine-123 and QZ-Leu are shown in Fig. 1. The binding free energy profiles of rhodamine-123 and QZ-Leu are shown in Fig. 2. It can be seen that the pathway via TM4/6 cleft is energetically favorable for both of the ligands moving from the inner leaflet of the membrane to the binding pocket of P-gp. In general, the binding free energy of rhodamine-123 decreased progressively from the lipid membrane to the region of TM4/6 cleft, began to increased upon entering the binding pocket, and then dropped slightly until reaching its bound state (Fig. 2). The binding free energy of rhodamine-123 (−24.2 KJ·mol−1) registered in the binding site is similar to those of many cationic substrates (verapamil, moxifloxacin, and daunorubicin), which were determined by ATPase and surface activity measurements [37]. By contrast, the binding free energy of QZ-Leu showed a continuous decreasing tendency along the access pathway, which indicates a spontaneous binding process (Fig. 2).
Fig. 1.
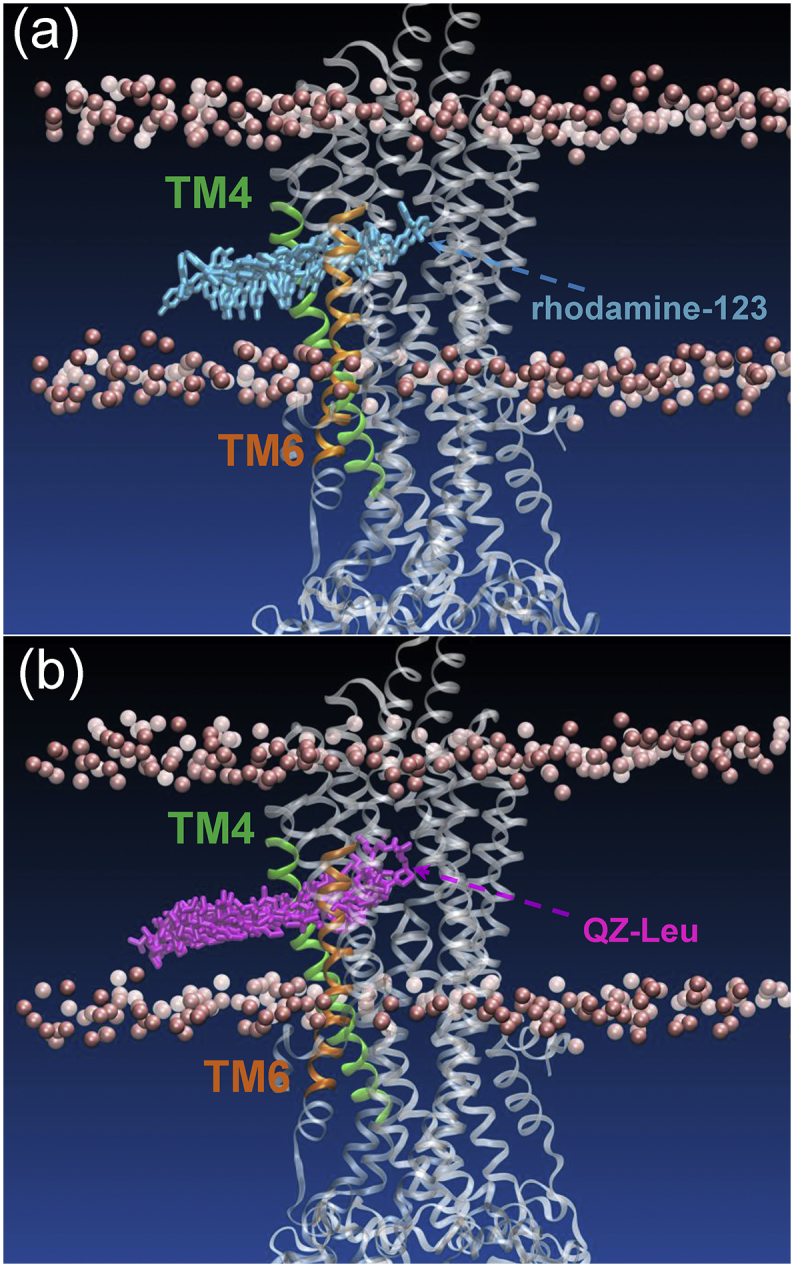
PNEB-optimized access pathways. The pathway is characterized by 16 snapshots of (a) rhodamine-123 and (b) QZ-Leu. TM4 is shown in green and TM6 in orange. The position of lipid membrane is indicated by phosphorus atoms in pink. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 2.

Free energy profiles of rhodamine-123 and QZ-Leu along the access pathways. The binding free energies were calculated based on 20 independent PNEB simulations. The region of TM4/6 cleft is indicated by gray shading, where the distance between the centroids of the ligand and residues in TM4/6 (Ala225 to His241, Ser345 to Gly356) was less than 6 Å.
From Fig. 2, it can be seen that when approaching TM4/6 cleft, the free energies of both ligands decreased significantly, and this tendency had been maintained during the whole TM4/6-gating process. That is to say, it is spontaneous for rhodamine-123 and QZ-Leu moving from the lipid membrane into the TM4/6 cleft.
Once away from the TM4/6 cleft to the binding pocket, rhodamine-123 was seemingly trapped in an energy well with a depth of −28.1 KJ·mol−1 (Fig. 2). The reason is largely due to the H-bond interactions and extensive hydrophobic interactions between rhodamine-123 and the residues of TM4, TM5 and TM6 (Fig. 3a). Xu et al. argued that H-bond interactions are important for the sensing and binding of P-gp substrates [37]. The residues lining the cleft, including Ser218 (TM4), Ile302, Tyr306 (TM5), Leu335, Ile336, Ala338 and Phe339 (TM6) have been shown experimentally to play an important role in the binding and transportation of P-gp substrates (rhodamine B, vinblastine, and verapamil) [40]. In addition, the location of this energy well overlaps with the putative R-site formed by TM4, TM5 and TM6 [[41], [42], [43], [44]]. So, it can be deduced that the R-site is accessible to the inner lipid leaflet via the TM4/6 cleft.
Fig. 3.
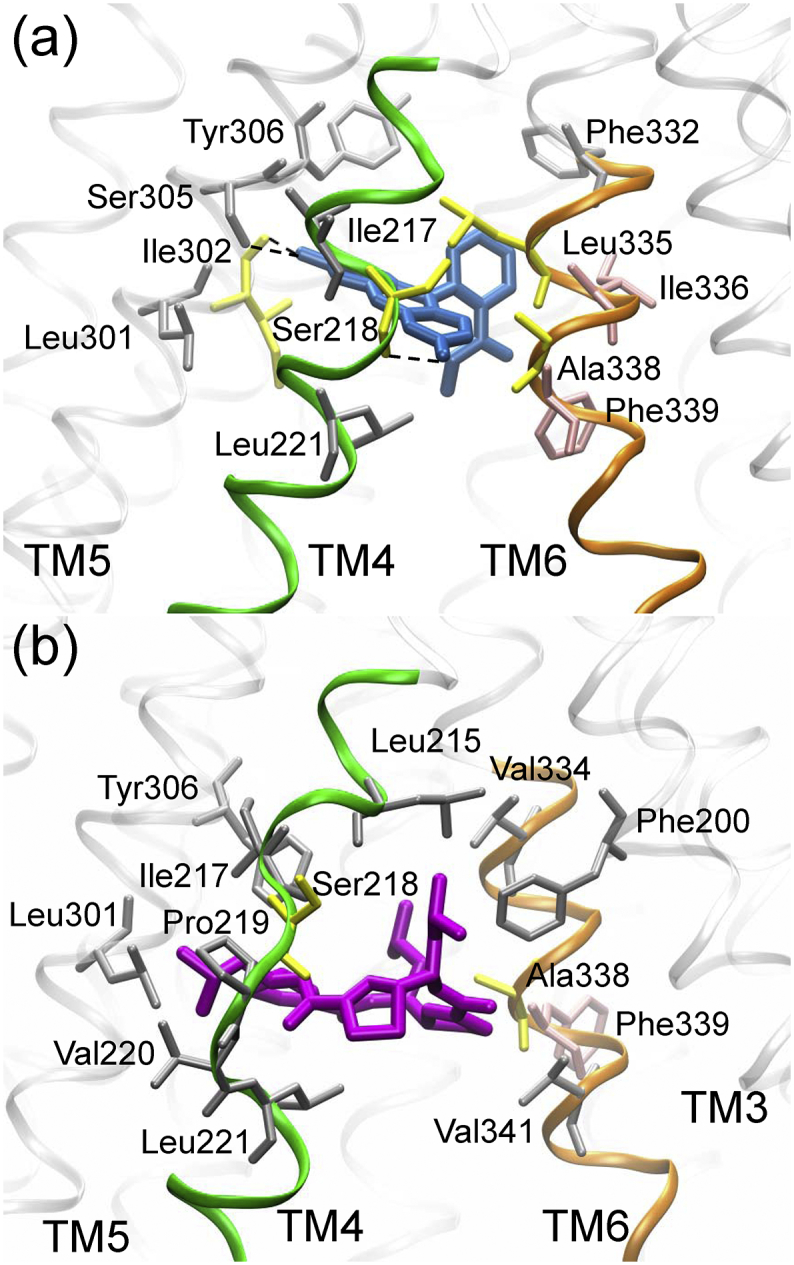
The interactions of (a) rhodamine-123 (blue) and (b) QZ-Leu (purple) with the residues in the region of TM4/6 cleft. The residues involved in the binding of rhodamine B and other substrates (vinblastine, verapamil) are shown in pink and yellow, respectively. Hydrogen-bonds are indicated by black dash lines. For clarity, hydrogen atoms are not shown. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
However, the binding free energy of rhodamine-123 tends to increase before reaching its binding site (Fig. 2). Recent researches have demonstrated a cooperativity between substrates and ATP in the process of P-gp transportation [[45], [46], [47]]. So, future researcher should pay adequate attentions to the influence of ATP on the substrate binding of P-gp.
In the case of QZ-Leu, the free energy had been decreased persistently since access into the TM4/6 cleft until reaching its binding site (Fig. 2), which indicates a spontaneous process for QZ-Leu binding. As expected, the relatively lower binding free energy of QZ-Leu (−52.2 KJ·mol−1) benefits to the occupation of substrate binding pocket [27]. Similar result was obtained for the inhibitor QZ59-RRR, which exhibited a lower free energy compared to the substrate daunorubicin registered in the binding pocket [48].
Fig. 4 shows the root-mean-square deviations (RMSDs) of TM4 and TM6 during the access processes of rhodamine-123 and QZ-Leu. It should be noted that the RMSD of TM4 increased significantly to a maximum (5.9 Å) upon QZ-Leu crossing TM4/TM6 cleft (Fig. 4b). However, no significant change in the RMSDs of TM6 was observed. The results indicate that TM4 may be of relatively larger flexibility in comparison with TM6. The available crystal structures of mouse and Cyanidioschyzon merolae P-gp [26,49] also showed a relatively weaker electron density of TM4 region. Mutation experiments demonstrated that the flexibility of TM4 is critical for substrate binding and transportation [49]. In this letter, the conformational plasticity of TM4 not only facilitated the entry of large QZ-Leu ligand into the cleft, but also fostered more hydrophobic interactions with the transmembrane domains (Fig. 3b), leading to a steep drop of the binding free energy (Fig. 2).
Fig. 4.

RMSDs of TM4 and TM6 in the binding process of (a) rhodamine-123 and (b) QZ-Leu derived from 20 independent PNEB simulations. The initial conformation of P-gp was used as a reference conformation for RMSD calculations. The region of TM4/6 cleft is indicated by gray shading.
Another interesting finding is that the benzyl group of Phe339 (TM6) rotated approximately 180 degrees during rhodamine-123 and QZ-Leu access into TM4/6 cleft (Fig. 5). This is reminiscent of the aromatic residues gating the access of ligands to the binding sites through the rotation of side chain and affording substrate selectivity in channel proteins and enzymes [50]. Early studies have also shown the important role of TM6 in determining substrate specificity of P-gp [51,52]. So, it can be speculated that Phe339 of TM6 may serve as an aromatic gate residue that contributes to the substrate specificity of P-gp.
Fig. 5.
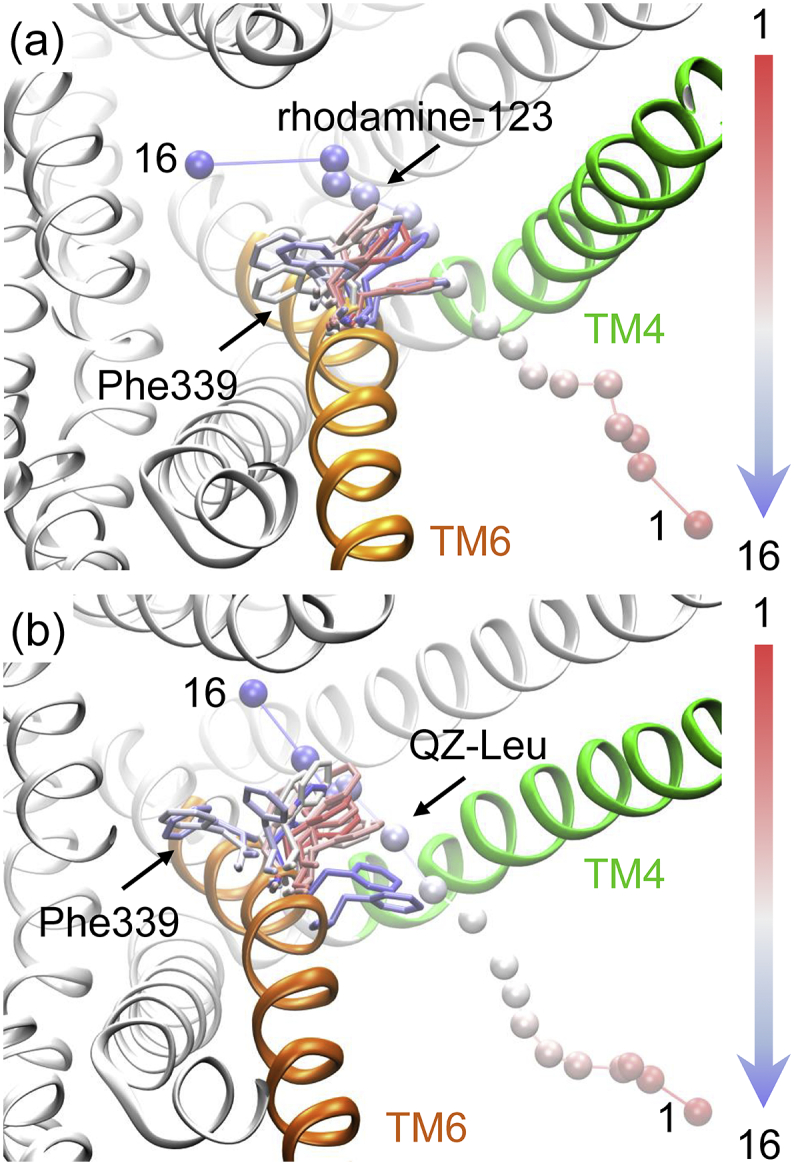
The rotation of benzyl group of Phe339 (TM6) during the access of (a) rhodamine-123 and (b) QZ-Leu into the TM4/6 cleft. The two ligands are represented as beads, and Phe339 is shown as stick.
In summary, the decreased free energy profiles of rhodamine-123 and QZ-Leu suggested that the access to the binding pocket of P-gp via TM4/6 cleft is energetically favorable for both ligands, and thus may be a spontaneous process. Besides, a significant conformational change of TM4 upon ligand passing through the TM4/6 cleft may provide a valuable insight into the flexibility of TM4, which has proved to be important for ligand binding and transportation. In addition, significant side-chain movements of Phe339 (TM6) was found during the access of rhodamine-123 and QZ-Leu. The function of Phe339, similar to a gating residue, need further experimental investigation. Overall, the results obtained in this letter support the flippase or hydrophobic vacuum cleaner model for P-gp transport.
In this paper, it is suggested that R-site substrates can enter into the binding pocket via TM4/6 cleft. It should be noted that our results cannot rule out other coexistent access pathways for P-gp substrates and inhibitors. Recently, Ferreira et al. have deduced that the H-site substrate colchicine can access into the binding pocket via TM10/12 cleft by using steered MD simulations [53]. Interestingly, the coexistence of two pseudo-symmetric translocation pathways in P-gp was recently confirmed by Parveen et al. [54], one of which was close to TM4/6 cleft and preferentially but non-exclusively taken by R-site substrates (e.g., rhodamine-123), whereas the other was close to TM10/12 cleft and taken by H-site substrates (e.g., propafenones). Together, these experimental and computational data provide a glimpse for the entrance gates, binding sites and translocation pathways in P-gp. However, the efflux mechanism of P-gp and other multidrug transporters is extremely complex and the exploration is still on the way.
Declaration of Interest
We confirm that there are no known conflicts of interest associated with this publication.
All named authors have read and approved this manuscript and that there are no other persons who satisfied the criteria for authorship but are not listed. The order of authors listed in the manuscript has been approved by all of us.
The manuscript is an original research paper, which has not been published previously. Also, this manuscript is not under consideration for publication elsewhere.
Conflicts of Interest
The authors declare no conflicts of interest regarding the publication of this article.
Acknowledgments
The authors acknowledge the support of the Fundamental Research Funds for the Central Universities (106112017CDJQJ238816), the Collaborative Fund of Science and Technology Agency of Luzhou Government and Southwest Medical University (2016LZXNYD-J19) and the Visiting Scholar Foundation of Key Laboratory of Biorheological Science, and Technology (Chongqing University), Ministry of Education (CQKLBST-2018-012).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2019.02.008.
Contributor Information
Hu Mei, Email: meihu@cqu.edu.cn.
XianChao Pan, Email: panxc@swmu.edu.cn.
Appendix A. Supplementary data
Supplementary material
References
- 1.Higgins C.F. Multiple molecular mechanisms for multidrug resistance transporters. Nature. 2007;446:749–757. doi: 10.1038/nature05630. [DOI] [PubMed] [Google Scholar]
- 2.Germann U.A., Pastan I., Gottesman M.M. P-glycoproteins: mediators of multidrug resistance. Semin Cell Biol. 1993;4:63–76. doi: 10.1006/scel.1993.1008. [DOI] [PubMed] [Google Scholar]
- 3.Kartner N., Riordan J.R., Ling V. Cell surface P-glycoprotein associated with multidrug resistance in mammalian cell lines. Science. 1983;221:1285–1288. doi: 10.1126/science.6137059. [DOI] [PubMed] [Google Scholar]
- 4.Kathawala R.J., Gupta P., Ashby C.R., Chen Z.S. The modulation of ABC transporter-mediated multidrug resistance in cancer: a review of the past decade. Drug Resist Updat. 2015;18:1–17. doi: 10.1016/j.drup.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Juliano R.L., Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 6.Loo T.W., Bartlett M.C., Clarke D.M. Simultaneous binding of two different drugs in the binding pocket of the human multidrug resistance P-glycoprotein. J Biol Chem. 2003;278:39706–39710. doi: 10.1074/jbc.M308559200. [DOI] [PubMed] [Google Scholar]
- 7.Lugo M.R., Sharom F.J. Interaction of LDS-751 and rhodamine 123 with P-glycoprotein: evidence for simultaneous binding of both drugs. Biochemistry. 2005;44:14020–14029. doi: 10.1021/bi0511179. [DOI] [PubMed] [Google Scholar]
- 8.Dey S., Ramachandra M., Pastan I., Gottesman M.M., Ambudkar S.V. Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc Natl Acad Sci U S A. 1997;94:10594–10599. doi: 10.1073/pnas.94.20.10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loo T.W., Bartlett M.C., Clarke D.M. Methanethiosulfonate derivatives of rhodamine and verapamil activate human P-glycoprotein at different sites. J Biol Chem. 2003;278:50136–50141. doi: 10.1074/jbc.M310448200. [DOI] [PubMed] [Google Scholar]
- 10.Shapiro A.B., Fox K., Lam P., Ling V. Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur J Biochem. 1999;259:841–850. doi: 10.1046/j.1432-1327.1999.00098.x. [DOI] [PubMed] [Google Scholar]
- 11.Martin C., Berridge G., Higgins C.F., Mistry P., Charlton P., Callaghan R. Communication between multiple drug binding sites on P-glycoprotein. Mol Pharmacol. 2000;58:624–632. doi: 10.1124/mol.58.3.624. [DOI] [PubMed] [Google Scholar]
- 12.Shapiro A.B., Ling V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur J Biochem. 1997;250:130–137. doi: 10.1111/j.1432-1033.1997.00130.x. [DOI] [PubMed] [Google Scholar]
- 13.Loo T.W., Clarke D.M. Identification of residues in the drug-binding site of human P-glycoprotein using a thiol-reactive substrate. J Biol Chem. 1997;272:31945–31948. doi: 10.1074/jbc.272.51.31945. [DOI] [PubMed] [Google Scholar]
- 14.Loo T.W., Clarke D.M. Identification of residues in the drug-binding domain of human P-glycoprotein. Analysis of transmembrane segment 11 by cysteine-scanning mutagenesis and inhibition by dibromobimane. J Biol Chem. 1999;274:35388–35392. doi: 10.1074/jbc.274.50.35388. [DOI] [PubMed] [Google Scholar]
- 15.Loo T.W., Clarke D.M. Identification of residues within the drug-binding domain of the human multidrug resistance P-glycoprotein by cysteine-scanning mutagenesis and reaction with dibromobimane. J Biol Chem. 2000;275:39272–39278. doi: 10.1074/jbc.M007741200. [DOI] [PubMed] [Google Scholar]
- 16.Loo T.W., Clarke D.M. Defining the drug-binding site in the human multidrug resistance P-glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. J Biol Chem. 2001;276:14972–14979. doi: 10.1074/jbc.M100407200. [DOI] [PubMed] [Google Scholar]
- 17.Loo T.W., Bartlett M.C., Clarke D.M. Permanent activation of the human P-glycoprotein by covalent modification of a residue in the drug-binding site. J Biol Chem. 2003;278:20449–20452. doi: 10.1074/jbc.C300154200. [DOI] [PubMed] [Google Scholar]
- 18.Loo T.W., Bartlett M.C., Clarke D.M. Transmembrane segment 7 of human P-glycoprotein forms part of the drug-binding pocket. Biochem J. 2006;399:351–359. doi: 10.1042/BJ20060715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loo T.W., Bartlett M.C., Clarke D.M. Transmembrane segment 1 of human P-glycoprotein contributes to the drug-binding pocket. Biochem J. 2006;396:537–545. doi: 10.1042/BJ20060012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loo T.W., Clarke D.M. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J Biol Chem. 2002;277:44332–44338. doi: 10.1074/jbc.M208433200. [DOI] [PubMed] [Google Scholar]
- 21.Loo T.W., Clarke D.M. The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. J Biol Chem. 1999;274:24759–24765. doi: 10.1074/jbc.274.35.24759. [DOI] [PubMed] [Google Scholar]
- 22.Loo T.W., Bartlett M.C., Clarke D.M. Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J Biol Chem. 2003;278:13603–13606. doi: 10.1074/jbc.C300073200. [DOI] [PubMed] [Google Scholar]
- 23.Loo T.W., Clarke D.M. Recent progress in understanding the mechanism of P-glycoprotein-mediated drug efflux. J Membr Biol. 2005;206:173–185. doi: 10.1007/s00232-005-0792-1. [DOI] [PubMed] [Google Scholar]
- 24.Aller S.G., Yu J., Ward A., Weng Y., Chittaboina S., Zhuo R. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–1722. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li J., Jaimes K.F., Aller S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014;23:34–46. doi: 10.1002/pro.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ward A.B., Szewczyk P., Grimard V., Lee C.W., Martinez L., Doshi R. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc Natl Acad Sci U S A. 2013;110:13386–13391. doi: 10.1073/pnas.1309275110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szewczyk P., Tao H., McGrath A.P., Villaluz M., Rees S.D., Lee S.C. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr D Biol Crystallogr. 2015;71:732–741. doi: 10.1107/S1399004715000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nicklisch S.C., Rees S.D., McGrath A.P., Gokirmak T., Bonito L.T., Vermeer L.M. Global marine pollutants inhibit P-glycoprotein: environmental levels, inhibitory effects, and cocrystal structure. Sci Adv. 2016;2 doi: 10.1126/sciadv.1600001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan X.C., Mei H., Qu S.J., Huang S.H., Sun J.Y., Yang L. Prediction and characterization of P-glycoprotein substrates potentially bound to different sites by emerging chemical pattern and hierarchical cluster analysis. Int J Pharm. 2016;502:61–69. doi: 10.1016/j.ijpharm.2016.02.022. [DOI] [PubMed] [Google Scholar]
- 30.Tan W., Mei H., Chao L., Liu T., Pan X., Shu M. Combined QSAR and molecule docking studies on predicting P-glycoprotein inhibitors. J Comput Aided Mol Des. 2013;27:1067–1073. doi: 10.1007/s10822-013-9697-8. [DOI] [PubMed] [Google Scholar]
- 31.Romsicki Y., Sharom F.J. The membrane lipid environment modulates drug interactions with the P-glycoprotein multidrug transporter. Biochemistry. 1999;38:6887–6896. doi: 10.1021/bi990064q. [DOI] [PubMed] [Google Scholar]
- 32.Higgins C.F., Gottesman M.M. Is the multidrug transporter a flippase? Trends Biochem Sci. 1992;17:18–21. doi: 10.1016/0968-0004(92)90419-a. [DOI] [PubMed] [Google Scholar]
- 33.Gottesman M.M., Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem. 1993;62:385–427. doi: 10.1146/annurev.bi.62.070193.002125. [DOI] [PubMed] [Google Scholar]
- 34.Shapiro A.B., Ling V. Transport of LDS-751 from the cytoplasmic leaflet of the plasma membrane by the rhodamine-123-selective site of P-glycoprotein. Eur J Biochem. 1998;254:181–188. doi: 10.1046/j.1432-1327.1998.2540181.x. [DOI] [PubMed] [Google Scholar]
- 35.Shapiro A.B., Ling V. Extraction of Hoechst 33342 from the cytoplasmic leaflet of the plasma membrane by P-glycoprotein. Eur J Biochem. 1997;250:122–129. doi: 10.1111/j.1432-1033.1997.00122.x. [DOI] [PubMed] [Google Scholar]
- 36.Jin M.S., Oldham M.L., Zhang Q., Chen J. Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature. 2012;490:566–569. doi: 10.1038/nature11448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xu Y., Egido E., Li-Blatter X., Muller R., Merino G., Berneche S. Allocrite sensing and binding by the breast cancer resistance protein (ABCG2) and P-glycoprotein (ABCB1) Biochemistry. 2015;54:6195–6206. doi: 10.1021/acs.biochem.5b00649. [DOI] [PubMed] [Google Scholar]
- 38.Bergonzo C., Campbell A.J., Walker R.C., Simmerling C. A partial nudged elastic band implementation for use with large or explicitly solvated systems. Int J Quantum Chem. 2009;109:3781–3790. doi: 10.1002/qua.22405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Case D.A.D., Cheatham T.E., III, Simmerling C.L., Wang J., Duke R.E., Luo R. University of California; San Francisco: 2012. Amber 12. [Google Scholar]
- 40.Loo T.W., Clarke D.M. Do drug substrates enter the common drug-binding pocket of P-glycoprotein through "gates"? Biochem Biophys Res Commun. 2005;329:419–422. doi: 10.1016/j.bbrc.2005.01.134. [DOI] [PubMed] [Google Scholar]
- 41.Pajeva I.K., Sterz K., Christlieb M., Steggemann K., Marighetti F., Wiese M. Interactions of the multidrug resistance modulators Tariquidar and Elacridar and their analogues with P-glycoprotein. ChemMedChem. 2013;8:1701–1713. doi: 10.1002/cmdc.201300233. [DOI] [PubMed] [Google Scholar]
- 42.Jabeen I., Wetwitayaklung P., Klepsch F., Parveen Z., Chiba P., Ecker G.F. Probing the stereoselectivity of P-glycoprotein-synthesis, biological activity and ligand docking studies of a set of enantiopure benzopyrano[3,4-b][1,4]oxazines. Chem Commun. 2011;47:2586–2588. doi: 10.1039/c0cc03075a. [DOI] [PubMed] [Google Scholar]
- 43.Jabeen I., Pleban K., Rinner U., Chiba P., Ecker G.F. Structure-activity relationships, ligand efficiency, and lipophilic efficiency profiles of benzophenone-type inhibitors of the multidrug transporter P-glycoprotein. J Med Chem. 2012;55:3261–3273. doi: 10.1021/jm201705f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jara G.E., Vera D.M., Pierini A.B. Binding of modulators to mouse and human multidrug resistance P-glycoprotein. A computational study. J Mol Graph Model. 2013;46:10–21. doi: 10.1016/j.jmgm.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 45.Marcoux J., Wang S.C., Politis A., Reading E., Ma J., Biggin P.C. Mass spectrometry reveals synergistic effects of nucleotides, lipids, and drugs binding to a multidrug resistance efflux pump. Proc Natl Acad Sci U S A. 2013;110:9704–9709. doi: 10.1073/pnas.1303888110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ledwitch K.V., Gibbs M.E., Barnes R.W., Roberts A.G. Cooperativity between verapamil and ATP bound to the efflux transporter P-glycoprotein. Biochem Pharmacol. 2016;118:96–108. doi: 10.1016/j.bcp.2016.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verhalen B., Dastvan R., Thangapandian S., Peskova Y., Koteiche H.A., Nakamoto R.K. Energy transduction and alternating access of the mammalian ABC transporter P-glycoprotein. Nature. 2017;543:738–741. doi: 10.1038/nature21414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma J., Biggin P.C. Substrate versus inhibitor dynamics of P-glycoprotein. Proteins. 2013;81:1653–1668. doi: 10.1002/prot.24324. [DOI] [PubMed] [Google Scholar]
- 49.Kodan A., Yamaguchi T., Nakatsu T., Sakiyama K., Hipolito C.J., Fujioka A. Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc Natl Acad Sci U S A. 2014;111:4049–4054. doi: 10.1073/pnas.1321562111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou H.X., McCammon J.A. The gates of ion channels and enzymes. Trends Biochem Sci. 2010;35:179–185. doi: 10.1016/j.tibs.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Loo T.W., Clarke D.M. Mutations to amino acids located in predicted transmembrane segment 6 (TM6) modulate the activity and substrate specificity of human P-glycoprotein. Biochemistry. 1994;33:14049–14057. doi: 10.1021/bi00251a013. [DOI] [PubMed] [Google Scholar]
- 52.Ma J.F., Grant G., Melera P.W. Mutations in the sixth transmembrane domain of P-glycoprotein that alter the pattern of cross-resistance also alter sensitivity to cyclosporin a reversal. Mol Pharmacol. 1997;51:922–930. doi: 10.1124/mol.51.6.922. [DOI] [PubMed] [Google Scholar]
- 53.Ferreira R.J., Ferreira M.J., Dos Santos D.J. Do drugs have access to the P-glycoprotein drug-binding pocket through gates? J Chem Theory Comput. 2015;11:4525–4529. doi: 10.1021/acs.jctc.5b00652. [DOI] [PubMed] [Google Scholar]
- 54.Parveen Z., Stockner T., Bentele C., Pferschy S., Kraupp M., Freissmuth M. Molecular dissection of dual pseudosymmetric solute translocation pathways in human P-glycoprotein. Mol Pharmacol. 2011;79:443–452. doi: 10.1124/mol.110.067611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material