

Abstract
Natural products (NPs) remain the most prolific resource for the development of small-molecule drugs. Here we report a new machine learning approach that allows the identification of natural products with high accuracy. The method also generates similarity maps, which highlight atoms that contribute significantly to the classification of small molecules as a natural product or synthetic molecule. The method can hence be utilized to (i) identify natural products in large molecular libraries, (ii) quantify the natural product-likeness of small molecules, and (iii) visualize atoms in small molecules that are characteristic of natural products or synthetic molecules. The models are based on random forest classifiers trained on data sets consisting of more than 265,000 to 322,000 natural products and synthetic molecules. Two-dimensional molecular descriptors, MACCS keys and Morgan2 fingerprints were explored. On an independent test set the models reached areas under the receiver operating characteristic curve (AUC) of 0.997 and Matthews correlation coefficients (MCCs) of 0.954 and higher. The method was further tested on data from the Dictionary of Natural Products, ChEMBL and other resources. The best-performing models are accessible as a free web service at http://npscout.zbh.uni-hamburg.de/npscout.
Keywords: natural products, natural product-likeness, machine learning, random forest, classification, similarity maps, visualization, molecular fingerprints, web service
1. Introduction
Natural products (NPs) continue to be the most prolific resource for drug leads [1,2,3,4]. A recent analysis found that over 60% of all small-molecule drugs approved between 1981 and 2014 are genuine NPs, NP analogs or their derivatives, or compounds containing an NP pharmacophore [5]. NPs are characterized by enormous structural and physicochemical diversity [6,7,8]. Some of the regions in chemical space covered by NPs are not, or only rarely, populated by synthetic molecules (SMs) [7,9]. The structural complexity of many NPs exceeds that of compounds found in conventional synthetic libraries for screening, in particular with respect to stereochemical aspects, molecular shape, and ring systems [10,11,12,13,14,15,16,17,18].
The primary bottleneck of NP research is the scarcity of materials for testing. In a recent study, we showed that the molecular structures of more than 250,000 NPs have been deposited in public databases, and that only approximately 10% of these are readily obtainable from commercial providers and other sources [19].
Given the fact that NPs exhibit a wide range of biological activities that are of immediate relevance to human health, new avenues that would make NP research more effective are being explored, in particular, research involving computational approaches [2]. For example, computational methods have been employed successfully for the identification of bioactive NPs [20,21,22] and their bio-macromolecular targets [23,24,25,26]. They have also been successfully utilized for the design of simple synthetic, bioactive mimetics of NPs [27,28,29]. In this context, computational methods for quantifying the NP-likeness of compounds can be valuable tools to guide the de novo generation of NP mimetics and optimize the NP-likeness of lead compounds. Such methods may also be useful for identifying genuine NPs in commercial compound libraries, which often also contain SMs [19]. This can be valuable in the context of library design and for the prioritization of compounds for experimental testing.
The best-known in-silico approach for identifying NPs is the NP-likeness score developed by Ertl et al. [30]. The NP-likeness score is a Bayesian measure that quantifies a compound’s similarity with the structural space of NPs based on structural fragments. As such, the model can identify sub-structures characteristic to NPs. The method has been re-implemented, with some modifications, in various platforms (e.g., [31,32,33]). Among them is the Natural-Product-Likeness Scoring System [31], which allows the calculation of the NP-likeness score (with some modifications). The Natural-Product-Likeness Scoring System also allows the use of customized data sets for training. An alternative approach for quantifying NP-Likeness, following a similar modeling strategy, but based on extended connectivity fingerprints (ECFPs), was reported by Yu [34]. Also a rule-based approach has been reported [35].
In this work, we present the development and validation of new machine learning models for the discrimination of NPs and SMs. To the best of our knowledge, these models are trained on the largest collection of known NPs that have been employed for the development of such classifiers. Among further developments, we present the utilization of similarity maps [36] for the visualization of atoms of a molecule, which are characteristic for NPs or SMs, according to the models.
2. Materials and Methods
2.1. Data Preparation
NPs were compiled from several physical and virtual NP databases (see Results for details). The chemical structures were parsed directly from SMILES notation, where available. Alternatively, chemical structures stored in chemical table files (e.g., SDF) were parsed with RDKit [37] and converted into SMILES. Minor components of salts were removed by the method described in ref. [38]. Any compounds with a molecular weight below 150 Da or above 1500 Da, and any compounds consisting of elements other than H, B, C, N, O, F, Si, P, S, Cl, Se, Br, or I were filtered. The “canonicalize” method, which was implemented in the “tautomer” class of MolVS [39], was used for neutralizing the molecular structures and merging tautomers. After the removal of duplicate SMILES (ignoring stereochemistry), the processed NP reference data set consisted of a total of 201,761 NPs.
SMs were compiled from the “in-stock” subset of ZINC [40,41]. In a first step, 500,000 compounds of ZINC were picked by random selection from the complete “in-stock” subset and pre-processed following the identical protocol used for the NP databases. After generating unique, canonicalized SMILES, any molecules present in the NP reference data set were removed from the SM data set (as determined by the comparison of canonicalized SMILES). Then, random sampling was used to compile a reference data set of SMs of identical size as the NP reference data set (i.e., 201,761 compounds).
The Dictionary of Natural Products (DNP) [42] and the ChEMBL database [43,44] were pre-processed following the identical protocol outlined for the NP and SM data sets. The ChEMBL sub-set of molecules, published in the Journal of Natural Products, was retrieved directly from ChEMBL [43,45]. The natural products subset of ZINC was downloaded from the ZINC website [46].
2.2. Principal Component Analysis
Fifteen two-dimensional molecular descriptors calculated with the Molecular Operating Environment (MOE) [47] were used for principle component analysis (PCA): MW (Weight), log P (log P (o/w)), topological polar surface area (TPSA), number of hydrogen bond acceptors (a_acc), number of hydrogen bond donors (a_don), number of heavy atoms (a_heavy), fraction of rotatable bonds (b_rotR), number of nitrogen atoms (a_nN), number of oxygen atoms (a_nO), number of acidic atoms (a_acid), number of basic atoms (a_base), sum of formal charges (FCharge), number of aromatic atoms (a_aro) and number of chiral centers (chiral), and number of rings (rings).
2.3. Model Building
Prior to model building, the preprocessed NP and SM reference data sets were merged, resulting in a total of 403,522 data records. The merged data set was then randomly split into a training set of 322,817 and a test set of 80,705 compounds (ratio of 4:1). In fingerprint space, structurally distinct molecules may have identical fingerprints. For this reason, de-duplication, based on fingerprints, was separately performed for all NPs and all SMs in the training data. Any fingerprints present in both the NP and SM subsets were removed, in order to avoid conflicting class labels. This procedure resulted in a training set of 156,119 NPs and 161,378 SMs represented by Morgan2 fingerprints, and in a training set of 108,393 NPs and 157,162 SMs represented by MACCS keys.
Morgan2 fingerprints (1024 bits) [48,49] and MACCS keys (166 bits) were calculated with RDKit, and 206 two-dimensional physicochemical property descriptors were calculated with MOE. Random forest classifiers (RFCs) were generated with scikit-learn [50,51] using default settings, except for “n_estimators”, which was set to “100”, and “class_weight”, which was set to “balanced”.
The NP-likeness calculator [30,31,52] was trained on atom signatures derived from the identical NP and SM data sets, used for training the RFCs. Subsequently, the NP-likeness score was calculated for each molecule in the test set, according to the atom signatures. All calculations used a signature height of 3, resulting in scores ranging from −3 to 3. Molecules with a score greater than 0.0 were labeled as NPs, and molecules with a score lower, or equal to 0.0 were labeled as SMs. NP class probabilities (and AUCs) were derived by normalizing these scores to a range from 0.0 to 1.0.
2.4. Similarity Maps
Similarity maps were computed with the RDKit [37] Chem.Draw.SimilarityMaps module based on RFCs derived from Morgan2 fingerprints (1024 bits).
3. Results
3.1. Compilation of Data Sets for Model Development
An NP reference data set of 201,761 unique NPs was compiled from 18 virtual NP libraries and nine physical NP databases. The reference data set is identical to that compiled as part of our previous work [8], with two amendments: First, the compounds of the DNP [42] were not included in the data set, as they serve as an external test set in this work, and second, the recently published Natural Products Atlas database [53] was added as a new data source. An overview of the NP data sources utilized in this work is provided in Table 1. The table also reports the number of molecules that are contained in the individual databases prior to, and after, data preprocessing. This is a procedure that includes the removal of salt components and stereochemical information, the filtering of molecules composed of uncommon elements, and with a molecular weight (MW) below 150 Da or above 1500 Da, and the removal of duplicate molecules (see Methods for details). An equal amount (i.e., 201,761) of synthetic organic molecules (SMs) was collected from the “in-stock” subset of ZINC [41] by random selection.
Table 1.
Size of the individual data sets prior to and after data preprocessing.
| Name 1 | Number of Molecules in SMILES Notation Successfully Parsed with RDKit | Number of Unique Molecules After Data Preprocessing | Scientific Literature and/or Online Presence |
|---|---|---|---|
| UNPD | 229,140 | 161,228 | [54,55] |
| TCM Database@Taiwan | 56,325 | 45,422 | [56,57] |
| NP Atlas | 20,018 | 18,358 | [53] |
| TCMID | 13,188 | 10,918 | [58,59] |
| TIPdb | 8838 | 7620 | [60,61,62] |
| Ambinter and Greenpharma NPs | 7905 | 6680 | [63,64] |
| AnalytiCon Discovery MEGx | 4315 | 4063 | [65] |
| NANPDB | 6841 | 3734 | [66,67] |
| StreptomeDB | 3990 | 3353 | [68,69] |
| NPs of PubChem Substance Database | 3533 | 2638 | [70,71] |
| NuBBE | 1856 | 1637 | [72,73] |
| Pi Chemicals NPs | 1783 | 1511 | [74] |
| NPCARE | 1613 | 1479 | [75,76] |
| NPACT | 1516 | 1376 | [77,78] |
| InterBioScreen NPs | 1359 | 1116 | [79] |
| AfroDb | 954 | 865 | [80,81] |
| TargetMol Natural Compound Library | 850 | 745 | [82] |
| HIM | 1284 | 641 | [83,84] |
| SANCDB | 623 | 588 | [85,86] |
| UEFS Natural Products | 493 | 469 | via ZINC [40,87] |
| p-ANAPL | 538 | 456 | [88] |
| NCI/NIH DTP NP set IV | 419 | 394 | [89] |
| HIT | 707 | 362 | [90,91] |
| AfroCancer | 388 | 352 | [92,93] |
| AfroMalariaDB | 265 | 250 | [94,95] |
| AK Scientific NPs | 242 | 177 | [96] |
| Selleck Chemicals NPs | 173 | 163 | [97] |
| NP data set TOTAL | - | 201761 |
1 UNPD: the Universal Natural Products Database; TCM Database@Taiwan: the Traditional Chinese Medicine Database@Taiwan; NP Atlas: the Natural Products Atlas; TCMID: the Traditional Chinese Medicine Integrated Database; TIPdb: the Taiwan Indigenous Plant Database; NANPDB: the Northern African Natural Products Database; StreptomeDB: Streptome Database; NuBBE: Nuclei of Bioassays, Ecophysiology and Biosynthesis of Natural Products Database; NPCARE: Database of Natural Products for Cancer Gene Regulation; NPACT: the Naturally Occurring Plant-based Anti-Cancer Compound-Activity-Target Database; AfroDb: NPs from African medicinal plants; HIM: the Herbal Ingredients in-vivo Metabolism Database; UEFS Natural Products: the natural products database of the State University of Feira De Santana; p-ANAPL: the Pan-African Natural Products Library; NCI/NIH DTP NP set IV: the NP (plated) set IV of the Developmental Therapeutic Program of the National Cancer Institute/National Institutes of Health; HIT, the Herbal Ingredients’ Targets Database; AfroCancer, the African Anticancer Natural Products Library; AfroMalariaDB, the African Antimalarial Natural Products Library.
3.2. Analysis of the Physicochemical Properties of Natural Products and Synthetic Molecules
Prior to model development, we compared the chemical space covered by the 201,761 unique NPs, and the equal number of unique SMs, using principal component analysis (PCA), based on 15 relevant physicochemical properties (see Methods for details). The score plot in Figure 1 shows that the chemical space of SMs is essentially a sub-space of NPs.
Figure 1.

Comparison of the chemical space covered by natural products (NPs) and synthetic organic molecules (SMs). The score plot is based on the principle component analysis (PCA) of all molecules in the data set, characterized by 15 calculated physicochemical properties. PCA was performed on the full data sets. For the sake of clarity, only a randomly selected 10% of all data points are reported in the score plot. The percentage of the total variance explained by the first two principal components is reported in the respective axis labels.
NPs have on average a higher MW than SMs (506 Da vs 384 Da) and a larger proportion of heavy compounds (38% vs. 10% of all molecules have a MW greater than 500 Da; Figure 2a). SMs have a narrower distribution of calculated log P values as compared to NPs (Figure 2b) but their averages are comparable (3.31 versus 3.25). SMs and NPs show clear differences in the entropy of element distributions in molecules, with NPs having, on average, a lower entropy than SMs (1.39 versus 1.63; Figure 2c). NPs tend to have more chiral centers (mean 6.66 vs. 0.75; Figure 2d), substantially fewer nitrogen atoms than SMs (mean 0.76 vs. 2.94; Figure 2e), and more oxygen atoms (mean 7.39 vs. 2.88; Figure 2f) [7,10,12,13,14,15,17].
Figure 2.


Distributions of key physicochemical properties among NPs and SMs: (a) Molecular weight; (b) log P (o/w); (c) entropy of the element distribution in molecules; (d) number of chiral centers; (e) number of nitrogen atoms; (f) number of oxygen atoms.
3.3. Model Development and Selection
Random forest classifiers [98] were trained on three different descriptor sets: 206 two-dimensional physicochemical property descriptors calculated with MOE [47], Morgan2 fingerprints (1024 bits) [48,49] calculated with RDKit [37], and MACCS keys (166 bits), also calculated with RDKit. Model performance was characterized utilizing the Matthews correlation coefficient (MCC) [99] and area under the receiver operating characteristic curve (AUC). The MCC is one of the most robust measures for evaluating the performance of binary classifiers, as it considers the proportion of all classes in the confusion matrix (i.e., true positives, false positives, true negatives, and false negatives). The AUC was used to measure how well the models are able to rank NPs early in a list.
As reported in Table 2, the models derived from any of the three descriptor sets performed very well. The AUC values, that were obtained during 10-fold cross-validation, were between 0.996 and 0.997; the MCC values were 0.950 or higher. No noticeable increase in performance was obtained by the further increase in the number of estimators (n_estimators) and the optimization of the maximum fraction of features considered per split (max_features; data not shown). Therefore, we chose to use 100 estimators, and the square root of the number of features, as the most suitable setup for model generation.
Table 2.
Performance of models derived from different descriptors or fingerprints.
| Test Method | Metric 1 | MOE Two-Dimensional Descriptors | Morgan2 Fingerprints (1024 Bits) | MACCS Keys | NP-Likeness Calculator |
|---|---|---|---|---|---|
| 10-fold cross-validation | AUC | 0.997 | 0.997 | 0.996 | / |
| MCC | 0.953 | 0.958 | 0.950 | / | |
| Independent test set | AUC | 0.997 | 0.997 | 0.997 | 0.997 |
| MCC | 0.954 | 0.960 | 0.960 | 0.959 |
1 AUC: area under the receiver operating characteristic curve: MCC: Matthews correlation coefficient.
3.4. Model Validation
In a first step, the performance of the selected models was tested on an independent test set. The AUC and MCC values, that were obtained for the selected models on this independent test set, are comparable with those obtained for the 10-fold cross-validation: AUC values were 0.997 for models based on any of the three types of descriptors and MCC values were 0.954 or higher.
Given the fact that the type of descriptor, used for model generation, did not have a substantial impact on model performance, we opted to select the model based on MACCS keys as the primary model for further experiments, because of its low complexity and good interpretability. This model achieved a very good separation of NPs and SMs for the independent test set, as shown in Figure 3a. Approximately 63% of all NPs were assigned an NP class probability of 1.0, whereas 51% SMs were assigned an NP class probability of 0.0. Only approximately 1% of all compounds were assigned values close to the decision threshold of 0.5 (i.e., between 0.4 and 0.6).
Figure 3.

Predicted NP class probabilities distributions for (a) the independent test set (stacked histogram), (b) the DNP (after the removal of any compounds present in the training set). Note that the y-axis is in logarithmic scale.
The model’s ability to identify NPs was also tested using the DNP as an external validation set. By definition, the DNP should consist exclusively of NPs. After the removal of any molecules present in the training data (based on canonicalized SMILES), the preprocessed DNP consisted of 60,502 compounds. Approximately 95% of these compounds were predicted as NPs by the model, demonstrating the model’s capacity to identify NPs with high sensitivity (Figure 3b).
3.5. Comparison of Model Performance with the NP-Likeness Calculator
We compared the performance of the model derived from MACCS keys to the NP-likeness calculator (based on the Natural-Product-Likeness Scoring System; see Introduction), which we trained and tested on the identical data sets used for the development of our models. On the independent test set, the NP-likeness calculator performed equally well as our model, with an AUC of 0.997 and an MCC of 0.959 (Table 2). Approximately 95% of all compounds of the DNP were classified as NPs (i.e., having assigned an NP-likeness score greater than 0; see Figure S1), which is comparable to the classification obtained with our model based on MACCS keys.
3.6. Analysis of Class Probability Distributions for Different Data Sets
In addition to the above experiments, we used the model based on MACCS keys for profiling the ChEMBL database and a subset thereof. The ChEMBL database [44] primarily contains SMs, and 87% of all compounds stored in ChEMBL were predicted as such (Figure 4a). Interestingly, 42,949 molecules (~3%) were assigned an NP class probability of 1.0, and therefore likely are NPs. This finding is in agreement with our previous study, which identified approximately 40,000 NPs in the ChEMBL database, by overlapping the database with a comprehensive set of known NPs [19].
Figure 4.

Predicted NP class probability distributions for (a) the ChEMBL database, (b) a subset of the ChEMBL database composed of molecules originating from the Journal of Natural Products, and (c) the natural products subset of ZINC. Note that the y-axis is in logarithmic scale.
A subset of the ChEMBL database containing molecules originating from the Journal of Natural Products [45] has been used as a source of genuine NPs to train models for the prediction of NP-likeness [31]. Our model based on MACCS keys predicts a small percentage of the molecules (less than 4%) in this data set as not NP-like (Figure 4b). Closer inspection of the compounds predicted as not NP-like reveals that these are, for example, SMs used as positive controls in biochemical assays. They include the drugs celecoxib, glibenclamide and linezolid, all of which are predicted with an NP class probability of 0.0. This experiment demonstrates that the classifiers can be used as powerful tools for the identification of NPs or SMs in mixed data sets with high accuracy.
A second example of a data set that by its name is assumed to consist exclusively of NPs is the natural products subset of ZINC [46]. The class probability distribution calculated for this subset however is similar to that obtained for the complete ChEMBL, indicating the presence of a substantial number of SMs (including NP derivatives and NP analogs) in this subset (Figure 4c): Only approximately 43% of all compounds in the NPs subset of ZINC were classified as NPs; around 23% were assigned an NP class probability of 1.0.
3.7. Analysis of Discriminative Features of Natural Products and Synthetic Molecules
The most discriminative features were determined, based on the feature_importances_ attributes computed with scikit-learn (see Methods for details). For the classifier based on MOE two-dimensional molecular descriptors, the three most important features were the number of nitrogen atoms (a large fraction of NPs has no nitrogen atom; see Figure 2e), the entropy of the element distribution in molecules (NPs have on average lower element distribution entropy than SMs; see Figure 2c), and the number of unconstrained chiral centers (NPs have on average more chiral centers than SMs; see Figure 2d). An overview of the ten most important features is provided in Table 3.
Table 3.
Feature importance for the random forest classifier based on MOE two-dimensional descriptors.
| Identifier Used by MOE | Feature Importance 1 | Description |
|---|---|---|
| a_nN | 0.103 | Number of nitrogen atoms. |
| a_ICM | 0.051 | Entropy of the element distribution in the molecule. |
| chiral_u | 0.045 | Number of unconstrained chiral centers. |
| GCUT_SLOGP_0 | 0.045 | Descriptor derived from graph distance adjacency matrices utilizing atomic contribution to log P. |
| SlogP_VSA0 | 0.044 | Surface area descriptor taking into account the contributions of individual atoms to log P. |
| chiral | 0.042 | Number of chiral centers. |
| GCUT_SLOGP_3 | 0.036 | Descriptor derived from graph distance adjacency matrices utilizing atomic contribution to log P. |
| a_nO | 0.025 | The number of oxygen atoms. |
| GCUT_PEOE_0 | 0.025 | Descriptor derived from graph distance adjacency matrices utilizing partial equalization of orbital electronegativities charges. |
| SlogP_VSA1 | 0.024 | Surface area descriptor taking into account the contributions of individual atoms to log P. |
1 From the feature_importances_ attribute of the classifier based on MOE two-dimensional descriptors. The higher, the more important the feature is.
For the classifier based on MACCS keys, the 15 most important features are reported in Figure 5. In agreement with the differences observed in the physiochemical property distributions of NPs versus SMs (see Analysis of the Physicochemical Properties of Natural Products and Synthetic Molecules), the most important MACCS keys describe the presence or absence of nitrogen atoms, such as key 161, matching molecules containing at least one nitrogen atom, key 142, matching molecules with at least two nitrogen atoms, and keys 117, 158, 122, 156, 75, 110, 133, 92 and 80, matching molecules containing specific nitrogen-containing substructures. Also several oxy gen-containing substructures are among the most important features, such as keys 139, 117, 110, 92.
Figure 5.

The 15 most relevant MACCS keys, sorted by decreasing feature importance. Above each diagram, the first line reports the index of the respective MACCS key and its SMARTS pattern. The second line reports the feature importance (feature_importances_ attribute). The figure was produced with SMARTSviewer [100,101].
3.8. Similarity Maps
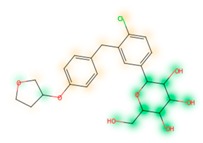
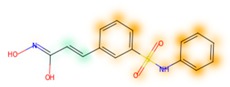
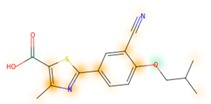
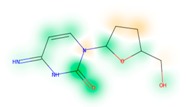
Similarity maps [36] allow the visualization of the atomic contribution of molecular fingerprints and can be extended to visualize the “atomic weights” of the predicted probability of the machine learning model. During several test runs with different Morgan fingerprint, radii, and bit vector lengths, we identified a radius of 2 and a bit vector length of 1024 bits as the most suitable setup for generating fine-grained similarity maps. The examples of similarity maps, generated with this descriptor, and the random forest approach, are reported in Table 4 for representative molecules, none of which have been part of model training. In this similarity maps, green highlights mark atoms contributing to the classification of a molecule as NP, whereas orange highlights mark atoms contributing to the classification of a molecule as SM. As expected, the similarity maps for the NP arglabin are mostly green, whereas for the synthetic drugs, bilastine and perampanel, are mostly orange. For NP derivatives and mimetics, the similarity maps are more heterogeneous and show green, as well as orange areas. The thrombin receptor antagonist vorapaxar is a derivative of the piperidine alkaloid himbacine. Vorapaxar shares a decahydronaphtho[2,3-c]furan-1(3H)-one scaffold with himbacine, but has the piperidine ring replaced by a pyridine, besides other modifications. The similarity map generated for vorapaxar shows that the model correctly identifies the decahydronaphtho[2,3-c]furan-1(3H)-one as NP-like, whereas it associates the modified areas with synthetic molecules. In the case of empagliflozin, which mimics the flavonoid phlorozin, the model correctly recognizes the C-glycosyl moiety as NP-like, whereas other atoms in the molecule are associated with synthetic molecules.
Table 4.
Examples of similarity maps generated by the NP classifier based on Morgan2 fingerprints.
| Similarity Map 1 | Name | Source 2 | NP Class Probability | Disease Indication | Year Introduced |
|---|---|---|---|---|---|
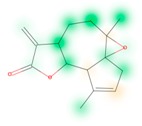
|
arglabin | N | 1.0 | anticancer | 1999 |
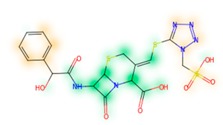
|
cefonicid sodium | ND | 0.34 | antibacterial | 1984 |
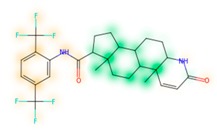
|
dutaseride | ND | 0.18 | benign prostatic hypertrophy | 2001 |
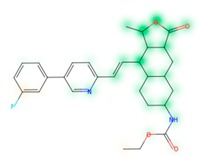
|
vorapaxar | ND | 0.30 | coronary artery disease | 2014 |
|
empagliflozin | S*/NM | 0.67 | antidiabetic (diabetes 2) | 2014 |
|
belinostat | S*/NM | 0.09 | anticancer | 2014 |
|
febuxostat | S/NM | 0.19 | hyperuricemia | 2009 |
|
zalcitabine | S* | 0.46 | antiviral | 1992 |
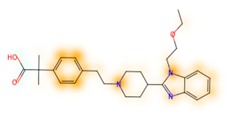
|
bilastine | S | 0.17 | antihistamine | 2011 |
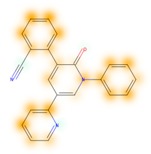
|
perampanel | S | 0.16 | antiepileptic | 2012 |
1 Green highlights mark atoms contributing to the classification of a molecule as NP, whereas orange highlights mark atoms contributing to the classification of a molecule as SM. 2 N: Unaltered NP; ND: NP derivative; S*: Synthetic drug (NP pharmacophore); S: Synthetic drug; NM: Mimic of NP. Definitions according to ref [5].
3.9. NP-Scout Web Service
A web service named “NP-Scout” is accessible free of charge via http://npscout.zbh.uni-hamburg.de/npscout. It features the random forest model, based on MACCS keys for the computation of NP class probabilities and the random forest model, based on Morgan2 fingerprints (with 1024 bits) for the generation of similarity maps.
Users can submit molecular structures for calculation, by entering SMILES, uploading a file with SMILES or a list of SMILES, or drawing the molecule with the JavaScript Molecule Editor (JSME) [102]. The results page (Figure 6) presents the calculated NP class probabilities and similarity maps of submitted molecules in a tabular format. The results can be downloaded in CSV file format. Calculations of the NP class probabilities and the similarity maps take few seconds per compound and approximately 15 min for 1000 compounds. Users may utilize a unique link provided upon job submission to return to the website after all calculations have been completed.
Figure 6.

Screenshot of the result page of NP-Scout.
4. Conclusions
In this work, we introduced a pragmatic machine learning approach for the discrimination of NPs and SMs and for the quantification of NP-likeness. As shown by validation experiments using independent and external testing data, the models reach a very high level of accuracy. An interesting and relevant new aspect of this work is the utilization of similarity maps to visualize atoms in molecules making decisive contributions to the assignment of compounds to either class. A free web service for the classification of small molecules and the visualization of similarity maps is available at http://npscout.zbh.uni-hamburg.de/npscout.
Acknowledgments
Gerd Embruch from the Center of Bioinformatics (ZBH) of the Universität Hamburg is thanked for his technical support with the web service.
Supplementary Materials
The following are available online at http://www.mdpi.com/2218-273X/9/2/43/s1, Figure S1: Distribution of calculated NP-likeness scores for the DNP (after removal of any compounds present in the training set).
Author Contributions
Conceptualization, Y.C. and J.K.; methodology, Y.C. and J.K.; software, Y.C., C.S., and S.H.; validation, Y.C.; formal analysis, Y.C; investigation, Y.C., C.S., and S.H.; resources, J.K.; data curation, Y.C.; writing—original draft preparation, Y.C., C.S., S.H., and J.K.; visualization, Y.C. and S.H.; supervision, J.K.; project administration, J.K.; funding acquisition, Y.C. and J.K.
Funding
Y.C. is supported by the China Scholarship Council, grant number 201606010345. C.S. and J.K. are supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), project number KI 2085/1-1. J.K. is also supported by the Bergens Forskningsstiftelse (BFS, Bergen Research Foundation), grant number BFS2017TMT01.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- 1.Cragg G.M., Newman D.J. Biodiversity: A continuing source of novel drug leads. J. Macromol. Sci. Part A Pure Appl. Chem. 2005;77:7–24. doi: 10.1351/pac200577010007. [DOI] [Google Scholar]
- 2.Rodrigues T., Reker D., Schneider P., Schneider G. Counting on natural products for drug design. Nat. Chem. 2016;8:531–541. doi: 10.1038/nchem.2479. [DOI] [PubMed] [Google Scholar]
- 3.Harvey A.L., Edrada-Ebel R., Quinn R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015;14:111–129. doi: 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- 4.Shen B. A new golden age of natural products drug discovery. Cell. 2015;163:1297–1300. doi: 10.1016/j.cell.2015.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newman D.J., Cragg G.M. Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016;79:629–661. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 6.Grabowski K., Baringhaus K.-H., Schneider G. Scaffold diversity of natural products: Inspiration for combinatorial library design. Nat. Prod. Rep. 2008;25:892–904. doi: 10.1039/b715668p. [DOI] [PubMed] [Google Scholar]
- 7.Ertl P., Schuffenhauer A. Cheminformatics analysis of natural products: Lessons from nature inspiring the design of new drugs. Prog. Drug Res. 2008;66:219–235. doi: 10.1007/978-3-7643-8595-8_4. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y., de Lomana M.G., Friedrich N.-O., Kirchmair J. Characterization of the chemical space of known and Readily Obtainable Natural Products. J. Chem. Inf. Model. 2018;58:1518–1532. doi: 10.1021/acs.jcim.8b00302. [DOI] [PubMed] [Google Scholar]
- 9.Chen H., Engkvist O., Blomberg N., Li J. A comparative analysis of the molecular topologies for drugs, clinical candidates, natural products, human metabolites and general bioactive compounds. Med. Chem. Commun. 2012;3:312–321. doi: 10.1039/C2MD00238H. [DOI] [Google Scholar]
- 10.Camp D., Garavelas A., Campitelli M. Analysis of physicochemical properties for drugs of natural origin. J. Nat. Prod. 2015;78:1370–1382. doi: 10.1021/acs.jnatprod.5b00255. [DOI] [PubMed] [Google Scholar]
- 11.Koch M.A., Schuffenhauer A., Scheck M., Wetzel S., Casaulta M., Odermatt A., Ertl P., Waldmann H. Charting biologically relevant chemical space: A structural classification of natural products (SCONP) Proc. Natl. Acad. Sci. USA. 2005;102:17272–17277. doi: 10.1073/pnas.0503647102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stratton C.F., Newman D.J., Tan D.S. Cheminformatic comparison of approved drugs from natural product versus synthetic origins. Bioorg. Med. Chem. Lett. 2015;25:4802–4807. doi: 10.1016/j.bmcl.2015.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wetzel S., Schuffenhauer A., Roggo S., Ertl P., Waldmann H. Cheminformatic analysis of natural products and their chemical space. CHIMIA Int. J. Chem. 2007;61:355–360. doi: 10.2533/chimia.2007.355. [DOI] [Google Scholar]
- 14.López-Vallejo F., Giulianotti M.A., Houghten R.A., Medina-Franco J.L. Expanding the medicinally relevant chemical space with compound libraries. Drug Discov. Today. 2012;17:718–726. doi: 10.1016/j.drudis.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Feher M., Schmidt J.M. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inf. Comput. Sci. 2003;43:218–227. doi: 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]
- 16.Clemons P.A., Bodycombe N.E., Carrinski H.A., Wilson J.A., Shamji A.F., Wagner B.K., Koehler A.N., Schreiber S.L. Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles. Proc. Natl. Acad. Sci. USA. 2010;107:18787–18792. doi: 10.1073/pnas.1012741107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Henkel T., Brunne R.M., Müller H., Reichel F. Statistical investigation into the structural complementarity of natural products and synthetic compounds. Angew. Chem. Int. Ed. Engl. 1999;38:643–647. doi: 10.1002/(SICI)1521-3773(19990301)38:5<643::AID-ANIE643>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 18.Lee M.L., Schneider G. Scaffold architecture and pharmacophoric properties of natural products and trade drugs: Application in the design of natural product-based combinatorial libraries. J. Comb. Chem. 2001;3:284–289. doi: 10.1021/cc000097l. [DOI] [PubMed] [Google Scholar]
- 19.Chen Y., de Bruyn Kops C., Kirchmair J. Data resources for the computer-guided discovery of bioactive natural products. J. Chem. Inf. Model. 2017;57:2099–2111. doi: 10.1021/acs.jcim.7b00341. [DOI] [PubMed] [Google Scholar]
- 20.Rupp M., Schroeter T., Steri R., Zettl H., Proschak E., Hansen K., Rau O., Schwarz O., Müller-Kuhrt L., Schubert-Zsilavecz M., et al. From machine learning to natural product derivatives that selectively activate transcription factor PPARγ. ChemMedChem. 2010;5:191–194. doi: 10.1002/cmdc.200900469. [DOI] [PubMed] [Google Scholar]
- 21.Maindola P., Jamal S., Grover A. Cheminformatics based machine learning models for AMA1-RON2 abrogators for inhibiting Plasmodium falciparum erythrocyte invasion. Mol. Inform. 2015;34:655–664. doi: 10.1002/minf.201400139. [DOI] [PubMed] [Google Scholar]
- 22.Chagas-Paula D.A., Oliveira T.B., Zhang T., Edrada-Ebel R., Da Costa F.B. Prediction of anti-inflammatory plants and discovery of their biomarkers by machine learning algorithms and metabolomic studies. Planta Med. 2015;81:450–458. doi: 10.1055/s-0034-1396206. [DOI] [PubMed] [Google Scholar]
- 23.Reker D., Perna A.M., Rodrigues T., Schneider P., Reutlinger M., Mönch B., Koeberle A., Lamers C., Gabler M., Steinmetz H., et al. Revealing the macromolecular targets of complex natural products. Nat. Chem. 2014;6:1072–1078. doi: 10.1038/nchem.2095. [DOI] [PubMed] [Google Scholar]
- 24.Rodrigues T., Sieglitz F., Somovilla V.J., Cal P.M.S.D., Galione A., Corzana F., Bernardes G.J.L. Unveiling (−)-englerin A as a modulator of L-type calcium channels. Angew. Chem. Int. Ed. Engl. 2016;55:11077–11081. doi: 10.1002/anie.201604336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Merk D., Grisoni F., Friedrich L., Gelzinyte E., Schneider G. Computer-assisted discovery of retinoid X receptor modulating natural products and isofunctional mimetics. J. Med. Chem. 2018;61:5442–5447. doi: 10.1021/acs.jmedchem.8b00494. [DOI] [PubMed] [Google Scholar]
- 26.Schneider P., Schneider G. De-orphaning the marine natural product (±)-marinopyrrole A by computational target prediction and biochemical validation. Chem. Commun. 2017;53:2272–2274. doi: 10.1039/c6cc09693j. [DOI] [PubMed] [Google Scholar]
- 27.Merk D., Grisoni F., Friedrich L., Schneider G. Tuning artificial intelligence on the de novo design of natural-product-inspired retinoid X receptor modulators. Commun. Chem. 2018;1:68. [Google Scholar]
- 28.Friedrich L., Rodrigues T., Neuhaus C.S., Schneider P., Schneider G. From complex natural products to simple synthetic mimetics by computational de novo design. Angew. Chem. Int. Ed. Engl. 2016;55:6789–6792. doi: 10.1002/anie.201601941. [DOI] [PubMed] [Google Scholar]
- 29.Grisoni F., Merk D., Consonni V., Hiss J.A., Tagliabue S.G., Todeschini R., Schneider G. Scaffold hopping from natural products to synthetic mimetics by holistic molecular similarity. Commun. Chem. 2018;1:44. [Google Scholar]
- 30.Ertl P., Roggo S., Schuffenhauer A. Natural product-likeness score and its application for prioritization of compound libraries. J. Chem. Inf. Model. 2008;48:68–74. doi: 10.1021/ci700286x. [DOI] [PubMed] [Google Scholar]
- 31.Jayaseelan K.V., Moreno P., Truszkowski A., Ertl P., Steinbeck C. Natural product-likeness score revisited: An open-source, open-data implementation. BMC Bioinform. 2012;13:106. doi: 10.1186/1471-2105-13-106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jayaseelan K.V., Steinbeck C. Building blocks for automated elucidation of metabolites: Natural product-likeness for candidate ranking. BMC Bioinform. 2014;15:234. doi: 10.1186/1471-2105-15-234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.RDKit NP_Score. [(accessed on 27 November 2018)]; Available online: https://github.com/rdkit/rdkit/tree/master/Contrib/NP_Score.
- 34.Yu M.J. Natural product-like virtual libraries: Recursive atom-based enumeration. J. Chem. Inf. Model. 2011;51:541–557. doi: 10.1021/ci1002087. [DOI] [PubMed] [Google Scholar]
- 35.Zaid H., Raiyn J., Nasser A., Saad B., Rayan A. Physicochemical properties of natural based products versus synthetic chemicals. Open Nutraceuticals J. 2010;3:194–202. doi: 10.2174/18763960010030100194. [DOI] [Google Scholar]
- 36.Riniker S., Landrum G.A. Similarity maps—A visualization strategy for molecular fingerprints and machine-learning methods. J. Cheminform. 2013;5:43. doi: 10.1186/1758-2946-5-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.RDKit Version 2017.09.3: Open-source cheminformatics software. [(accessed on 22 May 2018)]; Available online: http://www.rdkit.org.
- 38.Stork C., Wagner J., Friedrich N.-O., de Bruyn Kops C., Šícho M., Kirchmair J. Hit Dexter: A machine-learning model for the prediction of frequent hitters. ChemMedChem. 2018;13:564–571. doi: 10.1002/cmdc.201700673. [DOI] [PubMed] [Google Scholar]
- 39.MolVs Version 0.1.1. [(accessed on 12 July 2018)]; Available online: https://github.com/mcs07/MolVS.
- 40.Sterling T., Irwin J.J. ZINC 15-Ligand discovery for everyone. J. Chem. Inf. Model. 2015;55:2324–2337. doi: 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.ZINC “in-stock” subset ZINC15. [(accessed on 21 August 2018)]; Available online: http://zinc15.docking.org/
- 42.Dictionary of Natural Products. Chapman & Hall/CRC; London, UK: 2010. version 19.1. [Google Scholar]
- 43.Bento A.P., Gaulton A., Hersey A., Bellis L.J., Chambers J., Davies M., Krüger F.A., Light Y., Mak L., McGlinchey S., et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014;42:D1083–D1090. doi: 10.1093/nar/gkt1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.ChEMBL Version 24_1. [(accessed on 30 July 2018)]; Available online: https://www.ebi.ac.uk/chembl/
- 45.ChEMBL Version 23. [(accessed on 6 June 2017)]; Available online: https://www.ebi.ac.uk/chembl.
- 46.Natural products subset of ZINC ZINC15. [(accessed on 7 November 2018)]; Available online: http://zinc15.docking.org/substances/subsets/
- 47.Molecular Operating Environment (MOE) Chemical Computing Group; Montreal, QC, Canada: 2016. version 2016.08. [Google Scholar]
- 48.Morgan H.L. The generation of a unique machine description for chemical structures-A technique developed at Chemical Abstracts Service. J. Chem. Doc. 1965;5:107–113. doi: 10.1021/c160017a018. [DOI] [Google Scholar]
- 49.Rogers D., Hahn M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010;50:742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 50.Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V., et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011;12:2825–2830. [Google Scholar]
- 51.Scikit-Learn: Machine Learning in Python. version 0.19.1.
- 52.Natural Product Likeness Calculator Version 2.1. [(accessed on 5 October 2018)]; Available online: https://sourceforge.net/projects/np-likeness/
- 53.Natural Products Atlas. [(accessed on 20 August 2018)]; Available online: https://www.npatlas.org/
- 54.Gu J., Gui Y., Chen L., Yuan G., Lu H.-Z., Xu X. Use of natural products as chemical library for drug discovery and network pharmacology. PLoS ONE. 2013;8:e62839. doi: 10.1371/journal.pone.0062839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Universal Natural Products Database (UNPD) [(accessed on 17 October 2016)]; Available online: http://pkuxxj.pku.edu.cn/UNPD.
- 56.Chen C.Y.-C. TCM Database@Taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico. PLoS ONE. 2011;6:e15939. doi: 10.1371/journal.pone.0015939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.TCM Database@Taiwan. [(accessed on 17 October 2016)]; Available online: http://tcm.cmu.edu.tw.
- 58.Xue R., Fang Z., Zhang M., Yi Z., Wen C., Shi T. TCMID: Traditional Chinese medicine integrative database for herb molecular mechanism analysis. Nucleic Acids Res. 2013;41:D1089–D1095. doi: 10.1093/nar/gks1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Traditional Chinese Medicine Integrated Database (TCMID) [(accessed on 19 October 2016)]; Available online: www.megabionet.org/tcmid.
- 60.Lin Y.-C., Wang C.-C., Chen I.-S., Jheng J.-L., Li J.-H., Tung C.-W. TIPdb: A database of anticancer, antiplatelet, and antituberculosis phytochemicals from indigenous plants in Taiwan. Sci. World J. 2013;2013:736386. doi: 10.1155/2013/736386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tung C.-W., Lin Y.-C., Chang H.-S., Wang C.-C., Chen I.-S., Jheng J.-L., Li J.-H. TIPdb-3D: The three-dimensional structure database of phytochemicals from Taiwan indigenous plants. Database. 2014;2014:bau055. doi: 10.1093/database/bau055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taiwan Indigenous Plant Database (TIPdb) [(accessed on 19 October 2016)]; Available online: http://cwtung.kmu.edu.tw/tipdb.
- 63.Ambinter. [(accessed on 2 June 2017)]; Available online: www.ambinter.com.
- 64.GreenPharma. [(accessed on 2 June 2017)]; Available online: www.greenpharma.com.
- 65.AnalytiCon Discovery. [(accessed on 14 November 2017)]; Available online: www.ac-discovery.com.
- 66.Ntie-Kang F., Telukunta K.K., Döring K., Simoben C.V., A Moumbock A.F., Malange Y.I., Njume L.E., Yong J.N., Sippl W., Günther S. NANPDB: A resource for natural products from Northern African sources. J. Nat. Prod. 2017;80:2067–2076. doi: 10.1021/acs.jnatprod.7b00283. [DOI] [PubMed] [Google Scholar]
- 67.Northern African Natural Products Database (NANPDB) [(accessed on 5 April 2017)]; Available online: www.african-compounds.org/nanpdb.
- 68.Klementz D., Döring K., Lucas X., Telukunta K.K., Erxleben A., Deubel D., Erber A., Santillana I., Thomas O.S., Bechthold A., et al. StreptomeDB 2.0—An extended resource of natural products produced by streptomycetes. Nucleic Acids Res. 2015;44:D509–D514. doi: 10.1093/nar/gkv1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.StreptomeDB. [(accessed on 13 April 2017)]; Available online: http://132.230.56.4/streptomedb2/
- 70.Ming H., Tiejun C., Yanli W., Stephen B.H. Web search and data mining of natural products and their bioactivities in PubChem. Sci. China Chem. 2013;56:1424–1435. doi: 10.1007/s11426-013-4910-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Natural products subset PubChem Substance Database. [(accessed on 7 April 2017)]; Available online: http://ncbi.nlm.nih.gov/pcsubstance.
- 72.Pilon A.C., Valli M., Dametto A.C., Pinto M.E.F., Freire R.T., Castro-Gamboa I., Andricopulo A.D., Bolzani V.S. NuBBE: An updated database to uncover chemical and biological information from Brazilian biodiversity. Sci. Rep. 2017;7:7215. doi: 10.1038/s41598-017-07451-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Núcleo de Bioensaios, Biossíntese e Ecofisiologia de Produtos Naturais (NuBBE) [(accessed on 19 April 2017)]; Available online: http://nubbe.iq.unesp.br/portal/nubbedb.html.
- 74.PI Chemicals. [(accessed on 5 May 2017)]; Available online: www.pipharm.com.
- 75.Choi H., Cho S.Y., Pak H.J., Kim Y., Choi J.-Y., Lee Y.J., Gong B.H., Kang Y.S., Han T., Choi G., et al. NPCARE: Database of natural products and fractional extracts for cancer regulation. J. Cheminform. 2017;9:2. doi: 10.1186/s13321-016-0188-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Database of Natural Products for Cancer Gene Regulation (NPCARE) [(accessed on 20 February 2017)]; Available online: http://silver.sejong.ac.kr/npcare.
- 77.Mangal M., Sagar P., Singh H., Raghava G.P.S., Agarwal S.M. NPACT: Naturally Occurring Plant-based Anti-cancer Compound-Activity-Target database. Nucleic Acids Res. 2013;41:D1124–D1129. doi: 10.1093/nar/gks1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Naturally Occurring Plant-based Anti-cancer Compound-Activity-Target database (NPACT) [(accessed on 13 April 2017)]; Available online: http://crdd.osdd.net/raghava/npact.
- 79.InterBioScreen. [(accessed on 14 November 2017)]; Available online: www.ibscreen.com.
- 80.Ntie-Kang F., Zofou D., Babiaka S.B., Meudom R., Scharfe M., Lifongo L.L., Mbah J.A., Mbaze L.M., Sippl W., Efange S.M.N. AfroDb: A select highly potent and diverse natural product library from African medicinal plants. PLoS ONE. 2013;8:e78085. doi: 10.1371/journal.pone.0078085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.AfroDb. [(accessed on 18 October 2016)]; Available online: http://african-compounds.org/about/afrodb.
- 82.TargetMol. [(accessed on 17 May 2017)]; Available online: www.targetmol.com.
- 83.Kang H., Tang K., Liu Q., Sun Y., Huang Q., Zhu R., Gao J., Zhang D., Huang C., Cao Z. HIM-herbal ingredients in-vivo metabolism database. J. Cheminform. 2013;5:28. doi: 10.1186/1758-2946-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Herbal Ingredients In-Vivo Metabolism database (HIM) [(accessed on 13 April 2017)]; Available online: http://binfo.shmtu.edu.cn:8080/him.
- 85.Hatherley R., Brown D.K., Musyoka T.M., Penkler D.L., Faya N., Lobb K.A., Tastan Bishop Ö. SANCDB: A South African natural compound database. J. Cheminform. 2015;7:29. doi: 10.1186/s13321-015-0080-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.South African Natural Compound Database (SANCDB) [(accessed on 8 February 2017)]; Available online: http://sancdb.rubi.ru.ac.za.
- 87.UEFS Natural Products Catalog ZINC15. [(accessed on 26 May 2017)]; Available online: http://zinc15.docking.org.
- 88.Ntie-Kang F., Amoa Onguéné P., Fotso G.W., Andrae-Marobela K., Bezabih M., Ndom J.C., Ngadjui B.T., Ogundaini A.O., Abegaz B.M., Meva’a L.M. Virtualizing the p-ANAPL library: A step towards drug discovery from African medicinal plants. PLoS ONE. 2014;9:e90655. doi: 10.1371/journal.pone.0090655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Natural Products Set IV of the Developmental Therapeutic Program of the National Cancer Institute/National Institutes of Health. [(accessed on 20 October 2016)]; Available online: http://dtp.cancer.gov/organization/dscb/obtaining/available_plates.htm.
- 90.Ye H., Ye L., Kang H., Zhang D., Tao L., Tang K., Liu X., Zhu R., Liu Q., Chen Y.Z., et al. HIT: Linking herbal active ingredients to targets. Nucleic Acids Res. 2011;39:D1055–D1059. doi: 10.1093/nar/gkq1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Herbal Ingredients’ Targets database (HIT) [(accessed on 13 April 2017)]; Available online: http://lifecenter.sgst.cn/hit.
- 92.Ntie-Kang F., Nwodo J.N., Ibezim A., Simoben C.V., Karaman B., Ngwa V.F., Sippl W., Adikwu M.U., Mbaze L.M. Molecular modeling of potential anticancer agents from African medicinal plants. J. Chem. Inf. Model. 2014;54:2433–2450. doi: 10.1021/ci5003697. [DOI] [PubMed] [Google Scholar]
- 93.AfroCancer. [(accessed on 10 February 2017)]; Available online: http://african-compounds.org/about/afrocancer.
- 94.Onguéné P.A., Ntie-Kang F., Mbah J.A., Lifongo L.L., Ndom J.C., Sippl W., Mbaze L.M. The potential of anti-malarial compounds derived from African medicinal plants, part III: An in silico evaluation of drug metabolism and pharmacokinetics profiling. Org. Med. Chem. Lett. 2014;4:6. doi: 10.1186/s13588-014-0006-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.AfroMalariaDB. [(accessed on 10 February 2017)]; Available online: http://african-compounds.org/about/afromalariadb.
- 96.Natural products subset of AK Scientific AK Scientific. [(accessed on 19 April 2017)]; Available online: www.aksci.com.
- 97.Natural products of Selleck Chemicals Selleck Chemicals. [(accessed on 14 November 2017)]; Available online: www.selleckchem.com.
- 98.Breiman L. Random forests. Machine Learning. 2001;45:5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 99.Matthews B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 100.Schomburg K., Ehrlich H.-C., Stierand K., Rarey M. From structure diagrams to visual chemical patterns. J. Chem. Inf. Model. 2010;50:1529–1535. doi: 10.1021/ci100209a. [DOI] [PubMed] [Google Scholar]
- 101.SMARTSview. [(accessed on 30 November 2018)]; Available online: http://smartsview.zbh.uni-hamburg.de/
- 102.Bienfait B., Ertl P. JSME: A free molecule editor in JavaScript. J. Cheminform. 2013;5:24. doi: 10.1186/1758-2946-5-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.