

Abstract
Objectives
The Fragile X Mental Retardation Protein (FMRP) is a widely expressed RNA-binding protein involved in translation regulation. Since the absence of FMRP leads to Fragile X Syndrome (FXS) and autism, FMRP has been extensively studied in brain. The functions of FMRP in peripheral organs and on metabolic homeostasis remain elusive; therefore, we sought to investigate the systemic consequences of its absence.
Methods
Using metabolomics, in vivo metabolic phenotyping of the Fmr1-KO FXS mouse model and in vitro approaches, we show that the absence of FMRP induced a metabolic shift towards enhanced glucose tolerance and insulin sensitivity, reduced adiposity, and increased β-adrenergic-driven lipolysis and lipid utilization.
Results
Combining proteomics and cellular assays, we highlight that FMRP loss increased hepatic protein synthesis and impacted pathways notably linked to lipid metabolism. Mapping metabolomic and proteomic phenotypes onto a signaling and metabolic network, we predicted that the coordinated metabolic response to FMRP loss was mediated by dysregulation in the abundances of specific hepatic proteins. We experimentally validated these predictions, demonstrating that the translational regulator FMRP associates with a subset of mRNAs involved in lipid metabolism. Finally, we highlight that FXS patients mirror metabolic variations observed in Fmr1-KO mice with reduced circulating glucose and insulin and increased free fatty acids.
Conclusions
Loss of FMRP results in a widespread coordinated systemic response that notably involves upregulation of protein translation in the liver, increased utilization of lipids, and significant changes in metabolic homeostasis. Our study unravels metabolic phenotypes in FXS and further supports the importance of translational regulation in the homeostatic control of systemic metabolism.
Keywords: Fragile X mental retardation protein, RNA-binding protein, Translation, Metabolism, Glucose, Lipids
Graphical abstract
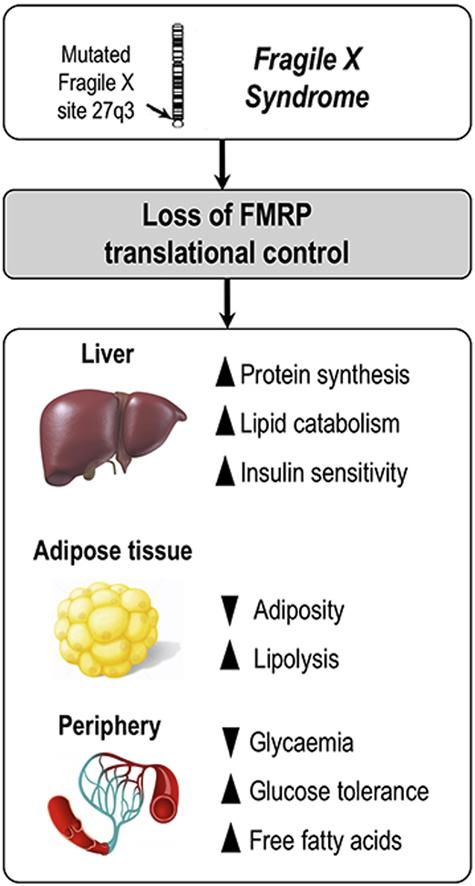
Highlights
-
•
Loss of the translational regulator FMRP impacts glucose and lipid homeostasis in mouse and human.
-
•
FMR1-deficiency modifies blood metabolic markers.
-
•
Loss of FMRP enhances the insulin response and lipolysis.
-
•
Loss of FMRP exaggerates hepatic protein synthesis.
-
•
FMRP controls the translation of key hepatic proteins involved in lipid metabolism.
1. Introduction
The Fragile X Mental Retardation Protein (FMRP) is an RNA-binding protein, which associates with polyribosomes to regulate mRNA translation together with its homologues FXR1P and FXR2P [1], [2], [3], [4]. A current model states that FMRP exerts repressive activity on translation by reversibly stalling ribosomes on its target mRNA [5], [6]. So far, FMRP functions and the mRNAs it targets have been mostly explored in the context of the central nervous system (CNS). Indeed, inactivation of the X-linked FMR1 gene, which encodes FMRP, is associated with neurodevelopmental defects, intellectual disability, and autism spectrum disorders in Fragile X Syndrome (FXS), a syndrome affecting 1:6,000 new-borns [7], [8]. Numerous studies have used the Fmr1-KO mouse models [9], [10] to show that loss of FMRP expression affects the translation of a wide range of mRNA, compromising brain translation homeostasis with deleterious consequences on synaptic plasticity and thus on behavior [4], [11].
Despite the wide expression of FMRP in peripheral tissues [12], [13], thus far, the consequences of its absence outside the CNS are only starting to be elucidated. In dfmr1-KO Drosophila, the loss of FMRP was shown to elevate insulin signaling in the brain and intestine, inducing developmental defects in those organs [14], [15], [16]. Another study in the Drosophila model of FXS, highlighted reduced whole-body carbohydrate and lipid stores, hypersensitivity to starvation, and altered mitochondrial functions [17]. Although no overt metabolic alterations was reported in the first generation FXS mouse model (Fmr1-KO1) [9], the Fmr1/Fxr2 double KO mouse displayed increased glucose tolerance and insulin sensitivity, reduced adiposity, and reduced circulating glucose [18]. Finally, independent clinical studies reported reduced levels of lipids, including cholesterol, in FXS patients compared to healthy controls [19], [20], [21]. These data supported possible functions for FMRP in the control of glucose and lipid metabolism, but mechanisms by which FMRP could impact metabolism and its mRNA targets in peripheral organs remain mostly unknown.
Here, we demonstrated that loss of FMRP in mice markedly impacted glucose and lipid metabolism. We further showed that loss of FMRP elevates hepatic protein synthesis and that FMRP likely controls the translation of key hepatic proteins involved in lipid metabolism. Finally, we provided clinical evidence that circulating metabolic markers were altered in FXS patients as compared to healthy controls.
2. Materials and methods
Detailed experimental procedures are available in the Supplemental Information Appendix.
2.1. Animal procedures
Fmr1-KO2 mice described in [10] were housed in a temperature (22–24 °C) and hygrometry (70–80%)-controlled room with a 12-h light–dark cycle (lights on at 07:00) and fed on standard chow (reference 4RF25, Mucedola). All the described experiments were performed on male littermates at 4-months of age, with the exception of initial blood sampling of fed animals for metabolic profiling (presented in Figure 1) that was performed on 4 to 6 months-old animals.
Figure 1.

Fmr1-deficiency modifies metabolic profiles in the fed and fasting state. (A) Orthogonal Partial Least Square Discriminant Analysis (OPLS-DA) of plasma 1H NMR spectra from fed Fmr1-KO and WT mice. PLS components (TPred1 & TYosc1) corresponding to a combination of the initial NMR spectral variables were computed for each individual. n = 18 WT, n = 19 KO. (B) Permutation testing to assess significance of the OPLS-DA model (n = 10,000 random iterations, p = 0.01). (C) Pseudo-spectrum representation of OPLS-DA model to highlight spectral regions discriminating Fmr1-KO from -WT samples. s. Positive and negative model coefficients respectively correspond to significantly higher or lower metabolite concentrations in Fmr1-KO animals as compared to WT. 1H NMR signals corresponding to glucose, aspartate, phosphocholine, the branched amino acids (BCAA) valine (1), isoleucine (2) and leucine (3), and broad resonances from CH3 methyl groups present in lipids are highlighted. (D) Fasting plasmatic levels of glucose in Fmr1-KO and WT animals. Data are means ± SEM; n = 11 WT, n = 12 KO; 2-tailed Student's T-test: ns. (E) Fasting plasmatic levels of triglycerides (TG) in Fmr1-KO and WT animals. Data are means ± SEM; n = 28 WT, n = 28 KO; 2-tailed Student's T-test: *, p < 0.05. (F) Fasting plasmatic levels of total cholesterol in Fmr1-KO and WT animals. Data are means ± SEM; n = 27 WT, n = 28 KO; 2-tailed Student's T-test: *, p < 0.05. (G) Fasting plasmatic levels of carnitine in Fmr1-KO and WT animals. Data are means ± SEM; n = 13 WT, n = 9 KO; 2-tailed Student's T-test: *, p < 0.05. (H) Fasting plasmatic levels of free fatty acids (FFA) in Fmr1-KO and WT animals. Data are means ± SEM; n = 26 WT, n = 25 KO; 2-tailed Student's T-test: *, p < 0.05. (I) Relative quantification of 1H NMR signal from acetone (2.22 ppm) in fasting Fmr1-KO and WT animals. Data are means ± SEM; n = 12 WT, n = 13 KO; 2-tailed Student's T-test: *, p < 0.05. (J) Relative quantification of 1H NMR signal from acetoacetate (2.27 ppm) in fasting Fmr1-KO and WT animals. Data are means ± SEM; n = 12 WT, n = 13 KO; 2-tailed Student's T-test: *, p < 0.05. (K) Fasting leptin in Fmr1-KO and WT animals. Data are means ± SEM; n = 21 WT, n = 18 KO; 2-tailed Student's T-test: *, p < 0.05. (L) Fasting insulin in Fmr1-KO and WT animals. Data are means ± SEM; n = 11 WT, n = 12 KO; 2-tailed Student's T-test: *, p < 0.05.
2.2. Clinical samples
25 fragile X patients and 29 sex- and age-matched healthy subjects were enrolled at the University of Antwerp (Antwerp, Belgium). Written informed consent was obtained from each participant or his legal guardian before research participation. The absence/presence of the Fragile X full mutation in the 5' UTR of FMR1 gene was confirmed in FXS patients by an accredited laboratory, using a CGG-repeat PCR and Southern Blotting on DNA isolated from blood. All patients had a number of CGG repeats above 200, yielding FMR1 gene inactivation (Pieretti et al., 1991). To avoid stress induced by fasting to Fragile X patients, individuals involved in the study were not advised to fast prior to venous blood sampling.
2.3. Metabolic profiling
For metabolome studies in mice, blood was sampled between 9 and 11a.m., either in nonfasted animals, i.e. fed ad libitum, or after an overnight fast. For FXS controls and patients, sera were obtained from nonfasted individuals. NMR experiments were carried out using a Bruker Avance spectrometer (Bruker GmbH, Rheinstetten, Germany) operating at 600 MHz as described previously [22]. Structural assignment was performed using data from literature, HMDB (http://www.hmdb.ca/), S-Base (Bruker GmbH, Rheinstetten, Germany) and in-house databases [23]. 1H NMR spectra were pre-processed and exported into Matlab for multivariate statistical analyses using orthogonal partial least square discriminant analysis (O-PLS-DA) as previously reported [24].
2.4. Blood chemistry and hormones measurements
Glucose was measured using a glucometer (AccuCheck Mobile, Roche). The determination of circulating TG, FFA, and total cholesterol was subcontracted to the Genotoul Anexplo Platform (Toulouse, France). Leptin and insulin were measured using dedicated ELISA kits (MesoScaleDiscovery). TG determination in liver was performed using a dedicated kit (Sigma Aldrich).
2.5. Insulin (ITT) and glucose tolerance tests (GTT)
These experiments were conducted according to the IMPReSS quidelines (International Mouse Phenotyping Resource of Standardized Screens). Mice were respectively fasted overnight (GTT) or 6 h (ITT) prior to i.p. injection of glucose (2 g/Kg, Sigma–Aldrich) or insulin (0.75 U/Kg, Humalog, Lilly) as per recommended procedures [25]. Glucose was measured in freely moving unrestrained animals using a glucometer (AccuCheck Mobile, Roche) in a drop of blood obtained after incision of the tip of the tail.
2.6. In vivo tissue insulin response measurement
Mice were fasted overnight then injected with insulin (0.75 U/Kg, Humalog, Lilly) or saline. Five minutes after injection, mice were sacrificed by cervical dislocation, and tissues were rapidly dissected and stored at −80 °C until further analyses. The western-blotting procedure and a list of antibodies and dilutions used are provided in Appendix.
2.7. Quantitative RT-PCR
Real-time PCR reactions procedures are detailed in Appendix. Primers used are available in Table S6.
2.8. Microcomputed tomography analysis and adipose tissue histology
Anesthetized animals were introduced in a SkyScan-1178 X-ray tomograph and analyzed as previously described [26]. Epididymal adipose tissue was fixed in 4% paraformaldehyde, paraffin-embedded, and stained with hematoxylin and eosin.
2.9. Lipolysis assays
For in vivo lipolysis experiments, 6 hrs-fasted Fmr1-KO and WT were injected intraperitoneally either with saline or isoproterenol at 1 mg/kg and blood sampled 15 min post injection. For ex vivo lipolysis experiments, 6 hrs-fasted Fmr1-KO and WT mice were sacrificed, and intra-abdominal white epididymal fat depots were cut in explants then treated with 1 μM isoproterenol or vehicle. Preparation of stromal vascular fraction (SVF) of epididymal adipose tissue was performed as described in [27]. Adipocytes derived from hMADS cells were obtained as described in [28], [29] and transfected with anti-FMR1 or control siRNA (Invitrogen) detailed in Appendix. Lipolysis experiments of SVF-derived adipocytes and hMADS adipocytes were performed as described in [27].
2.10. Indirect calorimetry
Indirect calorimetry experiments were performed using indirect calorimetry chambers (Phenomaster System, TSE) at the Anexplo Genotoul metabolic phenotyping platform (Toulouse, France).
2.11. Liver proteome analysis
Livers from 4 months-old overnight fasted animals were quickly collected after cervical dislocation, snapped frozen in liquid nitrogen and kept at −80 °C until further analysis. Proteins were digested by in-solution digestion and measured on an EASY-nLC 1200 coupled to an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). All acquired MS data were processed with the MaxQuant software suite [30], [31] version 1.5.2.8 against the complete mouse UniProt database (taxonomy ID10090). Comparison analysis for individual protein levels between genotype was run on normal rank-based inverse log-transformed label-free quantification (LFQ) values and individual FDR were calculated. GO analysis was performed using String (v10.5) [32] and Panther (v12.0) expression analysis tools [33], [34].
2.12. Puromycin-labeling of neosynthetized peptides
The hepatic cell line FL83B was transduced with a lentivirus expressing shFmr1 or shControl and FACS-sorted based on eGFP expression, as described in [35]. FMRP was tested using the m1C3 monoclonal antibody against FMRP in western-blotting [36] or the avian IgY#C10 polyclonal antibodies by immunofluorescence staining [6], [37], [38]. The puromycin-labeling assay was performed according to [39]. Puromycin (10 μg/mL) incorporation was performed in the presence or absence of cycloheximide (50 μg/mL) for 15 or 30 min. Proteins were visualized after protein transfer to nitrocellulose membranes by Ponceau staining and puromycin-labeled neosynthesized peptides were visualized by immunoblotting with anti-puromycin 12D10 antibody (Millipore) followed by HRP-coupled secondary antibodies and electroluminescence detection using a Fusion VX-Imager (Vilber). Puromycin signals were quantified in each lane by densitometric analysis using the ImageJ software, normalized to Ponceau signal in the corresponding lane and then adjusted to average signal in control cells at 15 min.
2.13. Metabolic network analysis
MetaboSignal R package [40], [41] was used to build the shortest paths network from FMRP to the metabolites of interest (Table S5), integrating the proteins dysregulated in the liver as extra nodes to the network (Table S6). Pivotal betweenness was then computed (Table S7).
2.14. Immunoaffinity (IA) capture of FMRP complexes in FL83B cells or liver extracts
FL83B cells or Fmr1-WT and -KO liver were homogenized in IA buffer (10 mM Tris pH 7.4, 150 mM NaCl, 1.25 mM MgCl2, 0.5% NP40, 1 mM DTT, 5 U/mL RNasin (Invitrogen)). The lysates were centrifuged at 10,000 g for 15 min at 4 °C. The collected supernatant was then raised to 400 mM NaCl and 30 mM EDTA [37]. For each IA assay, 500 μg of proteins from FL83B cells or liver extract was incubated in the presence of 60 μg of anti-FMRP IgY#C10 antibodies or non-immune IgY immobilized on 15 μL of anti-chicken IgY agarose beads (Gallus Immunotech Inc.). Samples were incubated overnight under rotation at 4 °C and washed 4 times with buffer containing 10 mM Tris pH 7.4, 400 mM NaCl, 10 mM EDTA, 0.5% NP40. 1/5th of each assay was used for immunoblot analyses using anti-FMRP mAb1C3 antibody [36] IA-captured mRNA were then phenol-extracted, precipitated with sodium acetate, and resuspended in water and subjected to reverse transcription (RT) using the SuperScript III system (Invitrogen). RT products were subjected to polymerase chain reaction (PCR), using a PCR Master Kit (Promega) and primers detailed in Table S8. PCR products were visualized on a 2.5% TAE agarose gel and amplicon size was verified using the BenchTop DNA ladder (Promega).
2.15. Statistics
Normality of data was assessed using Kolmogorov–Smirnov's test. To compare 2 groups, 2-tailed unpaired Student's T-test was used. For non-normal data, raw data were log-transformed to meet normality criteria prior to Student's T-test. If sample size n < 8 or if normality was not reached after log-transformation, data were analyzed using Mann & Whitney's non-parametrical U-test. Multiple group comparisons were performed ANOVAs and detailed statistics appear in Table S9. Post hoc comparisons were performed either using Šidák's correction for multiple comparison or Fisher's Least Significance Difference test (if less than 6 comparisons were performed), as stated in the legends. Statistical significance was set according to a two-tailed p-value (p) < 0.05. Statistical analysis was performed using GraphPad Prism version 6.00 for iOS (GraphPad Software, USA).
2.16. Study approvals
All animal studies were conducted in facilities accredited by legal authorities (Direction Départementale de Protection des Populations des Alpes-Maritimes, accreditation #C-06-152-5) using procedures approved by the Ministère de l'Enseignement Supérieur et de la Recherche (agreement #00788.01 and #05224.01). Samples and data collection from Fragile X patients and controls were approved by the medical ethics committee of the University of Antwerp in Belgium (agreement #B300201523589). The study was conducted in accordance with statutes and regulations regarding the protection of the right and welfare of human subjects' participation in biomedical research (World Declaration of Helsinki).
3. Results
3.1. Fmr1-deficiency alters plasmatic metabolites in FXS mouse model
To study the potential impact of Fmr1-deficiency on general metabolic homeostasis, we used proton nuclear magnetic resonance (1H NMR) spectroscopy to profile the plasma metabolome of 4-months old Fmr1-WT and -KO male littermates fed ad libitum. Supervised multivariate statistical modelling of 1H NMR data indicated that Fmr1-KO animals displayed a characteristic plasmatic metabolic signature, significantly distinct from controls (Figure 1A,B). In Fmr1-KO plasma, we identified a significant decrease in signals from glucose, paralleled by an increase in signals from branched chain amino acids, aspartate, and phosphocholine (Figure 1C). -CH3 resonances from fatty acid (FA) methyl groups at ∼0.9 ppm [42] were also significantly increased in Fmr1-KO animals compared to WT (Figure 1C). These results suggest that Fmr1-deficiency induces possible dysregulations of glucose and lipid metabolism observable in the fed state.
We then examined the levels of a number of metabolic markers after an overnight fast. Fasting glycemia appeared unaffected by Fmr1-deficiency as glycemia from Fmr1-WT and KO animals were both reached standard fasting glucose levels of 100 mg/dL (Figure 1D). In contrast, we observed reduced circulating levels of triglycerides (TG) and total cholesterol in fasted Fmr1-KO animals, accompanied by a reduction in plasma carnitine, a quaternary amine involved in lipid metabolism (Figure 1E–G). Conversely, we observed significant increases in free FA (FFA) and 1H NMR signals from the ketone bodies (KB) acetone and acetoacetate (Figure 1H–J). Finally, we highlighted that Fmr1-deficiency reduced the fasting circulating levels of the metabolic hormones leptin and insulin (Figure 1K,L). Collectively, these variations in metabolic markers both in the fasted and fed state pointed towards dysregulations in glucose and lipid homeostasis in Fmr1-KO mice.
3.2. Fmr1-deficiency enhances glucose tolerance and insulin response
To further explore these dysregulations, we challenged energy homeostasis in Fmr1-WT and -KO littermates by performing glucose and insulin tolerance tests (GTT and ITT, respectively). After an overnight or a 6 h fast, fasting glycemia was comparable for both genotypes (T0, Figure 2A,B). During the course of the GTT, there was a significant effect of genotype on glycemia over time (Figure 2A). Post injection of glucose, glycemia was significantly lower in Fmr1-KO animals at 30, 45 and 60 min, when compared to WT animals (Figure 2A). During ITT, there was also a significant effect of genotype on glycemia over time after insulin injection. At 90 and 120 min, glycemia of Fmr1-KO animals were significantly lower than in Fmr1-WT controls (Figure 2B). Cumulative glycemia calculated over the course of the GTT and ITT were also significantly decreased in Fmr1-KO animals (Figure 2C,D).
Figure 2.

Fmr1-deficiency improves glucose tolerance and insulin sensitivity in liver. (A) Glucose tolerance test (GTT) in Fmr1-KO and WT animals. Data are means ± SEM; n = 9 WT, n = 9 KO; 2-way ANOVA: p(Genotype) = 0.0034, p(Time) < 0.0001, p(Interaction) < 0.0001; Šidák's post hoc tests for genotype-wise comparisons: *, p < 0.05, ****, p < 0.0001. (B) Insulin tolerance test (ITT) in Fmr1-KO and WT animals. Data are means ± SEM; n = 8 WT, n = 8 KO; 2-way ANOVA: p(Genotype) = 0.0117, p(Time) < 0.0001, p(Interaction) = 0.0134; Šidák's post hoc tests for genotype-wise comparisons: **, p < 0.01. (C) Cumulative glycemia over the 3 h course of GTT. Data are means ± SEM; n = 9 WT, n = 9 KO; 2-tailed Student's T-test: **, p < 0.01. (D) Cumulative glycemia over the 3 h course of ITT. Data are means ± SEM; n = 8 WT, n = 8 KO; 2-tailed Student's T-test: *, p < 0.05. (E) Western-blot and densitometric analysis of insulin receptor (InsR) and Akt phosphorylation status in liver from Fmr1-KO and WT animals injected with saline or insulin. Densitometric analysis is presented as means ± SEM of phosphoproteins signals ratios relative to total protein, all signals being normalized to β-actin. InsR-Saline: n = 6 animals/group, InsR-Insulin: n = 5 animals/group; 2-way ANOVA: p(Genotype) = 0.0027, p(Treatment) < 0.0001, p(Interaction) = 0.7424; Fisher's LSD post hoc tests for genotype-wise comparisons: *, p < 0.05. Akt: n = 5 animals/group; 2-way ANOVA: p(Genotype) = 0.0215, p(Treatment) < 0.0001, p(Interaction) = 0.0924; Fisher's LSD post hoc tests for genotype-wise comparisons: **, p < 0.01.
We further investigated the insulin response at the molecular level in Fmr1-KO and WT liver. Injection of a bolus of insulin triggered activation and autophosphorylation of the insulin receptor (InsR) in the liver, leading to an increased phosphorylation at Tyr1146 and Tyr1150/1151 in both groups. In Fmr1-KO animals, the phosphorylation of InsR was increased both in basal conditions and upon insulin injection when compared to Fmr1-WT liver (Figure 2E). We then monitored the phosphorylation status of Akt at Ser473. Following insulin injection, the increase in Akt phosphosphorylation was significantly steeper in the Fmr1-KO liver (Figure 2E). These data suggested that loss of FMRP in the liver was accompanied by an increased response to insulin stimulation.
3.3. Fmr1-deficiency reduces adiposity and shifts metabolism towards a higher use of lipid as energy substrates
As several classes of circulating lipids were impacted in Fmr1-KO mice (Figure 1C,E–K) and leptin was reduced, we then sought to investigate the impact of Fmr1-deficiency on adipose tissue, the main tissue for lipid storage, as well as a site of expression for FMRP (Figure S1). We used μCT X-ray tomography and showed that Fmr1-deficiency differentially affected intra-abdominal adipose tissue (IAAT) and subcutaneous adipose tissue (SCAT). The volume of IAAT was significantly reduced by 53.4% in Fmr1-KO animals, whilst SCAT was not affected (Figure 3A). Further, fat pads of Fmr1-KO mice displayed smaller adipocytes, with a reduced average surface as compared to control animals (Figure 3B,C). Considering that adipocytes had a spherical shape, we extrapolated their volume from the adipocyte area measures and estimated that the mean adipocyte volume was decreased by 40.1% in Fmr1-KO animals (mean WT adipocyte volume = 1.76 ± 0.05 × 104 μm3 vs. mean KO adipocyte volume = 1.052 ± 0.17 × 104 μm3). This was within the range of the total WAT volume ratio (53.4%, Figure 3A) and suggests that the total number of adipocytes is likely preserved in the absence of FMRP.
Figure 3.

Fmr1-deficiency reduces adiposity, enhances lipolysis, and shifts metabolism towards a better utilization of lipids. (A) X-ray computerized tomography 3D reconstruction of intra-abdominal (IAAT) and subcutaneous (SCAT) adipose tissues and calculated volumes of IAAT and SCAT in Fmr1-WT and KO animals. Data are presented as means ± SEM; n = 14 WT, n = 14 KO; 2-way ANOVA: p(Genotype) < 0.0001; p(AT type) < 0.0001; p(Interaction) = 0.0004; LSD post hoc tests for genotype-wise comparisons: ****, p < 0.0001. (B) Representative sections of Fmr1-WT and KO IAAT stained with hematoxylin and eosin, black bars represent 100 μm. (C) Average adipocyte area in Fmr1-WT and KO IAAT. Data are presented as means ± SEM; n = 6 WT, n = 6 KO; 2-tailed Mann & Whitney U-test: *, p < 0.05. (D) Respiratory exchange rate (RER) follow-up over 24 h in 4 months-old Fmr1-KO and WT mice. Data are means ± SEM every 10 min; n = 8 WT, n = 10 KO. (E) Average RER over 24 h in Fmr1-KO and WT mice. Data are means ± SEM; n = 8 WT, n = 10 KO; 2-tailed Student's T-test: *, p < 0.05.
In parallel, we showed that Fmr1-KO and WT WAT express similar mRNA levels for the adipocyte precursor marker Pdgfra [43], suggesting that the adipocyte precursor pool is not impacted in WAT (Figure S2A). Also, FMRP expression increased upon differentiation of adipocytes derived from the stromal vascular fraction (SVF) of WAT (Figure S2B). However, analysis of SVF-derived Fmr1-KO adipocytes did not reveal clear adipogenesis defects when compared to Fmr1-WT adipocytes (Figure S2C). In addition, human multipotent adipose-derived stem (hMADS) cells repressing FMR1 did not display alterations in the course of differentiation or expression of the mature adipocyte markers PLIN1 and FABP4 (Figure S3A,B). Thus, the reduction in fat mass observed in Fmr1-KO animals appeared unlikely to be due to impaired adipogenesis but rather to reduced fat storage in adipocytes in vivo. We also observed a reduction in hepatic triglycerides storage in Fmr1-KO animals as compared to controls (Figure S4).
Reduced fat storage both in liver and WAT could reflect a higher consumption of lipids as energetic substrate in Fmr1-deficient animals. We therefore used indirect calorimetry chambers to measure circadian ingestive behavior, activity, O2 consumption, and CO2 production. No significant variations in drinking water intake, food consumption, or total cumulative activity were detected in Fmr1-KO animals over the course of indirect calorimetry recordings (Figure S5A–D). Furthermore, Fmr1-deficiency did not affect the energetic expenditure (EE) and its average over 24 h (Figure S5E,F). The respiratory exchange rate (RER), which provides information about metabolic substrate utilization (lipid or carbohydrate), was calculated by dividing the volume of CO2 produced (VCO2) over the volume of oxygen consumed (VO2) [44]. The RER profile of Fmr1-KO animals over 24 h appeared shifted towards lower RER values (Figure 3D). Furthermore, its average was significantly decreased in Fmr1-KO animals as compared to controls (Figure 3E), indicating that Fmr1-deficiency contributes to shift metabolism towards an enhanced utilization of lipid substrates as energetic substrate.
3.4. Fmr1-deficiency increases lipolysis in adipocytes
In white adipose tissue (WAT), fat accumulation is regulated by activation of β-adrenergic receptors, which elicits intracellular TG hydrolysis and release of glycerol and FFA to the general circulation. We monitored lipolysis in vivo in Fmr1-KO and WT littermates in basal conditions and upon systemic injection of isoproterenol, a pan β-adrenergic receptor agonist. Isoproterenol treatment induced an increase in peripheral plasma glycerol release in both Fmr1-WT and -KO mice. Independently of isoproterenol treatment, glycerol release was significantly increased in Fmr1-deficient animals (Figure 4A). We mirrored those results in WAT explants from Fmr1-KO and WT animals (Figure 4B). This suggests a constitutive overactivation of lipolysis in the absence of FMRP. Also, in Fmr1-KO SVF-derived adipocytes, there was a significant increase in glycerol release when isoproterenol was applied, suggestive of a higher sensitivity to β-adrenergic receptor agonists (Figure 4C). Finally, we performed time-course experiments of isoproterenol-induced lipolysis in hMADs transiently transfected with a siRNA that efficiently repressed FMR1 mRNA (Figure S3B). As a readout of lipolysis, in addition to glycerol release, we monitored intracellular TG and FFA release in the medium (Figure 4D). In both siFMR1 and siCtl-transfected cells, isoproterenol treatment impacted intracellular TG levels (Figure 4D). After an initial phase of intracellular TG depletion, de novo TG synthesis took over and likely accounted for the progressive recovery in TG levels, hindering the identification of a significant effect of FMR1-knockdown over the course of the experiment (Figure 4D). In contrast, when considering FFA and glycerol release induced by isoproterenol treatment, there was a significant impact of FMR1-knockdown (Figure 4D). By 120 min, siFMR1-transfected adipocytes had released significantly more glycerol and FFA than siCtl-transfected adipocytes. These data collectively suggest that loss of FMRP overactivates the β-adrenergic response and increases lipolysis, both in mouse and human adipocytes.
Figure 4.

Fmr1-deficiency increases lipolysis in murine and human adipocytes. (A) Glycerol release in serum as readout of lipolysis in Fmr1-KO and WT animals before and 15 min after injection of isoproterenol (Iso). Data are presented as means ± SEM; n = 13 WT, n = 12 KO; 2-way ANOVA: p(Genotype) = 0.0327; p(Treatment) < 0.0001; p(Interaction) = 0.4161; LSD post hoc tests for genotype-wise comparisons: *, p < 0.05. (B) Glycerol release as readout of lipolysis following treatment with Iso in ex-vivo culture supernatant of epididymal white adipose tissue (WAT) explants. Data were normalized to individual WAT explant weight and presented as means ± SEM; n = 8/condition; 3-way ANOVA: p(Genotype) < 0.0001, p(Treatment) < 0.0001, p(Time) < 0.0001, p(Genotype × Treatment) = 0.4884, p(Genotype × Time) < 0.0001, p(Treatment × Time) < 0.0001, p(Genotype × Treatment × Time) = 0.5818; Šidák's post hoc tests for genotype-wise comparisons: **, p < 0.01; p****, <0.0001. (C) Glycerol release as readout of lipolysis following treatment with Iso in vitro in SVF-derived adipocytes from Fmr1-WT and KO animals. Data were normalized to protein content and presented as means ratios relative to control WT – Iso ± SEM; n = 6 WT, n = 6 KO; 2-way ANOVA: p(Genotype) < 0.0205; p(Treatment) < 0.0001; p(Interaction) = 0.0102; LSD post hoc tests for genotype-wise comparisons: **, p < 0.01. (D) Intracellular TG and release of glycerol and FFA following isoproterenol-induced lipolysis in human hMADS cells differentiated into adipocytes, transiently transfected with anti-FMR1 (siFMR1) or control siRNA (siCtl). Data were normalized to protein content and presented as means ratios relative to siCtl – Iso ± SEM; n = 4/condition; 2-way ANOVA: p(siRNATG) = 0.7478, p(TimeTG)0.0049, p(InteractionTG) = 0.9339; p(siRNAGlycerol) = 0.0102, p(TimeGlycerol) < 0.0001, p(InteractionGlycerol) = 0.0048; p(siRNAFFA) = 0.0008, p(TimeFFA) < 0.0001, p(InteractionFFA) = 0.0081; LSD post hoc tests for siRNA-wise comparisons: **, p < 0.01; ***, p < 0.001.
3.5. The absence of FMRP leads to profound changes in the hepatic proteome and increases hepatic protein synthesis
Since FMRP is expressed in the liver (Figure S1), a key organ for the regulation of both glucose and lipid homeostasis, we reasoned that molecular changes in the hepatic proteome could help refine the molecular events underlying the broad metabolic effects driven by FMRP loss. We used quantitative Mass Spectrometry (MS) to characterize the liver proteome of Fmr1-KO and WT littermates (Table S1). The Volcano plot representing statistical significance against fold-of-change (FC KO vs WT) for the 2,087 detected proteins appeared dissymmetric (Figure 5A). Among the 307 proteins with a false discovery rate (FDR) ≤ 0.01 whose abundance was increased or decreased by at least 1.5-fold (absFC ≥ 1.5; Figure 5A), there was a shift towards higher FC with strongest changes in terms of abundance observed for upregulated proteins in the absence of FMRP. Further clustering analysis on these 307 proteins revealed that protein abundance patterns were consistent genotype-wise (Figure 5B).
Figure 5.

Loss of FMRP provokes profound changes in the hepatic proteome and enhances translation. (A) Volcano plot of statistical significance presented as -Log-10 transformed false-discovery rate (FDR) against fold-of-change for each quantified protein in Fmr1-KO and WT liver. n = 5 WT, n = 5 KO. Significantly upregulated proteins and downregulated proteins (FDR<0.01) appear respectively in red and blue. (B) Heatmap visualization and clustering analysis of significantly affected proteins whose abundance was increased or decreased by at least 1.5-fold (see list in SI Appendix, Table S2). n = 5 WT, n = 5 KO. Color code refers to the calculated z-score, missing values appear in black. (C) Selection of GO terms significantly enriched in the list of proteins presented in b and Table S2 (see SI Appendix) and associated adjusted p-value (Table S3, S4). (D) Immunofluorescence analysis of FMRP expression in the FL83B hepatic cell line transduced with a virus driving the expression of a control shRNA (shCtl) or an shRNA targeting Fmr1 (shFmr1) coupled to eGFP (green). FMRP was detected with IgY#C10 anti-FMRP antibodies (red) and the ribosomal protein S6 with anti-S6 antibodies (purple). Nuclei were counterstained with DAPI (blue) and merge images are shown in the last panel. The same exposure time was used for image captures of shFmr1- and shCtl transduced cells. (E) Translation monitoring assay using puromycin-labeled proteins in the shFmr1- or shControl (shCtl)- inactivated cells. Cells were labeled with puromycin for 15 and 30 min. Cell homogenates were analyzed by SDS-PAGE and proteins transferred to nitrocellulose membranes. Separated proteins were stained with Red Ponceau followed by immuno-blotting with an anti-puromycin antibody (α-Puro) that labels neosynthesized peptides. (F) Densitometric analysis of puromycin signal in (E). Data were normalized to total protein Ponceau signal, adjusted to average signal in shCtl cells at 15 min and presented as means ± cSEM; n = 6 independent measures/group, except for WT 30 min, n = 5; 2-way ANOVA: p(shRNA) < 0.0001, p(Time) < 0.0001, p(Interaction) = 0.4288; LSD post hoc tests for shRNA-wise comparisons: ***, p < 0.001; ****, p < 0.0001.
GO analysis of the 307 dysregulated proteins clearly pointed to selective enrichment in pathways linked to lipid metabolism, with the Fmr1-KO liver notably displaying increased levels of enzymes or transporters involved in the main routes of FA catabolism (Figure 5C, Table S2–S4). Consistent with this, there was a reduced abundance in proteins involved in lipid storage and formation of lipoprotein particles in the absence of FMRP (Figure 5C, Table S2–S4), in agreement with the significant reduction in hepatic TG content in Fmr1-KO liver (Figure S4). This supported that molecular changes in the liver in the absence of FMRP could participate to metabolic phenotypes linked to lipids in Fmr1-KO animals. In contrast to the numerous pathways linked to lipid metabolism highlighted with the GO analysis, only one pathway related to glucose was significantly enriched “starch and glucose metabolism” (Figure 5C, Table S3).
Besides clear overrepresentation of pathways involved in lipid metabolism, the primary enriched pathways involved ribosome, RNA translation, RNA-binding proteins, and amino-acyl-tRNA biosynthesis (Figure 5C, Table S2–S4). Notably, core ribosomal proteins and translation elongation factors were markedly upregulated in the absence of FMRP (Table S2–S4). We reasoned these changes could reflect a possible increase in hepatic translation in the absence of FMRP. We therefore monitored protein synthesis in the FL83B murine hepatocyte cell line by labeling neosynthesized polypeptides with puromycin [39]. We inactivated FMRP expression by lentiviral transduction of an shRNA directed against Fmr1. Transduced cells expressing the shFmr1 or control shCtl were selected based on their co-expression of eGFP. Immunofluorescence staining using the avian IgY#C10 antibodies revealed a strong reduction of FMRP expression in shFmr1 cells as compared to shCtl cells, while the signal from the ribosomal protein S6 remained unaffected (Figure 5D). Western blot analysis of FMRP expression confirmed that this repression is stable over passages in shFmr1-transduced cells (Figure S6A). Puromycin labeling of neosynthesized peptides was strongly decreased when the translation inhibitor cycloheximide was applied, showing the specificity of the labeling (Figure S6B). After both 15 min and 30 min of treatment, we observed a significant increase in puromycin incorporation in the FL83B-shFmr1 cell line as compared to the FL83B-shCtl cell line (Figure 5E,F). These data supported that FMRP loss exaggerated protein synthesis in hepatocytes, in line with the shift towards higher protein abundances in the Fmr1-KO liver and the significant enrichment in GO terms related to “Translation” or “Ribosome” highlighted in the study of the Fmr1-KO liver proteome (Figure 5A,C, Table S3, S4).
3.6. FMRP targets hepatic mRNAs linked to lipid homeostasis
To functionally connect FMRP to the observed metabolic phenotypes and identify the molecular pathways involved, we used a network-based approach integrating directed metabolic and signaling networks [40], [41]. We first compiled an extended list of metabolites deriving from the metabolic biomarkers identified in our initial metabolomics and biochemical measurements (Figure 1, Table S5) and imputed a number of interactions (Table S6). Notably, the network displays in the inner circle close around FMRP the putative interactions with the 307 proteins significantly dysregulated by at least 1.5-fold in the liver in its absence (Table S2). Then, to identify metabolic nodes forming functional paths between FMRP and its associated metabolites, we analyzed the topology of the resulting network (Figure 6A) and derived the relative importance of each node within the network using a measure of centrality (pivotal betweenness (PB) described in [45], Table S7). Among the 307 dysregulated proteins in the Fmr1-KO liver, 175 displayed PB distinct from 0 (Table S7), indicating that they are likely to contribute to the observed metabolic signature. The resulting network (Figure 6A) highlights a number of protein nodes dysregulated in the Fmr1-KO liver and connecting FMRP to the metabolites with single-step paths (Figure 6A,B).
Figure 6.

Metabolic network mapping links FMRP to metabolic dysregulations via specific mRNA targets involved in lipid catabolism. (A) Classes of circulating metabolites and hepatic proteins significantly dysregulated in Fmr1-KO animals were mapped onto the interactome. The resulting network allows connecting the causal protein FMRP to the downstream metabolic consequences of its absence. (B) Pivotal proteins of the network, Log2 fold-of-change (FC) in the Fmr1-KO vs WT liver, associated adjusted p-value (FDR) and normalized pivotal betweenness scores (PB). (C) Cell homogenates from FL83B hepatocytes (Input), immuno-affinity (IA) purified complexes, captured with control IgY (IA Ctl) or anti-FMRP IgY#C10 (IA C10) antibodies and corresponding post-IA flowthrough (FT) supernatants were separated by SDS-PAGE, transferred onto a nitrocellulose membrane and stained with Ponceau (upper panel). IgY heavy chains and light chains are indicated by an asterisk. Immunoblotting with anti-FMRP mAb1C3 antibody (lower panel). Note the depletion in FMRP load in the post-IA C10 flowthrough with IgY#C10 (FT C10), as compared to input and control IgY IA (FT Ctl). (D) RT-PCR analysis of mRNA associated with FMRP. Total RNA was extracted from the input cell lysate and IA preparations described in (C), and used as a template for RT-PCR. PCR products obtained from input, control IgY (IA Ctl) or IgY#C10 (IA C10) IA complexes as well as control blank PCR (Bk) performed in the absence of matrix were separated and visualized by agarose gel electrophoresis. The molecular weights (MW) of PCR amplicons are specified on the right side.
To further validate in vivo the network predictions and ascertain that FMRP could directly regulate the translation of some pivotal proteins, we tested whether FMRP interacted in hepatocyte with a series of mRNAs encoding proteins appearing in the network with PB distinct from zero and with functions directly involved in lipid and glucose metabolism (Figure 6A,B). FMRP-associated messenger ribonucleoparticles (mRNPs) from FL83B hepatocytes were efficiently trapped by immune-affinity (IA) capture, as shown by the enrichment in FMRP in the IA preparations and the concomitant strong depletion in FMRP signal in the post-IA supernatant (Figure 6C). Cpt1a, Slc16a1, Aldh3a2, Abcd3, and Tecr mRNA, as well as Fmr1 mRNA, itself an extensively validated mRNA target of FMRP [46], were detected by RT-PCR in IA-captured FMRP complexes and not recovered in the control fraction (Figure 6D). In contrast, the unrelated mRNA Actb, Ckdn1a, Hprt, and Gapdh or mRNAs encoding proteins with a null PB such as Plin3 were not detected (Figure 6D, Figure S6C). Also, the mRNAs encoding the pivotal proteins Agl, Gys2, or Apoa2 were not recovered in FMRP mRNP complexes, suggesting selectivity in FMRP mRNA recognition (Figure S6C). To further validate that the identified associations also occurred in vivo, we performed IA-capture experiments in liver homogenates obtained from Fmr1-WT and -KO mice and confirmed the presence of Cpt1a, Slc16a1, Aldh3a2, Abcd3 and Tecr mRNA in FMRP-containing mRNPs (Figure S7). These experiments suggest that FMRP associates with mRNA encoding pivotal enzymes and transporters in the network and likely controls their translation in the liver. This could contribute to the lipid-related metabotypes observed in Fmr1-KO animals.
3.7. FXS patients display reduced glycemia, insulinemia and increased circulating FFA
To further assess the clinical relevance of the identified metabolic effects induced by loss of FMRP in FXS mouse model, we collected sera samples in a cohort of 25 FXS patients, bearing FMR1 full-mutation (above 200 CGG repeats in the 5‘UTR of the gene) which leads to FMR1 gene inactivation and loss of FMRP [47], and 29 sex and age-matched controls (Figure S8A,B). Distributions of body mass indexes were not different between patients and controls (Figure S8C). Sera of patients and controls were collected randomly during the day, but the distribution of sample collection time was not differing between control and patient groups (Figure S8D). We then used 1H-NMR to profile the serum metabolome of controls and FXS patients. O-PLS-DA modelling of 1H-NMR data showed that FXS patients display a characteristic serum metabolic signature that is significantly different from controls (Figure 7A,B). Serum from FXS patients displayed a significant decrease in signals from glucose (Figure 7C,D), similarly to Fmr1-KO animals in the nonfasted state (Figure 1C). In FXS patients, there was also a significant decrease in signals from glutamine and alanine, paralleled by an increase in creatine signals (Figure 7C). Finally, FXS patients displayed a significant reduction in insulin levels and a significant increase in circulating FFA as compared to controls (Figure 7E,F).
Figure 7.

FMR1-deficiency in Fragile X Syndrome patients reduces glucose and insulin and increases circulating FFA. (A) OPLS-DA of 1H NMR spectra from sera of FXS patients and sex-, aged-, BMI-matched controls and computation of PLS scores maximizing the segregation of the groups (TPred1 & TYosc1). Sera were collected in the fed state. (B) Permutation testing to assess significance of the O-PLS-DA model presented in (A) (n = 10,000 random iterations, p = 0.026). (C) Pseudo-spectrum representation of OPLS-DA model coefficients to highlight spectral regions responsible for discrimination between FXS and control samples. Positive and negative model coefficients respectively correspond to significantly higher or lower metabolite concentrations in FXS patients as compared to controls. 1H NMR signals corresponding to glucose, glutamine, alanine and creatine are highlighted. (D) Relative quantification of the 1H NMR signal corresponding to glucose peak at 5.25 ppm in sera from controls and FXS patients. Data are means ± SEM; n = 28 controls, n = 24 FXS patients; 2-tailed Student's T-test on log-transformed data: *, p < 0.05. (E) Circulating levels of insulin in controls and FXS patients. Data are means ± SEM; n = 29 controls, n = 25 FXS patients; 2-tailed Student's T-test on log-transformed data: *, p < 0.05. (F) Circulating levels of FFA in controls and FXS patients. Data are means ± SEM; n = 28 controls, n = 25 FXS patients; 2-tailed Student's T-test on log-transformed data: *, p < 0.05.
4. Discussion
Here, we provide an in-depth metabolic phenotyping of the FXS mouse model and highlight the consequences of the loss of the translational regulator FMRP on metabolic homeostasis in mice and humans. We further highlighted the contributing molecular mechanisms, as we show that loss of FMRP increased hepatic synthesis of proteins notably involved in lipid metabolism.
4.1. The absence of the translational regulator FMRP dysregulates hepatic protein synthesis with consequences on systemic metabolism
In this study, we demonstrate that the absence of FMRP leads to an increase in protein synthesis, as supported by our proteomics data in the Fmr1-KO liver in combination with quantification of protein synthesis in Fmr1-deficient hepatocytes. This depicts FMRP as a regulator of hepatic translation, in agreement with its presence on polyribosomes from mouse liver [2]. Biological network integration of metabolomics and proteomics data enabled us to identify key mRNA targets of FMRP linked to the metabolic phenotypes observed in Fmr1-KO animals: Cpt1a, Slc16a1, Aldh3a2, Abcd3, and Tecr. Importantly, these mRNA were previously identified as putative FMRP targets in large-scale screenings [5], [48] and we have experimentally validated their association with FMRP and the overabundance of the corresponding proteins in the Fmr1-KO liver. These data support the fact that FMRP functions as a translational repressor in the liver for this subset of mRNAs and directly controls translation of the cognate protein. Our work indicates that loss of FMRP determines increased abundances of key enzymes or transporters cooperating in FA hepatic metabolism either through direct translational regulation, or indirectly possibly through compensatory or adaptive mechanisms. Although we cannot exclude the fact that the activity of these enzymes and transporters could be downregulated to compensate for their overabundance, dysregulated hepatic expression of these proteins could explain the variations in the circulating levels of a number of metabolites and contribute to the global metabolic changes observed in Fmr1-KO animals. Notably, in the absence of FMRP, overabundance of Cpt1a, the rate limiting enzyme of FA mitochondrial β-oxidation which transfers the acyl group of LCFA-CoA conjugates onto carnitine [49], could directly improve the mitochondrial uptake of long-chain fatty acids and their subsequent mitochondrial β-oxidation. This is in agreement with previous work showing that increase in CPT1A activity enhanced β-oxidation rates both in vitro and in vivo, reduced hepatocyte TG accumulation and secretion and increased KB formation [50], [51], in agreement with our findings in Fmr1-KO mice. In the absence of FMRP, the increased synthesis of the monocarboxylate transporter Slc16a1 [52] could also favour hepatic export of KB, the terminal breakdown products of mitochondrial β-oxidation, contributing to elevate the circulating levels of the KB acetone and acetoacetate in Fmr1-KO mice. Furthermore, overabundance of the FA transporter Abcd3 and of the aldehyde dehydrogenase Aldh3a2 could also be indicative of increased peroxisomal β-oxidation of lipids [53]. All these data suggest a direct link between loss of the translational regulator FMRP in liver and lipid homeostasis. FMRP is expressed in all peripheral tissues, except in adult muscle [13], [54]. Further studies are now required to understand the impact on translation of FMRP loss in extra-hepatic peripheral tissues with metabolic roles, and how it could disrupt their metabolic interplay.
4.2. The absence of FMRP perturbs key metabolic pathways
In the absence of FMRP, we also show that key metabolic signaling pathways are over-activated: the insulin-mediated control of glucose homeostasis and the β-adrenergic control of lipolysis. Excessive insulin signaling in the brain [14], [15] and in the intestine [16] was previously described in the dfmr1 FXS drosophila model and also associated with excessive Akt phosphorylation. Our work further extends these findings to the liver in the Fmr1-KO mouse model and suggests that FMRP is generally required for the fine-tuning of the response to insulin. Increased sensitivity to insulin in the liver could explain the improved glucose tolerance in Fmr1-KO animals, while the reduced circulating levels of insulin might result from a physiological adaptation to counteract excessive insulin signaling. Defective insulin signaling in other peripheral organs might participate to the glucose-related phenotypes we describe in the present study. In mice, intestinal gluconeogenesis is essential to maintain glycemia in the absence of hepatic glucose production [55]. Furthermore, bone is also a tissue exerting some control on metabolism, and notably on the insulin system [56]. Given the fact that FMRP is not expressed in adult muscle [13], [54], we anticipate that the consequences of FMRP loss in muscle tissue would only be indirect or secondary to the alterations we describe in the liver.
We also highlight that Fmr1-deficient experimental models display an enhancement of the β−adrenergic agonist-driven lipolytic response in WAT, which could underpin the reduced adiposity in Fmr1-KO animals. Further, the increased release of FFA from WAT could enhance the bioavailability of lipid substrates for hepatic β− and ω−oxidation and contribute to the shift towards a higher utilization of lipids substrates observed in Fmr1-KO mice using indirect calorimetry.
4.3. The Fragile X family of proteins and RNA-binding proteins have possible conserved roles in the control of metabolic homeostasis
We show that the absence of FMRP provokes profound changes in glucose and lipid metabolism in the Fmr1-KO2 mouse model, while no overt metabolic alterations were previously reported in the Fmr1-KO1 mouse model of FXS [18]. Besides differences in the breeding, housing or diet used for the Fmr1-KO1 and KO2 models, possible explanation for this could be the age of the animals studied and different experimental design for in vivo metabolic phenotyping. However, Lumaban & Nelson showed that the Fmr1/Fxr2 double knock out mouse exhibits metabolic abnormalities overlapping with our observations: enhanced glucose tolerance and insulin response, hypoglycemia, and reduced adiposity [18]. This suggests a conserved role for the Fragile X Related family of genes in the regulation of metabolic homeostasis. In keeping with this notion, a recent study indicates that the Drosophila FXS model displays anomalies in energy metabolism, with decreased whole-body glucose and lipid stores and impaired mitochondrial functions [17]. In addition, we show both in mouse and human that FMR1-deficiency impacts the circulating metabolome and notably reduces glucose and insulin levels. This suggests that FXS may also be accompanied by yet under reported modifications in insulin sensitivity and glucose homeostasis in humans, similar to that seen in our FXS mouse model and in FXS fly model [17]. We highlighted reduced levels of FFA in FXS patients; lower blood cholesterol was previously reported in FXS patients [19], [20], [21], and we now report it in the Fmr1-KO2 mouse model, suggesting that lipid homeostasis is generally impacted by FMR1-deficiency. Other translational regulators such as the translation initiation factors EIF6 [57] and EIF4BP1 [58] or the ribosomal protein S6 kinase p70S6K [59] have previously been described as modulators of metabolic phenotypes and notably of lipid homeostasis. In particular, similarly to Fmr1-KO mice, S6k1-KO and Eif4bp1-KO mice have reduced body fat mass, increased lipolysis, and decreased adipocyte size. Our study therefore strengthens the importance of RNA-binding proteins and translation regulators in the general control of metabolic homeostasis.
4.4. Clinical relevance of findings
Regarding the clinical relevance of our findings of reduced adiposity in FXS mouse model, there is a lack of large-scale epidemiological studies on adiposity in the general FXS population. Although severe obesity is reported in less than 10% of FXS patients, known as Prader Willi-like (PWL) [60], [61], obesity is not described in typical FXS patients [7], [62], such as the patients enrolled in our study. On one hand, obese FXS patients are likely to benefit at the behavioral and metabolic level from treatment with the anti-diabetic drug metformin, as recently shown in seven FXS cases [63]. On the other hand, we show that typical FXS patients are likely to present metabolic phenotypes distinct from type 2 diabetes (T2D) with reduced glycemia and insulinemia. One study even mentioned a lower incidence of T2D in FXS population [63]. In the same line, earlier studies have reported that FXS patients are not prone to hyperlipidemia since they display reduced cholesterol and TG [19], [20], [21], in agreement with our findings in FXS mouse model. Treatment with the cholesterol-lowering agent lovastatin improved behavioral outcome in FXS patients [64], but the authors also observed an unexpectedly strong diminution in TG and cholesterol levels after 3 months of treatment [20], [64]. This raises the issue that the possible untoward effects of metabolic drugs should be considered in clinical trials involving FXS patients not harbouring T2D features or hyperlipidemia.
4.5. Conclusions
Our study identifies metabolic phenotypes in FXS and supports a role for FMRP-mediated translation in the homeostatic control of systemic metabolism. Furthermore, this work underlines the importance to further investigate the metabolic roles of translational regulators, both in fundamental biology and from a clinical standpoint.
Author contributions
LD designed the project. AL, DP, JC, LMG, PBM, TG, BM, EZA, EWK, MED, and LD designed and carried out the experiments. AL, DP, JC, LMG, TG, BM, EZA, EWK, MED, and LD treated and interpreted data. FK and AVD provided human samples, contributed and interpreted clinical data. LD and MED wrote the manuscript.
Acknowledgements
We thank N. Durand and the IPMC animal facility for expert animal care. We acknowledge H. Van Esch for help with patient recruitment and are very grateful to the families and patients who have participated in this study. We thank the Anexplo phenotyping platform (Genotoul, Rangueil, Toulouse). LD thanks the generous support of FRAXA Research Foundation, Agence Nationale de la Recherche (ANR JCJC SVE6 MetaboXFra), Conseil Général 06, Fondation Jérôme Lejeune and the CNRS PICS program. LD and MED acknowledge the support of the Royal Society-CNRS International Exchange Program (IE120728). LD, BM and MED are grateful to the European Community 7th Framework Program under Coordinated Action NEURON-ERANET (grant agreement 291840). The MED lab is funded by METACARDIS (HEALTH-F4-2012-305312) and the UK Medical Research Council (MRC grant MR/M501797/1). RFK and AVD have received grants from FRAXA Research Foundation and Jérôme Lejeune Foundation. EWK was supported by grants from the CIHR and NSERC. JAJB and JLM thank Region Centre-Val de Loire (ARD2020 Biomedicaments–GPCRAb) and the Labex MabImprove for financial support and the Experimental Unit PAO #1297 (EU0028) of INRA Centre Val de Loire for animal housing.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2019.01.002.
Conflicts of interest
None declared.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Khandjian E.W., Corbin F., Woerly S., Rousseau F. The fragile X mental retardation protein is associated with ribosomes. Nature Genetics. 1996;12(1):91–93. doi: 10.1038/ng0196-91. [DOI] [PubMed] [Google Scholar]
- 2.Khandjian E.W., Huot M.E., Tremblay S., Davidovic L., Mazroui R., Bardoni B. Biochemical evidence for the association of fragile X mental retardation protein with brain polyribosomal ribonucleoparticles. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(36):13357–13362. doi: 10.1073/pnas.0405398101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stefani G., Fraser C.E., Darnell J.C., Darnell R.B. Fragile X mental retardation protein is associated with translating polyribosomes in neuronal cells. Journal of Neuroscience. 2004;24(33):7272–7276. doi: 10.1523/JNEUROSCI.2306-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Darnell J.C., Klann E. The translation of translational control by FMRP: therapeutic targets for FXS. Nature Neuroscience. 2013;16(11):1530–1536. doi: 10.1038/nn.3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darnell J.C., Van Driesche S.J., Zhang C., Hung K.Y., Mele A., Fraser C.E. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El Fatimy R., Davidovic L., Tremblay S., Jaglin X., Dury A., Robert C. Tracking the fragile X mental retardation protein in a highly ordered neuronal ribonucleoparticles population: a link between stalled polyribosomes and RNA granules. PLoS Genetics. 2016;12(7) doi: 10.1371/journal.pgen.1006192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kidd S.A., Lachiewicz A., Barbouth D., Blitz R.K., Delahunty C., McBrien D. Fragile X syndrome: a review of associated medical problems. Pediatrics. 2014;134(5):995–1005. doi: 10.1542/peds.2013-4301. [DOI] [PubMed] [Google Scholar]
- 8.Penagarikano O., Mulle J.G., Warren S.T. The pathophysiology of fragile X syndrome. Annual Review of Genomics and Human Genetics. 2007;8:109–129. doi: 10.1146/annurev.genom.8.080706.092249. [DOI] [PubMed] [Google Scholar]
- 9.Consortium, T.D.-B.F.X. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78(1):23–33. [PubMed] [Google Scholar]
- 10.Mientjes E.J., Nieuwenhuizen I., Kirkpatrick L., Zu T., Hoogeveen-Westerveld M., Severijnen L. The generation of a conditional Fmr1 knock out mouse model to study Fmrp function in vivo. Neurobiology of Disease. 2006;21(3):549–555. doi: 10.1016/j.nbd.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 11.Richter J.D., Bassell G.J., Klann E. Dysregulation and restoration of translational homeostasis in fragile X syndrome. Nature Reviews Neuroscience. 2015;16(10):595–605. doi: 10.1038/nrn4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davidovic L., Tremblay S., Gravel M., De Koninck P., Khandjian E.W. The fragile X syndrome: one protein missing and 1001 disoriented mRNA. Medical Science (Paris) 2006;22(1):41–46. doi: 10.1051/medsci/200622141. [DOI] [PubMed] [Google Scholar]
- 13.Khandjian E.W., Bardoni B., Corbin F., Sittler A., Giroux S., Heitz D. Novel isoforms of the fragile X related protein FXR1P are expressed during myogenesis. Human Molecular Genetics. 1998;7(13):2121–2128. doi: 10.1093/hmg/7.13.2121. [DOI] [PubMed] [Google Scholar]
- 14.Callan M.A., Clements N., Ahrendt N., Zarnescu D.C. Fragile X protein is required for inhibition of insulin signaling and regulates glial-dependent neuroblast reactivation in the developing brain. Brain Research. 2012;1462:151–161. doi: 10.1016/j.brainres.2012.03.042. [DOI] [PubMed] [Google Scholar]
- 15.Monyak R.E., Emerson D., Schoenfeld B.P., Zheng X., Chambers D.B., Rosenfelt C. Insulin signaling misregulation underlies circadian and cognitive deficits in a drosophila fragile X model. Molecular Psychiatry. 2017;22(8):1140–1148. doi: 10.1038/mp.2016.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luhur A., Buddika K., Ariyapala I.S., Chen S., Sokol N.S. Opposing post-transcriptional control of InR by FMRP and LIN-28 adjusts stem cell-based tissue growth. Cell Reports. 2017;21(10):2671–2677. doi: 10.1016/j.celrep.2017.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weisz E.D., Towheed A., Monyak R.E., Toth M.S., Wallace D.C., Jongens T.A. Loss of drosophila FMRP leads to alterations in energy metabolism and mitochondrial function. Human Molecular Genetics. 2018;27(1):95–106. doi: 10.1093/hmg/ddx387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lumaban J.G., Nelson D.L. The fragile X proteins Fmrp and Fxr2p cooperate to regulate glucose metabolism in mice. Human Molecular Genetics. 2015;24(8):2175–2184. doi: 10.1093/hmg/ddu737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berry-Kravis E., Levin R., Shah H., Mathur S., Darnell J.C., Ouyang B. Cholesterol levels in fragile X syndrome. American Journal of Medical Genetics. 2015;167A(2):379–384. doi: 10.1002/ajmg.a.36850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caku A., Seidah N.G., Lortie A., Gagne N., Perron P., Dube J. New insights of altered lipid profile in fragile X Syndrome. PLoS One. 2017;12(3) doi: 10.1371/journal.pone.0174301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lisik M.Z., Gutmajster E., Sieron A.L. Low levels of HDL in fragile X syndrome patients. Lipids. 2016;51(2):189–192. doi: 10.1007/s11745-015-4109-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dona A.C., Jimenez B., Schafer H., Humpfer E., Spraul M., Lewis M.R. Precision high-throughput proton NMR spectroscopy of human urine, serum, and plasma for large-scale metabolic phenotyping. Analytical Chemistry. 2014;86(19):9887–9894. doi: 10.1021/ac5025039. [DOI] [PubMed] [Google Scholar]
- 23.Dona A.C., Kyriakides M., Scott F., Shephard E.A., Varshavi D., Veselkov K. A guide to the identification of metabolites in NMR-based metabonomics/metabolomics experiments. Computational and Structural Biotechnology Journal. 2016;14:135–153. doi: 10.1016/j.csbj.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dumas M.E., Barton R.H., Toye A., Cloarec O., Blancher C., Rothwell A. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(33):12511–12516. doi: 10.1073/pnas.0601056103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ayala J.E., Samuel V.T., Morton G.J., Obici S., Croniger C.M., Shulman G.I. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Disease Models and Mechanisms. 2010;3(9–10):525–534. doi: 10.1242/dmm.006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beranger G.E., Pisani D.F., Castel J., Djedaini M., Battaglia S., Amiaud J. Oxytocin reverses ovariectomy-induced osteopenia and body fat gain. Endocrinology. 2014;155(4):1340–1352. doi: 10.1210/en.2013-1688. [DOI] [PubMed] [Google Scholar]
- 27.Pisani D.F., Beranger G.E., Corinus A., Giroud M., Ghandour R.A., Altirriba J. The K+ channel TASK1 modulates beta-adrenergic response in brown adipose tissue through the mineralocorticoid receptor pathway. The FASEB Journal. 2016;30(2):909–922. doi: 10.1096/fj.15-277475. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez A.M., Elabd C., Delteil F., Astier J., Vernochet C., Saint-Marc P. Adipocyte differentiation of multipotent cells established from human adipose tissue. Biochemical and Biophysical Research Communications. 2004;315(2):255–263. doi: 10.1016/j.bbrc.2004.01.053. [DOI] [PubMed] [Google Scholar]
- 29.Pisani D.F., Djedaini M., Beranger G.E., Elabd C., Scheideler M., Ailhaud G. Differentiation of human adipose-derived stem cells into "Brite" (brown-in-white) adipocytes. Frontiers in Endocrinology (Lausanne) 2011;2:87. doi: 10.3389/fendo.2011.00087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cox J., Hubner N.C., Mann M. How much peptide sequence information is contained in ion trap tandem mass spectra? Journal of the American Society for Mass Spectrometry. 2008;19(12):1813–1820. doi: 10.1016/j.jasms.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 31.Cox J., Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnology. 2008;26(12):1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 32.Jensen L.J., Kuhn M., Stark M., Chaffron S., Creevey C., Muller J. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Research. 2009;37(Database issue):D412–D416. doi: 10.1093/nar/gkn760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thomas P.D., Campbell M.J., Kejariwal A., Mi H., Karlak B., Daverman R. PANTHER: a library of protein families and subfamilies indexed by function. Genome Research. 2003;13(9):2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas P.D., Kejariwal A., Guo N., Mi H., Campbell M.J., Muruganujan A. Applications for protein sequence-function evolution data: mRNA/protein expression analysis and coding SNP scoring tools. Nucleic Acids Research. 2006;34(Web Server issue):W645–W650. doi: 10.1093/nar/gkl229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khalfallah O., Jarjat M., Davidovic L., Nottet N., Cestele S., Mantegazza M. Depletion of the fragile X mental retardation protein in embryonic stem cells alters the kinetics of neurogenesis. Stem Cells. 2017;35(2):374–385. doi: 10.1002/stem.2505. [DOI] [PubMed] [Google Scholar]
- 36.Devys D., Lutz Y., Rouyer N., Bellocq J.P., Mandel J.L. The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nature Genetics. 1993;4(4):335–340. doi: 10.1038/ng0893-335. [DOI] [PubMed] [Google Scholar]
- 37.El Fatimy R., Tremblay S., Dury A.Y., Solomon S., De Koninck P., Schrader J.W. Fragile X mental retardation protein interacts with the RNA-binding protein Caprin1 in neuronal RiboNucleoProtein complexes. PLoS One. 2012;7(6) doi: 10.1371/journal.pone.0039338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dury A.Y., El Fatimy R., Tremblay S., Rose T.M., Cote J., De Koninck P. Nuclear fragile X mental retardation protein is localized to cajal bodies. PLoS Genetics. 2013;9(10) doi: 10.1371/journal.pgen.1003890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmidt E.K., Clavarino G., Ceppi M., Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nature Methods. 2009;6(4):275–277. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- 40.Rodriguez-Martinez A., Ayala R., Posma J.M., Dumas M.E. Exploring the genetic landscape of metabolic phenotypes with MetaboSignal. Current Protocols in Bioinformatics. 2018;61(1) doi: 10.1002/cpbi.41. 14 14 11–14 14 13. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez-Martinez A., Ayala R., Posma J.M., Neves A.L., Gauguier D., Nicholson J.K. MetaboSignal: a network-based approach for topological analysis of metabotype regulation via metabolic and signaling pathways. Bioinformatics. 2017;33(5):773–775. doi: 10.1093/bioinformatics/btw697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ouldamer L., Nadal-Desbarats L., Chevalier S., Body G., Goupille C., Bougnoux P. NMR-based lipidomic approach to evaluate controlled dietary intake of lipids in adipose tissue of a rat mammary tumor model. Journal of Proteome Research. 2016;15(3):868–878. doi: 10.1021/acs.jproteome.5b00788. [DOI] [PubMed] [Google Scholar]
- 43.Berry R., Rodeheffer M.S. Characterization of the adipocyte cellular lineage in vivo. Nature Cell Biology. 2013;15(3):302–308. doi: 10.1038/ncb2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Even P.C., Nadkarni N.A. Indirect calorimetry in laboratory mice and rats: principles, practical considerations, interpretation and perspectives. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2012;303(5):R459–R476. doi: 10.1152/ajpregu.00137.2012. [DOI] [PubMed] [Google Scholar]
- 45.Davidovic L., Navratil V., Bonaccorso C.M., Catania M.V., Bardoni B., Dumas M.E. A metabolomic and systems biology perspective on the brain of the Fragile X syndrome mouse model. Genome Research. 2011;12:2190–2202. doi: 10.1101/gr.116764.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schaeffer C., Bardoni B., Mandel J.L., Ehresmann B., Ehresmann C., Moine H. The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. The EMBO Journal. 2001;20(17):4803–4813. doi: 10.1093/emboj/20.17.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pieretti M., Zhang F.P., Fu Y.H., Warren S.T., Oostra B.A., Caskey C.T. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66(4):817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 48.Ascano M., Jr., Mukherjee N., Bandaru P., Miller J.B., Nusbaum J.D., Corcoran D.L. FMRP targets distinct mRNA sequence elements to regulate protein expression. Nature. 2012;492(7429):382–386. doi: 10.1038/nature11737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miyazawa S., Ozasa H., Osumi T., Hashimoto T. Purification and properties of carnitine octanoyltransferase and carnitine palmitoyltransferase from rat liver. Journal of Biochemistry. 1983;94(2):529–542. doi: 10.1093/oxfordjournals.jbchem.a134384. [DOI] [PubMed] [Google Scholar]
- 50.Lin X., Yue P., Chen Z., Schonfeld G. Hepatic triglyceride contents are genetically determined in mice: results of a strain survey. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2005;288(6):G1179–G1189. doi: 10.1152/ajpgi.00411.2004. [DOI] [PubMed] [Google Scholar]
- 51.Stefanovic-Racic M., Perdomo G., Mantell B.S., Sipula I.J., Brown N.F., O'Doherty R.M. A moderate increase in carnitine palmitoyltransferase 1a activity is sufficient to substantially reduce hepatic triglyceride levels. American Journal of Physiology. Endocrinology and Metabolism. 2008;294(5):E969–E977. doi: 10.1152/ajpendo.00497.2007. [DOI] [PubMed] [Google Scholar]
- 52.Jackson V.N., Halestrap A.P. The kinetics, substrate, and inhibitor specificity of the monocarboxylate (lactate) transporter of rat liver cells determined using the fluorescent intracellular pH indicator, 2',7'-bis(carboxyethyl)-5(6)-carboxyfluorescein. Journal of Biological Chemistry. 1996;271(2):861–868. doi: 10.1074/jbc.271.2.861. [DOI] [PubMed] [Google Scholar]
- 53.Wanders R.J., Waterham H.R., Ferdinandusse S. Metabolic interplay between peroxisomes and other subcellular organelles including mitochondria and the endoplasmic reticulum. Frontiers in Cell and Developmental Biology. 2015;3:83. doi: 10.3389/fcell.2015.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Davidovic L., Sacconi S., Bechara E.G., Delplace S., Allegra M., Desnuelle C. Alteration of expression of muscle specific isoforms of the fragile X related protein 1 (FXR1P) in facioscapulohumeral muscular dystrophy patients. Journal of Medical Genetics. 2008;45(10):679–685. doi: 10.1136/jmg.2008.060541. [DOI] [PubMed] [Google Scholar]
- 55.Penhoat A., Fayard L., Stefanutti A., Mithieux G., Rajas F. Intestinal gluconeogenesis is crucial to maintain a physiological fasting glycemia in the absence of hepatic glucose production in mice. Metabolism. 2014;63(1):104–111. doi: 10.1016/j.metabol.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 56.Ferron M., Wei J., Yoshizawa T., Del Fattore A., DePinho R.A., Teti A. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 2010;142(2):296–308. doi: 10.1016/j.cell.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brina D., Miluzio A., Ricciardi S., Clarke K., Davidsen P.K., Viero G. eIF6 coordinates insulin sensitivity and lipid metabolism by coupling translation to transcription. Nature Communications. 2015;6:8261. doi: 10.1038/ncomms9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsukiyama-Kohara K., Poulin F., Kohara M., DeMaria C.T., Cheng A., Wu Z. Adipose tissue reduction in mice lacking the translational inhibitor 4E-BP1. Nature Medicine. 2001;7(10):1128–1132. doi: 10.1038/nm1001-1128. [DOI] [PubMed] [Google Scholar]
- 59.Carnevalli L.S., Masuda K., Frigerio F., Le Bacquer O., Um S.H., Gandin V. S6K1 plays a critical role in early adipocyte differentiation. Developmental Cell. 2010;18(5):763–774. doi: 10.1016/j.devcel.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nowicki S.T., Tassone F., Ono M.Y., Ferranti J., Croquette M.F., Goodlin-Jones B. The Prader-Willi phenotype of fragile X syndrome. Journal of Developmental and Behavioral Pediatrics. 2007;28(2):133–138. doi: 10.1097/01.DBP.0000267563.18952.c9. [DOI] [PubMed] [Google Scholar]
- 61.McLennan Y., Polussa J., Tassone F., Hagerman R. Fragile X syndrome. Current Genomics. 2011;12(3):216–224. doi: 10.2174/138920211795677886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hagerman R.J., Berry-Kravis E., Hazlett H.C., Bailey D.B., Jr., Moine H., Kooy R.F. Fragile X syndrome. Nature Reviews Disease Primers. 2017;3:17065. doi: 10.1038/nrdp.2017.65. [DOI] [PubMed] [Google Scholar]
- 63.Dy A.B.C., Tassone F., Eldeeb M., Salcedo-Arellano M.J., Tartaglia N., Hagerman R. Metformin as targeted treatment in fragile X syndrome. Clinical Genetics. 2017;93(2):216–222. doi: 10.1111/cge.13039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caku A., Pellerin D., Bouvier P., Riou E., Corbin F. Effect of lovastatin on behavior in children and adults with fragile X syndrome: an open-label study. American Journal of Medical Genetics, Part A. 2014;164A(11):2834–2842. doi: 10.1002/ajmg.a.36750. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.